Porphyrins And Bile Pigments
Porphyrins are cyclic compounds formed by four covalently-linked pyrrole rings. They are metabolic intermediates related to the pathway of haem synthesis. Bile pigments, on the other hand, are produced by catabolism of haem. The pathways of synthesis and catabolism of haem are described in this chapter, and the associated pathological states are highlighted.
After going through this chapter, the student should be able to understand:
Fundamental structure and general characteristics of porphyrins.
Metabolic pathway, enzymes and intermediates, regulation of biosynthesis of haem and biochemical defects in different type of porphyrias.
Pathway of haem catabolism that produces bilirubin; intermediates as well as the enzymes catalyzing various steps of bilirubin metabolism and the associated pathology.
I General Characteristics of Porphyrins
Structure of a pyrrole ring is shown in Figure 16.1 . The four pyrrole rings of the porphyrin are linked through methenyl (–CH =) bridges. This creates a cyclic structure, called cyclic tetrapyrrole. The pyrrole rings are named as I, II, III and IV; and the methenyl bridges as α, β, γ and δ. Since the tetrapyrrole contains alternate single and double bonds, structure is commonly referred to as the conjugated ring system (Fig. 16.2 ).
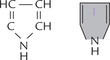
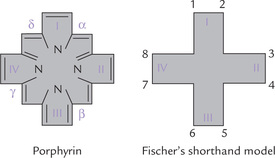
Fig. 16.2 Structures of porphyrin ring: I, II, III, IV are pyrrole rings, 1—8 are substituent positions and α, β, γ, δ are methenyl bridges.
The double bonds of the cyclic tetrapyrrole absorb visible light, and so porphyrins are coloured compounds. The red colour of haemoglobin is due to the porphyrin ring of its haem component. Porphyrins also fluoresce intense reddish pink colour in ultraviolet light. This property is also accounted by double bonds.
Each of the pyrrole rings of porphyrin contains a nitrogen atom which can link with a metal atom. This permits chelation of several metal atoms by the porphyrin ring. Joining of a metal with a porphyrin ring in this way yields metalloporphyrin. Physiologically, the metalloporphyrins containing iron are most important ones; they are termed iron-porphyrins. Most abundant of these is haem, which is present in haemoglobin, myoglobin, cytochromes, catalase, etc. Magnesium is present in chlorophyll, the metalloporphyrin that constitutes photosynthetic pigment of plants.
II Biological Significance of Porphyrins
In nature, the metalloporphyrins are linked with proteins to form conjugated proteins termed metalloporphyrino proteins. Most important of these are the iron-porphyrin containing haem-proteins, which participate in oxygen transport (haemoglobin), oxygen storage (myoglobin), electron transport (cytochromes), hydrogen peroxide inactivation (catalase), hydroxylation, oxygenation and other processes.
1. Haemoglobin is an oligomeric protein consisting of four subunits; each subunit contains a globin, a polypeptide, joined to a haem molecule. Haemoglobin is the principal transporter of oxygen in blood. It also carries some amount of carbon dioxide, and serves as an important blood buffer (Chapter 17).
2. Myoglobin, a monomeric protein, is structurally similar to a subunit of haemoglobin. It is present in muscles where it stores oxygen.
3. Cytochromes are electron transferring proteins that participate in oxidation-reduction reactions. Various types of cytochromes such as b, c, c1, and aa3 act in that sequence in electron transport chain (Chapter 14). Cytochrome P-450 participates in hydroxylation of aromatic and aliphatic compounds, such as steroids, alcohol, and many drugs.
4. Catalase is an iron-porphyrin enzyme present in animal cells, which catalyzes breakdown of hydrogen peroxide. The haem group forms a part of the active site of this enzyme.
Activity of catalase is minimal in plants where another iron-porphyrinoprotein, peroxidase, performs similar function.
5. Tryptophan pyrrolase is an iron-porphyrin enzyme that plays an important role in catabolism of tryptophan.
6. Erythrocruorins are iron-porphyrinoproteins occurring in blood and tissue fluids of some invertebrates. Their function corresponds to that of haemoglobin.
 Most of the haem in our body is in the oxygen transport protein, haemoglobin. Other haem-proteins, e.g. myoglobin, cytochrome, catalase, etc. participate in a broad range of biological processes.
Most of the haem in our body is in the oxygen transport protein, haemoglobin. Other haem-proteins, e.g. myoglobin, cytochrome, catalase, etc. participate in a broad range of biological processes.
It is important to note that the prosthetic group, haem, is performing different roles in different proteins. The biological role of haem in a given protein is dictated by the three-dimensional structure of the protein.
III Nomenclature
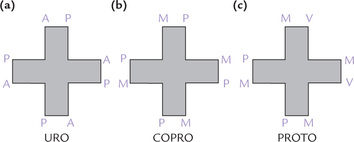
The porphyrins found in nature have side chains located at corners of the pyrrole rings. Figure 16.2 shows the possible sites of attachment that are denoted from 1 to 8. Different porphyrins vary in the nature and position of the side chains attached to these sites. The three most important porphyrins in humans are uroporphyrin (URO), coproporphyrin (COPRO), and protoporphyrin (PROTO).
• URO has four propionate and four acetate side chains.
• COPRO has four propionate and four methyl groups.
• PROTO has four methyl, two vinyl and two propionate groups (Fig. 16.3 ).
The side chain groups in porphyrins may be arranged in four different structural configurations (I to IV). In type I there is symmetric distribution of side chains, and in type III it is asymmetric (Fig. 16.4 ). For example, in uroporphyrin I, the acetate (A) and propionate (P) side chains of the pyrrole rings alternate regularly (1,3,5,7 and 2,4,6,8). In type III, on the other hand, there is asymmetric arrangement (1,3,5,8 and 2,4,6,7), meaning that the expected arrangement of side chains is reversed on ring IV. Only type I and type III series are found in nature and the porphyrins of the type III series are not only more abundant but also physiologically important in humans.
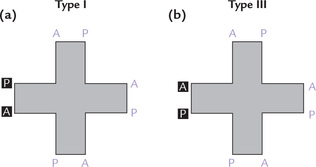
Fig. 16.4 Structure of (a) Uroporphyrin I with symmetric distribution of substituent groups, and (b) Uroporphyrin III with asymmetric distribution of substituent groups on the ring IV (A = acetate, P = propionate).
In case of PROTO, it is possible to arrange the two propionate, two vinyl and four methyl groups in 15 different configurations. Only the type IX isomer (Fig. 16.5 ) is produced in the human body.
IV Synthesis of Haem
Haem is a cyclic tetrapyrrole with a hexavalent iron (Fe2+) atom at the centre that is coordinately linked with four pyrrole nitrogens. Haem is the final product of the porphyrin synthetic pathway, which starts from two relatively simple precursors: glycine and succinyl CoA. The two major sites of the biosynthetic pathway are erythroid cells, which synthesize approximately 85% of the body’s haem groups, and the liver, which synthesizes most of the remainder.
Bone marrow and liver are the most important sites for the synthesis of haem, though some other tissues that synthesize haem-proteins can also produce haem in small amounts.
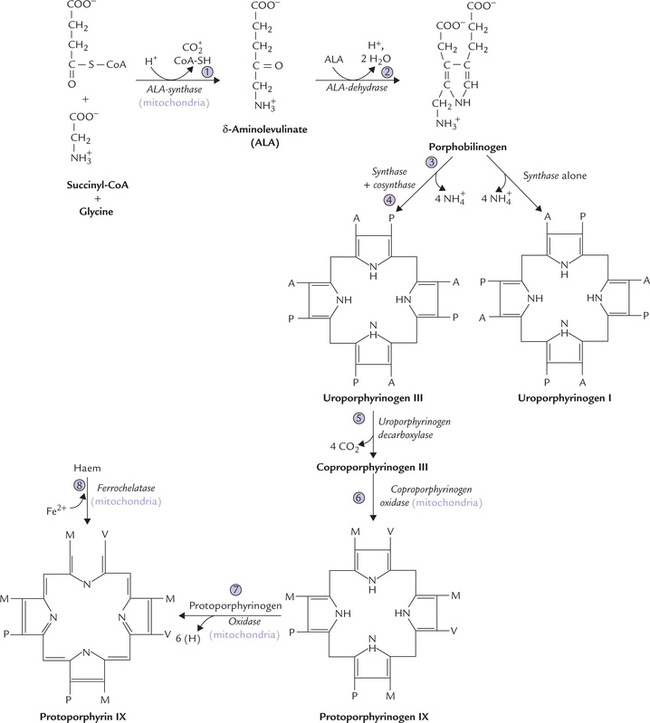
It requires participation of eight enzymes to synthesize haem. Of these enzymes, four (the first and the last three) are mitochondrial and the rest are cytosolic (Fig. 16.6 ). The reactions are all irreversible, occurring in 3 stages:
Stage I: Formation of Tetrapyrrole Ring
Step 1: Formation of δ-Aminolevulinic Acid
This step involves condensation of glycine and succinyl CoA to form δ-aminolevulinic acid (ALA). The reaction is catalyzed by δ-aminolevulinic acid synthase (ALA synthase), a mitochondrial enzyme, located on the matrix side of the inner mitochondrial membrane. Pyridoxal phosphate (PLP) is required as a coenzyme, hence anaemia may be manifested in PLP deficiency.
ALA synthase is an inducible, rate-limiting enzyme for the pathway. It is the principal regulated enzyme in the liver, as discussed later.
Step 2: Formation of Porphobilinogen
Two molecules of ALA are condensed by the cytosolic zinc containing ALA dehydratase to yield porphobilinogen. The enzyme is sensitive to inhibition by lead. This inhibition explains the elevation of ALA levels and anaemia in lead poisoning.
Step 3: Formation of Uroporphyrinogen III
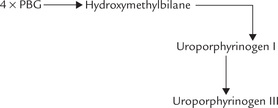
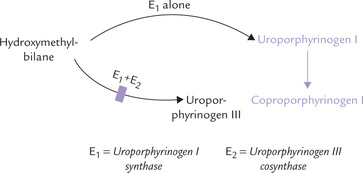
Condensation of four molecules of porphobilinogen yields the first cyclic tetrapyrrole, uroporphyrinogen III. It requires successive action of two enzymes: uroporphyrinogen I synthase (also called porphobilinogen deaminase) and uroporphyrinogen III cosynthase.
1. Uroporphyrinogen synthase catalyzes condensation of four PBG units to form the linear tetrapyrrole, hydroxymethylbilane, in which the acetate and propionate side chains of the pyrrole ring alternate regularly. This unstable intermediate may cyclize spontaneously to form uroporphyrinogen I.
2. Uroporphyrinogen III cosynthase rearranges the orientation of propionate and acetate side chains on one of the pyrrole rings (ring IV), to produce the asymmetric, physiological type III isomer of uroporphyrinogen.
Uroporphyrinogen III is the first compound containing a porphyrin-like ring structure in the haem biosynthetic pathway (Fig. 16.6). It is also a key intermediate in the synthesis of vitamin B12 in bacteria, and of chlorophyll in bacteria and plants.
Stage II: Processing of Uroporphyrinogen III to Protoporphyrin IX
Uroporphyrinogen III is processed to protoporphyrin IX by modification of its side chains and oxidation of porphyrinogen to porphyrin. It involves a series of reactions catalyzed by:
1. Uroporphyrinogen decarboxylase (step 5), decarboxylates all four acetate side chains (A) to form methyl (M) groups; the reaction product coproporphyrinogen III diffuses back into the mitochondrion.
2. Coproporphyrinogen oxidase (step 6), oxidatively decarboxylates two of the propionate side chains (P) from ring I and ring II to vinyl groups (V).
3. Protoporphyrinogen oxidase (step 7), oxidizes the methylene bridges (–CH2) linking the pyrrole rings to methenyl bridges (= CH–). Protoporphyrin IX is thus formed.
Stage III: Formation of Haem
Finally, formation of haem is accomplished by ferrochelatase (or haem synthase), which incorporates Fe2+ in the centre of the porphyrin ring. The enzyme is inhibited by lead.
Uroporphyrinogen III is processed to haem by decarboxylation of the acetate and propionate groups, oxidation of the porphyrinogen to the porphyrin, and chelation of iron in centre of the molecule.
The following points about this biosynthetic pathway are noteworthy.
1. Localization: The pathway takes place in most mammalian tissues except mature erythrocytes which do not contain mitochondria. Major sites of biosynthesis include liver, which synthesizes a number of haem-proteins (most prominently, the cytochrome P-450), and the erythrocyte-producing stem cells of the bone marrow, which are active in haemoglobin synthesis.
2. Polarity of intermediates: The initial intermediates of this pathway are polar. Uroporphyrinogen is strongly polar since all its side chains (four acetate and four propionate) are predominantly polar.
The subsequent intermediates become more and more non-polar.
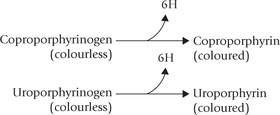
3. Porphyrinogens vs Porphyrins: The porphyrinogens are reduced porphyrins. They are not coloured because their pyrrole rings are connected by methy lene (–CH2) bridges, and the double bonds are not conjugated over the whole system. The porphyrinogens are the actual intermediates in the haem biosynthetic pathway. Porphyrinogens can be converted to the corresponding coloured porphyrins on exposure to light, when they lose hydrogen atoms.
4. Partly mitochondrial and partly cytosolic: The pathway is initiated in the mitochondria, and the first intermediate, ALA, diffuses into the cytosol where next few reactions, up to formation of coproporphyrinogen III takes place. The latter compound diffuses into the mitochondrion where the last three reactions occur (Fig. 16.6).
A Regulation of Haem Synthesis
The regulation revolves around the initial enzyme, ALA-synthase, and haem is the principal regulator. The regulatory effects in liver are multiple, as discussed here.
1. Repression mechanism: The ALA synthase has an unusually short biological half-life of 1 to 3 hours in the liver and its synthesis is suppressed very effectively by haem. Only free, non-protein bound haem can cause the repression. Haem (or its oxidation product haematin) activates a repressor protein that turns off ALA synthase biosynthesis at translation level.
Erythropoietin, a protein produced by the kidneys and found in larger than normal amounts in high-altitude dwellers, counteracts the effects of the repressor protein. Erythropoietin deficiency exists in chronic renal failure, which explains the observed anaemic condition in such patients.
2. Allosteric inhibition: The ALA synthase is allosterically inhibited by haem, haematin and haemin. The normal end product, haem, when present in excess, is oxidized to haematin, which contains a hydroxyl group attached to the Fe3+ atom.
Replacement of the hydroxyl group by a chloride ion produces haemin. In addition to acting as allosteric inhibitor of ALA synthase, haemin represses synthesis of ALA synthase and also inhibits transport of cytosolic ALA synthase into mitochondria, where it acts.
3. Others: Induction of ALA synthase is brought about by a variety of xenobiotics (environment pollutants), natural steroids and therapeutic drugs, such as barbiturates, phenytoin, griseofulvin, etc. These agents induce synthesis of cytochrome P-450. The latter belongs to a family of haem proteins that contain 65% of the total haem in the liver (Chapter 15). Increased synthesis of P-450 uses up the available haem, resulting in depletion of the small pool of free, unbound haem. The haem depletion leads to a derepression of ALA synthase.
The committed step of haem biosynthesis is the formation of ALA from succinyl CoA and glycine, and the enzyme catalyzing this step (ALA synthase) is subject to feedback inhibition by free, non-protein bound haem.
Thus, administration of phenobarbital and other such drugs is contraindicated in porphyric patients, because they cause an inappropriate induction of ALA synthase and consequent accumulation of the offending intermediates. This may precipitate an acute porphyric episode, as discussed later.

ALA synthase is stimulated by 5-β-dehydro steroids (e.g. testosterone) in their reduced form.
In erythroid cells, the regulatory enzymes and effectors are different (Box 16.1).
V Disorders of Porphyrin Metabolism: Porphyrias
Porphyrias are a group inherited and acquired disorders caused by abnormalities in the haem synthetic pathway (Fig. 16.7 ). They may affect the liver or the blood-forming tissues, and are characterized by excessive accumulation and excretion in urine/or faeces of porphyrins or porphyrin precursors. Porphyrins have a deep red or purple colour (Greek: porphyria = purple).
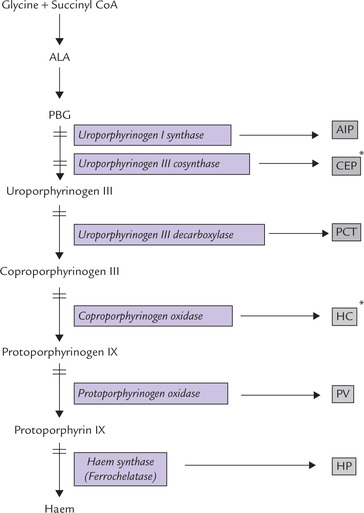
Fig. 16.7 Enzymatic blocks result in different types of inherited porphyrias (AIP = acute intermittent porphyria, CEP = congenital erythropoietic porphyria, PCT = porphyria cutanea tarda, HC = hereditary coproporphyria, PV = porphyria variegata, HP = hereditary protoporphyria. *CEP and HC are erythropoietic porphyrias, rest are hepatic porphyrias).
Unlike other disorders involving erythrocytes, anaemia does not usually dominate the clinical picture. The partial interruption of the haem biosynthetic pathway does not greatly diminish haem synthesis, but causes the intermediate metabolites to be present in excess. The excessive amount of porphyrins so formed are deposited in body tissues, to cause cutaneous, neurological or other clinical manifestations. Thus, excessive intermediate metabolites, and not diminished haem production, is the offending factor in porphyrias.
Porphyrias are caused by partial deficiency of one of the haem synthesizing enzymes other than ALA synthase. Any one of the enzymes from uroporphyrinogen I synthase to ferrochelatase may be affected.
Salient Features of Porphyrias
1. All porphyrias are characterized by accumulation and excretion of the intermediate porphyrins of the haem biosynthetic pathway.
2. ALA synthase, the key enzyme around which control of haem biosynthetics revolves, is derepressed in porphyrias because cellular haem levels are lower than normal.
3. The route of excretion of the accumulated porphyrins-urine or faeces-depends on their polarity. For example:
• Uroporphyrin with its eight carboxyl groups is most water-soluble and is excreted almost entirely in urine.
• Protoporphyrin, with only two carboxyl groups is excreted exclusively in faeces.
• Coproporphyrin has four carboxylic groups and is excreted by either route.
4. Level of metabolic block determines clinical features:
(a) Early block: If the metabolic block lies early in the pathway, so that synthesis of even the first tetrapyrrole ring does not occur, the defect may present as an acute pain crisis. This is because the accumulated metabolites (ALA and PBG) cause excitation of visceral pain fibres. ALA may block action of the inhibitory neurotransmitter (GABA) because of structural resemblance.
(b) Later block: When the metabolic block lies beyond the formation of the first tetrapyrrole ring, cutaneous manifestations are the predominant features. The porphyrins accumulated in the patient’s skin are responsible for these manifestations by the following sequence of events:
• The conjugated ring system of porphyrins has the property of absorbing light near 400 nm (this distinguishing absorption band is called the Soret band) and then emit intense red light.
• The emitted radiant energy generates superoxide radicals and other oxygen free radicals (OFRs).
• The OFRs so formed are capable of damaging biological membranes and cause the release of destructive enzymes from lysosomes (Chapter 27).
• The lysosomal enzymes in turn cause skin damage and scarring.
Treatment of porphyrias is aimed at alleviating the symptoms. Intravenous injection of haematin causes repression of ALA synthase, and therefore, prevents the accumulation of intermediates. In addition, the patient is advised to avoid sunlight and to increase dietary intake of free radical scavengers such as β-carotene and vitamin E.
The clinical manifestations of porphyrias are caused by abnormal accumulation of biosynthetic intermediates and may include abdominal pain, neuropsychiatric signs and cutaneous photosensitivitiy. According to the tissue in which the abnormality is expressed, hepatic and erythropoietic porphyrias can be distinguished.
Comparative properties of various types of porphyrias as listed in Table 16.1, depend on the molecular defect and the type of porphyrin that accumulates. Depending on the site where the enzyme deficiency is manifested, there are two major types: (a) erythropoietic porphyrias, and (b) hepatic porphyrias. (Porphyrias with both erythropoietic and hepatic abnormalities are also known.)
Table 16.1
Comparative properties of various types of porphyrias
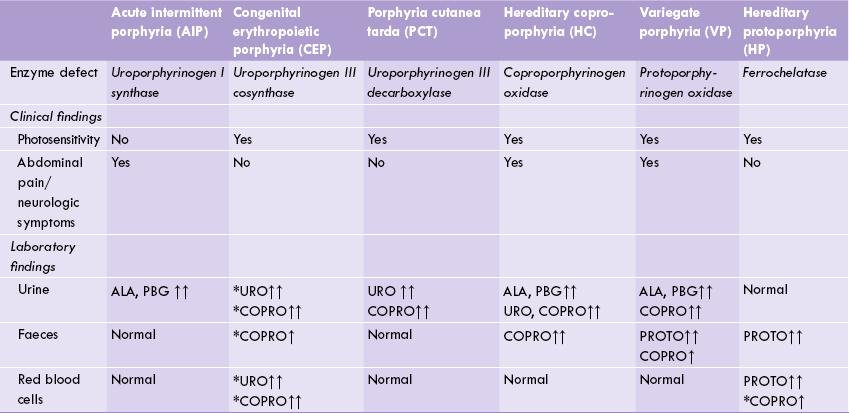
↑ = elevated; ↑ = highly elevated. *Type I isomers of uroporphyrin and coproporphyrin. Rest are type III isomers. HP and CEP are erythropoietic porphyrias, showing metabolic accumulation in RBC.
The two erythropoietic porphyrias (congenital erythropoietic porphyria and hereditary protoporphyria) are characterized by a build up of porphyrins in erythrocytes.
In hepatic porphyrias, liver is the primary organ affected and therefore the abnormality is expressed in this organ. Four types of hepatic porphyrias are known: acute intermittent porphyria, porphyria cutanea tarda, hereditary coproporphyria and porphyria variegate (Fig. 16.7).
All these disorders are inherited as autosomal dominant trait, except congenital erythropoietic porphyria, which is a genetically recessive disease.
A Acute Intermittent Porphyria (AIP)
It is one of the most bizarre disorders a physician ever encounters. The defective enzyme is uroporphyrinogen I synthase and the metabolites accumulated are ALA and PBG (Fig. 16.7). The disease is marked by intermittent attacks of abdominal pain (which need to be differentiated from various surgical causes) and neuropsychiatric symptoms.
Clinical Features and Laboratory Findings
Accumulation of ALA and PBG in pharmacological amounts has been implicated in clinical manifestations. These metabolites act on the nervous system to cause a variety of signs and symptoms:
• Abdominal pain is caused by excitation of visceral pain fibres; constipation and cardiovascular changes (including tachycardia and high blood pressure) are mediated by the autonomic nervous system.
• The involvement of peripheral nerves causes sensory or motor disturbances.
• Brain involvement is manifested by confusion, agitation or seizures.
• Excessive amounts of ALA and PBG are excreted in urine during and after acute attacks. However, the afflicted subject is not photosensitive because the metabolic block lies early in the pathway, and the porphyrin ring structure responsible for cutaneous photosensitivity is never formed (Case 16.1).
Urinary excretion of ALA and PBG increases during acute porphyric attacks, but the urine sample is colourless (ALA and PBG being colourless compounds). But it increases in colour intensity on standing because of photo-oxidation of PBG to porphobilin.
Treatment
General treatment guidelines for porphyrias were discussed earlier. An appropriate treatment for AIP consists of withdrawal of any offending drug and infusion of haematin; the latter represses synthesis of ALA synthase. A carbohydrate rich diet benefits the patients by repressing (by an unknown mechanism) the synthesis of ALA synthase.
An acute porphyric attack is precipitated by starvation and by drugs, e.g. barbiturates, phenytoin and other drugs that induce synthesis of cytochrome P-450, such drugs therefore must be avoided.
King George III, who ruled England during the American revolution (1773—76), and who was widely portrayed as having fits of madness, had attacks characteristic of AIP. Some of his precipitate actions were taken during acute (porphyric) episodes, which might have infuriated the colonists, ultimately leading to American independence.
B Congenital Erythropoietic Porphyria
Activity of uroporphyrinogen III cosynthase is severely depressed in this rare disorder, so that synthesis of type III isomers from hydroxymethylbilane is decreased. Instead, the hydroxymethylbilane is converted to uroporphyrinogen I, which, under the influence of uroporphyrinogen decarboxylase, may be converted to coproporphyrinogen I.
Both uroporphyrinogen I and coproporphyrinogen I are oxidized to their corresponding porphyrins, which are then accumulated in the tissues and excreted in the urine (Table 16.1). Their accumulation produces a pink to dark red colour in teeth, bones, and urine. Dark red urine (pot-wine appearance), and red-brown teeth (erythrodontia) are pathognomonic.
Extreme cutaneous sensitivity to light relates to accumulation in the skin of various type I porphyrins. Itching, burning, redness and swelling occur initially, and may lead to hyperpigmentation and scarring. The latter leads to a typical facial deformity, referred to as monkey faeces.
The deficient production of the type III isomer diminishes the regulatory effect on ALA synthase, which further increases levels of the type I isomers. Excessive amounts of porphyrins in erythrocytes may produce haemolysis.
C Porphyria Cutanea Tarda
It is the most common porphyria with an incidence of 1 in 25000 (the second commonest form of AIP has a much lower incidence—1 in 50000). It is mainly a skin disorder caused by a decreased activity of liver uroporphyrinogen III decarboxylase (Fig. 16.7). The patient’s urine contains large quantities of uroporphyrin III and some uroporphyrin I. Mild to severe photosensitivity and liver diseases are other clinical presentations. Acute episodes can be precipitated by alcohol intake or oestrogen therapy.
D Others
Enzyme defect in hereditary coproporphyria (HC), variegate porphyria (VP) and hereditary protoporphyria (HP) and their clinical features and laboratory findings are shown in Table 16.1. Some important features of these conditions are:
1. Clinical manifestations in HP are similar to AIP, though much less in severity.
2. VP (incidence 1 in 100,000) is especially prevalent among white population in South Africans.
3. Hereditary protoporphyria (HP), generally a benign condition, is characterized by accumulation of proto-porphyrin IX in maturing reticulocytes and young erythrocytes, and in liver; the latter may lead to severe liver damage. High levels of protoporphyrin are found in plasma, erythrocytes and faeces, but the urine is typically free of any porphyrin. When a smear of RBCs is exposed to fluorescent light, they exhibit red fluorescence.
Finally, it may be mentioned that porphyrias often make their appearance after puberty. This has been associated with the appearance of 5-ß-steroid reductase, as discussed (Case 2).
E Acquired Porphyrias
Porphyrias can occur in situations other than the inherited conditions described above, such as lead poisoning and iron deficiency.
1. Lead poisoning is the commonest cause of acquired porphyria. Lead inhibits the uptake of iron by immature red cells and has direct inhibitory effect on two enzymes of the porphyrin pathway: ALA dehydratase and ferrochelatase. Consequently there is:
• an increased excretion of ALA in urine (but not PBG);
• an increase in urine and fecal coproporphyrins and protoporphyrins.
2. Iron deficiency: Patients with iron deficiency can use zinc, instead of iron, as a substrate for ferrochelatase. Red cell lysates in such instances contain increased quantities of Zn-haemoglobin. In addition, red cells from iron deficient patients also contain increased amounts of protoporphyrin IX. Both Zn-haemoglobin and protoporphyrin IX determinations are therefore used in the diagnosis of iron deficiency anaemia.
Coproporphyrinuria: A small (less than twofold) increase in urinary coproporphyrin is most commonly caused by problems unrelated to haem synthesis, such as liver disease, acute illness or exposure to toxic chemicals.
The haem biosynthetic pathway is disrupted in porphyrias and related conditions, which can be caused by inherited enzyme deficiency (any one except ALA synthase), iron overload, or lead poisoning
Diagnosis of Porphyrias
Porphyrias can be diagnosed biochemically by (a) measurement of metabolites of the haem synthetic pathway, (b) assay of the relevant enzymes, and (c) DNA-based testing.
(a) The metabolite measurements are performed in plasma, urine, faeces and erythrocytes (Table 16.1). α-Aminolevulinic acid and porphobilinogen are quantified by colourimetric tests, and, uroporphyrin and coproporphyrin are determined fluorometrically. The most important test for diagnosis of acute porphyrias is the measurement of PBG in urine and plasma.
(b) Enzyme assay—only one enzyme of the synthetic pathway, uroporphyrinogen I synthase, is routinely measured by clinical laboratories. Its activity is decreased to about 50% of normal in the red cells of most individuals with acute intermittent porphyria, whether the disease is in latent or in an acute phase.
(c) DNA-based testing of porphyrias is more definitive than conventional biochemical tests. The test is possible now as all of the genes that encode enzymes of the haem synthetic pathway have been identified and their coding sequences determined. This has led to thwe discovery of mutations that cause each of the porphyrias.
VI Haem Breakdown
End products of haem breakdown are bile pigments: bilirubin and biliverdin. About 300 mg of bilirubin is produced per day, a major fraction of which (250 mg) is derived during turnover of haemoglobin. Another 50 mg of bilirubin is formed from other haem-containing proteins, such as myoglobin and cytochromes.
Lifespan of the erythrocyte is 120 days. Old cells are removed from blood circulation and degraded in the reticulo-endothelial system, liberating the haemoglobin. The protein portion of haemoglobin is hydrolyzed to its constituent amino acids, and channeled into the body’s amino acid pool. The iron so liberated is re-utilized, and the iron free porphyrin ring is converted to bilirubin (Fig. 16.8 ).
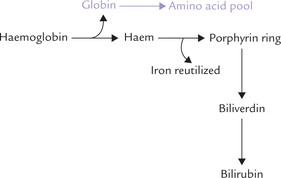
Fig. 16.8 Breakdown of haemoglobin in reticulo-endothelial cells of liver, spleen and bone marrow. (Total amount of haemoglobin in adult body is about 750 g and the daily turnover rate is around 6 g.)
A Bilirubin Metabolism
Production of bilirubin and its metabolism, and the abnormalities of bile pigment metabolism are described in the subsequent sections.
1. Bilirubin production: Bilirubin, the end product of haem degradation, is produced in reticulo-endothelial cells in two steps:
(a) The microsomal haem oxygenase system in the reticuloendothelial cells cleaves the porphyrin ring of haem (Fig. 16.9 ). Consequently, the cyclic tetrapyrrole ring structure of haem is converted to a linear tetrapyrrole. NADPH is required as a coenzyme in this step, and a ferrous iron and a carbon monoxide molecule are released. This results in the formation of biliverdin, a green pigment.
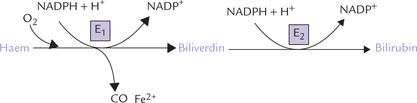
(b) Biliverdin is reduced to a red-orange pigment, bilirubin, by the enzyme biliverdin reductase; NADPH acts as donor of the reducing equivalents.
Changing colour of a bruise is due to appearance of biliverdin followed by bilirubin.
Further metabolism of bilirubin occurs in the liver and the intestine.
2. Transport of bilirubin to liver: Bilirubin forms a non-covalent complex with albumin in plasma, which is transported to the liver (Fig. 16.10 ). Formation of such a complex is necessary for the bilirubin transport since hydrophobic nature of bilirubin restricts free movement of the unbound-bilirubin in aqueous plasma. The complex has predominantly polar character (because of polar nature of albumin) which enables it to move freely in plasma.
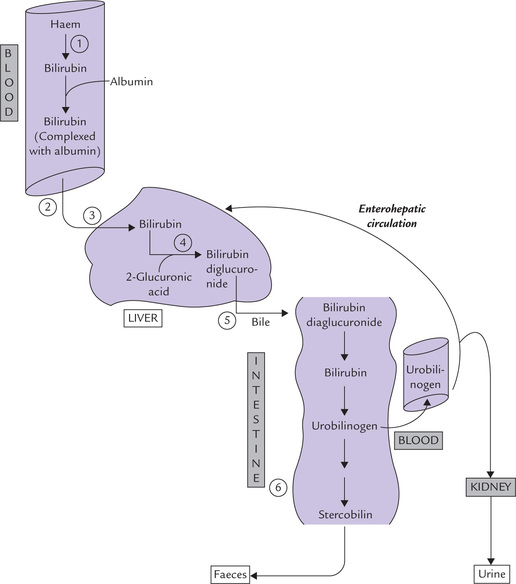
Fig. 16.10 Bilirubin metabolism. (1) Production in reticuloendothelial system. (2) Transport to liver. (3) Hepatic uptake. (4) Conjugation with glucuronic acid. (5) Excretion into bile. (6) Deconjugation and reduction in intestine.
On reaching the liver, bilirubin becomes free from its non-covalent association with albumin and enters the hepatocytes.
Albumin has two bilirubin-binding sites:
Bilirubin is first taken up by the high-affinity site, and only when all such sites are saturated, the excess bilirubin is bound to the low affinity site. In 100 ml plasma, albumin can take up to 20 mg bilirubin at its high affinity site. So, bilirubin associates with low affinity site only when serum bilirubin level crosses 20 mg/dL. Certain anionic drugs, such as sulphonamides, penicillin and salicylates can displace bilirubin from albumin, thus increasing the unbound-bilirubin concentration in plasma. This may have hazardous consequences in newborn infants because the unbound-bilirubin can cross the blood-brain barrier and enter the central nervous system. In the neurons, bilirubin has potential to cause neuronal tissue damage: the condition is known as bilirubin encephalopathy.
3. Hepatic uptake of bilirubin: When the albumin-bilirubin complex reaches the sinusoidal surface of the liver, the bilirubin is taken up by hepatocytes. The uptake is an active (energy-requiring) process, which requires mediation of the carrier protein. Within the hepatocyte, two proteins: ligand in Y and Z protein, bind the bilirubin.
4. Conjugation of bilirubin in hepatocytes: Inside the hepatocyte, two molecules of glucuronic acid are attached with bilirubin to form bilirubin diglucuronide, also called conjugated bilirubin (or direct bilirubin). UDP-glucuronic acid serves as activated donor of the glucuronic acid.
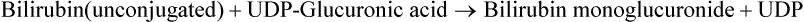
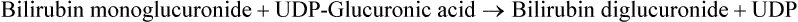
Both these steps are catalyzed by bilirubin glucuronyl transferase. Because glucuronic acid is highly polar, its attachment with bilirubin imparts highly polar characteristics to the latter, so that it can mix with bile and subsequently get excreted through the hepatobiliary route (Fig. 16.10).
5. Excretion of conjugated bilirubin into bile: The hepatocytes secrete the conjugated bilirubin into the bile canaliculi, from where it reaches the bile duct (Fig. 16.10). Since concentration of bilirubin in bile is very high, the above transport occurs against a steep concentration gradient, and therefore, requires input of energy. This step is ‘rate-limiting’ in the bilirubin metabolism. Being energy dependent, it is susceptible to impairment in the liver diseases.
The degradation of haem in reticulo-endothelial cells of the spleen or other organs involves oxidation to biliverdin first, which is reduced to bilirubin. The liver conjugates bilirubin to bilirubin diglucuronide and this product is actively secreted into bile.
6. Fate of conjugated bilirubin in intestine: The conjugated bilirubin enters the gut through bile, where it is acted upon by the intestinal bacteria.
• The first step is deconjugation by bacterial glucuronidase, which removes the polar glucuronate groups from the conjugated bilirubin.
• The free bilirubin so formed is further reduced to a colourless tetrapyrrole urobilinogen (URO).
• A part of urobilinogen (20%) is absorbed from the gut into the portal circulation. The rest remains within intestinal lumen, where it is reduced further.
• The reduction of the URO leads to the formation of mesobilinogen and stercobilinogen.
In small intestine, bilirubin is deconjugated and reduced to three substances, collectively called urobilinogens, that differ from each other in their degrees of oxidation.
The stercopbilinogen is mostly excreted in faeces (250–350 mg/day). Upon exposure to atmospheric air, it is oxidized to a coloured product, stercobilin. The characteristic brown colour of stools is accounted by stercobilin. Pale stools may, therefore, indicate biliary obstruction.
The enterohepatic circulation: The part of urobilinogen, which is absorbed into the portal circulation, can take two alternative routes:
(i) Some part of it is returned to the liver. Through hepatobiliary route, it reaches the intestine again. This is enterohepatic circulation, as depicted in Figure 16.10.
(ii) The remaining urobilinogen enters the systemic circulation and transported to kidneys. It is oxidized to a coloured product, urobilin, and excreted in urine. The normal colour of urine is accounted by the urobilin.
The reduced forms of bilirubin (urobilinogen, mesobilinogen and stercobilinogen) are colourless compounds. The oxidized forms—urobilin, mesobilin and stercobilin—are coloured (bile pigments). Colour of faeces becomes darker on standing because of further oxidation, which forms more of stercobilin. When intestinal bacterial flora is decreased by antibiotic therapy, bilirubin is not adequately reduced to urobilinogen. It is rather oxidized to biliverdin in large intestine, which imparts green colour to stools.
Plasma Bilirubin
The human bloodstream contains both unconjugated (albumin bound) and conjugated bilirubin. Their total concentration ranges from 0.2 mg/dL to 1.0 mg/dL, of which conjugated bilirubin accounts for 0.1–0.4 mg/dL.
The conjugated bilirubin is also known as direct reacting bilirubin because, being water-soluble, it gives purple colour immediately with Ehrlich’s diazo reagent. The unconjugated bilirubin, on the other hand, is water-insoluble and has to be extracted with methanol or ethanol for reacting with the diazo reagent. Therefore, it is also called indirect reacting bilirubin.
The conjugated bilirubin is termed direct bilirubin and the unconjugated bilirubin is called indirect bilirubin.
Normally, little, if any, conjugated bilirubin is found in the urine. But a 24-hour urine sample may contain 0.5–4.0 mg urobilin. Faeces normally contains 250–350 mg of stercobilinogen and stercobilin per day.
VII Jaundice
Rise in serum bilirubin level above normal (hyperbilirubinaemia), causes it to get deposited in tissues. A yellow discolouration of skin and sclera results and the condition is known as jaundice. Hyperbilirubinaemia exists when the plasma bilirubin concentration exceeds 1.0 mg/dL. Levels between 1.0–3.0 mg/dL indicate subclinical jaundice (latent jaundice). When the bilirubin level exceeds 3.0 mg/dL, it diffuses into tissues to produce yellowish discolouration of skin, sclera, conjunctiva and mucous membrane. This results in icterus (or clinical jaundice).
Elevation of serum bilirubin level, known as hyperbilirubinaemia, results in jaundice or icterus when it crosses 3 mg/dL.
Hyperbilirubinaemia occurs when there is imbalance between bilirubin production and excretion. There are three main reasons why this imbalance may occur:
1. In haemolysis, the increased haemoglobin breakdown produces bilirubin which overloads the hepatic conjugating mechanism.
2. In hepatic diseases there is important of the conjugating mechanism.
Based on these underlying causes, the jaundice is conventionally classified as haemolytic, hepatic and obstructive types, respectively.
A Haemolytic Jaundice
Liver has enormous capacity for conjugating bilirubin. In an adult, about 300 mg of bilirubin that is produced each day reaches liver, where it is conjugated. Liver is capable of handling about 10 times as much load of bilirubin (3g/day). However, in case of massive haemolysis, there is an increased bilirubin production to such an extent that the liver is not able to conjugate it fully. A portion of the bilirubin remains unconjugated, which results in rise in serum concentration of unconjugated bilirubin, i.e. unconjugated hyperbilirubinaemia). Some other common causes of increased rate of haemolysis are given in Table 16.2 . Unconjugated hyperbilirubinaemia, when rapidly rising in neonates, should be carefully monitored (discussed later).
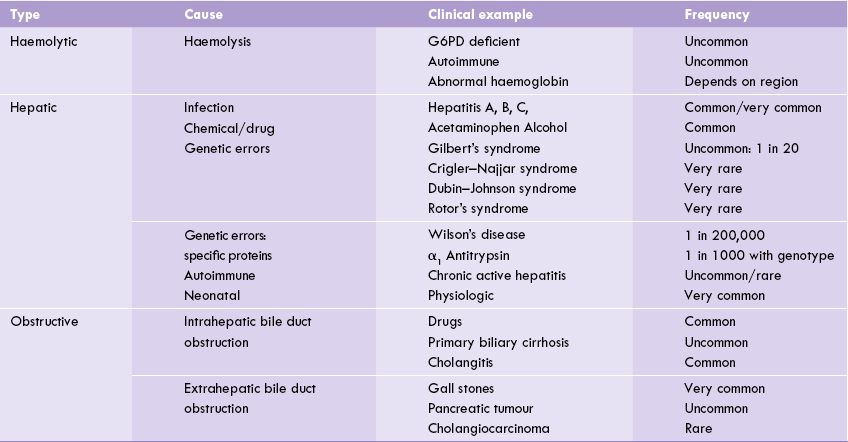
Hyperbilirubinaemia of haemolytic diseases rarely exceeds 3–4 mg/dL because of enormous capacity of healthy adult liver to metabolize bilirubin. The biliary excretion of conjugated bilirubin is increased in proportion to the severity of haemolysis. Levels of the compounds derived from conjugated bilirubin (urobilin in urine and stercobilin in faeces) are thereby elevated (Table 16.3 ).
Table 16.3
Comparative features of haemolytic, hepatic and obstructive jaundice
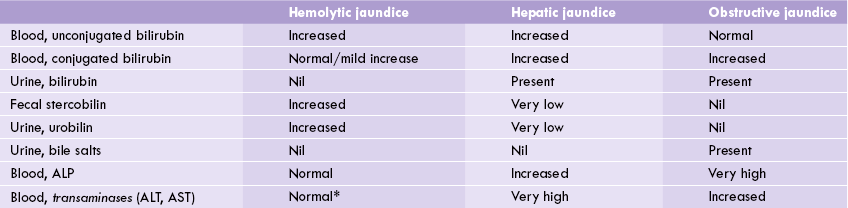
*AST increases in case of haemolysis because of its high concentration in red blood cells. However, activity of ALT, the other major transaminase, remains normal. ALT = alanine transaminase, AST = aspartate transaminase, ALP = alkaline phosphatase.
B Hepatic Jaundice
Jaundice occurs due to diminished functional capacity of liver. Various causes for the same are outlined in Table 16.2.
Plasma levels of both the conjugated and the unconjugated bilirubin rises in this type of jaundice (Table 16.3). This is referred to as a “biphasic rise”, the reasons for which are as below:
• The damaged hepatocytes have impaired capacity to take up the circulating bilirubin and to conjugate it. This leads to increased plasma level of free (unconjugated) bilirubin.
• The inflammatory oedematous cells compress intracellular biliary canaliculi and this produces an element of obstruction. Consequently, the conjugated bilirubin formed in the hepatocytes may leak out of the damaged cells into the blood circulation. The conjugated bilirubin, therefore, appears in blood circulation in increased amount.
Since less conjugated bilirubin is produced in hepatocytes, the amount that enters the gut is also less, leading to decreased formation of urobilinogen and stercobilinogen. As a result, urinary and faecal excretion of urobilin and stercobilin falls.
C Obstructive Jaundice
In this case, jaundice results due to obstruction of the intrahepatic or extrahepatic bile duct (Table 16.2), though the bilirubin-conjugating capacity of the liver is more or less intact. The obstruction blocks the passage of the conjugated bilirubin into the intestine. It, therefore, accumulates in the hepatocytes and subsequently overflows into the blood circulation. As a result, concentration of the conjugated bilirubin rises in plasma. Since conjugated bilirubin is polar in nature, it can be excreted into the urine. Therefore, in obstructive jaundice bile pigments can be detected in urine. This accounts for the yellow-brown colour of urine.
Since amount of conjugated bilirubin carried by bile into the intestine is decreased, urobilinogen formation also decreases correspondingly. As a result, urinary urobilin and fecal stercobilin excretions fall. These pigments may be totally absent in case of complete obstruction (Table 16.3). So the stools no longer have brown colour, but appear clay coloured. Liver function is unimpaired in acute cases but longstanding cholestasis causes irreversible liver damage.
Conjugated bilirubin is excreted in urine due to its polar nature (obstructive jaundice), but unconjugated bilirubin is not excreted (haemolytic jaundice) because (i) It is non-polar, and (ii) it is complexed with albumin. This explains presence of bile pigments in urine in obstructive and hepatic jaundice, but their absence in haemolytic jaundice.
D Physiologic Jaundice of the Newborn
In neonates, transient jaundice is common, particularly in premature infants, and it occurs due to immaturity of the enzymes involved in bilirubin conjugation. There is accumulation of unconjugated bilirubin in blood circulation, but the serum bilirubin level generally does not increase above 5 mg/dL. Normally, about 50% of the normal babies become jaundiced 48 hours after birth in this manner. The jaundice normally disappears by the second week of life. However, if it is prolonged for more than 2 weeks, the infant must be treated by phototherapy.
The unconjugated bilirubin is transferred from its loose association with albumin to other proteins, such as those in cell membranes. It is neurotoxic, and if levels rise too high (15–20 mg/dL) in neonates, permanent brain damage can occur. Since blood-brain barrier is not fully mature till the baby completes one year of life, the excess bilirubin can readily cross it to reach the brain. Within brain, bilirubin deposition in the basal ganglia, hippocampus, cerebellum, etc. can cause irreversible brain damage, a condition known as kernicterus kern: (German-nucleus). Fits, spasticity, and mental retardation are the predominant clinical features. It is often fatal and leaves behind permanent neurologic deficit if the baby survives.
Mild hyperbilirubinaemia (< 5 mg/dL) occurs in about 50% of newborn due to immature or sluggish hepatic enzymes, that cause bilirubin conjugation. It needs to be treated only when the serum bilirubin levels cross 15—20 mg/dL by phototherapy.
Role of Phototherapy and Phenobarbital
Treatment for neonatal jaundice by phototherapy involves putting the baby under an ultraviolet light lamp. This induces the isomerization of water-insoluble bilirubin (termed the Z-isomer) to the water soluble E-isomer. The latter can then be cleared by the kidneys. The administration of phenobarbital (to induce conjugating system of liver) is indicated when the bilirubin level stays dangerously high despite phototherapy.
Blood exchange transfusion is required in extreme cases, when sustained increase in serum bilirubin above 20 mg/dL occurs.
Haemolytic Disease of the Newborn
Any haemolytic condition in the neonatal period is potentially dangerous, but the most serious is erythroblastosis fetalis or “haemolytic disease of newborn”. It occurs because of rhesus incompatibility: when a Rh negative mother conceives an Rh-positive baby. The Rh antigen crosses placental barrier to enter maternal circulation to induce synthesis of maternal IgG antibody. These antibodies are transferred across placenta and enter the fetal circulation, where they destroy red cells.
In Rh incompatibility, the first child often escapes, but in the second pregnancy the maternal antibodies enter fetus in large quantities to cause extensive red cell destruction even before birth. Sometimes the child is born with severe haemolytic disease the erythroblastosis fetalis. This may require exchange transfusion immediately after birth or even in the fetus before birth.
E Genetic Causes of Jaundice
There are a number of genetic disorders, referred to as inherited hyperbilirubinaemia, that impair bilirubin conjugation or secretion. Gilbert’s syndrome, Crigler-Najjar syndrome, Dubin Johnson’s syndrome and Rotor syndrome are some commonly described disorders. All except Gilbert’s syndrome are extremely rare disorders; the latter affects 5% of the population.
Gilbert’s syndrome
It is an autosomal dominant trait characterized by mild unconjugated hyperbilirubinaemia that is harmless and asymptomatic. Some cases appear to involve a defect in hepatic uptake, but more commonly there is a modest impairment in conjugating enzyme, glucuronyl transferase, activity. There are no other signs, symptoms or laboratory abnormalities, and the prognosis is excellent.
Crigler—Najjar syndrome
This results due to complete absence or marked reduction in bilirubin conjugation system. Two types are known:
Type I: It is a rare variety in which there is a complete absence of the glucuronyl transferase activity, resulting in severe unconjugated hyperbilirubinaemia that presents at birth. Most affected infants die from bilirubin-induced neurological damage before reaching 1 year of age.
Type II: Patients with type II disease have a partial deficiency of glucuronyl transferase. The serum bilirubin concentration is lower than in type I, and neurological complications are unusual.
Inheritance is autosomal recessive for the type I disorder and autosomal dominant for type II.
Dubin—Johnson and Rotor syndrome
These syndromes impair the biliary secretion of conjugated bilirubin, and therefore, cause conjugated hyperbilirubinaemia, which is usually mild. The Dubin-Johnson syndrome is distinguished by the dark pigment that accumulates in hepatocytes. Inheritance is autosomal recessive and the prognosis is excellent.