Deoxyribonucleic Acid
Learning objectives
After reading this chapter you should be able to:
 Describe the composition and structure of DNA, based on the Watson–Crick model, including the concepts of directionality and complementarity in DNA structure.
Describe the composition and structure of DNA, based on the Watson–Crick model, including the concepts of directionality and complementarity in DNA structure.
Describe the packaging of DNA in the nucleus.
Explain how replication of DNA is achieved with high fidelity.
Discuss the enzymes involved, the activities at replication forks, and the structures and intermediates participating in the replication process.
Outline the mechanism by which replication is controlled in the eukaryotic cell.
Describe the types of damage to DNA and the mechanisms involved in DNA repair.
Describe the mechanism of action of AZT for treatment of AIDS.
Introduction
Cellular nucleic acids exist in two forms, deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). Approximately 90% of the nucleic acid within cells is RNA and the remainder is DNA. DNA is the repository of genetic information. This chapter deals with the structure of DNA, the manner in which it is stored in chromosomes in the nucleus, and the mechanisms involved in its biosynthesis and repair.
Structure of deoxyribonucleic acid
DNA is an antiparallel dimer of nucleic acid strands
DNA is composed of nucleotides containing the sugar deoxyribose. Deoxyribose is missing the hydroxyl group at the 2′-position. The chains of DNA are polymerized through a phosphodiester linkage from the 3′-hydroxyl of one ribose to the 5′-hydroxyl of the next ribose (Fig. 32.1A). Thus, DNA is a duplex linear polymer of deoxyribose 3′,5′-phosphate, with purine and pyrimidine bases attached to carbon-1′ of the deoxyribose subunit.
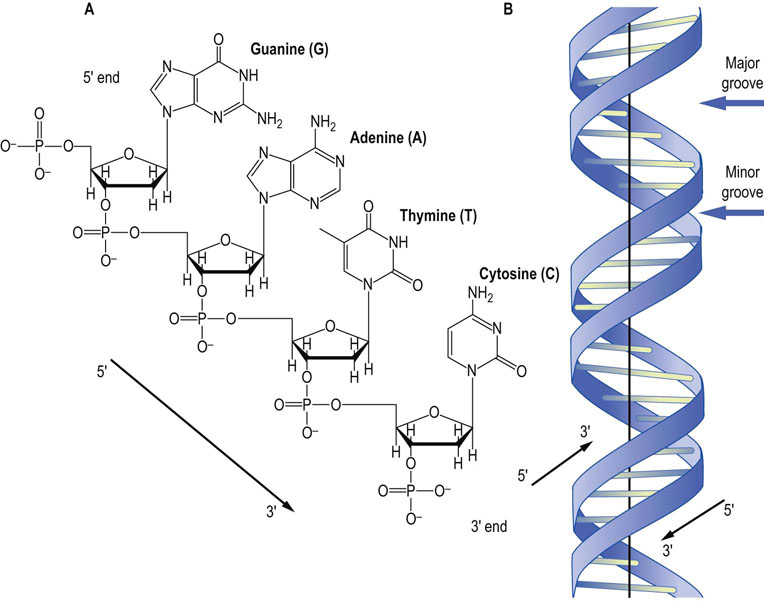
Fig. 32.1 Structure of DNA.
(A) A tetranucleotide sequence of DNA showing each of the nucleotides normally found in DNA. The deoxyribose sugars are missing the 2′-hydroxyl that is present in the ribose sugars found in RNA. By convention, DNA is read from the 5′ to 3′ end, so the sequence of this tetranucleotide is 5′-GATC-3′. (B) A graphic representation of the structure of B-DNA, the major form of DNA in the cell. The base pairs in the middle are aligned nearly perpendicular to the helical axis. The major groove and the minor groove are shown. Note that the strands are antiparallel.
Using X-ray diffraction photographs of DNA taken by Rosalind Franklin, James Watson and Francis Crick proposed a structure for DNA in 1953. This model proposed that DNA was composed of two intertwined complementary strands with hydrogen bonds holding the strands together (Fig. 32.1B). The basic simplicity of this structure was consistent with the observation that in all DNA the molar content of A is equal to that of T, and the molar content of G is equal to that of C. While some of the details of the model have been modified, the Watson–Crick hypothesis was rapidly accepted and its essential elements have remained unchanged since originally proposed.
Watson and Crick model of DNA
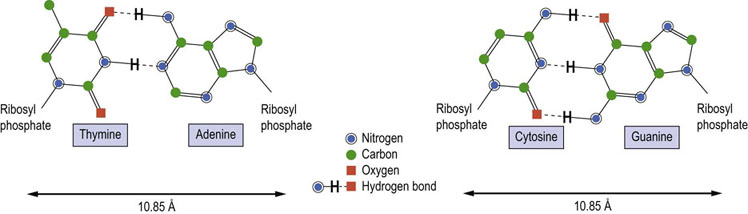
As originally presented by Watson and Crick, DNA is composed of two strands, wound around each other in a right-handed, helical structure with the base pairs in the middle and the deoxyribosyl phosphate chains on the outside. The orientation of the DNA strands is antiparallel, i.e. the strands run in opposite directions. The nucleotide bases on each strand interact with the nucleotide bases on the other strand to form base pairs (Fig. 32.2). The base pairs are planar and are oriented nearly perpendicular to the axis of the helix. Each base pair is formed by hydrogen bonding between a purine and a pyrimidine. Consistent with this pairing, Guanine forms three hydrogen bonds with cytosine, and adenine forms two with thymine. Because of the specificity of this interaction between purines and pyrimidines on the opposite strands, the opposing strands of DNA are said to have complementary structures. The composite strength of the numerous hydrogen bonds formed between the bases of the opposite strands and the hydrophobic interactions among the bases is responsible for the extreme stability of the DNA double helix. While the hydrogen bonds between strands are affected by temperature and ionic strength, stable complementary structures can be formed at room temperature with as few as 6–8 nucleotides.
Three-dimensional DNA
The three-dimensional structure of the DNA double helix is such that the deoxyribosyl phosphate backbones of the two strands are slightly offset from the center of the helix. Because of this, the grooves between the two strands are of different sizes. These grooves are termed the major groove and the minor groove (see Figs 32.1B). The major groove is more open and exposes the nucleotide base pairs. The minor groove is more constricted, being partially blocked by the deoxyribosyl moieties linking the base pairs. Binding of proteins to DNA occurs mostly in the major groove and is specific for the nucleotide sequence of DNA.
Alternative forms of DNA may help to regulate gene expression
Although the majority of DNA molecules in a cell exist in the B-form described above, alternative forms of DNA also exist. When the relative humidity of B-form DNA falls to less than 75%, the B-form undergoes a reversible transition into the A-form of DNA. In the A-form, the nucleotide base pairs are tilted 20° relative to the helical axis and the helix diameter is increased, compared to the B-form (Fig. 32.3). The A-form is observed when the DNA strands contain tracks of polypurine (and complementary polypyrimidine) residues. These regions do not efficiently bind histones and are therefore unable to form nucleosomes (see below), resulting in nucleosome-free (exposed) regions of DNA.
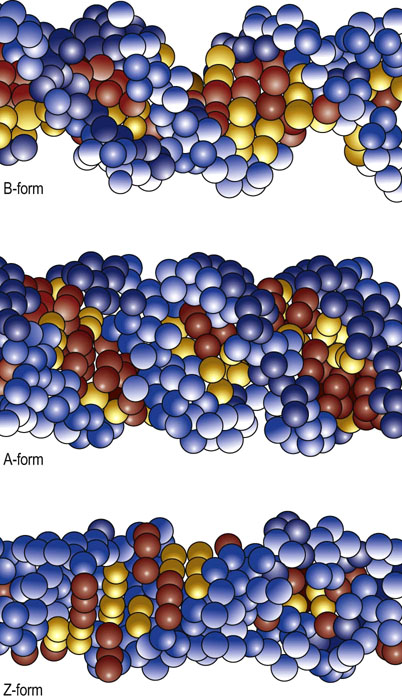
Fig. 32.3 The structures of different forms of DNA include the B-, A- and Z-forms.
The sugar phosphate backbone of the DNA strands is colored blue. The nucleotide bases forming the internal base pairs are yellow for pyrimidines (thymine and cytosine) and red for purines (adenine and guanine).
Another unique form of DNA exists when the sequence of nucleotides consists of alternating purine/pyrimidine stretches. This form, termed Z-DNA, is also favored at high ionic concentrations. In Z-DNA, the base pairs flip 180° relative to the sugar nucleotide bond. This results in a novel conformation of the base pairs relative to sugar phosphate backbones, yielding a form of DNA with a zigzag configuration (hence the name Z-DNA) along the sugar phosphate backbone. Surprisingly, this change in conformation leads to the formation of a left-handed DNA helix. While the Z-DNA form is favored at high ionic concentrations, it can also be induced at normal ionic concentrations by methylation of cytosine residues, a form of epigenetic modification of DNA (Chapter 35). Protein-binding interactions with these alternative forms of DNA, which are widely distributed in the genome, are involved in the regulation of gene expression.
The digital linear code (base pairing) in the DNA double helix has a significant component that acts by altering, along its length, the shape and stiffness of the molecule. In this way, one region of DNA is structurally differentiated from another, which provides another level of encoded information in three-dimensional space. These local shape and stiffness variations contribute superhelical structure and three-dimensional spatial interactions in DNA. It can be said that the super-helical density behaves as an analog regulatory mode as opposed to the more frequently accepted purely digital information content.
Separated DNA strands can reassociate to form duplex DNA
Complementary strands of DNA spontaneously hybridize to form helical structures
Because the DNA strands are complementary and are held together only by noncovalent forces, they can be separated into individual strands. This strand separation or denaturation of DNA is commonly induced by heating the solution. The dissociation is reversible and, on cooling, the complementary nucleotide sequences reassociate or reanneal to reform their original base pairs. This is the basis for one of the primary methods for DNA analysis, Southern hybridization (see Chapter 36). Because adenine and thymine interact through two hydrogen bonds and guanine and cytosine through three (see Fig. 32.2), AT-rich regions melt at lower temperatures than GC-rich regions in DNA. The denaturation of DNA can also be induced locally by enzymes or DNA-binding proteins. The promoter region of DNA contains a TATA sequence (the TATA box; see Fig. 35.1), an easily melted region of DNA that facilitates the unwinding of DNA during the early stages of gene expression (Chapter 35).
The human genome
The human genome contains 20,000–25,000 different protein coding genes spread over 23 chromosome pairs
Genes for specific proteins are unique DNA sequences that are present in single copies or at most only a few copies per genome. There are also several types of repeated DNA sequences within the genome. These are divided into two major classes: middle repetitive (<10 copies per genome) and highly repetitive (>10 copies per genome).
Some middle repetitive DNA consists of genes that specify transfer and ribosomal ribonucleic acids, which are involved in protein synthesis (Chapter 34), and histone proteins that are part of the nucleosome (below). Other middle repetitive DNA sequences have no known useful function but may participate in DNA strand association and chromosomal rearrangements during meiosis. The best-characterized highly repetitive sequence in humans is known as the Alu sequence. Between 300,000 and 500,000 Alu I repeats of about 300 base pairs are scattered throughout the human genome, comprising 3–6% of the total DNA. Individual repeats of the Alu sequence may vary by 10–20% in identity. Similar sequences are found in other mammals and in lower eukaryotes.
Satellite DNA
Satellite DNA was originally identified as a subfraction of DNA with a buoyant density slightly lower than that of genomic DNA because of its higher content of AT base pairs. It consists of clusters of short, species-specific, nearly identical sequences that are tandemly repeated hundreds of thousands of times. These clusters lack protein-coding genes and are found principally near the centromeres of chromosomes, suggesting that they may function to align the chromosomes during cell division to facilitate recombination. Because these repetitive sequences cover long stretches of chromosomes (100s to 1000s of kilobase pairs; kbp), determining the sequence of satellite DNA and sequencing the centromere region of DNA are major challenges to completing the non-coding sequence of eukaryotic genomes.
Mitochondrial DNA
The nucleus of eukaryotic cells contains the majority of the DNA in the cell – genomic DNA. However, DNA is also found in mitochondria and in plant chloroplasts, which is consistent with endosymbiont theories for the origins of these cellular organelles: namely, that they are parasites that adapted to intracellular life by symbiosis.
The mitochondrial genome is small in size, circular, and encodes relatively few proteins
In humans, the mitochondrial genome encodes 22 tRNAs, two rRNAs, and 13 mitochondrial proteins that are involved in the respiratory apparatus, including subunits of NADH dehydrogenase, cytochrome b, cytochrome oxidase and ATPase.
The remainder of the proteins that are found in mitochondria (about 1000) are produced from nuclear genes, synthesized in the cytoplasm on ‘free’ ribosomes (Chapter 34), then imported into the mitochondrion. This import process requires a special N-terminal mitochondrial-import sequence of about 25 amino acids in length that forms an amphipathic helix which interacts with transporter and chaperone proteins in the inner and outer mitochondrial membrane and matrix. Those few proteins that are encoded by the mitochondrial genome are synthesized in the mitochondrion, using machinery similar that used in the cytoplasm for synthesis of nonmitochondrial proteins (see Chapter 34).
DNA is compacted into chromosomes
Chromosomes are compact, highly organized forms of DNA
In eukaryotes, DNA is arranged in linear segments termed chromosomes. Each chromosome contains between 48 million and 240 million base pairs. The B-form of DNA has a contour length of 3.4 Å per base pair. Therefore, chromosomes have contour lengths of 1.6–8.2 cm, which is much larger than a cell. To fit within the nucleus, DNA is condensed >8000-fold into an organized structure. Interactions between DNA and mobile cations, such as Na+, Mg2+, and the polyamines such as spermidine, play an important role in the physical properties and biological function of DNA. Even in dilute solutions, approximately three out of four DNA charges are neutralized by a cation that is in some sense ‘bound’. This neutralization facilitates compaction of DNA into densely packaged chromatin and deformation of DNA by proteins.
Chromatin contains DNA, RNA and protein, plus inorganic and organic counterions
In the native chromosome, DNA is complexed with RNA and an approximately equal mass of protein. These DNA–RNA–protein complexes are termed chromatin. The majority of the proteins in chromatin are histones. Histones are a highly conserved family of proteins that are involved in the packing and folding of DNA within the nucleus. There are five classes of histones, termed H1, H2A, H2B, H3, and H4. They are all rich (>20%) in positively charged, basic amino acids (lysine and arginine). These positive charges interact with the negatively charged, acidic phosphate groups of the DNA strands to reduce electrostatic repulsion and permit tighter DNA packing.
Nucleosomes are the building-blocks of chromatin
The histone proteins associate into a complex termed a nucleosome (Fig. 32.4). Each of these complexes contains two molecules each of H2A, H2B, H3 and H4, and one molecule of H1. The nucleosome protein complex is encircled with about 200 base pairs of DNA that form two coils around the nucleosome core. The H1 protein associates with the outside of the nucleosome core to stabilize the complex. By forming nucleosomes, the packing density of DNA is increased about sevenfold.
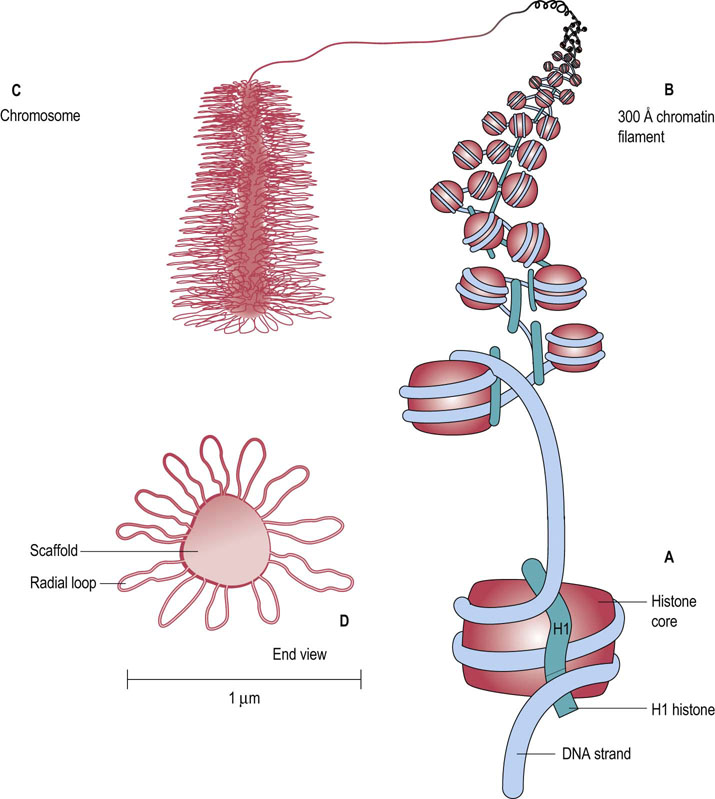
Fig. 32.4 Structures involved in chromosome packaging.
(A) The nucleosome core is composed of two subunits each of H2A, H2B, H3, and H4. The core is twice wrapped with DNA, and the H1 histone binds to the completed complex. (B) The 300 Å chromatin filament is formed by wrapping the nucleosomes into a spring-shaped solenoid. (C) The chromosome is composed of the 300 Å filaments, which bind to a nuclear scaffold, forming large loops of chromatin material. (D) The end view of a chromosome shows the central nuclear scaffold surrounded by the radial loops of chromatin. The diameter of a chromosome is about 1 µm.
The nucleosome particles themselves are also organized into other, more tightly packed structures, termed 300 Å chromatin filaments. These filaments are constructed by winding the nucleosome particles into a spring-shaped solenoid structure with about six nucleosomes per turn (see Fig. 32.4). The solenoid is stabilized by head-to-tail associations of the H1 histones. Finally, the chromatin filaments are compacted into the mature chromosome, using a nuclear scaffold. The scaffold is about 400 nm in diameter and forms the core of the chromosome. The filaments are dispersed around the scaffold to form radial loops about 300 nm in length. The final diameter of a chromosome is about 1 µm.
Telomeres
Telomeres are nucleoprotein complexes that cap the 3′ ends of the eukaryotic chromosomes. They are essential for cell viability. These structures consist of tandem repeats of short, G-rich, species-specific oligonucleotides. In humans, the repeated sequence is TTAGGG. Telomeres can contain as many as 1000 copies of this sequence. During the synthesis of telomeres, the enzyme telomerase, a ribonucleoprotein complex, adds preformed hexanucleotide repeats to the 3′-end of the chromosome, using its RNA as a template; there is no requirement for a DNA template. In human somatic cells telomeric DNA shortens in each cell division until it cannot exercise its end-protective functions, e.g. avoiding recognition of chromosome tips as double-stranded breaks. The shortening of telomeres after many cell replications has been linked to the development of cellular senescence. If telomeres turn out to be dysfunctional as a result of disproportionate shortening or defects in their intrinsic proteins, they trigger pathways that restrict proliferative life span. Telomere-based chromosome instability has been proposed as one driving force in oncogenesis.
The cell cycle in eukaryotes
Figure 32.5 shows the various phases of the growth and division of eukaryotic cells, known as the cell cycle. The G1 phase is a period of cell growth that occurs prior to DNA replication. The phase during which DNA is synthesized or replicated is termed the S phase. A second growth phase, termed G2, occurs after DNA replication but prior to cell division. The mitosis or M phase is the period of cell division. Following mitosis, the daughter cells either reenter the G1 phase or enter a quiescent phase termed G0, when growth and replication cease. The passage of cells through the cell cycle is tightly controlled by a variety of proteins termed cyclin-dependent kinases (see Chapter 42).
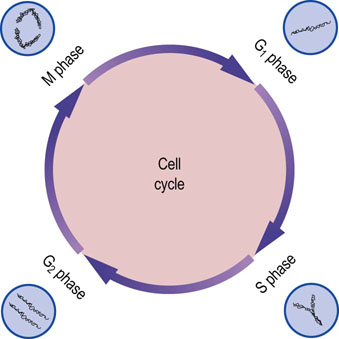
Fig. 32.5 Stages of the cell cycle.
G1 and G2 are growth phases that occur before and after DNA synthesis, respectively. DNA replication occurs during the S phase. Mitosis occurs during the M phase, producing new daughter cells that can reenter the G1 phase (compare Fig. 43.1).
DNA Replication
DNA is replicated by separating and copying the strands
For cells to divide, their DNA must be duplicated during the S phase of the cell cycle. The structure of the DNA double helix and its complementarity suggested the mechanism for DNA replication – strand separation followed by strand copying. The separated parent strands serve as templates for the synthesis of the new daughter strands. This method of DNA replication is described as semi-conservative – each replicated duplex, daughter DNA molecule contains one parental strand and one newly synthesized strand.
DNA replication
The site at which DNA replication is initiated is termed the origin of replication
In prokaryotes, a DNA-binding protein termed DnaA binds to repeated nucleotide sequences located within the origin. Binding of 20–30 DnaA molecules to the origin of replication induces unwinding, which separates the strands in an AT-rich region adjacent to the DnaA-binding sites. Next, the hexameric protein DnaB binds to the separated DNA strands. DnaB has helicase activity that catalyzes ATP-mediated unwinding of the DNA helix. DNA gyrase also participates in separation of the strands. As this complex continues unwinding the DNA strands in both directions from the origin of replication, single-stranded DNA-binding proteins coat the separated strands to inhibit their reassociation.
Once the strands are sufficiently separated, another protein, termed DNA primase, is added, resulting in the formation of a primosome complex at the replication fork. The primosome synthesizes short (n ≤ 10) RNA oligonucleotides complementary to each parental DNA strand. These oligonucleotides serve as primers for DNA synthesis. Once each RNA primer has been laid down, two DNA polymerase III complexes are assembled, one at each of the primed sites. Because of the unidirectional synthetic activity of the polymerase and the antiparallel nature of the two strands, the synthesis of DNA along the two strands is different (Fig. 32.6). The two daughter strands being synthesized are termed the leading strand and the lagging strand. In addition to its polymerase activity, one of the subunits of DNA polymerase III has a proofreading exonuclease activity, which corrects mismatches and assures fidelity in replication of DNA.
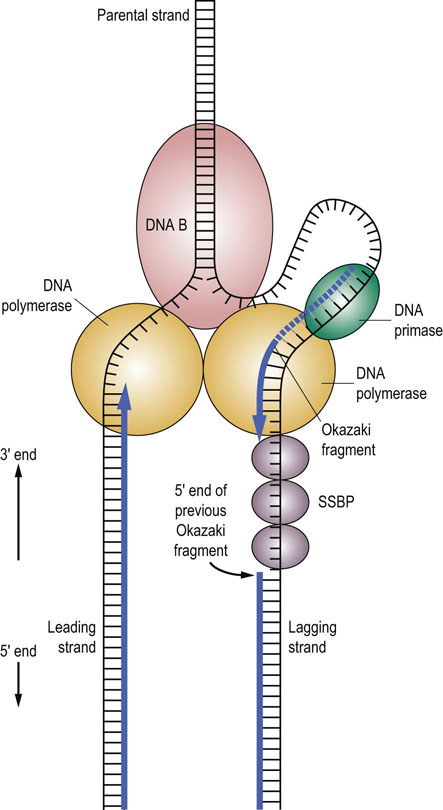
Fig. 32.6 DNA synthesis.
DNA synthesis occurs at a replication fork producing new strands termed the leading strand and the lagging strand. The ‘railroad tracks’ represent double-stranded DNA. Some of the enzymes involved in DNA synthesis are shown: DNA B (helicase), DNA primase, DNA polymerase, and single-strand DNA binding protein (SSBP). The leading strand is replicated in a continuous fashion. However, for the lagging strand, RNA primers are periodically added by the DNA primase along the strand. DNA polymerase III elongates these RNA primers to form Okazaki fragments. When the Okazaki fragment is complete, the DNA polymerase III on the lagging strand will shift to the next RNA primer to initiate another Okazaki fragment. The exonuclease activity of DNA polymerase I removes the RNA primers and replaces them with DNA. DNA ligase seals the gaps in the DNA strands to complete synthesis of the lagging strand.
DNA synthesis proceeds in opposite directions along the leading and lagging strands of the template DNA
DNA synthesis proceeds along the leading strand in a 5′ to 3′ direction, producing a single, long, continuous strand. However, because DNA synthesis adds new nucleotides only at the 3′-end of the elongating DNA strand, DNA polymerase III cannot synthesize the lagging strand in one long continuous piece as it does for the leading strand. Instead, the lagging strand is synthesized in small fragments, 1000–5000 base pairs in length, termed Okazaki fragments (see Fig. 32.6). The primosome remains associated with the lagging strand and continues periodically to synthesize RNA primers complementary to the separated strand. As DNA polymerase III moves along the parental DNA strand, it initiates the synthesis of Okazaki fragments at the RNA primers, elongating different fragments from each primer.
When the 3′-end of the elongating Okazaki fragment reaches the 5′-end of the previously synthesized Okazaki fragment, DNA polymerase III releases the template and finds another RNA primer further back along the lagging strand, synthesizing another Okazaki fragment. Eventually, the Okazaki fragments are joined by DNA polymerase I. This enzyme, which also has a role in DNA repair, has an exonuclease activity that permits it to remove and replace a stretch of nucleotides as it proceeds along a DNA template. During DNA replication, DNA polymerase I removes the RNA primer and replaces it with DNA. Finally, DNA ligase joins the lagging-strand DNA fragments to form a continuous strand.
Eukaryotes stringently regulate DNA replication
Eukaryotic DNA synthesis is remarkably similar to prokaryotic DNA synthesis. However, eukaryotes have many more origins of replication. These are activated simultaneously during the S phase of the cell cycle, permitting rapid replication of the entire chromosome. To insure that excess amounts of unfinished, replicating DNA do not accumulate, cells use a protein termed a licensing factor that is present in the nucleus prior to replication. Following each round of replication, this factor is inactivated or destroyed, preventing further replication until more licensing factor is synthesized later in the cell cycle.
Licensing factor is best understood in the yeast. In yeast there is a complex called the origin recognition complex (ORC), composed of six proteins. The ORC marks the origin of replication and remains bound to the origin throughout the cell cycle. It serves as a docking site for the other components that regulate DNA replication. Early events in DNA replication include the binding of several highly unstable proteins to ORC. These proteins include CDC6/18 and CDT1, which facilitate binding of a group of three additional proteins: MCM2, MCM3, and MCM5. Once the MCM proteins bind, the origin exists as a prereplication complex and is ‘licensed’ to enter S phase of the cell cycle. The initiation of DNA synthesis is triggered by the action of the CDC7 kinase together with other cyclin-dependent kinases. The activated MCM complex then participates in unwinding the replication origin and is thereby displaced from the origin. Following displacement, the origin forms a postinitiation complex and CDC6 is degraded, thereby preventing the reloading of the origin with additional licensing factor.
DNA repair
There are typically more than 10,000 modifications of DNA per cell per day
Because DNA is the reservoir of genetic information within the cell, it is extremely important to maintain the integrity of DNA. Therefore, the cell has developed multiple, highly efficient mechanisms for the repair of modified or damaged DNA.
 Clinical box AZT therapy for HIV infection
Clinical box AZT therapy for HIV infection
Human immunodeficiency virus (HIV) infection results in a profound weakening of the immune system that makes the patient susceptible to a range of bacterial, fungal, protozoal and viral superinfections. Kaposi's sarcoma may also develop; it is a cancer-like disease of blood vessels caused by infection with human herpesvirus-8 (HHV-8).
Effective treatments of the HIV viral infection rely on detailed knowledge of the viral life cycle. For the AIDS virus, the viral genome is RNA. In the infected cell, it is copied into a DNA form by a viral enzyme termed reverse transcriptase. Reverse transcriptase is an error-prone enzyme that does not have the proofreading capabilities of DNA polymerase III. One therapeutic approach for treatment of AIDS takes advantage of the enzyme's lack of specificity in choice of complementary substrates. Several important antiviral drugs are nucleotide analogues that inhibit reverse transcriptase, including AZT (azido-2′,3′-dideoxythymidine; Retrovir, zidovudine), ddC (2′,3′-dideoxycytidine; Hivid, zalcitabine), and 3TC (2′,3′-dideoxy-3′-thiacytidine; Epivir, lamivudine) (Fig. 32.7). AZT, for example, is metabolized in the body into the thymine triphosphate (TTP) analogue azido-TTP. The HIV reverse transcriptase misincorporates azido-TTP into the reverse-transcribed viral genome. The incorporation of azido-TTP into DNA blocks further chain elongation, because the 3′-azido group cannot form a phosphodiester bond with subsequent nucleoside triphosphates. The inability to synthesize DNA from the viral RNA template results in inhibition of viral replication The life cycle of HIV spans about 1.5 days from entry into a cell, replication, assembly, and release of new viral particles to infection of other cells. HIV lacks proofreading enzymes to correct errors occurring during conversion of its RNA into DNA via reverse transcription. Its short life cycle and high error rate cause the virus to mutate very hastily, causing large genetic variability of HIV. Most of the mutations are deleterious or bear no advantage, but some have a natural selection advantage to their parent and can allow them to evade the human immune system and antiretroviral drugs. The more active copies of the virus, the greater the possibility that one resistant to antiretroviral drugs will be made.
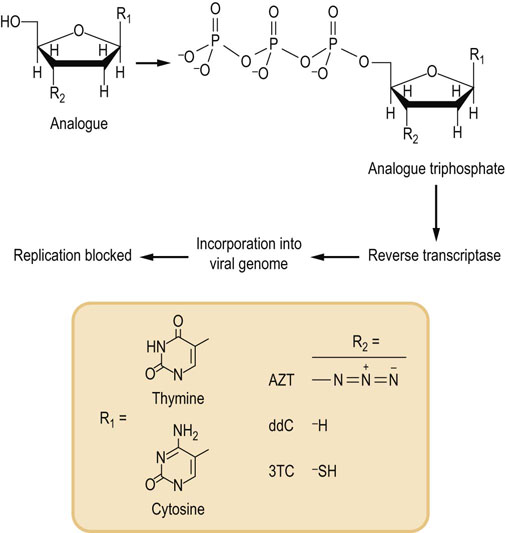
Fig. 32.7 Mechanism of action of antiretroviral chemotherapeutic agents.
This class of inhibitors includes several compounds with slightly different chemical structures in the nucleobase structure and in substitution at the 3′-carbon of the sugar ring. Structures of some of the most widely used drugs are shown. These compounds are metabolized to the triphosphate form via normal cellular metabolism (see Chapter 30). The triphosphate analogues are then incorporated into the viral genome by reverse transcriptase. This blocks viral DNA synthesis because the modified 3′ end R2 of the viral DNA molecule is not a substrate for additional rounds of DNA synthesis. AZT, azido-2′,3′-dideoxythymidine; ddC, 2′,3′-dideoxycytidine; 3TC, 2′,3′-dideoxy-3′-thiacytidine.
If antiretroviral therapy is incorrectly employed, these multi-drug resistant strains can turn out to be the dominant genotypes very promptly. Improper serial use of the reverse transcriptase inhibitors such as zidovudine, didanosine, zalcitabine, stavudine, and lamivudine may result in multi-drug resistant mutations.
DNA can be damaged by numerous types of endogenous and exogenous agents that cause nucleotide modifications, deletions, insertions, sequence inversions and transpositions. Some of this damage is secondary to chemical modification of DNA by alkylating agents (including many carcinogens), reactive oxygen species (Chapter 37) and ionizing radiation (ultraviolet or radioactive). Both the sugar and bases of DNA are subject to modification, yielding an estimated 10,000 to 100,000 modifications of DNA per cell per day. The nature of this damage is quite variable, including modification of single bases, single or double-strand breaks, and crosslinking between bases or bases and proteins. Oxidative damage is probably the most common form of DNA damage; it is increased in inflammation, by smoking, in aging and in age-related diseases, including atherosclerosis, diabetes and neurodegenerative diseases (Chapter 43). If not repaired, the accumulated damage will lead to permanent changes in the structure of DNA, setting the stage for loss of cellular functions, cell death or cancer.
Multiple enzymatic pathways repair a wide range of chemical modifications of DNA
Numerous chemical and environmental agents are known that produce specific chemical modification of the nucleotides in the DNA strand, leading to mismatches during DNA synthesis. After chromosomal replication, the resulting daughter strand contains a different DNA sequence (mutation) from the parent strand. Cells use excision repair to remove alkylated nucleotides and other unusual base analogues, thereby protecting the DNA sequence from mutations. The unmodified strand serves as the template for the repair process.
UV light produces thymine dimers: nucleotide excision repair
When short-wavelength ultraviolet (UV) light interacts with DNA, adjacent thymine bases undergo an unusual dimerization, producing a cyclobutylthymine dimer in the DNA strand (Fig. 32.8). The primary mechanism for repair of these intrastrand thymine dimers is an excision repair mechanism. An endonuclease, which appears to be specific for this type of modification, cleaves the dimer-containing strand near the thymine dimer, and a small portion of that strand is removed. DNA polymerase I, the same enzyme that is involved in DNA biosynthesis, then recognizes and fills in the resulting gap. DNA ligase completes the repair by rejoining the DNA strands.
Deamination: excision repair
Those nucleotides that contain amines, cytosine and adenosine, may spontaneously deaminate to form uracil or hypoxanthine, respectively. When these bases are found in DNA, specific N-glycosylases remove them. This produces base pair gaps that are recognized by specific apurinic or apyrimidinic endonucleases that cleave the DNA near the site of the defect. An exonuclease then removes the stretch of the DNA strand containing the defect. A repair DNA polymerase replaces the DNA and, finally, DNA ligase rejoins the DNA strand. This repair mechanism is also referred to as excision repair.
Depurination
Single base pair alterations also include depurination. The purine-N-glycosidic bonds are especially labile, so that an estimated 3–7 purines are removed from DNA per min per cell. Specific enzymes recognize these depurinated sites, and the base is replaced without interruption of the phosphodiester backbone.
Strand breaks
Single-stranded breaks are frequently induced by ionizing radiation. These are repaired by direct ligation or by excision repair mechanisms. Double-stranded breaks are produced by ionizing radiation and some chemotherapeutic agents. Otherwise, double-stranded ends of DNA are rare in vivo; they are found at the end of chromosomes and in some specialized complexes involved in gene rearrangement. A specialized enzyme system is designed to recognize and rejoin these ends but if the ends drift away from one another, the damage is not readily repaired.
Clinical box Xeroderma pigmentosum
Xeroderma pigmentosum (XP) is a group of rare, life-threatening, autosomal recessive disorders (incidence = 1/250,000) that are marked by extreme sensitivity to sunlight. Upon exposure to sunlight or ultraviolet radiation, the skin of XP patients erupts into pigmented spots, resembling freckles. Multiple carcinomas and melanomas appear early in life, exacerbated by sun exposure, and the majority of patients succumb to cancer before reaching adulthood.
XP is the result of a defect in repair of UV-induced thymine dimers in DNA. There are at least eight polypeptides (genes) involved in recognition, unwinding and excision repair of UV-induced thymine dimers. Patients with XP must avoid direct sunlight, fluorescent light, halogen light or any other source of ultraviolet light. An experimental form of protein therapy, currently undergoing clinical evaluation, involves application of a skin lotion containing the missing protein or enzyme. Ideally, this protein will enter the skin cells and stimulate the repair of UV-damaged DNA. However, protection occurs only where the lotion can be applied. For example, this treatment does not address the neurologic problems that affect about 20% of XP patients.
Mismatch repair
Errors that escape the proofreading activity of DNA polymerase III appear in newly synthesized DNA in the form of nucleotide mismatches. While readily repairable, the critical issue is identification of the strand to be repaired: which nucleotide strand is the daughter strand containing the error? In bacterial systems, mismatch repair is accomplished by post-replicative methylation of DNA at adenine residues in specific sequences spaced along the genome; methylation does not affect base pairing. Newly synthesized strands lack methylated adenine residues, so that the mismatch repair system enzymes scan the DNA, identify the mismatch, and then repair the unmethylated strand by excision repair. A similar approach is used to correct mismatches occurring during synthesis of mammalian DNA. Defects in mismatch repair are associated with hereditary nonpolyposis colon cancer, an autosomal dominant condition in humans.
8-Oxo-2′-deoxyguanosine
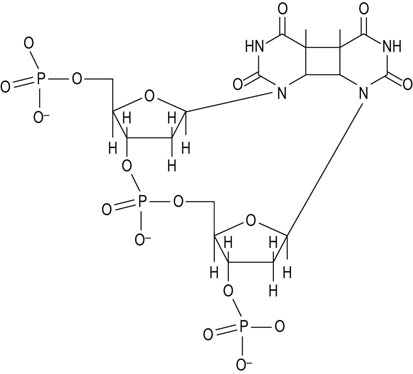
More than 20 different oxidative modifications of DNA have been characterized; the most studied is 8-oxo-2′-deoxyguanosine (8-oxoG) (Fig. 32.9). During the process of DNA replication, mismatches between the modified 8-oxoG nucleoside in the template strand and incoming nucleotide triphosphates result in G-to-T transversions, thereby introducing mutations into the DNA strand. Although excision repair mechanisms are effective, 8-oxoG, like other modified bases, may be reincorporated into DNA following excision.
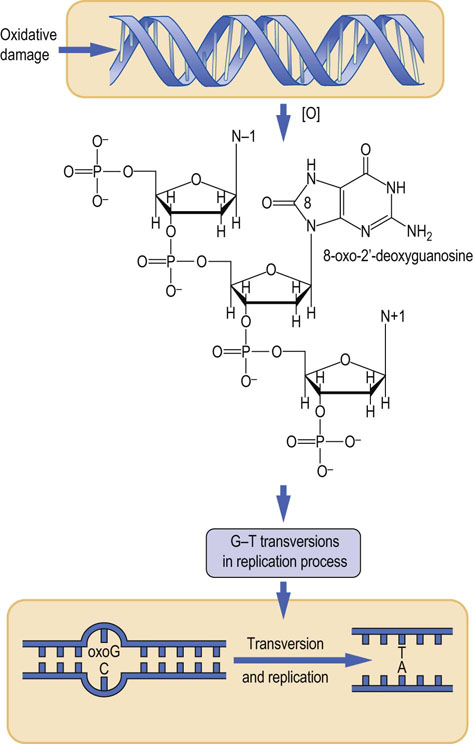
Fig. 32.9 Oxidative damage to DNA.
8-Oxo-2′-deoxyguanosine (oxoG) is an oxidative modification of DNA that causes mutations during replication of DNA. Replication of the strand containing oxoG frequently yields a pyrimidine A in the complementary strand, which, on further replication, yields an AT base pair, instead of the original GC base pair.
Recently, a mammalian protein, MTH1, was characterized that specifically degrades 8-oxo-dGTP, thereby preventing misincorporation of this altered nucleotide into DNA. Gene targeting was used to develop an MTH1 knockout mouse. Compared to the wild-type animal, the knockout showed a greater number of tumors in lung, liver, and stomach, illustrating the importance of this (and other) postrepair protection mechanisms.
In lung cells, inhalation of some particulate materials results in an increase in 8-oxoG levels. The inflammatory process may play a role in asbestos-induced formation of lung tumors. Smoking also induces oxidative damage and increases levels of DNA oxidation products in lungs, blood and urine. 8-Oxo-2′-deoxyguanosine is eliminated via renal filtration. Therefore, its urinary level is used as a sensitive biomarker for oxidative stress in many clinical studies (Chapter 43). A new competitive immunochromatography automatic analyzer that measures urinary 8-oxodG can be used as a point-of-care test for the assessment of oxidative stress.
 Clinical test box Ames test for mutagens
Clinical test box Ames test for mutagens
Mutagens are chemical compounds that induce changes in the DNA sequence. A large number of natural and man-made chemicals are mutagenic. To evaluate the potential to mutate DNA, the American biochemist Bruce Ames developed a simple test, using special Salmonella typhimurium strains that cannot grow in the absence of histidine (His- phenotype). These histidine auxotrophic strains contain nucleotide substitutions or deletions that prevent the production of histidine biosynthetic enzymes.
To test for mutagenesis, mutant bacteria are seeded on a culture medium lacking histidine; the suspected mutagen is added to the medium. The action of the mutagen occasionally results in the reversal of the histidine mutation, yielding a revertant strain that can now synthesize histidine and will grow in its absence. The mutagenicity of a compound is scored by counting the number of colonies that have grown, i.e. reverted to the His+ phenotype. There is a good correlation between results of the Ames mutagenicity test and direct tests of carcinogenic activity in animals.
Some chemicals (procarcinogens) are not mutagenic per se, but are activated to mutagenic compounds during metabolic processes, e.g. during drug detoxification in liver or kidney. Benzopyrene, for example, is not mutagenic but during its detoxification in liver, it is converted to diol epoxides which are potent mutagens and carcinogens. To provide sensitivity for detecting procarcinogens, the culture medium for the Ames test is supplemented with an extract of liver microsomes, a subfraction of tissue rich in smooth endoplasmic reticulum containing drug-metabolizing enzymes.
Clinical box Cancer treatment news: two new protein targets to counteract ‘relapse’ and ‘drug resistance’
Current cancer treatments such as ionizing radiation and chemotherapy target DNA. Their rationale is clear: these treatments disrupt the genome, and a balance is struck between preventing cancer cells from dividing and proliferating, while not irreversibly damaging healthy tissues. Nonetheless, cancer cells have a broad array of DNA repair mechanisms to limit injury. For this reason, DNA repair systems are targets of adjuvant therapy used to enhance sensitivity of cancer cells to DNA-targeted agents. Base excision repair and nucleotide excision repair are key mechanisms of DNA repair. There are two protein targets associated with the hallmark ‘relapse’ and ‘drug resistance’ phenomena seen during chemotherapy: Excision Repair Cross-Complementation Group 1 (ERCC1) and DNA polymerase beta. The former is a key player in nucleotide excision repair; the latter is the error-prone polymerase of base excision repair. Only a few ERCC1 inhibitors have been discovered, but more than 60 for DNA pol beta. The discovery of potent and tumor-specific inhibitors of these enzymes should improve current therapies where resistance develops, including bleomycin, alkylating agents and cisplatin.
Summary
The human genome is composed of DNA, an antiparallel, double-stranded helical polymer of deoxyribonucleotides, stabilized by hydrogen binding between complementary bases.
DNA is packaged in the chromosome in a highly organized, condensed structure known as chromatin.
Genetic information is replicated by a semi-conservative mechanism in which the parental strands are separated and both act as templates for daughter DNA.
The replication of DNA is a complex, stringently regulated process. DNA is essentially the only polymer in the body that is repaired, rather than degraded, following chemical or biological modification. Repair mechanisms generally involve excision of modified bases and replacement, using the unmodified strand as a template.
Frias, C, Pampalona, J, Genesca, A, et al. Telomere dysfunction and genome instability. Front Biosci. 2012; 17:2181–2196.
Kaneko, K, Kimata, T, Tsuji, S, et al. Measurement of urinary 8-oxo-7,8-dihydro-2-deoxyguanosine in a novel point-of-care testing device to assess oxidative stress in children. Clin Chim Acta. 2012; 413:1822–1826.
Ruemmele, FM, Garnier-Lengline, H. Why are genetics important for nutrition? Lessons from epigenetic research. Ann Nutr Metab. 2012; 60(Suppl 3):38–43.
Slijepcevic, P. DNA damage response, telomere maintenance and ageing in light of the integrative model. Mech Ageing Dev. 2008; 129:11–16.
Spry, M, Scott, T, Pierce, H, et al. DNA repair pathways and hereditary cancer susceptibility syndromes. Front Biosci. 2007; 12:4191–4207.
Travers, AA, Muskhelishvili, G, Thompson, JM. DNA information: from digital code to analogue structure. Philos Transact A Math Phys Eng Sci. 2012; 370:2960–2986.
Venter, JC. Genome sequencing anniversary: the human genome at 10 – successes and challenges. Science. 2011; 331:546–547.
Venter, JC, Adams, MD, Myers, EW. The sequence of the human genome. Science. 2001; 291:1304–1351.
Watson, JD. The double helix: a personal account of the discovery of the structure of DNA. New York: WW Norton; 1980.
Watson, JD. A passion for DNA. Genes, genomes and society. Oxford: Oxford University Press; 2000.
DNA interactive. www.dnai.org. [This is the best introduction to the subject].
DNA replication. http://www.freesciencelectures.com/video/dna-replication-process/.
DNA workshop. www.pbs.org/wgbh/aso/tryit/dna.
Graphics. www.accessexcellence.org/AB/GG/structure.html.
Mitochondrial DNA. www.mitomap.org.
National Human Genome Research Institut. www.genome.gov