Aging
Learning objectives
After reading this chapter you should be able to:
 Describe the relationship between aging and disease.
Describe the relationship between aging and disease.
Explain the Gompertz plot and how it describes the rate of aging of different species.
Differentiate between biological and chemical theories of aging.
Explain the precepts of the free radical theory of aging, including identification of characteristic oxidation products that accumulate in long-lived proteins with age.
Outline the evidence that the rate of mutation of DNA is an important determinant of the rate of aging.
Outline the evidence in support of the mitochondrial theory of aging and describe how this theory interfaces with the free radical theory of aging.
Describe the etiology and pathology characteristic of several diseases of accelerated aging.
Describe the effects of caloric restriction on the rate of aging of rodents.
Introduction
Aging may be defined as the time-dependent deterioration in function of an organism
While it has broad-based physiologic effects, aging is fundamentally the result of changes in cellular structure and function, biochemistry and metabolism (Table 43.1). The result of aging, even healthy aging, is increased susceptibility to disease and increased probability of death – the endpoint of aging. However, aging is not a disease. Diseases affect a fraction of the population; aging affects all of us, whether it is programmed or stochastic.
Table 43.1
Decline in biochemical and physiologic systems with age
| Biochemical | Physiologic |
| Basal metabolic rate | Lung expansion volume |
| Protein turnover | Renal filtration capacity (glomerular) |
| Glucose tolerance | Renal concentration capacity (tubular) |
| Reproductive capacity | Cardiovascular performance |
| Telomere shortening | Musculoskeletal system |
| Oxidative phosphorylation | Nerve conduction velocity |
| Endocrine and exocrine systems | |
| Immunological defenses | |
| Sensory systems (vision, audition) |
With the aging of the population, gerontology and geriatric medicine are becoming increasingly important. This chapter presents an overview on biochemical and physiologic changes associated with aging, in general, and with the aging of specific organ systems. It includes a review of current theories on aging (there are several theories and, in general, the more theories there are, the less we really understand about something) and concludes with a discussion of the relationship between cancer and aging and an update on approaches to lifespan extension.
Aging of complex systems
Excluding genetic defects, childhood disease and accidents, humans survive until about age 50 with limited maintenance requirements or risk of death; then we become increasingly frail and our death rate increases with time, reaching a maximum at about age 76. Our lifespan is affected by our genetics and our environmental exposure, and our death is usually attributable to failure of a critical organ system (cardiovascular, renal, pulmonary, etc.). The capacity of these interdependent physiologic systems usually declines as a linear function of age, leading to an exponential increase in our age-specific death rate (Fig. 43.1). Historically, improvements in health care and environment have resulted in ‘rectangularization’ of the survival curve – our mean lifespan has increased but without a significant effect on our maximum lifespan (see Fig. 43.1A).
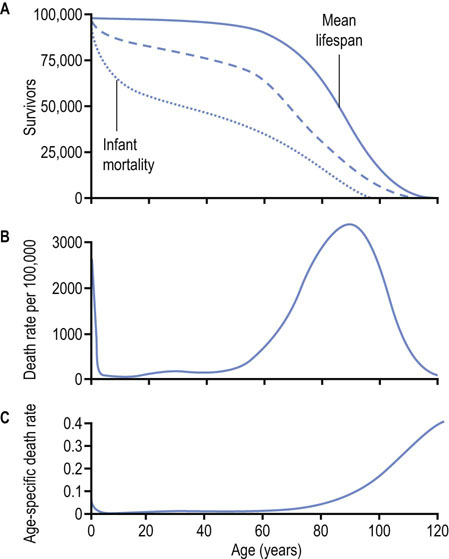
Fig. 43.1 Survival curve and death rate.
(A) Mean lifespan is defined as the age at which 50% of a population survives (or has died). The negative slope of the survival curve reaches a maximum at the mean lifespan of a species. The dotted line describes a survival curve in the Third World where infant mortality and disease significantly decrease mean lifespan. The dashed line describes the survival curve for the United States in the early part of the 20th century. The solid line applies to 21st-century Europe. (B) The death rate reaches a maximum at the mean lifespan. (C) The age-specific death rate, defined as the number of deaths per time at a given age, e.g. deaths per 100,000 persons of a specific age per year, increases exponentially with age. The lifespan or maximum lifespan potential (MLSP) is defined as the maximum age attainable by a member of the population, which is about 120 years for humans.
The Hayflick limit – replicative senescence
The replicative capacity of cells decreases with age
Differentiated cells from animals undergo only a limited number of cell divisions (population doublings) in tissue culture, unless they become transformed to cancer cells by mutation or infection with certain viruses. The number of potential cell divisions is greater in longer-lived animals, suggesting a relationship between cell division potential and longevity. Human neonatal fibroblasts will divide about 60 times, then enter a nondividing state, while fibroblasts from mice and rats, which have shorter lifespans, undergo fewer cell divisions in vitro. Cells from younger donors have greater replicative capacity and a greater number of cell divisions in cell culture, but the number of dividing cells decreases with age. This limited doubling capacity, described by Dr Leonard Hayflick, is known as the Hayflick limit. The relevance of the Hayflick limit to human aging is still debated – certainly, human cells retain some replicative capacity, even at advanced age, and major tissues, such as muscle and nerve, are largely postmitotic, i.e. not actively dividing. However, changes in the metabolism of senescent cells, including decreased responsiveness to hormones and the decline in their synthetic and degradative capacities, e.g. in the immunologic and reticuloendothelial systems, may affect our adaptability and susceptibility to stress and age-related diseases, placing limits on our lifespan.
Mathematical models of aging
In poikilotherms, the rate of aging is correlated with temperature, physical activity and metabolic rate
In the early 19th century, Gompertz observed that the age-specific death rate of humans increased exponentially after 35 years of age, and that human survival curves could be modeled by what is now known as the Gompertz equation (Fig. 43.2):
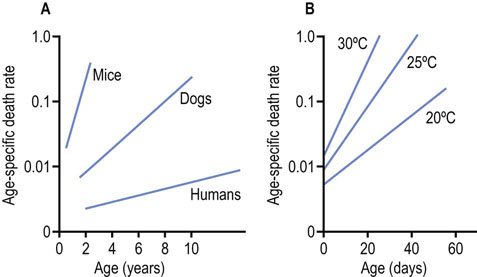
Fig. 43.2 Gompertz plots for humans and other species.
(A) Humans and other vertebrates. (B) Flies raised at various temperatures (adapted from the work of Professor RS Sohal).
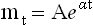
The term mt is the age-specific death rate at age t; α is the slope, the effect of time on the death rate; and A, the y-axis intercept, is the death rate at birth. The Gompertz–Makeham equation:
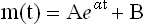
adds a constant, B, to correct for the age-independent death rate, e.g. as a result of infant mortality or accidents, and provides a better fit to actuarial data.
The Gompertz plots in Figure 43.2 illustrate the time-dependent changes in death rate for three different species of vertebrates and for flies raised at different temperatures. Shorter-lived mammals have a greater age-adjusted rate of death (α = slope), while the death rate for poikilotherms varies with ambient temperature – flies live longer when grown at lower temperatures. This observation has been interpreted as evidence for ‘rate of living’ or ‘wear and tear’ theories of aging. Flies, being more active at higher temperature, consume more energy and die more rapidly. Flies that are restrained, e.g. in a matchbox, rather than a large carboy, also live longer; wingless flies live longer; and male flies, segregated from females, also live longer. In each case, in small enclosures, without wings, and in the absence of the opposite sex, male flies are less active, have lower basal metabolic rates, and have longer mean and maximum lifespans. None of these strategies for lifespan extension is applicable to humans.
Theories of aging
Theories of aging can be divided into two general categories: biological and chemical
Biological theories treat aging as a genetically controlled event, determined by the programmed expression or repression of genetic information. Aging and death are seen as the orchestrated endstage of birth, growth, maturation and reproduction. Apoptosis (programmed cell death) and thymic involution are examples of genetically programmed events at the level of cells and organs, and the decline in the immunologic, neuroendocrine and reproductive systems may be seen, in a broader context, as evidence for action of a biological clock affecting the integrated functions of an organism. Biological theories attribute differences in lifespan to interspecies differences in genetics but also provide an explanation for the observation that there is a genetic component to longevity within a species, e.g. in families with a history of longevity. Differences in lifespan among species are also closely correlated with the efficiency of DNA repair mechanisms. Longer-lived species have more efficient DNA repair processes (Fig. 43.3). Numerous diseases of accelerated aging (progeria) also illustrate the importance of genetics and maintenance of the integrity of the genome during aging.
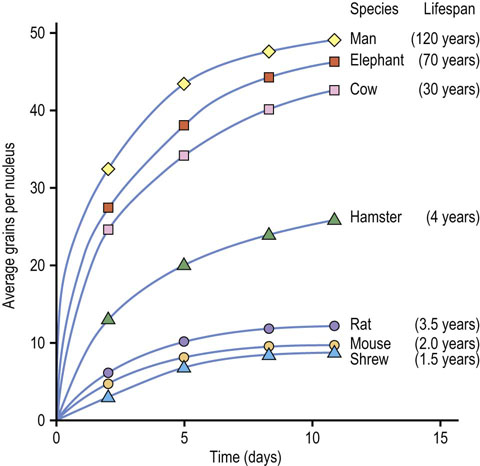
Fig. 43.3 Relationship between DNA repair activity and longevity.
Fibroblasts from various species were irradiated briefly, forming thymine dimers and thymine glycol (Chapter 32). The oxidized bases are removed and replaced by excision repair. DNA repair was assessed by the rate of incorporation of a [3H]thymidine tracer into DNA by autoradiography (adapted from Hart RW, Setlow RB: Correlation between deoxyribonucleic acid excision-repair and life-span in a number of mammalian species. Proc Natl Acad Sci USA 71:2169–2173, 1974).
Chemical theories of aging treat it as a somatic process resulting from cumulative damage to biomolecules. At one extreme, the error-catastrophe theory proposes that aging is the result of cumulative errors in the machinery for replication, repair, transcription, and translation of genetic information. Eventually, errors in critical enzymes, such as DNA and RNA polymerases or enzymes involved in the synthesis and turnover of proteins, gradually affect the fidelity of expression of genetic information and permit the accumulation of altered proteins. The propagation of errors and resultant accumulation of dysfunctional macromolecules lead eventually to the collapse of the system. Consistent with this theory, increasing amounts of immunologically detectable, but denatured or modified, functionally inactive enzymes accumulate in cells as a function of age.
More general chemical theories treat aging as the result of chronic, cumulative chemical (nonenzymatic) modification, insults or damage to all biomolecules (Table 43.2). Like rust or corrosion, the accumulation of damage with age gradually affects function. This damage is most apparent in long-lived tissue proteins, such as lens crystallins and extracellular collagens, which accumulate chemical modifications with age. These proteins gradually brown with age as a result of formation of a wide range of conjugated compounds with absorbance in the yellow-red region of the spectrum (Fig. 43.4); in the lens, they act as a filter, contributing to the loss of color vision with age. Highly modified crystallins, the major protein in the lens, gradually precipitate, leading to development of cataracts. Chemical damage to the integrity of the genome also occurs, but is more difficult to quantify because of the efficiency of repair processes that excise and repair modified nucleotides. As noted in Table 43.2, there are a number of silent consequences of DNA damage. This damage is primarily endogenous but is enhanced by xenobiotic and environmental agents.
Table 43.2
Age-dependent chemical changes in biomolecules
| Protein modification | DNA modification and mutation | Other |
| Crosslinking | Oxidation | Lipofuscin |
| Oxidation | Depurination | Inactive enzymes |
| Deamidation | Substitutions | |
| D-aspartate | Insertions and deletions | |
| Protein carbonyls | Inversions and transpositions | |
| Glycoxidation | ||
| Lipoxidation |
Long-lived proteins, such as lens crystallins and tissue collagens, accumulate damage with age. Modification and crosslinking of proteins occurs as a result of nonoxidative (deamidation, racemization) or oxidative (protein carbonyls) mechanisms or by reactions of proteins with products of carbohydrate or lipid peroxidation (glycoxidation, lipoxidation). Damage to DNA is often silent, i.e. modified forms of nucleotides may not accumulate, but the damage increases in the form of mutations resulting from errors in repair.
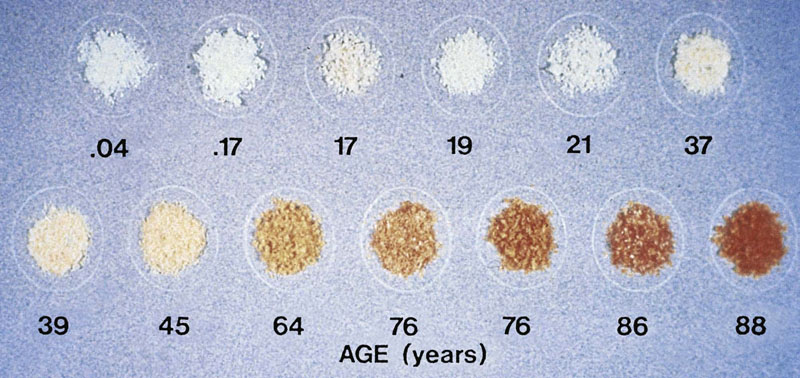
Fig. 43.4 Changes in costal cartilage with age.
Browning is a characteristic feature of the aging of proteins, not just in the lens, which is exposed to sunlight, but also in tissue collagens throughout the body. Crosslinking of proteins also increases with browning. Crosslinking contributes to the gradual insolubilization of lens protein with age. Crosslinking of articular and vascular collagens decreases the resilience of vertebral disks and compliance of the vascular wall with age. These changes in extracellular proteins are similar to changes induced by reaction of carbohydrates and lipids with protein during the cooking of foods, a process known as the Maillard or browning reaction. At one level, humans have been described as low-temperature ovens, operating at 37°C, with long cooking cycles (≈75 years). Many of the Maillard reaction products detected in the crust of bread and pretzels have been identified in human crystallins and collagens, and increase with age. (See also discussion of diabetic complications in Chapter 21.)
Organ system theories of aging incorporate various aspects of the above theories. These theories attribute aging to the failure of integrative systems, such as the immunologic, neurologic, endocrine or circulatory system. While they do not assign a specific cause, these theories integrate biological and chemical theories, acknowledging both genetic and environmental contributions to aging.
The free radical theory of aging
The free radical theory of aging is the most widely accepted theory of aging
The free radical theory of aging (FRTA) treats aging as the result of cumulative oxidative damage to biomolecules: DNA, RNA, protein, lipids, and glycoconjugates. From the viewpoint of the FRTA, longer-lived organisms have lower rates of production of reactive oxygen species (ROS; Chapter 37), better antioxidant defenses, and more efficient repair or turnover processes. While it is a chemical theory, the FRTA does not ignore the importance of genetics and biology in limiting the production of ROS, and the role of antioxidant and repair mechanisms. It also interfaces with other theories of aging, such as the rate of living theory (because the rate of generation of ROS is a function of the overall rate and/or extent of oxygen consumption) and the crosslinkage theory (because some products of ROS damage crosslink protein). Finally, as a chemical hypothesis, the FRTA does not exclude cumulative chemical damage, independent of ROS, such as racemization and deamidation of amino acids, but focuses on ROS as the primary source of damage and the fundamental cause of aging.
The FRTA is supported by the inverse correlation between basal metabolic rate (rate of oxygen consumption per unit weight) and maximum lifespan of mammals, and by evidence of increased oxidative damage to proteins with age. Protein carbonyl groups, such as glutamic and aminoadipic acid semialdehyde, formed by oxidative deamination of arginine and lysine, respectively, are formed in proteins exposed to ROS. The steady-state level of protein carbonyls in intracellular proteins increases logarithmically with age and at a rate inversely proportional to the lifespan of species. Protein carbonyls are also much higher in fibroblasts from patients with progeria (accelerated aging), e.g. Werner's or Hutchinson–Gilford syndromes, compared to age-matched subjects. Similar concentrations of protein carbonyls are also present in tissues of old rats and elderly humans, arguing that similar changes occur at old age in a range of organisms, regardless of the difference in their lifespans.
Figure 43.5 illustrates the accumulation of two relatively stable amino acid oxidation products in human skin collagen: methionine sulfoxide and ortho-tyrosine. These compounds are formed by different mechanisms involving different ROS (Chapter 37) and are present at significantly different concentrations in skin collagen, but increase in concert with age. Other amino acid modifications that accumulate in skin collagen with age include advanced glycoxidation and lipoxidation end products (AGE/ALEs), such as Ne-(carboxymethyl)lysine (CML) and pentosidine (Fig. 43.6 and see Fig. 21.19), and D-aspartate.
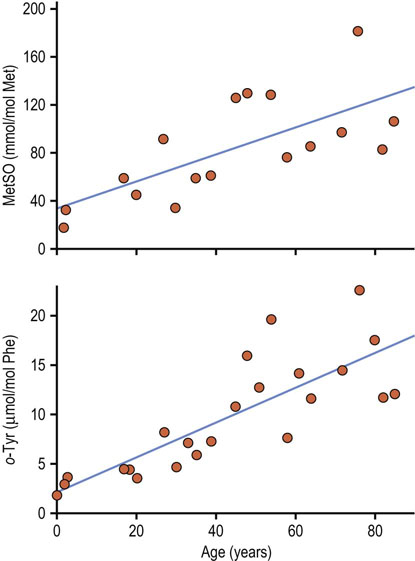
Fig. 43.5 Accumulation of amino acid oxidation products in human skin collagen with age.
Methionine is oxidized to methionine sulfoxide (MetSO) by HOCl or H2O2; ortho-tyrosine is a product of hydroxyl radical addition to phenylalanine (Phe). Despite a 100-fold difference in their rate of accumulation in collagen, levels of MetSO and o-tyrosine correlate strongly with one another, indicating that multiple ROS contribute to oxidative damage to proteins (adapted from Wells-Knecht MC et al: Age-dependent accumulation of ortho-tyrosine and methionine sulfoxide in human skin collagen is not increased in diabetes: evidence against a generalized increase in oxidative stress in diabetes. J Clin Invest 100:839–846, 1997).
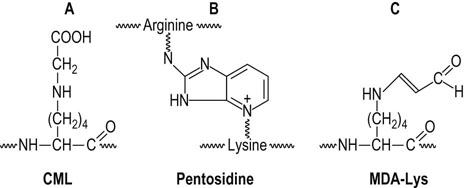
Fig. 43.6 Structure of major advanced glycoxidation and lipoxidation endproducts (AGE/ALEs).
(A) The AGE/ALE, Nε-(carboxymethyl)lysine (CML), which is formed during both carbohydrate and lipid peroxidation reactions. (B) The AGE pentosidine, a fluorescent crosslink in proteins. (C) The ALE, malondialdehyde-lysine (MDA-Lys), a reactive ALE that may proceed to form aminoenimine (RNHCH CHCHNR) crosslinks in proteins.
CHCHNR) crosslinks in proteins.
 Clinical box Progerias: accelerated aging resulting from defects in DNA repair
Clinical box Progerias: accelerated aging resulting from defects in DNA repair
Certain genetic diseases are considered models of accelerated aging (progeria). These monogenic diseases display many, but never all, of the features of normal aging; few progeric patients develop dementia or age-related pathologies, such as Alzheimer's disease. The progerias are sometimes described as caricatures of aging, but are useful models for understanding the aging process.
Werner's and Bloom's syndromes are autosomal recessive diseases caused by mutation of distinct DNA helicase genes which have specific roles in repair of damaged DNA. Patients with Werner's syndrome appear normal during childhood, but stop growing in their teens. They gradually show many symptoms of premature aging, including graying and loss of hair, thinning of skin, development of early cataracts, impaired glucose tolerance and diabetes, atherosclerosis and osteoporosis, and increased rates of cancer. Death usually occurs in their mid-40s from cardiovascular disease. Fibroblasts from Werner's patients divide only about 20 times in cell culture, compared to 60 for normal cells, and have higher levels of protein-bound carbonyl groups, an indicator of increased oxidative stress.
Bloom's syndrome is characterized by increased frequency of chromosomal breaks, dwarfism, photosensitivity and increased frequency of cancer and leukemia; death occurs typically in the mid-20s. Ataxia-telangiectasia, or fragile chromosome syndrome, is associated with increased loss of telomeres with cell division and deficiency in repair of double-strand DNA breaks. It is caused by a defect in a protein kinase involved in signal transduction, cell cycle control and DNA repair.
Hutchinson–Gilford syndrome is a severe, pediatric form of progeria. Patients have many of the symptoms of Werner's syndrome, but the symptoms appear at an earlier age and death usually occurs by the mid-20s. This syndrome is caused by a defect in a gene for lamin, a component of the nuclear envelope. Hutchinson–Gilford is one of several distinct syndromes associated with lamin mutations, which cause an increase in nuclear fragility and aberrant mRNA splicing; as in Werner's syndrome, cultured fibroblasts become prematurely senescent. These progeric diseases illustrate the importance of efficient repair of DNA for normal growth and aging.
D-aspartate is a nonoxidative modification of protein that is formed by spontaneous, age-dependent racemization of L-aspartate, the natural form of the amino acid in protein. The more rapid turnover of skin, compared to articular, collagen yields a lower rate of accumulation of D-aspartate in skin collagen with age and also explains the lower rates of accumulation of AGE/ALEs in skin versus articular collagen. AGE/ALEs are even higher in lens crystallins, which have the slowest rate of turnover among proteins in the body. Deamidation of asparagine and glutamine is another nonoxidative chemical modification that increases with age in proteins; it has been described primarily in intracellular proteins.
The rate of accumulation of these modifications depends on the rate of turnover of the collagens (Fig. 43.7) and is accelerated by hyperglycemia and hyperlipidemia in diabetes and atherosclerosis. The increase in AGE/ALEs and oxidative crosslinking of collagen is thought to impair the turnover and contribute to the thickening of basement membranes with age. Increased age-adjusted levels of AGE/ALEs in collagen are implicated in the pathogenesis of complications of diabetes and atherosclerosis. These products are also increased together in the brain in various neurodegenerative diseases, including Alzheimer's and Parkinson's disease and Creutzfeld–Jakob (prion) disease.
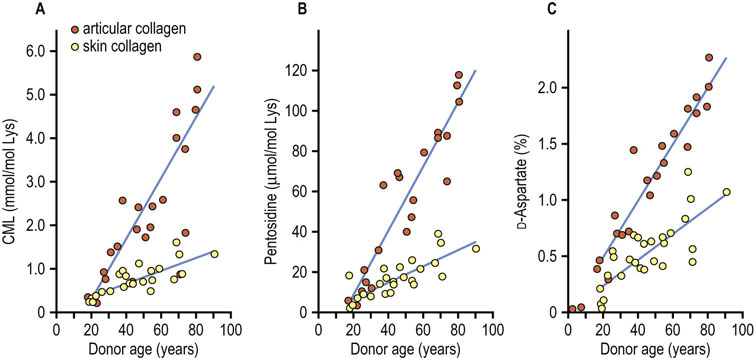
Fig. 43.7 Accumulation of advanced glycoxidation and lipoxidation end products (AGE/ALEs) and D-Aspartate in articular and skin collagens with age.
Nε-(carboxymethyl)lysine (CML) is formed by oxidative mechanisms from glycated proteins or reaction of glucose, ascorbate or lipid peroxidation products with protein. The fluorescent crosslink pentosidine is formed by oxidative reaction of glucose or ascorbate with proteins. D-Aspartate is formed nonoxidatively by racemization of L-aspartate residues in protein. Tissue levels of oxidative and nonoxidative biomarkers correlate with one another, and differences in their rates of accumulation in articular and skin collagen result from differences in rates of turnover of these collagens (adapted from Verzijl N et al: Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem 275:39027–39031, 2000).
The age pigment lipofuscin is a less well-characterized but characteristic biomarker of aging. It accumulates in the form of fluorescent granules, derived from lysosomes, in the cytoplasm of postmitotic cells at a rate that is inversely related to species lifespan. It is considered the accumulated, indigestible debris of reactions between lipid peroxides and proteins. Lipofuscin may account for 10–15% of the volume of cardiac muscle and neuronal cells at advanced age, and its rate of deposition in cardiac myocytes in cell culture is accelerated by growth under hyperoxic conditions. In flies, the rate of accumulation of lipofuscin varies directly with ambient temperature and activity, and inversely with lifespan, consistent with the effects of these variables on lifespan (see Fig. 43.2B).
In summary, there is a wide range of chemical modifications, both oxidative and nonoxidative, that accumulate in proteins with age. While attention is often focused on modification of protein, the real damage from free radicals and oxidative stress is at the level of the genome; if the DNA is not repaired correctly, the cell will die, its capacity may be impaired or the damage will be propagated. Damage to DNA accumulates not in the form of modified nucleic acids but as chemically ‘silent’ errors in repair – insertions, deletions, substitutions, transpositions and inversions of DNA sequences – that affect the expression and structure of proteins. Because repair is fairly efficient in humans compared to other animals, and the composition of DNA does not change on repair, mutations in DNA are not detectable in tissues by conventional analytic techniques. However, the presence of oxidized pyrimidines and purines in urine (see Figure 37.6) provides evidence of chronic oxidative damage to the genome.
Mitochondrial theories of aging
 Clinical box Biomarkers of oxidative stress and aging
Clinical box Biomarkers of oxidative stress and aging
Advanced glycoxidation and lipoxidation end products (AGE/ALEs) are formed by reaction of proteins with products of oxidation of carbohydrates and lipids (see Fig. 43.6). Some compounds, such as Nε-(carboxymethyl)lysine (CML), may be formed from either carbohydrates or lipids; others, such as pentosidine, are formed only from carbohydrates, and others, such as the malondialdehyde adduct to lysine, are formed exclusively from lipids. Carbohydrate sources of AGEs include glucose, ascorbate, and glycolytic intermediates; ALEs are derived from oxidation of polyunsaturated fatty acids in phospholipids. Lysine, histidine and cysteine residues are the major sites of AGE/ALE formation in protein. Over 30 different AGE/ALEs have been detected in tissue proteins, and many of these are known to increase with age. AGE/ALEs are useful biomarkers of the aging of proteins and their exposure to oxidative stress.
Clinical box Alzheimer's disease: oxidative stress in neurodegenerative disease
Alzheimer's disease (AD) is the most common form of progressive cognitive deterioration in the elderly. It is characterized microscopically by the appearance of neurofibrillary tangles and senile plaques in cortical regions of the brain. The tangles are localized inside neurons, and are rich in τ (tau) protein, which is derived from microtubules; it is hyperphosphorylated and polyubiquitinated. Plaques are extracellular aggregates, localized around amyloid deposits, formed from insoluble peptides derived from a family of amyloid precursor proteins. AD affects primarily cholinergic neurons, and drugs that inhibit the degradation of acetylcholine within synapses are a mainstay of therapy. A similar approach is used for preservation of dopamine in dopaminergic neurons in Parkinson's disease, i.e. by inhibiting the degradative enzyme monoamine oxidase.
Several studies have shown that both AGEs and ALEs are increased in tangles and plaques in the brain of AD patients, compared with age-matched controls. Other indicators of generalized oxidative stress in the AD brain include increased levels of protein carbonyls, nitrotyrosine and 8-OH-deoxyguanosine, all detected by immunohistochemical methods. The amyloid protein is toxic to neurons in cell culture and catalyzes oxidative stress and inflammatory responses in glial cells. Significant quantities of decompartmentalized, redox-active iron, a catalyst of Fenton reactions (Chapter 27) are also detectable histologically in the AD brain and can be removed reversibly (in vitro) by treatment with chelators, such as desferrioxamine. Based on these data, oxidative stress is strongly implicated in the development and/or progression of AD, and chelators are being evaluated clinically for treatment of AD. Epidemiologic studies also indicate that long-term treatment with nonsteroidal anti-inflammatory drugs, such as ibuprofen and acetaminophen, may reduce the risk of AD and delay its onset or slow its progression.
 Advanced concept box Aging of the circulatory system
Advanced concept box Aging of the circulatory system
The extracellular matrix of the aorta and major arteries becomes thicker and more highly crosslinked with age, contributing to both the decrease in elasticity and the ability of the endothelium to dilate blood vessels in response to physical and chemical stimuli. These changes occur naturally with age, independent of pathology, but may account for the increase in cardiovascular risk in the elderly. AGEs and ALEs are implicated in the crosslinking of the vascular extracellular matrix, explaining the age-adjusted increase in arterial crosslinking in diabetes and dyslipidemia. Increases in AGE/ALEs and protein crosslinking are also implicated in the altered filtration properties of the renal glomerular basement membrane in diabetes.
Mitochondrial DNA is particularly susceptible to oxidative damage
Mitochondrial theories of aging are a blend of biological and chemical theories, treating aging as the result of chemical damage to mitochondrial DNA (mtDNA). Mitochondria contain proteins specified by both nuclear and mitochondrial DNA but only 13 mitochondrial proteins are encoded by mitochondrial DNA. While this may seem trivial, these include essential subunits of the three proton pumps and ATP synthase. MtDNA is especially sensitive to mutations: mitochondria are the major site of ROS production in the cell (see Fig. 37.4), mtDNA is not protected by a sheath of histones, and mitochondria have limited capacity for DNA repair.
Mitochondrial diseases commonly involve defects of energy metabolism, including the pyruvate dehydrogenase complex, pyruvate carboxylase, electron transport complexes, ATP synthase, and enzymes of ubiquinone biosynthesis. These defects can be caused by mutations in both nuclear and mitochondrial DNA, but mtDNA suffers many more mutations than nuclear DNA. Such defects often result in the accumulation of lactic acid because of impaired oxidative phosphorylation, and may cause cell death, especially in skeletal (myopathies) and cardiac muscles (cardiomyopathies) and nerve (encephalopathies), all of which are heavily dependent on oxidative metabolism. The number of mitochondria and multiple copies of the mitochondrial genome in the cell may provide some protection against mitochondrial dysfunction as a result of mutation, but loss of fully functional mitochondria, and sometimes the number of mitochondria, is a characteristic feature of aging.
Genetic models of increased lifespan
Advanced concept box Telomeres: a clock of aging
Telomeres are the repetitive sequences at the ends of chromosomal DNA, typically thousands of copies of short, highly redundant, repetitive DNA, TTAGGG in humans (Chapter 32). DNA polymerase requires a double-stranded template for replication; RNA primers at the 5′-end of the template serve to initiate DNA synthesis. However, at the extreme ends of the chromosomes, DNA synthesis is restricted, because there are no sequences further upstream for DNA primase engagement. Therefore, each round of chromosome replication results in chromosome shortening. The enzyme telomerase is a reverse transcriptase containing an RNA with a sequence complementary to the telomere DNA. It functions to maintain the length of telomeres at the 3′-end of chromosomes. Telomerase is found in fetal tissues, adult germ cells and in tumor cells. but the somatic cells of multicellular organisms lack telomerase activity. This has led to the hypothesis that shortening of the telomere may contribute to the Hayflick limit and is involved in aging of multicellular organisms. Increased expression of telomerase in human cells results in elongated telomeres and an increase in the longevity of those cells by at least 20 cell doublings. Cells from individuals with premature aging diseases (progeria) also have short telomeres. In contrast, cancer cells, which are immortal, express an active telomerase activity. All of these observations suggest that the decrease in telomere length is associated with cellular senescence and aging. Knockout mice, in which the telomerase gene has been deleted, have chromosomes lacking detectable telomeres. These mice have high frequencies of aneuploidy and chromosomal abnormalities. The disease, autosomal dyskeratosis congenita, features a mutation in the telomerase locus, with inability of somatic cells to reconstitute their telomeres, and hence loss of epidermis and hematopoietic marrow. This disease has many of the characteristics of accelerated aging.
Advanced concept box Aging of muscle: damage to mitochondrial DNA
Old age is characterized by a general decrease in skeletal muscle mass (sarcopenia) and strength, as a result of a decrease in both the number of motoneurons and the number and size of myofibers. The fiber loss is accompanied by an increase in interstitial, fibrous connective tissue, and a reduction in capillary density, which limits the blood supply. The decrease in muscle mass and strength contributes to frailty and increased risk of mortality. The loss in skeletal muscle mass may also contribute to glucose intolerance in the elderly as a result of the decreasing mass of tissue available to take up glucose from blood.
One of the major changes in muscle biochemistry with age is an increase in the number of muscle cells with mitochondria deficient in cytochrome oxidase, which limits the muscle's ability to do work. As mitochondria become less efficient in oxidizing NADH, they become more reduced, and the accumulation of partially reduced ubiquinone (semiquinone) promotes the reduction of molecular oxygen, leading to increased superoxide production in older mitochondria (see Chapter 37). Under these conditions, when oxidative phosphorylation is impaired, cells appear to generate ATP primarily by glycolysis. NADH is also oxidized extramitochondrially, primarily by NADH oxidases in the plasma membrane, which produce hydrogen peroxide, but no ATP.

These changes are observed in both cardiac and skeletal muscle and appear to result from major, random deletions in mitochondrial DNA (25–75% of total mtDNA), which are then amplified by clonal expansion, leading to fiber atrophy and breakage. The muscle fiber is only as strong as its weakest link, so that small regions of fiber loss affect overall muscle capacity. Fortunately, sarcopenia can be delayed and partially reversed by resistance exercise, thus the emphasis on regular exercise among the elderly.
The effect of genetics on longevity is readily apparent in animal models
Different strains of mice vary by more than twofold in lifespan, and there are also significant differences in the lifespan of male and female mice of the same strain raised under identical conditions. Deficiencies in some hormones or defects in their receptors or postreceptor signaling pathways have a significant effect on mouse lifespan. Profound effects are observed in Ames and Snell dwarf mice. These mice have different pituitary defects, both resulting in negligible secretion of growth hormone (GH; stimulates IGF-1 secretion by liver), thyroid-stimulating hormone and prolactin (see Chapter 39). Their body weights are decreased as young adults by about 35% and their maximum lifespan increased by about 45% compared to littermates, but oddly they become obese with age. Similar effects on weight and lifespan are observed in mice with defects in GH or IGF-1 receptors or signal transduction. Many of these strains are fragile: Ames and Snell dwarfs are hypothyroid, hypoglycemic and hypoinsulinemic, and have low body temperature; they have impaired reproductive capacity, are more susceptible to infection, and require special housing conditions to maintain body temperature – but they live longer! Treatment of hypothyroidism in Snell dwarfs resulted in a restoration of a near normal lifespan, while hypophysectomy of young rats increases their maximum lifespan by 15–20%. Thus, three hormones that have a profound effect on metabolism and growth (growth hormone, IGF-1 (and insulin) and thyroxine) also have profound effects on lifespan.
In humans, the most significant genetic determinant of lifespan is sex: women live longer than men. Genetics accounts for an estimated 20–50% of the remaining variance in lifespan, the other 50–80% being attributed to environment and random developmental variations. It was estimated (in 2008) that there are at least 30 genes that have a significant effect on human lifespan. The cross-breeding of human populations and the many allelic combinations of these genes may obscure effects seen in inbred strains of worms or rodents. However, there is a recent report that Ashkenazi Jews who live past age 95 have a higher frequency of mutations in the gene for the IGF-1 receptor (IGF-1R). There are other genes or gene products that are associated with increased longevity in humans, e.g. variants in ApoE, ApoC3 and CETP. However, these genes seem to increase mean lifespan, probably by modulating the cardiovascular effects of dietary cholesterol, rather than cause an increase in maximum lifespan.
Anti-aging interventions – what works and what doesn't
Antioxidant supplements
Antioxidant supplements may improve health, but do not increase lifespan
Based on the FRTA, it seems reasonable to speculate that antioxidant supplementation should have an effect on longevity. In fact, however, there is no rigorous, reproducible experimental evidence that antioxidant supplements have any effect on maximum lifespan of humans or other vertebrates. At the same time, antioxidant supplements, most of which include vitamins, may improve health, particularly in persons with vitamin deficiencies. Thus, effects of antioxidant therapy on mean (and healthy) lifespan are not unexpected. Failure to affect maximum lifespan may result from the fact that there are so many mechanisms for production and control of free radicals and inhibiting or reversing damage to biomolecules. Many of these processes depend on the activity of enzymes that detoxify ROS or regenerate endogenous antioxidants. These enzymes, such as superoxide dismutase and glutathione peroxidase (Chapter 37), are induced in response to oxidative stress and may also be repressed during times of low oxidative stress. Thus, the body may respond to maintain a homeostatic balance between pro-oxidant and antioxidant forces (see Fig. 37.2), countering efforts to enhance antioxidant defenses. This response may be essential, for example, to maintain effective bactericidal activity during the respiratory burst accompanying phagocytosis.
Calorie restriction
Caloric restriction is the only regimen known to increase lifespan in animals
Calorie restriction (CR) is the only intervention that consistently extends maximum lifespan in a variety of species, including mammals, fish, flies, worms, and yeast. Reduction in total caloric intake is the essential feature of this intervention, i.e. the beneficial, life-extending effects are observed whenever CR is applied and regardless of dietary composition, although early and prolonged intervention has more impressive effects. As shown in Figure 43.8, CR leads to a significant increase in both the mean and maximum lifespan of laboratory rats, equivalent to extending human lifespan to about 180 years. Calorie-restricted rats have fewer muscle fibers lacking in cytochrome oxidase and decreased levels of deletions in muscle mitochondrial DNA. CR mice also have lower levels of inducible genes for hepatic detoxification, DNA repair and response to oxidative stress (heat shock proteins), suggesting a lower rate of oxidative stress and damage to proteins and DNA.
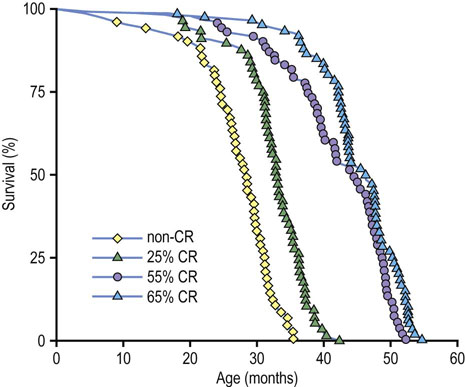
Fig. 43.8 Calorie restriction (CR) extends longevity of mice.
The non-CR group received food ad libitum. Other groups were restricted by 25%, 55% and 65% of the ad lib diet, starting at 1 month of age (adapted from Weindruch R et al: Retardation of aging in mice by dietary restriction. J Nutr 116:651–654, 1986).
Caloric restriction delays the onset of age-related diseases, including cancer (Fig. 43.9)
CR is the most potent, broad-acting cancer prevention regimen in rodents. It is argued that the extension of maximum lifespan by CR is achieved by delaying the onset of cancer. Long-lived animals are more efficient in protecting their genome and thereby delaying the onset of cancer, but CR may limit damage even more, preserving the integrity of the genome and thereby leading to a longer lifespan. While long-term CR has not been tested in humans, obesity, at the opposite end of the weight spectrum, is a pro-inflammatory state and is a risk factor for cancer in humans.
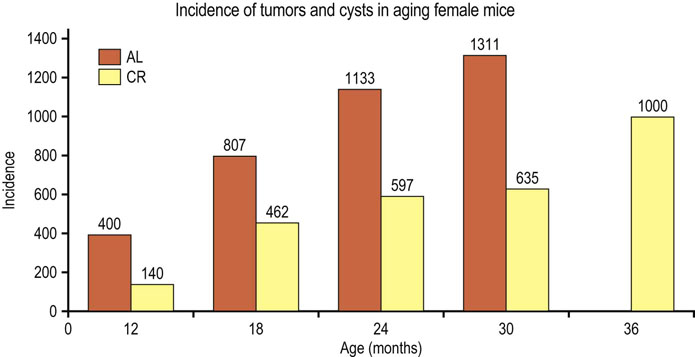
Fig. 43.9 Effect of calorie restriction (CR) on development of tumors in mice.
Over 1000 mice, 50:50 male and female, from four different genotypes were divided into two groups, one fed ad libitum (AL), the other receiving 60% of the caloric intake of the control group (CR), but with comparable intake of vitamins, minerals and micronutrients. Cohorts of animals were sacrificed at specified times and total lesions (tumors plus cysts) were measured. At 24 months, 51% AL and 13% CR mice had tumors. Note the absence of tumors in control mice at 36 months – all of the control mice are dead! CR extended the mean and maximum lifespan of the mice and also delayed the onset of cancer (adapted from Bronson RT, Lipman RD: Reduction in rate of occurrence of age related lesions in dietary restricted laboratory mice. Growth Dev Aging 55:169–184, 1991).
In CR experiments, it has been difficult to differentiate between the effects of dietary restriction on energy expenditure (rate of living) versus the reduction in body weight or adipose tissue mass that accompanies dietary restriction. FIRKO (adipose tissue (fat) insulin receptor knockout) mice have a 15–25% decrease in body mass, largely because of a 50% decrease in fat mass. However, these mice consume identical amounts of food per day as control littermates, actually more than the control animals when normalized to their body weight. They also have a 20% increase in lifespan, suggesting that the decrease in body or fat mass is more important than caloric intake in determining maximum lifespan potential. In another study, overexpression of the gluconeogenic enzyme PEPCK in skeletal muscle produced a leaner mouse, with 50% of body weight and 10% of fat mass, compared to controls. These mice were seven times as active and ate 60% more than control mice, but lived longer and had longer reproductive life. Overall, the decrease in body weight or adiposity during CR, rather than the decrease in food consumption, appears to have the greater effect on lifespan extension. One general outcome from these dietary and genetic experiments is that mitochondrial efficiency, measured as lower rates of ROS/ATP production, appears to be an important determinant of longevity.
Studies on CR in longer-lived, primate species have been under way since the 1980s. From these studies, there is clear evidence that monkeys on CR are more active and younger in appearance, have better insulin sensitivity and plasma lipid profiles and decreased risk for diabetes, have better overall cardiovascular and renal health, experience less age-related sarcopenia and brain atrophy, and have decreased risk for cancer, compared to age-matched normally fed animals. However, the evidence of lifespan extension is weak and still controversial (Fig. 43.10). Even if CR can be shown to extend lifespan in monkeys, it is unlikely that humans will be able to adopt the strict dietary control required for this regimen. However, similar improvements in health have been observed in shorter-term studies with humans, i.e. improvement in fasting glucose, insulin sensitivity and plasma lipid profile, and decreased blood pressure, compared to a matched control group. Understanding the biological mechanisms of the effects of CR may lead to alternative strategies that mimic CR and possibly extend, at least, the healthy lifespan of humans.

Fig. 43.10 Effects of calorie restriction in primates.
The animal on the left was raised with calorie restriction, while that on the right was fed a normal diet. A study from the University of Wisconsin in 2009 concluded that calorie restriction extended the lifespan of Rhesus monkeys, while another by the US National Institutes of Health in 2012, also with Rhesus monkeys, concluded there was no effect. Studies with other primate species are still in progress, so the issue is not fully resolved; however, it is clear in all studies that calorie restriction produces a healthier phenotype, with fewer age-related chronic diseases and decreased risk for cancer (with permission from the National Institutes of Health).
Summary
Aging is characterized by a gradual decline in the capacity of physiologic systems, leading eventually to failure of a critical system, then death.
At the biochemical level, aging is considered the result of chronic chemical modification of all classes of biomolecules.
According to the free radical theory of aging, ROS are the primary culprits, causing alterations in the sequence of DNA (mutations) and structure of proteins. Longevity is achieved by developing efficient systems to limit and/or repair chemical damage.
Caloric restriction is, at present, the only widely applicable mechanism for delaying aging and extending the mean, healthy, and maximum lifespan of species.
CR appears to work, in part, by inhibiting the production of ROS and limiting damage to biomolecules, delaying many of the characteristic features of aging, including cancer.
Baur, JA, Ungvari, Z, Minor, RK, et al. Are sirtuins viable targets for improving healthspan and lifespan? Nat Reviews. 2012; 11:443–461.
Cefalu, CA. Theories and mechanisms of aging. Clin Geriatr Med. 2011; 27:491–506.
Dai, DF, Rabinovitch, PS, Ungvari, Z. Mitochondria and cardiovascular aging. Circ Res. 2012; 110:1109–1124.
Ford, ES, Zhao, G, Tsai, J, et al. Low-risk lifestyle behaviors and all-cause mortality: finding from the National Health and Nutrition Examination Survey (NHANES) III Mortality Study. Am J Public Health. 2011; 101:1922–1929.
Jenny, NS. Inflammation in aging: cause, effect, or both? Discov Med. 2012; 13:451–460.
Kemnitz, JW. Calorie restriction and aging in nonhuman primates. ILAR J. 2011; 52:66–77.
Libert, S, Guarente, L. Metabolic and neuropsychiatric effects of calorie restriction and sirtuins. Annu Rev Physiol. 2013; 75:669–684.
Semba, RD, Nicklett, EJ, Ferrucci, L. Does accumulation of advanced glycation end products contribute to the aging phenotype? J Gerontol A Biol Sci Med Sci. 2010; 65:963–975.
Caloric restriction. www.crsociety.org.
www.pathguy.com/lectures/aging.htm
www.benbest.com/lifeext/aging.html
Progeria. www.progeriaresearch.org/index.html.
Telomeres. www.telomeres.net/.