Complex Carbohydrates
Glycoproteins
Learning objectives
After reading this chapter you should be able to:
 Describe the general structures of oligosaccharides (glycans) in various types of glycoproteins.
Describe the general structures of oligosaccharides (glycans) in various types of glycoproteins.
Outline the sequence of reactions involved in biosynthesis and processing of N-glycans to produce the various types of oligosaccharide chains.
Describe the role of N-glycans in protein folding, stability and cell–cell recognition.
Explain the importance of O-glycans in mucin function.
Describe how each of the monosaccharides involved in biosynthesis of N- and O-glycans is synthesized from glucose and activated for synthesis of glycoconjugates.
Distinguish lectins from other types of proteins and describe their role in physiology and pathology.
Describe several diseases that are associated with deficiencies in enzymes involved in synthesis, modification or degradation of complex carbohydrates.
Introduction
Glycoconjugates include glycoproteins, proteoglycans and glycolipids
Many mammalian proteins are glycoproteins, i.e. they contain sugars covalently linked to specific amino acids in their structure. There are two major types of sugar-containing proteins that occur in animal cells, generally referred to as glycoproteins and proteoglycans. Along with glycolipids, which are presented in the next chapter, all of these compounds are part of the group of sugar-containing macromolecules called glycoconjugates.
Glycoproteins (Fig. 27.1A) have short glycan chains; they can have up to 20 sugars but usually contain between three and 15 sugars. These oligosaccharides are highly branched, they do not have a repeating unit, and they usually contain amino sugars (N-acetylglucosamine or N-acetylgalactosamine), neutral sugars (D-galactose, D-mannose, L-fucose) and the acidic sugar sialic acid (N-acetylneuraminic acid). Glycoproteins generally do not contain uronic acids, acidic sugars that are a major part of the proteoglycans. Glycoproteins usually contain much smaller amounts of carbohydrate than of protein, typically from just a few percent carbohydrate to 10–15% sugar by weight.
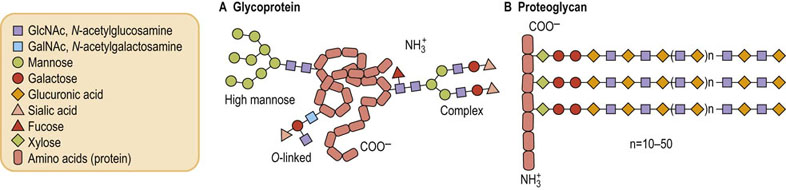
Proteoglycans (see Fig. 27.1B and Chapter 29) contain as much as 50–60% carbohydrate. In these molecules, the sugar chains are long, unbranched polymers that may contain hundreds of monosaccharides. These saccharide chains have a repeating disaccharide unit, generally made up of a uronic acid and an amino sugar.
Most proteins in cell surface membranes that function as receptors for hormones or participate in other important membrane-associated processes such as cell–cell interactions are glycoproteins. Many of the membrane proteins of the endoplasmic reticulum or Golgi apparatus, as well as those proteins that are secreted from cells, including serum and mucous proteins, are also glycoproteins. In fact, glycosylation is the major enzymatic modification of proteins in the body. Addition of sugars to a protein can occur either at the same time, and location, as protein synthesis is occurring in the endoplasmic reticulum, i.e. co-translationally, or following the completion of protein synthesis and after the protein has been transported to the Golgi apparatus, i.e. posttranslationally. The functions of the carbohydrate chains of the resulting glycoproteins are diverse (Table 27.1).

Structures and linkages
Sugars are attached to specific amino acids in proteins
Sugars can be attached to protein in either N-glycosidic linkages or O-glycosidic linkages. N-linked oligosaccharides (N-glycans) are widespread in nature and are characteristic of membrane and secretory proteins. The attachment of these oligosaccharides to protein involves a glycosylamine linkage between an N-acetylglucosamine (GlcNAc) residue and the amide nitrogen of an asparagine residue (Fig. 27.2A). The asparagine that serves as an acceptor of this oligosaccharide must be in the consensus sequence Asn-X-Ser (Thr) in order to be recognized as the acceptor by the oligosaccharide transferring enzyme (see below). However, not all asparagine residues, even those in this consensus sequence, become glycosylated, indicating that other factors such as protein conformation or other properties of the protein may be involved.
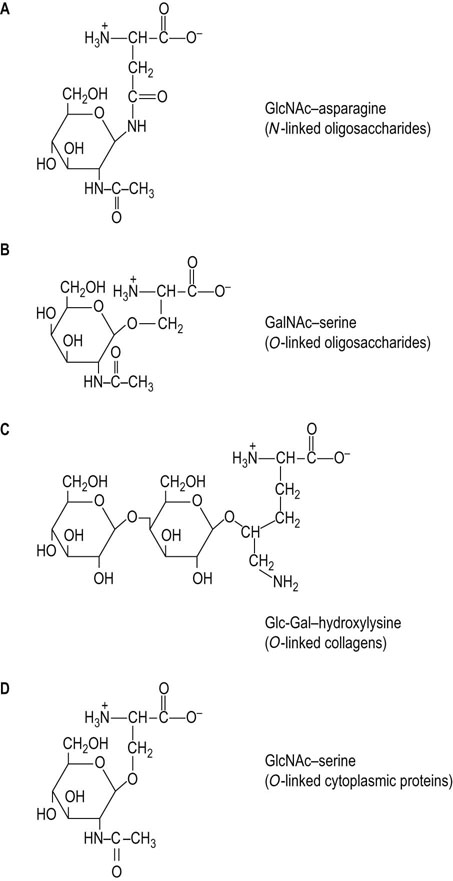
O-linked oligosaccharides (O-glycans) are most commonly found in proteins of mucous fluids, but also occur frequently on the same membrane and secretory proteins that contain the N-glycans. The O-glycans typically contain three or more sugars in linear or branched chains, attached to protein by a glycosidic linkage between an N-acetylgalactosamine (GalNAc) residue and the hydroxyl group of either a serine or threonine residue on the protein (Fig. 27.2B; see Fig. 27.4). There does not appear to be a consensus sequence of amino acids for O-linked glycosylation, so it is not known why some serines (or threonines) become glycosylated but others do not.
A glucosyl-galactose disaccharide is frequently linked to the hydroxyl group of hydroxylysine residues in the fibrous protein, collagen (Fig. 27.2C). Hydroxylysine is an uncommon amino acid, found only in collagens and proteins with collagenous domains. Hydroxylysine is not directly incorporated into protein as such, but is produced by posttranslational hydroxylation of lysine residues. Lysine hydroxylase requires vitamin C as a cofactor, which is why vitamin C is frequently used to expedite wound healing. Collagen is first synthesized in the cell as a precursor form called procollagen. Procollagens are usually synthesized as N-linked glycoproteins, but the N-glycan is removed as part of that peptide that is cleaved from procollagen during its maturation to collagen. Only the O-linked disaccharides remain on the mature collagen molecule. The less glycosylated collagens tend to form ordered, fibrous structures, such as occur in tendons, while the more heavily glycosylated collagens are found in meshwork structures, such as basement membranes in the vascular wall and renal glomerulus (see Chapter 29).
A single GlcNAc is attached to the hydroxyl group of serine or threonine residues on a number of cytoplasmic and nuclear proteins (Fig. 27.2D). This O-linked GlcNAc (O-GlcNAc) is linked to specific serine and threonine residues that become phosphorylated by protein kinases during hormonal stimulation or other signaling events. The enzyme that adds the GlcNAc is widespread, but how it is controlled is still not clear. There is a second enzyme that removes the GlcNAc from the serine and threonine residues, similar to the contrasting regulatory roles of protein kinases and phosphatases. The GlcNAc modification (O-GlcNAcylation) may represent a mechanism that allows the cells to block phosphorylation of specific serine and threonine residues on selected proteins, while allowing others to still be phosphorylated. Then, the GlcNAc can be removed under appropriate conditions to allow phosphorylation. The donor substrate of O-GlcNAcylation is UDP-GlcNAc (see below), which is derived from glucose. O-GlcNAcylation is dependent on the intracellular UDP-GlcNAc level, which is correlated with extracellular glucose concentration. O-GlcNAcylation of proteins involved in the insulin signaling pathway brings about insulin resistance in skeletal muscle, adipose tissue and pancreatic β-cells, which causes type 2 diabetes. O-GlcNAcylation also regulates transcription factors as well as the proteasome, which is involved in protein turnover (Chapter 34).
A novel class of O-linked glycans in which mannose is linked to serine or threonine residues is found on the muscle and nerve-specific protein dystroglycan. A typical O-mannose glycan consists of GlcNAc, galactose and sialic acid, which are attached to the core mannose in this order. O-mannose glycans serve as linkers between the intracellular cytoskeleton and the extracellular matrix to maintain myocyte function. In fact, deficiency in the biosynthesis of O-mannose glycans causes muscular dystrophy.
One other amino acid that can serve as a site for glycosylation is tyrosine. The only example of this linkage is in the protein glycogenin, found at the core of glycogen (see Chapter 13). Glycogenin is a self-glucosylating protein that initially attaches a glucose to the hydroxyl group of one of its tyrosine residues. The protein then adds a number of other glucoses to the protein-linked glucose to make an oligosaccharide which serves as the acceptor for glycogen synthase.
N-glycans have either ‘high-mannose’ or ‘complex’ structures built on a common core
Although there are a large number of different carbohydrate structures produced by living cells, most of the oligosaccharides on glycoconjugates have many sugars and glycosidic linkages in common. All N-glycans have oligosaccharide chains that are branched structures, having a common core of three mannose residues and two GlcNAc residues (Fig. 27.3A and B), but differing considerably beyond the core region to give high-mannose and complex types of chains. The reason for this similarity in structure is that the high-mannose type of N-linked oligosaccharide is the biosynthetic precursor for all other N-glycans. As indicated below (see Fig. 27.11), the oligosaccharide is initially assembled on a carrier lipid in the endoplasmic reticulum as a high-mannose structure, then transferred to protein. It may remain as a high mannose structure, especially in lower organisms, but in animals the oligosaccharide undergoes a number of processing steps in the endoplasmic reticulum and Golgi apparatus that involve removal of some mannoses and addition of other sugars. As a result, beyond the core region, the oligosaccharides give rise to a vast array of structures that are referred to as high-mannose (Fig. 27.3A), and complex chains (Fig. 27.3B).
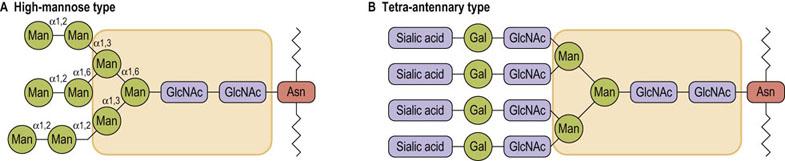
Fig. 27.3 Typical structures of high-mannose and complex, N-linked oligosaccharides.
The core structure (shaded area) is common to both structures. Asn, asparagine; Gal, galactose; GlcNAc, N-acetylglucosamine Man, mannose.
Complex oligosaccharides are so named because of their more complex sugar composition, including galactose, sialic acid and L-fucose. Complex chains have terminal trisaccharide sequences composed of sialic acid-galactose-GlcNAc attached to each of the branched core mannoses (Fig. 27.3A). L-Fucose may also be found attached to the core GlcNAc (Fig. 27.1A), the terminal galactose (Fig. 28.12) or the penultimate GlcNAc (Fig. 27.9). In common with sialic acid, fucose is usually a terminal sugar on oligosaccharides; that is, no other sugars are attached to it. Some of the complex oligosaccharides have two trisaccharide sequences, one attached to each of the branched core mannoses, and are therefore called biantennary complex chains, whereas others have three (triantennary) or four (tetra-antennary) of the trisaccharide structures (Fig. 27.3B). More than 100 different complex oligosaccharide structures have now been identified on various cell surface proteins, providing great diversity (microheterogeneity) as mediators of cellular recognition and chemical signaling events.
General structures of glycoproteins
A glycoprotein may have a single N-glycan chain or it may have several of these types of oligosaccharides. Furthermore, the N-glycans may all have identical structures or they may be quite different in structure. For example, the influenza virus coat glycoproteins, hemagglutinin and neuraminidase, are both glycoproteins which usually have seven N-glycan chains, of which five are biantennary complex chains and two are high-mannose structures. Thus, a range of related structures is commonly found in a single glycoprotein and, in fact, multiple different structures may also be found at a single site on a glycoprotein. Microheterogeneity of oligosaccharide structures results from incomplete processing on some chains during their biosynthesis (see Fig. 27.12). As a result, some of the oligosaccharides on a glycoprotein may be complete complex chains whereas others may be only partially processed. Such diversity in the processing of N-glycans is controlled by many factors. The protein structure near the N-glycosylation site can have some influence on the processing. In general, N-glycans exposed on the surface of a folded protein are vulnerable to the processing enzymes, whereas those shielded within the structure of the protein are inaccessible to the enzymes.
Many N-linked glycoproteins also contain O-glycans of the type shown in Figure 27.4. The number of O-glycans varies considerably depending on the protein and its function. For example, the low-density lipoprotein (LDL) receptor is present in plasma membranes of smooth muscle cells and fibroblasts, and functions to bind and endocytose circulating LDL, as a source of dietary cholesterol. The LDL receptor has two N-glycans that are located near the LDL-binding domain, and a cluster of O-glycans near the membrane-spanning region. As shown in Figure 27.5, this receptor has a membrane-spanning region of hydrophobic amino acids, an extended region of amino acids on the external side of the plasma membrane that contains the cluster of O-glycans, and a functional domain that is involved in binding LDL (see Chapter 18). Although the two N-glycans are near the functional domain, they do not play a role in binding LDL. Instead, their function appears to be in helping the protein to fold into the proper conformation in the endoplasmic reticulum so that it can be translocated to the Golgi apparatus. The negatively charged O-glycans, each having a sialic acid, are believed to function to keep the protein in an extended state, and to prevent it from folding back on itself.
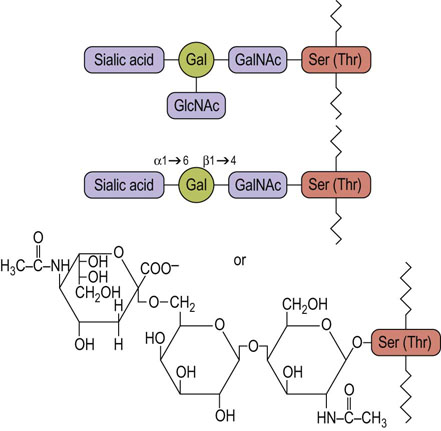
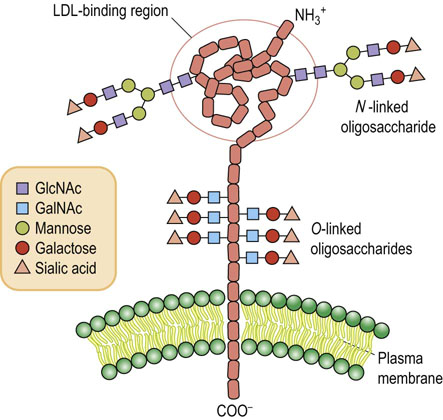
Fig. 27.5 Model of the low-density lipoprotein (LDL) receptor.
(See also Chapter 18 and compare Fig. 18.2.)
 Advanced concept box The structure of N-glycans depends on the enzyme complement of the cell
Advanced concept box The structure of N-glycans depends on the enzyme complement of the cell
The final structure of the N-glycan chain of a given glycoprotein is not coded in the genes for the proteins, but depends on the enzyme complement of the cell making that oligosaccharide. All cells appear to have the necessary enzymes to produce the lipid-linked saccharide precursor of the N-linked high-mannose chains, and can therefore glycosylate any membrane protein that has the appropriate asparagine in the right sequence and protein conformation. However, the glycosyltransferases and glycosidases involved in processing the oligosaccharide to its final complex structure are not so widely distributed, and a given glycosyltransferase may be present in one type of cell but not in another. For example, one cell type may have the GlcNAc transferase (GlcNAc T-IV or V) necessary to attach a second GlcNAc on the 2-linked α-mannoses to make a triantennary or tetra-antennary chain, whereas another cell may not have these GlcNAc transferases. Such a cell will only make biantennary chains. Enveloped viruses, such as the influenza virus or HIV, are examples of this phenomenon, since their N-glycan structures reflect that of the cell in which they are grown; viruses use the cellular machinery to make all of their structures, and therefore their glycoproteins will have carbohydrate structures characteristic of the infected cell. For the virus this is beneficial since their proteins will not be recognized as foreign proteins and will escape immune surveillance. In addition, it allows the virus to attach to host cell receptors and fuse with host cell membranes by interacting with host lectins. In the biotechnology industry, this means that, although a given protein will have the identical amino acid sequence regardless of cell type, it will have different oligosaccharide structures, depending on the cell in which it is expressed. These differences in carbohydrate structure may affect the conformation and functional properties of the protein and limit its use for protein or enzyme replacement therapy. In fact, many cells used to express ‘human’ proteins are bioengineered to contain the complement of enzymes needed to properly glycosylate the target protein.
Structure–function relationships in mucin glycoproteins
Mucins are glycoproteins that are secreted by epithelial cells lining the respiratory, gastrointestinal and genitourinary tracts. These proteins are very large in size with subunits having molecular weights of over one million daltons, and having as much as 80% of their weight as carbohydrate. Mucins are uniquely designed for their function, with about one-third of the amino acids being serines or threonines, and most of these being substituted with an O-glycan. Because most of these oligosaccharides carry a negatively charged sialic acid and these negative charges are in close proximity, they repel each other and prevent the protein from folding, causing it to remain in an extended state. Thus, the protein solution is highly viscous, forming a protective barrier on the epithelial surface, providing lubrication between surfaces and facilitating transport processes, such as the movement of food through the gastrointestinal system. There is a wide range of complex linear and branching oligosaccharide structures on mucins, including blood group antigens (see Chapter 28). Some oligosaccharides participate in interaction and binding to various bacterial cell surfaces. This property may play a significant role in bacterial sequestration and elimination, limiting colonization and infection.
Interconversions of dietary sugars
Cells can use glucose to make all the other sugars they need
Humans have a dietary requirement for some essential fatty acids and amino acids and vitamins (Chapter 11), but all the sugars that they need to make glycoconjugates can be synthesized from blood sugar, i.e. D-glucose. Figure 27.6 presents an overview of the sequence of reactions involved in interconversion of sugars in mammalian cells. All these sugar interconversion reactions involve sugar phosphates or sugar nucleotides.
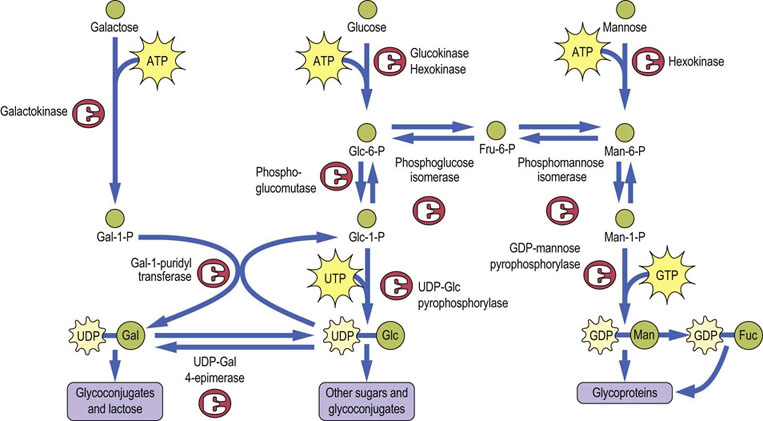
Formation of galactose, mannose and fucose from glucose
Glucose is phosphorylated by hexokinase (or glucokinase in liver) as it enters the cell. Glucose-6-phosphate (Glc-6-P) can then be converted by a mutase (phosphoglucose mutase) to form Glc-1-P, which reacts with uridine triphosphate (UTP) to form UDP-Glc, catalyzed by the enzyme UDP-Glc pyrophosphorylase (Fig. 27.6). This enzyme is a pyrophosphorylase, named for the reverse reaction in which the phosphate of Glc-1-P acts to cleave the pyrophosphate bond of UTP to form UDP-Glc and pyrophosphate (PPi); cleavage of the high energy bond of PPi by pyrophosphatase provides the driving force for the reaction. This is the same pathway used in liver and muscle for incorporation of glucose into glycogen. UDP-Glc is epimerized to UDP-galactose (UDP-Gal) by UDP-Gal 4-epimerase, providing UDP-Gal for synthesis of glycoconjugates.
Glc-6-P may also be converted to fructose-6-phosphate (Fru-6-P) by the glycolytic enzyme phosphoglucose isomerase, and Fru-6-P may be isomerized to mannose-6-phosphate (Man-6-P) by phosphomannose isomerase. The Man-6-P is then converted by a mutase (phosphomannose mutase) to Man-1-P, which condenses with GTP to form GDP-Man. GDP-Man is the form of mannose that is used in the formation of N-linked oligosaccharides and is also the precursor for the formation of activated fucose, GDP-L-fucose. Mannose is present in our diet in small amounts but it is not an essential dietary component, since it can be readily produced from glucose. However, dietary mannose can be phosphorylated by hexokinase to Man-6-P, and then enters metabolism through phosphomannose isomerase.
Metabolism of galactose
Although normal animal cells can make all the galactose they need from glucose, galactose is still an important component of our diet because it is one of the sugars that make up the milk disaccharide, lactose. The pathway of galactose metabolism requires three enzymes (see Fig. 27.6). Dietary galactose is transported to the liver where it is phosphorylated by a specific kinase, galactokinase, that attaches phosphate to the hydroxyl group on carbon-1, rather than carbon-6, to form galactose-1-phosphate (Gal-1-P). Humans lack a UDP-Gal pyrophosphorylase, so the conversion of Gal-1-P to Glc-1-P involves the participation of a sugar nucleotide, UDP-Glc. The enzyme Gal-1-P uridyltransferase catalyzes an exchange between UDP-Glc and Gal-1-P to form UDP-Gal and Glc-1-P (see Fig. 27.6). The UDP-Gal is used for synthesis of glycoconjugates, while the Glc-1-P can be converted to Glc-6-P by phosphoglucomutase, and thus the original galactose molecule enters glycolysis.
UDP-Glc is present in only micromolar concentrations in cells so that its availability for galactose metabolism would be quickly exhausted were it not for the presence of a third enzyme, UDP-Gal-4-epimerase. This enzyme catalyzes the equilibrium between UDP-Glc and UDP-Gal, providing a constant source of UDP-Glc during galactose metabolism. The reactions catalyzed by (1) galactokinase, (2) Gal-1-P uridyltransferase, and (3) UDP-Gal 4-epimerase are summarized below, illustrating the roundabout way by which galactose enters mainstream metabolic pathways.
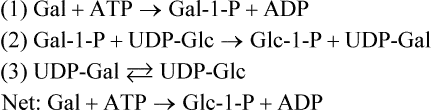
Metabolism of fructose
Fructose is a component of sucrose and also accounts for about half the sugar in high fructose corn syrup
Fructose can be metabolized by two pathways in cells as shown in Figure 27.7. It can be phosphorylated by hexokinase, an enzyme that is present in all cells; however, hexokinase has a strong preference for glucose as a substrate, and glucose, which is present at a concentration of about 5 mmol/L in blood, is a strong competitive inhibitor of the phosphorylation of fructose. The primary pathway of fructose metabolism, which is especially important in liver after a meal, involves the enzyme fructokinase. This enzyme is a very specific kinase that phosphorylates fructose at carbon-1 (rather than the 6-position like hexokinase) to give fructose-1-phosphate (Fru-1-P). The liver aldolase is called aldolase B, and it is different in substrate specificity from the muscle aldolase A, because aldolase B can cleave both Fru-1-P and Fru-1,6-P2, whereas aldolase A will only cleave Fru-1,6-P2. Thus, in liver, the products of fructose cleavage by aldolase B are dihydroxyacetone phosphate and glyceraldehyde (not glyceraldehyde-3-P).The glyceraldehyde must then be phosphorylated by triose kinase in order to be metabolized in glycolysis.
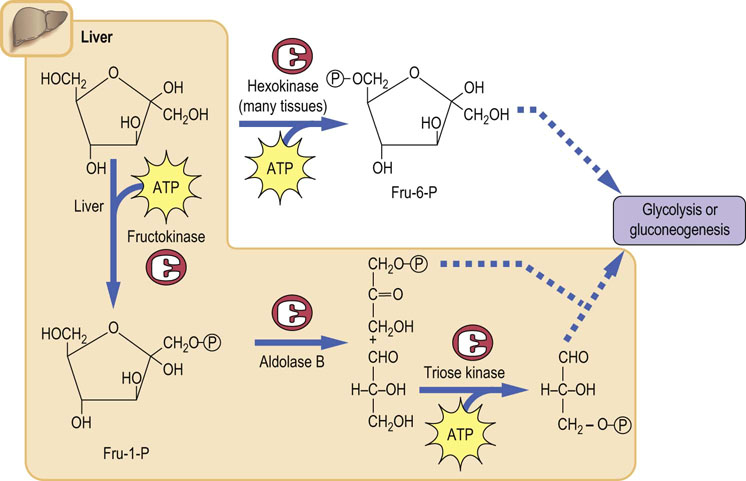
It should be noted that in the liver, fructose enters glycolysis at the level of the triose phosphate intermediates, rather than as Fru-6-P as in muscle. Thus, in liver, the ingested fructose is not subject to regulation by the usual control points for regulatory enzymes, hexokinase and phosphofructokinase. By circumventing these two rate-limiting steps, fructose provides a rapid source of energy in both aerobic and anaerobic cells. This is part of the rationale behind the development of high-fructose drinks such as Gatorade®. The significance of the fructokinase, as opposed to the hexokinase, pathway of fructose metabolism is indicated by the pathology of hereditary fructose intolerance (see Clinical Box below).
Advanced concept box Biosynthesis of lactose
Lactose synthase and α-lactalbumin.
Lactose (galactosyl-β1,4-glucose) is synthesized from UDP-Gal and glucose in mammary glands during lactation. Lactose synthase is formed by the binding of α-lactalbumin to the galactosyl transferase that normally participates in biosynthesis of N-linked glycoproteins. α-Lactalbumin, which is expressed only in the mammary glands during lactation, converts galactosyl transferase to lactose synthase by lowering the enzyme's Km for glucose by about three orders of magnitude, from 1 mol/L to 1 mmol/L, leading to preferential synthesis of lactose, rather than galactosylation of glycoproteins. α-Lactalbumin is the only known example of a ‘specifier’ protein that alters the substrate specificity of an enzyme.
 Clinical box Galactosemia: a baby who developed jaundice after breast-feeding
Clinical box Galactosemia: a baby who developed jaundice after breast-feeding
An apparently normal newborn baby began to vomit and develop diarrhea after breast-feeding. These problems, together with dehydration, continued for several days, when the child began to refuse food and developed jaundice indicative of liver damage, followed by hepatomegaly and then lens opacifications (cataracts). Measurement of glucose in the blood by a glucose oxidase assay (Chapter 3) indicated that the concentration of glucose was low, consistent with failure to absorb foods. However, glucose measured by a colorimetric method that measures total reducing sugar (i.e. any sugar that is capable of reducing copper) indicated that the concentration of sugar was quite high in both blood and urine. The reducing sugar that accumulated was eventually identified as galactose, indicating an abnormality in galactose metabolism known as galactosemia. This finding was consistent with the observation that, when milk was removed from the diet and replaced with an infant formula containing sucrose rather than lactose, the vomiting and diarrhea stopped, and hepatic function gradually improved.
The accumulation of galactose in the blood most often is the result of a deficiency in the enzyme Gal-1-P uridyl transferase (classic form of galactosemia), which prevents the conversion of galactose to glucose and leads to the accumulation of Gal and Gal-1-P in tissues. The accumulated Gal-1-P interferes with phosphate and glucose metabolism, leading to widespread tissue damage, organ failure and mental retardation. In addition, accumulation of galactose in tissues results in galactose conversion, through the polyol pathway, to galactitol, which in the lens results in osmotic stress and development of cataracts (compare diabetic cataracts, Chapter 21). Another form of galactosemia is caused by galactokinase deficiency, but in this case Gal-1-P does not accumulate and complications are milder.
Clinical box Hereditary fructose intolerence: a child who developed hypoglycemia after eating fruit
A child was brought into the emergency room suffering from nausea, vomiting and symptoms of hypoglycemia along with sweating, dizziness, and trembling. The parents indicated that these attacks occurred shortly after eating fruits (fructose) or candy (sucrose). As a result of these symptoms, the child was developing a strong aversion to fruits so the mother was providing a large supplementation of multivitamin preparations. The child was below normal weight, but he had not exhibited any of the above unusual symptoms during the period of time when he was breast-feeding. A series of clinical tests demonstrated some cirrhosis of the liver, and a normal glucose tolerance test. However, reducing substances were detected in the urine and these reducing substances did not react in the glucose oxidase test (i.e. they were not due to glucose). A fructose tolerance test was ordered using 3 g fructose/m2 of body surface area, given intravenously in a single and rapid push. Within 30 minutes, the child displayed symptoms of hypoglycemia. Blood glucose analysis confirmed this and revealed that the hypoglycemia was greatest after 60–90 min. Fructose concentrations reached a maximum (3.3 mmol/L) after 15 min and gradually decreased to zero by 3 h. Plasma phosphate concentration fell by 50% and tests for the enzymes alanine aminotransferase and aspartate aminotransferase indicated that they were elevated after about 90 min. The urine was also positive for fructose.
The results of a fructose tolerance test demonstrate the accumulation of fructose and its derivatives in blood and urine. The elevation of liver enzymes, alanine and aspartate aminotransferase, as well as jaundice and other symptoms indicate liver damage and suggest that Fru-1-P affects metabolism in a manner similar to that of Gal-1-P in galactosemia.
Other pathways of sugar nucleotide metabolism
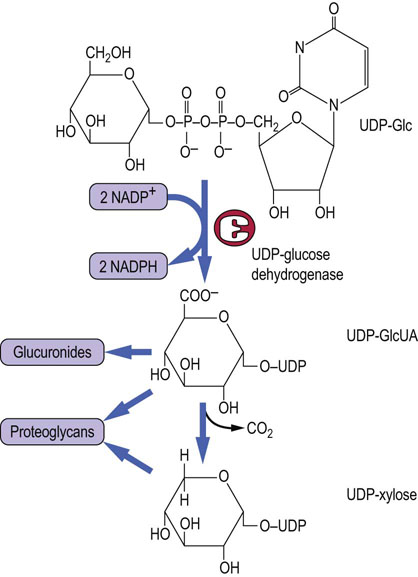
UDP-GlcUA
UDP-Glc is the precursor of a number of other sugars, such as glucuronic acid, xylose and galactose, which are required for proteoglycan and/or glycoprotein synthesis. The reactions that lead to the formation of these other sugars are outlined in Figures 27.6 and 27.8. A two-step oxidation by the enzyme UDP-Glc dehydrogenase leads to the formation of the activated form of glucuronic acid (UDP-GlcUA) (Fig. 27.8). This sugar nucleotide is the donor of glucuronic acid, both for the formation of proteoglycans (see Chapter 29) and for the detoxification and conjugation reactions that occur in the liver to remove bilirubin, drugs and xenobiotics (see Chapter 30). UDP-GlcUA is also the precursor of UDP-xylose, a pentose sugar nucleotide (Fig. 27.8). UDP-GlcUA undergoes a decarboxylation reaction that removes carbon-6 to form UDP-xylose, the activated form of xylose. Xylose is the linkage sugar between protein and glycan in proteoglycans (Figure 27.1B and Chapter 29). Xylose is also present on many plant glycoproteins, as part of their N-linked oligosaccharides, and is partly responsible for allergic reactions to peanut and nut proteins.
GDP-Man and GDP-Fuc
Guanosine diphospate-mannose (GDP-Man) is the donor substrate for most mannosyltransferases. As shown in Figure 27.6, it is produced from Man-6-P and is also the precursor to GDP-L-fucose (GDP-Fuc), which is the donor substrate for all fucosyltranferases. Fucose is a 6-deoxyhexose that is an important sugar participating in many recognition reactions in biological events, such as inflammatory response (Fig. 27.9). The conversion of GDP-Man to GDP-Fuc involves a complex series of oxidative and reductive steps, as well as epimerizations. Deficiency in the GDP-Fuc transporter that translocates GDP-Fuc from the cytosol into the Golgi lumen is associated with a defective inflammatory response and increased susceptibility to infection (Leukocyte adhesion deficiency II: LADII). Resulting depletion of GDP-Fuc in the Golgi lumen blocks the biosynthesis of the leukocyte recognition signal, sialyl Lewis-X structure (see Fig. 27.9).
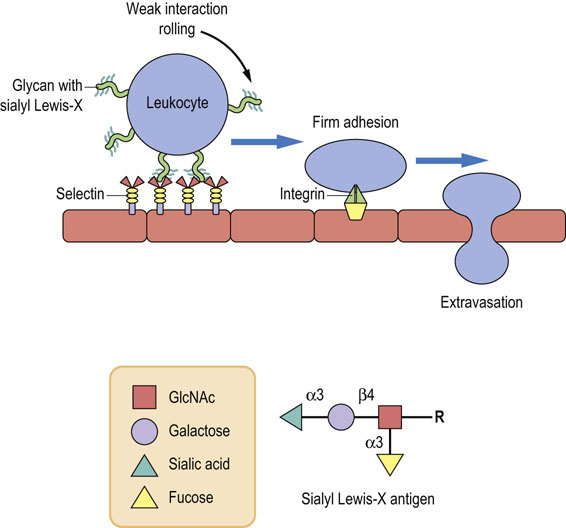
Fig. 27.9 Carbohydrate-dependent cell–cell interactions in inflammation.
Sialyl Lewis-X, a tetrasaccharide antigen that forms part of the membrane structure of leukocytes, is recognized by a carbohydrate-binding protein, selectin (Sel), on the surface of endothelial cells. The sialyl Lewis-X-selectin interaction mediates the initial, weak binding that results in rolling of the leukocytes along the endothelial monolayer. It facilitates the firm adhesion mediated by the protein–protein interactions leading to extravasation.
Advanced concept box Carbohydrate-dependent cell–cell interactions
An important example of carbohydrate-dependent cell–cell interactions occurs during inflammation. An injury, or infection, to the vascular endothelial cells elicits an inflammatory response that causes the release of cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1), from the damaged tissue. These cytokines attract leukocytes to the site of injury or infection to remove the damaged tissue or the invading organisms. Leukocytes must be able to stop or exit from the blood flow and attach to the injured tissue. They are able to do this because they have a carbohydrate ligand on their surface that is recognized by a lectin (carbohydrate-binding protein) that becomes exposed on the surface of the damaged endothelial cells. The carbohydrate ligand is a tetrasaccharide called the sialyl Lewis-X antigen (Fig. 27.9), which is a component of a glycoprotein or glycolipid on the surface of the leukocyte. The sialyl Lewis-X antigen is recognized by lectins, E-selectin and P-selectin, that are expressed on the surface of the endothelial cells by the stimulation of cytokines. Figure 27.9 schematically shows sequential events during vascular adhesion and extravasation of leukocytes to inflamed tissue. The sialyl Lewis-X–selectin interaction mediates the initial step of the leukocytes–endothelium interaction, which is described as tethering, followed by rolling of leukocytes along the endothelial cell surface. Although this carbohydrate–protein binding is weak and transient, it. is able to slow down the leukocytes circulating under a strong shear force in blood and promote the firm protein–protein interactions between the integrins and their receptors. Eventually, the leukocytes migrate through the endothelium into the underlying tissue.
On the other hand, interaction of L-selectin expressed on the lymphocytes with a sialyl Lewis-X-like oligosaccharide expressed on the endothelial cells of the high endothelial venules (HEV) enables lymphocytes circulating in the bloodstream to enter a lymph node by a similar mechanism. This process is called lymphocyte homing.
While these carbohydrate–protein interactions play a critical role in the immune system, they can be dangerous and life threatening under other circumstances. Some cancer cells utilize such a carbohydrate–protein interaction in their metastasis through the bloodstream. There is active research on developing drugs whose structure is similar to carbohydrates (glycomimetic) and will block the vascular adhesion of tumor cells and prevent metastasis.
This example of a protein–carbohydrate interaction is just one of many that occur in vivo. Each of these kinds of interactions involves a different lectin, each of which has a specific carbohydrate recognition site. Once the carbohydrate structure is known and the protein-binding site has been mapped, it may be possible for chemists to design compounds that mimic the carbohydrate structure. These synthetic compounds should bind at the carbohydrate-binding site of the lectin and block the natural interaction. One of the difficulties with this approach is that the individual binding interactions are weak and multiple cell–cell contacts are required; these may be difficult to block by small-molecule drugs. A second problem is that synthesis of specific oligosaccharides is difficult and expensive, and that large quantities may have to be injected into the blood for effective therapy.
Amino sugars
Fru-6-P is the precursor of amino sugars
Figure 27.10 shows the pathway of formation of GlcNAc, GalNAc and sialic acid. The initial reaction involves the transfer of an amino group from the amide nitrogen of glutamine to Fru-6-P to produce glucosamine-6-P (GlcN-6-P). An acetyl group is then transferred from acetyl-CoA to the amino group of the GlcN-6-P to form GlcNAc-6-P, which is converted to its activated form, UDP-GlcNAc, by sequential mutase and pyrophosphorylase reactions. In addition to its role as a GlcNAc donor, UDP-GlcNAc can also be epimerized to UDP-GalNAc. With few exceptions, all amino sugars in glycoconjugates are acetylated; thus they are neutral and do not contribute any ionic charge to the glycoconjugates.
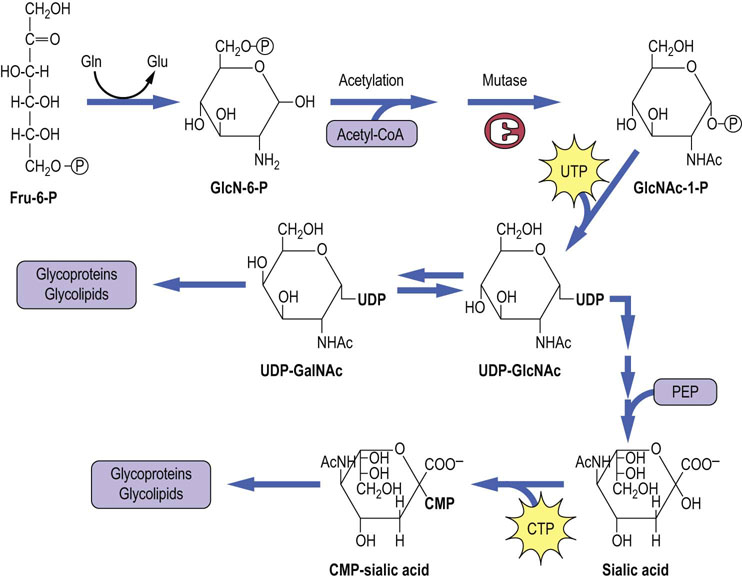
Sialic acid
UDP-GlcNAc is the precursor of N-acetylneuraminic acid (NeuAc), also referred to as sialic acid. Sialic acid, a 9-carbon N-acetylamino-ketodeoxyglyconic acid, is produced by the condensation of an amino sugar with phosphoenolpyruvate (see Fig. 27.10). Cytidine monophosphate neuraminic acid (CMP-NeuAc) is the activated form of sialic acid and is the sialic acid donor in biosynthetic reactions. CMP-sialic acid is the only nucleoside monophosphate sugar donor in glycoconjugate metabolism.
Biosynthesis of oligosaccharides
N-glycan assembly begins in the endoplasmic reticulum
The pathway of assembly of the N-glycans begins with the transfer of two GlcNAc residues to a membrane-bound lipid, called dolichyl phosphate. Mannose and glucose residues are added to build a lipid-linked oligosaccharide intermediate, which is transferred “en bloc” to protein in the lumen of the endoplasmic reticulum (Fig. 27.11). Dolichols are long-chain polyisoprenol derivatives usually having about 120 carbon atoms (about 22–26 isoprene units) with a phosphate group at one end. They are synthesized in membranes using the same machinery that is used to make cholesterol, but in contrast to cholesterol, the dolichols remain as long straight chains. The length of the chain requires it to snake through the phospholipid bilayer, providing a strong anchor for the growing oligosaccharide chain.
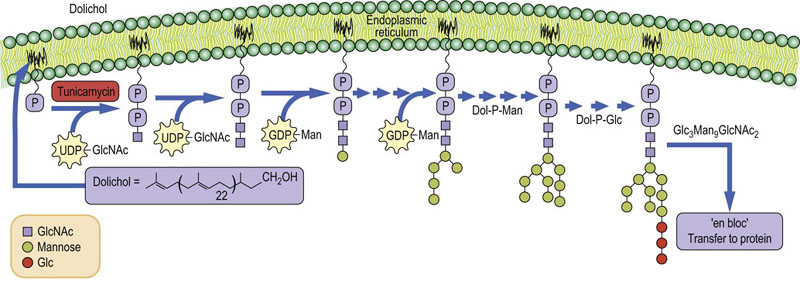
Fig. 27.11 Synthesis of N-linked oligosaccharides in the endoplasmic reticulum.
Tunicamycin is an inhibitor of the GlcNAc phosphotransferase that the catalyzes the first step in glycan synthesis. GlcNAc, N-acetylglucosamine; Dol, dolichol; Man, mannose.
The first sugar to be added to dolichyl-P from UDP-GlcNAc is GlcNAc-1-P by a GlcNAc-1-P transferase to produce dolichyl-P-P-GlcNAc. A second GlcNAc is linked to the first GlcNAc followed by addition of 4–5 mannose residues from GDP-Man. Dolichyl-P-Man and dolichyl-P-Glc serve as glycosyl donors for the remaining mannoses and the three glucoses residues. Each of the sugars is transferred by a specific glycosyltransferase located in or on the endoplasmic reticulum membrane. The glucoses are not found on any of the N-linked oligosaccharides on glycoproteins, but are removed by glucosidases in the endoplasmic reticulum. Why are they added in the first place? They serve two very important functions. First of all, the presence of glucoses on the lipid-linked oligosaccharide has been shown to expedite the transfer of oligosaccharide from lipid to protein – the transferring enzyme (oligosaccharide transferase) has a preference for oligosaccharides that contain three glucoses and transfers those oligosaccharides to protein much faster. Secondly, the glucoses are important in directing protein folding in the endoplasmic reticulum (below).
Intermediate processing continues in the endoplasmic reticulum (ER) and Golgi apparatus
In a series of trimming or pruning reactions (Fig. 27.12), all three glucoses are removed in the ER. The oligosaccharide may then remain as a high-mannose oligosaccharide or it may be further processed to a complex oligosaccharide structure. One or more mannoses may be removed in the ER, and the folded protein is then translocated to the Golgi apparatus where three or four additional mannoses may be removed to leave the core structure of three mannose and two GlcNAc residues. This core oligosaccharide is then elongated in the Golgi apparatus.
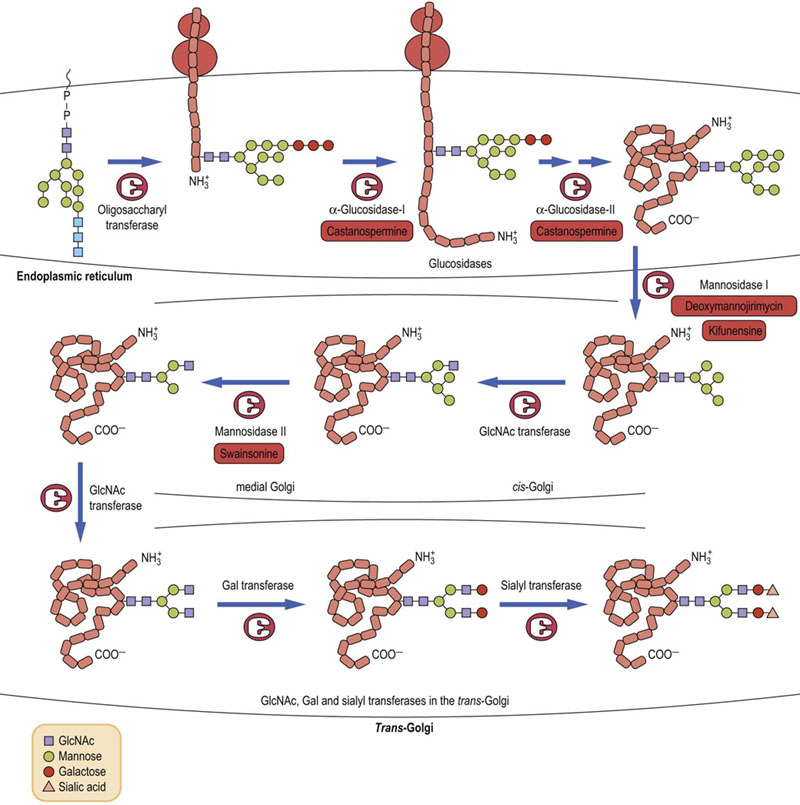
Final modifications of the N-linked oligosaccharides occur in the Golgi apparatus
After the pruning reactions in the ER and proper folding, the protein with one or more Man8GlcNAc2 oligosaccharides is transported to the Golgi apparatus where other modification reactions occur (see Fig. 27.12). Usually in the cis-Golgi, several other mannoses are removed by α-mannosidases to leave the core structure. Also in the cis-Golgi, GlcNAc residues are added to each of the mannoses. Then the protein enters the trans-Golgi fraction where the remaining sugars of the trisaccharide sequences, i.e. galactose, sialic acid and fucose, can be added to make a variety of different complex chains. The final structure of the oligosaccharide chains depends on the glycosyltransferases complement of the cell.
O-glycans
O-glycans are synthesized in the Golgi apparatus
In contrast to the biosynthesis of N-glycans, the synthesis of the O-glycans occurs only in the Golgi apparatus by the stepwise addition of sugars from their sugar nucleotide derivatives to the protein. No lipid intermediates are involved in O-glycan formation. Figure 27.13 outlines the stepwise sequence of reactions that are involved in the assembly of an oligosaccharide chain on salivary mucin. In this sequence, GalNAc is first transferred from UDP-GalNAc to serine or threonine residues on the protein by a GalNAc transferase in the Golgi apparatus. The resulting GalNAc-serine-protein serves as the acceptor for galactose and then sialic acid, transferred from their sugar nucleotides (UDP-Gal and CMP-sialic acid) by Golgi galactosyltransferases and sialyltransferases. Other Golgi glycosyltransferases are involved in the stepwise biosynthesis of more complex mucin oligosaccharides and in the synthesis of O-glycans in proteoglycans and collagens (see Chapter 29). There are more than 100 glycosyltransferases involved in glycoconjugate synthesis in a typical cell.
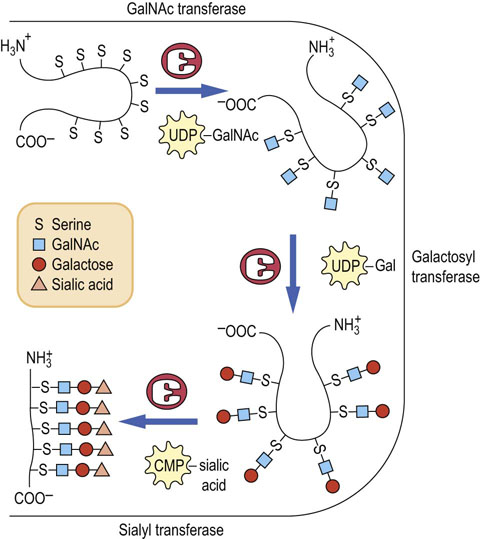
Fig. 27.13 Biosynthesis of O-linked oligosaccharides of mucins in the Golgi apparatus.
GalNAc, N-acetylgalactosamine.
Advanced concept box Inhibitors of glycoprotein biosynthesis
A number of inhibitors of the biosynthesis of N-glycans have been identified and these compounds have proven to be valuable reagents for studies on the role of specific carbohydrate structures in glycoprotein function. Tunicamycin is a glycoside antibiotic that inhibits the first step in the synthesis of N-glycans, i.e. the formation of dolichyl-PP-GlcNAc (see Fig. 27.11). Tunicamycin has varied effects on glycoprotein synthesis and on cells, from benign to profound. In some cases, the protein portion of the glycoprotein is synthesized, but without its carbohydrate it is misfolded, aggregates and is degraded in the cell. Thus, treatment of cells with tunicamycin frequently induces endoplasmic reticulum (ER) stress (see Chapter 34).
Other inhibitors inhibit specific steps in the processing pathway. Many are plant alkaloids that structurally resemble the sugars glucose and mannose, and inhibit the pruning glycosidases (see Fig. 27.12). Castanospermine inhibits the ER glucosidases, whereas kifunensine, deoxymannojirimycin and swainsonine each inhibit a different processing mannosidase. These drugs prevent the formation of complex chains and therefore are useful to evaluate structure–function relationships. Some compounds have been tested against HIV and against some cancers, and have shown positive inhibitory effects. However, they also have adverse effects on enzymes in normal cells and are therefore not usable for drug therapy. With more specific compounds, it may be possible to manipulate glycan structures for therapeutic purposes.
Functions of the oligosaccharide chains of glycoproteins
N-glycans have an important role in protein folding
Resident proteins in the endoplasmic reticulum, known as chaperones, assist newly synthesized proteins to fold into their proper conformations. Two of these chaperones, calnexin and calreticulin, bind to unfolded glycoproteins by recognition of high-mannose oligosaccharides that still contain a single glucose remaining on their structure, after the glucosidases have removed two of the three glucoses. Not all of the glycoproteins synthesized in the cell require assistance in folding but for those that do, the rate of folding is greatly accelerated by the chaperones. Incorrectly folded or unfolded proteins do not undergo normal transport to the Golgi apparatus and if they do not fold properly, they precipitate in the endoplasmic reticulum and are subsequently degraded by the ubiquitin–proteasome system in the cytoplasm.
Oligosaccharides containing Man-6-P target lysosomal enzymes to the lysosome
Lysosomes are subcellular organelles involved in the hydrolysis and turnover of many cellular organelles and proteins. They contain a variety of hydrolytic enzymes with acidic pH optima. Most of these lysosomal enzymes are N-linked glycoproteins that are synthesized and glycosylated in the endoplasmic reticulum and Golgi apparatus. The sorting of lysosomal enzymes occurs in the cis-Golgi. Proteins destined to be transported to the lysosomes contain a cluster of lysine residues that come together as a result of the protein folding into its proper conformation. As shown in Figure 27.14, this cluster of lysine residues serves as a docking site for an enzyme, GlcNAc-1-P transferase, that transfers a GlcNAc-1-P from UDP-GlcNAc to terminal mannose residues on the high-mannose chains of the lysosomal enzymes. A second enzyme, called an uncovering enzyme, then removes the GlcNAc, leaving the phosphate residues still attached to the mannoses on the high-mannose chains. The resulting Man-6-P residues on the high-mannose structure are now recognized by a Golgi protein called a Man-6-P receptor that directs the enzyme to the lysosomes. Thus, the Man-6-P residues are a targeting signal used by the cell to sort out those proteins that are destined to go to the lysosomes, and separate them from other proteins being synthesized in the Golgi apparatus. The Man-6-P receptor is also present on the cell surface, so that even extracellular enzymes that have this signal are endocytosed and transported to the lysosomes.
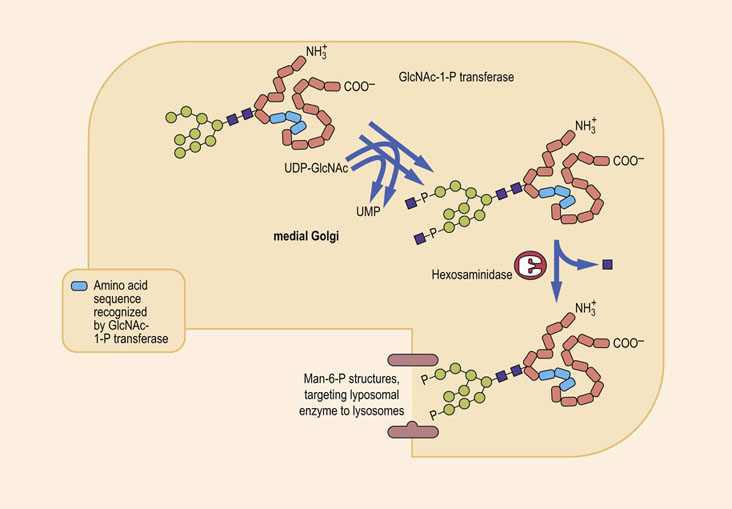
The oligosaccharide chains of glycoproteins frequently increase the solubility and stability of proteins
Clinical box Deficiencies in glycoprotein synthesis
The congenital disorders of glycosylation (CDG) are a recently described group of rare genetic diseases that affect the biosynthesis of glycoproteins. All patients show multisystem pathology, with severe involvement of the nervous system. Three distinct classes have been identified thus far and are characterized by a deficiency in the structure of the carbohydrate moiety of serum glycoproteins, lysosomal enzymes or membrane glycoproteins. The diagnosis of the disease is routinely made by electrophoresis of serum transferrin. In CDG, the transferrin contains less sialic acid and therefore the protein migrates more slowly. The decrease in sialic acid results from a defect in biosynthesis of the underlying oligosaccharide structure. While a change in the migration of serum transferrin indicates that the patient is suffering from one of the CDG, it does not identify the specific lesion. That can only be done by either characterizing the structure of the altered oligosaccharide chain(s) to determine what sugars or structures are missing, or doing a profile of key enzymes in the biosynthetic pathways, since the absence of any of these enzymes will affect the final oligosaccharide structure.
Because oligosaccharides are hydrophilic, they increase the solubility of proteins in the aqueous environment. Thus, most of the proteins that are secreted from cells are glycoproteins, including plasma proteins, excepting plasma albumin. These glycoproteins and enzymes generally also have high stability to heat, chemical denaturants, detergents, acids and bases. Enzymatic removal of the carbohydrate from many of these proteins greatly reduces their stability to stress. Indeed, when glycoproteins are synthesized in cells in the presence of glycosylation inhibitors, such as tunicamycin, which prevents the production and therefore the attachment of the N-glycan chain, many of these proteins become insoluble and form inclusion bodies in the cells as a result of incorrect folding and/or decreased hydrophilicity.
Sugars are involved in chemical recognition interactions with lectins
Clinical box I-cell disease
I-cell disease (mucolipidosis II) and pseudo-Hurler polydystrophy (mucolipidosis III) are rare inherited diseases that are caused by deficiencies in the machinery that targets lysosomal enzymes to lysosomes. Clinical presentation includes severe psychomotor retardation, coarse facial features and skeletal abnormalities; death usually occurs in the first decade. In cultured fibroblasts taken from patients suffering from mucolipidosis II, newly synthesized lysosomal enzymes are secreted into the extracellular medium rather than being targeted correctly to the lysosomes. Mesenchymal cells, especially fibroblasts, contain numerous membrane-bound vacuoles in the cytoplasm containing fibrillogranular material. These deposits are called inclusion bodies and this is the origin of the name I-cell disease.
I-cell disease results from a deficiency in synthesis of the targeting signal, Man-6-P residues on high-mannose oligosaccharides. The mutation is most commonly an absence of GlcNAc-1-P phosphotransferase, but defects in the uncovering enzyme also occur. It is likely that absence of the Man-6-P receptor protein would yield the same phenotype. In I-cell disease, the lysosomes, lacking the full spectrum of hydrolase enzymes, become engorged with indigestible substances.
N-glycans on the mammalian cell surface play critical roles in cell–cell interactions and other recognition processes. One cell may contain on its cell surface a carbohydrate-recognizing protein, known as a lectin, that binds to a specific oligosaccharide structure on the surface of the complementary cell. The interaction between these two chemical interfaces mediates a specific chemical recognition between the cells, and such a process is a key factor in fertilization, inflammation, infection, development and differentiation.
Carbohydrate–protein interactions are also important in non-self interactions. Many pathogens use this mechanism to recognize their target cells. E. coli, for example, and some other Gram-negative enteric bacteria have short hair-like projections called pili on their surfaces. These pili have mannose-binding lectins at their tip that can recognize and bind to high-mannose oligosaccharides on the brush border membranes of intestinal epithelial cells. This interaction allows the bacteria to be retained in the intestine. The influenza virus uses a hemagglutinin protein on its surface to bind to sialic acid residues on glycoproteins and glycolipids on the surfaces of target cells.
Variations in mucin structure appear to have a role in the specificity of fertilization, cell differentiation, development of the immune response, and virus infectivity. Glycoprotein ZP3, which is present on the zona pellucida of the mouse egg, functions as a receptor for sperm during fertilization. Enzymatic removal of O-glycans from ZP3 results in loss of sperm receptor activity, whereas removal of the N-glycans has no effect on sperm binding. The isolated O-glycans obtained from ZP3 also have sperm-binding activity and inhibit sperm–egg interaction and fertilization in vitro. Differences between the O-glycan structures of cytotoxic lymphocytes and helper cells involved in the immune response are also believed to be important in mediating cellular interactions during the immune response.
Advanced concept box Toxicity of ricin and other lectins
Lectins are found in a variety of foods, including beans, peanuts and dry cereals. Many plant lectins are toxic to animal cells. In edible plants, these may be less of a problem if the foods are cooked, since the lectins are denatured and therefore susceptible to intestinal proteases. On the other hand, lectins in uncooked plants are very resistant to proteases and can therefore cause serious problems. They bind to cells in the gastrointestinal tract, inhibiting enzyme activities, food digestion and nutrient absorption, and causing gastrointestinal distress and allergic reactions.
Ricin, produced by the castor bean plant, is among the most poisonous proteins known to man. These types of toxic lectins are usually composed of several subunits, one of which is the carbohydrate-recognizing or -binding site, while the other subunit is an enzyme that can catalytically inactivate ribosomes. Thus a single molecule of this catalytic subunit entering a cell can completely block protein synthesis in that cell. Other toxic lectins include modeccin, abrin and mistletoe lectin I.
Clinical box Changes in sugar composition and/or structure can be diagnostic markers of some types of cancer
Changes in glycosylation of both proteins and lipids have been consistently reported on cell surface carbohydrates of various types of cancer cells, including melanomas, ovarian cancer, and hepatocellular carcinoma. While these changes are not the cause of the disease, they are being evaluated as diagnostic tools for early detection of disease. Increased levels of the enzyme GlcNAc transferase V (the transferase involved in adding a second (branching) GlcNAc residue to a mannose residue to make a triantennary complex chain) is highly expressed in some transformed cells, resulting in increased branching and production of larger N-linked oligosaccharides. Changes in O-linked oligosaccharides have also been reported: for example, increased levels of sialyl Lewis-X antigen, which is thought to contribute to metastasis. Changes in the amount and sialylation of mucins are also associated with metastasis of lung and colon carcinoma cells and are being studied for their usefulness as diagnostic or prognostic biomarkers.
There is also evidence that changes in the level of fucose on some glycoproteins regulate the biological phenotype of cancer cells, and in fact, fucosylation of the protein α-fetoprotein (AFP-L3) has been used clinically as a marker for hepatocellular carcinoma.
The structure and composition of glycoproteins and glycolipids are altered in tumor cells, compared to normal cells. While these changes may not cause the cancer, they may have a significant effect on clinical outcome, e.g. if they limit leukocyte infiltration, assist in evading immune surveillance or facilitate metastasis. Analysis of oligosaccharide structures may be useful for early detection and diagnostic purposes and manipulation of oligosaccharide structure may prove useful in treatment of some cancers.
Summary
Glycosylation is the major posttranslational modification of tissue proteins.
Glycosylation is a multicompartment activity, involving sugar interconversions and activation in the cytosolic compartment, building of complex structures on lipid intermediates in the ER, and glycosylation and pruning reactions in the ER and Golgi apparatus. The outcome is an amazingly diverse range of oligosaccharide structures on proteins.
Sugars on glycoconjugates can serve a number of different functions, including:
modification of the physical properties of the protein (solubility, stability and/or viscosity);
aiding in the folding of the protein;
participating in the targeting of the protein to its proper location in the cell;
mediating cell–protein and cell–cell recognition during fertilization, development, inflammation and other processes.
A number of human diseases involve defects in sugar metabolism, including galactosemia and hereditary fructose intolerance, leukocyte adhesion deficiency, congenital disorders of glycosylation (CDG) and lysosomal storage diseases.
Asano, N. Sugar-mimicking glycosidase inhibitors: bioactivity and application. Cell Mol Life Sci. 2009; 66:1479–1492.
Etzioni, A. Genetic etiologies of leukocyte adhesion defects. Curr Opin Immunol. 2009; 21:481–486.
Kato, K, Kamiya, Y. Structural views of glycoprotein-fate determination in cells. Glycobiology. 2007; 17:1031–1044.
Martin, PT. Congenital muscular dystrophies involving the O-mannose pathway. Curr Mol Med. 2007; 7:417–425.
Schachter, H, Freeze, HH. Glycosylation diseases: Quo vadis? Biochim Biophys Acta. 2009; 1792:925–930.
Taylor, ME, Drickmer, K. Introduction to glycobiology, ed 2. New York: Oxford University Press; 2006.
Varki, A, Cummings, RD, Esko, JD, et al. Essentials of glycobiology, ed 2. New York: Cold Spring Harbor Press; 2009.
Zeidan, Q, Hart, GW. The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci. 2010; 123:13–22.
Congenital disorders of glycosylation. www.familyvillage.wisc.edu/lib_cdgs.htm.
Galactosemia. http://galactosemia.org/.
Glycoprotein biosynthesis. http://www.ccrc.uga.edu/~lwang/bcmb8020/N-glycans-A.pdf.
Glycoprotein topics. www.glycoforum.gr.jp/science/word/glycoprotein/GP_E.html.
Hereditary fructose intolerance. www.bu.edu/aldolase/HFI/.
I-cell disease. http://emedicine.medscape.com/article/945460-overview.
Plant lectins. www.ansci.cornell.edu/plants/toxicagents/lectins.html.