Chapter 30 NURSING MANAGEMENT: haematological problems
1. Describe the general clinical manifestations and complications of anaemia.
2. Outline the aetiologies, clinical manifestations, diagnostic findings, and nursing and collaborative management of iron-deficiency, megaloblastic and aplastic anaemias and anaemia of chronic disease.
3. Explain the nursing management of anaemia secondary to blood loss.
4. Outline the pathophysiology, clinical manifestations, and nursing and collaborative management of anaemia caused by increased erythrocyte destruction, including sickle cell disease and acquired haemolytic anaemias.
5. Outline the pathophysiology, and nursing and collaborative management of polycythaemia.
6. Outline the pathophysiology, clinical manifestations, and nursing and collaborative management of various types of thrombocytopenia.
7. Describe the types, clinical manifestations, diagnostic findings, and nursing and collaborative management of haemophilia and von Willebrand’s disease.
8. Explain the pathophysiology, diagnostic findings, and nursing and collaborative management of disseminated intravascular coagulation.
9. Outline the aetiology, clinical manifestations, and nursing and collaborative management of neutropenia.
10. Outline the pathophysiology, clinical manifestations, and nursing and collaborative management of myelodysplastic syndrome.
11. Compare and contrast the major types of leukaemia with regard to distinguishing clinical and laboratory findings.
12. Explain the nursing and collaborative management of acute and chronic leukaemias.
13. Compare Hodgkin’s lymphoma and non-Hodgkin’s lymphomas in terms of clinical manifestations, staging, and nursing and collaborative management.
14. Describe the pathophysiology, clinical manifestations, and nursing and collaborative management of multiple myeloma.
15. Describe the spleen disorders and related multidisciplinary care.
16. Outline the nursing management of the patient receiving transfusions of blood and blood components.
Anaemia
DEFINITION AND CLASSIFICATION
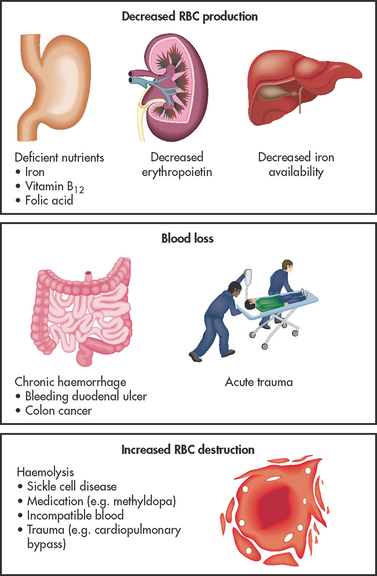
Anaemia is a deficiency in the number of erythrocytes (red blood cells [RBCs]), the quantity of haemoglobin and/or the volume of packed RBCs (haematocrit). It is a prevalent condition with many diverse causes, such as blood loss, impaired production of erythrocytes or increased destruction of erythrocytes (see Fig 30-1). Because RBCs transport oxygen, erythrocyte disorders can lead to tissue hypoxia. This hypoxia accounts for many of the signs and symptoms of anaemia. Anaemia is not a specific disease; it is a manifestation of a pathological process.
Anaemia is identified by a thorough history and physical examination, and then classified by laboratory review of the full blood count (FBC), reticulocyte count and peripheral blood smear. Once anaemia is identified, further investigation is done to determine its cause.1,2
Anaemia can result from primary haematological problems or it can develop as a secondary consequence of defects in other body systems. The various types of anaemia can be grouped according to either a morphological (cellular characteristic) classification or an aetiological (underlying cause) classification. Morphological classification is based on descriptive, objective laboratory information about erythrocyte size and colour. (The terms used in this classification system are explained in Ch 29.) Aetiological classification is related to the clinical conditions causing the anaemia, such as decreased erythrocyte production, blood loss or increased erythrocyte destruction (see Box 30-1). Although the morphological system is the most accurate means of classifying anaemia, it is easier to discuss patient care by focusing on the aetiology of the anaemia. Table 30-1 relates morphological classifications to various aetiologies.
BOX 30-1 Aetiological classification of anaemia
Decreased erythrocyte production
Increased erythrocyte destruction*
*Haemolytic anaemia.
TABLE 30-1 The relationship of morphological classification and aetiologies of anaemia
| Morphology | Aetiology |
|---|---|
| Normocytic, normochromic (normal size and colour) | Acute blood loss, haemolysis, chronic renal disease, chronic disease, cancers, sideroblastic anaemia, refractory anaemia, diseases of endocrine dysfunction, aplastic anaemia, sickle cell anaemia, pregnancy |
| Macrocytic, normochromic (large size, normal colour) | Vitamin B12 deficiency, folic acid deficiency, liver disease (including effects of alcohol abuse), postsplenectomy |
| Microcytic, hypochromic (small size, pale colour) | Iron-deficiency anaemia, thalassaemia, lead poisoning |
HEALTH DISPARITIES
• Sickle cell disease has a high incidence among African Americans.
• Thalassaemia has a high incidence among African Americans and people of Mediterranean origin.
• Tay-Sachs disease has the highest incidence in families of Eastern European Jewish origin, especially the Ashkenazi Jews.
• Pernicious anaemia has a high incidence among Scandinavians and African Americans.
CLINICAL MANIFESTATIONS
The majority of the clinical manifestations of anaemia are caused by the body’s response to tissue hypoxia. Specific manifestations can vary depending on the rate at which it has evolved, the severity of the anaemia and the presence of coexisting disease. Haemoglobin (Hb) levels are often used to determine the severity of anaemia. Mild states of anaemia (Hb 100–140 g/L) may exist without causing symptoms. If symptoms develop, it is because the patient has an underlying disease or is experiencing a compensatory response to activities such as heavy exercise. Symptoms include palpitations, dyspnoea and diaphoresis. In cases of moderate anaemia (Hb 60–100 g/L), the cardiopulmonary symptoms are increased and the patient may experience them while resting as well as with activity. The patient with severe anaemia (Hb <60 g/L) has many clinical manifestations involving multiple body systems, with fatigue and tiredness being the primary complaints (see Table 30-2).
TABLE 30-2 Clinical manifestations of anaemia
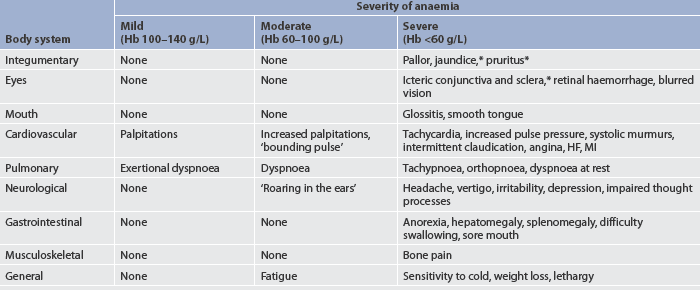
HF, heart failure; Hb, haemoglobin; MI, myocardial infarction.
Integumentary changes
Integumentary changes include pallor, jaundice and pruritus. Pallor results from reduced amounts of haemoglobin and reduced blood flow to the skin. Jaundice occurs when haemolysis of RBCs results in an increased concentration of serum bilirubin. Pruritus occurs because of increased serum and skin bile salt concentrations. In addition to the skin, the sclera of the eyes and mucous membranes should be evaluated for jaundice because they reflect the integumentary changes more accurately, especially in a dark-skinned individual.
Cardiopulmonary manifestations
Cardiopulmonary manifestations of severe anaemia result from additional attempts by the heart and lungs to provide adequate amounts of oxygen to the tissues. Cardiac output is maintained by increasing the heart rate and stroke volume. The low viscosity of the blood contributes to the development of systolic murmurs and bruits. In extreme cases or when concomitant heart disease is present, angina pectoris and myocardial infarction (MI) may occur if myocardial oxygen needs cannot be met. Heart failure, cardiomegaly, pulmonary and systemic congestion, ascites and peripheral oedema may develop if the heart is overworked for an extended period of time.
Gerontological considerations: anaemia
Modest changes in RBC mass occur in older adults. In healthy older men, there is a modest decline in haemoglobin of about 1 g/L between the ages of 70 and 88 years, in part due to the decreased production of androgens. Only a minimal decrease in haemoglobin occurs between these ages in healthy women (about 2 g/L).1 Vitamin B12 deficiency may occur in more than 20% of older people due to pernicious anaemia, insufficient dietary intake and malabsorption.3 Multiple comorbid conditions in older adults increase the likelihood of occurrence of many types of anaemia. Signs and symptoms of anaemia in the older adult may include pallor, confusion, ataxia, fatigue, worsening angina and heart failure. Unfortunately, anaemia may go unrecognised in the older adult because manifestations of anaemia may be mistaken as normal ageing changes or overlooked because of another health problem. By recognising signs of anaemia, the nurse can play a pivotal role in appropriate health assessment and related interventions for the older adult.4
 NURSING MANAGEMENT: ANAEMIA
NURSING MANAGEMENT: ANAEMIA
This section discusses general nursing management of anaemia. Specific care related to various types of anaemia is discussed later in this chapter.
Nursing assessment
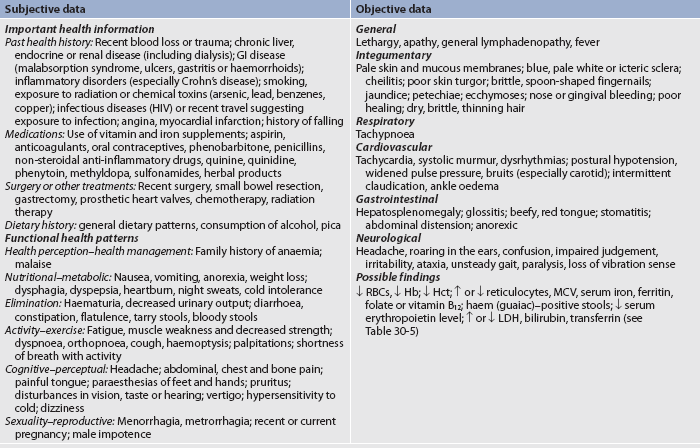
Subjective and objective data that should be obtained from the patient with anaemia are presented in Table 30-3.
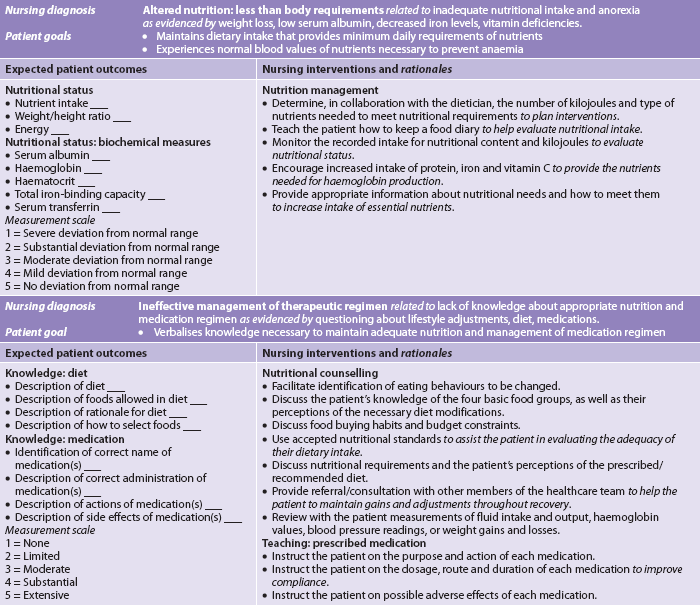
Nursing diagnoses
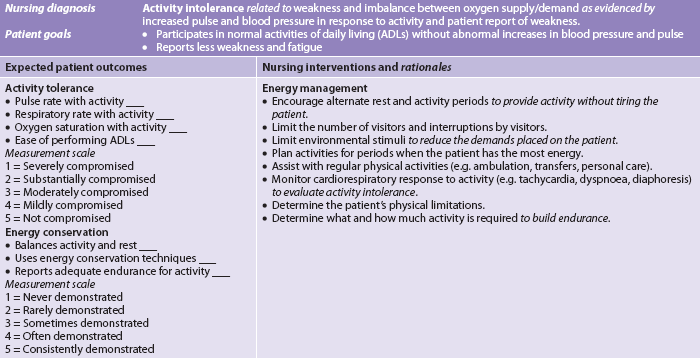
Nursing diagnoses for the patient with anaemia include, but are not limited to, those presented in NCP 30-1.
Planning
The overall goals are that the patient with anaemia will: (1) assume normal activities of daily living; (2) maintain adequate nutrition; and (3) develop no complications related to anaemia.
Nursing implementation
The numerous causes of anaemia necessitate different nursing interventions specific to the needs of the patient. Nevertheless, there are certain general components of care for all patients with anaemia that are presented in NCP 30-1.
Correcting the cause of the anaemia is ultimately the goal of therapy. Acute interventions may include blood or blood product transfusions, drug therapy (e.g. erythropoietin, vitamin supplements), volume replacement and oxygen therapy to stabilise the patient. Dietary and lifestyle changes (described with specific types of anaemia) can reverse some types of anaemia so that the patient can return to the former state of health. Ongoing assessment of the patient’s knowledge regarding adequate nutritional intake and compliance with safety precautions to prevent falls and injury and with drug therapies should be included in the plan of care.
ANAEMIA CAUSED BY DECREASED ERYTHROCYTE PRODUCTION
Normally, RBC production (termed erythropoiesis) is in equilibrium with RBC destruction and loss. This balance ensures that an adequate number of erythrocytes are available at all times. The normal life span of an RBC is 120 days. Three alterations in erythropoiesis may occur that decrease RBC production: (1) decreased haemoglobin synthesis may lead to iron-deficiency anaemia, thalassaemia and sideroblastic anaemia; (2) defective DNA synthesis in RBCs (e.g. vitamin B12 deficiency, folic acid deficiency) may lead to megaloblastic anaemias; and (3) diminished availability of erythrocyte precursors may result in anaemia of chronic disease and aplastic anaemia (see Box 30-1).
Iron-deficiency anaemia
Iron-deficiency anaemia, one of the most common chronic haematological disorders, is found in up to 30% of the world’s population. In Australia, it occurs in about 5–10% of people over age 45. In addition, those most susceptible to iron-deficiency anaemia are women in their reproductive years, vegetarians who eliminate all animal foods from their diet and athletes.5
Normally, adult males lose 1 mg of iron daily through faeces, sweat and urine and menstruating women lose 1.5 mg daily. The median total iron loss with pregnancy is about 500 mg—or almost 2 mg/day over the 280 days of gestation.6
AETIOLOGY
Iron-deficiency anaemia may develop from inadequate dietary intake, malabsorption, blood loss or haemolysis (normal iron metabolism is discussed in Ch 29). Dietary iron is adequate to meet the needs of men and older women, but it may be inadequate for those individuals who have higher iron needs (e.g. menstruating or pregnant women). Table 30-4 lists nutrients needed for erythropoiesis. Dietary guidelines are available to ensure adequate intake of nutrients needed for erythropoiesis.7
Nutrients needed for erythropoiesis
| Nutrient | Role in erythropoiesis | Food sources |
|---|---|---|
| Vitamin B12 | RBC maturation | Red meats, especially liver |
| Folic acid | RBC maturation | Green leafy vegetables, liver, meat, fish, legumes, wholegrains |
| Iron | Haemoglobin synthesis | Liver and muscle meats, eggs, dried fruits, legumes, dark green leafy vegetables, wholegrain and enriched bread and cereals, potatoes |
| Vitamin B6 | Haemoglobin synthesis | Meats (especially pork and liver), wheat germ, legumes, potatoes, cornmeal, bananas |
| Amino acids | Synthesis of nucleoproteins | Eggs, meat, milk and milk products (cheese, ice-cream), poultry, fish, legumes, nuts |
| Vitamin C | Conversion of folic acid to its active forms, aids in iron absorption | Citrus fruits, leafy green vegetables, strawberries, rockmelon |
RBC, red blood cell.
Malabsorption of iron may occur after certain types of gastrointestinal (GI) surgery and in malabsorption syndromes. Surgical procedures may involve removal or bypass of the duodenum (see Ch 42). As iron absorption occurs in the duodenum, malabsorption syndromes may involve disease of the duodenum in which the absorption surface is altered or destroyed.
Blood loss is a major cause of iron deficiency in adults. Two millilitres of whole blood contain 1 mg of iron. The major sources of chronic blood loss are from the GI and genitourinary (GU) systems. GI bleeding is often not apparent and therefore may exist for a considerable time before the problem is identified. Loss of 50–75 mL of blood from the upper GI tract is required for stools to appear black (melaena). The black colour results from the iron in the RBCs. Common causes of GI blood loss are peptic ulcer, gastritis, oesophagitis, diverticuli, haemorrhoids and neoplasia. GU blood loss occurs primarily from menstrual bleeding. The average monthly menstrual blood loss is about 45 mL and causes the loss of about 22 mg of iron. Postmenopausal bleeding can contribute to anaemia in a susceptible older woman.
Pregnancy contributes to iron deficiency because of the diversion of iron to the fetus for erythropoiesis, blood loss at delivery and lactation. In addition to anaemia of chronic renal failure, dialysis treatment may induce iron-deficiency anaemia because of the blood lost in the dialysis equipment and frequent blood sampling.
CLINICAL MANIFESTATIONS
In the early course of iron-deficiency anaemia, the patient may be free of symptoms. As the disease becomes chronic, any of the general manifestations of anaemia may develop (see Table 30-2). In addition, specific clinical symptoms may occur related to iron-deficiency anaemia. Pallor is the most common finding, and glossitis (inflammation of the tongue) is the second most common; another finding is cheilitis (inflammation of the lips). In addition, the patient may report headaches, paraesthesias and a burning sensation of the tongue, all of which are caused by lack of iron in the tissues.
DIAGNOSTIC STUDIES
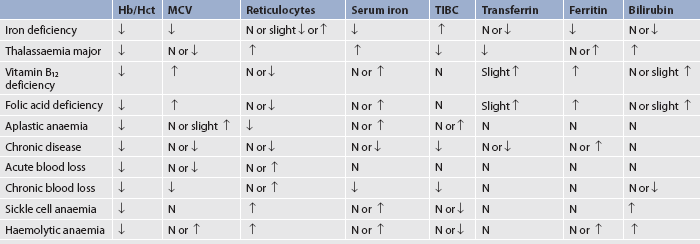
Laboratory abnormalities characteristic of iron-deficiency anaemia are presented in Table 30-5. Other diagnostic studies are done to determine the cause of the iron deficiency (e.g. stool guaiac [occult blood] test). Endoscopy and colonoscopy may be used to detect GI bleeding. A bone marrow biopsy may be done if other tests are inconclusive.
MULTIDISCIPLINARY CARE
The main goal of multidisciplinary care of iron-deficiency anaemia is to treat the underlying disease that is causing reduced intake (e.g. malnutrition, alcoholism) or absorption of iron. In addition, efforts are directed towards replacing iron (see Box 30-2). The patient should be taught which foods are good sources of iron (see Table 30-4). If nutrition is already adequate, increasing iron intake by dietary means may not be practical. Consequently, oral or occasionally parenteral iron supplements are used. If the iron deficiency is from acute blood loss, the patient may require a transfusion of packed RBCs and a thorough investigation of the source of blood loss.
BOX 30-2 Iron-deficiency anaemia
MULTIDISCIPLINARY CARE
Diagnostic studies
History and physical examination
RBC count, including morphology
Collaborative therapy
Identification and treatment of underlying cause
Ferrous sulfate or ferrous gluconate
Iron dextran, iron sucrose, sodium ferric gluconate IM or IV
Nutritional and diet therapy (see Table 30-4)
Transfusion of packed RBCs (symptomatic patient only)
Hb, haemoglobin; Hct, haematocrit; IM, intramuscular; IV, intravenous; RBCs, red blood cells.
Drug therapy
Oral iron should be used whenever possible because it is inexpensive and convenient. Many iron preparations are available, but intravenous iron is not often used except in those who are critically ill. The following factors should be considered in the administration of iron:
1. Iron is absorbed best from the duodenum and proximal jejunum. Therefore, enteric-coated or sustained-release capsules, which release iron further down in the GI tract, are counterproductive and expensive.
2. The daily dosage should provide 150–200 mg of elemental iron. This can be ingested in three or four daily doses, with each tablet or capsule of the iron preparation containing between 50 and 100 mg of iron (e.g. a 300 mg tablet of ferrous sulfate contains 60 mg of elemental iron).
3. Iron is best absorbed as ferrous sulfate (Fe2+) in an acidic environment. For this reason and to avoid binding the iron with food, iron should be taken about an hour before meals, when the duodenal mucosa is most acidic. Taking iron with vitamin C (ascorbic acid) or orange juice, which contains ascorbic acid, also enhances iron absorption. Gastric side effects, however, may necessitate ingesting iron with meals.
4. Undiluted liquid iron may stain the patient’s teeth; therefore, it should be diluted and ingested through a straw.
5. GI side effects of iron administration may occur, including heartburn, constipation and diarrhoea. If side effects develop, the dose and type of iron supplement may be adjusted. For example, many individuals who need supplemental iron cannot tolerate ferrous sulfate because of the effects of the sulfate base. However, ferrous gluconate may be an acceptable substitute. All patients should know that the use of iron preparations will cause their stools to become black because the GI tract excretes excess iron. Constipation is common, and the patient should be started on stool softeners and laxatives when started on iron.
In some situations, it may be necessary to administer iron parenterally. Parenteral use of iron is indicated for malabsorption, intolerance of oral iron, a need for iron beyond oral limits or poor patient compliance in taking the oral preparations of iron. Parenteral iron can be given intramuscularly (IM) or intravenously (IV). An iron dextran complex contains 50 mg/mL of elemental iron in 2 mL. Sodium ferrous gluconate and iron sucrose are alternatives, and may provide less risk of life-threatening anaphylaxis.8 IM iron solutions may stain the skin, so separate needles should be used for withdrawing the solution and injecting the medication. A Z-track injection technique should also be used.
NURSING MANAGEMENT: IRON-DEFICIENCY ANAEMIA
It is important to recognise groups of individuals who are at an increased risk for the development of iron-deficiency anaemia. These include premenopausal and pregnant women, people from low socioeconomic backgrounds, older adults and individuals experiencing blood loss. Dietary education and advice, with an emphasis on foods high in iron, is important for these groups. Supplemental iron is especially important for pregnant women. Appropriate nursing measures are presented in NCP 30-1. It is important to discuss with the patient the need for diagnostic studies to identify the cause of the anaemia. The haemoglobin and RBC counts are reassessed to evaluate the response to therapy. Compliance with dietary and drug therapy is emphasised. To replenish the body’s iron stores, the patient needs to continue to take iron therapy for 2–3 months after the haemoglobin level returns to normal. Patients who require lifelong iron supplementation should be monitored for potential liver problems related to iron storage.
Thalassaemia
AETIOLOGY
Another cause of decreased erythrocyte production is thalassaemia. Thalassaemia is a group of diseases that have an autosomal recessive genetic basis involving inadequate production of normal haemoglobin. Haemolysis also occurs in thalassaemia, but insufficient production of normal haemoglobin is the predominant problem. In contrast to iron-deficiency anaemia, in which haem synthesis is the problem, thalassaemia is due to an absent or reduced globulin protein. α-globin chains are absent or reduced in α-thalassaemia, and β-globin chains are absent or reduced in β-thalassaemia. Therefore, the basic defect of thalassaemia is abnormal haemoglobin synthesis.
Thalassaemia is commonly found in members of ethnic groups whose origins are near the Mediterranean Sea and equatorial or near-equatorial regions of Asia, the Middle East and Africa. An individual with thalassaemia may have a heterozygous or homozygous form of the disease. A person who is heterozygous has one thalassaemic gene and one normal gene and is said to have thalassaemia minor (or thalassaemic trait), which is a mild form of the disease. A homozygous person has two thalassaemic genes, causing a severe condition known as thalassaemia major.9 Patient information on thalassaemia is available from Thalassaemia Australia (see Resources on p 805).
CLINICAL MANIFESTATIONS
Thalassaemia major is a life-threatening disease in which growth, both physical and mental, is often retarded. The person who has thalassaemia major is pale and displays other general symptoms of anaemia (see Table 30-2). The symptoms develop in childhood by 2 years of age and can cause growth and development deficits. In addition, the person has pronounced splenomegaly and hepatomegaly. Jaundice from RBC haemolysis is prominent. As the bone marrow responds to the deficit of oxygen-carrying capacity of the blood, RBC production is stimulated and the marrow becomes packed with immature erythroid precursors that die. This stimulates further erythropoiesis, leading to chronic bone marrow hyperplasia and expansion of the marrow space. This may cause thickening of the cranium and maxillary cavity.
The patient with thalassaemia minor is frequently asymptomatic. The patient has mild-to-moderate anaemia with microcytosis (small cells) and hypochromia (pale cells).
MULTIDISCIPLINARY CARE
The laboratory abnormalities of thalassaemia major are summarised in Table 30-5. No specific drug or diet therapies are effective in treating thalassaemia. Thalassaemia minor requires no treatment because the body adapts to the reduction of normal haemoglobin. The symptoms of thalassaemia major are managed with blood transfusions or exchange transfusions in conjunction with IV deferoxamine (a chelating agent that binds to iron) to reduce the iron overloading (haemochromatosis) that occurs with chronic transfusion therapy.10 Transfusions are administered to keep the haemoglobin level at approximately 100 g/L. This level is low enough to maintain the patient’s own erythropoiesis without enlarging the spleen. Zinc supplementation may be needed (it is reduced with the chelation therapy) as well as ascorbic acid supplementation during the chelation therapy (increases urine excretion of iron; it should not be taken otherwise, as it increases the absorption of dietary iron). Iron supplements should not be given. Because RBCs are sequestered in the enlarged spleen, thalassaemia may be treated by splenectomy.
Hepatitis C is present in the majority of patients older than 25 years of age because they received blood transfusions before transfusions were screened for hepatitis C virus. Hepatitis C may result in cirrhosis and hepatocellular carcinoma. Cardiac complications from iron overload, pulmonary disease and hypertension also contribute to early death. Thus hepatic, cardiac and pulmonary organ function should be monitored and treated as appropriate. Endocrinopathies (hypogonadotrophic hypogonadism) and thrombosis may also be complications of the disease.
Although haematopoietic stem cell transplantation remains the only cure for patients with thalassaemia, the risk of this procedure may outweigh its benefits. With proper iron chelation therapy, patients are living longer.11,12
Megaloblastic anaemias
Megaloblastic anaemias are a group of disorders caused by impaired DNA synthesis and characterised by the presence of large RBCs. When DNA synthesis is impaired, defective RBC maturation results. The RBCs are large (macrocytic) and abnormal and are referred to as megaloblasts. Macrocytic RBCs are easily destroyed because they have fragile cell membranes. Although the overwhelming majority of megaloblastic anaemias result from vitamin B12 and folic acid deficiencies, this type of RBC deformity can also occur from suppression of DNA synthesis by drugs, from inborn errors of vitamin B12 and folic acid metabolism, and from erythroleukaemia (a malignant blood disorder characterised by a proliferation of erythropoietic cells in bone marrow). Two common forms of megaloblastic anaemia are vitamin B12 deficiency and folic acid deficiency (see Box 30-3).13
VITAMIN B12 DEFICIENCY
Normally, a protein termed intrinsic factor (IF) is secreted by the parietal cells of the gastric mucosa. IF is required for vitamin B12 (extrinsic factor) absorption. Therefore, if IF is not secreted, vitamin B12 will not be absorbed (vitamin B12 is normally absorbed in the distal ileum). There are many causes of vitamin B12 deficiency. The most common cause is pernicious anaemia, a disease in which the gastric mucosa is not secreting IF due to antibodies being directed against the gastric parietal cells and/or IF itself. Other causes of vitamin B12 deficiency include gastrectomy, gastritis, nutritional deficiency, chronic alcoholism and hereditary enzymatic defects of vitamin B12 utilisation (see Box 30-3).
Pernicious anaemia is a disease of insidious onset that begins in middle age or later (usually after age 40), with 60 years being the most common age at diagnosis. Pernicious anaemia occurs frequently in people of Northern European ancestry (particularly Scandinavians) and African Americans. In African Americans, the disease tends to begin early, occurs with higher frequency in women and is often severe.
AETIOLOGY
Vitamin B12 deficiency can occur in patients who have had GI surgery, such as gastrectomy; patients who have had a small bowel resection involving the ileum; and patients with Crohn’s disease, ileitis, diverticuli of the small intestine and/or chronic atrophic gastritis. Long-term use of metformin can also result in vitamin B12 deficiency. In these cases, the deficiency results from the loss of IF-secreting gastric mucosal cells or impaired absorption of vitamin B12 in the distal ileum. Vitamin B12 deficiency is also found in long-term users of histamine H2-receptor antagonists and proton pump inhibitors, and those who are strict vegetarians.13
Pernicious anaemia is caused by an absence of IF, from either gastric mucosal atrophy or autoimmune destruction of parietal cells. This results in a decrease of hydrochloric acid secretion by the stomach. An acid environment in the stomach is required for the secretion of IF.
CLINICAL MANIFESTATIONS
General symptoms of anaemia related to vitamin B12 deficiency develop because of tissue hypoxia (see Table 30-2). GI manifestations include a sore tongue, anorexia, nausea, vomiting and abdominal pain. Typical neuromuscular manifestations include weakness, paraesthesias of the feet and hands, reduced vibratory and position senses, ataxia, muscle weakness and impaired thought processes ranging from confusion to dementia. Because vitamin B12 deficiency anaemia has an insidious onset, it may take several months for these manifestations to develop.
Diagnostic studies
Laboratory data reflective of vitamin B12 deficiency anaemia are presented in Table 30-5. The RBCs appear large (macrocytic) and have abnormal shapes. This structure contributes to erythrocyte destruction because the cell membrane is fragile. Serum vitamin B12 levels are reduced. Serum folate levels are obtained, and if they are normal and vitamin B12 levels are low, this suggests that megaloblastic anaemia is due to a vitamin B12 deficiency. Because the potential for gastric cancer is increased in patients with pernicious anaemia, a gastroscopy and biopsy of the gastric mucosa may also be done.
Another means of assessing parietal cell function is by a Schilling test. After radioactive vitamin B12 is administered to the patient, the amount of vitamin B12 excreted in the urine is measured. An individual who cannot absorb vitamin B12 excretes only a small amount of this radioactive form. The same procedure may be followed with the parenteral administration of IF. Absorption of vitamin B12 when IF is added is diagnostic of pernicious anaemia.
Multidisciplinary care
Regardless of how much is ingested, the patient is not able to absorb vitamin B12 if IF is lacking or if there is impaired absorption in the ileum. For this reason, increasing dietary vitamin B12 does not correct the anaemia. However, the patient should be instructed on adequate dietary intake to maintain good nutrition (see Table 30-4). Parenteral administration of vitamin B12 is the treatment of choice. Without vitamin B12 administration, these individuals will die in 1–3 years. The dosage and frequency of vitamin B12 administration may vary. A typical treatment schedule consists of 1000 mg of vitamin B12 IM daily for 2 weeks and then weekly until the haematocrit is normal, then monthly for life. High-dose oral vitamin B12 is also available. As long as supplemental vitamin B12 is used, the anaemia can be reversed. However, if the person has had longstanding neuromuscular complications, they may not be reversible.
FOLIC ACID DEFICIENCY
Folic acid (folate) deficiency also causes megaloblastic anaemia. Folic acid is required for DNA synthesis leading to RBC formation and maturation. Common causes of folic acid deficiency include the following:
1. poor nutrition, especially a lack of leafy green vegetables, liver, citrus fruits, yeast, dried beans, nuts and grains
2. malabsorption syndromes, particularly small bowel disorders
3. drugs that impede the absorption and use of folic acid (e.g. methotrexate), antiseizure drugs (e.g. phenytoin, phenobarbitone) and others (e.g. trimethoprim, sulfasalazine)
5. haemodialysis, because folic acid is lost during dialysis.
The clinical manifestations of folic acid deficiency are similar to those of vitamin B12 deficiency. The disease develops insidiously and the patient’s symptoms may be attributed to other coexisting problems, such as cirrhosis or oesophageal varices. GI disturbances include dyspepsia and a smooth, beefy red tongue. The absence of neurological problems is an important diagnostic finding. This lack of neurological involvement differentiates folic acid deficiency from vitamin B12 deficiency.
The diagnostic findings for folic acid deficiency are presented in Table 30-5. In addition, the serum folate level is low (normal is 7–41 nmol/L), the serum vitamin B12 level is normal and the gastric analysis is positive for hydrochloric acid.
Folic acid deficiency is treated by replacement therapy. The usual dose is 1 mg per day by mouth. In malabsorption states, up to 5 mg per day may be required. The duration of treatment depends on the reason for the deficiency. The patient should be encouraged to eat foods containing large amounts of folic acid (see Table 30-4).
NURSING MANAGEMENT: MEGALOBLASTIC ANAEMIAS
Since there is a familial predisposition for pernicious anaemia, the most common type of vitamin B12 deficiency, patients who have a positive family history of pernicious anaemia should be evaluated for symptoms. Although disease development cannot be prevented, early detection and treatment can lead to reversal of symptoms. Signs and symptoms of other possible megaloblastic anaemias should also be considered and brought to the attention of the primary healthcare provider.
The nursing measures presented in the nursing care plan for the patient with anaemia (see NCP 30-1) are appropriate for the patient with vitamin B12 or folic acid deficiency anaemia. In addition to these measures, the nurse should ensure that injuries are not sustained because of the diminished sensations to heat and pain resulting from the neurological impairment. The patient must be protected from falling, burns and trauma. If heat therapy is required, the patient’s skin must be evaluated at frequent intervals to detect redness.
Ongoing care is focused on ensuring good patient compliance with treatment. There must also be careful follow-up to assess for neurological difficulties that were not fully corrected by adequate replacement therapy. Because the potential for cancer may be increased (e.g. gastric carcinoma is increased in patients with atrophic gastritis-related pernicious anaemia, alcohol increases the likelihood of oral and oesophageal cancers), the patient should have frequent and careful appropriate screenings.
Anaemia of chronic disease
Chronic inflammatory, autoimmune, infectious or malignant diseases can lead to anaemia of chronic disease. Anaemia of chronic disease is associated with an underproduction of RBCs and mild shortening of RBC survival. The RBCs are usually normocytic, normochromic and hypoproliferative. The anaemia is usually mild, but it can be more severe. This type of anaemia is primarily immune driven. The cytokines released by these conditions cause an increased uptake and retention of iron within macrophages. This leads to a diversion of iron from the circulation into storage sites, subsequent limitation of the availability of iron for erythroid progenitor cells, and iron-restricted erythropoiesis. For any chronic disease, there may also be additional factors. For example, with renal disease, the primary factor causing anaemia is decreased erythropoietin (EPO), a hormone made in the kidneys that stimulates erythropoiesis. With impaired renal function, less EPO is produced (see Ch 46).
Any condition that causes increased RBC destruction (e.g. autoimmune haemolysis) accompanied by the failure to augment erythropoiesis will contribute to anaemia. Myelosuppression and decreased erythropoiesis due to disease, medications (e.g. chemotherapy) or radiation will contribute to the anaemia of chronic disease. Human immunodeficiency virus (HIV) and its treatments, hepatitis, malaria and bleeding episodes are other contributors to this type of anaemia.14,15
Hypopituitary and hypothyroid states both lead to reduced tissue metabolism; therefore, tissue oxygen needs are diminished, leading to a reduced production of EPO by the kidneys. Adrenal dysfunction caused by either adrenalectomy or Addison’s disease also results in anaemia.
Anaemia of chronic disease must first be recognised and differentiated from anaemias of other aetiologies. Findings of elevated serum ferritin and increased iron stores distinguish it from iron-deficiency anaemia. Normal folate and vitamin B12 blood levels distinguish it from those types of anaemias. The best treatment of anaemia of chronic disease is correction of the underlying disorder. If the anaemia is severe, blood transfusions may be indicated, but they are not recommended for long-term treatment. EPO therapy is used for anaemia related to renal disease (see Ch 46) or anaemia related to cancer therapies (see Ch 15). Darbepoetin is a synthetic parenteral form of EPO. It has a longer duration of action than EPO. IV iron should only be administered, if necessary, to improve the response to therapy with EPO.
Aplastic anaemia
Aplastic anaemia is a disease in which the patient has peripheral blood pancytopenia (a decrease of all blood cell types—RBCs, white blood cells [WBCs] and platelets) and hypocellular bone marrow. The signs and symptoms range from a chronic condition managed with EPO or blood transfusions to a critical condition with haemorrhage and sepsis.
AETIOLOGY
The incidence of aplastic anaemia is low, affecting approximately 2 out of every 1 million persons.16 There are various aetiological classifications for aplastic anaemia, but they can be divided into two major groups: congenital and acquired (see Box 30-4).
1. Congenital aplastic anaemia is caused by chromosomal alterations. Approximately 30% of the aplastic anaemias that appear in childhood are inherited.
2. Acquired aplastic anaemia results from exposure to ionising radiation, chemical agents (e.g. benzene, insecticides, arsenic, alcohol), viral and bacterial infections (e.g. hepatitis, parvovirus, biliary tuberculosis) and prescribed medications (e.g. alkylating agents, antiseizure agents, antimetabolites, antimicrobials, gold). Approximately 70% of the acquired aplastic anaemias are idiopathic.17
CLINICAL MANIFESTATIONS
Aplastic anaemia can present abruptly (over days) or insidiously (over weeks to months) and can vary from mild to severe. Clinically the patient may have symptoms caused by suppression of any or all bone marrow elements. General manifestations of anaemia such as fatigue and dyspnoea, as well as cardiovascular and cerebral responses, may be seen (see Table 30-2). The patient with neutropenia (low neutrophil count) is susceptible to infection and may be febrile. Thrombocytopenia is manifested by a predisposition to bleeding (e.g. petechiae, ecchymosis, epistaxis).
DIAGNOSTIC STUDIES
The diagnosis is confirmed by laboratory studies. Because all marrow elements are affected, haemoglobin, WBC and platelet values are often decreased in aplastic anaemia. Other RBC indices are generally normal (see Table 30-5). The condition is therefore classified as a normocytic, normochromic anaemia. The reticulocyte count is low. Bleeding time is prolonged.
Aplastic anaemia can be further evaluated by assessing various iron studies. The serum iron and total iron-binding capacity (TIBC) may be elevated as initial signs of erythropoiesis suppression. Bone marrow biopsy, aspiration and pathological examination may be done for any anaemic state. However, the findings are especially important in aplastic anaemia because the marrow is hypocellular with increased yellow marrow (fat content).
NURSING AND COLLABORATIVE MANAGEMENT: APLASTIC ANAEMIA
Management of aplastic anaemia is based on identifying and removing the causative agent (when possible) and providing supportive care until the pancytopenia reverses. Nursing interventions appropriate for the patient with pancytopenia from aplastic anaemia are presented in the nursing care plan for the patient with anaemia (see NCP 30-1) and the nursing care plans for thrombocytopenia (see NCP 30-2) and neutropenia (see NCP 30-3). Nursing actions are directed at preventing complications from infection and haemorrhage.
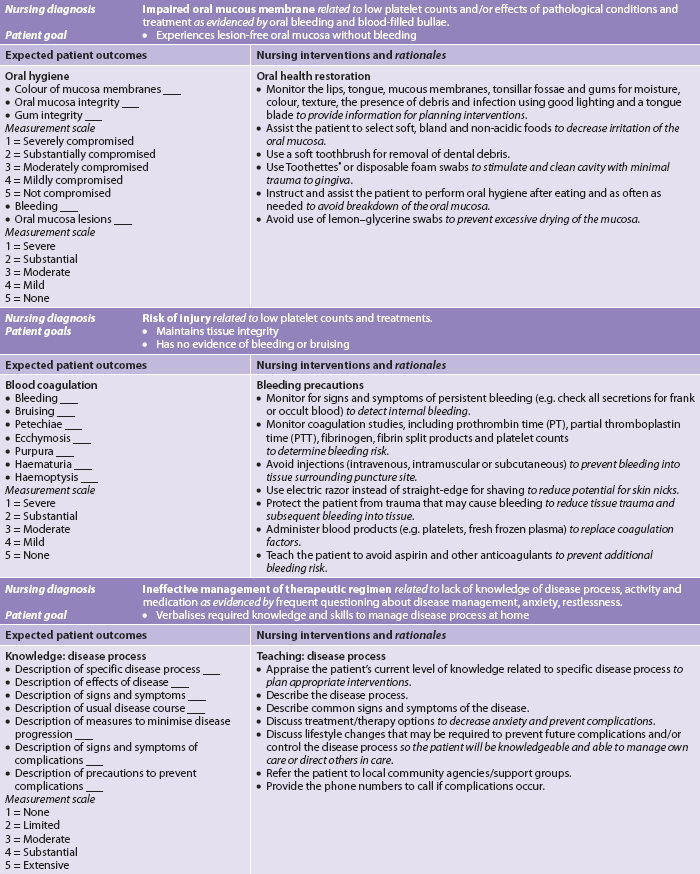

The prognosis of severe untreated aplastic anaemia is poor (approximately 75% fatal). However, advances in medical management, including haematopoietic stem cell transplant (HSCT) and immunosuppressive therapy with antithymocyte globulin (ATG) and cyclosporin or high-dose cyclophosphamide, have improved outcomes significantly. ATG is a horse serum that contains polyclonal antibodies against human T cells. It can cause anaphylaxis and a serum sickness. The rationale for this therapy is that idiopathic aplastic anaemia is considered a disorder resulting from upregulated cytotoxic T-cell subset actively targeting and destroying the patient’s own haematopoietic stem cells.18 (ATG and cyclosporine are discussed in Ch 13.)
The treatment of choice for adults less than 45 years of age who do not respond to the immunosuppressive therapy and who have a human leucocyte antigen (HLA)-matched donor is a HSCT. The best results occur in younger patients who have not had previous blood transfusions, as prior transfusions increase the risk of graft rejection. HSCT is discussed in Chapter 15.
For older adults or patients without an HLA-matched donor, the treatment of choice is immunosuppression with ATG or cyclosporin or high-dose cyclophosphamide. Although this therapy may be only partially beneficial, transfusions usually can be avoided.
ANAEMIA CAUSED BY BLOOD LOSS
The second major cause of anaemia, anaemia resulting from blood loss, may be caused by either acute or chronic problems.
Acute blood loss
Acute blood loss occurs as a result of sudden haemorrhage. Causes of acute blood loss include trauma, complications of surgery and conditions or diseases that disrupt vascular integrity. There are two clinical concerns in such situations. First, the sudden reduction in total blood volume can lead to hypovolaemic shock, which requires immediate intervention to prevent death. Second, if the acute loss is more gradual, the body maintains its blood volume by slowly increasing the plasma volume and constricting blood vessels. Although the circulating fluid volume is preserved, the number of RBCs available to carry oxygen is significantly diminished.
CLINICAL MANIFESTATIONS
The clinical manifestations of anaemia from acute blood loss are caused by the body’s attempts to maintain an adequate blood volume and meet oxygen requirements. Table 30-6 summarises the clinical manifestations of patients with varying degrees of blood volume loss. It is essential to understand that the clinical signs and symptoms the patient is experiencing are more important than the laboratory values. For example, an adult with a bleeding peptic ulcer who had a 750 mL haematemesis (15% of a normal total blood volume) within the past 30 minutes may have postural hypotension, but have normal values for haemoglobin and haematocrit. Over the ensuing 36–48 hours, most of the volume deficit will be repaired by the movement of fluid from the extravascular space into the intravascular space. Only at these later times will the haemoglobin and haematocrit reflect the blood loss.
TABLE 30-6 Clinical manifestations of acute blood loss
| Volume lost (%) | Clinical manifestations |
|---|---|
| 10 | None |
| 20 | No detectable signs of symptoms at rest, tachycardia with exercise and slight postural hypotension |
| 30 | Normal supine blood pressure and pulse at rest, postural hypotension and tachycardia with exercise |
| 40 | Blood pressure, central venous pressure and cardiac output below normal at rest; rapid, thready pulse and cold, clammy skin |
| 50 | Shock and potential death |
The nurse should be alert to the patient’s expression of pain. Internal haemorrhage may cause pain because of tissue distension, organ displacement and nerve compression. Pain may be localised or referred. In the case of retroperitoneal bleeding, the patient may not experience abdominal pain. Instead, they may have numbness and pain in a lower extremity secondary to compression of the lateral cutaneous nerve, which is located in the region of the first to third lumbar vertebrae. The major complication of acute blood loss is shock (see Ch 66). Any change in the level of consciousness or signs of agitation or confusion requires further immediate investigation.
DIAGNOSTIC STUDIES
When blood volume loss is sudden, plasma volume has not yet had a chance to increase, the loss of RBCs is not reflected in laboratory data and values may seem normal or high for 2–3 days. However, once the plasma is replaced by endogenous and exogenous means, the RBC mass is less concentrated. At this time, RBC, haemoglobin and haematocrit levels are low and reflect the blood loss.
MULTIDISCIPLINARY CARE
Multidisciplinary care is initially concerned with: (1) replacing blood volume to prevent shock; and (2) identifying the source of the haemorrhage and stopping the blood loss. IV fluids used in emergencies include solutions such as Hartmann’s. The amount of infusion varies with the solution used.
Once volume replacement is established, attention can be directed to correcting the RBC loss. The body needs 2–5 days to manufacture more RBCs in response to increased EPO. Consequently, blood transfusions (packed RBCs) may be needed if the blood loss is significant. In addition, if the bleeding is related to a platelet or clotting disorder, replacement of that deficiency should be addressed.
The patient may also need supplemental iron because the availability of iron affects the marrow production of erythrocytes. When anaemia results from acute blood loss, dietary sources of iron will probably not be adequate to maintain iron stores. Therefore, oral or parenteral iron preparations may be administered.
NURSING MANAGEMENT: ACUTE BLOOD LOSS
In the case of trauma, it may be impossible to prevent the situation leading to the blood loss. For the postoperative patient, the nurse carefully monitors the blood loss from various drainage tubes and dressings, monitors haemodynamic status and urine output and implements appropriate actions. The nursing care plan for the patient with anaemia resulting from acute blood loss will most likely include the administration of blood products (see p 797).
Once the source of haemorrhage is identified, blood loss is controlled, and fluid and blood volumes are replaced, the anaemia should begin to correct itself. There should be no need for long-term treatment of this type of anaemia.
Chronic blood loss
The sources of chronic blood loss are similar to those of iron-deficiency anaemia (e.g. bleeding ulcer, haemorrhoids, menstrual and postmenopausal blood loss). The effects of chronic blood loss are usually related to the depletion of iron stores and are usually considered as iron-deficiency anaemia. Management of chronic blood loss anaemia involves identifying the source and stopping the bleeding. Supplemental iron may be required. The nursing measures presented in NCP 30-1 are relevant to anaemia of chronic blood loss.
ANAEMIA CAUSED BY INCREASED ERYTHROCYTE DESTRUCTION
The third major type of anaemia is haemolytic anaemia, a condition caused by the destruction or haemolysis of RBCs at a rate that exceeds production. Haemolysis can occur because of problems intrinsic or extrinsic to the RBCs. Intrinsic haemolytic anaemias result from defects in the RBCs themselves caused by abnormal haemoglobin (e.g. sickle cells), enzyme deficiencies that alter glycolysis (glucose-6-phosphate dehydrogenase [G6PD] deficiency) or RBC membrane abnormalities. Intrinsic haemolytic anaemias are usually hereditary. More common are the acquired extrinsic haemolytic anaemias. In this type of anaemia the patient’s RBCs are normal, but damage is caused by external factors such as trapping of cells within the sinuses of the liver or spleen, antibody-mediated destruction, toxins or mechanical injury (e.g. prosthetic heart valves).
The two sites of haemolysis are classified as intravascular or extravascular. Intravascular destruction occurs within the circulation; extravascular haemolysis takes place in the macrophages of the spleen, liver and bone marrow. The spleen is the primary site of the destruction of RBCs that are old, defective or moderately damaged. Figure 30-2 indicates the sequence of events involved in extravascular haemolysis.
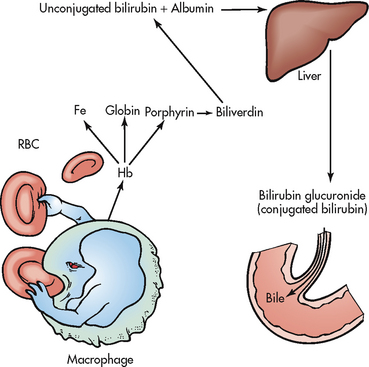
Figure 30-2 Sequence of events in extravascular haemolysis. Fe, iron; Hb, haemoglobin; RBC, red blood cell.
The patient with haemolytic anaemia manifests the general symptoms of anaemia and clinical manifestations specific to this type of anaemia (see Table 30-2). Jaundice is likely because the increased destruction of RBCs causes an elevation in bilirubin levels. The spleen and liver may enlarge because of their hyperactivity, which is related to macrophage phagocytosis of the defective erythrocytes.
In all causes of haemolysis a major focus of treatment is to maintain renal function. When an RBC is haemolysed, the haemoglobin molecule is released and filtered by the kidneys. The accumulation of haemoglobin molecules can obstruct the renal tubules and lead to acute tubular necrosis (see Ch 47).
Sickle cell disease
Sickle cell disease (SCD) is a group of inherited, autosomal recessive disorders characterised by the presence of an abnormal form of haemoglobin in the erythrocyte. (Autosomal recessive genetic disorders are discussed in Ch 13.) This abnormal haemoglobin, haemoglobin S (HbS), causes the erythrocyte to stiffen and elongate taking on a sickle shape in response to low oxygen levels. HbS results from substitution of valine for glutamic acid on the β-globin chain of haemoglobin. Because this is a genetic disorder, SCD is usually identified during infancy or early childhood. It is an incurable disease that is often fatal by middle age from renal and pulmonary failure and/or stroke.19
SCD has a high incidence among Americans: it affects more than 50,000 Americans and is predominant in African Americans, occurring in an estimated prevalence of about 1 in 400 live births. It also affects people of Mediterranean, Caribbean, South and Central American, Arabian and East Indian ancestry.20 Due to increased migration and diversity in the Australasian population, especially with African immigrants, healthcare professionals need to be aware of recognising and treating SCD. The importance of cultural awareness in treating SCD has been highlighted in one study.21
HEALTH DISPARITIES
Clinical implications
• Requires ongoing continuity of care and extensive patient education
• Sickle cell trait is the carrier state for sickle cell disease and represents a mild type of sickle cell disease; 1 in 10–12 African Americans and 1 in 25 Hispanics has sickle cell trait
• If both parents have the trait, 1 in 4 chance that the baby will have sickle cell disease
• Management of sickle cell disease should focus on the prevention of sickle cell crisis
• Genetic counselling is recommended for individuals with a family history of sickle cell disease; individuals should understand the risks of transmitting the genetic mutation
AETIOLOGY AND PATHOPHYSIOLOGY
Types of sickle cell disease
Types of SCD disorders include sickle cell anaemia, sickle cell–thalassaemia, sickle cell haemoglobin (HbC) disease and sickle cell trait. Sickle cell anaemia is the most severe of the SCD syndromes. This occurs when a person is homozygous for HbS; the person has inherited HbS from both parents. Sickle cell–thalassaemia and sickle cell HbC occur when a person inherits HbS from one parent and another type of abnormal haemoglobin (such as thalassaemia or HbC) from the other parent. Both of these forms of SCD are less common and less severe than sickle cell anaemia. Sickle cell trait occurs when a person is heterozygous for HbS (HbAS); the person has inherited HbS from one parent and normal haemoglobin (haemoglobin A) from the other parent. Sickle cell trait is typically a very mild to asymptomatic condition.
Sickling episodes
The major pathophysiological event of SCD is the sickling of RBCs. Sickling episodes are most commonly triggered by low oxygen tension in the blood. Hypoxia or deoxygenation of the RBCs can be caused by viral or bacterial infection, high altitude, emotional or physical stress, surgery and blood loss. Infection is the most common precipitating factor. Other events that can trigger or sustain a sickling episode include dehydration, increased hydrogen ion concentration (acidosis), increased plasma osmolality, decreased plasma volume and low body temperature. A sickling episode can also occur without an obvious cause.
Sickled RBCs become rigid and take on an elongated, crescent shape (see Fig 30-3). Sickled cells cannot easily pass through capillaries or other small vessels and can cause vascular occlusion, leading to acute or chronic tissue injury. The resulting haemostasis promotes a self-perpetuating cycle of local hypoxia, deoxygenation of more erythrocytes and more sickling. Circulating sickled cells are haemolysed by the spleen, leading to anaemia. Initially the sickling of cells is reversible with reoxygenation, but it eventually becomes irreversible due to cell membrane damage from recurrent sickling. Thus vaso-occlusive phenomena and haemolysis are the clinical hallmark of sickle cell disease.
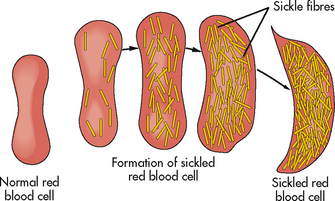
Figure 30-3 Sickle cell haemoglobin aggregates into long chains and alters the shape of the red blood cell.
Sickle cell crisis is a severe, painful, acute exacerbation of RBC sickling causing a vaso-occlusive crisis. As blood flow is impaired by sickled cells, vasospasm occurs, further restricting blood flow. Severe capillary hypoxia causes changes in membrane permeability, leading to plasma loss, haemoconcentration, the development of thrombi and further circulatory stagnation. Tissue ischaemia, infarction and necrosis eventually occur from lack of oxygen. Shock is a possible life-threatening consequence of sickle cell crisis due to severe oxygen depletion of the tissues and a reduction of the circulating fluid volume. Sickle cell crisis can begin suddenly and persist for days to weeks.
The frequency, extent and severity of sickling episodes are highly variable and unpredictable but are largely dependent on the percentage of HbS present. Individuals with sickle cell anaemia have the most severe form because the erythrocytes contain a high percentage of HbS.
CLINICAL MANIFESTATIONS
The effects of SCD vary greatly from person to person. Many people with sickle cell anaemia are in reasonably good health the majority of the time. However, they may have chronic health problems and pain due to organ tissue hypoxia and damage (e.g. involving the kidneys and/or liver). The typical patient is anaemic but asymptomatic except during sickling episodes. Clinical manifestations of chronic anaemia include pallor of mucous membranes, fatigue and decreased exercise tolerance. Since most individuals with sickle cell anaemia have dark skin, pallor is more readily detected by examining the mucous membranes. The skin may have a greyish cast. Because of the haemolysis, jaundice is common and patients are prone to gallstones (cholelithiasis).
The primary symptom associated with sickling is pain. During sickle cell crisis the pain is severe due to ischaemia of tissue. The episodes can affect any area of the body or several sites simultaneously, with the back, chest, extremities and abdomen being most commonly affected. The pain severity can range from trivial to excruciating. The pain can be described as deep gnawing or throbbing. Approximately half of pain episodes are accompanied by objective clinical signs such as fever, swelling, tenderness, tachypnoea, hypertension, nausea and vomiting.
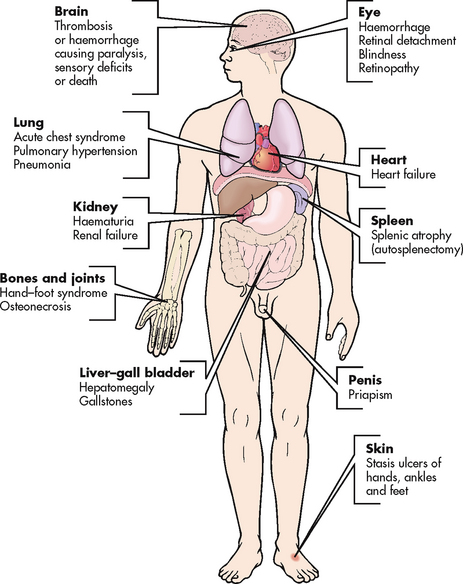
COMPLICATIONS
With repeated episodes of sickling there is gradual involvement of all body systems, especially the spleen, lungs, kidneys and brain. Organs that have a high need for oxygen are most often affected and form the basis for many of the complications of SCD (see Fig 30-4). Infection is a major cause of morbidity and mortality in patients with SCD. One reason for this is the failure of the spleen to phagocytise foreign substances as it becomes infarcted and dysfunctional (usually by 2–4 years of age) from the sickled red cells. The spleen becomes small because of repeated scarring, a phenomenon termed autosplenectomy. Pneumonia is the most common infection and is often of pneumococcal origin. Infections can be so severe that they can cause an aplastic and haemolytic crisis and gallstones. Acute chest syndrome is a term used to describe acute pulmonary complications that include pneumonia, tissue infarction and fat embolism. It is characterised by fever, chest pain, cough, pulmonary infiltrates and dyspnoea. Pulmonary infarctions may cause pulmonary hypertension, MI, heart failure and ultimately cor pulmonale. The heart may become ischaemic and enlarged, leading to heart failure. Retinal vessel obstruction may result in haemorrhage, scarring, retinal detachment and blindness. The kidneys may be injured due to the increased blood viscosity and the lack of oxygen and this can lead to renal failure. Stroke can result from thrombosis and infarction of cerebral blood vessels. Bone changes may include osteoporosis and osteosclerosis after infarction. Chronic leg ulcers can result from the hypoxia and are especially prevalent around the ankles. Priapism (persistent penile erection) may occur if penile veins become occluded.
DIAGNOSTIC STUDIES
A peripheral blood smear may reveal sickled cells and abnormal reticulocytes. The presence of sickle haemoglobin can be diagnosed by the sickling test, which uses RBCs (in vitro) and exposes them to a deoxygenation agent. Electrophoresis of haemoglobin readily identifies the presence of abnormal haemoglobin. DNA testing can be done, but is costly.
As a result of the accelerated RBC breakdown, the patient has characteristic clinical findings of haemolysis (jaundice, elevated serum bilirubin levels) and abnormal laboratory test results (see Table 30-5). Skeletal X-rays will demonstrate bone and joint deformities and flattening. Magnetic resonance imaging may be used to diagnose a stroke caused by blocked cerebral vessels from sickled cells. Doppler studies may be used to assess for deep vein thromboses. Other tests may be indicated, such as a chest X-ray, to diagnose infection or an enlarged heart.
NURSING AND COLLABORATIVE MANAGEMENT: SICKLE CELL DISEASE
Multidisciplinary care for the patient with SCD is directed towards alleviating the symptoms from the complications of the disease, minimising end-organ damage and promptly treating serious sequelae, such as acute chest syndrome, which can lead to immediate death.22 There is no specific treatment for the disease. Patients with SCD should be taught to avoid high altitudes, maintain adequate fluid intake and treat infections promptly. Pneumovax, Haemophilus influenzae, influenza and hepatitis immunisations should be administered. Chronic leg ulcers may be treated with bed rest, antibiotics, warm saline soaks, mechanical or enzyme debridement and grafting if necessary. Priapism is managed with pain medication and drugs such as nifedipine.
Sickle cell crises may require hospitalisation. Oxygen may be administered to treat hypoxia and control sickling. Rest is instituted to reduce metabolic requirements. Fluids and electrolytes are administered to reduce blood viscosity and maintain renal function. Transfusion therapy is indicated when an aplastic crisis occurs. These patients, like those with thalassaemia major, may require chelation therapy to reduce transfusion-produced iron overload.
Pain management in sickle cell crisis
CLINICAL PRACTICE
Situation
A 21-year-old African American male is admitted to the emergency department in sickle cell crisis with complaints of excruciating pain. He is known to several of the nurses and doctors in the department. One of the nurses remarks to you that it must be time for his ‘fix’ of narcotics.
Important points for consideration
• The experience of pain is subjective and experts in pain management agree that ‘pain is what the patient says it is’.
• Acute or chronic pain can have serious and debilitating physical and psychological effects for patients.
• Persons experiencing chronic pain have different physiological and psychological responses to pain and to pain medications.
• The nurse’s duty to prevent harm and provide benefit to patients is undermined when patient’s complaints of unrelieved pain are attributed to attention-seeking or drug-seeking behaviour.
• People with chronic pain are often stigmatised due to lack of knowledge on the part of healthcare providers or long-term use of narcotics or other pain medications.
• The nurse’s obligation to provide holistic individualised care to patients is compromised when stereotypes or myths interfere with the ability to accurately assess a patient’s problems.
Undertreatment of sickle cell pain is a major problem. Lack of understanding can lead healthcare professionals to underestimate how much pain these patients suffer. Some patients feel their reports of pain are not taken seriously.23 During an acute crisis, optimal pain control usually includes large doses of continuous (rather than as-needed) opioid analgesics along with breakthrough analgesia, often in the form of patient-controlled analgesia (PCA). (PCA is discussed in Ch 8.) Morphine is the drug of choice. As patients may be experiencing different types and sites of pain, a multimodal and multidisciplinary approach is often needed.24 Adjunctive measures, such as non-steroidal anti-inflammatory agents, antineuropathic pain medications (e.g. tricyclic antidepressants, antiseizure medications), local anaesthetics, nerve blocks, transcutaneous electrical nerve stimulation (TENS) and/or acupuncture may be used. After discharge, patients will often continue on oral opioid analgesics.
Infection is a frequent complication and must be treated. Pain is the most common symptom of patients with SCD seeking medical care. Patients with acute chest syndrome are treated with broad-spectrum antibiotics, oxygen therapy and fluid therapy. Because these patients have an increased need for folic acid, it is important for them to obtain daily supplementation. Blood transfusions should be used judiciously to treat a crisis. They have little, if any, role in treatment between crises. In general, iron therapy is not indicated.
Although many antisickling agents have been tried, hydroxyurea is the only one that has been shown to be clinically beneficial. However, the indication as to when to start it has yet to be determined.25 This drug increases the production of haemoglobin F (fetal haemoglobin), decreases the reactive neutrophil count, increases erythrocyte volume and hydration and alters the adhesion of sickle erythrocytes to the endothelium. The increase in Hb F is accompanied by a reduction in haemolysis, an increase in haemoglobin concentration and a decrease in sickled cells and painful crises.26
Haematopoietic stem cell transplantation (HSCT) is the only available treatment that can cure some patients with SCD. However, the selection of appropriate recipients, the scarcity of appropriate donors, the risk and its cost-effectiveness limit its use. (HSCT is discussed in Ch 15.) Recent advances in gene therapy technology provide some promise for the future treatment of SCD.27 (Gene therapy is discussed in Ch 13.)
Patient teaching and support are important in the long-term care of the patient with SCD. The patient and family must understand the basis of the disease and the reasons for supportive care. One source of information available to patients is Thalassaemia Australia (see Resources on p 805). The patient must be taught ways to avoid crises, which include taking steps to avoid dehydration and to reduce the chance of developing hypoxia—for example, by avoiding high altitudes and seeking medical attention quickly to counteract problems such as upper respiratory tract infections. Education on pain control is also needed because the pain during a crisis may be severe and often requires considerable analgesia. Minor pain episodes that are not associated with infection or other symptoms warranting medical attention can sometimes be managed at home.
Recurrent episodes of severe acute pain and unrelenting chronic pain can be profoundly disabling and depressing. Occupational therapists and physiotherapists can help the patient achieve optimum physical functioning and independence; a psychologist may be able to use cognitive behavioural therapy to help patients with SCD to cope with anxiety and depression. Because there are often these additional quality-of-life issues, the nurse can play an important role in ensuring that these needs are met through appropriate referrals. Clinical practice guidelines are available from the website of the Royal Children’s Hospital, Melbourne (see Resources on p 805).
Acquired haemolytic anaemia
Extrinsic causes of haemolysis can be separated into three categories: (1) physical factors; (2) immune reactions; and (3) infectious agents and toxins.
Physical destruction of RBCs results from the exertion of extreme force on the cells. Traumatic events causing disruption of the RBC membrane include haemodialysis, extracorporeal circulation used in cardiopulmonary bypass and prosthetic heart valves. In addition, the force needed to push blood through abnormal vessels, such as those that have been burned or affected by angiopathic disease (e.g. diabetes mellitus), may physically damage RBCs.
Antibodies may destroy RBCs by the mechanisms involved in antigen–antibody reactions. The reactions may be of an isoimmune or autoimmune type. Isoimmune reactions occur when antibodies develop against antigens from another person. Blood transfusion reactions typify this response, when the recipient’s antibodies haemolyse donor cells. Autoimmune reactions result when individuals develop antibodies against their own RBCs. Autoimmune haemolytic reactions may be idiopathic, developing with no prior haemolytic history as a result of the immunoglobulin IgG covering the RBCs, or secondary to other autoimmune diseases (e.g. systemic lupus erythematosus), leukaemia, lymphoma or medications (penicillin, indomethacin, phenylbutazone, quinidine, quinine and methyldopa).
Infectious agents foster haemolysis in four ways: (1) by invading the RBC and destroying its contents (e.g. parasites, such as in malaria); (2) by releasing haemolytic substances (e.g. Clostridium perfringens); (3) by generating an antigen–antibody reaction; and (4) by contributing to splenomegaly as a means of increasing removal of damaged RBCs from the circulation. Various agents may be toxic to RBCs and cause haemolysis. These haemolytic toxins involve chemicals such as oxidative drugs, arsenic, lead, copper and snake venom.
Laboratory findings in haemolytic anaemia are presented in Table 30-5. Treatment and management of acquired haemolytic anaemias involve general supportive care until the causative agent can be eliminated or at least rendered less injurious to the RBCs. Because a haemolytic crisis (where RBC destruction is greater than or exceeds RBC production) is a potential consequence, the nurse needs to be ready to institute appropriate emergency therapy. Supportive care may include administering corticosteroids and blood products or removing the spleen.
Haemochromatosis
Haemochromatosis is an autosomal recessive disease characterised by increased intestinal iron absorption and, as a result, increased tissue iron deposition (see the Health disparities box).28 It is the most common genetic disorder among Caucasians, with an incidence of 3–5 per 1000 people of European ancestry. It is one of the most common inborn errors of iron metabolism. The normal range for total body iron is 13–31 μmol/L. Individuals with haemochromatosis accumulate iron at a rate of 0.5–1.0 g each year and may exceed total iron concentrations of 250 μmol/L. Symptoms of haemochromatosis usually develop between 40 and 60 years of age. In addition to the primary genetic defect, haemochromatosis occurs secondary to diseases such as thalassaemia and sideroblastic anaemia (where bone marrow produces ringed sideroblasts [atypical, abnormal nucleated erythroblasts] rather than RBCs). It may also be caused by liver disease and multiple blood transfusions.29
HEALTH DISPARITIES
Genetic testing
• Genetic testing recommended for all first-degree relatives of people with the disease
• Useful diagnostic tests include serum iron concentration, total iron-binding capacity and transferrin saturation
• Liver biopsy, once considered the gold standard diagnostic test, is primarily used to quantify iron deposition and estimate the prognosis and extent of disease
Early symptoms are non-specific and include fatigue, arthralgia, impotence, abdominal pain and weight loss. Later, the excess iron accumulates in the liver and causes liver enlargement and eventually cirrhosis. Then other organs become affected, resulting in diabetes mellitus, skin pigment changes (bronzing), cardiac changes (e.g. cardiomyopathy), arthritis and testicular atrophy. Physical examination reveals an enlarged liver and spleen and pigmentation changes in the skin. Laboratory values demonstrate an elevated serum iron, TIBC and serum ferritin levels. Molecular testing for known genetic mutations is used clinically to confirm the diagnosis. If this testing is not definitive, a liver biopsy can quantify the amount of iron and establish the diagnosis.
The goal of treatment is to remove excess iron from the body and minimise any symptoms the patient may have. Iron removal is achieved by removing 500 mL of blood each week (known as venesection) for 2–3 years until the iron stores in the body are depleted. Then, less frequent removal of blood is needed to maintain iron levels within normal limits. Management of organ involvement (e.g. diabetes mellitus, heart failure) is the same as conventional treatment for these problems. Dietary modifications may also assist in the reduction of iron accumulation, such as avoidance of vitamin C and iron supplements, uncooked seafood and iron-rich foods. The most common causes of death are cirrhosis, liver failure, hepatic carcinoma and cardiac failure. With early diagnosis and treatment, life expectancy is normal. However, many cases go undetected and untreated. Those with haemochromatosis in Australasia can download information and contact others with the condition from the website of the Haemochromatosis Society Australia (see Resources on p 805).
Polycythaemia
Polycythaemia is the production and presence of increased numbers of RBCs. The increase in RBCs can be so great that blood circulation is impaired as a result of the increased blood viscosity (hyperviscosity) and volume (hypervolaemia).
AETIOLOGY AND PATHOPHYSIOLOGY
The two types of polycythaemia are primary polycythaemia, or polycythaemia vera, and secondary polycythaemia (see Fig 30-5). Their aetiologies and pathogenesis differ, although their complications and clinical manifestations are similar.
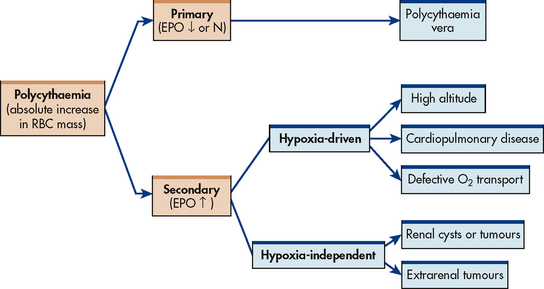
Figure 30-5 Differentiating between primary and secondary polycythaemia. EPO, erythropoietin; N, normal.
Polycythaemia vera is considered a chronic myeloproliferative disorder arising from a chromosomal mutation in a single pluripotent stem cell. Therefore, not only are RBCs involved but also WBCs and platelets, leading to increased production of each of these cell types. The disease develops insidiously and follows a chronic, vacillating course. The median age at diagnosis is 60 years old with a slightly male predominance. No strong evidence supports disease association with environmental exposure, although some links have been suggested. With this myeloproliferative disorder the patient has enhanced blood viscosity and blood volume and congestion of organs and tissues with blood. These patients have hypercoagulopathies that predispose them to clotting. Splenomegaly and hepatomegaly are common.
Secondary polycythaemia can be either hypoxia-driven or hypoxia-independent. In the former, hypoxia stimulates EPO production in the kidneys, which in turn stimulates erythrocyte production. The need for oxygen may be due to high altitude, pulmonary disease, cardiovascular disease, alveolar hypoventilation, defective oxygen transport or tissue hypoxia. EPO levels may return to normal once the haemoglobin is stabilised at a higher level. In this situation, secondary polycythaemia is a physiological response in which the body tries to compensate for a problem rather than a pathological response. (Hypoxia-driven secondary polycythaemia is discussed in the section on chronic obstructive pulmonary disease in Ch 28.) In hypoxia-independent secondary polycythaemia, EPO is produced by a malignant or benign tumour tissue. Serum EPO levels often remain elevated in these situations.
CLINICAL MANIFESTATIONS AND COMPLICATIONS
Circulatory manifestations of polycythaemia vera occur because of the hypertension caused by hypervolaemia and hyperviscosity. They are often the first symptoms and include subjective complaints of headache, vertigo, dizziness, tinnitus and visual disturbances. Generalised pruritus (often exacerbated by a hot bath) may be a striking symptom and is related to histamine release from an increased number of basophils. Paraesthesias and erythromelalgia (painful burning and redness of the hands and feet) may also be present. In addition, the patient may experience angina, heart failure, intermittent claudication and thrombophlebitis, which may be complicated by embolisation. These manifestations are caused by blood vessel distension, impaired blood flow, circulatory stasis, thrombosis and tissue hypoxia caused by the hypervolaemia and hyperviscosity. The most common serious acute complication is stroke secondary to thrombosis.30
Haemorrhagic phenomena caused by either vessel rupture from overdistension or inadequate platelet function may result in petechiae, ecchymoses, epistaxis or GI bleeding. Haemorrhage can be acute and catastrophic. Hepatomegaly and splenomegaly from organ engorgement may contribute to patient complaints of satiety and fullness. The patient may also experience pain from a peptic ulcer caused by either increased gastric secretions or liver and spleen engorgement. A ruddy complexion (plethora) may also be present. Hyperuricaemia is caused by the increase in RBC destruction that accompanies excessive RBC production. Uric acid is one of the products of cell destruction. As RBC destruction increases, uric acid production also increases, thus leading to hyperuricaemia. This problem may cause a form of gout.
DIAGNOSTIC STUDIES
The following laboratory manifestations are seen in the patient with polycythaemia vera: (1) elevated haemoglobin and RBC count with microcytosis; (2) low to normal EPO level (secondary polycythaemia will have a high level); (3) elevated WBC count with basophilia; (4) elevated platelets (thrombocytosis) and platelet dysfunction; (5) elevated leucocyte alkaline phosphatase, uric acid and vitamin B12 levels; and (6) elevated histamine levels. Bone marrow examination in polycythaemia vera shows hypercellularity of RBCs, WBCs and platelets. Splenomegaly is found in 90% of patients with polycythaemia vera but does not accompany secondary polycythaemia.
MULTIDISCIPLINARY CARE
Once the diagnosis of polycythaemia vera is made, treatment is directed towards reducing blood volume and viscosity and bone marrow activity. Phlebotomy is the mainstay of treatment. The aim of phlebotomy is to reduce the haematocrit and to keep it less than 0.45–0.48. Generally, at the time of diagnosis 300–500 mL of blood may be removed every other day until the haematocrit is reduced to normal levels. An individual managed with repeated phlebotomies eventually becomes deficient in iron, although this effect is rarely symptomatic. Iron supplementation should be avoided. Hydration therapy is used to reduce the blood’s viscosity. Myelosuppressive agents such as busulfan, hydroxyurea, melphalan and radioactive phosphorus may be given to inhibit bone marrow activity. However, due to their side effects, these treatments are usually reserved for patients at high risk for complications (e.g. thromboses). Newer, targeted therapies (targeting the JAK2 mutation) are being studied.30 Paroxetine or low-dose aspirin may be used adjunctively to alleviate symptoms of erythromelalgia. Interferon-α is of particular use in women of childbearing age or those with intractable pruritus. Allopurinol may reduce the number of acute gouty attacks.31
NURSING MANAGEMENT: POLYCYTHAEMIA
Polycythaemia vera is not preventable. However, because secondary polycythaemia is generated by any source of hypoxia, maintaining adequate oxygenation may prevent problems. Therefore, controlling chronic pulmonary disease, stopping smoking and avoiding high altitudes may be important.
When acute exacerbations of polycythaemia vera develop, the nurse has several responsibilities. Depending on the institution’s policies, the nurse may either assist with or perform the phlebotomy. Fluid intake and output must be evaluated during hydration therapy to avoid fluid overload (which further complicates the circulatory congestion) and underhydration (which can cause the blood to become even more viscous). If myelosuppressive agents are used, the nurse must administer the drugs as ordered, observe the patient and teach the patient about medication side effects.
Assessment of the patient’s nutritional status in collaboration with the dietician may be necessary to offset the inadequate food intake that can result from GI symptoms of fullness, pain and dyspepsia. Activities must be instituted to decrease thrombus formation. The relative immobility normally imposed by hospitalisation puts the patient at risk of thrombus formation. Active or passive leg exercises and ambulation when possible should be initiated.
Because of its chronic nature, polycythaemia vera requires ongoing evaluation. Phlebotomy may need to be done every 2–3 months, reducing the blood volume by about 500 mL each time. The nurse must evaluate the patient for the development of complications.
Although the incidence is low, myelofibrosis and leukaemia develop in some patients with polycythaemia vera (10% and 5%, respectively). These may be caused by the chemotherapeutic drugs used to treat the disease, or they may be secondary to a disorder in the stem cells that progresses to erythroleukaemia. The major cause of morbidity and mortality from polycythaemia vera is related to thrombosis (e.g. stroke).
PROBLEMS OF HAEMOSTASIS
The homeostatic process involves the vascular endothelium, platelets and coagulation factors, which normally function together to arrest haemorrhage and repair vascular injury. (These mechanisms are described in Ch 29.) Disruption in any of these components may result in bleeding or thrombotic disorders.
Three major disorders of haemostasis discussed in this section are: (1) thrombocytopenia (low platelet count); (2) haemophilia and von Willebrand’s disease (inherited disorders of specific clotting factors); and (3) disseminated intravascular coagulation.
Thrombocytopenia
AETIOLOGY AND PATHOPHYSIOLOGY
Thrombocytopenia is a reduction of platelets below 150 × 109/L. Acute, severe or prolonged decreases from this normal range can result in abnormal haemostasis that manifests as prolonged bleeding from minor trauma to spontaneous bleeding without injury.
Platelet disorders can be inherited (e.g. Wiskott-Aldrich syndrome), but the vast majority are acquired. The causes of acquired disorders include autoimmune diseases, increased platelet consumption, splenomegaly, marrow suppression (infectious or drug-mediated) and bone marrow failure (see Box 30-5). Most acquired abnormalities occur following ingestion of certain foods, herbs or drugs (see Box 30-6). Although some agents are directly myelosuppressive (e.g. chemotherapy, ganciclovir), the usual mechanism of thrombocytopenia caused by foods, herbs or drugs is accelerated platelet destruction caused by drug-dependent antibodies. Antibodies attack the platelets when the offending agent binds to a platelet surface glycoprotein. A careful review of the patient’s history helps distinguish these causes from one of the causes mentioned below. For example, quinine is in tonic water and in many herbal preparations. In addition, some drugs can affect platelet aggregation. Aspirin doses as low as 81 mg (a baby aspirin) can alter platelet aggregation. Normal function is restored with the generation of newly formed platelets.
BOX 30-5 Causes of thrombocytopenia
Acquired
Non-immune
– Thrombotic thrombocytopenic purpura
– Disseminated intravascular coagulopathy
Turbulent blood flow (haemangiomas, abnormal cardiac valves, intraaortic balloon pumps)
BOX 30-6 Food, drug and herbal causes of thrombocytopenia*
• Non-steroidal anti-inflammatory drugs: ibuprofen, indomethacin, naproxen
• Antibiotics: penicillins, cephalosporins, sulfonamides
• Other anti-infectives: rifampicin, ganciclovir, amphotericin
• Analgesics: aspirin and aspirin-containing drugs, paracetamol
• Antipsychotics and antiseizures: haloperidol, valproate, lithium
• Platelet glycoprotein inhibitors: abciximab
• Histamine H2 antagonists: cimetidine, ranitidine
• Spices: ginger, cumin, turmeric, cloves
• Vitamins: vitamin C, vitamin E
• Herbs: angelica, bilberry, evening primrose, feverfew, garlic, ginger, ginkgo biloba, ginseng, goldenseal
• Quinine compounds: tonic water, china bark, peruvian bark, yellow cinchona
*List is not all-inclusive.
Immune thrombocytopenic purpura
The most common acquired thrombocytopenia is a syndrome of abnormal destruction of circulating platelets termed immune thrombocytopenic purpura (ITP). It was originally termed idiopathic thrombocytopenic purpura because its cause was unknown. However, it is now known that ITP is an autoimmune disease. In ITP, platelets are coated with antibodies. Although these platelets function normally, when they reach the spleen, the antibody-coated platelets are recognised as foreign and are destroyed by macrophages. In addition, it is now known that decreased platelet production and the presence of infection, such as Helicobacter pylori or viral infection, may contribute to this disorder.32
Platelets normally survive 8–10 days, but in ITP they survive only 1–3 days. Chronic ITP occurs most commonly in women between 20 and 40 years of age. Chronic ITP has a gradual onset, and transient remissions occur.
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura (TTP) is an uncommon syndrome characterised by haemolytic anaemia, thrombocytopenia, neurological abnormalities, fever (in the absence of infection) and renal abnormalities; not all features are always present in all patients. Because it is almost always associated with haemolytic–uraemic syndrome (HUS), it is often referred to as TTP–HUS. The disease is associated with enhanced agglutination of platelets, which form microthrombi that deposit in arterioles and capillaries. In most cases, the cause is due to the deficiency of a plasma enzyme (ADAMTS13) that usually breaks down the von Willebrand (vWF) clotting factor into normal size (vWF is the most important protein mediating platelet adhesion to damaged endothelial cells). Without the enzyme, unusually large vWF multimers attach to activated platelets, thereby promoting platelet aggregation.
TTP is seen primarily in adults between 20 and 50 years of age, with a slight female predominance. The syndrome may be idiopathic (thought to be due to an autoimmune disorder against ADAMTS13), but may be due to certain drug toxicities (e.g. chemotherapy, cyclosporin, quinine, oral contraceptives, valaciclovir), pregnancy, infection or the result of a known autoimmune disorder such as systemic lupus erythematosus or scleroderma. TTP is a medical emergency because bleeding and clotting occur simultaneously.33
Heparin-induced thrombocytopenia and thrombosis syndrome
One of the risks associated with the broad and increasing use of heparin is the development of the life-threatening condition called heparin-induced thrombocytopenia and thrombosis syndrome (HITTS), also called white clot syndrome or HIT. HITTS can be mild (type I) or severe (type II). An estimated 8–17% of patients on heparin therapy develop HITTS.34 A lower incidence is seen in patients receiving porcine preparations of heparin as compared with other types. The major clinical problem of HITTS is venous thrombosis; arterial thrombosis can also develop. Deep vein thromboses and pulmonary emboli most commonly result as a complication of the thromboses. Additional complications may include arterial vascular infarcts resulting in skin necrosis, stroke and end-organ damage, such as to the kidneys. There are rarely symptoms of bleeding because the platelet count rarely drops below 60 × 109/L.
Type I HITTS is usually of no clinical consequence. In type II HITTS, platelet destruction and vascular endothelial injury are the two major responses to what is believed to be an immune-mediated response to heparin. Platelet factor 4 (PF4) binds to heparin; this complex then binds to the platelet surface, leading to further platelet activation and release of more PF4, thus creating a positive feedback loop. Antibodies are created against this complex and they are removed prematurely from circulation, leading to thrombocytopenia and platelet–fibrin thrombi. Platelet aggregation also induces heparin to be neutralised. Thus more heparin is required to maintain therapeutic activated partial thromboplastin times.
CLINICAL MANIFESTATIONS
Many patients with thrombocytopenia are usually asymptomatic. The most common symptom is bleeding, usually mucosal or cutaneous. Mucosal bleeding may be manifest as epistaxis and gingival bleeding, and large bullous haemorrhages may appear on the buccal mucosa due to the lack of vessel protection by the submucosal tissue. Bleeding into the skin is manifested as petechiae, purpura or superficial ecchymoses (see Fig 30-6). Petechiae are small, flat, pinpoint red or reddish brown micro-haemorrhages. When the platelet count is low, RBCs may leak out of the blood vessels and into the skin to cause petechiae. When petechiae are numerous, the resulting reddish skin bruise is called purpura. Larger purplish lesions caused by haemorrhage are termed ecchymoses. Ecchymoses may be flat or raised; pain and tenderness are sometimes present.
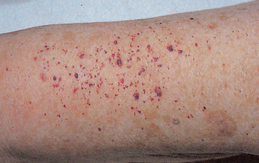
Figure 30-6 Acute idiopathic thrombocytopenic purpura commonly manifests with purpuric lesions of this kind, although they may often be more widespread by the time medical attention is sought.
Source: Forbes CD, Jackson WF. Color atlas and text of clinical medicine. 3rd edn. London: Mosby; 2003.
Prolonged bleeding after routine procedures such as venipuncture or IM injection may also indicate thrombocytopenia. Because the bleeding may be internal, the nurse must also be aware of manifestations that reflect this type of blood loss, including weakness, fainting, dizziness, tachycardia, abdominal pain and hypotension.
The major complication of thrombocytopenia is haemorrhage. The haemorrhage may be insidious or acute, and internal or external. It may occur in any area of the body, including the joints, retina and brain. Cerebral haemorrhage may be fatal in persons with ITP. Insidious haemorrhage may be first detected by discovering the anaemia that accompanies blood loss.
Because thrombocytopenia can be accompanied by vascular thromboses in some of these disorders, signs and symptoms of vascular ischaemic problems can manifest (see Ch 37). For example, subtle confusion, headache or even serious manifestations such as seizures and coma due to TTP-related thrombosis may be seen. Signs and symptoms may be subtle, so astute and thorough assessment of the patient is essential.
DIAGNOSTIC STUDIES
The platelet count is decreased in cases of thrombocytopenia. Any reduction below 150 × 109/L may be termed thrombocytopenia. However, prolonged bleeding from trauma or injury does not usually occur until the platelet count is less than 50 × 109/L. When the count drops below 20 × 109/L, spontaneous, life-threatening haemorrhages (e.g. intracranial bleeding) can occur. Platelet transfusions are generally not recommended until the count is below 20 × 109/L unless the patient is actively bleeding. Examination of the peripheral blood smear may assist in distinguishing acquired disorders such as ITP and TTP from congenital disorders, the latter of which may be indicated by abnormally sized platelets.
Laboratory tests that assess secondary haemostasis or coagulation, such as the prothrombin time (PT) and activated partial thromboplastin time (APTT), can be normal even in severe thrombocytopenia. If they are elevated, this may point towards disseminated intravascular coagulation. Bone marrow examination is done to rule out production problems as the cause of thrombocytopenia (e.g. leukaemia, aplastic anaemia, other myeloproliferative disorders). Specific assays, such as ITP-positive antigen specific assay or 14C-serotonin release assay for HIT, can be done to assist with the diagnosis. In TTP, testing for deficiency of ADAMTS13 is not widely clinically available, so an increase of lactate dehydrogenase (LDH) is used to help establish the diagnosis. Bone marrow analysis is done if other tests are inconclusive, especially in older patients due to a higher suspicion of an underlying bone marrow disorder. When destruction of circulating platelets is the aetiology, bone marrow analysis shows megakaryocytes (precursors of platelets) to be normal or increased, even though circulating platelets are reduced. The absence or decreased numbers of megakaryocytes on bone marrow biopsy is consistent with thrombocytopenia due to decreased bone marrow production (e.g. aplastic anaemia). Special blood analyses using flow cytometry and other techniques can detect antiplatelet antibodies as the source of destruction.
The nurse needs to closely monitor the platelet count and other coagulation studies. In addition, the nurse should monitor the patient’s haemoglobin and haematocrit levels and observe the patient for cardiopulmonary distress and other manifestations of anaemia. When thrombocytopenia occurs with anaemia characterised by altered RBC morphology, including spherocytes (small, globular, completely haemoglobinated erythrocytes), fragmented cells (schistocytes) and pronounced reticulocytosis, a diagnosis of TTP should be suspected. These findings are partially a result of intravascular fibrin deposition causing a ‘slicing’ of RBCs. In TTP, thrombocytopenia may be severe, but coagulation studies are normal.
MULTIDISCIPLINARY CARE
Multidisciplinary care of thrombocytopenia varies depending on the aetiology of the thrombocytopenia. Discussion of management strategies for these different aetiologies follows (see Box 30-7).
MULTIDISCIPLINARY CARE
Collaborative therapy
Immune thrombocytopenic purpura
Immunosuppressives (e.g. rituximab, cyclosporin, cyclophosphamide, azathioprine, mycophenolate mofetil)
Thrombotic thrombocytopenic purpura
Plasmapheresis (plasma exchange)
Immune thrombocytopenic purpura
Multiple therapies are used to manage the patient with ITP. If the patient is asymptomatic, therapy may not be utilised unless the patient’s platelet count is less than 30 × 109/L. Corticosteroids (e.g. prednisolone) are used initially to treat ITP because of their ability to suppress the phagocytic response of splenic macrophages. This alters the spleen’s recognition of platelets and increases the life span of the platelets. In addition, corticosteroids depress antibody formation. Corticosteroids also reduce capillary fragility and bleeding time. The mechanism of action for this response is poorly understood. If there are neurological manifestations related to intracranial bleeding, high-dose methylprednisolone can be administered IV. Methylprednisolone has also been used when patients are resistant to prednisolone.
Splenectomy is indicated if the patient does not respond to prednisolone initially or requires unacceptably high doses to maintain an adequate platelet count. Approximately 80% of patients benefit from splenectomy, resulting in a complete or partial remission.35 The effectiveness of splenectomy is based on four factors. First, the spleen contains an abundance of the macrophages that sequester and destroy platelets. Second, structural features of the spleen enhance antibody-coated platelets and macrophage interaction. Third, some antibody synthesis occurs in the spleen; thus antiplatelet antibodies decrease after splenectomy. Fourth, the spleen normally sequesters approximately one-third of the platelets, so its removal increases the number of platelets in circulation. Post-splenectomy refractory patients should be evaluated for an accessory spleen and have subsequent medical therapies tailored to the individual.
High doses of IV immunoglobulin (IVIG) and a component of IVIG, anti-Rh (D) (anti-D, WinRho), may be used in the patient who is unresponsive to corticosteroids or splenectomy. These agents work by competing with the antiplatelet antibodies for macrophage receptors. They effectively raise the platelet count, but the beneficial effects may be temporary.
Danazol, an androgen, is often used along with steroids in some patients. Although the mechanism is not totally understood, it is suggested that danazol increases CD4 T cells, thereby reducing the immune response. Immunosuppressive therapy used in refractory cases includes rituximab, cyclophosphamide, azathioprine, cyclosporin and mycophenolate mofetil. High-dose cyclophosphamide and combination chemotherapy is third-line therapy.
Platelet transfusions may be used to increase platelet counts in cases of life-threatening haemorrhage. Platelets should not be administered prophylactically because of the possibility of antibody formation. The usual indication for administering platelets is a platelet count <10 × 109/L or if there is bleeding or prior to a procedure. Several studies indicate that the risk of bleeding may not be substantial until the count is <5 × 109/L. ABO compatibility is not a necessary prerequisite for platelet transfusions. However, after multiple platelet transfusions, a patient may develop anti-HLA antibodies to the transfused platelets.
By using lymphocyte typing to match HLA types of the donor and the recipient, multiple platelet transfusions can be given with fewer complications. In addition, the patient may be premedicated with an antihistamine (e.g. diphenhydramine) and hydrocortisone to decrease the possibility of reacting to platelet transfusions. Aminocaproic acid, an antifibrinolytic agent, may be used for severe bleeding. Aspirin and aspirin-containing compounds should be avoided in the patient with thrombocytopenia.
Thrombotic thrombocytopenic purpura
TTP may be treated in a variety of ways. The first step is to treat the underlying disorder (e.g. infection) or remove the causative agent, if identified. If untreated, TTP usually results in irreversible renal failure and death. Corticosteroids are used initially. Plasma exchange or plasmapheresis (see Ch 13) may be needed to aggressively reverse the process.33 This reverses platelet consumption by supplying the appropriate vWFs and enzyme (ADAMTS13) and removing the large vWF molecules binding with platelets. Treatment should be continued daily until the patient’s platelet count normalises and haemolysis has ceased. Splenectomy, corticosteroids, dextran (antiplatelet agent) and vincristine or vinblastine have also been used with success. Recently, immunosuppressive therapies such as cyclophosphamide and rituximab have been used. The administration of platelets is generally contraindicated because this may lead to new vWF–platelet complexes and increased clotting.
Heparin-induced thrombocytopenia and thrombosis syndrome
Heparin must be discontinued when HITTS is first recognised, which is usually when the patient’s platelet count has fallen 50% or more from its baseline or a thrombus forms while the patient is on heparin therapy. Heparin flushes for vascular catheters should also be stopped.
To maintain anticoagulation, the patient should be started on a direct thrombin inhibitor. Warfarin may be started when the platelet count has reached 100 × 109/L. If the clotting is severe, the most commonly used treatment modalities are plasmapheresis to clear the platelet-aggregating IgG from the blood, protamine sulfate to interrupt the circulating heparin, thrombolytic agents to treat the thromboembolic events and surgery to remove clots. Platelet transfusions are not effective because they may enhance thromboembolic events. Patients who have had HITTS should never be given heparin or low-molecular-weight heparin. This should be clearly marked on the patient’s medical record.
Acquired thrombocytopenia from decreased platelet production
The management of acquired thrombocytopenia is based on identifying the cause and treating the disease or removing the causative agent. If the precipitating factor is unknown, the patient may receive corticosteroids. Platelet transfusions are given if life-threatening haemorrhage develops. Splenectomy is not used because the spleen is not contributing to this type of thrombocytopenia.
Often, acquired thrombocytopenia is caused by another underlying condition (e.g. aplastic anaemia, leukaemia) or therapy used to treat another problem. For example, in acute leukaemia all blood cell types may be depressed. Additionally, the patient may receive chemotherapeutic drugs that cause bone marrow suppression. If the patient can be adequately supported throughout the course of chemotherapy-induced thrombocytopenia, the thrombocytopenia will also resolve.
Since thrombopoietin (a haematopoietic growth factor that stimulates the bone marrow to make platelets) was discovered there has been much progress in the development of recombinant thrombopoietin to be used for therapy.36
NURSING MANAGEMENT: THROMBOCYTOPENIA
Nursing assessment
Subjective and objective data that should be obtained from a patient with thrombocytopenia are presented in Table 30-7.
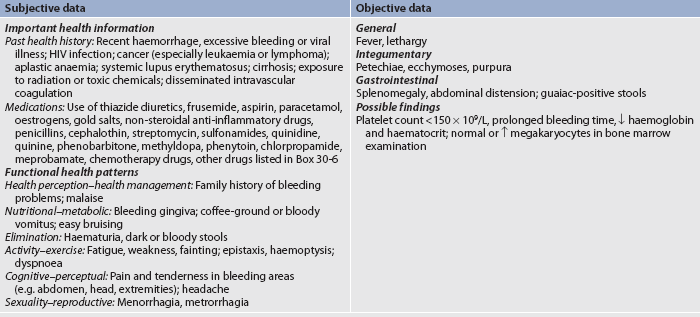
Nursing diagnoses
Nursing diagnoses for the patient with thrombocytopenia may include, but are not limited to, those presented in NCP 30-2.
Planning
The overall goals are that the patient with thrombocytopenia will: (1) have no gross or occult bleeding; (2) maintain vascular integrity; and (3) manage home care to prevent any complications related to an increased risk of bleeding.
Nursing implementation
Health promotion
It is important for the nurse to discourage excessive use of over-the-counter medications known to be possible causes of acquired thrombocytopenia. Many medications contain aspirin as an ingredient. Aspirin reduces platelet adhesiveness, thus contributing to bleeding.
The nurse should also encourage patients to have a complete medical evaluation if manifestations of bleeding tendencies (e.g. prolonged epistaxis, petechiae) develop. In addition, the nurse must observe for early signs of thrombocytopenia in the patient receiving cancer chemotherapy drugs.
Acute intervention
The goal during acute episodes of thrombocytopenia is to prevent or control haemorrhage (see NCP 30-2).5 In the patient with thrombocytopenia, bleeding is usually from superficial sites; deep bleeding (into muscles, joints and abdomen) usually occurs only when clotting factors are diminished. It is important to emphasise that a seemingly minor nose bleed or new petechiae may indicate potential haemorrhage and the healthcare provider should be notified. Bleeding from the posterior nasopharynx may be difficult to detect because the blood may be swallowed. If an IM or SC injection is unavoidable, the use of a small-gauge needle and application of direct pressure for at least 5–10 minutes after injection is indicated, or application of an ice pack may be helpful. The patient needs to understand the importance of adherence to self-care measures that reduce the risk of bleeding (see Box 30-8).
PATIENT & FAMILY TEACHING GUIDE
This instruction sheet explains precautions you should take to protect yourself when your platelet count is low. Please make sure to ask your healthcare provider specific precautions you should take that relate to your bleeding risk factors.
• Notify your healthcare provider of any manifestations of bleeding. These include:
• Ask your healthcare provider regarding restrictions in your normal activities, such as vigorous exercise, lifting weights etc. Generally, walking can be done safely and should be done with sturdy shoes or slippers. If you are weak and at risk of falling, get help or supervision when getting out of bed.
• Do not blow your nose forcefully; gently pat it with a tissue if needed. For a nosebleed, keep your head up and apply firm pressure to the nostrils and bridge of your nose. If bleeding continues, place an ice bag over the bridge of your nose and the nape of your neck. If you are unable to stop a nosebleed after 10 min, call your healthcare provider.
• Do not bend down with your head lower than your waist.
• Prevent constipation by drinking plenty of fluids and do not strain when having a bowel movement. Your healthcare provider may prescribe a stool softening agent. Do not use a suppository, enema or rectal thermometer without the permission of your healthcare provider.
• Shave only with an electric razor; do not use blades.
• Do not pluck your eyebrows or other body hair.
• Do not puncture your skin, such as getting tattoos or body piercing.
• Avoid using any medication that can prolong clotting, such as aspirin. Other medications and herbs can have similar effects. If you are unsure about any medication, ask your healthcare provider or pharmacist about it in relation to your thrombocytopenia.
• Use a soft bristle toothbrush to prevent injuring the gums. Flossing is also usually safe if it is done gently using thin tape floss. Do not use alcohol-based mouthwashes as they can dry your gums and increase bleeding.
• Women who are menstruating should keep track of the number of pads that are used per day. When you start using more pads per day than usual or bleed for more days, notify your healthcare provider. Do not use tampons; use sanitary pads only.
• Ask your healthcare provider before you have any invasive procedures done, such as a dental cleaning, manicure or pedicure.
In a woman with thrombocytopenia, menstrual blood loss may exceed the usual amount and duration. Counting sanitary pads used during menses is another important intervention to detect excess blood loss: 50 mL of blood will completely soak a sanitary pad. Suppression of menses with hormonal agents may be indicated during predictable periods of thrombocytopenia to reduce blood loss from menses (e.g. during chemotherapy and HSCT).
The proper administration of platelet transfusions is an important nursing responsibility. Platelet concentrates, derived from fresh whole blood, can increase the platelet level effectively. Centrifuging 500 mL of whole blood derives 1 unit of platelets, a yellow liquid that is usually 30–50 mL in volume. Platelet concentrates from multiple units of blood (usually from 6–8 different donors) can be pooled together for a single administration. The degree of increase or increment from a pooled platelet product varies widely and is usually measured by performing a platelet count within 1 hour following the transfusion.
Platelet transfusions can also be prepared by pheresing or removing the platelets from a single donor. This may be indicated when HLA-matched platelets are needed, especially for patients requiring multiple platelet transfusions. In this procedure, blood is removed from the donor, the platelets are removed and the rest of the blood is reinfused into the donor. This procedure results in 200–400 mL of platelets and plasma. Once acquired from a donor, platelets can be stored at room temperature for 1–5 days. Gentle agitation of the bag is useful to prevent the platelets from adhering to the plastic. In a severely immunocompromised patient, these products are also irradiated to further ensure WBC removal and prevent the complication of graft-versus-host disease (see Ch 13).
Again, it should be noted that many of these disorders may be accompanied by vascular clotting and appropriate assessment and management should be taken (see Ch 37).
Ambulatory and home care
The patient with ITP who is receiving corticosteroids should be monitored frequently for their response to therapy. If the ITP is reversed by splenectomy, there is usually no recurrence. The person with acquired thrombocytopenia must be taught to avoid causative agents when possible (see Box 30-7). If the causative agents cannot be avoided (e.g. chemotherapy), the patient should learn to avoid injury or trauma during these periods and to detect the clinical signs and symptoms of bleeding caused by thrombocytopenia (see Box 30-6). Patients with either ITP or acquired thrombocytopenia should have planned periodic medical evaluations to assess their status and to intercede in situations in which exacerbations and bleeding are likely to occur.
Haemophilia and von Willebrand’s disease
Haemophilia is a sex-linked recessive genetic disorder caused by defective or deficient coagulation factor (see the Health disparities box and Fig 13-3). (Sex-linked genetic disorders are discussed in Ch 13.) In Australia approximately 2000 males have haemophilia,37 and recent studies suggest that around 300 New Zealand residents have haemophilia.38 The two major forms of haemophilia, which can occur in mild-to-severe forms, are haemophilia A (classic haemophilia, factor VIII deficiency) and haemophilia B (Christmas disease, factor IX deficiency). Von Willebrand’s disease is a related disorder involving a deficiency of the von Willebrand coagulation protein. Factor VIII is synthesised in the liver and circulates as a complex with von Willebrand factor.
Haemophilia A is the most common form of haemophilia; it makes up approximately 80% of all cases. The incidence of haemophilia A is approximately 1 in 5000–10,000 males; haemophilia B is seen in 1 in 30,000–50,000 males. Von Willebrand’s disease is considered the most common congenital bleeding disorder in humans, with estimates as high as 1 or 2 in 100. However, because this disease can exist in mild-to-severe forms, life-threatening haemorrhage in patients is rare.36 The deficiencies and inheritance patterns of these three forms of inherited coagulopathies are compared in Table 30-8.
TABLE 30-8 Comparison of types of haemophilia
| Disorder | Deficiency | Inheritance pattern |
|---|---|---|
| Haemophilia A | Factor VIII | Recessive sex-linked (transmitted by female carriers, displayed almost exclusively in men) |
| Haemophilia B | Factor IX | Recessive sex-linked (transmitted by female carriers, displayed almost exclusively in men) |
| von Willebrand’s disease | VWF, various factor VIII deficiencies and platelet dysfunction | Autosomal dominant, seen in both sexes recessive (in severe forms of the disease) |
VWF, von Willebrand Factor.
HEALTH DISPARITIES
Clinical implications
• Female carriers will transmit the genetic defect to 50% of their sons, and 50% of their daughters will be carriers.
• Males with haemophilia will not transmit the genetic defect to their sons, but all of their daughters will be carriers.
• Female haemophilia can occur if a male with haemophilia mates with a female carrier. However, this is a rare situation.
• Clinical manifestations of haemophilia A and B are very similar.
• Replacement therapy is available for factors VIII and IX (see Table 30-10).
CLINICAL MANIFESTATIONS AND COMPLICATIONS
Clinical manifestations and complications related to haemophilia include: (1) slow, persistent, prolonged bleeding from minor trauma and small cuts (see Figs 30-7 and 30-8); (2) delayed bleeding after minor injuries (the delay may be several hours or days); (3) uncontrollable haemorrhage after dental extractions or irritation of the gingiva with a hard-bristle toothbrush; (4) epistaxis, especially after a blow to the face; (5) GI bleeding from ulcers and gastritis; (6) haematuria from GU trauma and splenic rupture resulting from falls or abdominal trauma; (7) ecchymoses and subcutaneous haematomas; (8) neurological signs, such as pain, anaesthesia and paralysis, which may develop from nerve compression caused by haematoma formation; and (9) haemarthrosis (bleeding into the joints) (see Fig 30-9), which may lead to joint injury and deformity severe enough to cause crippling (most commonly in the knees, elbows, shoulders, hips and ankles).
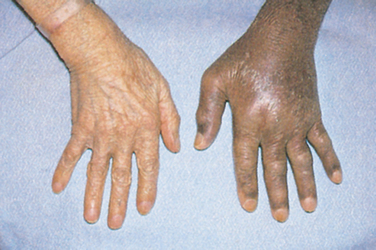
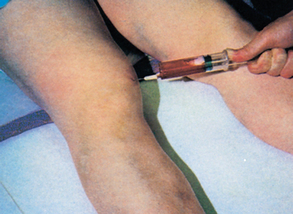
In children these symptoms may lead to the diagnosis. In adults, these developments may be the first sign of a newly diagnosed mild form of the disease that escaped detection through a childhood free of major injuries, dental procedures or surgeries. All clinical manifestations relate to bleeding, and any bleeding episode in persons with haemophilia may lead to a life-threatening haemorrhage.
Historically, haemophilia was a disease of childhood because of early death from complications. In the early 1900s the average life expectancy was 11 years. By the 1970s, advances in treatment enabled persons with haemophilia to have an average life expectancy of 68 years. Unfortunately, the acquired immunodeficiency syndrome (AIDS) epidemic and the HIV contamination of blood products in the 1980s caused the majority of haemophilia deaths in the late 1980s. However, longer-term survival (up to 72 years old) is now being observed because of improved preparation of replacement products, improved screening of blood donor populations and use of recombinant replacement factors.39
DIAGNOSTIC STUDIES
Laboratory studies are used to determine the type of haemophilia present. Any factor deficiency within the intrinsic system (factors VIII, IX, XI or XII or vWF) will yield the laboratory results presented in Table 30-9.
TABLE 30-9 Laboratory results in haemophilia
| Test | Comments |
|---|---|
| Prothrombin time | No involvement of extrinsic system |
| Thrombin time | No impairment of thrombin–fibrinogen reaction |
| Platelet count | Adequate platelet production |
| Partial thromboplastin time | Prolonged because of deficiency in any intrinsic clotting system factor |
| Bleeding time | Prolonged in von Willebrand’s disease because of structurally defective platelets; normal in haemophilia A and B because platelets not affected |
| Factor assays | Reduction of factor VIII in haemophilia A, VWF in von Willebrand’s disease; reduction of factor IX in haemophilia B |
VWF, von Willebrand factor.
MULTIDISCIPLINARY CARE
The goals of multidisciplinary care are to prevent and treat bleeding. Multidisciplinary care for persons with haemophilia or von Willebrand’s disease requires the provision of preventative care, the use of replacement therapy during acute bleeding episodes and as prophylaxis, and the treatment of the complications of the disease and its therapy.
Replacement of deficient clotting factors is the primary means of supporting the patient with haemophilia. In addition to treating acute crises, replacement therapy may be given before surgery and dental care as a prophylactic measure. Examples of replacement therapy are listed in Table 30-10. Fresh frozen plasma, once commonly used for replacement therapy, is rarely used today.
For mild haemophilia A and certain subtypes of von Willebrand’s disease, desmopressin acetate (also known as DDAVP), a synthetic analogue of vasopressin, may be used to stimulate an increase in factor VIII and vWF. This drug acts on endothelial cells to cause the release of vWF, which subsequently binds with factor VIII, thus increasing their concentration. It can be administered intravenously, subcutaneously or by intranasal spray. Beneficial effects (e.g. decreased bleeding time) of DDAVP, when administered IV, are seen within 30 minutes and can last for more than 12 hours. Because the effect of DDAVP is relatively short-lived, the patient must be closely monitored and repeated doses may be necessary. It is an appropriate therapy for minor bleeding episodes and dental procedures. The intranasal form may be indicated for home therapy for some patients with mild-to-moderate forms of the disease.40
Antifibrinolytic therapy (tranexamic acid) inhibits fibrinolysis by inhibiting plasminogen activation in the fibrin clot, thereby enhancing clot stability. These agents are useful therapeutic adjuncts to stabilise clots in areas of increased fibrinolysis, such as the oral cavity, and in patients with difficult episodes of epistaxis and menorrhagia. They are contraindicated in patients with haematuria as lack of fibrinolysis in the kidneys could lead to renal failure.
Complications of treatment of haemophilia include the development of inhibitors to factors VIII or IX, transfusion-transmitted infectious disorders, allergic reactions and thrombotic complications with the use of factor IX because it contains activated coagulation factors. Patients with vWF may also develop alloantibody against vWF, the infusion of which could cause life-threatening anaphylaxis; thus replacement factors for these patients should be devoid of vWF. Because of the improved virus-detecting processes and donor screening practices, the risk of HIV and hepatitis B and C transmission is greatly reduced.
The most common difficulties with acute management are starting factor replacement therapy too late and stopping it too soon. Generally, minor bleeding episodes should be treated for at least 72 hours. Surgery and traumatic injuries may need support for 10–14 days. Because of the short half-life of the factors, regular intermittent or continuous infusions have been used to manage bleeding episodes or expected traumatic procedures. With chronic use, development of inhibitors to the factor products has occurred and requires individualised expert patient management by the healthcare team.
Designated treatment centres have been established in Australia, New Zealand and many other countries to provide multidisciplinary care of haemophilia and related disorders. The multidisciplinary team provides optimal chronic disease management.
Gene therapy has been used on an experimental basis to treat haemophilia. These clinical trials have involved: (1) removing cells from the patient and genetically modifying them to secrete factor VIII or IX; and (2) injecting vectors with the genes for factors VIII and IX.40 (Gene therapy is discussed in Ch 13.)
NURSING MANAGEMENT: HAEMOPHILIA
Nursing implementation
Health promotion
Because of the hereditary nature of haemophilia, referral for genetic counselling is essential when considering preventative measures. This is especially important because many persons with haemophilia live into adulthood. Reproductive concerns and long-term effects are issues that the nurse should include in the patient’s care plan.
Acute intervention
Interventions are related primarily to controlling bleeding and include the following:
1. The topical bleeding is stopped as quickly as possible by applying direct pressure or ice, packing the area with Gel foam or fibrin foam, and applying topical haemostatic agents such as thrombin.
2. The specific coagulation factor is administered to raise the patient’s level of the deficient coagulation factor. The patient is monitored for signs and symptoms, such as hypersensitivity.
3. When joint bleeding occurs, in addition to administering replacement factors, it is important to totally rest the involved joint to prevent crippling deformities from haemarthrosis. The joint may be packed in ice. Analgesics (e.g. codeine) are given to reduce severe pain. However, aspirin and aspirin-containing compounds should never be used. As soon as bleeding ceases, it is important to encourage mobilisation of the affected area through range-of-motion exercises and physiotherapy. Weight bearing is avoided until all swelling has resolved and muscle strength has returned.
4. It is important to manage any life-threatening complication that may develop as a result of haemorrhage or side effects from coagulation factors or other medications, such as hyponatraemia by desmopressin. Examples include nursing interventions to prevent or treat airway obstruction from haemorrhage into the neck and pharynx, as well as early assessment and treatment of intracranial bleeding.
Ambulatory and home care
Home management is a primary consideration for the patient with haemophilia because the disease follows a progressive, chronic course. The quality and length of life may be significantly affected by the patient’s knowledge of the illness and how to live with it. The patient and family can be referred to the Haemophilia Foundation (see Resources on p 805) to encourage contact with other individuals who are dealing with the problems of haemophilia.37,38 The nurse must provide ongoing assessment of the patient’s adaptation to the illness. Psychosocial support and assistance should be readily available as needed.
Most of the long-term care measures are related to patient teaching. The patient with haemophilia must be taught to recognise disease-related problems and to learn which problems can be resolved at home and which require hospitalisation. Immediate medical attention is required for severe pain or swelling of a muscle or joint that restricts movement or inhibits sleep, and for a head injury, a swelling in the neck or mouth, abdominal pain, haematuria, melaena and skin wounds in need of suturing.
Daily oral hygiene must be performed without causing trauma. Understanding how to prevent injuries is another consideration. This is no easy task; there are many potential sources of trauma. The patient can learn to participate in non-contact sports (e.g. golf) and wear gloves when doing household chores to prevent cuts or abrasions from knives, hammers and other tools. The patient should wear a medical alert tag to ensure that healthcare providers know about the haemophilia in case of an accident.
The patient needs information about routine follow-up care and their compliance with scheduled visits must be assessed. A reliable person can be taught to self-administer some of the factor replacement therapies at home.
Evaluation
The overall expected outcomes for the patient with haemophilia are similar to those for the patient with thrombocytopenia and are presented in NCP 30-2.
Disseminated intravascular coagulation
Disseminated intravascular coagulation (DIC) is a serious bleeding and thrombotic disorder. It results from abnormally initiated and accelerated clotting. Subsequent decreases in clotting factors and platelets ensue, which may lead to uncontrollable haemorrhage. The term disseminated intravascular coagulation can be misleading because it suggests that blood is clotting. However, the paradox of this condition is characterised by the profuse bleeding that results from the depletion of platelets and clotting factors. DIC is always caused by an underlying disease or condition. The underlying problem must be treated for the DIC to resolve.
AETIOLOGY AND PATHOPHYSIOLOGY
DIC is not a disease; it is an abnormal response of the normal clotting cascade stimulated by a disease process or disorder. The diseases and disorders known to predispose a patient to DIC are listed in Box 30-9. DIC can occur as an acute, catastrophic condition, or it may exist at a subacute or chronic level. Each condition may have one or multiple triggering mechanisms to start the clotting cascade. For example, tumours and traumatised or necrotic tissue release tissue factors into circulation. Endotoxin from Gram-negative bacteria activates several steps in the coagulation cascade.