Diseases of Bone and Joints (Non-neoplastic and Non-infectious Disorders of Bone, Skeletal Dysplasias/Dysostoses, Constitutional Bone Disorders)
 . Group I: Defects in Extracellular Structural Proteins
. Group I: Defects in Extracellular Structural Proteins
. Diseases of Bone of Questionable Etiology
. Diseases of Temporomandibular Joint
. Development Disturbances of Temporomandibular Joint
. Traumatic Disturbances of Temporomandibular Joint
. Inflammatory Disturbances of Temporomandibular Joint
The diseases of bone to be considered in this chapter do not include specific infections, neoplasms or other recognized injuries restricted to the jaws, but constitute a group of generalized skeletal diseases which frequently manifest involvement of the maxilla or mandible and therefore the use of the term ‘diseases’ should be viewed with caution, but is adapted here more in a general context. The category of diseases described here may be better called constitutional bone disorders due to the reasons mentioned. Bone is a dense calcified tissue which is specifically affected by a variety of diseases that often cause it to react in a dynamic fashion. Some of these diseases involve the entire bony skeleton, while others affect only a single bone. It is characteristic for certain of these conditions to follow a strict Mendelian pattern of heredity, although sometimes a specific disease will be inherited in one case and apparently not in another.
The maxilla and mandible, like other bones, suffer from both the generalized and the localized forms of skeletal diseases. Although the basic reactions are the same, the peculiar anatomic arrangement of teeth embedded partially in bone, through which the bone may be subjected to an unusual variety of stresses, strains and infections, often produces a modified response of bone to the primary injury.
Skeletal dysplasias are a heterogeneous group of disorders, which result in disproportionate short stature. The nomenclature of these disorders remains confusing. In an attempt to develop uniformity, an international nomenclature and classification was proposed in 1969 and then updated many times later. In the 1992 revision, the classification was based on radiodiagnostic and morphologic criteria. In the 1997 revision, the groups of disorders were rearranged based on current etiopathogenetic information regarding the gene and/or protein defect in these disorders (Table 17-1). In the 2001 revision, the term dysostoses was incorporated in the nomenclature. All these revisions merely reflect the complexity of skeletal-genetic phenotypes. Over the recent years the accumulation of knowledge on genes and proteins responsible for genetic disorders of the skeleton has been unprecedented. A molecular pathogenetic classification of skeletal dysplasias based on the structure and function of the causative gene and protein was recently proposed (Table 17-2).
Table 17.1
International nosology and classification of genetic disorders of bone —2006
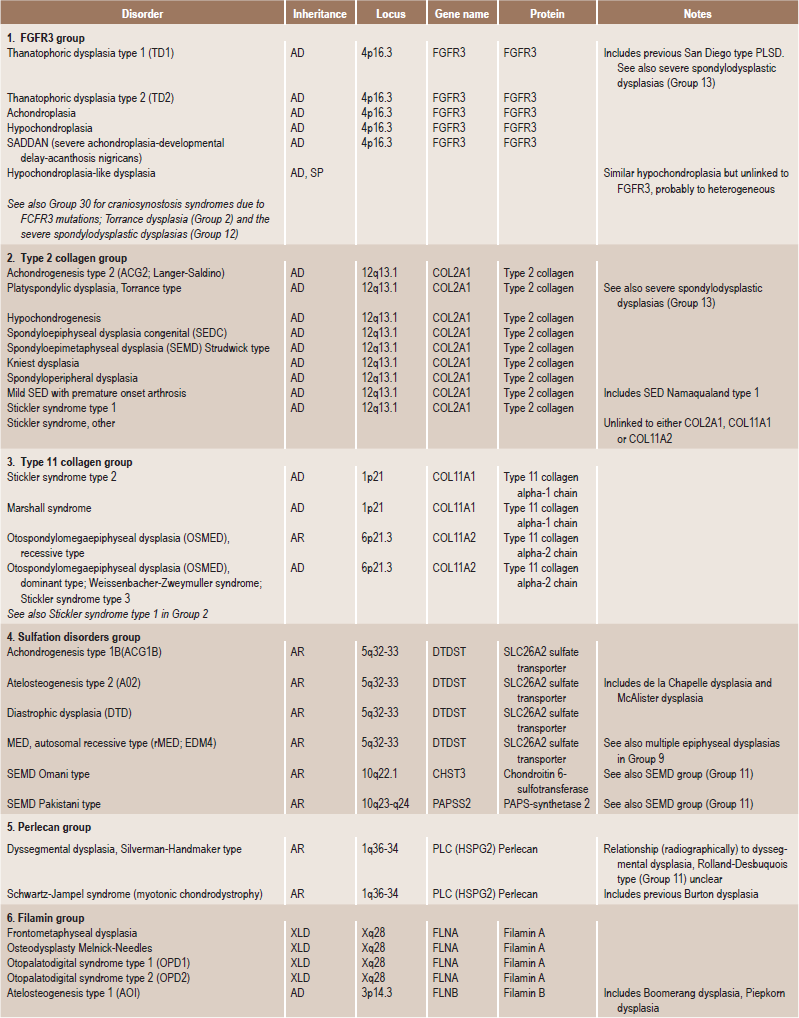
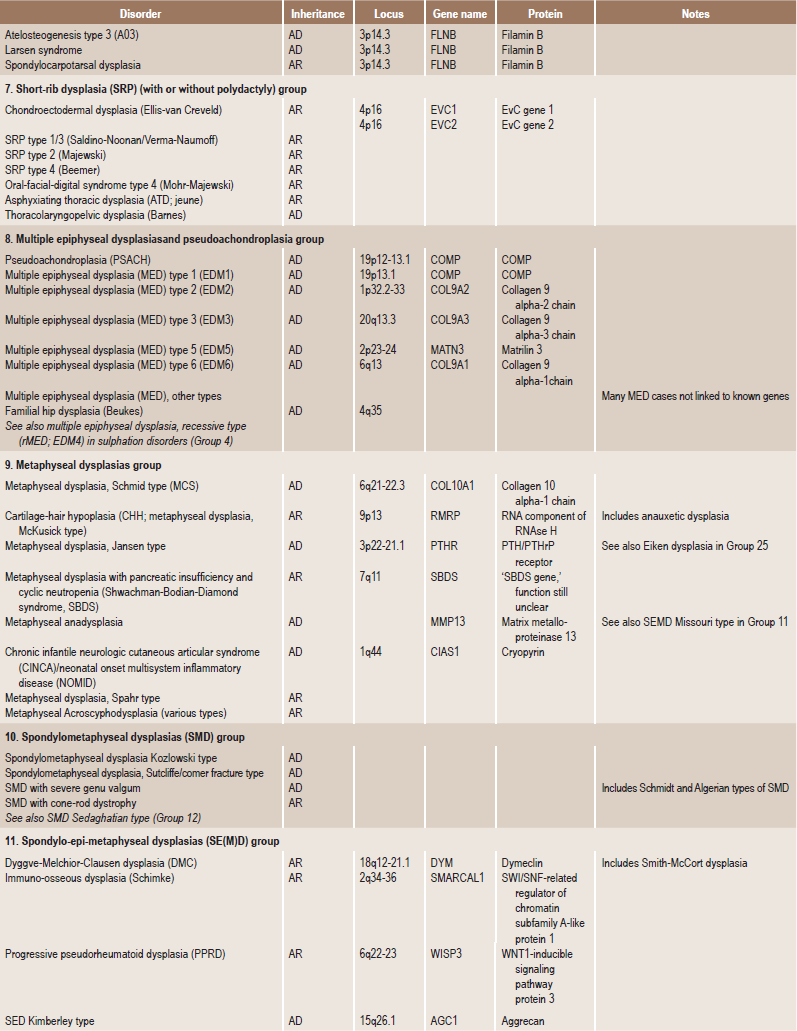
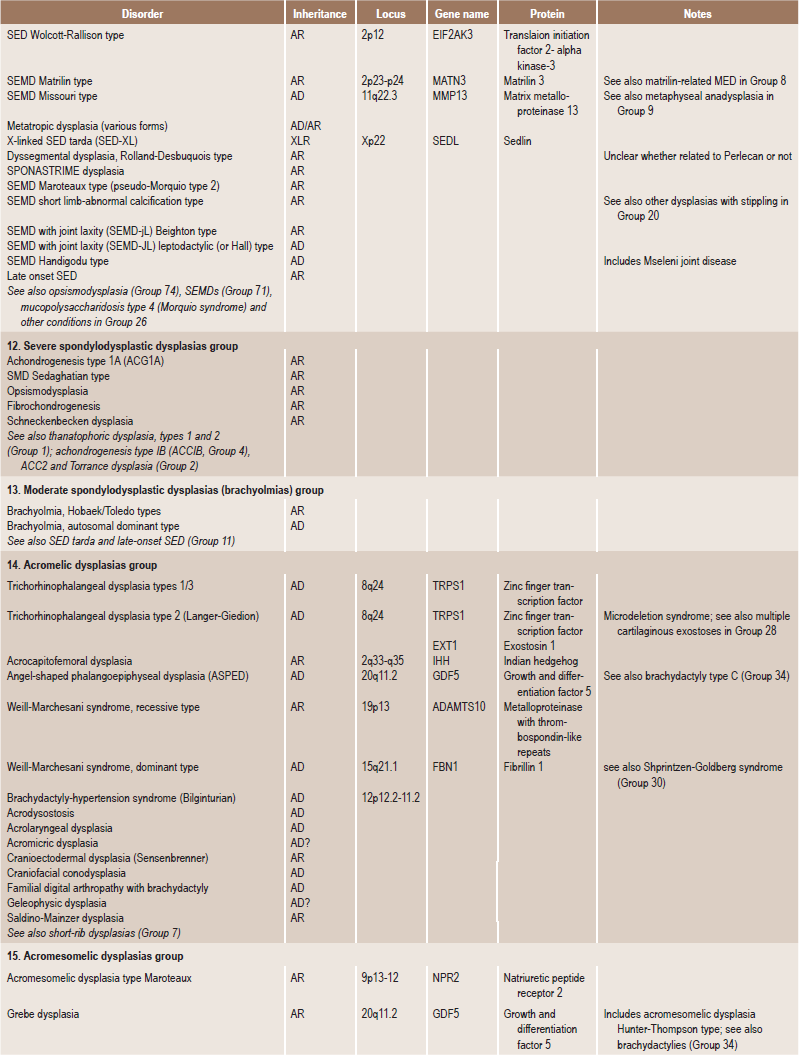
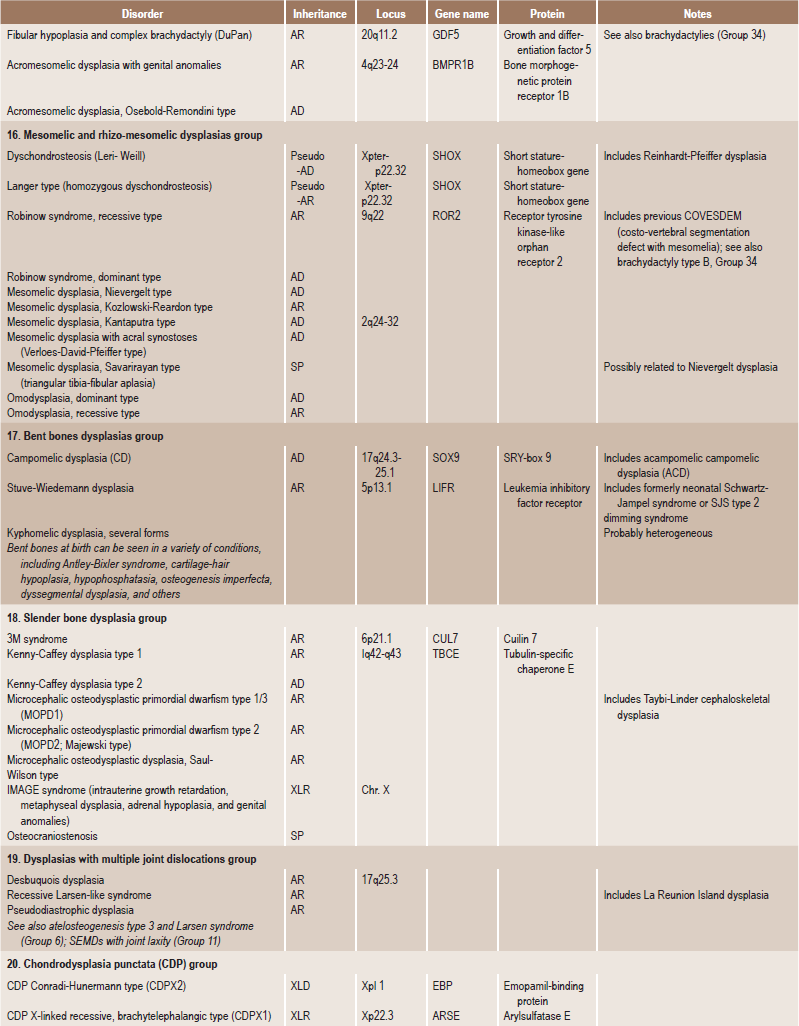
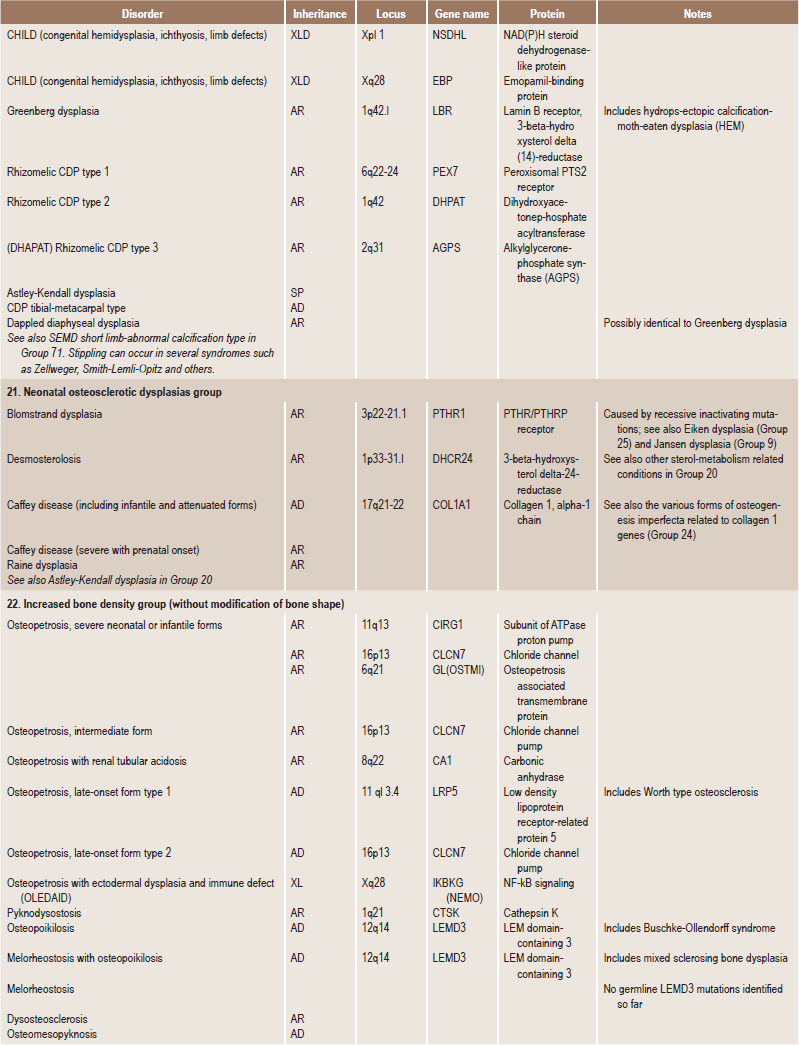
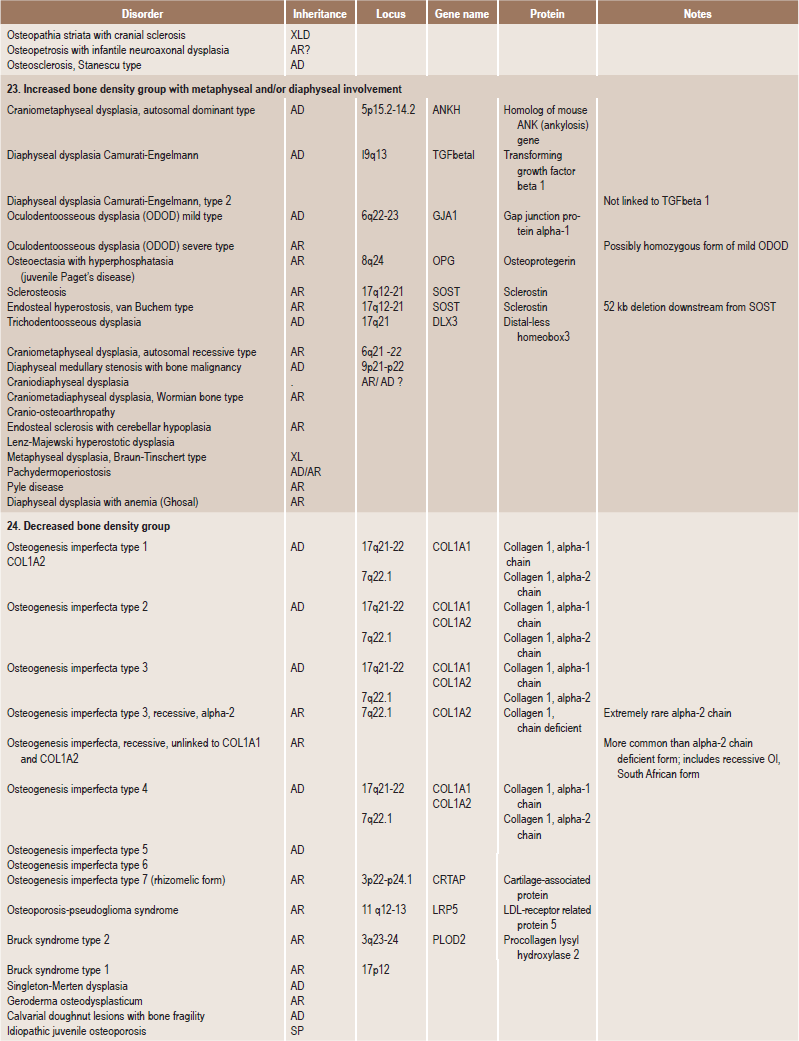
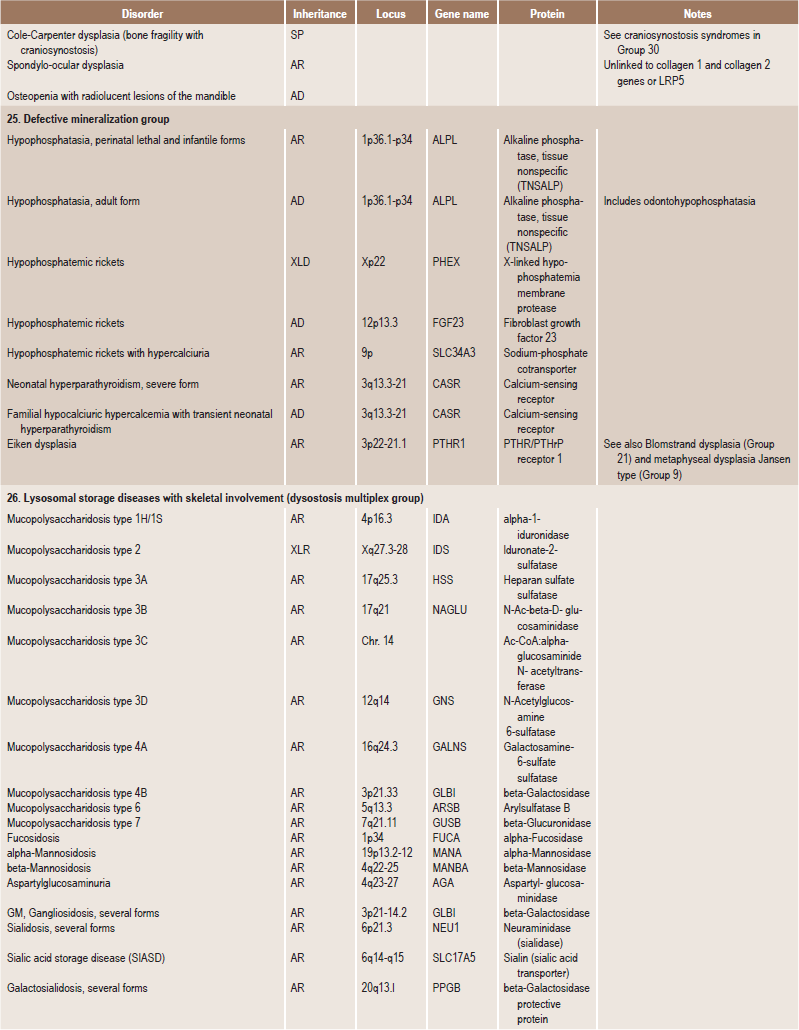
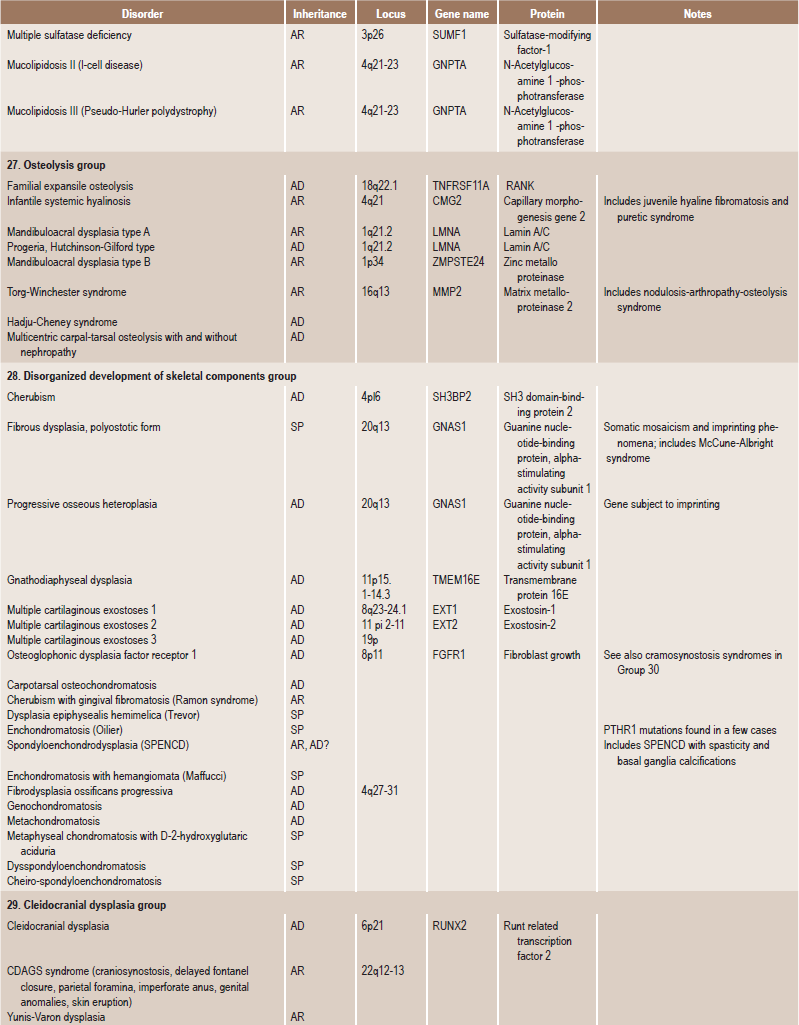
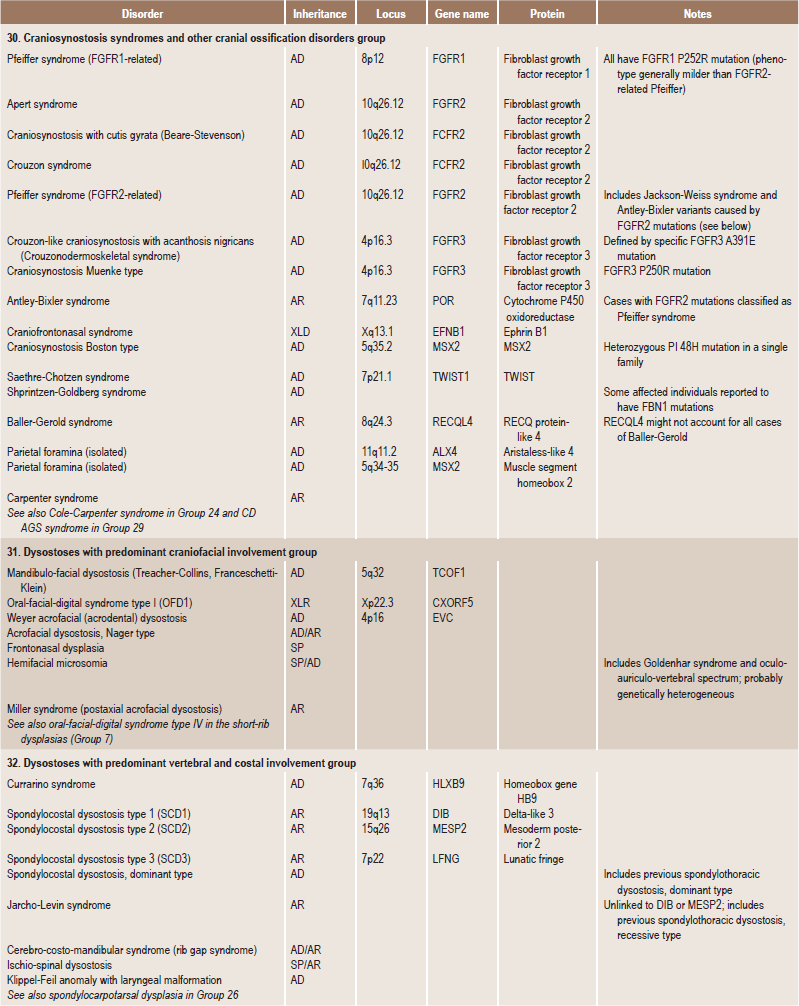
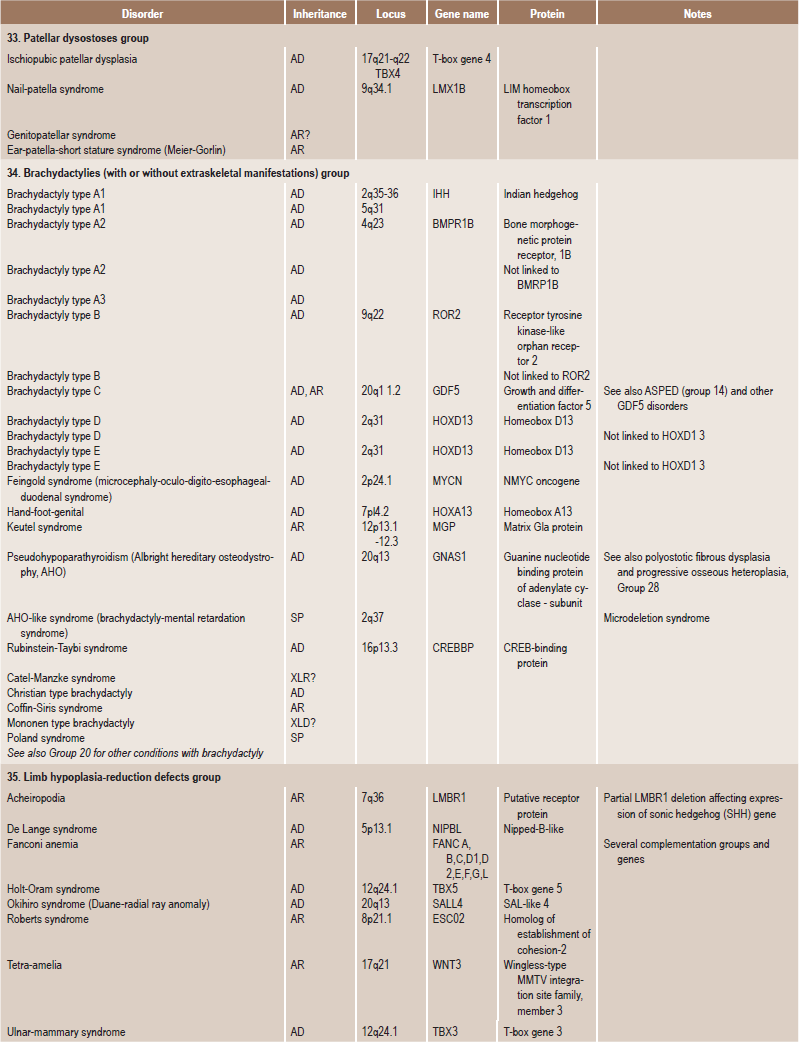
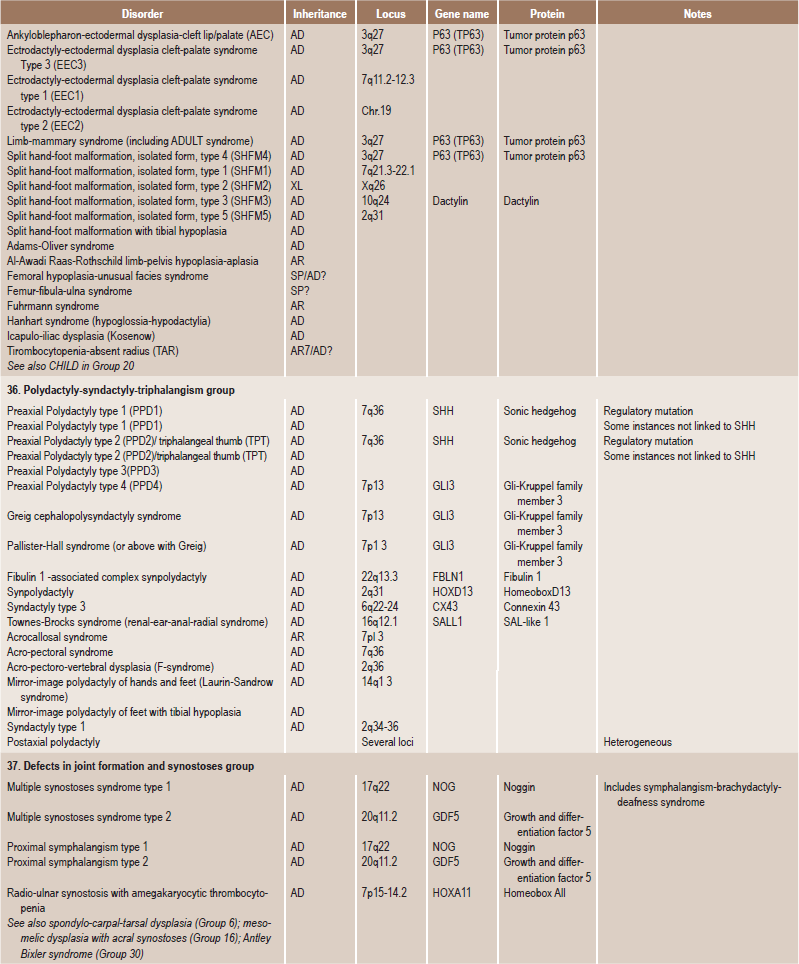
AD, Autosomal dominant; AR; autosomal recessive; SP, sporadic, XL; x linked; XID, X linked dominant; XLR, X linked recessive.
The article was published in Taybi and Lachman’s Radiology of Syndromes, Metabolic Disorders and Skeletal Dysplasias (5th edition), Ralph S Lachman: International Nosology and Classification of Genetic
Disorders of Bone-2006, pages 1322–36, Copyright Elsevier, 2007.
Table 17-2
Molecular-pathogenetic classification of genetic disorders of the skeleton
| Gene or protein | Inheritance | Clinical phenotype |
| Group 1: Defects in extracellular structural proteins | ||
| COL1A1, COL1A2 (collagen 1 α1, α2 chains) | AD | Family: Osteogenesis imperfecta |
| COL2A1 (collagen 2 α1 chain) | AD | Family: Achondrogenesis 2, hypochondrogenesis, congenital spondylepiphyseal dysplasia (SEDC), Kniest, Stickler arthro-ophthalmopathy, familial osteoarthritis, other variants |
| COL9A1, COL9A2, COL9A3 (collagen 9 α1, α2, α3 chains) | AD | Multiple epiphyseal dysplasia (MED; two or more variants) |
| COL 10A1 (collagen 10 α1 chain) | AD | Metaphyseal dysplasia Schmid |
| COL 11A1, COL 11A2 (collagen 11 α1, α2 chains) | AR, AD | Oto-spondylo-megaepiphyseal dysplasia (OSMED): Stickler (variant), Marshall syndrome |
| COMP (cartilage oligometic matrix protein) | AD | Pseudoachondroplasia, multiple epiphyseal dysplasia (MED, one |
| MATN3 (matrilin-3) | AD | Multiple epiphyseal dysplasia (MED; one variant) |
| Perlecan | AR | Schwartz-Jampel type 1; dyssegmental dysplasia |
| Group 2: Defects in metabolic pathways (including enzymes, ion channels, and transporters) | ||
| TNSALP (tissue nonspecific alkaline phosphatase) | AR, AD | Hypophosphatasia (several forms) |
| ANKH (pyrophosphate transporter) | AD | Craniometaphyseal dysplasia |
| DTDST/SLC26A2 (diastrophic dysplasia sulfate transporter) | AR | Family: achondrogenesis 1B, atelosteogenesis 2, diastrophic dys-plasia, recessive multiple epiphyseal dysplasia (rMED) |
| PAPSS2, phosphoadenosine-phosphosulfate-synthase 2 | AR | Spondylo-epi-metaphyseal dysplasia Pakistani type |
| TCIRGI, osteoblast proton pump subunit | AR | Severe infantile osteopetrosis |
| CIC-7 (chloride channel 7) | AR | Severe osteopetrosis |
| Carboanhydrase II | AR | Osteopetrosis with intracranial calcifications and renal tubular acidosis |
| Vitamin K-epoxide reductase complex | AR | Chondrodysplasia punctata with vitamin K-dependent coagulation defects |
| MGP (matrix Gla protein) | AR | Keutel syndrome (pulmonary stenosis, brachytelephalangism, cartilage calcifications and short stature) |
| ARSE (arylsulfatase E) | XLR | X-linked chondrodysplasia punctata (CDPXI) |
| 3-β-hydroxysteroid dehydrogenase | XLD | CHILD syndrome |
| 3-β-hydroxysteroid D(8)D(7)-isomerase | XLD | X-linked chondrodysplasia punctata, Conradi-Hunermann type (CDPX2); Child syndrome |
| PEX7 (peroxisomal receptor/importer) | AR | Rhizomelic chondrodysplasia punctata 1 |
| DHAPAT (Dihydroxyacetonphosphate-acyltransferase, peroxisomal enzyme) | AR | Rhizomelic chondrodysplasia punctata 2 |
| Alkyl-dihydroxydiacetonphosphate synthase (AGPS; peroxisomal enzyme) | AR | Rhizomelic chondrodysplasia punctata 3 |
| Group 3: Defects in folding and degradation of macromolecules | ||
| Sedlin (endoplasmic reticulum protein with unknown function) | XR | X-linked spondyloepiphyseal dysplasia (SED-XL) |
| Cathepsin K (lysosomal proteinase) | AR | Pyknodysostosis |
| Lysosomal acid hydrolase and transporters (sulfatase, glycosidase, translocase, etc.) | AR, XLR | Lysosomal storage disease: mucopolysaccharidoses, oligosacchari-doses, glycoproteinoses (several forms) |
| Targeting system of lysosomal enzymes (GlcNAc-1-phosphotransferase) | AR | Mucolipidosis II (I-cell disease), mucolipidosis III |
| MMP2 (matrix metalloproteinase 2) | AR | Torg type osteolysis (nodulosis arthropathy and osteolysis syndrome |
| Group 4: Defects in hormones and signal transduction mechanisms | ||
| 25-α-hydroxycholecalciferol-1-hydroxylase | AR | Vitamin D-dependent rickets type 1 (VDDR1) |
| 1, 25-α-dihydroxy-vitamin D3 receptor | AR | Vitamin D-resistant rickets with end-organ unresponsiveness to vitamin D3 (VDDR 2) |
| CASR (calcium ‘sensor’/receptor) | AD | Neonatal severe hyperparathyroidism with bone disease (if affected fetus in unaffected mother); familial hypocalciuric hypercalcemia |
| PTH/PTHrP receptor | AD (activating mutations) | Metaphyseal dysplasia Jansen |
| AR (inactivating mutation) | Lethal dysplasia Blomstrand | |
| GNAS1 (stimulatory Gs alpha protein of adenylate cyclase) | AD | Pseudohypoparathyroidism (Albright hereditary osteodystrophy and osteodystrophy and several variants) with constitutional haploin-sufficiency mutations; McCune-Albright syndrome with somatic mosaicism for activating mutations |
| PEX proteinase | XL | Hypophosphatemic rickets, X-linked semidominant type (impaired cleav age of FGF23) |
| FGF23, fibroblasts growth factor 23 | AD | Hypophosphatemic rickets, autosomal dominant type (resistance to PEX cleavage) |
| FGFR 1 (fibroblast growth factor receptor 1) | AD | Craniosynostosis syndromes (Pfeiffer, other variants) |
| FGFR 2 | AD | Craniosynostosis syndromes (Apert, Crouzon, Pfeiffer; several variants) |
| FGFR 3 | AD | Thanatophoric dysplasia, achondroplasia, hypochondroplasia, SADDAN: craniosynostosis syndromes (Crouzon with acanthosis nigricans, Muenke nonsyndromic craniosynostosis) |
| ROR-2 (‘orphan receptor tyrosine kinase’) | AR | Robinow syndrome |
| TNFRSF11A (receptor activator of under factor kB; RANK) | AD | Familial expansile osteolysis |
| TGFβ1 | AD | Diaphyseal dysplasia (Camurati-Engelmann) |
| CDMP1 (cartilage-derived morphogenetic protein 1) | AR | Acromesomelic dysplasia Grebe/Hunter-Thompson |
| AD | Brachydactyly type C | |
| Noggin (‘growth factor,’ TGF antegonist) | AD | Multiple synostosis syndrome; synphalangism and hypoacusis syndrome |
| DLL3 (delta-like 3, intercellular signaling) | AR | Spondylocostal dysostosis (one form) |
| IHH (Indian hedgehog signal molecule) | AD | Brachydactyly A1 |
| C7orf2 (orphan receptor) | AR | Acheiropodia |
| SOST (sclerosin; cystine knot secreted protein) | AR | Sclerosteosis, van Buchem disease |
| LRPS (LDL receptor-related protein 5) | AR | Osteoporosis-pseudoglioma syndrome |
| WISP 3 (growth regulator/growth factor) | AR | Progressive pseudorheumatoid dysplasia |
| Group 5: Defects in nuclear proteins and transcription factors | ||
| SOX9 (HMG-type DNA binding protein/ transcription factor) | AD | Compomelic dysplasia |
| GII3 (zinc finger gene) | AD | Greig cephalopolysyndactyly, polydactyly type A and others, Pallister-Hall syndrome |
| TRPS 1 (zine-finger gene) | AD | Tricho-rhino-phalangeal syndrome (types 1–3) |
| HVC (leucine-zipper gene) | AR | Chondroectodermal dysplasia (Ellis-van Creveld) |
| TWIST (helix-loop-helix transcription factor) | AD | Craniosynostosis Saethre-Chotzen |
| P63 (p53 related transcription factor) | AD | EEC syndrome, Hay-Wells syndrome, Limb-mammary syndrome, split hand-split foot malformation (some forms) |
| CBFA-1 (core binding factor A1; runt-type transcription factor) | AD | Cleidocranial dysplasia |
| LXM1B (LIM homeodomain protein) | AD | Nail-patella syndrome |
| DLX3 (distal-less 3 homeobox gene) | AD | Trichodentoosseous syndrome |
| HOXD 13 (homeobox gene) | Ad | Synpolydactyly |
| MSX2 (homeobox gene) | AD (gain of function) | Craniosynostosis, Boston type |
| AD (loss of function) | Parietal foramina | |
| ALX4 (homeobox gene) | AD | Parietal foramina (cranium bifidum) |
| SHOX (short stature-homeobox gene) | Pseudo-autosomal | Leri-Weill dyschondrosteosis, idiopathic short stature |
| TBX3 (T-box 3, transcription factor) | AD | Ulnar-mammary syndrome |
| TBX5 (T-box 5, transcription factor) | AD | Holt-Oram syndrome |
| EIF2AK3 (transcription initiation factor kinase) | AR | Wolcott-Rallison syndrome (neonatal diabetes mellitus and spondy-loepiphyseal dysplasia) |
| NEMO (NFkB essential modulator; kinase activity) | LX | Osteopetrosis, lymphedema, ectodermal dysplasia and immunode-ficiency (OLEDAID) |
| Group 6: Defects in oncogenes and tumor suppressor genes | ||
| EXT1, EXT2 (exostosin-1, exostosin-2; heparan-sulfate polymerases) | AD | Multiple exostoses syndrome type 1, type 2 |
| SH3BP2 (a-Abl-binding protein) | AD | Cherubism |
| Group 7: Defects in RNA and DNA processing and metabolism | ||
| RNAse MRP-RNA component | AR | Cartilage-hair-hypoplasia |
| ADA (adenosine deaminase) | AR | Severe combined immunodeficiency (ACID) with (facultative) metaphyseal changes |
Adapted from Superti-Furga A, Bonafe L, Rimoin DL. Molecular pathogenetic classification of genetic disorders of the skeleton. Am J Med Genet 2001; 106: 282–93.
Definitions
Osteochondrodysplasias refer to abnormalities of cartilage or bone growth and development. This term denotes a generalized disorder of the skeletal system encompassing multiple bones at the time of presentation.
Dysostoses refer to malformations of individual bones, single or in combination, and does not refer to a generalized disorder of the skeleton. Many disorders that were previously referred to as dysostoses are now listed with the osteochondro- dysplasias, since they are due to mutations of genes associated with dysplasias, and therefore, of a more generalized nature.
The clinical evaluation should start with a complete medical history that includes previous milestones of growth. Since skeletal dysplasias may become apparent at various ages, study of growth points since birth may help to narrow the differential diagnosis. The family history should include information about other affected family members and possible consanguinity. Parents should be examined for evidence of disproportionate stature or other evidence of a skeletal dysplasia. Physical examination should focus on anthropometric measurements. The osteochondrodysplasias are generalized disorders of the skeleton, which usually result in disproportionate short stature. A disproportionate body habitus may not be readily appreciated unless anthropometric measurements (i.e. arm span, upper to lower segment ratio, etc.) are carefully obtained. This assessment may help to determine if the disproportionate shortening affects primarily the trunk or the limbs [the proximal (rhizomelic), middle (mesomelic) or distal segment (acromelic)].
The next step in the evaluation of disproportionate short stature is to obtain a full set of skeletal radiographs including views of the skull, spine, pelvis, extremities, hands and feet. Attention should be paid to the specific parts of the skeleton that are involved, the location of the lesion within each bone (epiphysis, metaphysis, diaphysis) and the recognition of unique patterns of abnormal skeletal ossification. Review of radiographs taken at different ages or before and after puberty may be helpful, because the radiographic features of many of these disorders may change with age.
Group I: Defects in Extracellular Structural Proteins (Based on Molecular Pathogenetic Classification of Genetic Disorders of Skeleton)
Osteogenesis Imperfecta (‘Brittle bones’, fragilitas ossium, osteopsathyrosis, Lobstein’s disease)
Osteogenesis imperfecta (OI) is a serious disease, the molecular pathogenesis of which is being elucidated and it bears a superficial relatedness to dentinogenesis imperfecta (refer Chapter 1, section on dentinogenesis imperfecta), a milder condition affecting mesodermal tissues. It is a condition resulting from abnormality in the type I collagen, which most commonly manifests as fragility of bones. Although osteogenesis imperfecta is generally recognized as representing a hereditary autosomal dominant characteristic, autosomal recessive and nonhereditary types also occur.
Four types of osteogenesis imperfecta exist (Table 17-3), based on the classifications of Sillence et al (1979).
Table 17-3
Clinical types of osteogenesis imperfecta
Osteogenesis imperfecta, type I
Osteogenesis imperfecta tarda
Osteogenesis imperfecta with blue sclerae
Gene map locus 17q21.31–q22, 7q22.1
Osteogenesis imperfecta congenita; type II
Osteogenesis imperfecta congenita, neonatal lethal
Vrolik type of osteogenesis imperfecta
Gene map locus 17q21.31–q22, 7q22.1
Osteogenesis imperfecta, progressively deforming, with normal sclerae: type III
Gene map locus 17q21.31–q22, 7q22.1
Osteogenesis imperfecta, type IV
Osteogenesis imperfecta with normal sclerae
Gene map locus 17q21.31–q22
Researchers have defined three more types of osteogenesis imperfecta (type V, type VI, and type VII), but the genetic causes have not yet been identified.
Type I collagen fibers are found in bones, organ capsules, fascia, cornea, sclera, tendons, meninges, and dermis. Structurally, this protein is composed of a left-handed helix formed by intertwining of pro-a1 and pro-a2 chains. Mutations in the loci coding for these chains (COL1A1 on band 17q21 and COL1A2 on band 7q22.1, respectively) cause osteogenesis imperfecta. Qualitative defects (abnormal collagen I molecule) and quantitative defects (decrease in production of normal collagen I molecules) both exist in its causation.
Osteogenesis imperfecta is an inherited disorder. Type I is autosomal dominant, type II is autosomal dominant with new mutation, type III is autosomal dominant with new mutation (rarely recessive forms also are observed), and type IV which is autosomal dominant.
Clinical Features
The chief clinical characteristic of osteogenesis imperfecta is the extreme fragility and porosity of the bones, with an attendant proneness to fracture. The fractures heal readily, but the new bone is of a similar imperfect quality.
The age of onset of symptoms varies depending on the type of OI with fractures in type I and IV occurring during infancy and type II in utero. In type III, half the cases present fracture in utero, and other half in the neonatal period. No known differences based on gender exist. Prenatal screening by ultrasound during the second trimester shows bowing of long bones, fractures, limb shortening, and decreased skull echogenicity.
A second characteristic clinical feature of osteogenesis imperfecta is the occurrence of pale blue sclerae. The sclerae are abnormally thin, and for this reason the pigmented choroid shows through and produces the bluish color. However, the appearance of blue sclera is not confined to this disease since it may also be seen in osteopetrosis, fetal rickets, Turner syndrome, Paget’s disease, Marfan syndrome, and EhlersDanlos syndrome, as well as in normal infants. While the blue sclerae are a prominent sign in this disease, they are not invariably present. In a series of 42 patients reported by Bauze and his associates, 12 of the patients had white sclerae, and these were generally found in the older patients with the more severe disease and earlier onset of fractures.
In a thorough review of osteogenesis imperfecta and dentinogenesis imperfecta by Winter and Maiocco, the following additional signs and symptoms were described as being characteristic of osteogenesis imperfecta: deafness due to otosclerosis, abnormalities of the teeth (identical to those of dentinogenesis imperfecta, or ‘hereditary opalescent dentin’), laxity of the ligaments, a peculiar shape of the skull and an abnormal electrical reaction of the muscles.
Many patients with osteogenesis imperfecta also have a tendency towards capillary bleeding although no specific blood dyscrasia or defect has been demonstrated.
Physical features can vary depending on the type. It forms the basis for Sillence classification.
Type I: Osteogenesis imperfecta
This is the most common and mildest form. In subtype A, dentinogenesis imperfecta is absent, while in subtype B, dentinogenesis imperfecta is present. Symptoms of both subtypes include blue sclera, in utero fractures in 10% of patients (fractures are more common during infancy), mild-to-moderate bone fragility with frequency of fractures decreasing after puberty, kyphoscoliosis, hearing loss, easy bruising and short stature.
Type II: Osteogenesis imperfecta
Osteogenesis imperfecta type II exhibits extreme bone fragility and frequent fractures. In utero fractures are present in 100% of cases. Many are stillborn, and 90% die before four weeks of age. Blue sclera may be present. Hearing loss is not common to type II OI. Dentinogenesis imperfecta may be present along with small nose, micrognathia and short trunk.
Type III: Osteogenesis imperfecta
Type III is associated with dentinogenesis imperfecta, sclera of variable hue, limb shortening and progressive deformities, triangular facies with frontal bossing and pulmonary hypertension. In utero fractures occur in 50% of cases. The remaining half of the cases have fractures in the neonatal period. No hearing loss has been reported in this type.
Type IV: Osteogenesis imperfecta
In subtype A, dentinogenesis imperfecta is absent, while in subtype B, dentinogenesis imperfecta is present. Symptoms of both subtypes include normal sclera, normal hearing, fractures that begin in infancy (in utero fractures are rare) and mild angulation and shortening of long bones. Bleeding diathesis have not been reported in this type.
Oral Manifestations
Osteogenesis imperfecta is basically a disturbance of mesodermal tissues, particularly the calcified tissues. When a widespread congenital disturbance in bone formation exists, it is only logical to expect a concomitant disturbance in dentin formation. The large head size, frontal and temporal bossing, and exaggerated occiput create a greater percentage of class III malocclusions. Anterior and posterior cross bites and open bites are also frequent. These conditions seem to be caused by maxillary hypoplasia rather than mandibular hyperplasia. A surprising large number of impactions and ectopic teeth have been reported. In permanent dentition, OI patients often have unerupted first and second molars, a condition which is rare in the general population. These abnormalities have no relation to the existence of dentinogenesis imperfecta. Dentinogenesis imperfecta represents the disturbance in tooth formation associated with OI, and is one of the most significant clinical patterns of OI. It can be the only abnormality noted at times amongst the spectra of clinical manifestations. Therefore, clinical and radiological evaluations of the dentition may be the only affirmative component in the diagnosis of a questionable case of OI.
Radiographic Features
The radiographic hallmarks of osteogenesis imperfecta include osteopenia, bowing, angulation or deformity of the long bones, multiple fractures, and wormian bones (sutural bone) in the skull.
Histologic Findings
The bones in patients with osteogenesis imperfecta exhibit thin cortices, sometimes being composed of immature spongy bone, while the trabeculae of the cancellous bone are delicate and often show microfractures (Fig. 17-1). Osteoblastic activity appears retarded and imperfect, and for this reason the thickness of the long bones is deficient. The basic defect appears to lie in the organic matrix with failure of fetal collagen to be transformed into mature collagen. Qualitative defects (abnormal collagen I molecule) and quantitative defects (decrease in production of normal collagen I molecules) both exist. There is some evidence that the progressive intermolecular cross-linkage of adjacent collagen molecules, which is an essential characteristic of normal collagen maturation, is defective in this disease. Calcification proceeds normally. Defective microvascular system and decreased collagen fibril diameter have also been observed. The length of the long bones is usually normal unless multiple fractures have caused undue shortening.
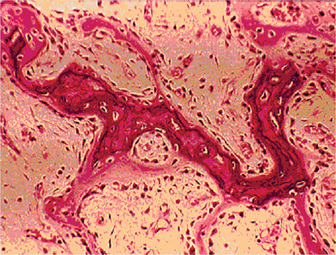
Figure 17-1 Osteogenesis imperfecta (OI).
The typical microscopic changes of OI can be seen in a section of a long bone of a severely affected child. The bone cortex is thin and porous. The bone trabeculae are thin, delicate, and widely separated. Many osteoblasts and osteocytes are present, but the formation and organization of osteoid is deficient. There is less bone tissue than normal and most of it is woven or nonlamellar bone with collagen fibers of small size and random distribution. The woven bone has an increase in basophilic ground substance (shown by blue staining in H and E sections). (Courtesy of Dr Robert C Mellors)
Treatment and Prognosis
There is no known treatment for osteogenesis imperfecta. No medical therapy is involved, other than the treatment of infections when they occur. The prognosis varies from relatively good to very poor. In type IA, life expectancy is similar to that of general population; type II, most patients die within the first year of life. A slight decrease in life expectancy has been observed in the other types.
Marfan Syndrome (Marfan-Achard syndrome, arachnodactyly)
Marfan syndrome is a spectrum of disorders caused by a heritable genetic defect of connective tissue that has an autosomal dominant mode of transmission, one of the more famous instances being that of president Abraham Lincoln. The defect itself has been isolated to FBN1 gene on chromosome 15, bands q15–q23, which codes for the connective tissue protein, fibrillin. Abnormalities in this protein cause a myriad of distinct clinical problems, of which the musculoskeletal, cardiac, and ocular problems predominate.
Several investigators studied various molecules that are found in the extracellular matrix over many years in attempts to elucidate the cause of Marfan syndrome. These included collagen, elastin, hyaluronic acid, and more recently, fibrillin. Several point mutations have now been identified in the fibrillin gene, most of which affect cysteine residues within the microfibril. These mutations are thus thought to cause defective fibrillin to be produced. Fibrillin’s structure and function are altered by abnormal protein folding due to the alteration of bonding between cysteine residues, which in turn causes defective microfibril production (conformational protein change).
Clinical Features
The estimated incidence of Marfan syndrome ranges from 1 in 5,000 to 1 in 10,000 births which includes stillbirths. The wide variation in the sites of mutations noticed in the fibrillin gene causes the varied phenotypic manifestations of this syndrome. Several other diseases present similar to Marfan syndrome, making it exceedingly difficult to determine the exact incidence. The skeleton typically displays multiple deformities including arachnodactyly, dolichostenomelia (i.e. long limbs relative to trunk length), and thoracolumbar scoliosis. The shape of the skull and face is characteristically long and narrow, and commonly suggests the diagnosis of the disease (Figs. 17-2, 17-3). Other features of the disease include hyperextensibility of joints with habitual dislocations, kyphosis and flat feet. In the cardiovascular system, aortic dilation, aortic regurgitation, and aneurysms are the most worrisome clinical findings. Mitral valve prolapse requiring valve replacement can occur as well. Ocular findings include myopia, cataracts, retinal detachment, and superior dislocation of the lens.

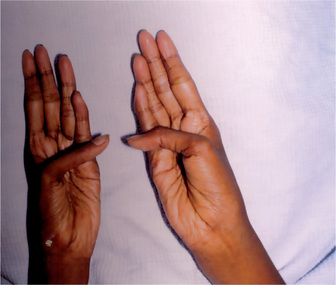
Oral Manifestations
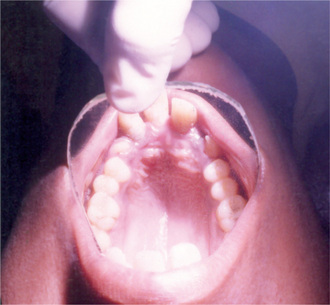
According to Baden and Spirgi, who have reviewed the oral manifestations of this disease, a high, arched palatal vault is very prevalent and may be a constant finding. Bifid uvula is also reported as well as malocclusion. In addition, multiple odontogenic cysts of the maxilla and mandible have occasionally been reported, most recently by Oatis and his coworkers. One additional finding sometimes present, is temporomandibular dysarthrosis (Fig. 17-4).
Radiographic Features
Skull radiographs (AP and lateral) may demonstrate a high arched palate, increased skull height, and an enlarged frontal sinus.
Treatment and Prognosis
There is no specific treatment for this condition. Recent strides in the management of the cardiovascular manifestations of Marfan syndrome have led to a significant decrease in morbidity and mortality. Patient longevity now approaches that of persons without Marfan syndrome, although cardiovascular compromise is still the most common cause of patient death.
Achondrogenesis
Marco Fraccaro first described achondrogenesis in 1952. By the 1970s, researchers concluded that achondrogenesis was a heterogeneous group of chondrodysplasias lethal to neonates; achondrogenesis type I (Fraccaro-Houston-Harris type) and type II (Langer-Saldino type) were distinguished on the basis of radiological and histological criteria. In 1983, a new radiological classification of achondrogenesis (types I-IV) by Whitley and Gorlin was adopted in the McKusick catalog.
Etiology
Type IA is an autosomal recessive disorder with an unknown chromosomal locus. Type IB is an autosomal recessive disorder resulting from mutations of the DDST (diastrophic dysplasia sulfate transporter) gene, which is located at 5q32– q33. Type II is an autosomal dominant type collagenopathy resulting from mutations in the COL2A1 (collagen 2 αl chain) gene, which is located at 12q13.1–q13.3. Different mutations in the gene encoding type II collagen (COL2A1) cause achondrogenesis type II as well as other type II collagenopathies (e.g. spondyloepiphyseal dysplasias, hypochondrogenesis).
Clinical Features
Achondrogenesis type I results in still-birth more frequently than type II. Males and females are affected equally. Achondrogenesis is detected prenatally or at birth because of typical clinical, radiographic, histological, and molecular findings.
In achondrogenesis type I, the craniofacial features include a disproportionately large head, soft skull, sloping forehead, convex facial plane, flat nasal bridge, occasionally associated with a deep horizontal groove, small nose, often with anteverted nostrils, long philtrum, retrognathia, increased distance between lower lip and lower edge of chin and double chin appearance (often). In achondrogenesis type II, the features seen are a disproportionately large head, large and prominent forehead, flat facial plane, flat nasal bridge, small nose with severely anteverted nostrils, normal philtrum (often), micrognathia (Fig. 17-5). The differential diagnoses include achondroplasia, hypophosphatasia, osteogenesis imperfecta and thanatophoric dysplasia.
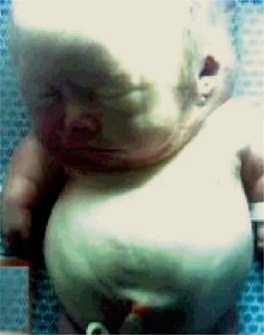
Figure 17-5 An infant with achondrogenesis type II.
Note the disproportionately large head, large and prominent forehead, flat facial plane, flat nasal bridge, small nose with severely anteverted nostrils, micrognathia, extremely short neck, short and flared thorax, protuberant abdomen, and extremely short upper extremities.
Radiographic Features
The radiographic features may vary, and no single feature is consistently noticed. Distinction between type IA and type IB on radiographs is not always possible. Degree of ossification is age dependent, and caution is needed when comparing radiographs at different gestational ages.
Histologic Findings
Achondrogenesis type IA, has a normal cartilage matrix. No collagen rings are present around the chondrocytes. Vacuolated chondrocytes, intrachondrocytic inclusion bodies (periodic acid-Schiff stain [PAS] positive, diastase resistant), extraskeletal cartilage involvement, enlarged lacunas, and woven bone are all present.
Achondrogenesis type IB, has a cartilage matrix that shows coarsened collagen fibers that are particularly dense around the chondrocytes, forming collagen rings.
Achondrogenesis type II, has slightly larger than normal and grossly distorted (lobulated and mushroomed) epiphyseal cartilage. There is severe disturbance in endochondral ossification and hypercellular reserve cartilage with large, primitive mesenchymal (ballooned) chondrocytes with abundant clear cytoplasm. The cartilaginous matrix is markedly deficient.
Hypophosphatasia
Initially recognized by Rathbun in 1948, hypophosphatasia is a rare inherited metabolic disease of decreased tissue nonspecific alkaline phosphatase and defective bone mineralization. Varying widely in its clinical presentation, it has been subdivided into five categories known as perinatal, infantile, childhood, adult, and odontohypophosphatasia. The different clinical forms have different modes of presentation, history, and inheritance.
Etiology
Patients with hypophosphatasia have defects in mineralization of bone due to TNSALP (tissue nonspecific alkaline phosphatase) deficiency. A mutation in the gene coding for tissue nonspecific alkaline phosphatase is believed to be the cause of hypophosphatasia. The gene, designated ALPL, is located at band 1p36.1–34.
Clinical Features
Hypophosphatasia affects all age groups; however, the severity of the disease differs with age. Males and females are affected equally. Hypophosphatasia occurs in all races. The perinatal form is considered lethal, while the infantile form has a mortality rate of 50%. Individuals with the other forms can reach adulthood, although often with increased morbidity. Patients with the childhood form often have rachitic deformities, and those with the adult type have increased morbidity from poorly healing stress fractures. All patients are affected by premature loss of dentition.
The perinatal form has the most severe manifestations. It is usually diagnosed at birth, and the infant rarely survives for more than a few hours. Death is due to respiratory failure. Marked hypocalcification of the skeletal structures is observed.
Patients with the infantile form may appear normal at birth; however, the clinical signs of hypophosphatasia appear during the first six months. This form also has respiratory complications due to rachitic deformities of the chest. Despite the presence of an open fontanelle, premature craniosynostosis is a common finding that may result in increased intracranial pressure. Hypercalcemia is also present and increased excretion of calcium may lead to renal damage.
Skeletal deformities, such as dolichocephalic skull and enlarged joints, a delay in walking, short stature, and a waddling gait accompany the childhood form. A history of fractures and bone pain usually exists as well. Premature loss of dentition is common with the incisor teeth often being the first affected.
The adult form presents during middle age. The first complaint may be foot pain, which is due to stress fractures of the metatarsals. Thigh pain, due to pseudofractures of the femur, may also be a presenting symptom. Upon obtaining an in-depth history, many of these patients will reveal that they had premature loss of deciduous teeth.
The only physical finding in the odontohypophosphatasic form is the premature loss of teeth.
Oral Manifestations
The earliest manifestation of the disease may be loosening and premature loss of deciduous teeth, chiefly the incisors. There are varying reports of gingivitis; however it does not seem to be a consistent feature of the disease. The differential diagnoses include achondrogenesis, osteogenesis imperfecta, rickets and thanatophoric dysplasia.
Radiographic Features
The childhood form is characterized by rachitic deformities. Upon radiologic examination of the metaphysis, evidence of radiolucent projections from the epiphyseal plate into the metaphysis is present. This is not found in other types of rickets. Radiographic findings are normal for patients with odontohypophosphatasia. Dental radiographs generally reveal hypocalcification of teeth and the presence of large pulp chambers, as well as alveolar bone loss; however these findings have not been consistently reported.
Histologic Findings
Histologic examination of the skeleton will reveal rachitic abnormalities of the growth plates such as failure of cartilage calcification. Both osteoclasts and osteoblasts appear morphologically normal, but the latter lack membrane associated alkaline phosphatase (ALP) activity on histochemical testing. This disrupts incorporation of calcium into the matrix. The long bones characteristically exhibit an increased width of proliferating cartilage with widening of the hypertrophic cell zone, irregularity of cell columns, irregular penetration of the cartilage by marrow with persistence of numerous cartilage islands in the marrow, and formation of large amounts of osteoid which is inadequately calcified. These findings are indistinguishable from those in true rickets. Histological examination of the teeth reveals a decrease in cementum, which varies with the severity of the disease. This is presumably as a result of failure of cementogenesis, so that there is no sound functional attachment of the tooth to bone by periodontal ligament. This lack of attachment is thought to account for the early spontaneous exfoliation of the deciduous teeth. The pulp chamber also appears to be enlarged. The incisors tend to be the most affected. Bone biopsy findings are normal for patients with odontohypophosphatasia.
Treatment and Prognosis
Currently, no medical therapy is available. Various treatments have been attempted including zinc, magnesium, cortisone, plasma, and enzyme replacement therapy. The results have been inconsistent. Orthopedic surgical involvement may be necessary in patients with hypophosphatasia. The perinatal form is considered lethal. The infantile form is thought to be fatal in approximately 50% of patients. Longevity studies have not been conducted for the infantile and childhood forms. Individuals with the adult and odontohypophosphatasia forms are believed to have normal lifespans.
Osteopetrosis (Marble bone disease, Albers-Schönberg disease, osteosclerosis fragilis generalisata)
Osteopetrosis is a rare hereditary bone disease of heterogeneous pathophysiology in which failure of osteoclastic bone resorption leads to increased bone mass. However, the bone has poor mechanical properties. A German radiologist, AlbersSchönberg, first described osteopetrosis in 1904.
Etiology
The primary underlying defect in all types of osteopetrosis is failure of the osteoclasts to resorb bone. This results in thickened sclerotic bones, which have poor mechanical properties. Increased bone fragility results from a failure of the collagen fibers to augment bone matrix and from defective remodeling of woven bone to compact bone. Heterogeneous molecular or genetic defects can result in impaired osteoclastic function. The exact molecular defects or sites of these mutations largely are unknown.
Clinical Features
Three distinct forms of the disease are based on age and clinical features. These are adult onset, infantile, and intermediate. Other rare forms have been described (e.g. lethal, transient, postinfectious). The infantile and intermediate types have an autosomal recessive mode of transmission, while the adult onset type shows autosomal dominant inheritance. If untreated, infantile osteopetrosis usually results in death by the first decade of life due to severe anemia, bleeding, or infection. Adult patients with osteopetrosis are usually asymptomatic and have good longterm survival rates.
Infantile osteopetrosis (also called malignant osteopetrosis) is diagnosed early in life. Failure to survive and growth retardation are symptoms. Bony defects occur. Nasal stuffiness due to mastoid and paranasal sinus malformation is often the presenting feature of infantile osteopetrosis. Cranial nerve entrapment neuropathies occur due to failure of the foramina in the skull to widen completely. Manifestations include deafness, proptosis, and hydrocephalus. Dentition might be delayed. Osteomyelitis of the mandible is common due to a deficient blood supply. Bones are fragile and can fracture easily. Defective osseous tissue tends to replace bone marrow, which can cause bone marrow failure with resultant pancytopenia. Patients might have anemia, easy bruising and bleeding (due to thrombocytopenia), and recurrent infections (due to inherent defects in the immune system). Extramedullary hematopoiesis might occur with resultant hepatosplenomegaly, hypersplenism, and hemolysis. Other manifestations include sleep apnea and blindness due to retinal degeneration.
Adult osteopetrosis (also called benign osteopetrosis) is diagnosed in late adolescence or adulthood. Approximately one half of the patients are asymptomatic, and the diagnosis is made incidentally (often in late adolescence because radiological abnormalities start appearing only in childhood) or is based on family history. Other patients might present with osteomyelitis or fractures. Many patients have bone pain. Bony defects are common and include cranial nerve entrapment neuropathies (e.g. with deafness, with facial palsy), carpal tunnel syndrome, and osteoarthritis. Bones are fragile and might fracture easily. Approximately 40% of patients have recurrent fractures. Osteomyelitis of the mandible occurs in 10% of patients. Bone marrow function is not compromised. Other manifestations include visual impairment due to retinal degeneration and psychomotor retardation. Physical findings are related to bony defects and include short stature, frontal bossing, a large head, nystagmus, hepatosplenomegaly, and genu valgum in infantile osteopetrosis. The differential diagnoses include hypoparathyroidism, myeloproliferative disease, Paget’s disease, pseudohypoparathyroidism and lead toxicity.
Oral Manifestations
The jaws are involved in the same manner as the other bones in the body, and the oral manifestations have been reviewed by Kaslick and Brustein. However, a clear distinction has usually not been made as to the type of the disease present, benign or malignant. The medullary spaces of the jaws are remarkably reduced in both dominant and recessive osteopetrosis so that there is a marked predilection for the development of osteomyelitis should infection gain entrance to the bone. This is a complication of dental extraction which has been reported frequently and discussed by Dyson. Similar findings were noted by Bjorvatn and his associates in four children with the malignant form of the disease. They stressed the necessity of administering large doses of antibiotics to control the recurring infection, which even then did not prevent the progressive osseous destruction. Fracture of the jaw during tooth extraction, even when the extraction is performed without undue force, may also occur because of the fragility of the bone. It has been reported that the teeth are of defective quality, enamel hypoplasia, microscopic dentinal defects and arrested root development all having been described. However, this may not be true in the benign dominant form of the disease. It is also reported that the teeth are especially prone to dental caries. Since dental findings have been recorded in so few cases, this observation is difficult to evaluate. An additional rather constant finding is retardation of tooth eruption due to the sclerosis of bone.
Radiographic Features
Radiographic features are usually diagnostic. Because the disease is a heterogeneous group of disorders, the findings vary depending on the subtype. Patients usually have generalized osteosclerosis. Bones may be uniformly sclerotic, but alternating sclerotic and lucent bands may be noted near the ends of long bones (Fig. 17-6). The bones might appear club like or show an appearance of a bone within bone (endobone). The entire skull is thickened and dense, especially at the base. Sinuses are small and underpneumatized. Vertebrae are extremely radiodense. They may show alternating bands, known as the ‘rugger-jersey’ sign. Radiographs may show evidence of fractures or osteomyelitis. When the jaws are affected, the density of the bone may be such that the roots of the teeth are nearly invisible on the dental radiograph.
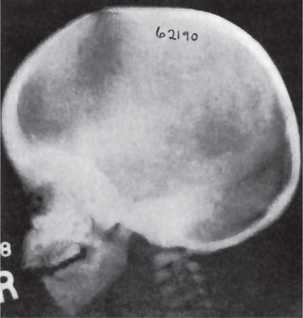
Laboratory Findings
The patients manifest a myelophthisic anemia due to the displacement of hematopoietic marrow tissue by bone. Hypocalcemia can occur and cause rickets if it is severe enough. Parathyroid hormone (PTH) is often elevated (secondary hyperparathyroidism). Acid phosphatase and creatinine kinase (CK-BB) levels are increased due to increased release from defective osteoclasts.
Histologic Features
Bone biopsy is not essential for diagnosis because radiographs are usually diagnostic. Osteopetrosis is characterized by the endosteal production of bone with an apparent concomitant lack of physiologic bone resorption (Fig. 17-7). Osteoblasts are prominent, but osteoclasts are seldom found in significant numbers in tissue sections. The predominance of bone formation over resorption typically leads to the persistence of cartilaginous cores of bony trabeculae long after their replacement should have occurred in endochondral bones. The trabeculae themselves are disorderly in arrangement, and the marrow tissue present is usually fibrous.
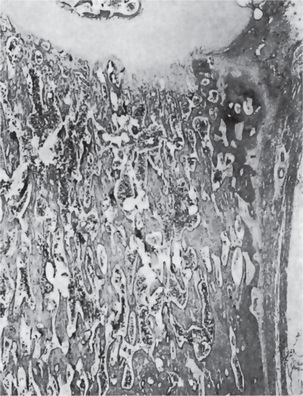
Figure 17-7 Osteopetrosis.
A photomicrograph of a long bone showing replacement of the marrow by endosteal bone. (Courtesy of Dr Frank Vellios)
It has been reported by Johnston and his associates; however, that adult patients with benign osteopetrosis do not appear to have a deficiency in osteoclastic activity but rather an abnormality in the type and structure of bone. They found osteoblastic and osteoclastic activity with prominent remodeling of bone. However, by polarized light, the bone was found to be markedly deficient in collagen matrix fibrils and these seldom crossed from one osteon to another. This deficiency of fibrils could account for the tendency for fracture in these patients.
Treatment and Prognosis
Infantile osteopetrosis warrants treatment due to the adverse outcome associated with the disease. Calcitriol appears to help by stimulating dormant osteoclasts, and thus, stimulating bone resorption. Erythropoietin can be used to correct anemia. Corticosteroids have been used with the hope of stimulating bone resorption and treating the anemia. Treatment with gamma interferon has been shown to produce long-term benefits. Adult osteopetrosis requires no treatment by itself, though complications of the disease might require intervention. No specific medical treatment exists for the adult type. If untreated, infantile osteopetrosis usually results in death by the first decade of life due to severe anemia, bleeding, or infections. Patients fail to survive, have growth retardation, and increased morbidity. Prognosis can change remarkably in some patients after bone marrow transplantation. Patients with adult osteopetrosis have good long-term survival rates.
Chondrodysplasia Punctata
Chondrodysplasia punctata is a rare congenital syndrome caused by a peroxisomal dysfunction and was first described in 1914. It is one of the four syndromes of the peroxisome biogenesis disorders resulting from anomalous enzymatic function of the metabolism of the fatty acids. It has been defined as erratic cartilage calcification during growth which produces the heterogeneous group of disorders that result in small ossification centers in the epiphyseal cartilage of the long bones and spine, skin lesions, cataracts, craniofacial dysmorphism, joint contractures, and cardiac malformation. In surviving children, abnormal growth leads to dysmorphism, kyphoscoliosis, limb shortness, and luxation of the hip.
Autosomal Dominant Type (Nonrhizomelic, nonlethal type, dysplasia epiphysealis congenita, stippled epiphyses, chondrodysplasia punctata dominant type, chondrodysplasia epiphysealis punctata, chondrodystrophia calcificans congenita, Conradi-Hunermann syndrome)
Autosomal dominant type is the most common of all chondrodysplasia punctata; most are new mutations. An autosomal dominant inheritance is observed with a male: female ratio of 3:1.
Major Diagnostic Criteria
Asymmetric head, frontal bossing; flat nasal bridge; dysplastic auricles; mongoloid palpebral fissures; hypertelorism; high arched palate.
Asymmetric mild shortening of all long bones; bowing; stippled epiphysis; vertebral scoliosis, clefting; or wedging; flexion contracture of the joints; clubfoot or valgus deformity.
Associated findings such as mild mental retardation and postaxial polydactyly has been rarely described. Complex congenital cardiac disease and central nervous system anomalies have also been reported.
Radiographic Features
Mild shortening of all long bones with multiple epiphyseal punctate calcific deposits in the infantile cartilaginous skeleton, which may or may not be seen by ultrasound after 14 weeks. Vertebral body deformities and scoliosis can also be seen. Stippling of the proximal humerus may also help to identify the condition.
Pyknodysostosis
The disorder was first described and named by Maroteaux and Lamy in 1962. Andren et al, simultaneously and independently delineated this syndrome. The features are deformity of the skull (including wide sutures), maxilla and phalanges (acroosteolysis), osteosclerosis, and fragility of bone. Pyknodysostosis is inherited as an autosomal recessive trait. The locus for the dysplasia has been mapped to chromosome 1q21. Mutations in this region lead to cathepsin K deficiency. Cathepsin K is a cysteine protease that is highly expressed in osteoclasts. The estimated prevalence of pyknodysostosis is 1 per million.
Clinical Features
This dysplasia is characterized by a short-limbed stature. There is hypoplasia or absence of the lateral portion of the clavicles, and hypoplasia of the terminal phalanges of the digits (termed acro-osteolysis), leading to short, stubby hands with large finger nails. The skull has widened sutures and persistent open fontanels, even into adulthood. The mandible is small, and the angle of the mandible is obtuse, leading to a very small chin. The nose is protuberant. The teeth are delayed in appearance and disordered when present.
Radiographic Features
Radiographs show generalized osteosclerosis. The medullary canal is always present, but it is small and irregular. The sclerotic bone has a propensity to fracture, with fractures generally occurring in the lower extremities. Bone formation and resorption are simultaneously diminished. MRI studies have shown the cortex to be of normal thickness, whereas the space within the medullary canal was limited as a result of the increase in trabecular bone. Bone scan reveals increased uptake.
Histologic Features
Microscopic examination of bone biopsy specimens are similar to those in osteopetrosis. Meredith and associates (1978) proposed that normal osteoblasts and osteoclasts fail to respond as they should to the demands of stress on the bone. Although osteoclast are present, they do not appear to function properly in resorbing bone. At fracture sites, all cellular elements of fracture repair are present.
The differential diagnosis includes osteopetrosis. Unlike osteopetrosis, pyknodysostosis does not lead to aplastic anemia, because the medullary canal is partially preserved. Cleidocranial dysostosis may be considered because of the hypoplasia of the clavicles; however, osteosclerosis is not seen in cleidocranial dysostosis.
Treatment and Prognosis
Orthopedic treatment consists of fracture care. Life expectancy is normal. Chronic osteomyelitis of the jaw occurs frequently and is resistant to standard forms of treatment.
The types of MPS linked to specific enzyme deficiencies are listed below; some have been assigned an enzyme commission (EC) number.
• MPS type I-H (Hurler syndrome): Alpha-L-iduronidase deficiency (EC 3.2.1.76)
• MPS type I-S (Scheie syndrome, formerly MPS type V): Alpha-L-iduronidase deficiency
• MPS type I-H/S (Hurler-Scheie syndrome): AlphaL-iduronidase deficiency
• MPS type II, mild (Hunter syndrome, mild form): L-sulfoiduronate sulfatase deficiency
• MPS type II, severe (Hunter syndrome, severe form): L-sulfoiduronate sulfatase deficiency (EC 3.1.6.13)
• MPS type III-A (Sanfilippo syndrome type A): Heparan sulfate sulfamidase deficiency (EC 3.1.6.14)
• MPS type III-B (Sanfilippo syndrome type B): N-acetyl-alpha-D-glucosaminidase deficiency (EC 3.2.1.50)
• MPS type III-C (Sanfilippo syndrome type C): AcetylCoA: alpha-glucosamide N-acetyltransferase deficiency (EC 2.3.1.3)
• MPS type III-D (Sanfilippo syndrome type D): N-acetyl-alpha-D-glucosamine-6-sulfatase deficiency (EC 3.1.6.14)
• MPS type IV-A (Morquio syndrome, classic form): N-acetylgalactosamine-6-sulfatase (gal-6-sulfatase) deficiency (EC 3.1.6.4)
• MPS type IV-B (Morquiolike syndrome): Betagalactosidase deficiency (EC 3.2.1.23)
• MPS type VI (Maroteaux-Lamy syndrome, mild form): N-acetylgalactosamine-4-sulfatase (arylsulfatase B) deficiency
• MPS type VI (Maroteaux-Lamy syndrome, severe form): N-acetylgalactosamine-4-sulfatase (arylsulfatase B) deficiency (EC 3.1.6.1)
• MPS type VII (Sly syndrome): Beta-glucuronidase deficiency (EC 3.2.1.31)
Mucopolysaccharidoses Types I–VII (Lysosomal storage disease)
Mucopolysaccharidoses (MPS) are a group of lysosomal storage diseases, each of which is produced by an inherited deficiency of an enzyme involved in the degradation of acid mucopolysaccharides (now called glycosaminoglycans [GAG]). These diseases are autosomal recessive, except for MPS type II, which is X-linked.
Etiology
Glycosaminoglycans (GAG) are long, linear polysaccharide molecules composed of repeating dimers, each of which contains a hexuronic acid (or galactose in the case of keratan sulfate) and an amino sugar. The large proteoglycan molecules made up of protein cores and GAG branches are secreted by cells and constitute a significant fraction of the extracellular matrix of the connective tissue. The turnover of these molecules depends on their subsequent internalization by endocytosis, their delivery to the lysosomes, and their digestion by lysosomal enzymes. The enzyme deficiencies lead to the accumulation of mucopolysaccharides in the lysosomes of the cells in the connective tissue and to an increase in their excretion in the urine.
The enzyme synthesis is controlled at the following gene loci:
• 4p16.3 (Hurler syndrome, Scheie syndrome)
• 6q24.3 (Morquio syndrome): The deficiency of enzymes in Morquio syndrome type A or type B leads to the accumulation of keratan sulfate and chondroitin-6-sulfate in the connective tissue, the skeletal system, and the teeth
Clinical Features
Onset usually occurs in early childhood. Skeletal findings include dwarfism, with rather characteristic radiologic changes of the hands and the lumbar vertebral column; stiff articulations; and coarse facies. Patients with Hurler syndrome usually die by the time they are aged 5–10 years. The life expectancy of patients with Scheie syndrome may be nearly normal. They can live until the fifth or sixth decade of life, and they can have healthy offspring. As for patients with Hunter and Sanfilippo syndrome, death usually occurs by the time of puberty. In the classic form of Morquio syndrome, long-term survival is rare, with death occurring in persons aged 20–40 years. In patients with the severe form of Maroteaux-Lamy syndrome, death usually occurs by early adulthood.
Differential diagnoses include Gaucher disease, NiemannPick disease, syphilis, osteogenesis imperfecta, vitamin D-resistant rickets, nephrogenic osteopathy, spondyloepiphysial dysplasia, metaphysial dysplasia.
Rickets
Rickets is an entity that commonly affects children leading to decreased mineralization at the level of the growth plates with resultant growth retardation and delayed skeletal development. Osteomalacia is found in adults which affects trabecular bone, and results in undermineralization of osteoid. By definition, rickets is found only in children prior to the closure of the growth plates, while osteomalacia occurs in persons of any age. The term rickets is said to have been derived from the ancient English word ‘wricken’, which means to bend. In several European countries, rickets is also termed English disease, which appears to stem from the turn of the 19th century in England when rickets was endemic in larger cities.
Etiology
Rickets results either from a deficiency or abnormal metabolism of vitamin D or from abnormal metabolism or excretion of inorganic phosphate. Histologic changes are seen at the level of the growth plates, or more specifically, at the level of the hypertrophic zone, where an increased number of disorganized cells is found. The increased number of cells results in increased width and thickness of the hypertrophic zone (rachitic metaphysis).
In most developing countries, rickets is seldom seen, supposedly due to high exposure to sunlight. An exception occurs in groups of women who are rarely allowed to leave the house (largely for religious reasons) or who must wear veils when they do. Since these women may have low vitamin D levels, their babies are at a higher risk of developing rickets. When patients receive adequate treatment, no mortality is associated with this disease; however concomitant diseases, such as pneumonia, tuberculosis, and enteritis, occur with a higher frequency and may cause death. Boys and girls are affected equally with rickets. There is a form of genetic rickets, called X-linked hypophosphatemic rickets, in which some children, often girls, may be only moderately affected, although girls with X-linked hypophosphatemic rickets can have rickets symptoms that are just as severe as those in boys. By definition, rickets occurs only in children whose growth plates have not closed. The growth plates close at the end of puberty, at approximately age of 17 years in females and age of 19 years in males. Premature neonates are especially at risk because their requirements for vitamin D, calcium, and phosphate are higher than the requirements in full-term neonates (for details, refer to Chapter 15 on Oral Aspects of Metabolic Disease).
Hyperparathyroidism
The parathyroid glands regulate serum calcium and phosphorus levels by its secretion and maintenance within physiological limits of its hormone, parathyroid hormone (PTH). Under normal conditions, the rate of secretion of parathyroid hormone is inversely proportional to the serum calcium level. Secretion of PTH is mainly controlled through the interaction of calcium with specific calcium-sensing receptors on the membrane of parathyroid cells. Hyperparathyroidism is a syndrome of hypercalcemia resulting from excessive release of parathyroid hormone. Most cases of hyperparathyroidism are discovered accidentally when hypercalcemia is noted during a routine serum chemistry examination. In most patients, symptoms are mild at the time of presentation and resolve with surgical correction of the disorder.
Etiology
In 85% of affected persons, primary hyperparathyroidism results from an adenoma in a single parathyroid gland. Hypertrophy of the parathyroid glands causes hyperparathyroidism in 15% of patients. Parathyroid malignancies account for a small number of hyperparathyroidism cases. Hyperparathyroidism is common in patients with type I and type II multiple endocrine neoplasia (MEN) and in patients who received radiation therapy to the head and neck during childhood for benign diseases. Also, a syndrome of familial hyperparathyroidism has been observed. Secondary hyperparathyroidism occurs when the parathyroid glands become hyperplastic after long-term stimulation to release PTH in response to chronically low serum calcium. Chronic renal failure, rickets, and malabsorption syndromes are the most frequent causes. In secondary hyperparathyroidism, high levels of PTH do not cause hypercalcemia because the primary problem makes calcium unavailable. With long-term hyperstimulation, the glands eventually function autonomously and continue to produce high levels of parathyroid hormone even after the chronic hypocalcemia has been corrected. Hypercalcemia caused by autonomous parathyroid function after long-term hyperstimulation is referred to as tertiary hyperparathyroidism.
Clinical Features
Of the endocrine disorders, only diabetes mellitus and hyperthyroidism occur more frequently than hyperparathyroidism. Hereditary hyperparathyroidism occurs most frequently as part of a syndrome of multiple endocrine neoplasia (MEN). MEN 1 consists of hyperparathyroidism with tumors of the pituitary and pancreas. MEN 2A consists of hyperparathyroidism, pheochromocytoma, and medullary carcinoma of the thyroid. Although hyperparathyroidism can occur at any age, it is most common in the fifth and sixth decades of life. Prevalence is higher in females than in males, with a male-to-female ratio of approximately 1 : 2. At least one half of patients with hyperparathyroidism are asymptomatic. Manifestations of hyperparathyroidism may be subtle, and the disease may run a benign course for many years. Less commonly, hyperparathyroidism may worsen abruptly and cause severe hypercalcemic complications (e.g., profound dehydration, coma). This is referred to as hypercalcemic parathyroid crisis.
The skeletal and neuromuscular changes manifest as bone pain and/or tenderness, muscle fatigue, weakness and spontaneous fractures; nonspecific myalgias, osteoporosis, osteopenia, cystic bone lesions, vertebral collapse, chondrocalcinosis and pseudogout can develop. Patients have a tendency to develop pancreatitis and/or pancreatic calcification and peptic ulcer disease which can result in abdominal distress, constipation, vomiting, anorexia and weight loss. Neuropsychiatric illness and altered mental status such as anxiety, depression, psychosis and apathy have been reported. Signs of hypertension and congestive heart failure may be apparent.
Radiographic Features (Refer to Chapter 15)
Laboratory Findings
The diagnosis of hyperparathyroidism is made by demonstrating elevated parathyroid hormone levels in the setting of high serum calcium. Almost all other causes of hypercalcemia suppress the release of parathyroid hormone, which is measured by radioimmunoassay. Other findings include elevated serum chloride levels, decreased serum phosphate level (less than 2.5 mg/dl (0.81 mmol/L), decreased serum carbon dioxide, hyperchloremic metabolic acidosis, increase in urine cyclic adenosine monophosphate (cAMP).
Histologic Features (Refer to Chapter 15)
Treatment and Prognosis
The emergency management of hyperparathyroidism is focused on the treatment of the hypercalcemia. Specifically, the goal of treatment is to reduce the calcium level to below 11.5 mg/dl, less than the level in which most patients have resolution of hypercalcemia-induced symptoms.
Hypoparathyroidism
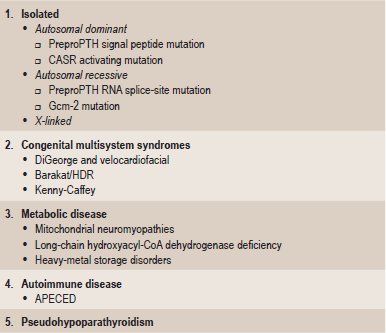
Primary hypoparathyroidism is caused by a group of heterogeneous conditions in which hypocalcemia and hyperphosphatemia occur as a result of deficient parathyroid hormone (PTH) secretion. This most commonly results from surgical excision of, or damage to, the parathyroid glands. However, genetic forms of hypoparathyroidism due to decreased secretion of PTH are known (Table 17-4).
Clinical Features
The signs and symptoms of hypoparathyroidism include evidence of latent or overt neuromuscular hyperexcitability due to hypocalcemia. The effect may be aggravated by hyperkalemia or hypomagnesemia, but there is wide variation in the severity of the symptoms. Patients may complain of circumoral numbness, paresthesia of the distal extremities or muscle cramping which can progress to carpopedal spasm or tetany. Laryngospasm or bronchospasm and seizures may also occur. Other less specific manifestations include fatigue, irritability, and personality disturbance. Patients with chronic hypocalcemia may have calcification of the basal ganglia or more widespread intracranial calcification, detected by skull X-ray or CT scan. Also seen are extrapyramidal neurological symptoms (more often with intracranial calcification), subcapsular cataracts, band keratopathy, and abnormal dentition.
In hypoparathyroidism, serum calcium concentrations are decreased and serum phosphate levels are increased. Serum PTH is low or undetectable (the important exception is PTH resistance—pseudohypoparathyroidism—discussed below). Usually, serum 1,25(OH)2D is low, but alkaline phosphatase activity is normal. Despite an increase in fractional excretion of calcium, intestinal calcium absorption and bone resorption are both suppressed. The renal filtered load of calcium is decreased, and the 24-hour urinary calcium excretion is reduced; nephrogenous cyclic AMP excretion is low and renal tubular reabsorption of phosphate is elevated. After parenteral administration of biologically active PTH, plasma and urinary cyclic AMP and inorganic phosphate excretion increase — a test (the EllsworthHoward test) that differentiates hypoparathyroidism from pseudohypoparathyroidism. Hypoparathyroidism is a feature common to various kinds of inherited disorders. Although familial occurrences are reported, sporadic cases are common. Autoimmune hypoparathyroidism can occur as an isolated endocrine condition or with other glandular deficiencies in a pluriglandular autoimmune syndrome, and it can occur as a congenital hypoplasia/aplasia with or without other congenital anomalies such as lymphedema, nephropathy, nerve deafness or cardiac malformation. It also occurs as an isolated finding.
Autoimmune parathyroid gland ablation or destruction
Antibodies directed against parathyroid tissue have been detected in over 30% of patients with isolated hypoparathyroid disease, and over 40% of patients having hypoparathyroidism combined with other endocrine deficiencies. It remains to be seen whether the autoantibodies are of primary or secondary importance in these cases.
Pseudohypoparathyroidism
Several clinical disorders characterized by end-organ resistance to PTH have been described collectively by the term pseudohypoparathyroidism (PHP). They are associated with hypocalcemia, hyperphosphatemia, and increased circulating PTH, but target tissue unresponsiveness to the hormone manifests as a lack of increased cAMP excretion in response to PTH administration.
Treatment
The goal of treatment in hypoparathyroid state is to raise the serum calcium sufficiently to alleviate acute symptoms and prevent the complications of chronic hypocalcemia. The calcium concentration required for this purpose is generally the low-normal range. Acute or severe symptomatic hypocalcemia is best treated with intravenous calcium infusion. Initial doses of 2–5 millimoles of elemental calcium as the gluconate salt can be given over a 10–20 minute period, followed by 2 millimoles elemental calcium per hour as a maintenance dose, to be adjusted according to symptoms and biochemical response.
Fibrous Dysplasia
Fibrous dysplasia is a skeletal developmental anomaly of the bone-forming mesenchyme that manifests as a defect in osteoblastic differentiation and maturation. Virtually any bone in the body can be affected. It is a nonhereditary disorder of unknown cause.
Etiology
The exact cause of fibrous dysplasia is not known. The condition is not believed to be hereditary. Fibrous dysplasia is usually caused by a mutation in the GNAS1 gene (20q13.2). The GNAS1 (guanine nucleotide-binding protein, α-stimulating activity polypeptide) gene encodes a G-protein that stimulates the production of cAMP. The mutation results in a continuous activation of the G-protein leading to overproduction of cAMP in affected tissues. This results in a hyperfunction of affected endocrine organs, frequently giving rise to precocious puberty, hyperthyroidism, growth hormone and cortisol overproduction. Secondly, there is an increased proliferation of melanocytes resulting in large café-au-lait spots with irregular margins as opposed to the regular outlined café-au-lait spots in neurofibromatosis. Thirdly, cAMP is thought to have an effect on the differentation of osteoblasts leading to fibrous dysplasia. In fibrous dysplasia, the medullary bone is replaced by fibrous tissue, which appears radiolucent on radiographs, with the classically described ground-glass appearance. Trabeculae of woven bone contain fluid-filled cysts that are embedded largely in collagenous fibrous matrix, contributes to the generalized hazy appearance of the bone.
Clinical Features
The following three disease patterns are recognized:
The initial manifestations of fibrous dysplasia are most commonly found in persons aged 3–15 years. Two-thirds of patients with polyostotic disease are asymptomatic before they are aged 10 years. With monostotic disease, patients as old as 20 or 30 years are asymptomatic. No specific racial predilection exists. The incidence is equal in males and females. Clinical findings of increasing pain and an enlarging soft tissue mass suggest malignant change.
Approximately 70–80% of fibrous dysplasias are monostotic. This form most frequently occurs in the rib (28%), femur (23%), tibia, craniofacial bones (10–25%), and humerus, in decreasing order of frequency. This form may present with pain or a pathologic fracture in patients aged 10–70 years. The degree of bone deformity is relatively less severe compared with that of the polyostotic type. No clearly documented evidence supports the conversion from the monostotic form to the polyostotic form.
Approximately 20–30% of fibrous dysplasias are polyostotic. Polyostotic fibrous dysplasia more frequently involves the skull and facial bones, pelvis, spine, and shoulder girdle. The sites of involvement are the femur, tibia, pelvis, ribs, skull and facial bones, upper extremities, lumbar spine, clavicle, and cervical spine in decreasing order of frequency. The dysplasia may be unilateral or bilateral, and it may affect several bones of a single limb or both limbs with or without axial skeleton involvement. Although the polyostotic variety tends to occur in a unilateral distribution, involvement is asymmetric and generalized when disease is bilateral.
Two-thirds of patients are symptomatic before they are 10 years of age. Often, the initial symptom is pain in the involved limb associated with a limp, spontaneous fracture, or both. In one series, pathologic fracture was present in 85% of polyostotic fibrous dysplasias. Leg-length discrepancy of varying degrees occurs in about 70% of patients with limb involvement. The structural integrity of the bone is weakened, and the weight-bearing bones become bowed. The curvature of the femoral neck and proximal shaft of the femur markedly increase causing a Shepherd’s crook deformity, which is a characteristic sign of the disease. Overgrowth of adjacent soft tissues may be present. Two apparently separate types of polyostotic fibrous dysplasia are described:
• Fibrous dysplasia involving a variable number of bones, although most of the skeleton is normal, accompanied by pigmented lesions of the skin or ‘café-au-lait’ spots (Jaffe’s type).
• An even more severe fibrous dysplasia involving nearly all bones in the skeleton and accompanied by pigmented lesions of the skin, and in addition, endocrine disturbances of varying types (Albright’s syndrome).
This pattern of the disease occurs in 10–
25% of patients with the monostotic form and in 50% with the polyostotic form. It also occurs in an isolated craniofacial form. In the isolated variety, no extracranial lesions are present. Sites of involvement most commonly include the frontal, sphenoid, maxillary, and ethmoidal bones. The occipital and temporal bones are less commonly affected. Hypertelorism, cranial asymmetry, facial deformity, visual impairment, exophthalmos, and blindness may occur because of involvement of orbital and periorbital bones. Involvement of the sphenoid wing and temporal bones may result in vestibular dysfunction, tinnitus, and hearing loss. When the cribriform plate is involved, hyposmia or anosmia may result.
This is an entirely different entity having microscopic similarity more with giant cell lesions than fibrous dysplasia, and is an autosomal disorder of variable penetrance. The gene for cherubim was mapped to chromosome 4p16. The gene mutated was identified as SH3BP2 within this locus. It is believed that mutations in the gene may lead to pathologic activation of osteoclasts and disruption of jaw development. Regression may occur after adolescence. The jaw is broad and protruding. Involvement of the maxilla and the mandible is symmetric.
The only significant laboratory abnormality is an elevated alkaline phosphatase level. Differential diagnoses include enchondroma and enchondromatosis, eosinophilic granuloma, fibrous cortical defect and nonossifying fibroma, giant cell tumor, central hemangioma, hyperparathyroidism, primary neurofibromatosis type 1 and Paget’s disease.
Radiographic Features
The usual appearance of fibrous dysplasia in long and short tubular bones includes a lucent lesion in the diaphysis or metaphysis, with endosteal scalloping and with or without bone expansion and the absence of periosteal reaction. The lucent lesion has a thick sclerotic border and is called the rind sign. Among skull and facial bones the frontal bone is involved more frequently than the sphenoid, with obliteration of the sphenoid and frontal sinuses. Single or multiple, symmetric or asymmetric, radiolucent or sclerotic lesions in the skull or facial bones may be present. Most commonly, maxillary and mandibular involvement has a mixed radiolucent and radiopaque pattern, with displacement of the teeth and distortion of the nasal cavities.
Oral Manifestations
The oral manifestations of polyostotic fibrous dysplasia are related to the severe disturbance of the bony tissue. One-third of the polyostotic patients in the series of Van Horn and his associates had lesions in the mandible. The occurrence of maxillary lesions was not mentioned, although Harris and his group stated that maxillary and mandibular involvement was not rare.
There may be expansion and deformity of the jaws, and the eruption pattern of the teeth is disturbed because of the loss of normal support of the developing teeth. The endocrine disturbance also may alter the time of eruption of the teeth. A classic case with involvement of the maxilla has been reported by Church. In this instance there was no intraoral pigmentation, although it has been reported to occur.
Histologic Features
The lesions are composed of fibrillar connective tissue within which are numerous trabeculae of coarse, woven immature bone, irregular in shape but evenly spaced, showing no relation to functional patterns. The osteocytes are quite large, and collagen fibers of these trabeculae can often be seen extending out into the fibrous tissue. Bone formation by stellate osteoblasts can be observed, although rows of cuboidal osteoblasts lined up on the surfaces of trabeculae are absent (osteoblastic riming). These trabeculae typically have wide osteoid seams. Osteoclastic activity may be seen where the calcification of osteoid extends to the surface of the trabeculae.
Treatment and Prognosis
Treatment is usually conservative and primarily to prevent deformity. Any underlying endocrine disturbances should be treated. In upper extremity lesions, more than 80% respond to nonsurgical management. No specific medical treatment exists for the bone disease, although early evidence suggests that vitamin D and bisphosphonates (after epiphyseal closure) may be helpful in ameliorating pain and possibly in reconstituting lesions with normal bone. Surgical therapy with curettage and replacement of the bone defect with autograft or allograft usually results in resorption of the graft at the surgical site. Use of allograft or cortical autograft usually delays this conversion, as it is more resistant to resorption and replacement by dysplastic bone.
Of special concern is malignant degeneration and metabolic changes in patients with fibrous dysplasia. The estimated frequency of malignant transformation is 0.4–1% in fewer than 50 reported cases. The interval from the diagnosis of fibrous dysplasia to the development of malignancy varies and is usually years or decades. Most often, skull and facial bones undergo malignant change in monostotic disease, whereas femoral and facial bones undergo malignant change in polyostotic disease. Osteosarcoma and fibrosarcoma are the most common tumors. Chondrosarcomas occur less frequently. Radiographic features suggestive of malignant degeneration include a rapid increase in the size of the lesion and a change from a previously mineralized bony lesion to a lytic lesion. Clinical findings of increasing pain and an enlarging soft tissue mass suggest malignant change.
Monostotic Fibrous Dysplasia of the Jaws
Monostotic fibrous dysplasia, though less serious than polyostotic fibrous dysplasia, is of greater concern to the dentist because of the frequency with which the jaws are affected. Nearly every bone has, at one time or another, been reported involved. In a series of 67 cases of monostotic fibrous dysplasia, Schlumberger found the following distribution:
| Ribs | 29 cases |
| Femur | 9 cases |
| Tibia | 8 cases |
| Maxilla | 7 cases |
| Calvarium | 5 cases |
| Mandible | 2 cases |
| Humerus | 2 cases |
| Ulna | 2 cases |
| Vertebra | 1 case |
| Pelvis | 1 case |
| Fibula | 1 case |
There is now evidence to indicate; however, that the incidence of jaw lesions is proportionately far greater than this study would indicate. It is now recognized that some cases of jaw lesions which in the past were diagnosed under a variety of other names are now embraced by the term ‘fibrous dysplasia’. As an example, certain cases of so-called central giant cell tumors of the jaws have been found upon reevaluation to be classifiable as fibrous dysplasia. This has been emphasized particularly by Jaffe, Lichtenstein and Portis and by Waldron. In past years the designation ‘ossifying fibroma’ (q.v.) was a common one for a certain group of jaw lesions which occurred with considerable frequency. Many authorities now view at least some of these lesions as a type of monostotic fibrous dysplasia. Another lesion of bone, the nonosteogenic fibroma, also is considered by some investigators to be a form of fibrous dysplasia. The clinical term ‘leontiasis ossea’ has often been applied to cases of fibrous dysplasia which affect the maxilla or facial bones and give the patient a leonine appearance. Thus it can be appreciated that fibrous dysplasia of bone has come to include a number of lesions once described by other terms. Although investigators differed as to the desirability of inclusion of certain bony lesions in this group, the trend in the past few years had been to recognize monostotic fibrous dysplasia as an entity with considerable clinical and histologic variation, probably dependent upon the stage or phase of the disease.
In contrast; however, it has been suggested that this trend to classify many fibro-osseous lesions of the jaws under the term ‘fibrous dysplasia’ may be unfortunate, and many pathologists have now reverted again to the ‘purist’ idea that fibrous dysplasia does represent a specific entity with welldefined microscopic and radiographic features. This would man that there are certain fibro-osseous lesions of the jaws which would not be designated as fibrous dysplasia, and until more knowledge of the true nature of the lesions accumulates, some workers have simply classified them as ‘fibro-osseous lesions’, after first being certain that they do not represent some specific entity.
Clinical Features
Monostotic fibrous dysplasia of the jaws occurs with apparently equal predilection for males and females, although some reports show a mild predominance of females. It is more common in children and young adults than in older persons. The mean age of occurrence in the 69 patients reported by Zimmerman and his associates was 27 years, while in 53 patients with craniofacial fibrous dysplasia reported by Gardner and Halpert, the mean age was 34 years.
The first clinical sign of the disease is a painless swelling or bulging of the jaw. The swelling usually involves the labial or buccal plate, seldom the lingual aspect, and when it involves the mandible it sometimes causes a protuberant excrescence of the inferior border. There may be some malalignment, tipping or displacement of the teeth due to the progressive expansile nature of the lesion, and tenderness may ultimately develop. The mucosa is almost invariably intact over the lesion.
Fibrous dysplasia of the maxilla is an especially serious form of the disease since it has a marked predilection for occurrence in children and is almost impossible to eradicate without radical, mutilating surgery (Fig. 17-8). These lesions are not well circumscribed, commonly extend locally to involve the maxillary sinus, the zygomatic process and the floor of the orbit, and even extend back toward the base of the skull. Severe malocclusion and bulging of the canine fossa or extreme prominence of the zygomatic process, producing a marked facial deformity, are typical sequelae of this disease in this location need not be truly monostotic, but neither is it usually classified as a polyostotic type. It has sometimes been referred to as craniofacial fibrous dysplasia, since it does affect the craniofacial complex and is so characteristic in its clinical and radiographic features that it closely resembles a distinct entity (Fig. 17-9). This form of the disease has been described in detail by Waldron and Giansanti and by Eversole and his associates.
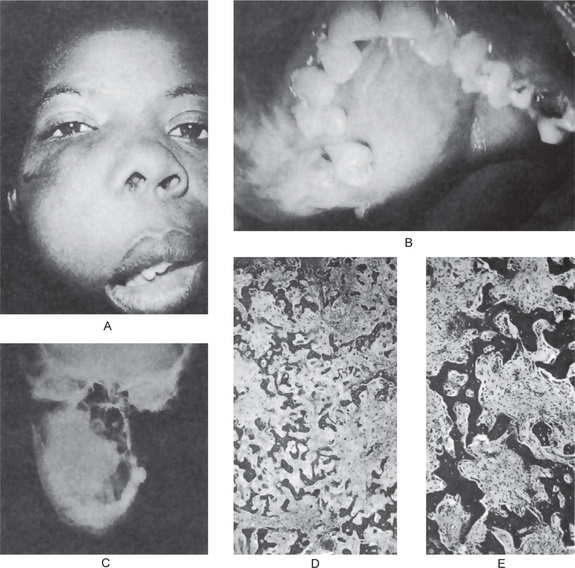
Radiographic Features
The radiographic appearance of fibrous dysplasia of the jaw is extremely variable (Fig. 17-10). There are three basic patterns which may be seen. In one type, the lesion is generally a rather small unilocular radiolucency or a somewhat larger multilocular radiolucency, both with a rather well-circumscribed border and containing a network of fine bony trabeculae. In the second type, the pattern is similar except that increased trabeculation renders the lesion more opaque and typically mottled in appearance. The third type of quite opaque with many delicate trabeculae gives a ‘ground-glass’ or ‘peau d’orange’ appearance to the lesion. This latter type characteristically is not well circumscribed but instead blends into the adjacent normal bone. Any of the three types may be found in either maxilla or mandible. In all types, generally the cortical bone becomes thinned because of the expansile nature of the growth, but seldom is this bony plate perforated, or is periosteal proliferation obvious. The roots of teeth in the involved areas may be separated or moved out of normal position but only occasionally exhibit severe resorption. In some cases, the bone appears so opaque that the roots of teeth may be indistinct or not visible.
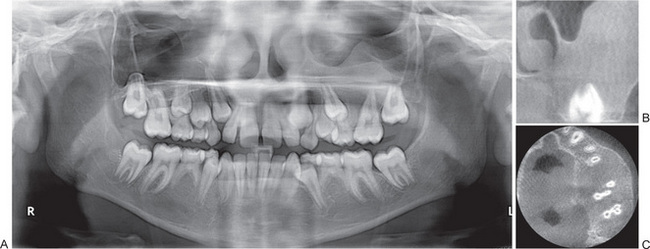
Figure 17-10 Monostotic fibrous dysplasia of bone.
Fibrous dysplasia in the left maxillary molar region. (A) Panoramic radiograph shows anterior displacement of the left maxillary second premolar and displacement of the floor of the sinus. (B) Coronal section of the same patient shows grainy alveolar bone and displacement of the floor of the sinus. (C) Axial section shows significant buccal-palatal expansion of the alveolar bone in the premolar-molar region. Area of fibrous dysplasia does not have clear demarcation.
It is of interest that, in craniofacial fibrous dysplasia, there is characteristic radiographic thickening of the base of the skull.
Histologic Features
There is considerable microscopic variation in cases of monostotic fibrous dysplasia of the jaws. The lesion is essentially a fibrous one made up of proliferating fibroblasts in a compact stroma of interlacing collagen fibers (Fig. 17-11A, B). Irregular trabeculae of bone are scattered throughout the lesion with no definite pattern of arrangement. Characteristically, some of these trabeculae are C-shaped, or as described by one author, Chinese character-shaped. These trabeculae are usually coarse woven bone but may be lamellar, although not as well organized as normal lamellar bone. The relationship of osteoblasts and osteoclasts to the trabeculae is similar to that seen in the polyostotic form of the disease. Large lesions may show variation from area to area and sometimes present a greater bony reaction around the periphery of the lesion than in the central portion.
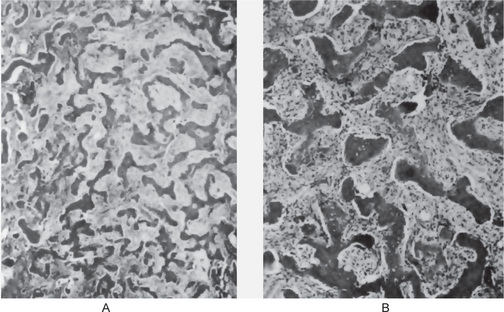
Some of the earlier literature dealing with this disease suggested that it represents a permanent maturation arrest in the woven bone stage and proposed that lesions demonstrating lamellar bone transformation should not be diagnosed as fibrous dysplasia. However, it is generally well accepted now, particularly on the basis of the work of Waldron and Giansanti, that lesions of fibrous dysplasia of the jaws, especially the craniofacial type, will mature over a period of time and the lesional tissue may show lamellar bone.
McCune-Albright Syndrome (Polyostotic fibrous dysplasia)
McCune-Albright syndrome or polyostotic fibrous dysplasia (PFD) is defined as the association of polyostotic fibrous dysplasia, precocious puberty, café-au-lait spots, and other endocrinopathies due to hyperactivity of various endocrine glands. Fuller Albright first described this syndrome in 1937. McCune-Albright syndrome has been shown to be due to a postzygotic activating mutation of the GS alpha gene in the affected tissues. The GS alpha subunit is the component of the G-protein complex, which couples hormone receptors to adenylate cyclase (the intracellular second messenger) in a submembrane site. It then mediates the cellular effects of hormone binding.
Clinical Features
Precocious puberty associated with the condition is gonadotrophin-independent. Among the endocrine disturbances described in association with Albright syndrome are:
Some severely affected patients may present with associated hepatic, cardiac, and GI dysfunction (i.e. elevated hepatic transaminases, GI polyposis, and cardiomyopathy).
Cutaneous pigmentation is the most common extraskeletal manifestation in fibrous dysplasia and occurs in more than 50% of cases of the polyostotic form. Cutaneous pigmentation in polyostotic fibrous dysplasia is ipsilateral to the side of bony lesions, a feature that differentiates this disease from pigmentation in neurofibromatosis. The pigmented macules or café-au-lait spots (Fig. 17-12) are related to increased amounts of melanin in the basal cells of the epidermis. They tend to be arranged in a linear or segmental pattern near the midline of the body, usually overlying the lower lumbar spine, sacrum, buttocks, upper back, neck, and shoulders. Similar lesions may occur on the lips and oral mucosa. Pigmentation may occur at birth, and in fact, they occasionally precede the development of skeletal and endocrine abnormalities.
The association of fibrous dysplasia and intramuscular myxoma is a rare disease known as Mazabraud’s syndrome. Both lesions tend to occur in the same anatomical region. The relationship between fibrous dysplasia and myxoma remains unclear, whereas an underlying localized error in tissue metabolism has been proposed to explain this occasional coexistence. Patients with soft tissue myxomas should be thoroughly examined for fibrous dysplasia. The greater risk of sarcomatous transformation in fibrous dysplasia with Mazabraud’s syndrome has been reported.
Only a few cases of malignant transformation of skeletal lesions have been described in the setting of McCune-Albright syndrome. Malignancies described in this include:
• OsteosarcoMcCune-Albright syndrome (most common)
• ChondrosarcoMcCune-Albright syndrome
These malignancies occur most commonly in the setting of therapeutic irradiation exposure. Females may have a greater risk for breast cancer, probably due to their prolonged exposure to elevated estrogen levels. The underlying GS alpha gene mutation also may play a role in this. For the same reasons, these patients also appear to be at an increased risk of thyroid and secondary osseous malignancies. Hypophosphatemic rickets is another potential complication that may worsen the bone disease associated with polyostotic fibrous dysplasia. While on vitamin D and phosphorus supplements, patients with McCune-Albright syndrome and hypophosphatemic rickets must be monitored closely for hypercalcemia and secondary hyperparathyroidism.
Laboratory Findings
There are no consistent significant changes in the serum calcium or phosphorus, although the serum alkaline phosphatase level is sometimes elevated. Premature secretion of pituitary follicle-stimulating hormone has been reported, as well as moderately elevated basal metabolic rate.
Histologic Findings
The bone affected by polyostotic fibrous dysplasia has areas of fibrous metaplasia within flat and tubular bones. The basic anomaly in fibrous dysplasia lesions is a progressively expanding fibrous lesion of bone-forming mesenchyme. The lesions typically expand concentrically from the medullary cavity outwards (i.e. towards the cortex). The bony lesions are well defined, although invariably not encapsulated. The lesions are rich in spindle-shaped fibroblasts, with a swirled appearance within the marrow space and erratically arranged ‘tongues’ of woven bone. Islands of cartilaginous tissue also may be interspersed within the lesions. Some parts of the affected bones may have cystic lesions lined by multinucleated giant cells akin to ostitis fibrosa cystica (of severe hyperparathyroidism) but with a paucity of osteoblasts.
Treatment and Prognosis
McCune-Albright syndrome is a multisystem condition with a host of variable presentations. Management often is challenging and requires a multidisciplinary approach. Apart from the small subgroup of patients that has increased mortality and those who develop malignancies, McCune-Albright syndrome is not associated with a significantly increased mortality risk. Deformities associated with polyostotic fibrous dysplasia result in variable degrees of morbidity, ranging from mild to very severe.
Cherubism (Familial fibrous dysplasia of jaws, disseminated juvenile fibrous dysplasia, familial multilocular cystic disease of jaws, familial fibrous swelling of jaws)
An autosomal dominant fibro-osseous lesion of the jaws involving more than one quadrant that stabilizes after the growth period, usually leaving some facial deformity and malocclusion.
Cherubism, a non-neoplastic hereditary bone lesion that is histologically similar to central giant cell granuloma, affects the jaws of children bilaterally and symmetrically, usually producing the so-called cherubic look (Fig. 17-13). The disease was first described in 1933 by Jones, who called it familial multilocular disease of the jaws. The term ‘cherubism’, was introduced by Jones and others to describe the clinical appearance of affected patients. According to the WHO classification, cherubism belongs to a group of non-neoplastic bone lesions affecting only the jaws. It is a rare, benign condition with autosomal dominant inheritance, and it is one of the very few genetically determined osteoclastic lesions in the human body. It appears to have 100% penetrance in males and only 50–70% penetrance in females. There is great variation in the clinical expression. Although the condition is known to be hereditary, in some cases there has been no detectable family history, and although it usually occurs bilaterally, there have also been cases of unilateral involvement, perhaps because of incomplete penetrance or new mutations. Some investigators believe that cherubism arises from the mutation of a nonsex-linked gene responsible for the development of the jaw bones. Typically, the jaw lesions of cherubism remit spontaneously when affected children reach puberty, but the reason for this remission is unknown. The reduction in osteoclast formation caused by sex steroids and the increase in plasma concentrations of estradiol and testosterone at puberty both suggest that the genetic defect responsible for the localized increase in osteoclasts in cherubism is overridden and normalized by the increased synthesis of sex steroids. The gene related to cherubism was located on chromosome 4p16.3.
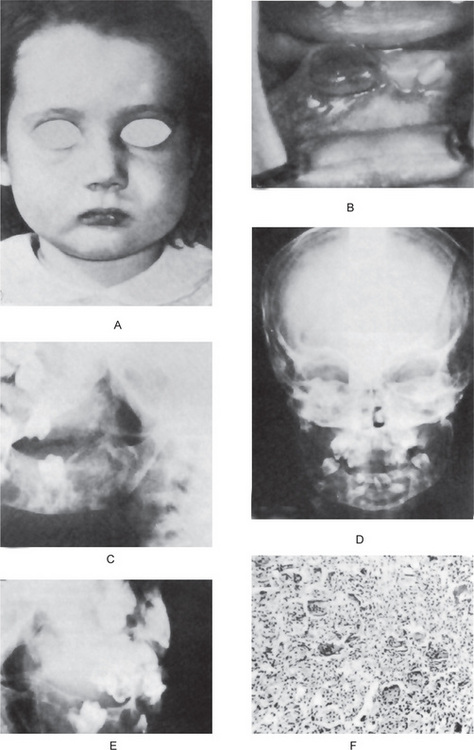
Figure 17-13 Cherubism.
The patient has a cherubic appearance owing to the expansion of the jaws (A). Occasionally the mucosa will be perforated by the underlying bony lesion (B). The bilateral involvement of the mandible is seen in the lateral jaw and skull radiographs (C, D, E) where there has been serious destruction of bone. A biopsy of the bone lesion reveals a cellular fibrous mass with many interspersed multinucleated giant cells (F). (Courtesy of Dr Ralph E McDonald: Am J Dis Child, 89: 354, 1955)
Genetics
Mutations in the SH3BP2 (SH3 domain binding protein 2) gene have been identified in about 80% of people with cherubism. In most of the remaining cases, the genetic cause of the condition is unknown.
The SH3BP2 gene provides instructions for making a protein whose exact function is still unclear. The protein plays a role in transmitting chemical signals within cells, particularly cells involved in the replacement of old bone tissue with new one (bone remodeling) and certain immune system cells.
Mutations in the SH3BP2 gene lead to the production of an overactive version of this protein which is expected to disrupt critical signaling pathways in cells associated with the maintenance of bone tissue and some immunologic effecter cells. The overactive protein likely causes inflammation in the jaw bones and triggers the production of osteoclasts, which cause breakage of bone tissue while remodeling. A combination of bone loss and inflammation likely underlies the cyst-like growths characteristic of cherubism. Atleast 11 mutations in the SH3BP2 gene have been identified in people with cherubism.
Clinical Features
Affected children are normal at birth and are without clinically or radiographically evident disease until 14 months to 3 years of age. At that time, symmetric enlargement of the jaws begins. Typically, the earlier the lesion appears, the more rapidly it progresses. The self-limited bone growth usually begins to slow down when the patient reaches five years of age, and stops by the age of 12–15 years. At puberty the lesions begin to regress. Jaw remodeling continues through the third decade of life, at the end of which the clinical abnormality may be subtle. The signs and symptoms depend on the severity of the condition and range from clinically or radiographically undetectable features to grotesquely deforming mandibular and maxillary overgrowth with respiratory obstruction and impairment of vision and hearing. The jaw lesions are usually painless and symmetric and have florid maxillary involvement. The lesions, which are firm to palpation and nontender, most commonly involve the molar to coronoid regions, the condyles always being spared, and are often associated with cervical lymphadenopathy. Enlargement of the cervical lymph nodes contributes to the patient’s full-faced appearance and is said to be caused by lymphoid hyperplasia with fibrosis. The lymph nodes become enlarged before the patient reaches 6 years of age, decrease in size after the age of 8 years and are rarely enlarged after the age of 12 years. Intraoral swelling of the alveolar ridges may occur. When the maxillary ridge is involved, the palate assumes a V shape. A rim of sclera may be visible beneath the iris, giving the classic ‘eye to heaven’ appearance.
Oral Manifestations
Numerous dental abnormalities have been reported, such as agenesis of the second and third molars of the mandible, displacement of the teeth, premature exfoliation of the primary teeth, delayed eruption of the permanent teeth, and transpositions and rotation of the teeth. In severe cases, tooth resorption occurs. Although cherubism was initially described as a familial disease affecting the jaws, cases without any apparent hereditary origin have been reported. In a few cases, cherubism has been described as being connected with other diseases and conditions such as Noonan’s syndrome, a lesion in the humerus, gingival fibromatosis, psychomotor retardation, orbital involvement and obstructive sleep apnea. The deciduous dentition may be shed prematurely, beginning as early as 3 years of age. The permanent dentition is often defective, with absence of numerous teeth and displacement and lack of eruption of those present. The oral mucosa is usually intact and of normal color.
Grading System
Arnott (1978) suggested the following grading system for the lesions of cherubism: grade I is characterized by involvement of both mandibular ascending rami, grade II by involvement of both maxillary tuberosities as well as the mandibular ascending rami, and grade III by McCuneAlbright syndrome involvement of the whole maxilla and mandible except the coronoid process and condyles.
Radiographic Features
Radiologically, cherubism is characterized by bilateral multilocular cystic expansion of the jaws. Early lesions occur in the posterior body of the mandible and the ascending rami. Maxillary lesions may occur at the same time but escape early radiographic detection because of overlap of the sinus and nasal cavities. Displacement of the inferior alveolar canal has been reported. The presence of numerous unerupted teeth and the destruction of the alveolar bone may displace the teeth, producing a radiographic appearance referred to as floating tooth syndrome (Fig. 17-14). With adulthood, the cystic areas in the jaws become re-ossified, which results in irregular patchy sclerosis. There is a classic (but nonspecific) ground glass appearance because of the small, tightly compressed trabecular pattern.
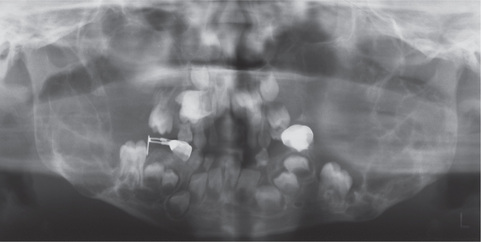
Histologic Features
Histologic examination of the lesions usually reveals numerous multinucleated giant cells. These multinucleated cells show strong positivity for tartrate-resistant acid phosphatase, which is characteristic of osteoclasts. The collagenous stroma, which contains a large number of spindleshaped fibroblasts, is considered unique because of its waterlogged, granular nature. Numerous small vessels are present, and the capillaries exhibit large endothelial cells and perivascular cuffing. The eosinophilic cuffing appears to be specific to cherubism. However, these deposits are not present in many cases, and their absence does not exclude the diagnosis of cherubism. Older, resolving lesions of cherubism show an increase in fibrous tissue, a decrease in the number of giant cells and formation of new bone. The microscopic findings seldom permit a specific diagnosis of cherubism in the absence of clinical and radiological information.
The differential diagnoses of cherubism consist of giant cell granuloma of the jaws, osteoclastoma, aneurysmal bone cyst, fibrous dysplasia and hyperparathyroidism.
Treatment
As Laskin (1985) stated, “the treatment of cherubism should be based on the known natural course of the disease and the clinical behavior of the individual case”. Therefore, surgery to correct the jaw deformities of cherubism is rarely indicated. If necessary, surgery is usually undertaken after puberty, when the remission phase of the lesions have been reached, unless esthetic considerations or severe functional problems justify earlier treatment. Although exacerbation has sometimes been reported after surgery, it is believed that surgery ultimately accelerates the involution process.
Vitamin D-resistant Rickets (Familial hypophosphatemic rickets, refractory rickets, phosphate diabetes)
Since the early 20th century, ultraviolet radiation or vitamin D ingestion has been recognized as a cure for nutritional rickets, although certain forms of rachitic diseases have remained refractory to this therapy. Study of these refractory cases has revealed low serum phosphate concentration as a common factor. Familial occurrence of this condition led to the diagnosis of familial hypophosphatemic rickets. Treatment with vitamin D produced no change in the rachitic state of these patients, even at rather high doses, leading to the term vitamin D-resistant rickets.
Etiology
Several of the most vexing questions about the underlying mechanism causing the clinical phenotype of X-linked hypophosphatemia remain unanswered. Great strides have been made in recent years, particularly with the cloning of the mutant gene known as PEX. This gene, found on the X chromosome, is thought to produce a currently unknown hormone involved in phosphate regulation. The gene for hypophosphatemic rickets has been localized. The X-linked dominant form of the disease is attributed to mutations in the PEX gene, located at Xp22.1. The gene locus for autosomal dominant hypophosphatemic rickets has been located on chromosome 12p13. The pathogenesis of this disorder is clear; phosphate wasting at the proximal tubule level is the basis of the affected individual’s inability to establish normal ossification. This phenomenon is secondary to defective regulation of the sodium-phosphate cotransporter in the epithelial cell brush border. Normal phosphate reabsorption in response to 1,25-dihydroxycholecalciferol (calcitriol) provides clear evidence that the sodium-phosphate cotransporter is capable of proper function and is not intrinsically defective.
Clinical Features
As in all genetic disorders, the disease is present from conception. Affected newborns are of normal weight, but infants may show growth retardation. Intellectual development is unaffected. Although serum phosphate levels are depressed similarly in affected males and females, the degree of bone involvement is substantially less severe in heterozygous females. All hemizygous males are clinically affected. Widened joint spaces and flaring at the knees may become apparent in children by their first birthday, particularly in boys. When a child begins to stand and walk, bowing of the weight-bearing long bones quickly becomes clinically evident. Dentition may be absent or delayed in very young children due to abnormal tooth formation; older children may experience multiple dental abscesses.
Laboratory Findings
Laboratory evaluation of rickets begins with assessment of serum calcium, phosphate, and alkaline phosphatase levels. In hypophosphatemic rickets, calcium levels may be within or slightly below reference ranges; alkaline phosphatase levels are significantly above reference ranges. Serum phosphate levels must be carefully evaluated in the first year because the concentration reference range for infants (5.0–7.5 mg/dl) is high compared to adults (2.7–4.5 mg/dl). Hypophosphatemia can be missed easily in a baby. Serum parathyroid hormone level is within reference ranges to slightly elevated, while calcitriol level is low or in the lower reference range. Most importantly, urinary loss of phosphate is above reference ranges.
Radiographic Features
In all cases of rickets, the study of choice is radiography of the wrists, knees, ankles, and long bones. No pathognomonic sign on X-ray distinguishes hypophosphatemic rickets from other variants of rickets.
Oral Manifestations (Refer to Chapter 15)
Histologic Features (Refer to Chapter 15)
Treatment and Prognosis
Treatment can be administered safely on an outpatient basis, although serum calcium concentrations must be monitored periodically and carefully. Conscientious follow-up is essential. The usual vitamin D preparations are not useful for treatment in this disorder because they lack significant 1-alpha-hydroxylase activity. Original treatment protocols advocated vitamin D at levels of 25,000–50,000 U/d (at the lower limit of toxic dosage), which placed the patient in jeopardy of frequent hypercalcemic episodes. Now more widely available, calcitriol substantially diminishes but does not eliminate this risk. Amiloride and hydrochlorothiazide are administered to enhance calcium reabsorption and to reduce the risk of nephrocalcinosis. Surgical care involves osteotomy to realign extremely distorted leg curvatures in children whose diagnosis was delayed or whose initial treatment was inadequate. Skull deformity may require treatment for synostosis. Spontaneous abscesses often require periodic dental procedures. Apart from the short stature of most affected adults, the prognosis for a normal lifespan and normal health is good.
Craniosynostosis Syndromes
Craniosynostosis consists of premature fusion of one or more cranial sutures, often resulting in an abnormal head shape. It may result from a primary defect of ossification (primary craniosynostosis), or more commonly, from a failure of brain growth (secondary craniosynostosis). Simple craniosynostosis is a term used when only one suture fuses prematurely. Complex or compound craniosynostosis is used to describe premature fusion of multiple sutures. When children with craniosynostosis, usually complex, also display other body deformities, this is termed syndromic craniosynostosis.
Etiology
Multiple theories have been proposed for the etiology of primary craniosynostosis, but the most widely accepted is a primary defect in the mesenchymal layer ossification in the cranial bones. Secondary craniosynostosis typically results from systemic disorders such as endocrine disorders, hypothyroidism, hypophosphatemia, vitamin D deficiency, renal osteodystrophy, hypercalcemia, and rickets; hematologic disorders that cause bone marrow hyperplasia (e.g. sickle cell disease, thalassemia) and inadequate brain growth, including microcephaly and its causes.
The syndromic causes appear to result from genetic mutations responsible for fibroblast growth factor receptors 2 and 3. A gene locus for single suture craniosynostosis has not been identified. Primary craniosynostosis results when one or more sutures fuse prematurely, skull growth can be restricted perpendicular to the suture. If multiple sutures fuse while the brain is still increasing in size, intracranial pressure can increase. Secondary craniosynostosis is more frequent than the primary type, and results from early fusion of sutures due to primary failure of brain growth. Since brain growth drives the bony plates apart at the sutures, a primary lack of brain growth allows premature fusion of all the sutures. Intracranial pressure usually is normal, and surgery seldom is needed. Typically, failure of brain growth results in microcephaly. Intrauterine space constraints may play a role in the premature fusion of sutures in the fetal skull. This has been demonstrated in coronal craniosynostosis.
Clinical Features
Craniosynostosis may be evident at birth or in infancy from craniofacial abnormalities. It is equally distributed in both genders. It may become evident later when the child exhibits neurodevelopmental delays. Typically, careful examination alone can make the diagnosis. Craniosynostosis sometimes is associated with sporadic craniofacial syndromes such as Crouzon, Apert, Chotzen, Pfeiffer, or Carpenter syndromes. In this context, facial features, typically craniofacial abnormalities, suture ridging, and early closure of fontanels, suggest the diagnosis. Raised intracranial pressure is rare with fusion of a single suture. Intracranial pressure may be elevated in primary multiple suture craniosynostosis, such as cloverleaf skull and the syndromic synostoses. Signs include sun-setting eyes, papilledema, vomiting, and lethargy. Differential diagnoses include benign skull tumors, hydrocephalus, mental retardation, neural tube defects, syringomyelia, thyroid disease and torticollis.
Radiographic Features
Skull X-ray with anteroposterior, lateral, and Waters’ views show prematurely fused sutures which are easily identified by the absence of sutures and associated ridging of the suture line. Sutures either are not visible or have evidence of sclerosis.
Treatment and Prognosis
In the past 30 years, a better understanding of the pathophysiology and management of craniosynostosis has developed. Currently, surgery is usually cosmetic for infants with fusion of one or two sutures that result in a misshapen head. For infants with microcephaly (i.e. secondary craniosynostosis), surgery usually is not required. Surgery typically is indicated for increased intracranial pressure or for cosmetic reasons. Patients with primary craniosynostosis must be monitored after surgery. In secondary craniosynostosis, prognosis is dependent upon underlying etiology.
Craniofacial Dysostosis (Crouzon disease or syndrome)
The craniosynostosis syndromes constitute a group of conditions each characterized by premature craniosynostosis occurring in association with a variety of other abnormalities. These may or may not occur with syndactyly, anomalies of the hands and feet. The most common of the craniosynostotic syndromes occurring without syndactyly is craniofacial dysostosis, or Crouzon disease. The most common one occurring with syndactyly is the Apert syndrome, which is otherwise similar to Crouzon disease. Crouzon syndrome, described in 1912 as one of the varieties of craniofacial dysostosis, is caused by premature obliteration and ossification of two or more sutures, most often coronal and sagittal. Some authors connect those syndromes as one, calling it Crouzon-Apert syndrome, but symptomatologic differentiation makes classification difficult. Acanthosis nigricans is the main dermatologic manifestation of Crouzon syndrome.
Dysplasias of the skeleton (including craniofacial dysostosis) are caused by the malformations of the mesenchyme and ectoderm. The unknown teratogenic factors are taken into account. Dysplasias are inherited in an autosomal dominant pattern. Mutation of the fibroblast growth factor receptor (FGFR)-2 gene could be responsible for Crouzon syndrome. Moreover, the mutation in the transmembrane region of FGFR3 was detected in this syndrome.
Clinical Features
Although there is considerable individual variation in the appearance of patients with craniofacial dysostosis, the signs are all basically due to early synostosis of the sutures. Facial deformity is observed at birth, followed — with time — by other features of the syndrome. Coronal and sagittal sutures are obliterated; fontanels remain not obliterated and pulsating for a long time. Lateral and anteroposterior flattening of the acrocranium is observed, growing only at the vertical axis. Anteroposterior diameter is smaller than transverse diameter. The forehead is high and wide. Wide face and hypoplastic maxilla producing pseudoprognathism are observed. Deviation of the nasal septum, narrowed or obliterated anterior nares, and wide beaked nose are present. Hypertelorism, divergent squint, eyelid seems antimongoloid, and upper eyelid mimicking ‘frog face’ are observed. The upper lip is shortened and sometimes cleaved. Progressing optic nerve atrophy leads to vision impairment because of the intracranial hypertension. Impairment of hearing indicates disorders of the middle ear. Malocclusion, malposed teeth, and dysphasia are noted. Short stature and no physiologic spinal curvature are observed. The skin usually is dark. Syndromic acanthosis nigricans appears in the axillary fossa, the angle of the mouth, and on the lips in children. Patients report headache. Convulsions often occur; mental retardation is frequently observed (Fig. 17-15).

Radiographic Features
Skull, spine, and hand radiography is usually necessary to confirm the diagnosis. Skull radiography reveals the following: obliterated sutures (mostly coronal, sagittal); shallow eye sockets (exophthalmos); shortened anterior cranial fossa; underdeveloped lateral nasal sinuses; tympanic membranes fixed obliquely, narrowed external auditory canals, and small pyramids with symptoms of sclerosis; On spine radiography, the presence of bifid spinous process is possible, and slight symptoms of achondroplasia may be visible. Radiographic examination of the metacarpal bones and fingers reveals slight achondroplasia.
Treatment and Prognosis
A neurosurgical procedure is recommended in cases of intracranial hypertension leading to further optic atrophy. The surgery is difficult, and the procedure must be considered and undertaken in stages. Plastic surgery of the face could be of great help. It is one of the few syndromes where the cosmetic results of the surgery can be strikingly effective. These patients may ultimately come to lead a relatively normal life.
Mandibulofacial Dysostosis (Treacher Collins-Franceschetti syndrome)
The mandibulofacial dysostosis syndrome encompasses a group of closely related defects of the head and face, often hereditary or familial in pattern, following an irregular form of dominant transmission. A historic review of the disease was made by Pavsek, who, in addition to reporting an additional case, summarized the embryologic faults of the conditions.
The occurrence of Treacher Collins syndrome is in the range of 1 in 25,000 to 1 in 50,000 live births. Inheritance is autosomal dominant: males and females are equally affected; inheritance may be from one parent; multiple generations are affected. The gene for Treacher Collins syndrome was mapped to chromosome 5q32–q33.1. More than 50 Treacher Collins syndrome families have been analyzed. DNA diagnosis can be performed indirectly by linkage analysis in a family with more than one affected member, with greater than 95% accuracy if relevant DNA markers are informative within the family.
Clinical Features
Wide variations in the clinical expression of this syndrome are recognized, ranging from a complete, typical form manifesting all abnormalities listed below through incomplete, abortive, and atypical forms. The important clinical manifestations of the disease are:
• Antimongoloid palpebral fissures with a coloboma of the outer portion of the lower lids, and deficiency of the eyelashes (and sometimes the upper lids).
• Hypoplasia of the facial bones, especially of the malar bones and mandible.
• Malformation of the external ear, and occasionally of the middle and internal ears.
• Macrostomia, high palate (sometimes cleft) and abnormal position and malocclusion of the teeth.
• Blind fistulas between the angles of the ears and the angles of the mouth.
• Atypical hair growth in the form of a tongue-shaped process of the hairline extending towards the cheeks.
• Other anomalies such as facial clefts and skeletal deformities (Fig. 17-16).
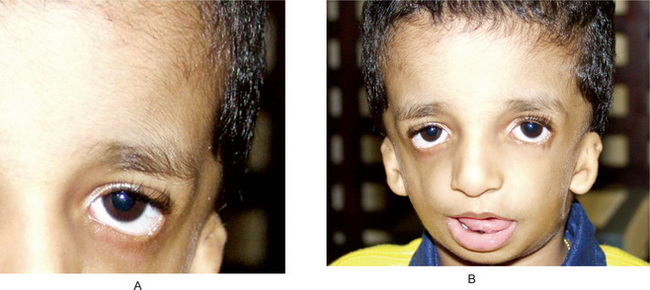
The characteristic faces of the patients have often been described as being bird like or fish like in nature.
The syndrome is thought to result from a retardation or failure of differentiation of maxillary mesoderm at and after the 50 mm stage of the embryo. The fact that the teeth of the upper jaw are usually unaffected, and ordinarily are present by the sixth week, is further evidence of retardation or arrest of differentiation at or after the second month of fetal life. The first visceral arch of the visceral mesoderm also advances secondarily to form the mandible, and again retardation occurs on the same basis.
A disease that has sometimes been confused with mandibulofacial dysostosis because of certain clinical features in common is hemifacial microsomia (also known as oculoauriculovertebral dysplasia or Goldenhar syndrome). However, hemifacial microsomia is sporadic in the vast majority of cases, although familial cases have been reported. In addition as the name implies, this disease is unilateral and has been suggested to be related to an abnormality in the vascular supply of the head. It has been discussed in detail by Gorlin and his associates.
Radiographic Features
As Pavsek pointed out, the bodies of both malar bones tend to be grossly and symmetrically underdeveloped in mandibulofacial dysostosis. There may be agenesis of the malar bones with nonfusion of the zygomatic arches, as well as absence of the palatine bones. Cleft palate may be visible on the radiograph. There is usually hypogenesis, and sometimes agenesis of the mandible. The paranasal sinuses are grossly underdeveloped. The auditory ossicles are often absent, and the cochlea and vestibular apparatus may be deficient. The cranial vault is normal in most instances.
Pierre Robin Malformation (Pierre Robin syndrome, Robin sequence, Pierre Robin anomalad, Robin complexes, Pierre Robin malformation complex)
Robin sequence, previously known as Pierre Robin syndrome and Pierre Robin anomalad, consists of three essential components which include:
• Micrognathia or retrognathia
• Glossoptosis, often accompanied by airway obstruction. (The tongue is not actually larger than normal, but because of the small mandible, the tongue is large for the airway and therefore causes obstruction. Rarely, the tongue is smaller than normal).
Robin sequence occurs as an isolated defect, as part of a recognized syndrome, or as part of a complex of multiple congenital anomalies. The condition is named after the French dental surgeon Pierre Robin (1867–1950).
Etiology
Three pathophysiological theories exist to explain the occurrence of Pierre Robin sequence:
1. The mechanical theory. This is the most accepted theory. Initially, mandibular hypoplasia occurs between the 7th and 11th week of gestation. This keeps the tongue high in the oral cavity, causing a cleft in the palate by preventing the closure of the palatal shelves. This theory explains the classic inverted U-shaped cleft and the absence of an associated cleft lip. Oligohydramnios could play a role in the etiology since the lack of amniotic fluid could cause deformation of the chin and subsequent impaction of the tongue between the palatal shelves.
2. The neurological maturation theory. A delay in neurological maturation has been noted on electromyography of the tongue musculature, the pharyngeal pillars, and the palate, as has a delay in hypoglossal nerve conduction. The spontaneous correction of the majority of cases with age supports this theory.
3. The rhombencephalic dysneurulation theory. In this theory, the motor and regulatory organization of the rhombencephalus is related to a major problem of ontogenesis.
Clinical Features
This heterogeneous birth defect has a prevalence of approximately 1 per 8,500 live births. The maleto-female ratio is 1:1. Micrognathia is reported in the majority of cases (91.7%). The mandible has a small body, obtuse gonial angle, and a posteriorly located condyle. The mandibular hypoplasia; however, resolves and the child attains a normal profile by the age of five to six years. Glossoptosis is noted in 70–85% of reported cases. Macroglossia and ankyloglossia are relatively rare findings, noted in 10–15% of reported cases. The combination of micrognathia and glossoptosis may cause severe respiratory and feeding difficulty in the newborn. Obstructive sleep apnea may also occur. The prevalence of cleft palate varies from 14–91%. It can affect the soft and hard palate and is usually U-shaped (80%) or V-shaped (Fig. 17-17). Occasionally, it may present as a bifid or double uvula or as an occult submucous cleft. Velopharyngeal insufficiency is usually more pronounced in these patients than in those with isolated cleft palate.
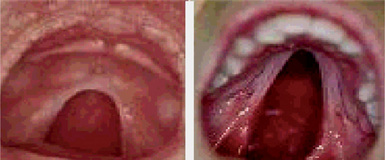
Other associated anomalies are also seen which include; otitis media, hearing loss, nasal deformities, dental and philtral malformations. Anomalies involving the musculoskeletal system are the most frequent systemic anomalies (noted in 70–80% of cases). They include syndactyly, dysplastic phalanges, polydactyly, clinodactyly, hyperextensible joints, and oligodactyly in the upper limbs. Central nervous system (CNS) defects such as language delay, epilepsy, neurodevelopmental delay, hypotonia, and hydrocephalus may occur.
Treatment and Prognosis
A multidisciplinary approach is required to manage the complex features involved in the case of these children. Treatment is prioritized according to the severity of airway compromise followed by the extent of feeding difficulties. Infants with pronounced micrognathia may experience severe respiratory distress or failure to thrive. Surgical intervention is necessary in these cases.
Apert Syndrome (Acrocephalosyndactyly)
Apert syndrome is named after the French physician who described the syndrome acrocephalosyndactylia in 1906. It is a rare autosomal dominant disorder characterized by craniosynostosis, craniofacial anomalies, and severe symmetrical syndactyly (cutaneous and bony fusion) of the hands and feet. It probably is the most familiar and best-described type of acrocephalosyndactyly.
Etiology
More than 98% of cases of Apert syndrome are caused by specific missense substitution mutations (i.e. Ser252Trp, Ser252Phe, Pro253Arg) involving fibroblast growth factor receptor 2 (FGFR2), which maps to chromosome bands 10q25–q26. The remaining cases are due to mutations in or near exon 9 of FGFR2. Fibroblast growth factor receptor 2 (FGFR2) mutations lead to an increase in the number of precursor cells that enter the osteogenic pathway. Ultimately, this leads to increased subperiosteal bone matrix formation and premature calvaria ossification during fetal development. The order and rate of suture fusion determine the degree of deformity and disability. The evidence that syndactyly of Apert syndrome could be a keratinocyte growth factor receptor (KGFR)-mediated effect has also been reported.
Clinical Features
Apert syndrome is detected in the neonatal period due to craniosynostosis and associated findings of syndactyly in the hands and feet. Asians have the highest reported prevalence (22.3 per million live births). No gender predilection is seen. Craniostenosis is present and most commonly involves the coronal sutures, resulting in acrocephaly, brachycephaly, flat occiput, and high prominent forehead. Large late-closing fontanels and a gaping midline defect are seen. Patients have apparent low-set ears with occasional conductive hearing loss. Eyes exhibit down-slanting palpebral fissures, hypertelorism, shallow orbits, proptosis and exophthalmos. The nose has a markedly depressed nasal bridge. It is short and wide with a bulbous tip, parrot-beaked appearance, and choanal stenosis or atresia (Fig. 17-18A, B).
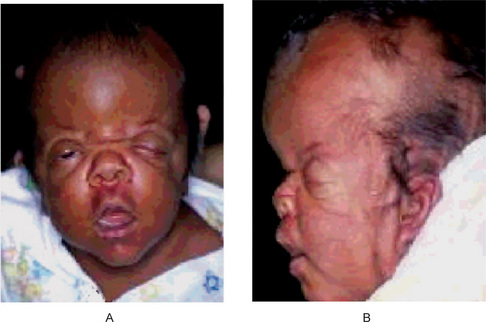
Figure 17-18 Apert syndrome.
(A) An infant with Apert syndrome is shown. Note the characteristic ocular hypertelorism, down-slanting palpebral fissures, proptotic eyes, horizontal groove above the supraorbital ridge, break of the continuity of eyebrows, depressed nasal bridge, and short wide nose with bulbous tip. (B) In this profile, turribrachycephaly, high prominent forehead, proptosis, depressed nasal bridge, short nose, and low-set ears are prominent. (Courtesy of Dr Harold Chen)
The jaw shows a prominent mandible, maxillary hypoplasia, drooping angles of the mouth, high arched palate, bifid uvula, cleft palate, crowded upper teeth, malocclusion, delayed and ectopic eruption, shovel-shaped incisors, supernumerary teeth, V-shaped maxillary dental arch, and bulging alveolar ridges.
Syndactyly involves the hands and feet with partial-tocomplete fusion of the digits, often involving second, third, and fourth digits. These often are termed mitten hands and sock feet (Fig. 17-19). In severe cases, all digits are fused, with the palm deeply concave and cup-shaped and the sole supinated (Fig. 17-20). Intelligence varies from normal to subnormal mentality. Malformations of the CNS may be responsible for most cases. Papilledema and optic atrophy with loss of vision may be present in cases of subtle increased intracranial pressure. Hyperhidrosis is commonly seen. Cardiovascular manifestations like atrial septal defect, patent ductus arteriosus, ventricular septal defect and pulmonary stenosis are present. Gastrointestinal, genitourinary and respiratory symptoms may be present in a small percentage of cases.
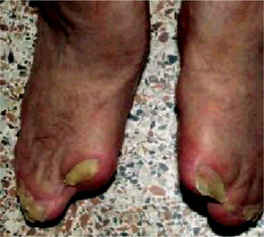
Figure 17-19 Apert syndrome.
Note the sock appearance of the feet with syndactyly involving the second, third, fourth, and fifth toes. The patient also has contiguous nail beds (synonychia). (Courtesy of Dr Harold Chen).
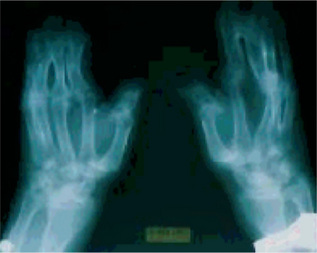
Figure 17-20 Apert syndrome.
Note osseous syndactyly involving the second, third, fourth, and fifth fingers; multiple synostosis involving distal phalanges and proximal fourth and fifth metacarpals; symphalangism of interphalangeal joints; shortening and radial deviation of distal phalanx; and delta-shaped deformity of proximal phalanx of the thumbs.
Thanatophoric Dysplasia
Thanatophoric dysplasia (TD) is the most common form of skeletal dysplasia that is lethal in the neonatal period. It is an autosomal dominant disorder resulting from sporadic de novo mutations in the FGFR3 gene. Characteristics of TD include severe shortening of the limbs, a narrow thorax, macrocephaly, and a normal trunk length. It is divided into two clinically defined subtypes. TD type 1, the most common subtype, features a normally shaped skull and curved long bones (shaped like a telephone receiver) with the femurs affected most. TD type 2 features a cloverleafshaped skull and straight femurs.
Clinical Features
A macrocephalic head with a frontal bossing, a flattened nasal bridge, and proptotic eyes has been observed. In TD2, a cloverleaf-shaped skull resulting from premature closure of the cranial sutures. Narrow thorax with small ribs, micromelic limbs with brachydactyly, protuberant abdomen, hydrocephalus and other cerebral parenchymal abnormalities are seen. Characteristic orodental abnormalities are not described associated with this syndrome.
Achondroplasia (Chondrodystrophia fetalis)
Achondroplasia is a common nonlethal form of chondrodysplasia. It is transmitted as an autosomal dominant trait with complete penetrance. De-novo mutations cause 75–80% of cases.
Etiology
Achondroplasia is caused by mutations in the gene for fibroblast growth factor receptor-3 (FGFR3). The gene has been mapped to band 4p16.3. The common mutations cause a gain of function of the FGFR3 gene, resulting in decreased endochondral ossification, inhibited proliferation of chondrocytes in growth plate cartilage, decreased cellular hypertrophy, and decreased cartilage matrix production.
Clinical Features
Frequency is believed to be 1 case per 15,000–40,000 births worldwide (Fig. 17-21). Cardinal features include short stature, rhizomelic shortening of the arms and legs, a disproportionately long trunk, trident hands, midfacial hypoplasia, prominent forehead (frontal bossing), thoracolumbar protuberance, true megalencephaly, and characteristic limitation of joint motion. The incongruous appearance of the achondroplastic dwarf is in contrast to that of the pituitary dwarf, and the incongruity becomes more pronounced as he/she approaches adulthood and later life, chiefly because of the disproportionate size of the head in relation to the remainder of the body. Despite their misshapen appearance, achondroplastic dwarfs are of normal intelligence. Often they are also endowed with unusual strength and agility, characteristics which have led some to adopt the occupation of professional wrestler.

Oral Manifestations
The maxilla is often retruded because of restriction of growth of the base of the skull, and the retrusion may produce a relative mandibular prognathism (Fig. 17-22). The resultant disparity in size of the two jaws produces an obvious malocclusion. The dentition itself is usually normal, although congenitally missing teeth with disturbance in the shape of those present have been reported.
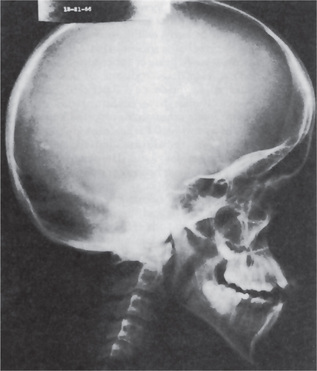
Radiographic Features
Radiographs of the skull, spine, and extremities reveal the characteristic features. A lateral skull radiograph demonstrates midface hypoplasia, enlarged calvaria, frontal prominence, and shortening of the base of the skull. The size of the foramen magnum is diminished. The long bones are shorter than normal, and there is thickening or mild clubbing of the ends. The epiphyses generally appear normal, but may close either early or late. The bones at the base of the skull fuse prematurely. Except for the retrusion of the maxilla and the malocclusion between the two jaws, there are no changes in the jawbones.
Histologic Features
The abnormality seen in the bone of patients with achondroplasia is failure of endochondral ossification. Intramembranous and periosteal ossification are undisturbed. Histologic studies have shown disarray of the chondrocytes, with loss of columnation and loss of normal chondrocyte proliferation. Fibrous tissue is present in the zone of provisional calcification, but bone trabeculae present are irregular. Because endochondral growth is affected, the orderly longitudinal growth of bone is disrupted, resulting in stunting of the bone. Intramembranous ossification is normal, leading to normal clavicles and skull. Because the width of the long bones is a product of intramembranous periosteal ossification, these bones are of normal diameter.
Treatment and Prognosis
There is no treatment for achondroplasia. There may be delay in motor milestones but speech is normal. The frequent middle ear infections and dental crowding require attention. If the patient survives the first few years of life, the chances are excellent that he/she will have the life expectancy of a normal person.
Robinow Syndrome
Around 1969, Dr Meinhard Robinow identified a new syndrome, which was unreported before. He named it fetal face syndrome, based upon his views of the facial features of an eight-month old fetus. Later the name was changed to ‘Robinow’ syndrome.
Two types of this syndrome have been described, dominant and recessive. The dominant type is the most common and the parents are usually not carriers of the dominant gene that produces the syndrome. In the recessive type, which is the rarer of the two, both parents carry the recessive gene (but are not affected) and have a 25% chance of producing an affected offspring. The recessive form is caused by mutation in ROR2 gene. This gene is located in chromosome 9q22 and works in cartilage and bone formation. The gene responsible for the dominant form has not been established yet, but genes related to ROR2 are being studied as candidates.
Clinical Features
The features described refer to both types of this syndrome. The patients usually have most of these signs in varied proportions.
Mild to moderate short stature (dwarfism), short lower arms (mesomelic brachymelia), small hands with clinodactyly usually of the fifth finger (abnormal lateral or medial bending of one or more fingers or toes) and brachydactyly (abnormally short fingers or toes) and small feet.
Hyperostosis Corticalis Generalisata (van Buchem’s disease, hyperphosphatasemia tarda, endosteal hyperostosis, autosomal recessive)
This disease of bone, described by van Buchem and his associates in 1955, appears to represent an excessive deposition of endosteal bone throughout the skeleton in a pattern suggestive of a hereditary condition with an autosomal recessive characteristic. The disease gene has been mapped to chromosome 17q11.2.
Clinical Features
The disease is usually not discovered until adult life, and in nearly all reported cases, has been a chance finding. The facial appearance of these patients may be altered and this may be the reason that they seek professional advice. Such a case has been reported by Dyson. The face may appear swollen, particularly with widening at the angles of the mandible and at the bridge of the nose. Some patients also have loss of visual acuity, loss of facial sensation, some degree of facial paralysis and deafness, all due to cranial nerve involvement through closure of foramina. Intraorally, there is sometimes overgrowth of the alveolar process. Most patients, except for the facial appearance, appear normal and are free of symptoms, including bone tenderness.
Radiographic Features
A skeletal survey will reveal increased density of many bones of the body, although some bones, such as those of the hands and feet, may be unaffected. The skull also exhibits diffuse sclerosis, as may the jaws.
Chondroectodermal Dysplasia (Ellis-van Creveld syndrome)
Ellis-van Creveld is an extremely rare form of dysplasia first described by Ellis and van Creveld in 1940. It is characterized by four components: chondrodysplasia; polydactyly; ectodermal dysplasia affecting the hair, teeth, and nails; and congenital heart failure (Fig. 17-23). The dysplasia is one of the short-rib polydactyly syndromes. As its name implies, this syndrome affects both mesodermal and ectodermal tissues. The prevalence of Ellis-van Creveld dysplasia is 0.1 per million.
Ellis-van Creveld syndrome is inherited as an autosomal recessive disorder. The locus gene has been mapped to chromosome 4p16.1.
Clinical Features and Oral Manifestations (Refer to Chapter 19)
Cleidocranial Dysplasia (Marie and Sainton’s disease, Scheuthauer-Marie-Sainton syndrome, mutational dysostosis)
A congenital disorder of bone formation manifested with clavicular hypoplasia or agenesis with a narrow thorax, which allows approximation of the shoulders in front of the chest. Delayed ossification of the skull, excessively large fontanels, and delayed closing of the sutures are prominent features of this disorder. The fontanels may remain open until adulthood, but the sutures often close with interposition of wormian bones. Bossing of the frontal, parietal, and occipital regions give the skull a large globular shape with small face. The characteristic skull abnormalities are sometimes referred to as the Arnold head named after the descendants of a Chinese who settled in South Africa and changed his name to Arnold. More than 100 additional anomalies may be associated, including wide pubic symphysis, dental abnormalities, short middle phalanges of the fifth finger, delayed skeletal maturation, hearing deficiency, and mild mental retardation in some cases.
The syndrome is familial and is transmitted as an autosomal dominant trait. Several chromosome abnormalities have been reported to be associated with this syndrome, including rearrangement of long arm of chromosome 8(8q22) and the long arm of chromosome 6. Mutations in the core-binding factor alpha-1 (CBFA1) gene, located on chromosome 6p21, have been shown to be the cause of cleidocranial dysplasia.
Clinical Features
Cleidocranial dysplasia is characterized by abnormalities of the skull, teeth, jaws and shoulder girdle as well as by occasional stunting of the long bones. In the skull the fontanels often remain open or at least exhibit delayed closing, and for this reason tend to be rather large. The sutures also may remain open and wormian bones are common. The sagittal suture is characteristically sunken, giving the skull a flat appearance. Frontal, parietal, and occipital bones are prominent and the paranasal sinuses are underdeveloped and narrow. Based on the cephalic index, the head is brachycephalic, or wide and short, with the transverse diameter of the skull being increased. A variety of other skull abnormalities are sometimes present. Additional abnormalities include calvarial thickening in the supraorbital part of the frontal bone, squamous part of the temporal bone and the occipital bone; occasional absence of the parietal bones, faulty development of the foramen magnum, and dysplasia of the paranasal sinuses. An excellent review and discussion of the varied clinical findings in cleidocranial dysplasia has been published by Kalliala and Taskinen.
The defect of the shoulder girdle, from which the condition derives a portion of its name, ranges from complete absence of clavicles, in about 10% of cases, to partial absence or even a simple thinning of one or both clavicles (Figs. 17-24 A, 17-25). Because of this clavicular disturbance, the patients have an unusual mobility of the shoulders and may be able to bring their shoulders forward until they meet in the midline (Fig. 17-24B). Defects of the vertebral column, pelvis and long bones, as well as bones of the digits, are also relatively common. Thus cleidocranial dysplasia, once thought to be a disease involving only membranous bones, is now recognized as affecting the entire skeleton. In addition, changes outside the skeleton, such as anomalous muscles, have been reported, but these may be secondary to the bony involvement.
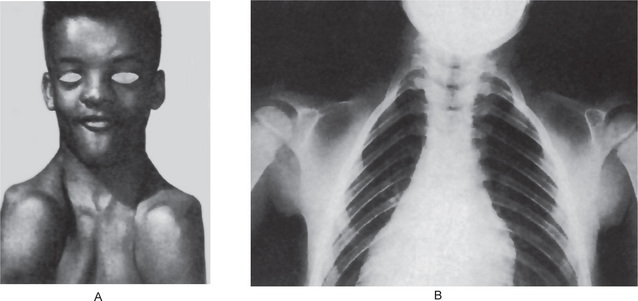
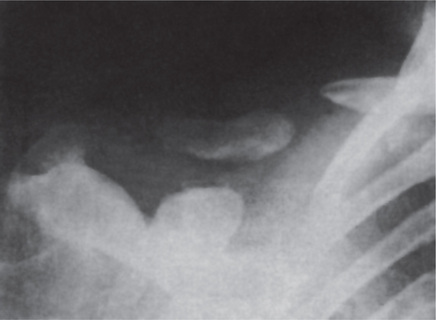
Oral Manifestations
Patients with cleidocranial dysplasia characteristically exhibit a high, narrow, arched palate, and actual cleft palate appears to be common. The maxilla is almost invariably reported to be underdeveloped and smaller than normal in relation to the mandible. However, Davis has reported that in a series of patients studied by cephalometric analysis all showed that the maxilla was of normal size and the position was either normal or anteriorly positioned in all cases. In addition, 70% of the affected patients had larger mandibles than those of controls, which suggests that patients with cleidocranial dysplasia have enlarged mandibles rather than small maxillae, as reported in the literature. These findings remain to be confirmed. The lacrimal and zygomatic bones are also reported to be underdeveloped.
One of the outstanding oral findings is prolonged retention of the deciduous teeth and subsequent delay in eruption of the succedaneous teeth. Sometimes this delay in tooth eruption is permanent. The roots of the teeth are often somewhat short and thinner than usual and may be deformed.
In addition, Rushton reported that there is absence or paucity of cellular cementum on the roots of the permanent teeth, and this may be related to the failure of eruption so frequently seen. This has also been studied by Smith, who confirmed the absence of cellular cementum on both deciduous and permanent teeth. A surprising and unexplained feature was the absence of this cementum on the erupted teeth in both dentitions, with no increased thickening of the primary acellular cementum. The manner of anchorage of periodontal fibers and the maintenance of periodontal ligament width are also not understood in this disease. Furthermore, it is characteristic for numerous unerupted supernumerary teeth to be found by radiographic examination (Fig. 17-26). Crypt formation around impacted teeth, and ectopic teeth have been reported.
Figure 17-26 Cleidocranial dysplasia.
There are numerous unerupted and supernumerary teeth. (Courtesy of Dr Wilbur C Moorman)
These are most prevalent in the mandibular premolar and incisor areas. Interestingly, partial anodontia has also been recorded in this condition but is rare.
Radiographic Examination
Reveals the widely patent anterior fontanel and sutures with wormian bones in cranium. The clavicles typically are reduced to single or double fragments on each side with middle part being deficient. Frequently the changes are asymmetric. Marked delay in ossification of pelvic bones especially pubic and ischial bones is regulary observed. Spina bifida occulta is observed in the cervical and upper thoracic levels. Hands and feet demonstrate various anomalies including shortening and broadening of carpal, metacarpal, tarsal, metatarsal bones.
Treatment and Prognosis
There is no specific treatment of cleidocranial dysplasia, although care of the oral conditions is important. The retained deciduous teeth should be restored if they become carious, since their extraction does not necessarily induce eruption of the permanent teeth. However, in recent years, there has been increasing use of a multidisciplinary approach to treatment of these patients, utilizing the pedodontist, the orthodontist, and the oral surgeon. It has been found, as in the case reviewed by Hutton and his associates, that the permanent teeth do have the potential to erupt and that correct timing of surgical procedures for uncovering teeth and orthodontic repositioning can give excellent functional results. Life expectancy is normal. Complications may arise during delivery in cases with narrow pelvis.
Tricho-dento-osseous Syndrome
The tricho-dento-osseous (TDP) syndrome is a hereditary condition which chiefly involves the hair, teeth, and bones. Individuals with this syndrome are born with a full head of kinky hair, which sometimes tends to straighten with age. Nails are thin and likely to peel or fracture. The sweat glands are developed normally. The chief bony abnormality in patients with the condition are bones which are found to be more dense than normal. In some families, the skull bones are excessively thick. These abnormalities are of no clinical significance and should not cause individuals with this syndrome any problem. They are, however, helpful in making the diagnosis. There is no evidence that people who have this condition are shorter or taller than normal.
Teeth may become infected and dental abscesses are common during the first few years of life. They have thin, pitted and yellow-brown enamel. On dental X-ray, large pulp chambers (taurodontia) are found. In addition, teeth may remain unerupted for long giving the condition of partial anodontia. Intelligence is normal, as is life span. The condition is inherited as an autosomal dominant disorder. Prenatal diagnosis for this syndrome is not yet possible. The disorder has been mapped to locus 17q21.3–q22.
There may be three distinct types of TDO syndrome that have similar but not identical characteristics. Some researchers suggest that these variants may be differentiated mainly by whether the calvaria and/or long bones exhibit abnormal hardening (sclerosis), thickening, and/or density. Other symptoms also vary among the three types.
Down Syndrome (Down’s syndrome, trisomy 21 syndrome, mongolism, congenital acromicria syndrome)
Down syndrome is a frequent form of mental retardation associated with characteristic morphologic features (mongolism) and many somatic abnormalities due to a number of chromosomal aberrations. The characteristic clinical features that discriminate the syndrome from other mental deficiencies were first described by John Langdon Down in 1866.
Three cytogenetic variants cause Down syndrome: trisomy 21, chromosomal translocation, and mosaicism. Trisomy 21 accounts for nearly 95% of all patients with Down syndrome. It is now generally accepted that there are at least three forms of Down syndrome: one in which there is the typical trisomy 21 with 47 chromosomes (accounting for about 95% of cases); another termed the translocation type, in which there appear to be only 46 chromosomes, although the extra chromosome material of number 21 is translocated to another chromosome of G or D group, either 21/22 translocation or 21/21 translocation (about 3% of cases); and another that is the result of chromosomal mosaicism (about 2%) (Fig. 17-27). Children with the translocation type of Down syndrome are more commonly born to mothers over 30 years of age. The incidence of mongolism in subsequent siblings may be greatly increased in such instances. Mothers over 40 years rarely have translocation mongoloids. In contrast, the risk of having an affected child of the typical trisomy 21 type is approximately one in 2,000 live births in women under 30 years of age but rises dramatically to one in 50 live births in women over 45 years of age.
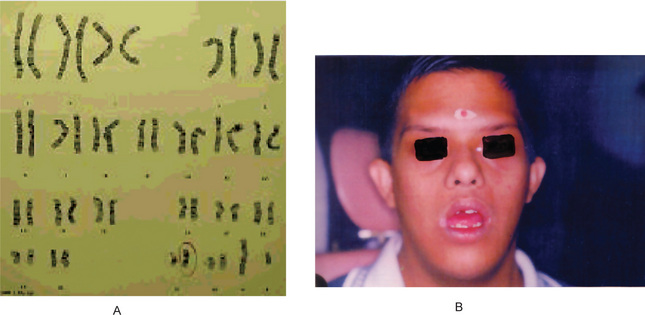
Figure 17-27 Down syndrome.
(A) A G-banded karyotype showing trisomy 21 of ISO chromosome arm 21 q type [46 × 4i (q 10)]. (B) Frontal view of a patient with Down syndrome. (Courtesy of Dr Harold Chen)
Clinical Features
Down syndrome is the most common autosomal abnormality and occurs in approximately 1 per 700 live births. Down syndrome accounts for the majority of mentally handicapped children in the preschool age category. It has been reported in people of all races. Both genders are affected equally. Characteristic morphologic features of mongolism can be recognized immediately at birth, but they are obvious in children older than one year. The major features of Down syndrome are mental retardation which can be mild to severe with an intelligence quotient (IQ) of 25–50; characteristic head appearance; small head (brachycephaly), flat facies with increased interocular distance (hypertelorism), depressed nasal bridge, flat occiput, and broad short neck. Narrow, upward and outward slanting of the palpebral fissures, medial epicanthal folds, strabismus, cataract and retinal detachment are the ocular anomalies. Small and misshapen ears with anomalies of the folds are observed. Skeletal anomalies include short stature; broad and short hands, feet, and digits; short curved fifth finger (dysplasia of the midphalanx), clinodactyly of the fifth finger; dysplasia of the pelvis; joint laxity; a wide gap between the first and second toes; and atlanto-occipital instability. Muscle hypotonia in newborns with decreased response to normal stimuli has been reported.
Protuberant abdomen (with or without an umbilical hernia), hypogenitalism, hypospadia, cryptorchism, and delayed and incomplete puberty. Congenital defects of the heart, or endocardial defects (40%), duodenal atresia, Hirschsprung disease, polydactylia, and syndactylia are reported. Other features include recurrent respiratory infections, leukemia (1%), epilepsy (10%), hypothyroidism (3%), and presenile dementia.
Oral Manifestations
Small mouth with protrusion of the tongue (macroglossia) with difficulty in eating and speaking, scrotal tongue, hypoplasia of the maxilla, delayed tooth eruption, partial anodontia, enamel hypoplasia, juvenile periodontitis, and cleft lip or palate (rare) are noticed commonly. Fissuring and thickening of the lips and angular cheilitis are frequent and gets increased in incidence and severity with age. Cheilitis occurs with greater frequency in children with Down syndrome than in unaffected persons. It is explained by mechanical factors, trauma, actinic influence, atopy, avitaminosis, or low-grade infections (candidiasis). A fissured tongue (plicated or scrotal) occurs in as many as 80% of children with Down syndrome, but it affects about 5% of the general population. Geographic tongue occurs in 11.3% of patients with Down syndrome. Juvenile periodontitis is a feature of Down syndrome, and its incidence among the various age groups parallels the occurrence of cheilitis but without significant correlation.