Amino Acids, Peptides And Proteins
Proteins are the most abundant macromolecules in living cells. The name protein means “first” or “foremost”. The body proteins constitute 50% or more of dry weight of the living cell and exhibit enormous diversity; hundreds of different types of proteins are usually found in a single cell. All proteins are made up of a set of building block molecules, called amino acids, which are covalently linked to one another. Peptides are the short amino acid sequences linked covalently.
After going through this Chapter the student should be able to understand:
• Basic structure, classification, general characteristics and acid-base properties of amino acids, application of these properties for separation of individual amino acids in a mixture.
• General structure and properties of peptides, formation and characteristics of peptide bond, and role of some biologically active peptides in the body.
• Classification, various levels of organization of proteins; primary, secondary, tertiary and quaternary structures; and methodologies for separation of proteins.
I Amino Acids
Amino acids in free form are found in small amounts in the living systems, but the bulk exists as constituent units of proteins. More than 300 amino acids have been identified in nature, but all body proteins are constructed from a basic set of only 20 amino acids, which are specified by genetic code. These DNA coded amino acids, called primary amino acids, are regarded as alphabet of protein structure. It is amazing that all body proteins, which display high structural diversity, could be made up of such a limited number of amino acids.
 Proteins and peptides are formed from a string of amino acids linked together by peptide bonds. The latter term refers to amide bond between two amino acids.
Proteins and peptides are formed from a string of amino acids linked together by peptide bonds. The latter term refers to amide bond between two amino acids.
A Composition
Amino acid are structures that have an amine and an acid group. Amino acids have a central carbon, the α-carbon, that has bonds to an amino group, a carboxylic acid, a hydrogen and a variable side chain designated as R group (Fig. 4.1 ). Because of their position on the α-carbon, the carboxyl and amino groups are called the α-carboxyl and α-amino groups, respectively. The side chain R defines chemical nature and structure of different amino acids; for example, it is –CH3 in alanine, –CH2OH in serine, CH2COOH in aspartic acid, and –H in glycine. Thus, R group represents the variable portion which differs from one amino acid to another.
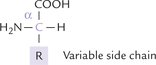
B Stereochemistry
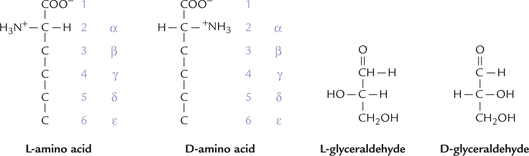
The α-carbon has four different groups attached to it and so is a chiral or asymmetric carbon Hence, there are two possible enantiomers, L and D, which represent nonsuperimposable mirror images and are optically active. Nearly all amino acids occurring in proteins are of the L-form. D-amino acids are rare in nature although they do occur in some bacterial products. This is opposite of sugars which nearly always occur as the D isomer. Note that the L designation has nothing to do with the way an amino acid rotates the polarized light; for example L-leucine rotates polarized light 10.4° to the left, whereas L-arginine rotates polarized light 12.5° to the right (the enantiomers of these compounds rotate polarized light to the same degree but in the opposite direction). The designation (D and L) is purely structural, being based on the structure of L-glyceraldehyde (Fig. 4.2 ).
The α-carbon of all amino acids is asymmetric, except for glycine (Fig. 4.3 ), which, therefore, lacks the mirror image pair of enantiomers and is not optically active.
Numbering of carbons in amino acids: The various carbon atoms are assigned sequential letters in Greek alphabets, beginning with the carbon next to the carboxyl group (Fig. 4.2). Standard numbering schemes are also used; for example the carbon atoms in Figure 4.2 are labelled 1–6 and α through ε.
C Classification
The 20 amino acids in proteins encoded by DNA are shown in Table 4.1 ; each can be designated by a three-letter abbreviation and one-letter symbol. They are grouped as per the classification schemes based on:
Table 4.1
| Primary amino acid | Three-letter abbreviation | One-letter symbol |
| Alanine | Ala | A |
| Arginine | Arg | R |
| Asparagine | Asn | N |
| Aspartic acid | Asp | D |
| Cysteine | Cys | C |
| Glutamine | Gln | Q |
| Glutamic acid | Glu | E |
| Glycine | Gly | G |
| Histidine | His | H |
| Isoleucine | IIe | I |
| Leucine | Leu | L |
| Lysine | Lys | K |
| Methionine | Met | M |
| Phenylalanine | Phe | F |
| Proline | Pro | P |
| Serine | Ser | S |
| Threonine | Thr | T |
| Tryptophan | Trp | W |
| Tyrosine | Tyr | Y |
| Valine | Val | V |
Based on Polarity and Charge on R Groups
The properties of an amino acid are, in large part, dependent on its side chain (–R), which uniquely defines each of the 20 amino acids. Based on polarity and charge on the side chain, the primary amino acids are allocated among three different subgroups (Table 4.2 ).
Table 4.2
Amino acids according to polarity and charge of their R groups (side chains) at pH 7
| Non-polar R groups | Polar but uncharged R groups | Charged polar R groups |
| Alanine | Serine | Negatively charged |
| Valine | Threonine | Aspartic acid |
| Leucine | Glycine | Glutamic acid |
| Isoleucine | Asparagine | Positively charged |
| Phenylalanine | Glutamine | Lysine |
| Tryptophan | Tyrosine | Arginine |
| Methionine | Cysteine | Histidine |
| Proline |
Non-polar R Groups
Eight amino acids are classified as having non-polar side chains (R group). In proteins these amino acid residues are usually buried in the hydrophobic interior of the biomolecule and are out of contact with water. Alanine, valine, leucine and isoleucine have aliphatic hydrocarbon side chains ranging in size from methyl group for alanine to isomeric butyl groups for leucine and isoleucine (Fig. 4.4 ). Alanine is the most abundant amino acid in most proteins. Phenylalanine with its phenyl moiety and tryptophan with its indole group contain aromatic side chains, and (together with the aliphatic amino acids) contribute to the internal hydrophobic interactions of the proteins. They are also responsible for the ultraviolet absorption of most proteins, discussed later. Methionine has a thiol ether side chain. Proline differs from other amino acids in that its side chain pyrrolidine ring includes both the α-carbon and the α-amino group. Chemically speaking proline is not an α-amino (–NH2) acid but rather an α-imino (–NH) acid.
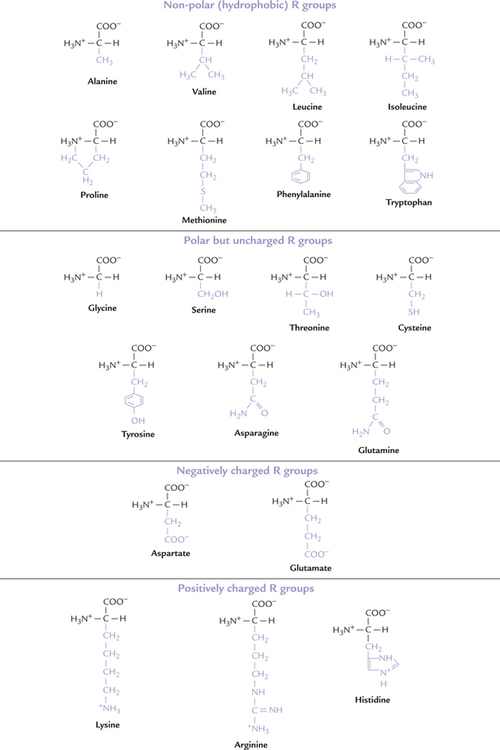
Uncharged Polar R Groups
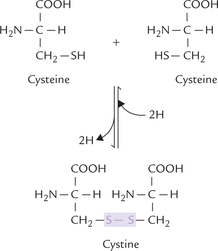
Side chains of these amino acids are uncharged, and they have polar groups (–OH, –SH, –NH, C = O) that can hydrogen bond to water. Serine and threonine, for example, bear hydroxylic groups of different sizes and glycine has hydrogen for its R group. Asparagine and glutamine have amide-bearing side chains of different sizes. Tyrosine has a phenolic group (and like phenylalanine and tryptophan is aromatic). Cysteine has a thiol group that can form disulphide bond with another cysteine through oxidation of the two thiol groups to form cystine (Fig. 4.5 ).
Charged Polar R Groups
Five amino acids have charged side chains (Fig. 4.4), having acidic or basic groups. These groups can assume respectively, negative or positive charge at physiologic pH values.
1. The basic amino acids have a positive charge on their R group at physiological pH values (because the R groups are protonated). Lysine has a primary amino group attached to the terminal ε -carbon of the side chain (i.e. butylammonium side chain), which has pK' of11. Arginine is the most basic amino acid (pK' = 13) and its guanidine group exists as a protonated guanidinium ion at pH 7.0. Histidine, which carries an imidazoline ring as the side chain functions as a general acid-base catalyst. This is because with its pK' = 6.0, it ionizes within the physiological pH range. In contrast to lysine and arginine, which are fully charged at the physiological pH, histidine is only partially charged; its side chains being weakly basic.
2. The acidic amino acids, have negative charge at physiologic pH, e.g. glutamic acid and aspartic acid. Both contain carboxylic acids on their side chains and are ionized at pH 7.0, and as a result, carry negative charges on their (β- and γ-carboxyl groups, respectively. In their ionized state, they are referred to as aspartate and glutamate (asparagine and glutamine are, respectively, the amides of aspartic acid and glutamic acid).
The allocation of the 20 amino acids among the three different subgroups is somewhat arbitrary. The following examples illustrate this point:
• Tryptophan with its heterocyclic aromatic R group may be thought of as uncharged polar amino acid (not uncharged non-polar amino acid as in Table 4.2).
• Glycine with its smallest R group might as well be classified as non-polar amino acid.
• Side chains of tyrosine and cysteine are ionizable, particularly at higher pH values, and so they might be classified as charged polar amino acids.
Based on Structure of Side Chain
Structures of side chain R groups of the 20 primary amino acids have been shown in Figure 4.4, which forms the basis of their classification into the following subgroups:
Branched chain amino acids: Valine, leucine, isoleucine Sulphur containing amino acids: Methionine, cysteine Amide group containing amino acids: Asparagine, glutamine Hydroxy amino acids: Serine, threonine, tyrosine Simple amino acids: Glycine and alanine
• Aromatic amino acids: Phenylalanine, tyrosine, tryptophan
• Heterocyclic amino acids: Histidine, tryptophan
Tryptophan finds place in two subgroups (aromatic and heterocyclic), which again shows arbitrary nature of the classification schemes.
Based on Catabolic Fate of the Amino Acid
From the viewpoint of their catabolic fate, amino acids may be divided into three categories: those that can give rise to glucose and glycogen, those that can give rise to ketone bodies and those that can give rise to both. They are called the glucogenic, the ketogenic, and both glucogenic and ketogenic amino acids, respectively. Examples of amino acids of each category are given later (Chapter 13).
Based on Body’s Ability to Synthesize the Amino Acid
Two major subclasses are recognized—essential and non-essential amino acids.
Essential amino acids cannot be endogenously synthesized, and therefore, their dietary intake is essential. Examples are leucine, isoleucine, methionine, phenylalanine, lysine, tryptophan, valine and threonine. The nonessential amino acids, on the other hand, can be endogenously synthesized in adequate quantities so as to meet the body’s requirements. Examples include alanine, aspartate, asparagine, glutamate, glutamine, tyrosine, serine, proline, glycine, and cysteine.
Two amino acids, histidine and arginine, are called the semi-essential amino acids (Chapter 13).
Specific Roles of Some Side Chains
Properties of the amino acids are determined by the functional groups in their side chains and by the noncovalent interactions that the latter can form. In addition, the functional groups may perform some specific roles, thereby imparting special properties to proteins.
1. The hydroxyl groups of serine and threonine exist at the catalytic sites of certain kinds of enzyme, e.g. the enzymes that regulate energy metabolism and fuel storage in the body. They can be phosphorylated, hence their charge changes from neutral to negative, which changes the shape and function of the enzyme proteins. Thus, these (hydroxyl) groups are important in the regulation of activities of these enzymes (Chapter 5).
2. Thiol group of a cysteine can form a disulphide bond with another cysteine through the oxidation of the two thiol groups (Fig. 4.5). The dimeric compound so formed, called cystine, is important in cross-linking the adjacent polypeptide chains.
3. Amide bearing side chain of asparagine forms covalent linkage with oligosaccharide unit (called N-glycosidic linkage) in glycoproteins. The –OH groups of serine and threonine also similarly form linkage with reducing end of oligosaccharide, called O-glycosidic linkage (Chapter 5).
4. Aromatic side chains are responsible for the ultraviolet absorption of most proteins, which have absorption maxima between 275 nm and 285 nm. Tryptophan has greater absorption in this region than the other two aromatic amino acids. Since nearly all proteins contain aromatic amino acids, the amount of light absorbed at 280 nm by a protein is used as an indirect measure of protein concentration.
5. Hydroxyl group of tyrosine is subject to phosphorylation in some enzymes, which is important in the regulation of metabolic pathways.
6. Imidazole group of histidine is important in the buffering activities of proteins.
7. Cyclic pyrrolidine group of proline introduces bend in the peptide chain.
8. Butylammonium side chain of lysine binds with the co-enzymes pyridoxal phosphate and biotin.
9. R group of glycine provides little stearic hindrance because of its small size, so that proteins can bend or rotate easily wherever glycine forms part of their structure.
D Non-standard Amino Acids
In addition to the 20 primary amino acids found in protein, some “non-standard amino acids” are present in small amounts in specialized structures. They may also occur in free or combined states and independently play a variety of biological roles.
1. Non-standard amino acids found in proteins: These amino acids are produced by specific modification of a primary amino acid residue after the polypeptide chain has been synthesized. They are important, and in most cases, essential for the function of the protein. Hydroxylation, methylation, acetylation, carboxylation and phosphorylation are some common modifications, though more elaborate modifications are found in some amino acid residues. Some examples of modified amino acids are: hydroxylysine and hydroxyproline in collagen, methylhistidine in muscle proteins, phosphoserine in casein and certain enzymes, ’γ-carboxyglutamic acid in prothrombin and other calcium-binding proteins, and pyroglutamic acid as the N-terminal amino acid in thyrotropin and several proteins.
2. Non-standard amino acids not found in proteins: Termed non-protein amino acids, they occur in free state in cells, e.g. ornithine and citrulline that are intermediates of urea cycle. The most abundant amino acid in human organism, taurine, also occurs in a free state in bile and plays an important role in fat digestion and absorption.
3. Biologically active amino acids: These amino acids are used for functions other than protein synthesis. Examples: (a) as chemical messengers for communication between cells, e.g. glycine, dopamine (tyrosine derivative), γ-aminobutyric acid (glutamate derivative; refer to Box 4.1), (b) as local mediator of allergic reactions, e.g. histamine (histidine derivative), and as a hormone, e.g. thyroxine (another tyrosine derivative).
4. D-Amino acids: They are components of bacterial polypeptides that are widely distributed as constituents of bacterial cell walls. They are also found in many bacterially produced peptide antibiotics, including valinomycin and gramicidin A.
E Acid-Base Properties of Amino Acids
Amphoteric Nature
An amino acid is capable of acting as both an acid (i.e. proton donor) and a base (i.e. proton acceptor). Such substances are termed as amphoteric substances. The amphoteric nature of amino acids is because of presence of the following ionizable groups:
Each Amino Acid can Exist in Three Charged States: Positive, Neutral, Negative
The three charged states of an amino acid are represented in Figure 4.6 . These are the diprotonated form (positively charged, cationic), the deprotonated form (negatively charged, anionic) and the dipolar form (neutral, zwitterion). The form in which an amino acid exists at a given time is determined by relative values of the following two parameters:
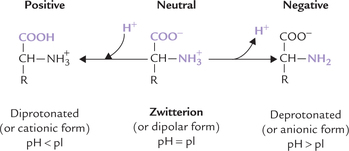
Isoelectric pH (pI) of an amino acid is defined as the pH value at which carries equal number of positive and negative charges, so that net charge on the molecule is nil. Predominant form of an amino acid at its pI is zwitterion (in German Zwitter means “hermaphrodite”).
For a simple amino acid such as glycine, the pI is halfway between the pK' values of the two ionizable groups, and so calculated by averaging the two pK' values.
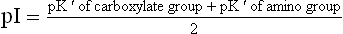
The pK' values for the carboxylate and the amino groups being 2.4 and 9.8 respectively, the pI is 6.1.
The pI of acidic amino acids is halfway between the pK' of two acidic (carboxylate) groups, and the pI of basic amino acids is half-way between the pK' of two basic (amino) groups.
Acidic amino acids thus have low pI values, basic amino acids have high pI values, and neutral amino acids have pI values near 6.0 (Table 4.3 ).
Table 4.3
Isoelectric pH (pI) values of some primary amino acids. At its pI an amino acid is neutral

When the pH of the surrounding medium is lowered below the pI value of an amino acid (i.e. concentration of protons raised), its carboxyl group accepts a proton to produce the protonated form. This form of amino acid carries net positive charge.
Conversely, elevation of pH (i.e. low concentration of protons) results in loss of proton from the anionic form.
As a result, overall charge on the amino acid molecule becomes negative.
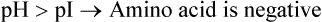
Amino acids are zwitterionic structures, carrying both a positive and a negative charge. The pH value at which the number of positive charges equals the number of negative charges is isoelectric pH of the amino acid.
Titration Curve of a Monoamino-monocarboxylic Acid (Alanine)
The titration curve of alanine proceeds in the same way as that of a weak acid. There is, however, an important difference: the titration curve of this amino acid proceeds in two distinct stages. This is because an amino acid molecule, when fully protonated, is capable of donating two protons. Each of the two stages of the titration corresponds to loss of a proton (Fig. 4.7 ). In the first stage of titration, dissociation of the carboxyl group occurs while the second stage involves dissociation of the amino group.
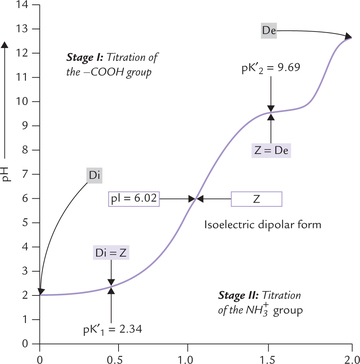
Fig. 4.7 The titration curve of 0.1 M alanine. The ionic species (Z = dipolar form, or zwitterion Di = diprotonated form, De = deprotonated form) predominating at various pH values are shown in boxes (pK’1 = pK' of the α-carboxyl group; pK’2 = pK' of the α-amino group; pI = Isoelectric pH).
Stage I: Dissociation of the carboxyl group: At low pH both the carboxyl group as well as the amino group of the amino acid are protonated, and hence, the predominant ionic species is the diprotonated (positive) form. As the pH is raised by addition of NaOH, the COOH group donates its proton to neutralize the hydroxyl group of the alkali (i.e. sodium hydroxide). Thus, formation of the carboxyl (COO-) ion occurs at the expense of COOH group, so that some diprotonated form is converted to the dipolar (zwitterion) form. At the end of this stage of titration, dissociation of carboxylate group is complete and the diprotonated form is almost completely converted to the dipolar (Z) form.
Midway through this stage, the following relations exist:
1. Concentrations of the Z form and the diprotonated form are equal.
2. The pH value of the solution equals the pK' value of carboxyl group (pK' is the negative log of the dissociation constant of the carboxyl group).
The solution has buffering power in this zone, as shown by flat shape of the titration curve. Normally, a system acts as buffer in the pH zone of pK' ± 1. Since pK' of the carboxyl group (pK’1 of alanine is 2.34, it acts as a buffer in the pH zone of2.34 ± 1 (i.e. from pH range of1.34 to 3.34).
Stage II: Dissociation of the amino group: Dissociation of proton from the amino group of alanine occurs in this stage, resulting in the formation of the deprotonated (negative) form from the Z form (Fig. 4.11 ).
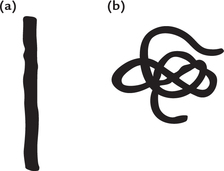
Fig. 4.11 Classification of proteins based on their shape. (a) Fibrous protein, (b) Globular protein.
Midway through this stage, dissociation of amino group is half complete, and the following relations exist:
The solution has buffering power at this stage as evidenced by a flat zone in the titration curve. Since pK' of the amino group is 9.69, the second buffering zone of alanine lies in the pH zone of 9.69 ± 1 (i.e. from 8.69 to 10.69).
The following observations about the titration curve are noteworthy.
• Alanine has two buffering zones, the first in the pH range of 2.34 ± 1, and the second in the pH range of 9.69 ± 1. Since both lie beyond the physiological pH range, alanine lacks any buffering power in the body.
• At the end of the first stage of titration, alanine exists predominantly in the Z form. The pH value corresponding to this point is isoelectric pH (pI). It lies midway between the pK' values of the two ionizable groups.
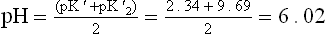
where, pK’1 and pK’2 = pK' values of α-carboxyl and the α-amino groups respectively, pI = isoelectric pH.
At this pH, both the amino group and the carboxyl groups of the amino acid are ionized, but the overall charge on the molecule in nil.
Titration Curve of Basic Amino Acids
These amino acids have a third ionizable amino, group in side chain. Since this group can reversibly gain or lose a proton, the basic amino acids have an additional third phase of titration. Accordingly, there are three regions of buffering; one region each corresponding to loss of a proton from the carboxyl group, the amino group, and the side–CHain. The pK' value of the imidazole group of histidine is 6.0, and therefore, it can serve as a buffer in pH range of 6.0 ± 1, which is very close to the physiological pH. Hence, histidine has the capacity to provide buffering action at the body pH.
F Separation of Amino Acids
A number of techniques are available for the separation of a given amino acid from a mixture. The important ones discussed here are (paper) chromatography, electrophoresis, ion exchange chromatography, etc.
Chromatography
(Greek: chroma means colour; graphin means to write): It was discovered by a Polish scientist, Tswett, in 1901, who used this technique for separating solubilized plant pigments on solid adsorbents. In most modern chromatographic procedures, a mixture of substances to be fractionated is dissolved in a liquid (the mobile phase) and percolated on an inert support medium on which separation occurs (the stationary phase). The migrating solutes interact with the stationary phase and are differentially retarded; the retarding force depends on the properties of each solute. The solutes having higher affinity for the stationary phase are retarded more than those having relatively higher affinity for the mobile phase. The substances with different rates of migration are thereby separated.
The chromatographic procedures for separating amino acids (or proteins) are based on such interactions between the amino acids in the solution and the stationary phase. Strip of chromatography paper may be used as the inert support medium (i.e. paper chromatography), or alternately, a thin layer of silica spread on a glass plate (i.e. thin layer chromatography or TLC) is employed. a small quantity of the amino acid mixture is placed on a dry piece of chromatography paper (or silic-gel coated plate in case of TLC) and a solvent is allowed to permeate on it from one end (Fig. 4.8 ). The solvent is usually a mixture of organic solvents and water. As the solvent advances on the paper (or plate), the amino acids also travel with it. Different amino acids travel at different speeds, because each one has a different partition coeffident between the mobile phase (i.e. the solvent) and the stationary phase (i.e. the paper). In other words, the amino acid having higher affinity for the solvent moves faster than the one having lower affinity. As a result, the amino acids are separated from one another as the solvent ascends (in ascending chromatography) or descends (in descending chromatography) along the stationary phase.
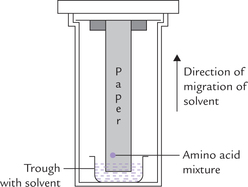
The amino acids thus separated are located by spraying the paper with ninhydrin. Resolution may be improved by a second run at right angle to the first one. This is referred to as two-dimensional separation. The two dimensional separation may also be performed using chromatography and electrophoresis at right angle; the latter procedure has considerable diagnostic utility (Case 17.1).
Electrophoresis
In electrophoresis, proteins dissolved in a buffer solution at a particular pH is placed in an electric field. Depending on the relationship of the buffer pH to its pI, a given protein moves towards the cathode (–), anode ( + ), or remains stationary (pH = pI).
Ion-exchange chromatography
In this technique preparative separation of amino acids by charge occurs on ionexchange resins, which are packed in a column. Negatively charged resins bind cations and retard their movement, whereas positively charged resins retard movement of anions.
A detailed account of the latter two techniques is given later in this chapter.
II Peptides
A Structure
Short sequences of amino acids, linked covalently, are known as peptides. The covalent linkage, termed peptide bond, is formed by reaction of the amino group of one amino acid with the carboxyl group of other (Fig. 4.9 ). The dipeptide so formed retains a carboxyl and an amino group, which are free to form peptide linkages with other amino acids. In this fashion, peptides of various lengths may be formed. On the basis of number of amino acids they contain, shorter peptides are termed di-, tri-, tetr-peptides, and so on. Peptides consist of a few (3 to approximately 50) amino acids, polypeptides contain up to 100 amino acids, and the polyamide chains larger than this are proteins.
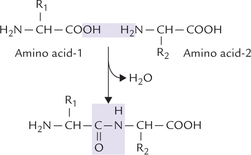
The peptide has two ends: the amino terminal end, and the carboxyl terminal end. The amino acid present at the amino terminal end has a free amino group, and the amino acid at the C-terminal end possesses a free carboxyl group. The amino acid sequence of a peptide (primary structure) is always listed from the N-terminal; therefore, it is Cys-Al-Tyr-Gly-Leu for the pentapeptide shown (Fig. 4.10 ). Note that the formation of peptide bond involves removal of water from the carboxyl group of one amino acid and the amino group of the other (Fig. 4.9). Therefore, the constituent amino acids of a peptide, having lost a portion of their carboxyl group and the amino group, can no longer be termed amino acids. Instead they are called the amino acid residues.
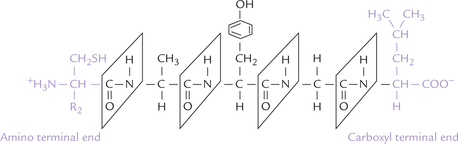
B Reactions of Peptides
Acid-Base Behaviour
The acid-base behaviour of a peptide is a function of its N-terminal amino group, the C-terminal carboxyl group, and those R groups which can ionize. For example, a peptide having high content of dicarboxylic amino acids (glutamate and aspartate) has acid-like behaviour at the physiological pH. Conversely, a peptide with high content of lysine, arginine or histidine tends to have baselike behaviour at physiological pH.
Hydrolytic Cleavage
A peptide can be cleaved to its constituent amino acids by hydrolysis of the peptide linkages. Boiling with strong acids or bases brings about the hydrolysis. All the available peptide bonds can be cleaved by this treatment, irrespective of nature of the constituent amino acids.
Proteolytic Cleavage
Certain enzymes, namely trypsin and chymotrypsin, can also bring about hydrolysis of peptide bonds. Accordingly The peptide is shown in the ionized state in which it occurs at they are referred to as proteolytic, the word meaning “protein dissolving”. Their action is highly specific because a given enzyme attacks only certain specific regions of the peptide chain.
C Functions
Peptides are formed as intermediates during partial hydrolysis of the long polypeptides chains. In addition, some naturally occurring peptides are found in the living matter (Table 4.4 ).
Table 4.4
Some important biologically active peptides in humans
| Peptides | Number of amino acid residues | Biological effect |
| ThyroTropin-releasing hormone | 3 | Stimulates anterior pituitary to release thyroid stimulating hormone |
| GIuTarhione | 3 | First line of defense against oxidative stress |
| Enkephalins | 5 | Binds with certain brain receptors to alleviate pain (i.e. opiate-like activity) |
| Oxytocin | 9 | Stimulates uterine contractions |
| Bradykinin | 9 | Inhibits inflammatory reactions in tissues |
| Vasopressin | 9 | Secreted from posterior pituitary it causes kidney to retain water from urine |
| Linle gastrin | 10 | Secreted by mucosal cells in stomach; causes parietal cells of stomach to secrete the acid |
| Glucagon | 10 | Pancreatic hormone involved in glucose homeostasis (hyperglycaemic) |
| Substance P | 10 | Neurotransmitter |
| Gramicidin | 10 | An antibiotic, toxic to many microorganisms |
Some of these peptides possess enormous biological activities; for example thyrotropin-releasing hormone (TRH) is a tripeptide released from the hypothalamus.
Pyroglutamate-Histidine-Proline: The TRH regulates the secretion of the thyroid stimulating hormone (TSH) from the anterior pituitary. The N-terminal glutamate residue is in the pyro form in which its γ-carboxyl group is covalently joined to its α-amino group via amide linkage. The C-terminal amino acid, proline, is amidated and thus also not free.
Another peptide of biological significance is glutathione, comprising glutamate, cysteine and glycine. The glutamate forms an amide bond with cysteine through its γ-carboxyl group, rather than its α-carboxyl group and so reffered to as pseudopeptide linkage.
γ-Glu-Cys-Gly: Glutathione acts as a co-enzyme in many reactions, e.g. for transhydrogenases and peroxidases, and participates in the second phase of the xenobiotic metabolism. When it is oxidized, many highly reactive and toxic compounds are reduced; one of the most important features of glutathione is to reduce hydrogen peroxide to water (Chapter 27).
The enkephalins are analgesi-inducing agents that are produced in the central nervous system (the word enkephalin means “in the head”). They bind with specific receptors in the brain cells, the receptors are the same to which the synthetic opiates like morphine, heroin and other addictive opiates bind. Therefore, the enkephalins are commonly referred to as endogenous opiates.
It is noteworthy, that it is the sequence of the amino acids that gives the peptides such intense biological effects while the constituent amino acids by themselves are devoid of any such activity.
D Characteristics of Peptide Bonds
Some characteristics of peptide bonds are as follows.
Partial Double Bond Character
The peptide bond is conventionally described as a single bond (C–N), but it does not show the rotational freedom expected of a single bond. This rigidity is explained by the fact that the double bond between the C and the is actually shared across the bond between the C and the N. Therefore, the peptide bond is actually resonance hybrid of two electron isomers:
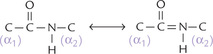
Its “real” structure is halfway between these two forms. Thus, the peptide bond between the C and the N has partial characteristic of double bond and so does not rotate.
This is supported by spectroscopic measurements and X-ray diffraction studies. These studies show that C to N bond length (1.33 Å) of a peptide bond is approximately half-way between that of C–N single bond (~1.45 Å) and C = N double bond (~1.25 Å).
Trans-planar
The two atoms of peptide bond (C and N) have four substituents: a hydrogen, an oxygen, and two α-carbon atoms (see Fig. 4.10; an exception is the peptide bond formed by nitrogen of proline). All the substituents are fixed in the same plane and the O and the H are in the trans (opposite) positions.
Hydrogen Bonding
The sharing of electrons between the C and the N results in unpairing of electrons between the C and the O. Thus, O has a slightly negative charge (δ– ). The electron of the H atom spends most of its time in the bond between the N and the H, which gives the H atom a slightly positive charge (δ+ ). This means that these atoms, by virtue of positive or negative charges, are able to participate in hydrogen bonding.
φ and Angles
In addition to the peptide bond, the polypeptide backbone contains bonds between the a1-carbon and the peptide bond carbon, and between the α-carbon and the peptide bond nitrogen. These two being single bonds show free rotation. Rotation around the nitrogen-and-α- carbon bond is measured as the φ (phi) angle and rotation around the peptide carbon and α-carbon bond as the ψ (psi) angle.
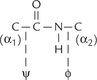
III Proteins
Proteins are not only the most abundant biomolecules but are also centre of action in biological processes. They exhibit enormous diversity with regard to their structures. In general, structures of proteins consist of polypeptides, which are long, unbranched chains; each chain contains usually more than 100 amino acid residues. The chains containing fewer residues than that are simply called peptides.
A Size
Proteins contain between 100 and 2000 amino acid residues. The mean molecular mass of an amino acid residue is about 110 dalton units (Da). Therefore, the molecular mass of most proteins is between 11,000 and 220,000 Da. The largest known polypeptide chain belongs to the 26,926 residue titin, a giant (2990 kD) protein that helps to arrange the muscle fibres. Some proteins consist of a single polypeptide; others are formed from two or more polypeptides held together by covalent bonds or by noncovalent interactions.
B Functions
Range and scope of biological functions of proteins are very vast. Some important functions performed by proteins are as follows: (a) act as biological catalysts, called enzymes, (b) provide the structural framework of cells and tissues, (c) act as transport media in bloodstream for a variety of substances, such as lipids and oxygen, (d) act as hormones or regulatory proteins for controlling various biological processes, (e) perform mechanical work, such as in skeletal muscle contraction and the pumping of heart, (f) serve as essential nutrients, (g) act as antibodies in the bloodstream to provide natural defense against invading pathogens and play vital role in blood clotting mechanism, (h) regulate gene expression on chromosomes; and play important role in food digestion respiration, vision, etc. These functions are summarized in Table 4.5 .
Table 4.5
Biological functions of body proteins
| Protein | Example |
| Enzyme | Amylase, ribonuclease, trypsin |
| Structural proteins | Collagen, elastin, keratin, fibroin |
| Transport proteins | Haemoglobin, myoglobin, lipoproteins, serum albumin, membrane transporters |
| Hormonal proteins | Insulin, growth hormone, corticotropin |
| Regulatory proteins | Repressors and inducers |
| Contractile or motile proteins | Actin, myosin, tubulin |
| Nutrient and storage proteins | Gliadin (wheat), ovalbumin (egg), casein (milk) |
| Defense proteins | Antibodies |
| Blood proteins | Fibrinogen, thrombin |
| Toxin protein | Snake venoms |
| Vision protein | Rhodopsin |
Specialized functions are performed by certain proteins, which cannot be easily classified in one of the above categories. For example:
• Antifreeze protein is present in blood of some arctic and antarctic fishes. It lowers the freezing point of water, thereby saving aqueous plasma from freezing.
• Reasilin is the major component of the wing hinges of some insects. This protein has extraordinary elastic properties.
Biological functions of a given protein are same in different species. For example, haemoglobin serves as oxygen transporter in humans, mammals, birds, insects, etc. (refer to Box 4.2).
Studies have been conducted in homologous proteins obtained from different species, including mammals, fishes, reptiles, amphibians, birds, insects, plants and fungi. These studies intend to examine the biological significance of sequence homology. In one such study, cytochrome C, a mitochondrial protein of 100 amino acids (MW 125,000) and an essential component of electron transport chain, was studied in about 60 different species.
• At about 27 positions, identical amino acid residues were present (i.e. invariant residues). This suggests that these residues are important determinants of the biological activity of cytochrome C.
• The amino acid residues in other positions showed variation: the greater the phylogenic difference between the species, more the variation. Such information is helpful in getting insight into the evolutionary process.
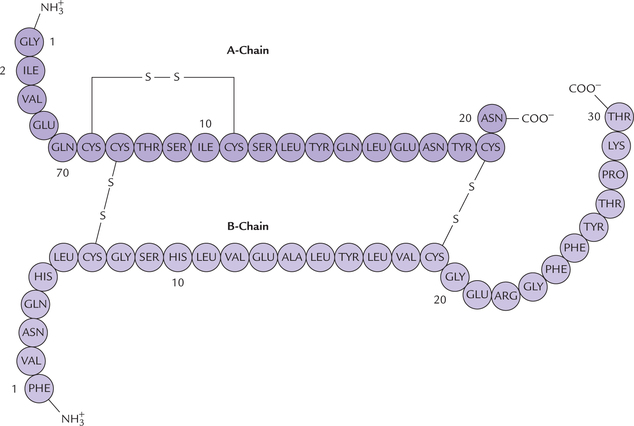
Human insulin (a peptide of 51 amino acids) also exhibits sequence homology with the insulins obtained from other mammals (Table 4.6 ). For instance, human insulin and porcine insulin have identical amino acids in 50 positions, only at position B30 the amino acid is different. It is threonine in human insulin but alanine in porcine insulin. This permits use of porcine insulin for the treatment of diabetic patients. However, prolonged use of porcine insulin may result in side effects, which can be avoided by using modified insulins (Case 4.1).
Table 4.6
Variation in positions of A8, A9 and B30 of insulin in different species
| A–CHain (8, 9, 10) | B–CHain position (30) | |
| Human | Thr-Ser-Ile | Thr |
| Porcine | Thr Ser IIe | Ala |
| Bovine | Al-Ser-Val | Ala |
| Dog | Thr-Ser-IIe | Ala |
In bovine insulin, 8th and 10th amino acids are different in a chain also.
C Classification
Proteins can be classified in different ways, based on the following criteria:
Based on Shape
Based on their three-dimensional shape (i.e. conformation), the proteins are divided into two classes:
Fibrous Proteins
The polypeptide chains of these proteins extend along a longitudinal axis without showing any sharp bends of folding (Fig. 4.11). Simplicity of structure of these proteins is responsible for their mechanical properties. They are essential components of several proteins, including collagen and elastin (for details see Chapter 5).
Globular Proteins
Polypeptide chain of a globular protein is tightly folded and packed into a compact structure (Fig. 4.11). The hydrophilic groups of the constituent amino acids are exposed to the exterior, so that these proteins can diffuse through aqueous systems. Most enzymes, transport proteins, nutrient proteins, antibodies and hormones are globular.
Based on Structural Components
Two major classes of proteins are recognized depending on the nature of their structural components.
Simple Proteins
Simple proteins comprise solely of amino acids; no other chemical group is present in them. Pancreatic ribonuclease is an example.
Conjugated Proteins
Conjugated proteins contain a polypeptide group as well as a non-polypeptide group. In these proteins, the non-polypeptide group is called the prosthetic group, and the polypeptide is called the apoprotein. a prosthetic group is often employed when the biological function of the protein requires a functional group that is not available in any of the 20 amino acids. For example, in oxygen transport proteins, haemoglobin and myoglobin, binding of oxygen occurs with the iron-porphyrin group. The prosthetic group is sugar in glycoproteins (Greek: glycos means sweet), lipid in lipoproteins, and a metal in metalloproteins (Table 4.7 ).
Table 4.7
Conjugated proteins and their prosthetic groups
| Class | Prosthetic group | Example |
| Lipoproteins | Lipids | Chylomicron |
| Glycoproteins | Carbohydrates | γ-Globulin of blood |
| Phosphoproteins | Phosphate groups | Casein of milk |
| Haemproteins | Haem (iron porphyrin) | Myoglobin |
| Flavoproteins | Flavin nucleotides | Succinate dehydrogenase |
| Metalloproteins | Iron | Ferritin |
| Zinc | Alcohol dehydrogenase | |
| Calcium | Calmodulin |
D Protein Structure
Every protein has a unique three-dimensional structure, which is referred to as its native conformation. Protein conformation is complex and is best analyzed by considering it in terms of the following organizational levels; primary, secondary, tertiary, and quaternary (Fig. 4.12–d ).
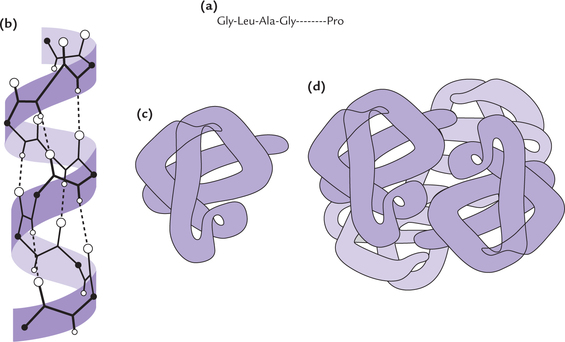
Fig. 4.12 Levels of protein structure. (a) Primary structure refers to the amino acid sequence in a polypeptide chain, (b)Secondary structure is the local spatial arrangement of the polypeptides backbone atoms, (c) Tertiary structure refers to overall folding pattern of the entire polypeptide chain, (d) Quaternary structure is formed by interactions in a non-covalent manner between different polypeptide subunits of a protein
Primary Structure
Primary structure of proteins refers to the specific sequence of amino acids in a polypeptide chain. The amino acids are covalently linked to one another, e.g. Al-Gly-Ser-Leu (Fig. 4.12a).
Thus, the polypeptide chains having different sequences of amino acids are said to differ with respect to their primary structure. it is noteworthy that it is the sequence of amino acids, and not the composition, that determines the primary structure. Thus, two polypeptide chains having identical set of amino acids, but arranged in different sequences, are said to have different primary structures. Such sequence variation is the most important element of protein diversity; for instance, a polypeptide with 100 amino acids can have 20100 different amino acid sequences.
The covalent backbone of protein, called its primary structure, is specified by the amino acid sequence. The most important element of proteins diversity is variability of the amino acid sequence.
Primary structure of insulin is shown in Figure. 4.13 . The specific information that determines the final three dimensional form adapted by a protein is inherent in its primary structure which determines the function of a protein (Case 4.2). a slight change in the primary structure of a protein (e.g. modification of a single amino acid residue) results in loss of its biological activity, in sickle cell anaemia, change in primary structure results in deranged function of the affected protein (Case 17.1).
Secondary Structure
The secondary structure refers to the local spatial arrangement of a polypeptides backbone atoms (without regard to the conformations of its side chains). It includes mainly the regional bends and local folding. The α-helix and (β-pleated sheets are some examples of the secondary structures that are commonly encountered in some proteins or portion of proteins.
α-Helix
It is a spiral structure consisting of a polypeptide chain that is coiled around a longitudinal axis in a helical fashion (Fig. 4.14a ). The α-helical structure was first defined by Linus Pauling from his studies on fibrous proteins. However, short stretches of α-helix are found in the globular proteins as well. a vast variety of proteins contain the α-helical structure. They constitute almost the entire dry weight of hair, wool, feathers, and scales and form much of the outer layer of skin.
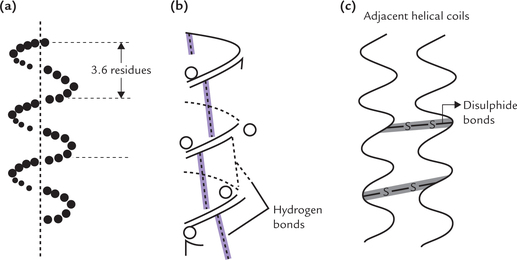
Fig. 4.14 The α-helical conformation. (a) A right handed helix with 3.6 residues per turn, (b) Intra-chain hydrogen bonding between the nth and (n + 4)th residues of α-helix, (c) Cysteine cross links between adjacent α-helical coils in α-keratin.
Characteristics
The polypeptide chain is coiled around the longitudinal axis to form the right handed helix (i.e. the coils turn in a clockwise fashion around the axis). Each turn of the α-helix contains 3.6 amino acid residues. The peptide bonds linking the successive amino acids are coiled inside and their R groups extend outward from the central longitudinal axis. This arrangement minimizes the stearic interference of the R groups with each other.
Forces stabilizing the α-helix
Two types of linkages stabilize the α-helix. These are hydrogen bonds and disulphide bonds.
• Hydrogen bonds: The hydrogen bonds are formed between the peptide bonds. Each peptide bond C = O is hydrogen bonded to the peptide bond N–H, four amino acid residues ahead of it. Each C = O and each N–H in the main chain is thus involved in hydrogen bonding (Fig. 4.14b).
• Disulphide bonds: Because the polypeptide chain is rich in cysteine residues, disulphide cross-links may form between the cysteine residues of adjacent helical coils. This enhances strength of the structure (Fig. 4.14c).
Amino acids not compatible with the α-helix
The following amino acids are not found in the α-helix because they disrupt the conformation.
Proline: The cyclic R group of proline is not geometrically compatible with the right-handed helical conformation. When incorporated in this structure, it causes the chain to bend sharply so that the regular helical conformation is disrupted. The helix-breaking property of proline has a significant effect on conformation of body proteins, including enzymes. Alteration of conformation may cause changes in biological activity of the affected protein (Case 4.3).
Amino acids with bulky R groups: Amino acids with bulky R groups, namely valine, leucine and isoleucine are too large to be accommodated in large numbers within the compact α-helix.
Amino acids with charged R groups: These amino acids (aspartate, glutamate, arginine and lysine) interfere with formation of α-helix because of electrostatic attraction or repulsion between the charged R groups.
β-Pleated Sheet
In 1951, the same year Pauling proposed α-helix, Pauling and Corney reported existence of a different type of secondary structure, the β sheet.
Characteristics
This structure is found in many fibrous proteins (e.g. silk fibroin) and some globular proteins. Features of β-pleated and α-helical conformation are given in Table 4.8 . The β-sheet consists of two or more polypeptide chains in an extended conformation, i.e. each of the chains is almost fully extended into a zigzag (Fig. 4.15 ). This is in contrast to the α-helix, where the chain is coiled around a central axis. The structure is called pleated because it resembles a series of pleats.
Table 4.8
Comparative features of α-helix and β-pleated sheet structure
| α-Helix | β-Pleated | |
| Hydrogen bonds | Intr-CHain, between n and (n +4) residues. Parallel to helix axis | Inter-CHain perpendicular to chain axis |
| Residues | Small or uncharged residues, such as alanine, leucine, and phenylalanine (most common); proline never found | Alanine, glycine and serine are common |
| Covalent cross linking | Inter–CHain disulphide | None |
| Chain direction and aggregation | Right-handed helical chains | Antiparallel chains |
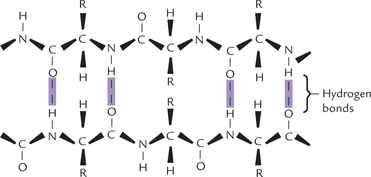
Stabilizing forces
Like the α-helix, the β-sheet uses the full hydrogen-binding capacity of the polypeptide backbone. In β-sheets, however, hydrogen bonds occur between neighbouring polypeptide chains (inter–CHain hydrogen bonds) rather than one chain, as in α-helix. The hydrogen bonds form between the peptide-bond C = O and N–H groups of polypeptides that lie side by side.
The interacting polypeptides are aligned either parallel or antiparallel. In the parallel arrangement, the N-terminals of the interacting polypeptide are together, whereas in antiparallel arrangement, the N-terminal and the C-terminal ends of the β-strands alternate. Sometimes the chain folds upon itself, as in the case of globular proteins. intr-CHain hydrogen bonds form between the adjacent folds in these proteins.
Tertiary Structure
The secondarily ordered polypeptide chain of the globular protein tends to fold onto itself (Fig. 4.12c) in aqueous solutions so that the protein molecule assumes a spherical or globular shape. The term tertiary structure refers to the overall folding pattern of the polypeptide. it not only describes folding of the secondary structural elements into a compact globular or spherical shape, but also depicts location of each atom in the protein. This includes geometric relationship between distant segments of the primary structure and the positional relationship of the side chains with one another. This is in contrast to the secondary structure, which refers to spatial relationship between side chains of the successively adjacent amino acids.
Globular conformation involves tight folding. This is evidenced by the fact that actual dimensions of a globular protein in its native conformation are much smaller than the approximate dimensions if it occurred as an α-helix or in β-pleated conformation (Fig. 4.16 ). Finally the hydrophobic side chains of the amino acids tend to aggregate towards the interior in globular proteins, whereas the hydrophilic groups strongly prefer to be on the exterior, exposed to water. Thus, globular proteins have a hydrophobic core but a hydrophilic surface, which enables them to interact with aqueous surroundings.
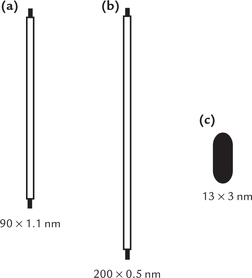
Fig. 4.16 Comparative dimensions of bovine serum albumin if it existed in (a) α-Helical form, (b) β-Pleated conformation, (c) Native globular conformation.
Folding of Proteins and Chaperones
Polypeptide chain of a given amino acid sequence (i.e. primary structure) always folds in the same characteristic fashion, suggesting that the information for protein folding is inherent in the primary structure itself. This is in line with the Anfinsens law stating that the secondary, tertiary and quaternary structures are determined by the primary structure. indeed, protein folding starts, and often makes considerable headway, while translation is still in progress (Chapter 22).
In addition to the primary structure, certain other factors also determine the overall folding pattern. This is evident from the fact that in some cases, following denαturation of protein (i.e. unfolding) by temperature or pH alterations, the native conformation is not regained (by the unfolded protein) even when the original temperature or pH is restored.
It is now known that a group of specialized proteins called “chaperones” are required for the native folding of many proteins. The chaperones are also called polypeptide chain binding (PCB) proteins. Though they aid in folding during synthesis, they are not responsible for the stability of the final product. Chaperones bind to hydrophobic patches and stabilize intermediates during folding. They prevent aggregation of incompletely folded proteins by precluding adventitious contacts between exposed hydrophobic regions. Defective interaction of a polypeptide with the corresponding chaperone may cause disease (Case 7.2).
The energy requirements of chaperones can be substantial, and more than 120 ATP molecules can be expended during the folding of a single polypeptide chain. Many chaperones are heat shock or stress proteins, but they primarily serve the aforementioned function under physiological conditions.
Domains
Long polypeptides that contain more than 200 residues fold into two or more globular clusters known as domains. The domains are connected by more extended part of the polypeptide chain, which gives these polypeptides a bior multi-lobal appearance. Each subunit of the enzyme glyceraldehyde 3-phosphate dehydrogenase, for example, has two domains connected by more extended part of the polypeptide chain.
The globular proteins have hydrophilic surface and hydrophobic core. Large proteins form domains. Folding of domains and their final arrangement in the polypeptide is the another way in which the tertiary structure is defined.
Many domains are structurally independent units, each having characteristics of a small globular protein. Generally, domains are formed from between 40 to 400 amino acid residues (mean size = 200 amino acid residues). Different domains may have different biological functions.
Forces Stabilizing the Tertiary Structure
Tertiary structure mostly represents a state of lowest energy and imparts greatest stability for the polypeptide in question. Five kinds of interactions, cooperate to stabilize it (Fig. 4.17 ). These are:
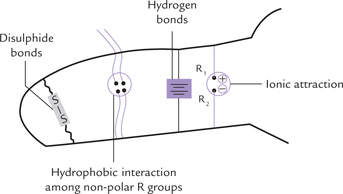
Fig. 4.17 Representation of interactions that stabilize the tertiary structure of globular proteins.
1. Hydrophobic interactions: These are the major cohesive forces in determining the native protein structure, and arise due to tendency of non-polar substances to minimize their contact with water. In water soluble globular proteins, the non-polar side chains aggregate in the interior hydrophobic core (whereas the hydrophilic side chains occupy surface), and this provides much of the driving force for protein folding.
2. Hydrogen bonds: These are formed by sharing of a hydrogen atom between one atom that has a hydrogen atom (donor) and another atom that has a lone pair of electrons (acceptor). The donors are –NH (peptide, imidazole), -OH (serine), and –NH2 (lysine, arginine), and the accepting groups are COO- (aspartate, glutamate), C = O (peptide) and S-S (disulphide). The hydrogen bonds make only a minor contribution to protein stability.
3. Ionic interactions: The association between two ionic side chains of opposite charge (e.g. + ve lysine or arginine; and –ve Glu or Asp) is known as an ion pair or salt bridge. Though the electrostatic attraction between the members of an ion pair is strong, these interactions contribute little to the stability of the tertiary structure. This is because of their relatively small number.
4. Van der Waals interactions and London dispersion forces: These are very short range interactions that occur between the tightly packed aliphatic side–CHains in the interior of the protein. They are weak and develop only when atoms are packed very closely to each other. They serve to bring the atoms closer especially in the hydrophobic core in the interior, squeezing out gaps, if any, and thereby, contribute enormously towards stability of the native protein structure.
5. Covalent cross linkages: Cysteine residues, when present on the adjacent loops of the polypeptide(s), form covalent cross links (i.e. disulphide bonds). These linkages, however, do not establish the higher-order structure; they only stabilize the structure that has been formed by non-covalent interactions.
Quaternary Structure
The proteins, discussed so far consisted of a single polypeptide chain. But several other proteins consist of two, three or more subunits (i.e. oligomeric proteins). The subunit chains may be structurally identical or different. Haemoglobin consists of four polypeptide chains of two different types—α and β. Number, size and shape of component polypeptide chains and their spatial relationship with one another is called the quaternary structure of the protein (Fig. 4.12d).
Non-covalent interactions, such as hydrophobic interactions, hydrogen bonds, electrostatic bonds, etc., hold the subunits together in some proteins, but others are stabilized by interchain disulphide bonds.
Different subunits may cooperate with one another (i.e. subunit interaction); for example, binding of oxygen to one of the subunits of the haemoglobin is followed by transfer of the ligand-binding information to other subunits, which in turn respond by increasing their affinity for oxygen (Chapter 17).
The formation of secondary, tertiary and quaternary structures of protein follows the thermodynamic principles. In a polypeptide chain, the primary structure determines the higher levels of organization in such a way that the final form represents the lowest energy stage. Therefore, the quaternary structure is the most stable of all the possible conformations. Simply stated, it represents the state of least energy.
E Protein Denaturation
The term denaturation refers to disruption of the higher order structure of the protein. a number of agents are known, which act by breaking the physical bonds in a protein without splitting any peptide linkages. As a result, the well-ordered, neatly folded polypeptide is converted to a messy tangle work, called a random coil, and the process is protein denaturation. Note that only the non-covalent interactions that maintain the higher order structure are disrupted, but the covalent bonds (including peptide bonds) are left intact. Thus, the primary structure is not altered during denaturation, but denaturation may completely disrupt secondary, tertiary and quaternary structures (e.g. denatured oligomeric protein dissociates into subunits, each with a random coil conformation). Denaturation may be partial in which only certain aspects of the secondary and/or tertiary structures are broken.
Denaturation is always accompanied by a loss of biologic functions, e.g. enzymes are inactivated, antibodies fail to act with antigens and toxins lose their diseaseproducing effects. This is because biological functions of proteins depend on their higher order structures. Denaturation is generally irreversible boiled egg does not regain its original form when kept in cold. Under well-controlled laboratory conditions, renaturation is possible for some small proteins, e.g. ribonuclease is denatured by 8M urea, but when urea is removed by dialysis, the renaturation occurs and the enzyme activity is restored.
Changes in Physical Properties
A number of physical parameters of the protein change because of partial or complete denaturation:
1. Solubility: Water-soluble proteins become insoluble, the boiling of an egg is a familiar example of this phenomenon.
2. Precipitation: As the protein becomes less symmetric, it exerts resistance to various types of movements, and in many cases it precipitates.
3. Optical rotation: a more negative optical rotation results specific rotation of native proteins is between —20° and —40°, whereas that of denatured proteins is near –100°.
4. The sedimentation rate and diffusion rate fall and the intrinsic viscosity increases.
Agents Causing Denaturation
Proteins can be denatured by many treatments:
1. Heat: Most proteins are denatured at temperatures between 50°C and 80°C; at these temperatures the non-covalent interactions, which are individually very weak, are readily broken.
2. Detergents and organic solvents: They serve to disrupt hydrophobic interactions. Being non-polar, they insert themselves between the side chains of hydrophobic amino acids.
3. Strong acids and bases: They disrupt the intramolecular salt bonds by changing the charge pattern of the protein. in a strong acid, the protein loses its negative charges; and in a strong base, it loses its positive charges; both deprive the protein of the salt bonds.
4. 8M Urea or 6M guanidine hydrochloride: Both are hydrophilic agents with high hydrogen binding potentials. They can denature proteins by disrupting the hydrogen bonds between the protein and the surrounding water molecules. Normally the water molecules assume an “ordered” position at an aqueous/ non-polar interface, which is reduced by these agents, and consequently the hydrophobic interactions within the protein are weakened.
5. Heavy metal ions: They have high affinity for carboxylate groups, and for the sulphydryl groups in many proteins.
6. Trichloroacetic acid (TCA): It is of special importance in laboratories because it can rapidly denature and precipitate proteins, which can then be removed by centrifugation.
7. Physical agents: X-Rays, ultraviolet rays, high pressure, vigorous shaking and surface tension also cause denaturation.
F Precipitation Reactions of Proteins
Solubility of proteins depends on pH and salt concentration. Precipitation (of proteins) occurs when their solubility decreases. Unlike denaturation, precipitation is reversible and does not cause permanent loss of the protein’s activity.
1. Salting-in and -out: Salts at moderate concentration may cause increased protein solubility (salting-in) and at high concentration may decrease the solubility resulting in protein precipitation (salting-out).
(i) Salting-in: It is shown in Figure 4.18b . The protein molecules have multiple salt bonds among them, which cause them to aggregate. As a result they are insoluble (Fig. 4.18a). In 5% NaCl, salt ions bind to surface charges of protein molecules, thereby preventing intermolecular salt bonds. This causes increased solubility.
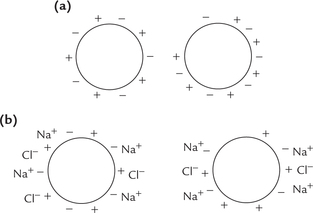
Fig. 4.18 Effects of salt on protein solubility. (a) Protein in distilled water. Salt bonds between protein molecules cause the molecule aggregate. The protein becomes insoluble, (b) Salting-in: Protein in 5% sodium chloride. Salt ions bind to the surface charges of the protein molecules, thereby preventing intermolecular salt bonds.
Thus higher solubility at a moderate concentration (1–10% salt) is because the salt ions shield the protein’s multiple ionic charges, thereby weakening the attractive forces between different protein molecules.
(ii) Salting-out: Excess salt, however, precipitates the proteins because most of the water molecules become tied up in forming the hydration shells around the salt ions. Effectively, so many salt ions are hydrated that there is significantly less bulk water available to dissolve other substances, including proteins. Ammonium chloride, one of the most soluble salts, is the most commonly used reagent for salting out the plasma proteins.
2. Precipitation by organic solvents: The water-miscible organic solvents like ethanol or acetone, because of their high affinity for water, tie up most of the available water molecules. Thus, like salts they cause precipitation of proteins, without denaturing them.
3. Precipitation by altering pH: The pH value also affects water solubility—a protein is least soluble at its isoelectric pH when equal numbers of positive and negative charges exist and so the protein molecules have maximal opportunity to form intermolecular salt bonds (Fig. 4.19 ). At pH values higher or lower than pI, the interactions are mainly repulsive. The pH may be adjusted to approximate the isoelectric point (pI) of the desired protein to cause its precipitation.
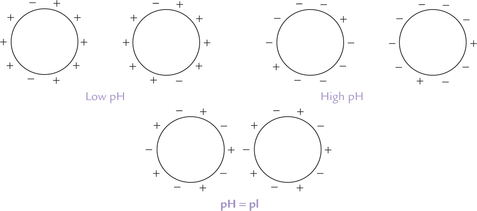
Fig. 4.19 The effect of pH on protein solubility: Formation of intermolecular salt bonds is favoured at the isoelectric point; at pH values greater or less than the pl, the electrostatic attractions between the molecules get weakened.
4. Precipitation by heavy metals: Heavy metals, such as lead, cadmium, and mercury bind to negative groups (carboxylate) or sulphydryl groups in many proteins, thus decreasing their solubility. Because of their affinity for functional groups, these metals tend to precipitate normal proteins of the gastro-intestinal tract, which explains their toxicity.
G Separation and Purification of Proteins
To study and use a protein it is necessary to separate it from a biologic fluid and purify it. Purification is also necessary for proteins that are used for therapeutic purposes, such as insulin and clotting factor VIII used to treat haemophiliacs. Several techniques are available for this purpose which use fundamental properties of proteins, such as solubility, molecular size, molecular charge and selective binding of proteins to specific substances (i.e. ligands). Generally, a combination of these techniques is employed to separate a given protein from other proteins and molecules. In clinical laboratories, protein separation is routinely carried out for diagnostic purpose. For example, separation of enzyme proteins (by electrophoresis) is useful for the diagnosis of various disorders (Case 6.5). Similar techniques are employed for therapeutic and research purposes as well.
The aim of protein purification is to isolate one particular protein from all the others in the sample mixture. The purification techniques exploit solubility, molecular size, charge or/and specific binding affinity of the protein of interest.
Separation on the Basis of Protein Solubility
Protein solubility varies with salt concentration of the medium. Addition of salts to the medium results in decreased solubility of some proteins, an effect called salting out. The salt concentration at which a protein precipitates differs from one protein to another. For example, globulins precipitate at half saturation of ammonium sulphate (or 22% sodium sulphate) but full saturation of ammonium sulphate (or 28% sodium sulphate) is required for precipitating albumin.
Separation on the Basis of Molecular Size Dialysis
Proteins can be separated from solute particles by a process termed dialysis which uses the fact that protein molecules are much larger (MW > 10,000) than solute particles. The mixture of protein and solute is put in a cellophane bag, which is made of a semi-permeable membrane. This membrane contains pores which allow the passage of smaller solute particles, but not of protein molecules (Fig. 4.20 ). When the bag is suspended in stirring buffer solution, the low-molecular weight solute particles gradually pass out.
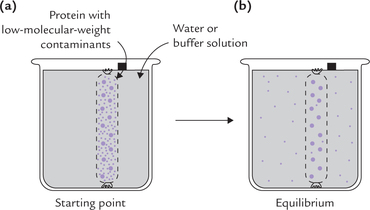
Fig. 4.20 (a) The dialysis bag containing the protein mixture and small solute particles is suspended in water, (b) Magnified view of a portion of the semi-permeable dialysis bag showing outward movement of solute particles.
The removal of low molecular weight waste products from blood of patients with renal failure is based on the same principle; the process called haemodialysis. The patient’s blood is passed along a semi-permeable membrane through which the waste products (e.g. urea) are removed, but plasma proteins and blood cells are retained. The blood, however, is dialyzed against a solution with physiologic concentration of inorganic ions and nutrients, and not against any buffer.
Proteins can be separated from small molecules by dialysis through a semi-permeable membrane which has pores that allow solute particles to pass through but not proteins.
Gel Filtration (Column Chromatography)
Particles of different sizes can be separated from one another by gel filtration, also called, size-exclusion or molecular-sieve chromatography. The equipment consists of a column, which is packed with a beaded hydrophilic material such as Sephadex gel. Sephadex is a dextran derivative, obtained from bacterial cell walls. a gel bead contains numerous pores. The pore size is such that the smaller proteins can penetrate the porous gel beads, while the larger proteins bypass the beads altogether (Fig. 4.21 ). Having access to gels internal volume, the small proteins take a longer and tortuous route through the column. Therefore, they are retarded on the column with respect to the larger protein that cannot enter the porous gel beads. Thus, the latter traverse the column more rapidly and emerge earlier in the column effluent than the smaller ones. The proteins of intermediate size have partial access to gel’s internal volume, and therefore, emerge in between, before the larger proteins but after the smaller proteins.
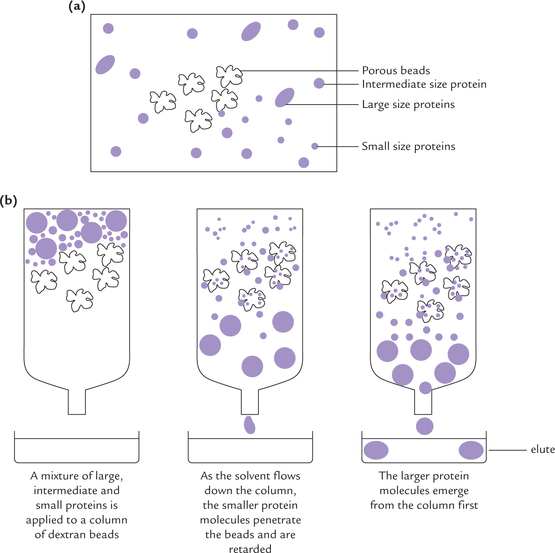
Fig. 4.21 Gel filtration (column chromatography). (a) Structure of a bead, (b) Process of separation.
Fractions of a few milliliters (each fraction called an elute) are collected from the bottom of the column; the process is called elution. Each of these elutes would contain different size particle.
Ultracentrifuge
Proteins of different molecular sizes can be separated by high-speed centrifugation into different components. Rate at which a protein moves in a centrifugal field (i.e. sedimentation) depends on its size; larger proteins sediment faster than the smaller ones. This method can separate those proteins which have large differences in their molecular size.
Separation on the Basis of Molecular Charge
Differences in the nature and degree of electric charges carried by various proteins at a given pH forms the basis of their separation by several techniques, e.g. ion-exchange chromatography and electrophoresis. Difference in charges is due to difference in acid-base behaviour of these proteins; which in turn is due to different isoelectric pH values of the proteins (Table 4.3).
Ion-exchange Chromatography
Preparative separation of proteins can be achieved on the basis of charge-differences by this technique. The method takes advantage of differential affinity between the following:
• Charged ions or molecules in the sample mixture.
• Charged groups that have been immobilized on ion exchange resins.
The resins are packed in chromatography column (Fig. 4.22 ). They are cross-linked polymers (e.g. cellulose or agarose) containing charged groups as part of their structure. The charged groups most commonly attached are diethylaminoethyl (DEAE) group and carboxymethyl (CM) group:
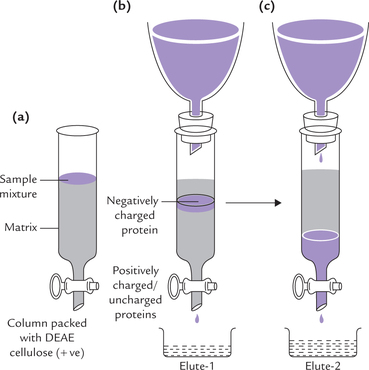
Fig. 4.22 Ion exchange chromatography. (a) Sample mixture consisting of positively charged, negatively charged and neutral proteins in a small volume of buffer applied at the top of the column, (b) The positively charged and neutral proteins pass through the column and are collected in elute-1. Flow of the negatively charged proteins is retarded, (c) The negatively charged proteins, earlier attached to the positive matrix, are made to pass through the column by applying a high-salt/low pH elution buffer.
DEAE groups are protonated and have a positive charge associated with a negatively charged counterion, such as chloride; this system is called DEAE-cellulose chromatography (Fig. 4.23 ). If a protein solution at a specific pH is applied to this column:
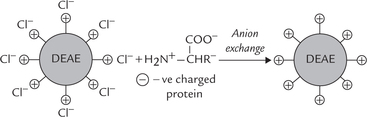
Fig. 4.23 DEAE cellulose chromatography (anion exchange). The protonated groups ( + ve) on DEAE associated with chloride counterions (— ve). The negatively charged proteins exchange  with chloride and get bound to resin.
with chloride and get bound to resin.
• The negatively charged proteins replace the chloride ions and associate with the positively charged DEAE groups. This is anion exchange.
• The positively charged or uncharged proteins have relatively low affinity for the anion exchanger than the negatively charged proteins. They, therefore, flow faster and emerge from the column earlier (Fig. 4.22a).
• In this way, the negatively charged proteins get separated from the uncharged/or the +ve proteins (Fig. 4.22b). They (–ve proteins) can be subsequently eluted (washed through the column) by changing elution conditions, such as applying a buffer that has a higher salt concentration, or at a pH that reduces the affinity with which the resin binds the protein (Fig. 4.22c).
CM groups with negative charge are associated with counterion Na+. (Fig. 4.24 ). In this case, positively charged proteins exchange with Na+ and get bound to the resin (i.e. cation exchange), whereas the negative ones are eluted faster.
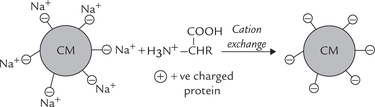
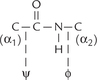 attach with carboxy methyl groups (negative) and get bound to resin.
attach with carboxy methyl groups (negative) and get bound to resin.High Performance Liquid Chromatography (HPLC)
HPLC resembles the conventional chromatographic techniques where the solution of proteins is passed through special resins that have attached side groups. These side groups can react with the protein sample ionically or hydrophobically. However, there is one important differences HPLC is carried out under high pressure (5000–10,000 pounds per square inch) in the chromatographic matrix. The matrix consists of specially fabricated 3–300 μm-diameter glass or plastic beads, which are coated with a uniform layer of chromatographic material. This greatly improves the speed, resolution, and reproducibility of the separation.
Electrophoresis
This process uses an electric field to drive the movement of charged particles. It is used for the separation of a wide variety of polyelectrolytes (carry positive and negative charges), including amino acids, polypeptides, proteins (and DNA).
At pH values greater than the isoelectric point (pI), the protein carries an excess of negative charge and moves to the anode; at pH values less than pi, it carries positive charge and moves towards the cathode; and at isoelectric point the net charge is zero and it does not move. if for example, an alkaline pH is used (greater than pI of most proteins) the serum proteins are negatively charged and move towards the anode, but are separated according to their net charge (Fig. 4.25 ). a porous support such as paper, cellulose acetate foil, starch gel, or other carrier material is commonly used to minimize diffusion and convection. Electrophoresis is the most common method of protein separation in the clinical laboratory, used for separation of plasma proteins and isoenzymes and for the detection of structurally abnormal proteins. Case 5.1 illustrates diagnostic utility of electrophoresis in a bone disorder.
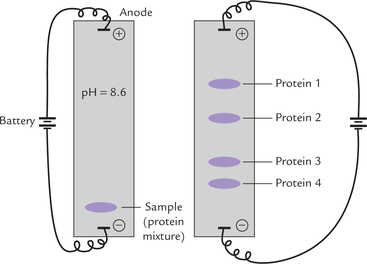
Fig. 4.25 Protein separation by electrophoresis on a wet cellulose acetate foil. At pH = 8.6, all proteins are negatively charged and move towards anode. They are separated into various bands according to their net charge.
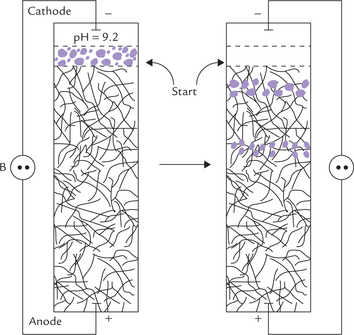
Polyacrylamide gel electrophoresis (PAGE)
Electrophoresis can be performed in a cross-linked polyacrylamide gel with characteristic pore size (Fig. 4.26 ). Small molecules can move in the gel, but the larger ones “get stuck” in the gel and so their movement is retarded. The molecular separation is, therefore, based on sieving effects as well as electrophoretic mobility.
SDS-PAGE
Under pH conditions at which all proteins move to the same pole, they are better separated according their molecular mass rather than their charge. To accomplish this, the above method (PAGE) is used in the presence a strong detergent (e.g. sodium dodecyl sulphate; SDS). It causes denaturation by interfering with the hydrophobic interactions that normally stabilize proteins; the denatured proteins assume a rod-like shape. The large negative charges that the SDS imparts mask the protein’s intrinsic charge. The net result is that SDStreated proteins have similar shapes and charge-to-mass ratio. SDS-PAGE, therefore, separates proteins by gel filtration effect, i.e. according to molecular mass. The method is performed in presence of a reducing agent such as mercaptoethanol, which cleaves the disulphide bonds.
Isoelectric focusing
It is a variant of electrophoretic technique, that has extremely high resolution. It is carried out on a gel that contains a gradient of pH. The pH gradient is created across the supporting medium and electrophoresis of the protein mixture carried out through this gradient. a charged protein migrates through the gradient till it reaches a pH region of the gradient equal to its pI value. At this point it stops moving (net charge on the molecule being zero) and can be visualized. An extremely sensitive technique, isoelectric focusing can separate proteins that differ by as little as 0.0025 pI values.
Separation on the Basis of Affinity Binding
Affinity Chromatography
This is the only method of protein purification that exploits a property related to the function of the protein, namely the ligand-binding ability. This is a definite advantage over exploiting small differences in the physicochemical properties between proteins in the other chromatographic methods.
The column is packed with a chromatographic material that carries an immobilized ligand (Fig. 4.27 ).
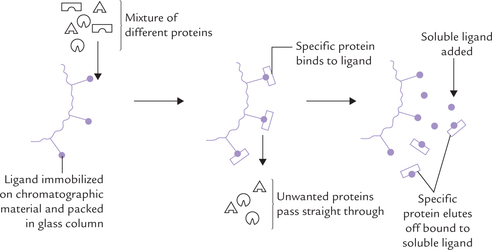
The mixture of proteins is poured over the column. As it percolates down, the protein molecules with an appropriate ligand-binding site gets bound to the immobilized ligand, whereas the other proteins are washed through the column with the buffer. The bound protein can then be recovered in highly purified form by changing the elution conditions. (Generally, it is washed with solution having a high concentration of soluble-ligands.)
Precipitation by Antibodies
For separating a given protein from a protein mixture, antibodies are prepared against it. When these antibodies are added to the protein mixture, they form antigenantibodies complexes with protein of interest. These complexes are large in size, and so can be separated from the rest of mixture by centrifugation. Since it is necessary to create complexes of sufficiently large size, the antiglobulin (anti IgG) is further added, so that the large triple complex (of antigen-antibody-anti IgG) is formed.
Separation on the Basis of Hydrophobicity
In this technique, called hydrophobic interaction chromatography, separation of protein mixtures is accomplished on the basis of protein hydrophobicity. The proteins and the matrix material packed in the column interact hydrophobically. The matrix material is lightly substituted with acetyl or phenyl groups and the nonpolar groups on the surface of proteins interact with them by the hydrophobic interactions. Subsequently, the bound proteins are eluted by decreasing salt concentration, increasing concentration of detergent (which disrupts hydrophobic interactions), or by causing changes in pH.
H Determination of Amino Acid Sequence of Peptides and Proteins*
The primary structure of proteins can be determined by chemical methods.
• The preliminary steps (steps 1 and 2) provide clue to protein structure (e.g. size and amino acid composition of the protein), and
• The subsequent steps determine the exact amino acid sequence.
Step 1: The size of the protein
The molecular size of the protein is estimated usually by SDS-PAGE (sodium dodecyl sulphate-polyacrylamide gel electrophoresis), as discussed earlier. Since all proteins are covered with negative charges of SDS, they tend to be linear in shape and their movement in electric field solely depends on their size.
Step 2: Amino acid composition of the protein
It is determined by complete hydrolysis of the polypeptide, followed by the analysis of the liberated amino acids. The hydrolysis is accomplished by acid hydrolysis of the peptide linkages in 6N–HCl for 24–72 hours at 100°–110°C. This treatment, however, destroys tryptophan. Alkaline hydrolysis is, therefore, used to liberate this amino acid. The amino acids so liberated are then separated by chromatographic methods and determined quantitatively with suitable reagents. Modern amino acid analyzers can completely analyze a protein digest containing as little as 1 pmol of each amino acid in less than an hour.
Step 2 reveals the protein’s amino acid composition, but not the amino acid sequence. it is, however, useful because the ratio of the amino acids can be compared with that from known proteins and this may give a clue to its structure.
Step 3: Determination of N and C terminal amino acids
The N-terminal amino acid is covalently bound to dansyl chloride. The tagged protein is then hydrolyzed to its constituent amino acids by acid hydrolysis. Because only the N-terminal amino acid is dansylated, it is identified by separating it out by column chromatography and comparing against dansylated standards.
The C-terminal amino acid is determined by treating intact polypeptide with hydrazine, which reacts hydrolytically at the carbonyl grouping (C–O) at each peptide bond (resulting in acyl derivatives of each residue). However, it cannot react with the C-terminal, which has carboxyl group (rather than a peptide linkage). The amino acids are then analyzed by column chromatography and compared with standards. The amino acid without hydrazine is the C-terminal amino acid.
Step 4: Sequence analysis
It is performed by Edman degradation method on automatic machines called, cyclic sequenators. This method sequentially removes one residue at a time from the amino end of a peptide. Phenyliosthiocyanate reacts with the N-terminal amino group of the peptide to form a phenylthiocarbamyl derivative in alkaline solution. It is released by gentle acid hydrolysis and identified by HPLC. The second amino acid now becomes the N-terminal amino acid and the cycle can be repeated by returning to alkaline conditions.
Only about 20–25 amino acids can be sequenced in this way. The large polypeptides, however, must be fragmented first. a number of fragmenting agents break specific peptide linkages, among which are digestive endopeptidases, trypsin and chymotrypsin.
• Trypsin cleaves only those peptide bonds in which carboxyl group is contributed by basic amino acids, lysine or arginine.
• Chymotrypsin cleaves only the bonds on the carboxyl side of the aromatic amino acid residues.
• Cyanogen bromide breaks those peptide bond in which methionine contributes the carboxyl groups.
• The short fragments so generated are more manageable in sequencing studies and the amino acid sequences in each fragment are then determined, as discussed above.
Step 5: Reconstructing the protein’s sequence
After individual peptide fragments have been sequenced, their order in the original polypeptide must be elucidated. For this a second round of protein cleavage with a different endopeptidase is carried out. This generates an overlapping set of peptide fragments. These fragments are then separated and sequenced. The sequences of the overlapping peptides are then used, much as one would fit parts of a jigsaw puzzle, to obtain the whole protein structure.
Sequencing by Molecular Biology Techniques
This is an entirely different and relatively simple approach based on the fact that genes encode amino acid sequences of polypeptides; the base sequence of the gene predicts the amino acid sequence.
Step 1
The protein is partially purified and the first eight (or ten) amino acids at the N-terminal end are sequenced using an automatic sequenator.
Step 2
These eight amino acids are compared with an international protein/DNA sequence library available on the Internet, e.g. GenBank. If lucky, a match occurs in terms of molecular weight, which means that the protein (or a closely similar protein) has already been isolated and sequenced. Further sequencing may not be necessary in this case.
Step 3
If no match is found, then a cDNA molecule that codes for the N-terminal amino acid sequence is made. This is called cDNA primer.
Step 4
Using the cDNA as a probe, the RNA that codes for the protein is extracted from the cells. The two readily hybridize (attach together) because their sequences are complementary.
Step 5
The DNA complementary to the rest of the RNA sequence is then synthesized by polymerase chain reaction (Chapter 25). This results in the synthesis of large amounts of DNA that codes for the protein.
Step 6
The DNA is then sequenced using techniques described in Chapter 21 and, using the universal genetic code, and the protein sequence is derived from the DNA sequence.
This process sounds complicated but offers advantage that very little of the protein is needed and the protein does not have to be fully purified. Full purification is especially difficult in case of insoluble proteins (e.g. membrane proteins). Moreover, sequencing DNA is a relatively simple procedure.
I Determination of Higher Order Structure of Proteins+
X-Ray Crystallography
This method is based on the scattering of the beam of x-rays passed through a protein crystal by high mass proton cores. The scattered x-rays form a diffraction pattern which is captured on film (or collected by an electronic device) in the form of an array of dots, from which bond lengths, folding and coiling and the overall three dimensional structure in proteins may be deduced. Because this pattern indicates the relative positions of atoms in the molecule, a careful study and analysis of it permits modelling of the 3-dimensional structure of the crystalline protein. Pauling and Corey were pioneers in interpreting such protein diffraction pattern to determine the structures of α-helix and β-pleated sheet.
Ultraviolet Light Spectroscopy
Proteins absorb ultraviolet radiation with two absorption maxima: one at 190 nm is caused by peptide bonds, and the other at 280 nm is caused by the aromatic side chains of tyrosine and tryptophan (aromatic character of purine and pyrimidine bases also causes absorbance peak at 260 nm for nucleic acids).
Nuclear Magnetic Resonance (NMR)
Since mid 1980s advances in NMR spectroscopy have permitted the determination of the three-dimensional structure of proteins (and nucleic acids) in aqueous solution. The sample of protein is placed in a magnetic field so that the spin of protons is aligned. Radiofrequency pulses are applied, which cause excitation of protons. The excited protons then emit signals, whose frequency depends on the molecular environment of these protons. interpretation of these signals helps to construct the three-dimensional protein structure. The technique is useful for the proteins that fail to crystallize. However, utility of this technique is limited to a size of less than 30 kD.
Computer Based Modelling
A number of proteins have been modelled till now, and based on them computer programs have been developed to predict the most likely tertiary structure from the primary amino acid sequence.
IV Chemical Synthesis of Proteins
Using chemical means, protein chemists have synthesized several small proteins and peptides in laboratory. The basic mechanism involves fixing of the carboxyl group of the last amino acid on the resin, followed by addition of other amino acids sequentially. Steps must be taken to assure that carboxyl and amino groups that are not participating in peptide bond formation must be blocked, and groups that are to participate in bond formation must be activated (for example, by PCl5). Though synthesis of several polypeptides with more than 100 amino acids has been made possible, production of only small oligopeptides is cost effective. This limitation is deplorable because several naturally occurring proteins, such as hormones, and blood-clotting factor are useful therapeutic agents, and it is easier and cheaper to synthesize them rather than isolate and purify them from biological sources.
Exercises
Essay type questions
1. What do you mean by the term conformation as applied to a protein? Describe the various orders of structure of proteins.
2. Define secondary and tertiary structures. Mention the bonds that are responsible for maintaining these structures.
3. Give two examples each of fibrous and globular proteins. Describe the structural and functional relationship with respect to any one protein.
4. Describe the separation techniques that exploit the following molecular properties: charge, polarity, size and specificity.
5. Why are proteins called amphoteric molecules? Explain why histidine, but not alanine, can act as buffer at physiologic pH.
*This topic relates to higher level learning and it is meant only for postgraduates. Undergraduates or MBBS students may refer to it for higher learning.
+This topic relates to higher level learning and it is meant only for postgraduates. Undergraduates or MBBS students may refer to it for higher learning.