Oxygen Transporters: Haemoglobin And Myoglobin
Molecular oxygen is sparingly soluble in body fluids. This limits the amount that can be transported in physical solution to less than 30 g per day, whereas the body requires approximately 500 g of molecular oxygen per day. An effective oxygen transport is made possible by haemoglobin, the oxygen-binding protein, present in the erythrocytes. Because of the oxygen-binding properties of this protein, blood can dissolve approximately 70 times more oxygen than the plasma free of haemoglobin can do.
Just as haemoglobin is the oxygen-binding protein in erythrocytes, the myoglobin binds oxygen in muscle tissue. It permits storage of oxygen in muscles. The stored oxygen is released when it is required, for example, during vigorous muscular exercise. Binding of oxygen to haemoglobin and myoglobin is called oxygenation.
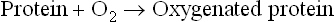
It is a non-enzymatic, reversible process. Therefore, oxygen, when available in plenty, binds to the oxygen-binding proteins. Conversely, oxygen is released from these proteins when it is scarce. None of the constituent amino acids of these proteins has any binding affinity for molecular oxygen. Therefore, the oxygen-binding proteins have to employ a non-protein component to serve as the oxygen-binding site. This component is haem in haemoglobin as well as in myoglobin.
In this chapter, general characteristics and oxygen-binding properties of haemoglobin and myoglobin are described. After going through this chapter, the student should be able to understand:
• General properties of haemoglobin, such as structure of haem, organization of various globin chains, positioning of iron in the tetrapyrrole ring and mode of binding of oxygen; structural and functional peculiarities of various types of haemoglobins.
• Behaviour of haemoglobin as an allosteric oxygen binder, sigmoid shape of the oxygen saturation curve of haemoglobin, Bohr effect, and role of 2,3-BPG in oxygen off-loading.
• Aetiology and oxygen-binding peculiarities in various types of haemoglobinopathies.
• Dxygen-binding properties of myoglobin and its suitability to store oxygen in muscles.
I Haemoglobin
Haemoglobin is a tetrameric metalloprotein consisting of four polypeptide chains, each with its own haem. In addition to carrying oxygen from lungs to the peripheral tissues, it carries carbon dioxide from actively metabolizing tissues to lungs. Haemoglobin possesses buffering property as well.
Upon oxygenation, the haemoglobin is converted to oxyhaemoglobin. This is not a simple metal-gas interaction, but is a dynamic process subject to various allosteric controls. Binding of other ligands such as carbon dioxide (and H+) to haemoglobin is also regulated by allosteric mechanism. The allosteric mechanism is very well understood in case of haemoglobin, and discussed in detail in this chapter.
A Basic Structure
Humans have several types of haemoglobins. All types are made up of two α chains and two non-α chains (β, δ or γ) with a total molecular weight of approximately 65,000. These chains, collectively termed globin polypeptides, differ in primary structures but have similar secondary and tertiary structures. The differences in tertiary structure among them are critical to the functional properties of each. A non-covalently bound haem group with a molecular weight of 616 is associated with each chain (Fig. 17.1 ). It should be noted that all globin polypeptides are homologous proteins, having arisen from the same ancestral gene.
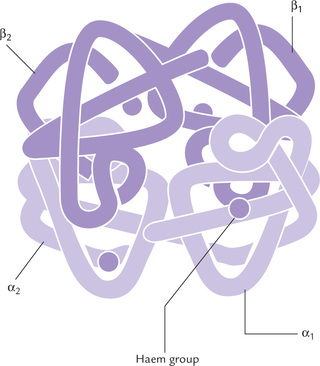
Fig. 17.1 Structure of HbA molecule consisting of two α-chains and two β-chains, each with a non-covalently bound haem group.
Globin chains
The globin chains present in the major adult haemoglobin, called HbA, are α and β, and its subunit structure is α2β2. The minor adult haemoglobin (HbA2) and fetal haemoglobin (HbF) also have two α-chains but instead of the β-chains they have δ-chains and γ-chains respectively (Fig. 17.2 ).
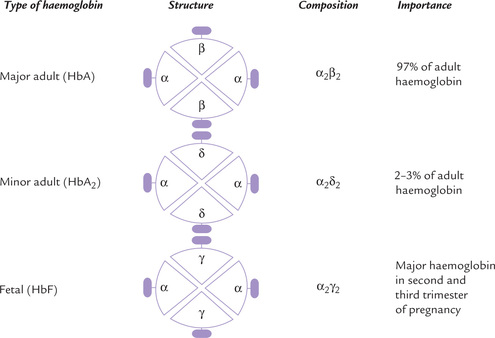
Fig. 17.2 Subunit structure and importance of various haemoglobin types. A subunit consists of a polypeptide chain and its haem group (  = haem,
= haem,  = polypeptide chain).
= polypeptide chain).
The α-chain has 141 amino acids (MW 15,750) and the β-chain has 146 amino acids (MW 16,500). Like the β-chains, the other two non- α-chains (δ and γ) also have 146 amino acids and are structurally related.
The β- and δ-chain differ in 10 and the β- and γ-chains in 39 of their 146 amino acids. The α- and β-chains are not as closely related: they are identical only in 64 of their amino acids.
Secondary and tertiary structures of globin polypeptides
The secondary structure of all globin polypeptides consists largely of α-helical segments and short stretches of various bends and interhelical segments. α-chain has 7, and β-chain has 8 helical segments, labelled as A (N-terminal) through H. They are organized into a tightly packed, nearly spherical, globular tertiary structure, shown in Figure 17.1.
Like most soluble proteins, haemoglobin has a relatively hydrophilic surface and a hydrophobic interior. Polar amino acids are located almost exclusively on the exterior surface of the globin polypeptides, and contribute to their remarkably high solubility in cytoplasm of erythrocytes (5.2 mmol/L). This permits building up of a high intracellular concentration of Hb within red cells, which appear as “bags” filled with Hb. Amino acids having both polar and non-polar side chains, such as tyrosine, threonine and tryptophan, are oriented with their polar functions toward the protein’s exterior. Hydrophobic amino acids are buried within the interior, where they stabilize the folding of the polypeptide and binding of the iron-porphyrin prosthetic group.
 Polar amino acids, located towards the exterior, make haemoglobin highly soluble, whereas the non-polar amino acids are buried in the interior and influence folding.
Polar amino acids, located towards the exterior, make haemoglobin highly soluble, whereas the non-polar amino acids are buried in the interior and influence folding.
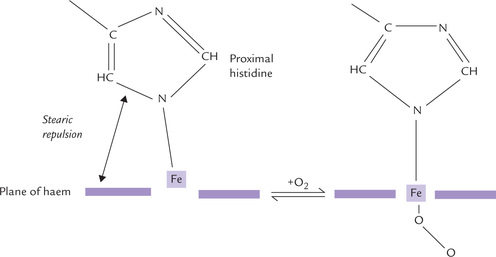
The only exception to this general distribution of amino acid residues in globins are the two histidine residues, termed proximal- and distal-histidines, which are oriented perpendicular to and on either side of the planar haem prosthetic group. They create a hydrophobic pocket in which the haem-group resides.
Haem group
A haem group is associated with each globin chain in the hydrophobic interior (tucked between the E and the F helices). Haem consists of a porphyrin ring system with various side chains: methyl, vinyl and propionate (Chapter 16). A ferrous iron, fixed in the centre of the ring through complexation to the nitrogen atoms of the four pyrrole rings, is functionally the most important element (Fig. 17.3 ).
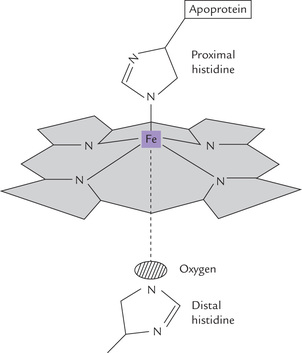
The ferrous iron has an octahedral geometry and can be attached to six ligands:
• Four of these ligands are the pyrrole nitrogen atoms of the porphyrin ring system.
• Fifth is an imidazole nitrogen of the so called proximal histidine.
• The sixth ligand is molecular oxygen bound between the iron and the imidazole side chain of distal histidine.
In deoxyhaemoglobin, the iron is coordinated with only five ligands since oxygen is not present.
Proximal and disfai histidine
They are located on either side of the haem prosthetic group. Side chains of both these histidines are oriented perpendicular to the planar haem group. The proximal histidine is bound to iron, and the distal histidine guards the entrance on the opposite site (Fig. 17.3). Their presence ensures that the immediate environment of the haem iron is the same (i.e. hydrophobic).
The ferrous iron in haem
The haem iron occurs in the ferrous state in haemoglobin (and myoglobin). During oxygen binding, it is not oxidized to the ferric form; it becomes oxygenated but not oxidized. Oxidation to the ferric state converts the haemoglobin to methaemoglobin, which is functionally inactive. It can no longer carry oxygen. Oxidation to ferric form imparts a net positive charge (haem is neutral), therefore, a negative counter ion must be associated with the molecule. If the counter ion is Cl” then the compound is haemin; if it is OH” then the compound is haematin.
B Quaternary Structure
Haemoglobin is a tetramer of four subunits, each subunit consisting of a polypeptide chain with a non-covalently bound haem group. Packing of these subunits relative to one another is referred to as the quaternary structure, as shown earlier in Figure 17.1.
The four polypeptide chains are visualized as comprising two identical dimers, each dimer having an α- and β-chain. The dimers are designated (αβ)1 and (αβ)2, where the numbers refer to dimer 1 and dimer 2. Two chains within a dimer are held together tightly by the ionic bonds and the hydrophobic interactions, which prevent their movement relative to each other. However, the two dimers are linked with each other by weak polar bonds, so that movement at the interface of these two occurs more freely.
C Allosteric Effects
Oxygen-binding characteristics of haemoglobin change by the following effectors:
• pO2: The partial pressure of oxygen (pO2).
• pCO2: The partial pressure of carbon dioxide (pCO2).
These effectors are collectively called allosteric (“other site”) modulators because their interaction at one site of haemoglobin molecule influences the binding characteristics of some other site within the same molecule.
The ligand-binding information is communicated withir the haemoglobin molecule from one subunit to another Such subunit interaction makes the haemoglobin wel adapted for integrating the transport of oxygen, carbon dioxide and H+.
Binding of Oxygen: Positive Cooperativity
Binding affinity of haemoglobin for oxygen changes with the state of oxygenation. As the first oxygen molecule binds with one of the haem groups, the binding affinity of the haemoglobin for the second oxygen molecule is enhanced. As a result, the second oxygen molecule binds much more easily. The same process is repeated for the third and fourth oxygen molecules.
Effectively, binding of oxygen with one of the haem groups of haemoglobin increases the oxygen affinities of the remaining haem groups. This effect is called positive cooperativity. It indicates a cooperative interaction between different subunits of haemoglobin (Box 17.1) because the ligand-binding information is transmitted from one subunit to another.
These characteristics are shown by the sigmoid (S-shaped) curve of oxygen dissociation of haemoglobin (Fig. 17.4 ). This curve describes the fractional saturation of the haem groups at various oxygen partial pressures. The initial flat region reflects low oxygen affinity of the deox-ygenated haemoglobin for the first oxygen. With increasing oxygen partial pressure, however, the first haem group gets oxygenated. This facilitates subsequent binding of oxygen molecules. Therefore, the curve becomes steeper, indicating that subsequent oxygen molecules are bound with much higher affinity. In fact, the fourth oxygen molecule binds haemoglobin 300 times as tightly as the first; the binding affinities of the second and the third oxygen molecules are intermediate between those of the first and the last.
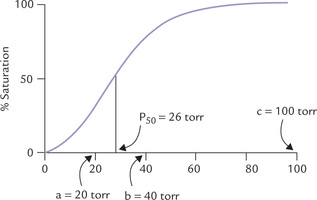
Fig. 17.4 The sigmoid curve of oxygen dissociation of haemoglobin. The arrows indicate: a = pO2 in capillaries of working muscles; b = pO2 in capillaries of resting muscles; c = pO2 in alveoli of lungs, P50 indicates 50% saturation of haemoglobin when pO2 equals 26 torr.
Oxygen binding to a haem group in haemoglobin increases the oxygen affinities of the remaining haem groups; this effect is positive cooperativity and it leads to sigmoid oxygen-binding curve.
The oxygen dissociation curve is characterized by P50 value, the oxygen pressure at which the haemoglobin is 50% saturated with oxygen. Its value is 26 torr. It should be noted that in proteins that are not allosteric and do not show coooperative oxygen-binding kinetics, have a simple hyperbolic curve with low P50; this is what is observed with myoglobin (see Fig. 17.14).
Significance of Positive Cooperativity
The cooperative binding of oxygen enhances the efficiency of haemoglobin as an oxygen transporter. Without positive cooperativity, an 81-fold increase of the pO2 would be required to raise the oxygen saturation from 10% to 90%. In case of haemoglobin, however, about fivefold increase is sufficient to do the same. This means that haemoglobin can rapidly bind oxygen in lungs (where pO2 is high) and then readily liberate it in the tissue capillaries (where pO2 is low).
This may be seen from the oxygen dissociation curve:
• Haemoglobin is 96% saturated with oxygen in lungs (pO2 = 100 torr).
• In resting muscle (pO2 = 40 torr) it is only about 64% saturated (Fig. 17.4). Thus, it delivers 32% (96% minus 64%) of its oxygen content to the resting muscle.
• In exercising muscles, further fall in pO2 occurs because of rapid oxygen consumption. Since haemoglobin is only about 20% saturated at this partial pressure (Table 17.1 ), further liberation of oxygen occurs.
Table 17.1
Per cent saturation of haemoglobin at different partial pressures of oxygen
| pO2 (toor) | Per cent saturation of Hb |
| 100 (in alveoli) | 96 |
| 40 (in resting muscles) | 64 |
| 20 (in working muscles) | 20 |
| 10 (in vigorously exercising muscles) | 10 |
• Likewise, in the vigorously exercising muscles (pO2 = 10 torr), where oxygen need is maximum, the saturated haemoglobin may release up to 90% of its oxygen content.
Note
Torr is a unit of pressure equal to that exerted by a column of mercury 1 mm high at O°C and standard gravity (1 mmHg). This unit is named after Evangelista Torricelli, the inventor of mercury barometer.
Bohr Effect
The oxygen affinity of haemoglobin exhibits exquisite sensitivity to pH; the affinity decreases as the pH becomes more acidic (Fig. 17.5 ). This is known as the ‘Bohr effect’ after the discoverer Christian Bohr, father of Neil Bohr, the atomic physicist.
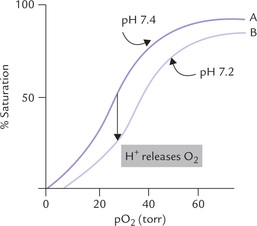
Fig. 17.5 Effect of pH on the oxygen affinity of haemoglobin. The decreased affinity in response to fall in pH is reflected by rightward shift of the oxygen dissociation curve.
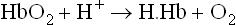
Closely related to the Bohr effect is the ability of carbon dioxide to alter the oxygen affinity of Hb. Like the negative allosteric effect of H+, increasing levels of CO2 decrease affinity for oxygen. Thus, there exists an inverse relation of oxygen binding affinity of haemoglobin vis-á-vis binding of CO2 and H+.
Decreased affinity of haemoglobin for oxygen is reflected as rightward shift of the oxygen dissociation curve. It represents oxygen-offloading.
Effect of CO2 on oxygen affinity is mediated via [H+] because in solution there exists the equilibria:
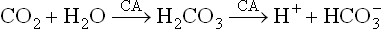
These reactions are enhanced in the red blood cells through the action of carbonic anhydrase (CA). Thus, rise in pCO2 induces a decrease in pH, which then causes a loss of oxygen from oxyhaemoglobin.
Significance of Bohr Effect
Bohr effect changes the oxygen-binding characteristics of haemoglobin according to the requirements of the tissue. Since an increase in pCO2 and a decrease in pH are both characteristics of actively metabolizing cells, these cells promote the release of oxygen from oxyhaemoglo-bin. This is desirable since the actively metabolizing tissues require an adequate supply of oxygen.
A closer look at the curves A and B (Fig. 17.5) will illustrate this point further. At tissue pO2 = 40 torr, haemoglobin will be saturated ~64% at physiologic pH (curve A), and ~50% at acidic pH (curve B). In lungs the haemoglobin is 96% saturated with oxygen. Thus, for curve A, 96 — 64% = 32% of the oxygen bound to haemoglobin would be released to the tissues, whereas for curve B the rate of delivery is 96 — 50% = 46%. Thus, a rightward shift of the curve provides a mechanism whereby additional oxygen can be supplied to tissues.
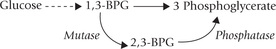
Effect of 2,3-bisphosphoglycerate (2,3-BPG)
This compound is an alternative intermediate in the glyco-lytic pathway, and serves as the third important modulator of oxygen affinity (others are oxygen and carbon dioxide/ protons). It is formed in the human erythrocytes from 1,3-bisphosphoglycerate (1,3-BPG), where it is present at a concentration of approximately 5 mM, roughly equimo-lar with haemoglobin. Like H+ and CO2, 2,3-BPG is a negative allosteric effector of oxygen binding to haemoglobin: it causes a marked increase in P50, thereby causing unloading of oxygen from oxyhaemoglobin. Indeed, if it were not for the high erythrocyte concentration of 2,3-BPG, the oxygen saturation curve of Hb would approach that of myoglobin.
In hypoxic states, production of 2,3-BPG is increased which enhances release of oxygen from oxyhaemoglobin (Fig. 17.6 ).
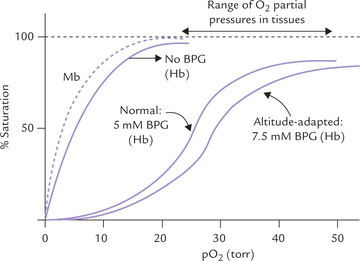
Fig. 17.6 Effect of 2,3-BPG on the oxygen-binding affinity of haemoglobin (Hb). Oxygen binding by myoglobin (Mb) is also shown (---).
Rapoporf-Luebering Cycle
It is a shunt pathway in RBCs, that bypasses the glycerophosphate kinase reaction (step 6) of glycolysis. As diagrammed above, the 2,3-BPG is produced from 1,3-BPG, an intermediate of glycolysis, which is hydrolyzed to 3-phosphoglyc-erate. This is useful not only in oxygen-offloading, but also in dissipation of energy, as described earlier in Box 9.1.
Clinical Significance of 2,3-BPG
BPG has an indispensable physiological function. As noted, arterial blood, haemoglobin is ~96% saturated, and in venous blood (pO2 = ~40 torr) it is only 64% saturated, and so unloads 32% (96-64%) of its bound oxygen in passing through capillaries. In the absence of BPG, little of this oxygen would be released, since haemoglobin’s oxygen affinity will be increased, thus shifting the oxygen dissociation curve significantly towards the left (Fig. 17.6). The erythrocyte concentration of BPG is responsive to various physiologic and pathologic conditions: it increases when there is deprivation of oxygen, including lung diseases, severe anaemia and during adaptation to high altitude. This enhances unloading of oxygen in the tissues, thereby ensuring adequate oxygenation.
2,3-BPG accounts for oxygen-binding peculiarity of the fetal haemoglobin, discussed later.
Role in Stored Blood
2,3-BPG plays an important role in blood that has been stored for transfusion. The concentration of 2,3-BPG shows a drastic fall in the red blood cells from 2.2 mM to one-tenth of this concentrations within a few days of storage. This causes increased affinity of Hb for oxygen, so that the transferred blood cannot effectively release O2 to peripheral tissues. This problem is solved by addition of inosine (ribose + purine base). Haemoglobin releases a significant amount of bound oxygen while passing through capillaries of metabolizing tissues. 2,3-BPG enhances this release to the medium, which is readily transported into red cells (2,3-BPG cannot move across cell membrane). Inside the cell, ribose moiety of inosine is split off from purine base and the former is metabolized (via a section of the HMP shunt and glycolysis) to produce 2,3-BPG.
Haemoglobin responds to body requirements and alters its binding properties accordingly. It can sense changes in the concentration of any of its four ligands—oxygen, carbon dioxide, H+ and 2,3-BPG. The message is transmitted between different subunits through change in the conformation of the molecule.
Interactions between different ligands, such as BPG and oxygen, are called heterotropic effects and interactions between identical ligands, as in the cooperativity of oxygen binding, are called homotropic effects.
D Molecular Mechanism of Allosteric Effects*
The allosteric effects depend on transmission of molecular signal from one subunit of the protein to another. The mechanisms underlying these effects were elucidated following the discovery that the subunits of haemoglobin can take on either one of the two conformations: the R (relaxed) and T (tense) form. The overall interactions between the subunits in the T form are stronger than in the R form, as discussed below.
The Tense (T) Form
The deoxyhaemoglobin conformation is called the tense (T) or taut form, in which the subunits are held together by eight salt bonds between polypeptides in addition to a number of hydrogen bonds and other non-covalent interactions.
The Relaxed (R) Form
It is a less constrained form produced by tearing away of some of the salt linkages that stabilize the T form. Binding of oxygen to the T form causes rotation of an aβ pair relative to the other aβ pair by 15 degrees. This brings about rupture of some of the salt linkages to produce the R form.
The binding affinity of the R form for oxygen is 150 - 300 times greater than that of the T form. Hence, the R form has greater tendency for getting oxygenated. Thus, any allosteric modulator that favours formation of the R form (i.e. T to R shift) favours oxygen binding. Conversely, the allosteric modulators favouring R to T shift increase oxygen liberation.
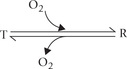
With this information, it is now possible to understand the mechanism of oxygenation, the Bohr effect and to appreciate role of protons at molecular level.
Mechanism of Positive Cooperativity in Oxygenation
Binding of oxygen to haemoglobin causes rupture of some of the salt linkages, thus favouring the T to R shift. Since the binding affinity of the R form for oxygen is higher than that of the T form, it accepts oxygen molecules readily. Thus, T → R transition explains why binding of the first oxygen enhances the binding of subsequent oxygen molecules to the other haem groups in the haemoglobin molecule.
Haemoglobin has two conformational states: the T state (conformation of deoxyhaemoglobin) and the R state (conformation of oxyhaemoglobin). Oxygen binding to a haem group induces a T to R transition, which in turn induces con-formational change in the entire haemoglobin molecule.
How does oxygen binding cause rupture of salt linkages for T to R transition? Detailed information about the molecular mechanisms responsible for this has been elucidated by comparative X-ray analysis. In the deoxygenated state, the iron atom is about 0.6Å out of the haem plane, due in part to stearic repulsion between the proximal histidine and the nitrogen atoms of the prophyrin ring (Fig. 17.7 ). Oxygen binding induces certain changes in the haem’s electronic state, which shortens the Fe-porphyrin bonds by ~0.1 Å. Consequently, the iron atom moves into the centre of the haem plane, where it can more tightly bind oxygen, and in doing so it drags the covalently linked proximal histidine (F8 His) and its attached F helix. A subtle conformational change is thereby induced, which strains other non-covalent bonds elsewhere in the subunit causing some of them to break. This is beginning of a series of reactions leading to the quaternary structure transition from the deoxy-T form to oxy-R form.
In defining transition from T to R form, a number of models have been developed (Box 17.2).
Mechanism of Bohr Effect
The principal residues involved in Bohr effect are the N-terminal amino groups of the α-chains, the C-terminal carboxy groups of β-chains and the imidazole side chains of His122α. They interact with protons to acquire more positive charge. The protonated, positively charged residues then form electrostatic interactions with negatively charged residues, thus converting the R forms into T form and thereby releasing oxygen.
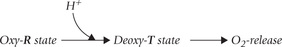
For every equivalent of oxygen released from haemoglobin, about 0.3 equivalents of [H+] is bound. The bound H+ are called Bohr protons. They are released in the lungs, where pO2 is high and pCO2 is low, and the T form changes back to R form.
Binding of Bohr protons to certain amino acid residues favours formation of new bonds, thus causing R → T transition and off-loading of oxygen from oxy-haemoglobin.
Effect of CO2
Lowering of oxygen-binding affi nity of haemoglobin by carbon dioxide is effected by the spontaneous covalent binding of CO2 with the N-terminal group (valine) of each of the globin polypeptides to give arbamino complexes.
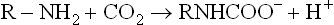
Deoxyhaemoglobin because of its higher pI (7.6) has a greater affi nity for CO2 than oxyhaemoglobin (pI = 6.7). When the CO2 concentration is high, as it is in the capillaries, the deoxy-T state is favoured, stimulating haemoglobin to release its bound oxygen. The H+ produced in the above reaction further promotes oxygen release through the Bohr effect. Clearly, effects of H+ and CO2 are often tightly associated, both promoting oxygen release from oxyhaemoglobin.
It may be re-emphasized that like the pH effect, the CO2 effect ensures that oxygen is released in actively metabolizing tissues, where it is most needed. On return to the lungs, blood is exposed to low pCO2 and by law of mass action the carbamination reaction is reversed and T to R conversion occurs, therefore binding of oxygen is again favoured.
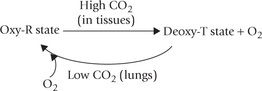
Mechanism of Oxygen Off-loading by BPG
The oxygen unloading effect of 2,3-BPG is accounted by the fact that it binds tightly to the deoxy-T state of haemoglobin but only weakly to the oxy-R state. The result is stabilization of the T conformation, which has low oxygen-binding affinity. Moreover, the presence of 2,3-BPG also lowers intracellular pH (6.95). This dual role leads to the observed effect of oxygen unloading. How does 2,3-BPG stabilize the deoxy-T state? The X-ray structure of a BPG-deoxyhaemoglobin complex shows that BPG binds in the central cavity formed among the four subunits of Hb. The binding site for BPG is created at one end of this cavity by multiple positive charges, contributed by several residues, e.g. 2nd histidine, 143rd histidine, 82nd lysine in β -chain. BPG interacts electrostatically with these and the interaction in turn stabilizes the complex between the effector (BPG) and the Hb in its deoxy-T state.
II Haemoglobin
A Fetal Haemoglobin (HbF)
Fetal haemoglobin differs from the adult haemoglobin in having γ-polypeptide chain, a variant of the β-chain (subunit composition α2γ2; see Fig. 17.2). While it accounts for less than 1% total adult haemoglobin in adults, HbF predominates in fetus during the second and third trimesters of gestation. Its synthesis starts by seventh week of gestation and at birth it accounts for about 75% haemoglobin. There is rapid post-natal decline and within 4 months after birth HbF is almost completely replaced by HbA (Fig. 17.8 ).
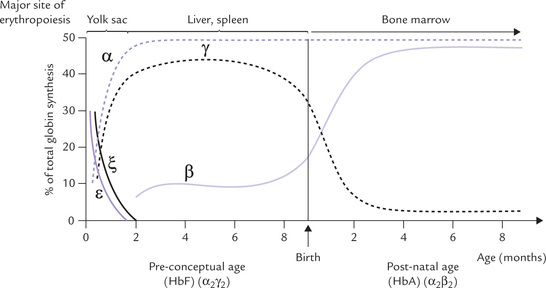
Fig. 17.8 Human haemoglobin polypeptide chains as a function of time during gestation and post-natal development.
Since the γ-chain differs from β-chain in 39 of the residues, the physicochemical properties of the HbF isoform are different: its electrophoretic mobility is slower and the deoxy HbF has higher solubility than HbA. The most striking difference between the two is: HbF has lower affinity for 2,3-BPG (because the residue 143 of the β -chain, which is histidine in HbA but is replaced by serine in HbF; the absence of this histidine eliminates a pair of interactions that stabilize the BPG-deoxyhaemoglobin complex). As a result, affinity of HbF for oxygen (P50 = 19 torr, vs 26 torr for HbA) is much higher and formation of the oxygenated R form is favoured. The oxygen dissociation curve in fetus consequently is shifted to left, a direct benefit of which is that there is a more efficient transplacental transfer of oxygen from maternal HbA to fetal HbF.
HbF is elevated up to 15-20% in individuals with mutant adult HbS, such as sickle cell disease where it has a compensatory effect. Children with anaemia and β -thalassaemia also show such compensatory response.
B Embryonic Haemoglobins
The non-α-chains present in embryonic haemoglobins are the embryonic chains, i.e. epsilon (ε) and zeta (ξ). Expression of these chains is limited to early embryonic stage, from 3rd to 8th week of gestation (Fig. 17.8). The embryonic haemoglobin Gower-2 has two alpha and two epsilon chains, and the haemoglobin Gower-1 comprises only embryonic chains.
C Minor Adult Haemoglobin (HbA2)
It is a normal variant of adult haemoglobin, having δ-chains instead of β (subunit composition α2δ2; Fig. 17.2). In β-thalassaemia, concentration of this form of haemoglobin is increased as a compensatory measure. Its pI value is 7.4, which is higher than that of HbA. Therefore, it shows slower anodic mobility on electrophoresis.
III Haemoglobin Derivatives
Haemoglobin derivatives are formed by:
• Change in oxidation state of haemoglobin, e.g. meth-aemoglobin, a non-functional oxidized form of haemoglobin.
• Combination of different ligands with the haem part of the haemoglobin molecule. For example, carboxy haemoglobin is formed by binding of carbon monoxide to the haem iron; sulph-Hb, by its binding with hydrogen sulphide.
Haemoglobin derivatives can be detected by their characteristic absorption spectra.
A Methaemoglobin
The haem iron of haemoglobin is present in the ferrous state. Its oxidation to the ferric form produces haemin from haem, and the haemoglobin molecule is now called methaemoglobin. Methaemoglobin is ineffective as an oxygen transporter.
Normally, about 1% of the circulating haemoglobin occurs in form of methaemoglobin. Certain chemicals cause rapid ferrous to ferric conversion, resulting in production of methaemoglobin. Examples include organic and inorganic nitrites, aniline dyes, and aromatic nitro compounds. The methaemoglobin production by these agents leads to conditions, collectively referred to as acquired methaemoglobinaemias. In contrast to the acquired forms, the congenital methaemoglobinaemia results mostly because of defect in the cytochrome b5 reductase system, as discussed below.
Methaemoglobin is a non-functional oxidized form of haemoglobin formed by oxidizing agents that oxidize the ferrous haem iron to the ferric state.
Since the methaemoglobin formation results in loss of oxygen carrying capacity, it is hazardous and must be prevented. Erythrocytes have certain defensive mechanisms to prevent excessive formation of methaemoglobin. They are described below.
• Ascorbic acid and glutathione are reducing substances which destroy many oxidizing radicals, thus preventing them to react with haemoglobin (Chapter 27).
• The methaemoglobin that is spontaneously produced can be reduced back to haemoglobin by the methaemoglobin reductase system present within the RBCs (Fig. 17.9 ).
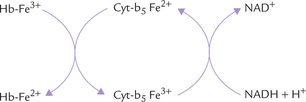
This enzyme system uses NADH as the reductant. Cytochrome b5 and cytochrome b5 reductase are the other essential components of this system. Impaired function of this system leads to congenital methaemo-globinaemia (Case 17.1).
• The polypeptide chains of haemoglobin contain a protected pocket each between the E and the F helices, in which the haem molecule is tucked in. These pockets offer protection against oxidation of haem. This is evident from the fact that when the haem molecule is dissociated from the polypeptide chain, it rapidly gets oxidized to haemin.
The distal histidine residues appear to play an important role of creating a protective environment for haem. Replacement of this histidine by a tyrosine residue, for example, favours production of methaemoglobin.
Defect in the cytochrome b5 reductase system, or structured abnormality in the polypeptide chain(s), result in congential methaemoglobinaemia. It is a mild condition and does not manifest clinically under normal circumstances. However, use of certain drugs, as mentioned earlier, results in excessive production of methaemoglobin and the oxygen deficiency symptoms appear.
Treatment of methaemoglobinaemia involves conversion of the ferric iron back to the ferrous state. Methylene blue is commonly used for this purpose.
B Carboxy Haemoglobin
The toxic effects of carbon monoxide (CO) were explained first by John Scott Haldane on the basis of competition between carbon monoxide and oxygen for binding the haem iron. In fact, the affinity of carbon monoxide for Hb is 200 times more than that of oxygen.
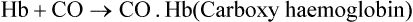
Since CO.Hb is unsuitable for oxygen transport, its binding renders the haemoglobin functionally inactive (Case 17.2).
C Glycated Haemoglobins
These are the haemoglobin derivatives that are produced by non-enzymatic post-translational modification (e.g. glycation) of the β-chains. They make up about 4-6% of the total haemoglobin in normal red blood cells. Being present in very small amounts, the glycated haemoglobins are not pathological, but rather serve a useful purpose of monitoring patient’s compliance in diabetic state or even in diagnosis of diabetes (Chapter 15).
D Sulfhaemoglobin
It is a greenish compound produced by covalent attachment of sulphur to the porphyrin ring (not iron atom). Sulfhaemoglobin cannot combine with oxygen. Exposure to sulphur containing compounds, either occupationally or from air pollution, or depsone (used to treat leprosy) can produce this compound. In poisoning by phenace-tin, acetanilide or sulfanilamides, sulfhaemoglobin is produced. These drugs produce methaemoglobin also, so that sulfhaemoglobin and methaemoglobin appear together in these patients.
E Cyanmethaemoglobin
Haemoglobin cannot combine with cyanide, but methae-moglobin reacts with cyanide forming cyanmethaemoglobin (cyanmet-Hb). This property is used as a treatment modality in cyanide poisoning. First, the patient is given nitrite plus sodium thiosulphate, which converts haemoglobin to met-Hb. The latter then traps cyanide as cyanmet-Hb, which is a non-toxic compound.
IV Haemoglobinopathies
Haemoglobinopathies are a family of disorders resulting due to mutations in genes for the haemoglobin chains. The mutation may cause synthesis of defective globin chain or may even stop the synthesis altogether. More than 400 different mutations have been described, which result in as many structural variants of the haemoglobin polypeptide chains. Two types of defects arise due to these mutations: the qualitative defects and the quantitative defects.
Qualitative defects lead to production of structurally abnormal haemoglobin molecules. These defects involve structural alteration in the polypeptide chain of the haemoglobin, leading to functional impairment. Replacement, addition and deletion of the amino acids are some common structural alterations.
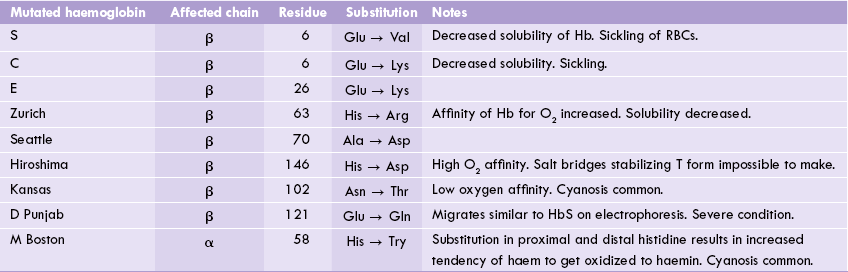
A number of qualitative defects (over 300) have been identified, which may affect either of the two types of chains. For example, in sickle cell anaemia, the β-chain is affected, whereas in HbM Boston the α-chain undergoes structural alteration (Table 17.2 ).
Quantitative defects result in synthesis of the normal haemoglobin molecules, but in insufficient quantities. Prime example of this type is the thalassaemias in which either the α- or β-chains are underproduced. Rarely both these types of defects occur together in a single disorder.
A partial listing of haemoglobinopathies is given in Table 17.2. By convention, the haemoglobinopathies are usually named with a capital letter (e.g. haemoglobin S), a geographic location (e.g. haemoglobin Seattle), or both (e.g. haemoglobin M Boston or D Punjab).
Diagnostic Analysis of Normal and Mutant Haemoglobins
All significant Hb variants and many of the more common mutants, including HbS and HbC, can be separated by electrophoresis. The process is carried out at pH 8.6 at which these proteins are negatively charged, and therefore, move towards anode.
Change in amino acid composition of a globin chain may produce a haemoglobin with a different electropho-retic mobility. By examining the position and relative amount of the haemoglobin bands, it is possible to diagnose the most common haemoglobinopathies. For example, in sickle cell anaemia, the predominant haemoglobin (85-95% of the total) is haemoglobin S (HbS); its position on electrophoretogram is shown in Figure 17.10 . The HbS has lower anodic mobility because of removal of a negative charge by the sickle cell mutation (Glu →.Val). Likewise, any mutant exhibiting a net gain or loss of charged residues when compared with HbA can be detected by this method.
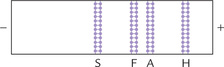
The volume of blood sample required is less than 100 μ1, making this technique acceptable for even neonates. Quantification is performed by scanning densitome-try. Another method, isoelectric focusing, also based on differences in pI, separates the proteins rapidly in a pH gradient generated by an electric field (see Chapter 4).
A Sickle Cell Anaemia
It is the most commonly occurring haemoglobinopathy caused by an inherited structural abnormality in the β globin polypeptide. Originally described in 1910 by James Herrick, the disease was the first inherited disorder shown to arise from a specific amino acid change in a protein. Since then it has been so extensively studied, biochemically and biophysically, that it has virtually become a paradigm of a molecular disease.
Molecular Defect
The sickle cell haemoglobin (HbS) arises out of a substitution of the glutamic acid residue in position 6 of the β-chain with the valine residue (β6Glu → Val). As noted earlier, this substitution can be traced to an A → T trans-version in the β-chain gene and results in formation of an abnormal haemoglobin of subunit composition a2βs2 (Fig. 17.11 ). Both, homozygous and heterozygous forms of disease have been described. The heterozygous form is also referred to as sickle cell trait.
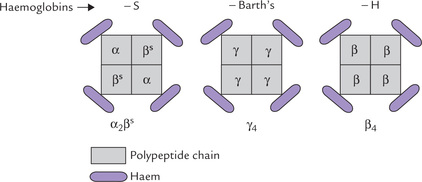
Fig. 17.11 Structure and subunit composition of different haemoglobins: HbS, HbH and Hb Barth’s. HbS is the predominant haemoglobin type in sickle cell anaemia, whereas HbH and Hb Barth’s occur in thalassaemia.
Sticky Patches and Tubular Polymer Formation
Replacement of a negatively charged glutamic acid with a hydrophobic valine changes surface properties of the molecule, especially its solubility. It becomes less soluble in water in its deoxygenated form, although the oxygen-binding affinity remains unaffected. In a chain comprising of 146 amino acids, a single substitution may appear minor, but its effects are far from trivial. This is because the substituted valine forms a hydrophobic pocket, the so called sticky patch, on the surface of the molecule which is reactive to the nearby Hb molecules.
The X-ray structure of deoxyhaemoglobin S has revealed that one mutant valine side chain in each HbS molecule fits into a sticky patch on the surface of another haemoglobin molecule. This intermolecular contact allows HbS to form linear polymers of deoxy HbS. These polymers distort the RBCs into sickle shape, which in highly vulnerable to lysis.
The hydrophobic pocket on surface of the β-subunit is not seen in case of oxyhaemoglobin, therefore oxyhae-moglobin S cannot polymerize. This also explains why danger of polymerization is greatest in hypoxic conditions.
Clinical Features
Sickle cell anaemia is a severe-haemolytic, painful, debilitating, and often fatal disease characterized by erythrocytes of distorted shapes. The stiff polymers form precipitate in the erythrocyte, which causes distortion of shape of the RBCs into sickle shape (i.e. sickling). At first the sick-ling is reversible, but after some time the oxygenation-deoxygenation cycle permanently deforms the shape of the erythrocyte. The deformed cells increase the viscosity of blood and cause obstruction to blood flow in small vessels and capillaries. The resulting vascular occlusion may result in tissue death (infarction). Painful bone and joint infarctions are common; multiple renal infarcts can lead to renal failure; and many patients are crippled by recurrent CNS infarctions.
Recurrent attacks of sever pain in extremities, bones or abdomen, known as painful crisis or sickling crisis are experienced by most patients. Others may experience aplastic crisis (bone marrow failure), or sequestration crisis where sudden trapping of erythrocytes in the enlarged spleen occurs; the latter is associated with high mortality. Renal failure, cerebrovascular accidents, cardiac failure and liver diseases are the other important causes of death.
Haemoglobin S forms rigid fibres in the deoxy form within the erythrocyte and distorts its shape into sickle shape. The patient suffers from recurrent episodes of haemolytic and painful vaso-occlusive crisis.
Interestingly, the disease follows a milder course in many HbS homozygotes because they express relatively high levels of fetal haemoglobin, which contains γ-instead of defective β-chain.
People with sickle cell trait (heterozygous form with about 40% HbS) also form insoluble fibres, though at a lower rate. They are asymptomatic in normal circumstances. Their cells exhibit sickling only when pO2 is low, such as at high altitudes or in chronic lung disorders. Therefore, sickle cell trait is a relatively benign condition as such not producing any clinical symptoms and compatible with a normal life span. The red cells of these patients contain approximately 70% HbA and 30% HbS (Case 17.3).
Diagnosis
Sickling test
A blood smear is exposed to low oxygen tension by adding a reducing agent, e.g. sodium dithio-nite. The red cells respond by changing to sickle shape. This is detected microscopically.
Electrophoresis
Electrophoresis remains the mainstay of diagnosis of sickle cell anaemia. Electrophoretic separation is possible because the sickle cell mutation (Glu → Val) removes a negative charge from the β-chain resulting in a decreased anodal mobility (Fig. 17.10).
Treatment
1. Initial management aims at avoiding hypoxia and dehydration and administration of cyanate, which increases the oxygen affinity by covalent modification of the amino termini of the globin polypeptides.
2. An alternative approach, which involves manipulations to increase the synthesis of fetal haemoglobin, has been found useful. Hydroxyurea has been found to increase the fraction of cells containing fetal haemoglobin, although the mechanism whereby it acts is not known. Fetal Hb is useful because of its high O2 affinity.
3. Repeated blood transfusions are required in severe anaemic cases.
Protection from Tropical Malaria
HbS gene occurs in India, Mediterranean region and Saudi Arabia. In some parts of Africa, up to 40 per cent of the population carries the sickle gene. These areas have highest prevalence of malaria as well. Presence of the sickle gene has a survival value since the native Africans with sickle cell genes show a much lower susceptibility to malaria. The mosquito borne parasite that invades the red blood cells finds less favorable conditions for its growth in the sickled cell compared to normal cells. Moreover, increased lysis cause premature destruction of the parasite.
HbC Disease
The HbC is a haemoglobin with a β6Glu → Lys substitution. Like HbS it is found in black population and is related to sickle cell anaemia. Its symptoms, however, are less severe. Presence of HbC also appears to create a less hospitable environment for the malaria, perhaps even causing premature destruction of the parasitized cells.
B Thalassaemias
Thalassaemias are a group of disorders characterized by insufficient production of structurally normal α- or β-chains. High prevalence of thalassaemias has been reported among the people from the regions around the Mediterranean Sea (thalassa is the Greek word for sea).
Insufficient production of one of the globin chains of haemoglobin results in lack of coordination between the synthesis of the α- and the β-chains. Normally, the subunit synthesis is coordinated in such a way that each newly synthesized β-chain readily pairs with an α-chain; and conversely, each newly synthesized α-chain pairs with a β-chain. The lack of coordination results in:
• Formation of insoluble aggregates by the polypeptide chains. The aggregaetes are damaging to the cell and reduce its lifespan.
• Impairment of the haemoglobin synthesis, so that the erythrocytes are small and poorly filled with haemoglobin.
Molecular Defect
Underlying cause of the underproduction of a globin chain is a gene mutation. A number of mutations have been reported, including gene deletion, gene substitution, or deletions of one to several nucleotides in the DNA.
Types of Thalassaemias
Depending on which chain is affected, the thalassaemias are divided into two major classes: α- and β-thalassaemias. In α-thalassaemia relative or absolute deficiency of the α-chain occurs, whereas in β-thalassaemia the β-chains are affected. Heterozygous forms of α- or β-thalassaemia are called thalassaemia minor, and the homozygous forms are termed thalassaemia major.
In thalassaemia minor, anaemia may be present but is very mild. In thalassaemia major, severe anaemia develops.
The thalassaemias are caused by underproduction of either the α- or the β-chain. The α-thalassaemia is caused most often by large deletions, and many different mutations can cause β-thalassaemias.
α-Thalassaemias
Genes for the α-chains are clustered on chromosome 16. Genome of an individual contains four copies (two copies from each parent) of the α-globin genes; two copies are located on each of the chromosome 16. Most patients with α-thalassaemia have large deletions that remove one or both of the α-chain genes from a chromosome. Figure 17.12 shows the possible deletion types. Four types of α-thalassaemias corresponding to deletion of one, two, three, or all four of the α-chain genes occur:
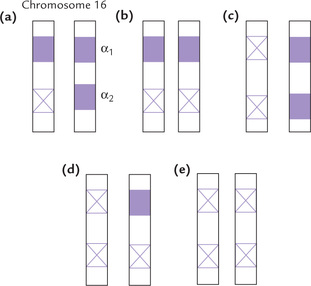
Fig. 17.12 One or more genes for the α-chains are deleted in α-thalassaemias. (a) Silent carrier, (b and c), α-Thalassaemia trait, (d) Haemoglobin H disease, (e) Haemoglobin Barth’s disease.
1. Silent carrier state (Fig. 17.12a) is due to deletion of one gene. It is a haematologically normal state; no clinical manifestations are observed in this condition.
2. α-Thalassaemia trait involves deletion of two genes from the same or different chromosomes (Fig. 17.12b and c). This results in mild physical manifestations, such as mild anaemia and slightly enlarged spleen.
3. Haemoglobin H disease involves deletion of three genes (Fig. 17.12d). The condition is characterized by mild to moderate haemolytic anaemia.
4. Homozygous α-thalassaemia (Fig. 17.12e) with four deletions is always lethal, either in utero or at birth. The condition is also called hydrops foetalis or haemoglobin Barth’s disease (Box 17.3).
β-Thalassaemias
Synthesis of β-chains decreases or stops altogether in the β-thalassaemias. In contrast to large deletions in α-thalassaemia, most patients with β-thalassaemia have single base substitutions. More than 90 such substitutions are known. Few cases of β-thalassaemia are caused by large deletions.
Normally, there are only two genes responsible for the β -globin chains, one from each parent. Each copy of chromosome 11 has one gene for β-globin chain. The mutation in β-thalassaemia may involve a single chromosome (i.e. the heterozygous state, called β-thalassaemia minor) or it is a homozygous state, involving both the chromosomes (called β-thalassaemia major).
1. β-Thalassaemia minor is benign and require any treatment. There is almost 50% decrease in β-chain synthesis. A compensatory increase in production of the δ- and the γ-chains occurs. These chains combine with the α-chains. Thus an increase in the amount of haemoglobin A2(α2δ2) and F(α2γ2) occurs, which is a distinguishing feature of β-thalassaemias.
β -Thalassaemia minor is mostly asymptomatic and life expectancy is also normal since some amount of β-chains are normally synthesized by these individuals.
2. β-Thalassaemia major is the homozygous state, which is most severe among all congenital haemolytic anaemias. It is also called Cooley's anaemia. The affected individuals require regular blood transfusions for survival.
Clinical features of β-thalassaemia major
Since the β-chain begins to be synthesized at a later stage in fetal gestation, the physical manifestations of β-thalassaemia become apparent only after birth. The affected infants are normal at the time of birth because of abundance of fetal haemoglobin, but a severe anaemia develops during the first year of life and the haemoglobin level in most infants falls below 5 gm/dL. The marrow responds to anaemia and becomes over-active. Consequently, a massive expansion of the red bone marrow occurs, leading to facial deformities (“chipmuk facies”). Extramedullary erythro-poiesis is seen in liver and spleen in some patients.
Treatment
Untreated, the patients with thalassaemia major suffer recurrent infections and severe anaemia. With regular blood transfusions, life expectancy is approximately 20 years of age. Although blood transfusion is life saving, the cumulative effect of the procedure is iron overload, a syndrome known as haemosiderosis. It has a high mortality, usually between the ages of 15 and 25 years. For this reason, the patients are treated not only with regular blood transfusions but also with desferrioxamine, an iron chelator that is best administered through a subcutaneous infusion pump. Desferrioxamine forms a soluble iron complex that can be excreted by the kidneys. Bone marrow replacement therapy has evolved as a highly successful mode of treatment since the last decade or so. It has improved life expectancy significantly.
V Myoglobin
Myoglobin is a relatively small molecule (MW 167,00), consisting of a single polypeptide chain, attached non-covalently to a haem molecule (Fig. 17.13 ). It is mainly located in cardiac and skeletal muscles, where it serves as a reservoir for oxygen. In addition, it transports oxygen to the mitochondria. The haem group is responsible for the deep brown colour of myoglobin (and of haemoglobin as well). Myoglobin is particularly abundant in muscles of diving mammals such as whale and seal, whose muscles are so rich in myoglobin that they are brown coloured. Storage of oxygen by muscle myoglobin permits these animals to remain submerged in water for long periods.
A Basic Structure
The single polypeptide chain of myoglobin has 153 amino acid residues. About 75% of the chain is in a α-helical conformation. There are eight major α-helical segments, referred to as A, B, C, ... H. Four of the these segments are terminated by proline residues, whose rigid R group does not fit within the straight stretch of α-helix. The non-helical segments lie in between these helical portions.
The polypeptide chain is so compactly folded that there is no space in its interior. Most of the hydrophobic R groups are in the interior of the molecule, hidden from water. They form a clustered structure stabilized by hydrophobic interactions. In contrast, the charged amino acids are located almost exclusively towards the exterior where they form hydrogen bonds with the surrounding aqueous medium.
The compact structure of myoglobin is stabilized by hydrogen and ionic bonds as well as by the hydrophobic interactions between the hydrophobic R groups.
The polypeptide chain of myoglobin is structurally similar to the individual polypeptide chains of the haemoglobin molecule. Thus, myoglobin provides a simpler model for the study of complex oxygen-binding properties of haemoglobin.
B Oxygen-binding Characteristics of Myoglobin
Myoglobin has a very high affinity for oxygen; it is 50% saturated with oxygen at a partial pressure of just 1-2 torr. At about 20 torr, it is over 95% saturated. Binding of oxygen to myoglobin is directed by mass action of oxygen. When oxygen is plentiful, formation of oxygenated myoglobin occurs and when oxygen is very scarce, dissociation of the oxygenated myoglobin occurs.
The oxygen dissociation curve of myoglobin is a simple hyperbolic curve (Fig. 17.14 ) in contrast to the sigmoid shape of the oxygen dissociation curve of haemoglobin. The curve is a molecular adaptation for its storage function.
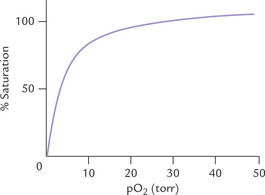
• High affinity of myoglobin for oxygen permits myoglobin to store oxygen even at the relatively lower partial pressure (40 torr) that exists in resting muscles.
• In exercising muscles, where the oxygen partial pressure falls to about 20 torr, myoglobin is still over 95% saturated. It unloads very little oxygen which makes it suitable for its role as a reservoir of oxygen.
• It is only during severe physical exercise—when pO2 within the muscles falls to a low level (< 5 torr)—that myoglobin releases a significant proportion of its stored oxygen.
The myoglobin has high oxygen affinity, but does not show Bohr effect, cooperative effect and 2,3-BPG effect.
Myoglobin is a monomeric haem-containing muscle protein that reversibly binds a single oxygen molecule. Unlike haemoglobin it lacks allosteric properties, but has far greater affinity for oxygen.
Properties of haem are different in haemoglobin, myoglobin and other haem-containing proteins (Box 17.4).
VI Anaemias
In our country, anaemia is one of the most common medical problem. In this condition, the haemoglobin concentration is reduced (normal is 13-16 g/dL in males; and 12-15 g/dL in females).
The commonest cause of anaemia in India is iron deficiency. For clinical manifestation of the iron deficiency anaemia (see Case 19.1). The causes of anaemia are given below:
Decreased production of erythrocytes
Defective synthesis of haemoglobin may lead to decreased production of erythrocytes. A number of cofactors and mineral elements are required for this process, including copper, iron, ascorbic acid, pyridoxal phosphate, and folic acid. Nutritional deficiency of these factors therefore results in anaemia.
Enhanced destruction of erythrocytes
Under normal circumstances a balance exists between the production and the destruction of erythrocytes. Integrity of the erythro-cyte structure is affected in a number of disorders, both intracorpuscular and extracorpuscular. These disorders result in excessive destruction of the cells (haemolysis), resulting in (haemolytic) anaemia. Some of the common conditions that lead to haemolytic anaemia are:
(a) Intracorpuscular defects are haemoglobinopathies caused by enzyme deficiencies such as deficiency of glucose-6-phosphate dehydrogenase or pyruvate kinase (Case 10.1).
(b) Extracorpuscular defects such as infections (malaria or coxsackie virus infection), drug exposure (quinine or methyl dopa), autoimmune haemolysis or mismatched blood transfusion.
Exercises
Essay type questions
1. Draw a diagram of the sigmoidal- and hyperbolic-oxygen dissociation curves of haemoglobin and myoglobin respectively. Describe how haemoglobin effectively delivers oxygen to myoglobin in muscles.
2. Explain the structural and functional differences between haemoglobin and myoglobin.
3. Explain the structural basis for cooperative oxygen binding to haemoglobin. What is the physiological relevance of the Bohr effect?
4. How do BPG, carbon dioxide and pH value affect oxygen binding to haemoglobin?
Write short notes on
Case 17.1 Shortness of breath and cyanosis in a 1-year-old child
A 1-year-old had frequent episodes of headache, exertional dyspnoea and cyanosis. The venous blood sample, drawn for biochemical investigations, was chocolate-brown in color but turned red on shaking. Addition of a few drops of 10 per cent potassium cyanide to the blood resulted in rapid production of a bright red colour. A number of biochemical tests were performed as an initial screen; no abnormality was, however, detected.
Physical examination ruled out any cardiac or pulmonary disease and there was no history of any recent exposure to any drugs or chemicals. Oxygen therapy was started but the cyanosis was not as promptly alleviated with it, as would be expected in a normal subject.
Defective (or diminished) action of some enzyme protein was suspected and further investigations were conducted for measuring intracellular levels of some major proteins of eryth-rocytes. When antibodies to the enzyme cytochrome b5 reductase were added, the precipitation was less than that expected in a normal subject. It was thereby concluded that the level of this enzyme protein within the erythrocytes was low.
Case 17.2 Oxygen insufficiency following inhalation of automobile exhaust
A 48-year-old car mechanic was brought to the hospital emergency in a disoriented state. Earlier, he had felt confusion, throbbing headache and chest pain. These symptoms seemed to have made sudden appearance, for he had appeared his normal self at the time he reported for duty early in the morning.
On examination, his blood pressure was 108/70 mmHg, pulse was 112/min, and respiration was shallow and fast (34/min). Analysis of the blood sample showed increased concentration of carboxy-haemoglobin (39.6%).
The patient was put on oxygen therapy with a diagnosis of sub-acute carbon monoxide poisoning. Hyperbaric oxygen was given, to which he responded well.
Q.1. What is the rationale behind arriving at the above diagnosis? What further tests are required to confirm this diagnosis?
Q.2. What are the various factors that lead to tissue hypoxia in case of carbon monoxide poisoning?
Q.3. What is the rationale behind starting oxygen therapy immediately? Why was hyperbaric oxygen given?
Q.4. Has the smoking habit of the patient influenced development of the present condition?
Q.5. In spite of oxygen insufficiency, the patient does not present with any signs of cyanosis. Give reason.
Case 17.3 A 10-year-old boy with breathlessness, pallor and persistent tiredness
A 10-year-old boy was brought to casualty with fever and breathlessness. Earlier he was seen by a general practitioner who had prescribed antibiotics with a diagnosis of upper respiratory tract infection. Presently the child appeared out of breath and was complaining of aches and pains, and tiredness. On examination, he was clinically anaemic, icteric and showed pallor and signs of retarded growth and development. His blood pressure was 98/68 mmHg and pulse was 100 beats per minute, with wide pulse pressure and hyperdynamic precordium. The sclera was yellow, abdomen was distended and the spleen was enlarged. Urine was analyzed by the staff-nurse in the side lab; presence of abnormally large amounts of urobilinogen was reported.
The child was admitted for the treatment of respiratory tract infection and for further investigations. Emergency blood sample was sent to the biochemistry and haematol-ogy laboratory. A sputum sample was also analyzed. The antibiotic was changed when pneumococci were isolated from the sputum sample. The boy responded well to the new antibiotic and the fever rapidly subsided. However, most other symptoms persisted as before. Results of some of the blood tests are as here.
| Test | Patient’s reports | Reference range |
| Haemoglobin | 5.2 g/dL | 13-16g/dL |
| Red Blood cells | 2 (×1012)/L | 4.5-6.5 × 1012/L |
| Platelets | 190,000/μL | 150,000-400,000/μL |
| Serum | ||
| Bilirubin | 4.8 mg/dL | 0.1-1.0 mg/dL |
| Alanine transaminase | 48 U/L | 10-40 U/L |
| Alkaline phosphatase | 90 U/L | 40-100U/L |
| Lactate dehydrogenase | 386U/L | 100-300 U/L |
| Sodium | 138 mmol/L | 132-144 mmol/L |
| Potassium | 5.5 mmol/L | 3.6-5.0 mmol/L |
The van den Bergh test for water soluble (conjugated) and water insoluble (unconjugated) bilirubin in the serum sample showed elevation of unconjugated bilirubin level. Bilirubin was not detected in the urine sample. However, the urine contained large amount of urobilinogen (i.e. urobiliru-binuria), as stated above.
A fresh blood smear contained a few crescent-shaped cells. However, after 24-hour incubation in a sealed wet smear, nearly all the red cells assumed the shape of sickles. An increased reticulocyte count was also reported in this smear.
Electrophoresis of haemoglobin was performed to detect haemoglobin structural variant, if any. It revealed presence of HbS. Based on these tests, the child was diagnosed as having sickle cell anaemia.
Peptide maps (or finger prints) of the trypsin digestion of the haemoglobin of this child were obtained and compared with that of a normal healthy child of the same age group. The two were found to be remarkably different.
Q.1. How does the above diagnosis explain clinical features of the child?
Q.2. Paper electrophoresis of the haemoglobin obtained from the child’s parents was also carried out; the results are shown below.
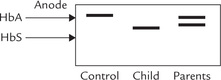
Interpret the results and discuss whether the child’s parents also have clinical abnormalities like him?
Q.3. Comment on the quantitative blood picture. What are other biochemical features of this disorder?
Q.4. State principle of the peptide finger print test. How does the finger print test of this child differ from that of a normal subject? Give reason for your answer.
*This topic relates to higher level learning and it is meant only for postgraduates. Undergraduates or MBBS students may refer to it for higher learning.