Disturbances of free water, electrolytes and acid–base balance
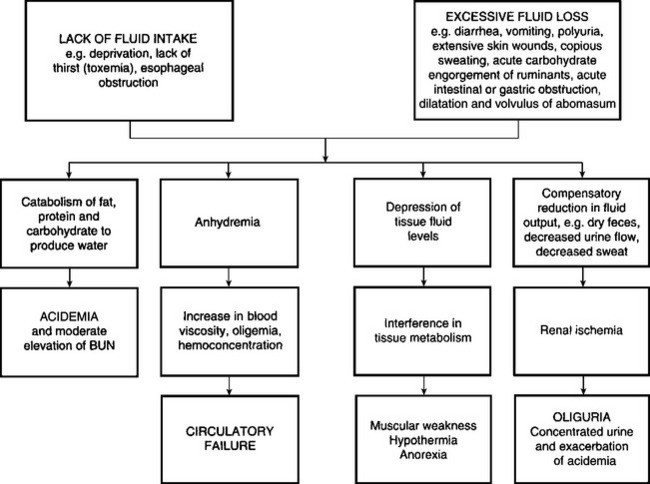
There are many diseases of farm animals in which there are disturbances of body fluids (free water), electrolytes and acid– base balance. A disturbance of body water balance in which more fluid is lost from the body than is absorbed results in reduction in circulating blood volume and in dehydration of the tissues. In contrast, the rapid ingestion of large quantities of water can lead to overhydration (water intoxication).
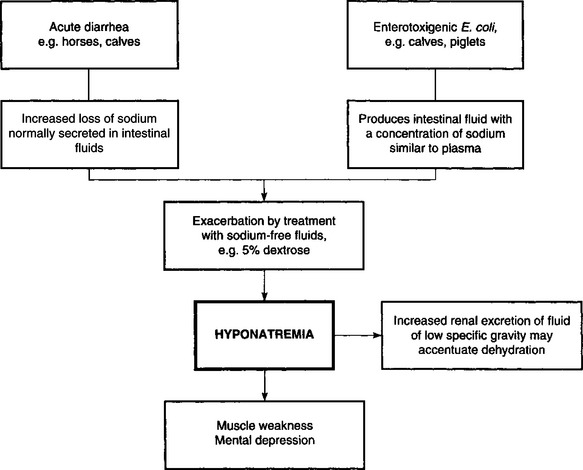
Electrolyte imbalances occur commonly as a result of loss of electrolytes, shifts of certain electrolytes or relative changes in concentrations due to loss of water. Common electrolyte imbalances include hyponatremia, hypokalemia, hypocalcemia and hypochloremia.
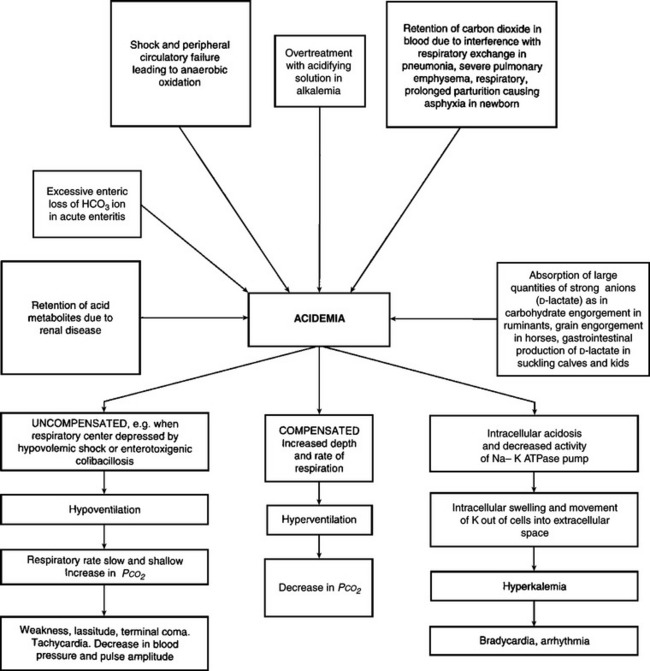
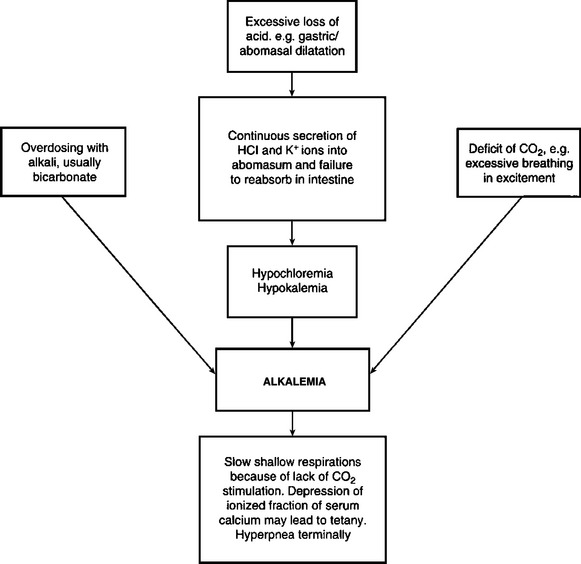
Acid–base imbalances, either acidemia or alkalemia, occur as a result of the addition of acid and depletion of alkali reserve, or the loss of acid with a relative increase in alkali reserve.
Under most conditions, the above disturbances of fluid and electrolyte balance will occur simultaneously, in varying degrees, depending on the initial cause. Each major abnormality will be described separately here with emphasis on etiology, pathogenesis, clinical pathology and treatment. However, it is important to remember that actual disease states in animals in which treatments with fluids and electrolytes are contemplated are rarely caused by single abnormalities. In most cases it is a combination of dehydration together with an electrolyte deficit, and often without a disturbance of the acid–base balance, that necessitates treatment.
DEHYDRATION
ETIOLOGY
There are two major causes of dehydration:
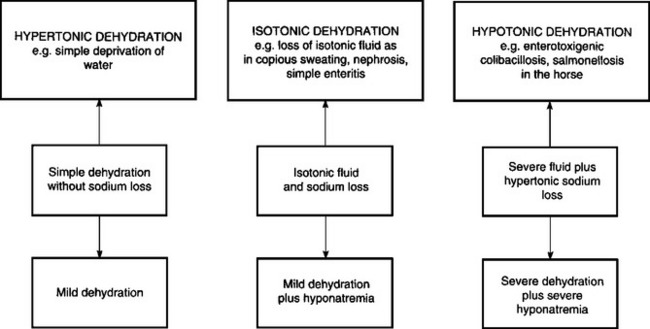
Deprivation of water, a lack of thirst due to toxemia, and the inability to drink water as in esophageal obstruction, are examples of dehydration due to inadequate water intake. The most common cause of dehydration is when excessive fluid is lost. Diarrhea is the most common reason for excessive fluid loss, although vomiting, polyuria and loss of fluid from extensive skin wounds or by copious sweating may be important in sporadic cases. Severe dehydration also occurs in acute carbohydrate engorgement in ruminants, acute intestinal obstruction and diffuse peritonitis in all species, and in dilatation and volvulus of the abomasum. In most forms of dehydration, deprivation of drinking water being an exception, the serious loss, and the one that needs correction, is not the fluid but the electrolytes (Fig. 2.1).
The ability to survive for long periods without water in hot climates represents a form of animal adaptation that is of some importance. This adaptation has been examined in camels and in Merino sheep. In the latter, the ability to survive in dry, arid conditions depends on a number of factors, including insulation, the ability to carry water reserves in the rumen and extracellular fluid space, the ability to adjust electrolyte concentrations in several fluid locations, the ability of the kidney to conserve water and the ability to maintain the circulation with a lower plasma volume. Dehydrated mammals in hot environments can save water by reducing the rate of panting and sweating and regulating body temperature above hydrated levels. Sweating is a significant avenue of evaporative heat loss in goats when they are hydrated and exposed to high ambient temperatures above 40°C.
Observations of drinking behavior of cattle transported to the abattoir indicate that those animals that had been sold in livestock markets prior to arrival at the abattoir are more thirsty and more tired than cattle sent directly from farms.1 This indicates inadequate water intake and dehydration.
PATHOGENESIS
Two factors are involved in the pathogenesis of dehydration:
The initial response to negative water balance is the withdrawal of fluid from the tissues and the maintenance of normal blood volume. The fluid is drained primarily from the intracellular and interstitial fluid spaces. Essential organs including the central nervous system, heart and skeleton contribute little and the major loss occurs from connective tissue, muscle and skin. The loss of fluid from the interstitial and intracellular spaces results in loss of skin elasticity, dryness of the skin and mucosa, and a reduction and retraction of the eyeball (enophthalmia) due to reduction in the volume of the postorbital fat deposits. In the goat, total body water may be reduced as much as 44% before death occurs.
The secondary response to continued negative water balance is a reduction in the fluid content of the blood causing a reduction in circulating blood volume (volume depletion) and an increase in the concentration of the blood (hemoconcentration). Because of the hemoconcentration, there is an increase in the viscosity of the blood, which impedes blood flow and may exacerbate peripheral circulatory failure. The loss in circulating blood volume also contributes to the mental depression of dehydrated animals, which is also due to varying degrees of acidemia and toxemia depending on the cause of the dehydration. In deprivation of water and electrolytes or in deprivation of water alone or inability to consume water in an otherwise normal animal (e.g. esophageal obstruction), the dehydration is minimal because the kidney compensates effectively by decreasing urine output and increasing urine osmolality. In addition, water is preserved by reduced fecal output and increased absorption, which results in dehydration of the contents of the rumen and large intestine, which in turn results in dry, scant feces.
In calves with acute diarrhea there is increased fecal output of water compared to normal calves but the total water losses are not much greater than in normal calves. In the diarrheic calf the kidney compensates very effectively for fecal water loss, and the plasma volume can be maintained if there is an adequate oral fluid intake. Urine excretion decreases, the urine becomes progressively more concentrated and the renal insufficiency may accentuate pre-existing acidemia and electrolyte imbalance, hence the importance of restoring renal function. The newborn calf is able to concentrate urine at almost the same level as the adult. This illustrates the importance of oral fluid and electrolyte intake during diarrhea to compensate for continuous losses. However, it is possible for metabolic acidosis to occur in diarrheic calves and goat kids that are not dehydrated.2,3
Goats are more sensitive to water deprivation during pregnancy and lactation than during anestrus. Water deprivation for 30 hours causes a marked increase in the plasma osmolality and plasma sodium concentration in pregnant and lactating goats. Pregnant and lactating goats drink more than goats in anestrus.
The dehydration in horses used for endurance rides is hypotonic, in which both sodium and water are lost through sweating. This may account for the lack of thirst in some dehydrated horses with the exhaustion syndrome. Weight losses of 10–15 kg/h may occur in horses exercising in high environmental temperatures exceeding 32°C (89°F) and a horse weighing 450 kg can lose 45 L of fluid in a 3-hour ride.
Dehydration exerts important effects on tissue metabolism. There is an increase in breakdown of fat, then carbohydrate and finally protein, to produce water of metabolism. The increased endogenous metabolism under relatively anaerobic conditions results in the formation of acid metabolites and the development of metabolic acidosis. Urine formation decreases because of the restriction of renal blood flow and this, together with the increased endogenous metabolism, causes a moderate increase in blood levels of nonprotein nitrogen.4 The body temperature may increase slightly initially – dehydration hyperthermia – because of insufficient fluid to maintain the loss of heat by evaporation. The onset of sweating in steers after exposure to high environmental temperatures has been shown to be delayed by dehydration.
Dehydration may cause death, especially in acute intestinal obstruction, vomiting and diarrhea, but it is chiefly a contributory cause of death when combined with other systemic states, such as acidosis, electrolyte imbalances, toxemia and septicemia.
CLINICAL FINDINGS
The first and most important clinical finding in dehydration is dryness and wrinkling of the skin, giving the body and face a shrunken appearance. The eyes recede into the sockets and the skin subsides slowly after being picked up into a fold. The dehydration is usually much more marked if water and electrolyte losses have been occurring over a period of several days. Peracute and acute losses may not be obvious clinically because major loss will have occurred from the intravascular compartment and only minor shifts have occurred from the interstitial spaces. Sunken eyes and inelastic skin are not remarkable clinical findings of dehydration in the horse.
The best indicator of hydration status in calves has been demonstrated to be the degree of recession of the eye into the orbit. Hydration status is assessed by gently rolling the lower eyelid out to its normal position and estimating the distance of eye recession in millimeters. This distance is multiplied by 1.7 to provide an estimate of the degree of dehydration as a percentage of euhydrated body weight.5 The second best indicator of hydration status in calves is the elasticity of the skin of the neck and lateral thorax, which are assessed by pinching the skin between the fingers, rotating the skin fold 90° and noting the time required after release of the skin fold for the skin fold to disappear (normally <2 s). The elasticity of the skin fold on the upper or lower eyelid is a poor indicator of hydration status in calves and is not recommended. The best methods for assessing hydration status in adult cattle and other large animals has not been determined but it is likely that eye recession and skin tent duration in the neck region provide the most accurate and sensitive methods for estimating hydration status.
In diarrheic calves, the severity of dehydration, hypothermia and metabolic acidosis are associated with the degree of mental depression.6 The combined effects of acidemia and dehydration also contribute to hypothermia.
Loss of body weight occurs rapidly in dehydration and muscular weakness and inappetence or anorexia are common. In horses deprived of water for 72 hours there is a mean body weight loss of about 15%, and 95% of the animals have a urine specific gravity of 1.042, a urine osmolality of 1310 mosmol/kg and a urine osmolality/serum osmolality ratio of 4:14. Prerenal azotemia also develops.
The degree of thirst present will depend on the presence or absence of other diseases causing an inflammatory response or endotoxemia. In primary water deprivation, dehydrated animals are very thirsty when offered water. In dehydration secondary to enteritis associated with severe inflammation, acidemia and electrolyte imbalance, there may be no desire to drink. Horses that become dehydrated in endurance rides may refuse to drink and the administration of water by oral intubation and enemas may be necessary. In cattle on pasture and deprived of water for up to 9 days and then given access to water, there will be staggering, falling, convulsions and some death – signs similar to salt poisoning in pigs. Experimental restriction of the water intake in lactating dairy cattle for up to 4 days may reduce milk yield by 75% and decrease body weight by 14%. A 10% reduction in water intake causes a drop in milk production that may be difficult to detect. Behavioral changes are obvious: cows spend considerable time licking the water bowls. In cold climates, cattle are often forced to eat snow as a source of water. The snow must be soft enough so that it can be scooped up by the cattle and 3–5 days are necessary for the animals to adjust to the absence of water and become dependent on snow. During this time there is some loss of body weight. Lactating ewes relying on snow as a source of free water reduce their total water turnover by approximately 35%.
 H+ + A−
H+ + A−