MULTIDISCIPLINARY CARE
The GOLD is the main evidence base and, together with systematic reviews and meta-analyses from the Cochrane Database, is used by the Thoracic Society of Australia and New Zealand and the Australian Lung Foundation to inform the development of guidelines for the management of COPD. The GOLD report lists a summary of recommended treatment at each stage (see Table 28-12).3 The Australian and New Zealand guidelines place the emphasis on the role of non-pharmacological interventions and self-management and are titled COPD-X after the key components: Confirm diagnosis, Optimise function, Prevent deterioration, Develop a self-management plan and manage eXacerbations.1 The primary goals of the guidelines are to:
• effect changes in clinical practice based on sound evidence
• shift the emphasis from a predominant reliance on pharmacological treatment of COPD to a range of interventions that include patient education, self-management of exacerbations and pulmonary rehabilitation.
TABLE 28-12 Therapy at each stage of COPD

Source: Adapted from global Initiative for Chronic Obstructive lung Disease. Global strategy for diagnosis, management, and prevention of COPD (p 54). Available at www.goldcopd.org/guidelineitem.asp?l1=2&l2=1&intId=989, accessed 22 January 2011.
Despite the prevalence and serious consequences of COPD, much can be done to improve quality of life and exercise capacity and reduce morbidity and mortality for people with COPD. The majority of people with COPD are treated as outpatients and are hospitalised only for acute exacerbations and complications such as respiratory failure, pneumonia and chronic heart failure.
Environmental or occupational irritants should be evaluated for their possible negative effect and ways to control or avoid them should be determined. For example, aerosol hairsprays and smoke-filled rooms should be avoided. The patient with COPD should have an influenza vaccination annually in the autumn and a pneumococcal vaccination is recommended for patients with a reduced respiratory reserve.1
Exacerbations of COPD should be treated as soon as possible, especially if the patient is in the severe stages of COPD. Often the best indication of the presence of a respiratory infection is the increasing quantity, viscosity or purulence of sputum. Some patients are given a 7–10-day supply of antibiotics and are instructed to begin taking them at the first signs of a change in sputum. The most common antibiotics given for outpatients are macrolides (e.g. azithromycin), doxycycline and cephalosporins (e.g. cefpodoxime). If the patient has failed prior antibiotic therapy or is hospitalised, common antibiotics are amoxicillin/clavulanate (Augmentin) or respiratory fluoroquinolone.3,42
Source: Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease. Available at www.goldcopd.com, accessed 22 January 2011.
EVIDENCE-BASED PRACTICE

Smoking cessation
Cessation of cigarette smoking in the early stages is the most significant factor in slowing the progression of the disease. After discontinuation of smoking, the accelerated decline in pulmonary function slows and pulmonary function usually improves. Normally individuals after the age of 35 years lose approximately 20–25 mL (as measured by FEV1) of lung function per year as measured by spirometry. Persons with COPD who continue to smoke lose approximately 50 mL per year. With the cessation of smoking the loss can fall to almost non-smoking levels at 35 mL per year. Thus the sooner the smoker stops, the less pulmonary function is lost and the sooner the symptoms decrease, particularly cough and sputum production. (Smoking cessation techniques are discussed in Ch 10.)
Drug therapy
Medications for COPD can reduce or abolish symptoms, increase the capacity to exercise, improve overall health and reduce the number and severity of exacerbations. Presently no drug modifies the decline of lung function with COPD. Bronchodilator drug therapy decreases airway resistance and dynamic hyperinflation of the lungs, which results in reducing the degree of breathlessness.1 Although patients with COPD do not respond to bronchodilator therapy as dramatically as those with asthma, a reduction in dyspnoea and an increase in FEV1 are usually achieved. The inhaled route of medication is preferred and given on an as-required or regular basis. Medications are given in a stepwise fashion, stepping up but usually not stepping down as in asthma, because in COPD there are probably continual symptoms (see Table 28-12).
Reducing risk factors for COPD
Source: Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Available at www.goldcopd.com, accessed 22 January 2011.
EVIDENCE-BASED PRACTICE
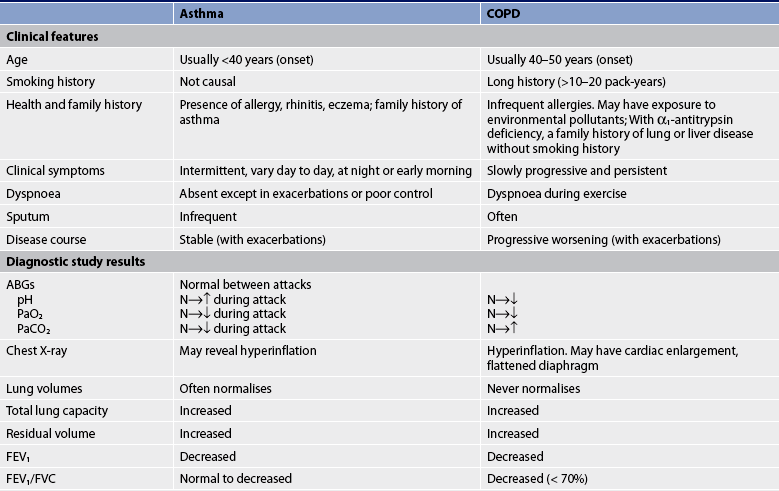
Bronchodilator medications commonly used are β2-adrenergic agonists, anticholinergic agents and methylxanthines (see Table 28-4). The choice of bronchodilator depends on the availability and the patient’s response. However, when the patient has mild COPD or intermittent symptoms, a short-acting bronchodilator is used as needed. Short-acting bronchodilators increase exercise tolerance. Salbutamol or ipratropium may be used as a single agent, but combining bronchodilators improves their effect and decreases the risk of adverse effects, compared to the use of a single agent. These two agents can be nebulised together or delivered by one MDI.
As symptoms persist or moderate stages of COPD develop, a long-acting bronchodilator is used in addition to a short-acting bronchodilator (see Table 28-4). Salmeterol is a widely used long-acting β2-adrenergic agonist and, unlike in drug therapy for asthma, it can be used in COPD as monotherapy. Formoterol is another long-acting β2-agonist. Tiotropium, a long-acting anticholinergic, can be used for daily therapy of bronchospasm and dyspnoea in COPD. Tiotropium also improves bronchodilation, results in less dyspnoea, improves quality of life and decreases the number of COPD exacerbations when compared to ipratropium.47
The use of long-acting theophylline in the treatment of COPD is controversial as it interacts with many drugs. Although it has some action as a mild bronchodilator in the patient with partial reversibility of airflow obstruction, its main value may be to improve contractility of the diaphragm and decrease diaphragmatic fatigue.
Inhaled corticosteroid therapy may be beneficial for moderate-to-severe COPD (stage III or IV). However, it does not appear to help patients with mild COPD. Inhaled corticosteroid therapy combined with long-acting β2-adrenergic agonists (e.g. fluticasone/salmeterol) is more effective than the single drug therapy. Some patients are on triple therapy with salmeterol/fluticasone (Advair) and tiotropium (Spiriva).48 Oral corticosteroids should not be used for long-term therapy in COPD but are effective in the short term for exacerbations.
Oxygen therapy
Oxygen (O2) therapy is frequently used in the treatment of COPD and other problems associated with hypoxaemia. Long-term O2 therapy improves survival, exercise capacity, cognitive performance and sleep in hypoxaemic patients.3 O2 is a colourless, odourless, tasteless gas that constitutes 20.95% of the atmosphere. Administering supplemental O2 raises the partial pressure of O2 (PO2) in inspired air. Used clinically it is considered a drug but, for reimbursement purposes, it is considered durable medical equipment.
Indications for use
Goals for O2 therapy are to reduce the work of breathing, maintain the PaO2 and/or reduce the workload on the heart, keeping the SaO2 >90% during rest, sleep and exertion or PaO2 >8 kPA (60 mmHg). O2 is usually administered to treat hypoxaemia caused by: (1) respiratory disorders such as COPD, cor pulmonale, pneumonia, atelectasis, lung cancer and pulmonary emboli; (2) cardiovascular disorders such as myocardial infarction, arrhythmias, angina pectoris and cardiogenic shock; and (3) central nervous system disorders such as overdose of opioids, head injury and disordered sleep (sleep apnoea). In COPD, level A evidence supports the use of long-term O2 therapy for patients who have SaO2 levels of 88% or less on air.1 There is limited evidence for the effectiveness of short-term use and since it is expensive, it must be prescribed with caution. In Australia and New Zealand, O2 therapy for COPD receives government funding as long as a respiratory physician is the prescriber.49
Methods of administration
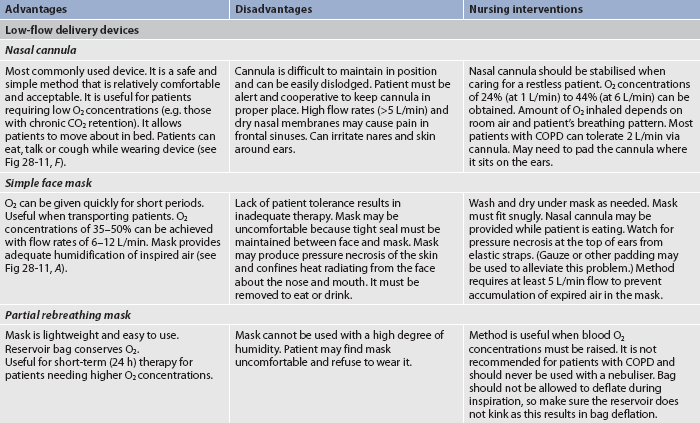
O2 administration aims to supply the patient with adequate O2 to maximise the O2-carrying ability of the blood. There are various methods of O2 administration (see Table 28-13 and Figs 28-11 to 28-14). The method selected depends on factors such as the fraction of inspired O2 (FiO2) and the mobility of the patient, the humidification required, patient cooperation, comfort, cost and available financial resources.
TABLE 28-13 Methods of oxygen administration
ABGs, arterial blood gases; COPD, chronic obstructive pulmonary disease.
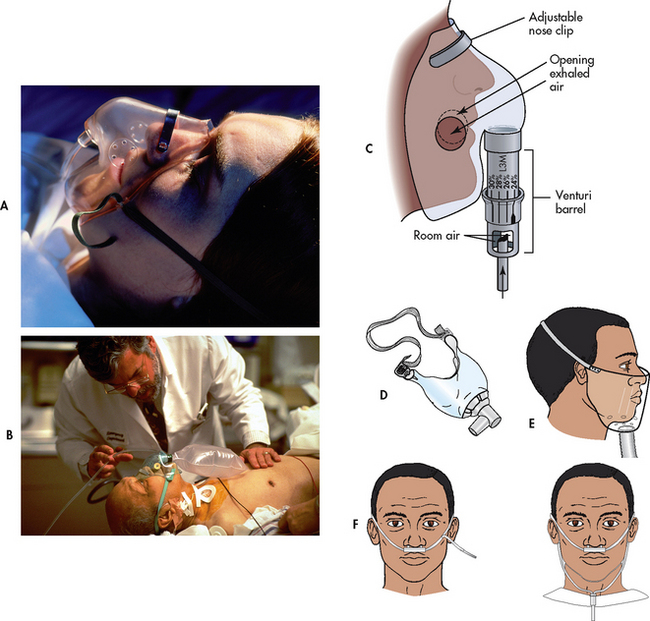
Figure 28-11 Methods of oxygen administration. A, Simple face mask. B, Plastic face mask with reservoir bag. C, Venturi mask. D, Tracheostomy mask. E, Face tent. F, Standard nasal cannulae.

O2 delivery systems are classified as low- or high-flow systems. Most methods of O2 administration use low-flow devices that deliver O2 in concentrations that vary with the person’s respiratory pattern. In contrast, the Venturi mask is a high-flow device that delivers fixed concentrations of O2 independent of the patient’s respiratory pattern. With the Venturi mask, O2 is delivered to a small jet (Venturi device) in the centre of a wide-based cone (see Fig 28-11, C). Air is entrained (pulled through) openings in the cone as O2 flows through the small jet. The mask has large vents through which exhaled air can escape. The degree of restriction or the narrowness of the jet determines the amount of entrainment and dilution of pure O2 with room air and thus the concentration of O2. Nasal cannulae can be used short term. Mechanical ventilators are another example of a high-flow O2 delivery system. Because room air is mixed with O2 in low-flow systems, the percentage of O2 delivered to the patient is not as precise as with high-flow systems.
Humidification and nebulisers
O2 obtained from cylinders or wall systems is dry. Dry O2 has an irritating effect on mucous membranes and dries secretions. Therefore it is important that O2 be humidified when administered, either by humidification or nebulisation when flow rates are higher than 4 L/min. A common device used for humidification when the patient has a catheter, cannula or low-flow mask is a bubble-through humidifier. This is a small plastic jar filled with sterile distilled water that is attached to the O2 source by means of a flow meter. O2 passes into the jar, bubbles through the water and then goes through tubing to the patient’s catheter, cannula or mask. The purpose of the bubble-through humidifier is to restore the humidity conditions of room air. However, the need for bubble-through humidifiers at flow rates between 1 and 4 L/min is controversial when humidity in the environment is adequate. The amount of flow at this level depends on patient comfort.
Another means of administering humidified O2 is via a nebuliser. It delivers particulate water mist (aerosols) with nearly 100% humidity. The humidity can be raised by heating the water, which increases the ability of the gas to hold moisture. Heated (37°C) and humidified (100%) gas is required when the upper airway is bypassed in acute care. However, patients with established tracheotomies do not always require 100% humidity. When nebulisers are used, large-size tubing should be employed to connect the device to a face mask or T-bar. If small-size tubing is used, condensation can occlude the flow of O2.
Vapotherm can deliver high flows (15–20 L/min) of warm humidified air (either sterile air or O2) to the patient through a nasal or transtracheal cannula using technology to warm and saturate the gas stream. Preliminary findings in COPD patients in pulmonary rehabilitation suggest increases in exercise tolerance with Vapotherm high-flow therapy.35
Complications
Combustion
O2 supports combustion and increases the rate of burning. This is why it is important that smoking be prohibited in the area in which O2 is being used. A ‘no smoking’ sign should be prominently displayed on the patient’s door. The patient should also be cautioned against smoking cigarettes when O2 prongs or cannulae are in place.
Carbon dioxide narcosis
The two chemoreceptors in the respiratory centre that control the drive to breathe are CO2 and O2. Normally, CO2 accumulation is the major stimulant of the respiratory centre. Over time COPD patients develop a tolerance for high CO2 levels (the respiratory centre loses its sensitivity to the elevated CO2 levels). Theoretically, for these individuals the ‘O2 drive’ to breathe is hypoxaemia. Thus there has been concern regarding the dangers of administering O2 to COPD patients and wiping out their drive to breathe. This has been a pervasive myth but is not regarded as a serious threat. In fact, not providing adequate O2 to these patients is much more detrimental. Although O2 administration should be titrated to the lowest effective dose, many patients who have end-stage chronic obstructive disease require high-flow rates and higher concentrations for survival. They may, in fact, exhibit higher than normal levels of CO2 in their blood, but this is of little concern. What is important is careful, ongoing assessment when providing O2 to these patients.
It is critical to start O2 at low-flow rates until ABGs can be obtained. ABGs are used as a guide to determine what FiO2 level is sufficient and can be tolerated. The patient’s mental status and vital signs should be assessed before starting O2 therapy and frequently thereafter.
Oxygen toxicity
Pulmonary oxygen toxicity may result from prolonged exposure to high levels of O2 (PaO2). The development of O2 toxicity is determined by patient tolerance, exposure time and effective dose. High concentrations of O2 damage alveolar–capillary membranes, inactivate pulmonary surfactant, cause interstitial and alveolar oedema and decrease compliance. These individuals develop acute respiratory distress syndrome (ARDS; see Ch 67).3
Early manifestations of O2 toxicity are reduced vital capacity, cough, substernal chest pain, nausea and vomiting, paraesthesia, nasal stuffiness, sore throat and malaise. The later stages of O2 toxicity affect the alveolar–capillary gas exchange unit, causing oedema and production of copious sputum. The end stage of O2 toxicity is progressive fibrosis of the lungs. Prevention of O2 toxicity is important for the patient who is receiving O2. The amount of O2 administered should be just enough to maintain the PaO2 within a normal or acceptable range for the patient. ABGs should be monitored frequently to evaluate the effectiveness of therapy and to guide the tapering of supplemental O2. A safe limit of O2 concentrations has not yet been established. All levels above 50% and used for longer than 24 hours should be considered potentially toxic. Levels of 40% and below may be regarded as relatively non-toxic and may not result in the development of significant O2 toxicity if the exposure period is short.
Absorption atelectasis
Normally nitrogen, which constitutes 79% of the air that is breathed, is not absorbed into the bloodstream. This prevents alveolar collapse. When high concentrations of O2 are given, nitrogen is washed out of the alveoli and replaced with O2. If airway obstruction occurs, the O2 is absorbed into the bloodstream and the alveoli collapse. This process is called absorption atelectasis.
Infection
Infection can be a major hazard of O2 administration. Heated nebulisers present the highest risk. The constant use of humidity supports bacterial growth, with the most common infecting organism being Pseudomonas aeruginosa. Disposable equipment that operates as a closed system should be used. Each hospital has a policy stating the required frequency of equipment changes based on the type of equipment used at that particular institution.
Chronic oxygen therapy at home
Improved prognosis and enhanced quality of life have been noted in patients with COPD who receive long-term O2 therapy to treat hypoxaemia.1,49 The improved prognosis results from preventing progression of the disease and subsequent cor pulmonale. The benefits of long-term continuous O2 therapy include improved neuropsychological function, increased exercise tolerance, decreased haematocrit and reduced pulmonary hypertension. It also improves sleep, may reduce nocturnal arrhythmias and may extend life.35
Some patients believe they will become ‘addicted’ to O2 and are very reluctant to use it. They need to be educated that it is not ‘addictive’ and that it needs to be used for the prescribed times during the day because of the positive effects on the heart, lungs and brain. The need for long-term O2 therapy should be evaluated when the patient’s condition has stabilised. The goal of O2 therapy is to maintain SaO2 >90% during rest, sleep and exertion.
Short-term home O2 therapy (1–30 days) may be indicated for the patient in whom hypoxaemia persists after discharge from the hospital. For example, the patient with underlying COPD who develops a serious respiratory tract infection may continue to have clearing of the infection after completion of antibiotic therapy and discharge from the hospital. This patient may demonstrate continued hypoxaemia for 4–6 weeks after discharge. It is important to measure the patient’s oxygenation status by pulse oximetry 2–3 months after an acute episode to determine if the O2 is still warranted.
Desaturation only during exercise or sleep suggests consideration of O2 therapy specifically under those conditions. Patients may receive O2 only during exercise or sleep, or at both times. The need for O2 during these periods should be evaluated with oximetry. (Pulse oximetry is discussed in Ch 25.) Sleep disordered breathing may be seen in some of these patients and they will require a full sleep study.
Periodic re-evaluations are necessary for the patient who is using chronic supplemental O2. The recommendation is generally that the patient should be re-evaluated 1–2 months after commencing O2 therapy and then annually or more frequently depending on the clinical condition.49
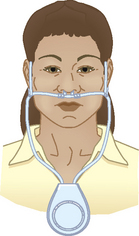
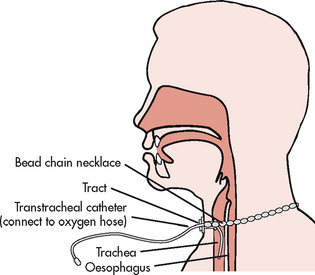
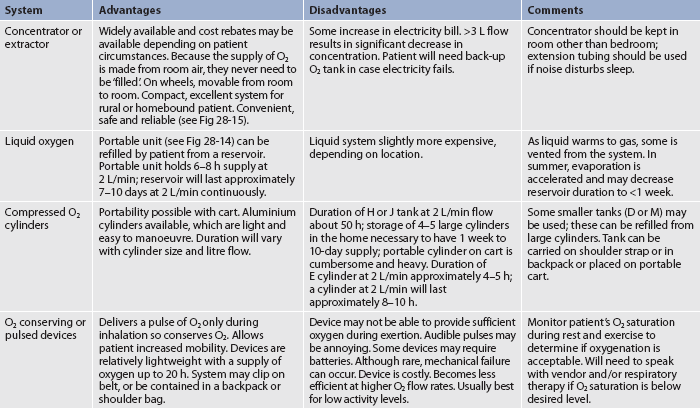
Nasal cannulae, either regular or the O2-conserving type (see Table 28-13 and Figs 28-11 and 28-14), are usually used to deliver O2 from a central source in the home. The source may be a liquid O2 storage system, compressed O2 in tanks or an O2 concentrator or extractor, depending on the patient’s home environment and activity level (see Table 28-14). The concentrator is the least expensive option. The patient can use extension tubing (up to 15 m) without adversely affecting the O2 flow delivery to increase mobility in the home, provided that the flow meter is the back pressure-compensated type. Small portable systems, such as liquid O2 or, more commonly, an O2 concentrator, may be provided for the patient who remains active outside the home (see Fig 28-15).

Reservoir cannulae operate on the principle of storing O2 in a small reservoir during exhalation. The O2 is then delivered to the patient during the subsequent inhalation, similar to a bolus effect. The reservoir cannulae can reduce flow requirements by approximately 50%. Another type that fits onto the frame of spectacles is also available and is less visible on the face.
It is important that patients who require home O2 systems are taught how to use the system, how to care for it and how to recognise when the supply is running low and needs to be reordered. A patient and family teaching guide for the use of O2 at home is presented in Box 28-8. Care must also be taken to ensure that the patient or carer notifies their electricity provider if the system is dependent on electricity to operate.
PATIENT & FAMILY TEACHING GUIDE
Decreasing risk of fire injuries
• Post ‘no smoking’ warning signs in home where they can be seen
• Do not use electric razors, portable radios, open flames, wool blankets or mineral oils in the area where oxygen is in use
A good resource is: The Australian Lung Foundation. Home oxygen treatment. Available at www.lungfoundation.com.au/lung-information/patient-educational-material/getting-started-on-home-oxygen, accessed 22 January 2011.
The patient who uses home O2 should be encouraged to remain active and travel normally. If travel is by car, arrangements can be made for O2 to be available at the destination point. O2 supply companies can often assist in these arrangements. If a patient wishes to travel by bus, train or aeroplane, the patient should inform the appropriate people when reservations are made that O2 will be needed for travel. If there is a potential for the patient to become hypoxic, oxygen needs for flying can be determined via a hypoxia inhalation test or through a mathematical formula. Portable oxygen concentrators are a ready source of renewable O2 and can be available by recharging at home or in a DC (e.g. car) power supply. These systems are widely approved by airlines for in-flight use. The patient should contact the specific airline to determine the particular accommodations and policies for in-flight O2.
Surgical therapy for COPD
Three different surgical procedures have been used in severe COPD, although none provides any surgical advantage.1
Lung volume reduction surgery (LVRS) reduces lung volume and improves lung and chest wall mechanics.50 The goal is to reduce the size of the hyperinflated emphysematous lungs by removing much of the diseased tissue. This can decrease airway obstruction and increase room for the remaining healthy lung tissue to function more effectively. There is evidence of improvements in exercise capacity among those having LVRS compared with medical therapy, but overall the procedure is associated with increased mortality and negligible gain.51
Bullectomy is used for carefully selected patients with emphysematous COPD who have large bullae (>5 cm).1 The bullae are usually resected via thoracoscope. The procedure has resulted in improved lung function and reduction in dyspnoea,3 and it has the greatest success in patients who have very large cysts that are preventing expansion of adjacent apparently normal lung.52
Lung transplantation is appropriate for some carefully selected patients with advanced COPD.53 Although single-lung transplantation is the most commonly used technique, bilateral transplantation can be performed and evidence suggests that this may be the future preferred technique due to the improved longer term outcomes. However, rejection and the effects of immunosuppressive therapy remain obstacles for these patients. (Lung transplantation is discussed in Ch 27.)
Breathing retraining
The patient with COPD develops an increased respiratory rate with a prolonged expiration to compensate for obstruction to airflow resulting in dyspnoea. In addition, the accessory muscles of breathing in the neck and upper part of the chest are used excessively to promote chest wall movement. These muscles are not designed for long-term use and as a result the patient experiences increased fatigue. Breathing exercises can assist the patient during rest and activity (e.g. lifting, walking, stair climbing). The main types of breathing exercises are: (1) pursed-lip breathing; and (2) diaphragmatic breathing.
The purpose of using pursed-lip breathing (PLB) is to prolong exhalation and thereby prevent bronchiolar collapse and air trapping. PLB is simple and easy to teach and learn and gives the patient more control over breathing, especially during exercise and periods of dyspnoea (see Box 28-9). Patients should be taught to use ‘just enough’ positive pressure with the pursed lips, because excessive resistance may increase the work of breathing. The issue of whether and how extensively pursed-lip breathing affects dyspnoea is still questioned, but current evidence seems to support its use to improve the breathing of patients with COPD.54–56
BOX 28-9 Guidelines for pursed-lip breathing
PATIENT & CAREGIVER TEACHING GUIDE
Teach the patient how to do pursed-lip breathing using the following guidelines.
1. Use PLB before, during and after any activity causing you to be short of breath.
2. Inhale slowly and deeply through the nose.
3. Exhale slowly through pursed lips, almost as if whistling.
4. Relax your facial muscles without puffing your cheeks—like whistling—while you are exhaling.
5. Make breathing out (exhalation) three times as long as breathing in (inhalation).
6. The following activities can help you get the ‘feel’ of PLB:
7. Practice 8–10 repetitions of PLB three or four times a day.
Source: Adapted from Guidelines for the diagnosis and management of asthma. Expert Panel Report 3. National Asthma Education and Prevention Program, National Heart, Lung, and Blood Institute; 2007.
Diaphragmatic (abdominal) breathing focuses on using the diaphragm instead of the accessory muscles of the chest to: (1) achieve maximum inhalation; and (2) slow the respiratory rate. However, there is some controversy regarding its effectiveness for COPD patients.57 For patients with severe COPD, diaphragmatic breathing may result in hyperinflation because of increased fatigue and dyspnoea and abdominal paradoxical breathing (the inward movement of the abdomen and the outward movement of the upper chest during inspiration) rather than with normal chest wall motion (the outward movement of the abdomen and upper chest simultaneously during inspiration).
Pursed-lip breathing slows the respiratory rate and is much easier to learn than diaphragmatic breathing. In the setting of extreme acute dyspnoea when the patient is hospitalised for infection or heart failure, it is important to focus on helping the patient slow the respiratory rate by using the principles of pursed-lip breathing.
Airway clearance
Many patients with COPD and conditions such as cystic fibrosis and bronchiectasis retain secretions and require help to adequately clear their airways. Airway clearance techniques (ACTs) loosen mucus and secretions so that they can be cleared by coughing. A variety of techniques can be used to achieve airway clearance. No one technique is really better than another and it is largely a matter of patient preference.58,59 Respiratory therapists (RTs), physiotherapists and nurses are involved in performing these techniques. ACTs are often used with other treatments. Typically, the patient will receive bronchodilator therapy via an inhaled device (e.g. nebulisation) before ACT, then an ACT, followed by effective coughing (e.g. huff coughing).
Effective coughing
Many patients with COPD have developed ineffective coughing patterns that do not adequately clear their airways of sputum. In addition, they fear they may develop spastic coughing, resulting in increased dyspnoea. Guidelines for effective coughing are presented in Box 28-10. Huff coughing is an effective technique that the patient can be taught easily. The main goals of effective coughing are to conserve energy, reduce fatigue and facilitate removal of secretions. This technique clears secretions with less change in pleural pressure and less likelihood of bronchial collapse. Before coughing the nurse should ensure that the patient is breathing deeply from the diaphragm. The nurse should place the patient’s hands on the lower, lateral chest wall and then ask the patient to breathe deeply through the nose; the nurse should feel the patient’s hands move outwards, which represents a breath from the diaphragm.
BOX 28-10 Guidelines for effective coughing
PATIENT & FAMILY TEACHING GUIDE
1. Patient assumes a sitting position with head slightly flexed, shoulders relaxed, knees flexed and forearms supported by pillow and, if possible, with feet on the floor.
2. Patient then drops head and bends forwards while using slow, pursed-lip breathing to exhale.
3. Sitting up again, patient uses diaphragmatic breathing to inhale slowly and deeply.
4. Patient repeats steps 2 and 3 three to four times to facilitate mobilisation of secretions.
5. Before initiating a cough, patient should take a deep abdominal breath, bend slightly forwards and then huff cough (cough three to four times on exhalation). Patient may need to support or splint thorax or abdomen to achieve a maximum cough.
Chest physiotherapy
Chest physiotherapy is indicated for the patient with excessive bronchial secretions who has difficulty clearing them (e.g. because of cystic fibrosis, bronchiectasis). It consists of postural drainage, percussion and vibration (see Box 28-11). Postural drainage uses the principle of gravity to assist in bronchial drainage. Percussion and vibration are manual or mechanical techniques used to augment postural drainage and are used after the patient has assumed a postural drainage position to assist in loosening the mobilised secretions (see Fig 28-16). Postural drainage, percussion and vibration may assist in bringing secretions into larger, more central airways. Effective coughing is then necessary to help raise these secretions.
BOX 28-11 Steps in chest physiotherapy
1. Perform procedure 1 h before meals or 1–3 h after meals.
2. Administer bronchodilator (if nebulised or metered dose inhaler is ordered) approximately 15 min before procedure.
3. Collect equipment such as tissues, emesis basin, paper bag and pillows.
4. Help patient to assume correct position for postural drainage based on findings from X-ray, auscultation, palpation and percussion of chest. If patient’s tolerance is limited, start with the lower lung fields. Position should be maintained for 5–15 min to mobilise secretions via gravity.
5. Observe patient during treatment to assess tolerance. Particularly observe breathing and colour changes, especially duskiness in face.
6. Have patient take several deep abdominal breaths.
7. Percuss appropriate area for 1–2 min keeping the patient’s face in full view.
8. Vibrate the same area while the patient exhales four to five deep breaths.*
9. Assist patient to cough while assuming same position. Splinting with towel or hands may be necessary to aid in effective coughing. Patient may have to assume sitting position to generate enough airflow to expel secretions. (Coughing productively may be a long waiting process that may occur 30 min after procedure.) Suction may be necessary if coughing is not effective.
10. Repeat percussion, vibration and coughing until patient no longer expectorates mucus.
11. Repeat same procedure in all necessary positions.
12. After procedure, help patient to assume a comfortable position, assist with oral hygiene and discard used tissues.
13. Monitor for hypoxaemia if patient has any respiratory difficulty during the procedure.
14. Evaluate and chart effectiveness of treatment by amount of sputum produced and the results of auscultation. Also chart patient tolerance.
*If using an electronic vibrator, use for periods of 5–20 min in each position according to the patient’s tolerance.
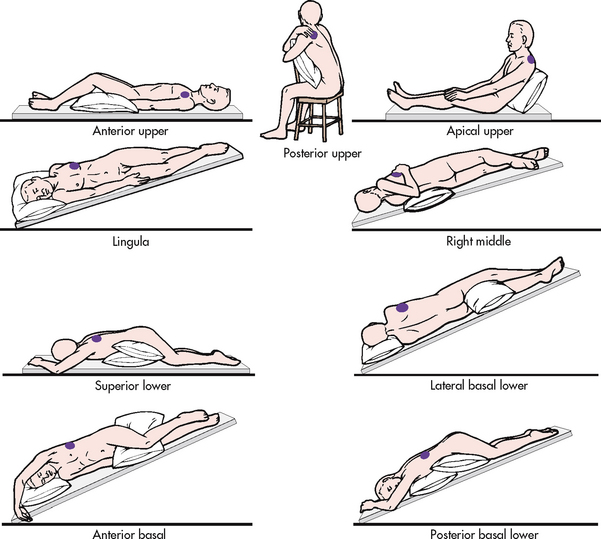
Figure 28-16 Representative positions for postural drainage. Shaded areas in each drawing indicate the segment of the lung in which drainage is promoted.
Chest physiotherapy should be performed by an individual who has been properly trained. Contraindications for chest physiotherapy include situations in which there is head, neck, chest or back instability and/or injuries; anatomical deformities; severe spasticity; mental limitations; or the patient cannot tolerate the position for other reasons. Complications associated with improperly performed chest physiotherapy include fractured ribs, bruising, hypoxaemia and discomfort to the patient. Chest physiotherapy may not be beneficial and may be stressful for some patients.
Postural drainage
Postural drainage involves the use of positioning techniques that drain secretions from specific segments of the lungs and bronchi into the trachea. The lungs are divided into five lobes, with three on the right side and two on the left side. There are 18 segments in the lungs, which can be drained by 18 positions. Figure 28-16 shows the modified postural drainage positions most often used in clinical practice.
The purpose of various positions in postural drainage is to drain each segment towards the larger airways. The postural drainage positions used depend on the areas of involved lung, determined by patient assessment (including the patient’s preference), chest X-rays and chest auscultation. For example, some patients with left lower lobe involvement will require postural drainage of only the affected region, whereas patients with cystic fibrosis may require postural drainage of all segments.
Aerosolised bronchodilators and hydration therapy are frequently administered before postural drainage. The chosen postural drainage position is maintained for 5–15 minutes. The degree of slope can be obtained with pillows, blocks, books or a tilt board. The frequency and choice of postural drainage positions depend on the location of retained secretions and patient tolerance to dependent positions. A common order is two to four times a day. In acute situations, postural drainage may be performed as frequently as every 1–2 hours. The procedure should be planned to occur and be completed at least 1 hour before meals or 3 hours after meals.
There are beds available that can rotate and percuss in various postural drainage positions, and these are quite effective. Some positions for postural drainage (e.g. Trendelenburg) should not be performed on the patient with chest trauma, haemoptysis, heart disease or head injury and in other situations where the patient’s condition is not stable.
Percussion
Percussion is performed in the appropriate postural drainage position with the hands in a cup-like position (see Fig 28-17). The hands are cupped and the fingers and thumbs are closed. The cupped hand should create an air pocket between the patient’s chest and the hand. Both hands are cupped and used in an alternating rhythmic fashion. Percussion is accomplished with flexion and extension of the wrists. If it is performed correctly, a hollow sound should be heard. The air-cushion impact facilitates the movement of thick mucus. A thin towel should be placed over the area to be percussed or the patient may choose to wear a T-shirt or hospital gown.
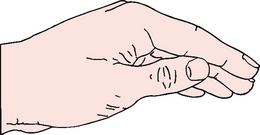
Vibration
Vibration is accomplished by tensing the hand and arm muscles repeatedly and pressing mildly with the flat of the hand on the affected area while the patient slowly exhales a deep breath. The vibrations facilitate movement of secretions to larger airways. Mild vibration is tolerated better than percussion and can be used in situations where percussion may be contraindicated. Commercial vibrators are available for hospital and home use.
Airway clearance devices
Various airway clearance devices are available to mobilise secretions. They are easier to tolerate than chest physiotherapy and take less than half the time of conventional chest physiotherapy sessions.40,41 These devices include the Flutter mucus clearance device, the TheraPEP therapy system, the ThAIRaphy vest and Acapella.
Flutter mucus clearance device
The Flutter mucus clearance device is a hand-held device that provides positive expiratory pressure (PEP) treatment for patients with mucus-producing conditions (see Fig 28-18). The Flutter has a mouthpiece, a high-density stainless steel ball and a cone that holds the ball. When the patient exhales through the Flutter, the steel ball moves, which causes vibrations in the lungs and loosens mucus. It helps move mucus up through the airways to the mouth where the mucus can be expectorated. Although the Flutter valve is mostly used in patients with cystic fibrosis, it has been used effectively in patients with COPD and bronchiectasis who have excessive secretions.
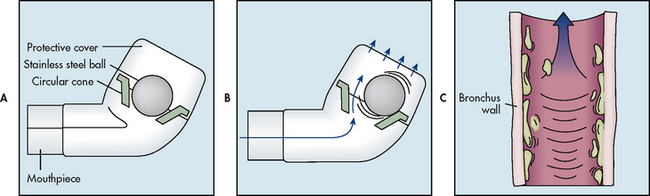
Figure 28-18 The Flutter mucus clearance device is a small hand-held device that provides positive expiratory pressure therapy. It is used to facilitate the removal of mucus from the lungs. A, It consists of a hard plastic mouthpiece, a plastic perforated cover and a high-density stainless steel ball resting in a circular cone. B, The Flutter effect occurs during expiration. Before exhalation, the steel ball blocks the conical canal of the Flutter. During exhalation, the position of the steel ball is the result of an equilibrium between the pressure of the exhaled air, the force of gravity on the ball and the angle of the cone where the contact with the ball occurs. As the steel ball rolls and moves up and down, it creates an opening and closing cycle, which repeats itself many times throughout each exhalation. The net result is that vibrations occur in the airways, resulting in the ‘fluttering’ sensation. C, These vibrations loosen mucus from the airway walls and facilitate their movement up the airways.
TheraPEP therapy system
The TheraPEP therapy system also provides sustained PEP and simultaneously delivers aerosols so the patient can inhale and exhale through it. The device comprises a mouthpiece attached to tubing connected to a small cylindrical resistor and a pressure indicator. The pressure indicator provides visual reinforcement about the pressure the patient needs to hold in an exhalation to receive the PEP. The device is available in Australia and New Zealand and a description and photo are available on the TheraPEP therapy system website (see Resources on pp 727–728).
ThAIRaphy vest
The ThAIRaphy vest delivers high-frequency chest compression via an inflatable vest with hoses connected to a high-frequency pulse generator. The pulse generator delivers air to the vest, which vibrates the chest. The high-frequency airwaves clear all lobes of the lungs. The vest has been found to be more effective than conventional chest physiotherapy in clearing mucus and it can be used without the aid of another person. The unit is light, quiet and portable.
Acapella
Acapella is a small hand-held device that combines the benefits of PEP therapy and airway vibrations of the Flutter valve to mobilise pulmonary secretions (see Fig 28-19). It works by using oscillating vibrations that travel to the lung, shaking free mucus plugs that the patient can then cough up. It can be used in virtually any setting as the patient is free to sit, stand or recline. It improves clearance of secretions, is easier to tolerate and quicker than conventional chest physiotherapy and facilitates opening of the airways.

Nutritional therapy
Approximately one-third of COPD patients are underweight, experiencing loss of muscle mass and cachexia, especially in the severe stages of the disease.60 Weight loss is a predictor of poor prognosis and increased frequency of COPD exacerbations.61 Weight gain after nutritional support can decrease the mortality risk. The cause of weight loss is not entirely known. Eating becomes an effort because of dyspnoea, which occurs as a result of the energy expended to chew, the reduction of airflow while swallowing and O2 desaturation. Therefore weight loss and muscle wasting are likely. Weight loss is also thought to occur because systemic inflammation causes the metabolism to increase. This hypermetabolism could explain why some individuals with COPD lose weight despite having an adequate nutritional intake.
Patients with COPD should try to keep their BMI between 21 and 25 kg/m2. Being either overweight or underweight can be a problem with COPD. The dietician can serve as a good resource to determine what supplements are the best for patients.
To decrease dyspnoea and conserve energy, patients should rest for at least 30 minutes before eating, use the bronchodilator before meals and select foods that can be prepared in advance. They should eat five to six small, frequent meals to avoid feelings of bloating and early satiety. A full stomach puts pressure on the diaphragm and decreases lung movement. Liquid, blended or commercial diets may be helpful. Foods that require a great deal of chewing should be avoided or served in another manner (e.g. grated, pureed). Cold foods may give less of a sense of fullness than hot foods. Exercises and treatments should be avoided for at least 1 hour before and after eating. The exertion involved in the preparation and eating of food is often fatiguing. The use of frozen foods and a microwave oven may help conserve patient energy in food preparation.
Many patients with COPD feel bloated and experience early satiety when eating. This can be attributed to swallowing air while eating, the side effects of medications (especially corticosteroids and theophylline) and the abnormal position of the diaphragm relative to the stomach in association with hyperinflation. Intestinal gas-forming foods, such as cabbage, brussel sprouts and beans, should be avoided.
Underweight patients with emphysematous COPD have a greater than normal nutritional requirement for protein and kilojoules. A high-energy, high-protein diet is recommended and can be divided into five or six small meals a day. High-protein, high-energy nutritional supplements may be offered between meals. Ice-cream added to these supplements can help increase kilojoules. Drinking low-fat milk rather than whole milk may cause less mucus production. (Nutritional supplements are discussed in Ch 39.) Non-protein energy should be divided evenly between fat and carbohydrate while not overfeeding the patient.62 In most cases just getting the patient to eat adequate amounts of any foods can be difficult. If the patient has O2 prescribed, use of supplemental O2 by nasal prongs (cannulae) while eating may also be beneficial because eating expends energy. Fluid intake should be at least 3 L per day unless contraindicated for other medical conditions, such as heart failure. Fluids should be taken between meals (rather than with them) to prevent excess stomach distension and to decrease pressure on the diaphragm. Sodium restriction may be indicated if there is accompanying heart failure.
In contrast, some patients with COPD are obese, which also causes dyspnoea. These patients may have increased appetite if on oral corticosteroids and may have little mobility. Advice about controlling food portions and increasing their exercise may be needed.