A summary of the haematological examination and extending the haematological examination
TEXT BOX 22.1 The haematological examination: a suggested method
Weight (normal, reduced, increased)
Bruising (thrombocytopenia, scurvy etc)
Rashes and infiltrative lesions (lymphoma)
Scratch marks (myeloproliferative diseases, lymphoma)
Palmar crease pallor (anaemia)
Arthropathy (haemophilia, secondary gout, drug treatment etc)
3. Epitrochlear and axillary nodes
Sclera—jaundice, pallor, conjunctival suffusion (polycythaemia)
Mouth—gum hypertrophy (monocytic leukaemia etc), ulceration, infection, haemorrhage (marrow aplasia etc.); atrophic glossitis, angular stomatitis (iron, vitamin deficiencies)
5. Cervical nodes (sitting up)
7. Abdomen (lying flat) and genitalia
Vasculitis (Henoch-Schönlein purpura—buttocks, thighs)
Ulceration (e.g. haemoglobinopathies)
Neurological signs (subacute combined degeneration, peripheral neuropathy)
Fundi (haemorrhages, infection etc)
This will be a targeted examination during follow-up consultations but should be completed in full for the first visit if haematological disease is suspected or known.
Position the patient as for a gastrointestinal examination. Make sure he or she is fully undressed, in stages and with a gown for women. Look for bruising, pigmentation, cyanosis, jaundice and scratch marks (due to myeloproliferative disease or lymphoma). Also note the racial origin of the patient.
Pick up the patient’s hands. Look at the nails for koilonychia (spoon-shaped nails, which are rarely seen today and indicate iron deficiency) and the changes of vasculitis. Pale palmar creases may indicate anaemia (typically the haemoglobin level has to be lower than 70 g/L). Evidence of arthropathy may be important (e.g. rheumatoid arthritis and Felty’s syndrome, recurrent haemarthroses in bleeding disorders, secondary gout in myeloproliferative disorders).
Examine the epitrochlear nodes. Note any bruising. Remember, petechiae are pinhead haemorrhages, while ecchymoses are larger bruises.
Go to the axillae and palpate the axillary nodes. There are five main areas: central, lateral (above and lateral), pectoral (most medial), infraclavicular and subscapular (most inferior).
Look at the face. Inspecting the eyes, note jaundice, pallor or haemorrhage of the sclerae, and the injected sclerae of polycythaemia. Examine the mouth. Look for peri-oral telangiectasias. Note gum hypertrophy (e.g. from acute monocytic leukaemia or scurvy), ulceration, infection, haemorrhage, atrophic glossitis (e.g. from iron deficiency, or vitamin B12 or folate deficiency) and angular stomatitis. Look for tonsillar and adenoid enlargement (Waldeyer’s ring).
Sit the patient up. Examine the cervical nodes from behind. There are eight groups: submental, submandibular, jugular chain, supraclavicular, posterior triangle, postauricular, preauricular and occipital. Then feel the supraclavicular area from the front. Tap the spine with your fist for bony tenderness (caused by an enlarging marrow e.g. in myeloma or carcinoma). Also gently press the sternum, clavicles and shoulders for bony tenderness.
Lay the patient flat again. Examine the abdomen. Focus on the liver and spleen. Feel for para-aortic nodes. Do not forget to feel the testes in males and to perform a rectal and pelvic examination (for tumour or bleeding). Spring the hips for pelvic tenderness. Palpate the inguinal nodes. There are two groups: along the inguinal ligament and along the femoral vessels.
Examine the legs. Note particularly leg ulcers. Examine the legs from a neurological aspect, for evidence of vitamin B12 deficiency or peripheral neuropathy from other causes. Remember, hypothyroidism can cause anaemia and neurological disease.
Finally, examine the fundi, look at the temperature chart and test the urine.
Extending the haematological physical examination
HAEMATOLOGY TESTS
Diagnosis of haematological disease depends on the history, physical examination and thoughtful testing. While haematology is a test-heavy specialty, the ultimate diagnosis is still guided by the clinical setting.
Key tests to consider include:
• Complete blood count (CBC) or full blood count (FBC)—for example, to identify anaemia and the likely cause (which must be confirmed by more specific testing), or to identify white cell disorders (e.g. leukaemia) or platelet disorders (low: thrombocytopenia; or high: thrombocytosis).
• Peripheral blood smear—an extension of the physical examination to identify disease (e.g. target cells in liver disease, burr cells in renal disease, teardrop cells in bone marrow infiltration—see below).
• Reticulocyte count—low if there are red-cell production or maturation defects, or high if red cell survival is short (as occurs with blood loss).
• Tests for excess bleeding—for example, prothrombin time (International Normalised Ratio, INR) is increased with vitamin K deficiency or warfarin therapy.
• Tests for excess clotting (such as a history of multiple deep venous thromboses)—for example, Factor V Leiden mutation, anti-thrombin III deficiencies, protein C or S deficiency, anti-phospholipid antibodies.
• Special tests—for example, JAK2 mutation and polycythaemia rubra vera, Philadelphia chromosome and chronic myelogenous leukaemia, immunoelectrophoresis of serum and urine for multiple myeloma (M component), tests for haemolysis (including Coombs test—antibody-mediated red-cell damage).
• Bone marrow—for example, to identify the type of leukaemia, excess plasma cells (multiple myeloma).
EXAMINATION OF THE PERIPHERAL BLOOD FILM
This is a simple and useful clinical investigation. A properly made peripheral blood film is one of the simplest, least invasive and most readily accessible forms of ‘tissue biopsy’ and can be a very useful diagnostic tool in clinical medicine. An examination of the patient’s blood film can: (1) assess whether the morphology of RBCs, WBCs and platelets is normal; (2) help characterise the type of anaemia; (3) detect the presence of abnormal cells and provide clues about quantitative changes in plasma proteins—e.g. paraproteinaemia; and (4) help make the diagnosis of an underlying infection, malignant infiltration of the bone marrow or primary proliferative haematological disorder.
The following pages present illustrated examples of some clinical problems diagnosed by examination of the blood film (see Figures 22.2 to 22.14).
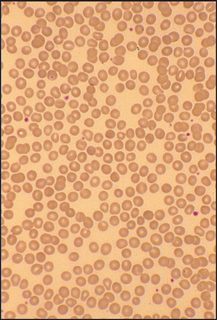
Figure 22.2 Spherocytic anaemia
Hereditary spherocytosis or autoimmune haemolytic anaemia. The numerous RBCs that are small, round and lack central pallor are spherocytes (the big RBCs are probably reticulocytes).
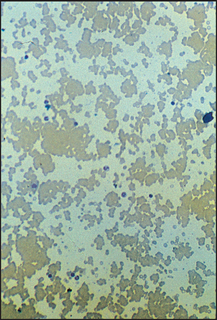
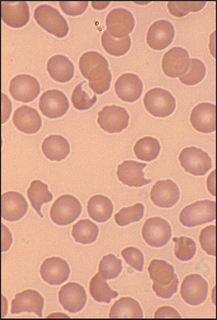
Figure 22.4 Microangiopathic haemolysis (e.g. disseminated intravascular coagulation) Frequent fragmented (bitten) red cells.
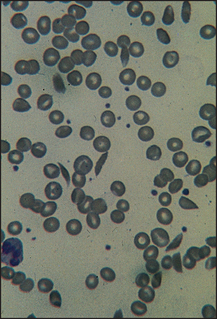
Figure 22.5 Sickle cell anaemia Film shows several sickle-shaped cells with target cells probably secondary to the ‘autosplenectomy’ that occurs in this disease.
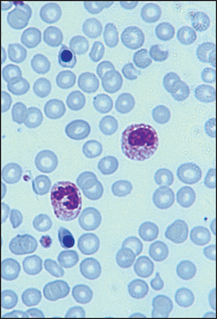
Figure 22.6 Leucoerythroblastic film Film indicative of bone marrow infiltration. Shows circulating nucleated RBCs and immature WBCs.
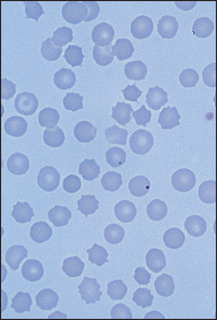
Figure 22.7 Postsplenectomy picture Film shows several Howell-Jolly bodies, target cells and crenated cells.
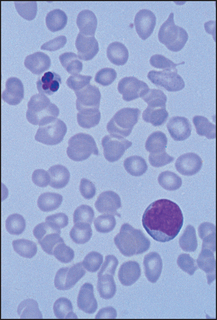
Figure 22.8 Myelofibrosis Film shows a dysplastic nucleated RBC, frequent tear-drop poikilocytes and a primitive granulocyte.
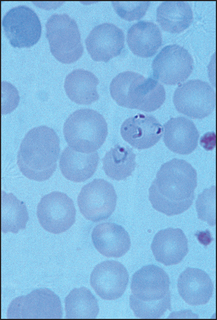
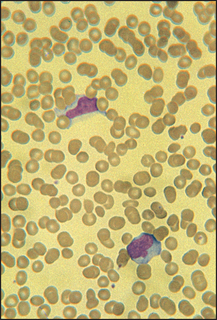
Figure 22.10 Viral illness (e.g. infectious mononucleosis) Film shows two atypical or ‘switched-on’ lymphocytes.
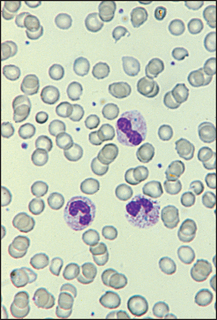
Figure 22.11 Bacterial infection (e.g. pneumonia, infective endocarditis) The WBC in the centre is a band form with prominent ‘toxic’ granules.
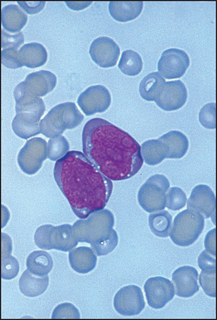
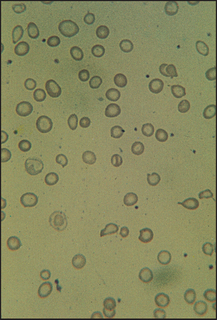
Figure 22.13 Iron-deficiency anaemia RBCs show varying shape and size; they are generally hypochromic.
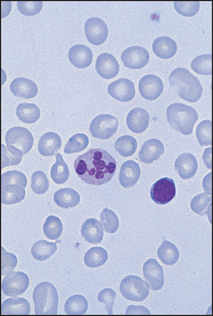
Figure 22.14 Megaloblastic anaemia RBCs are macrocytic with many oval forms and the neutrophil is hypersegmented.
ANAEMIA
Anaemia is a reduction in the concentration of haemoglobin below 135 g/L in an adult man and 115 g/L in an adult woman. Anaemia is not a disease itself but results from an underlying pathological process (see List 22.1). It can be classified according to the blood film. Red blood cells (RBCs) with a low mean cell volume (MCV) appear small (microcytic) and pale (hypochromic). Those with a high MCV appear large and round or oval-shaped (macrocytic). Alternatively, the RBCs may be normal in shape and size (normochromic, normocytic) but reduced in number.
Signs of a severe anaemia of any cause include pallor, tachycardia, wide pulse pressure, systolic ejection murmurs due to a compensatory rise in cardiac output and cardiac failure if myocardial reserve is reduced. There may be signs of the underlying cause.
PANCYTOPENIA
Signs
There may be clinical evidence of anaemia, leucopenia (reduced numbers of white blood cells [WBCs] resulting in susceptibility to infection) and thrombocytopenia (petechiae and bleeding)—a deficiency in all three bone marrow cell lines. If confirmed on a blood count, this condition is called pancytopenia.
Causes
• Aplastic anaemia: severe hypoplasia of the erythroid, myeloid and platelet precursor cell lines in the bone marrow, resulting in a bone marrow that is fatty and empty of cells. The causes are listed in List 22.1; 50% have no cause identified.
• Marrow infiltration by leukaemia, lymphoma, carcinoma, myeloma, myelofibrosis or granulomata.
• Other: acute leukaemia (subleukaemic phase), pernicious anaemia, hypersplenism, systemic lupus erythematosus, folate deficiency, paroxysmal nocturnal haemoglobinuria (PNH).
ACUTE LEUKAEMIA
Leukaemia is a neoplastic proliferation of one of the blood-forming cells. Acute leukaemia presents with marrow failure from progressive infiltration of the marrow with immature cells. The course is rapidly fatal without treatment. Acute leukaemia can be divided into two main types: acute lymphoblastic leukaemia and acute myeloid (monocytic) leukaemia.
General signs of acute leukaemia
Pallor (anaemia), fever (which usually indicates infection secondary to neutropenia) and petechiae (thrombocytopenia) are all due to bone marrow failure. Weight loss, muscle wasting (hypercatabolic state) and localised infections (e.g. of the tonsils or perirectal region, due to leucopenia) also occur.
Signs of infiltration of the haematopoietic system
These include: (1) bony tenderness, due to infiltration or infarction; (2) lymphadenopathy (slight to moderate, especially in acute lymphoblastic leukaemia); (3) splenomegaly (slight to moderate, occurs especially in acute lymphoblastic leukaemia; the spleen may be tender due to splenic infarction); and (4) hepatomegaly (slight to moderate).
Signs of infiltration of other areas
There may be: (1) tonsillar enlargement (especially in acute lymphoblastic leukaemia); (2) swelling or bleeding of the gums, especially in monocytic leukaemia; (3) pleural effusions; (4) nerve palsies, involving the spinal nerve roots or the cranial nerves; or (5) meningism due to infiltration of the meninges, especially in acute lymphoblastic leukaemia.
CHRONIC LEUKAEMIA
This is a haematological malignancy in which the leukaemic cell is at first well differentiated. These have a better prognosis untreated than acute leukaemia. There are two main types: chronic myeloid leukaemia and chronic lymphocytic leukaemia.
Signs of chronic myeloid leukaemia
Chronic myeloid leukaemia is one of the myeloproliferative disorders. There is an expanded granulocytic mass in the bone marrow, liver and spleen.
General signs may include pallor (anaemia due to bone marrow infiltration) and secondary gout (common).
Haematopoietic system signs include massive splenomegaly and moderate hepatomegaly. (Note: Lymphadenopathy is usually a sign of blast transformation.)
Signs of chronic lymphocytic leukaemia
There may be tiredness and pallor. Recurrent acute infections occur.
Haematopoietic system signs include marked or moderate lymphadenopathy and moderate hepatosplenomegaly.
Other abnormalities may include a Coombs’a test—positive haemolytic anaemia, herpes zoster skin infections and nodular infiltrates. Patients may note a hypersensitivity to insect bites before the diagnosis is made.
MYELOPROLIFERATIVE DISEASE
This is a group of disorders of the haematopoietic stem cell. These include polycythaemia rubra vera, primary myelofibrosis, chronic myeloid leukaemia and essential thrombocythaemia. Overlapping clinical and pathological features occur in these disorders. Therefore, patients may have signs of one or more of the conditions. Any of them may progress to acute myeloid leukaemia.
Polycythaemia
This is an elevated haemoglobin concentration and can result from an increased RBC mass or a decreased plasma volume. Polycythaemia rubra vera results from an autonomous increase in the RBC production. Patients with polycythaemia often have a striking ruddy, plethoric appearance. To examine a patient with suspected polycythaemia, assess for both the manifestations of polycythaemia rubra vera and other possible underlying causes of polycythaemia (see List 22.2).
Look at the patient and estimate the state of hydration (dehydration alone can cause an elevated haemoglobin due to haemoconcentration). Note whether there is a Cushingoid (page 367) or virilised (page 375) appearance. Cyanosis may be present because of an underlying condition such as cyanotic congenital heart disease or chronic lung disease. Look for nicotine staining (smoking). All these diseases can result in secondary polycythaemia.
Inspect the patient’s arms for scratch marks; post-bathing pruritus occurs in polycythaemia rubra vera, possibly due to basophil histamine release. Take the blood pressure: very rarely a phaeochromocytoma will cause secondary polycythaemia and hypertension.
Examine the patient’s eyes. Look for injected conjunctivae. Fundal hyperviscosity changes—including engorged, dilated retinal veins and haemorrhages—may be present. Inspect the tongue for central cyanosis.
Examine the cardiovascular system for signs of cyanotic congenital heart disease and the respiratory system for signs of chronic lung disease. Assess the abdomen carefully for splenomegaly, which occurs in 80% of cases of polycythaemia rubra vera but does not usually occur with the other causes of polycythaemia. There may be evidence of chronic liver disease or hepatocellular carcinoma, which may cause secondary polycythaemia. Palpate for the kidneys and perform a urinalysis. In women palpate the uterus. Polycystic kidney disease, hydronephrosis, renal carcinoma and uterine fibromas can all rarely cause secondary polycythaemia.
Inspect the patient’s legs for scratch marks, gouty tophi (see Figure 25.6 on page 331) and arthropathy, as well as for signs of peripheral vascular disease. In polycythaemia rubra vera, secondary gout occurs due to the increased cellular turnover resulting in hyperuricaemia. Peripheral vascular disease occurs in polycythaemia rubra vera because of thrombosis (as there is increased platelet adhesiveness and accelerated atherosclerosis) and slowed circulation due to hyperviscosity.
Look for cerebellar signs, which may be due to the presence of a cerebellar haemangioblastoma, a very rare cause of secondary polycythaemia. Examine the central nervous system for signs of a stroke due to thrombosis.
Primary myelofibrosis
This is a clonal haematopoietic stem cell disorder with fibrosis as a secondary phenomenon. Gradual replacement of the marrow by fibrosis and progressive splenomegaly characterise the disease.
• General signs include pallor (anaemia occurs in most patients eventually) and petechiae (in 20% of patients, due to thrombocytopenia).
• Haematopoietic system signs include splenomegaly (in almost all cases, and often to a massive degree—there may also be a splenic rub due to splenic infarction), hepatomegaly (occurs in 50% of patients and can be massive) and lymphadenopathy (very uncommon).
• Other signs are bony tenderness (uncommon) and gout (occurs in 5% of patients).
Essential thrombocythaemia
This is a sustained elevation of the platelet count above normal without any primary cause.
• General signs include spontaneous bleeding and thrombosis.
• Haematopoietic system signs include splenomegaly.
• Causes of thrombocytosis (platelet count more than 450 × 109/L) include: (1) following haemorrhage or surgery; (2) postsplenectomy; (3) iron deficiency; (4) chronic inflammatory disease; and (5) malignancy.
• Causes of thrombocytosis (platelet count more than 800 × 109/L) include: (1) myeloproliferative disease; and (2) secondary to recent splenectomy, malignancy or marked inflammation occasionally.
LYMPHOMA
Lymphoma is a malignant disease of the lymphoid system. There are two main clinicopathological types: Hodgkin’s disease (with the characteristic Reed-Sternbergb cell) and non-Hodgkin’s lymphoma. Signs of lymphoma depend on the stages of the disease. Hodgkin’s disease often presents in stage I or II, while non-Hodgkin’s lymphoma usually presents in stage III or IV.
Staging of lymphoma using Ann Arbor classification:
Stage I Disease confined to a single lymph node region or a single extra-lymphatic site (IE)
Stage II Disease confined to two or more lymph node regions on one side of the diaphragm
Stage III Disease confined to lymph nodes on both sides of the diaphragm with or without localised involvement of the spleen (IIIS), other extra-lymphatic organ or site (IIIE), or both (IIIES)
Stage IV Diffuse disease of one or more extra-lymphatic organs (with or without lymph node disease)
For any stage, a = no symptoms and b = fever, weight loss greater than 10% in 6 months or night sweats.
Signs of Hodgkin’s disease
1. Lymph node enlargement (see Figure 22.15): discrete, rubbery, painless, large and superficial nodes, often confined to one side and one lymph node group.
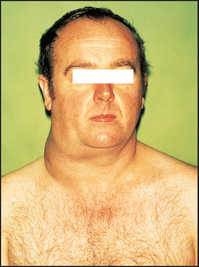
Figure 22.15 Cervical lymph node enlargement in a patient with lymphoma (From Mir MA. Atlas of clinical diagnosis, 2nd edn. Edinburgh: Saunders, 2003, with permission.)
2. Weight loss and fever with or without infection (reduced cell-mediated immunity) suggest a poor prognosis.
3. Splenomegaly and hepatomegaly. Splenomegaly does not always indicate extensive disease.
4. Organ infiltration occurs with late disease. Look especially for signs of: (a) lung disease, such as a pleural effusion; (b) bone pain or pathological fractures (rare); (c) spinal cord or nerve compression (rare); and (d) nodular skin infiltrates (rare).
Signs of non-Hodgkin’s lymphoma
1. Lymph node enlargement: often more than one site is involved and Waldeyer’s ring is more commonly affected.
2. Hepatosplenomegaly is common.
3. Systemic signs (e.g. weight loss or fever) are less common.
4. Signs of extra-nodal spread are more common.
5. The disease may sometimes arise at an extra-nodal site (e.g. the gastrointestinal tract).
MULTIPLE MYELOMA
This is a disseminated malignant disease of plasma cells.
General signs
There may be signs of anaemia (due to bone marrow infiltration or as a result of renal failure), purpura (due to bone marrow infiltration and thrombocytopenia) or infection (particularly pneumonia).
Bony tenderness and pathological fractures may be present. Weight loss may be a feature.
Skin changes include hypertrichosis, erythema annulare, yellow skin and secondary amyloid deposits.
There may be signs of spinal cord compression, or mental changes (due to hypercalcaemia).
Look for signs of chronic kidney disease (which may be due to tubular damage from filtered light chains, uric acid nephropathy, hypercalcaemia, urinary tract infection, secondary amyloidosis or plasma cell infiltration).
HAEMATOLOGICAL IMAGING
CT and ultrasound are often used to help determine the presence of splenomegaly or lymphadenopathy (see Figures 22.16 and 22.17).
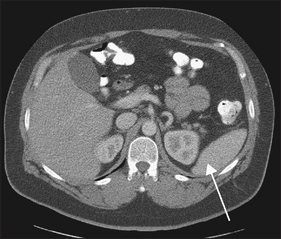
Figure 22.16 CT scan of the abdomen. A normal-sized spleen is visible (arrow). (From Halpert RD. Gastrointestinal imaging: the requisites, 3rd edn. Mosby, 2006.)