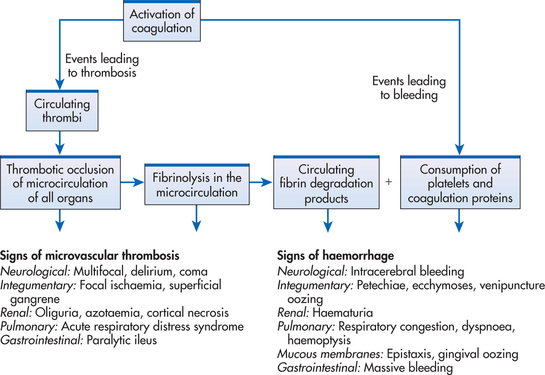
Figure 30-10 The sequence of events that occur during disseminated intravascular coagulation (DIC).
Tissue factor is released at the site of tissue injury and by some malignancies, such as leukaemia, and causes normal coagulation mechanisms to be enhanced. Abundant intravascular thrombin, the most powerful coagulant, is produced (see Fig 30-10). It catalyses the conversion of fibrinogen to fibrin and enhances platelet aggregation. There is widespread fibrin and platelet deposition in capillaries and arterioles, resulting in thrombosis; this can lead to multiorgan failure. In addition, clotting inhibitors, such as antithrombin III (AT III) and protein C, are depressed. This excessive clotting activates the fibrinolytic system, which in turn breaks down the newly formed clot, creating fibrin split products (fibrin degradation products). These products have anticoagulant properties and inhibit normal blood clotting. Ultimately with fibrin split products accumulating and clotting factors being depleted, the blood loses its ability to clot. Therefore, a stable clot cannot be formed at injury sites. This situation predisposes the patient to haemorrhage.
Chronic and subacute DIC are most commonly seen in patients with long-standing illnesses such as malignant disorders or autoimmune diseases. Occasionally these patients have subclinical disease manifested only by laboratory abnormalities. However, the clinical spectrum ranges from easy bruising to haemorrhage and from hypercoagulability to thrombosis.
There is no well-defined sequence of events in acute DIC. Bleeding in a person with no previous history or obvious cause should be questioned because it may be one of the first manifestations of acute DIC. Other non-specific manifestations can include weakness, malaise and fever.
Both bleeding and thrombotic manifestations occur in DIC. Bleeding manifestations of DIC are multifactorial (see Fig 30-10) and result from consumption and depletion of platelets and coagulation factors, as well as clot lysis and formation of fibrin split products that have anticoagulant properties.
Bleeding manifestations include integumentary manifestations, such as pallor, petechiae, purpura (see Fig 30-11), oozing blood, venipuncture site bleeding, haematomas and occult haemorrhage; respiratory manifestations such as tachypnoea, haemoptysis and orthopnoea; cardiovascular manifestations, such as tachycardia and hypotension; GI manifestations, such as upper and lower GI bleeding, abdominal distension and bloody stools; urinary manifestations, such as haematuria; neurological changes, such as vision changes, dizziness, headache, changes in mental status and irritability; and musculoskeletal complaints, such as bone and joint pain.
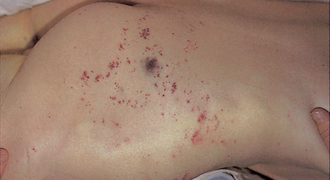
Figure 30-11 Disseminated intravascular coagulation resulting from staphylococcal septicaemia. Note the characteristic skin haemorrhage on the buttock, ranging from small purpuric lesions to larger ecchymoses.
Source: Forbes CD, Jackson WF. Color atlas and text of clinical medicine. 3rd edn. London: Mosby; 2003.
Thrombotic manifestations are a result of fibrin or platelet deposition in the microvasculature (see Fig 30-10) and include integumentary changes, such as cyanosis, ischaemic tissue necrosis (e.g. gangrene) and haemorrhagic necrosis; respiratory changes, such as tachypnoea, dyspnoea, pulmonary emboli and acute respiratory distress syndrome (ARDS); cardiovascular changes, such as electrocardiogram (ECG) changes and venous distension; GI changes, such as abdominal pain and paralytic ileus; and kidney damage and oliguria, leading to failure.
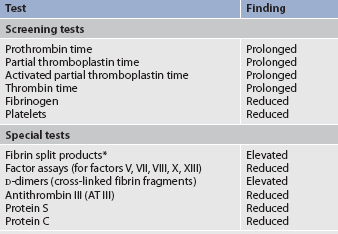
Tests used to diagnose acute DIC and their findings are listed in Table 30-11. As more clots are made in the body, more breakdown products from fibrinogen and fibrin are formed (fibrin split products) and they work in three ways to interfere with blood coagulation. First, the fibrin split products coat the platelets and interfere with platelet function. Second, they interfere with thrombin and thereby disrupt coagulation. Third, they attach to fibrinogen, which interferes with the polymerisation process necessary to form a stable clot. A much more specific test is the D-dimer assay. D-dimer, a specific polymer resulting from the breakdown of fibrin (and not fibrinogen), is a specific marker for the degree of fibrinolysis. In general, tests that measure raw materials needed for coagulation (e.g. platelets, fibrinogen) are reduced, and values that measure times to clot are prolonged. Fragmented erythrocytes (schistocytes), indicative of partial occlusion of small vessels by fibrin thrombi, may be found on blood smears.
It is important to diagnose DIC quickly, stabilise the patient if needed (e.g. oxygenation, volume replacement), institute therapy that will resolve the underlying causative disease or problem, and provide supportive care for the manifestations resulting from the pathology of DIC itself. The treatment of DIC remains controversial and under investigation as researchers attempt to determine the most suitable means of managing this dangerous syndrome. Consequently it is imperative that nurses maintain an ongoing awareness of current modes of therapy and that each patient is managed on the basis of their particular clinical manifestations. Diagnosing and treating the primary disease process is essential to the resolution of DIC.
Depending on its severity, a variety of different methods are used to provide supportive and symptomatic management of DIC (see Fig 30-12). First, if chronic DIC is diagnosed in a patient who is not bleeding, no therapy for DIC is necessary. Treatment of the underlying disease may be sufficient to reverse the DIC (e.g. antineoplastic therapy when DIC is caused by malignancy). Second, if the patient with DIC is bleeding, therapy is directed towards providing support with necessary blood products while treating the primary disorder. The blood products are administered cautiously on the basis of specific component deficiencies to patients who have serious bleeding, are at high risk of bleeding (e.g. surgery) or require invasive procedures. Blood product support with platelets, cryoprecipitate and fresh frozen plasma is usually reserved for a patient with life-threatening haemorrhage. The concern is that one is adding ‘fuel to the fire’ of already activated coagulation. However, it may be the only method to avoid fatal haemorrhage in some patients. Therapy will stabilise a patient, prevent exsanguination or massive thrombosis and permit institution of definitive therapy to treat the underlying cause. In general, platelets are given to correct thrombocytopenia if the platelet count is less than 20 × 109/L or less than 50 × 109/L with bleeding. Cryoprecipitate replaces factor VIII and fibrinogen, and is given if the fibrinogen is below 100 mg/L. Fresh frozen plasma replaces all clotting factors except platelets and provides a source of anti-thrombin.
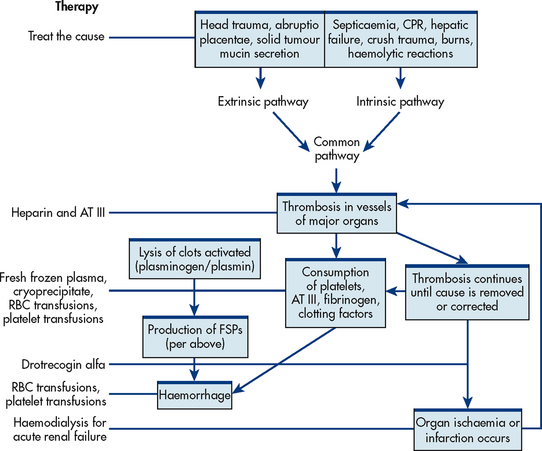
Figure 30-12 Intended sites of action for therapies in disseminated intravascular coagulation. AT III, antithrombin III; CPR, cardiopulmonary resuscitation; FSPs, fibrin split products; RBC, red blood cell.
A patient with manifestations of thrombosis is often treated by anticoagulation with heparin or low-molecular-weight heparin. However, the use of heparin in the treatment of DIC remains controversial. A recombinant human-activated protein C has been shown to have both anticoagulant and anti-inflammatory effects and has reduced the relative risk of death due to sepsis. However, because it causes increased bleeding, it is contraindicated in patients with a platelet count less than 30 × 109/L. Aminocaproic acid is generally contraindicated because of its ability to inhibit fibrinolysis and enhancement of thrombosis.42–44
Chronic DIC does not respond to oral anticoagulants, but it can be controlled with long-term use of heparin. Some patients with indolent (inactive and slowly developing) tumours and severe, chronic DIC may need continuous infusion of heparin with portable pumps.
 NURSING MANAGEMENT: DISSEMINATED INTRAVASCULAR COAGULATION Nursing diagnoses
NURSING MANAGEMENT: DISSEMINATED INTRAVASCULAR COAGULATION Nursing diagnosesNursing diagnoses for the patient with DIC may include, but are not limited to, the following:
• ineffective tissue perfusion (cerebral, cardiopulmonary, renal, GI and peripheral) related to bleeding and sluggish or diminished blood flow secondary to thrombosis
• acute pain related to bleeding into tissues and diagnostic procedures
• decreased cardiac output related to fluid volume deficit and hypotension
• anxiety related to fear of the unknown, disease process, diagnostic procedures and therapy.
Nursing implementationThe nurse must be alert to the possible development of DIC and especially to the precipitating factors listed in Box 30-9. This may be difficult because the nurse is focusing on the complex care often required by the primary problem that precipitated the DIC. It is important to remember that because DIC is secondary to an underlying disease, appropriate care for managing the causative problem must be provided while providing supportive care related to the manifestations of DIC.41
Appropriate nursing interventions are essential to the survival of the patient with acute DIC. Astute, ongoing assessment, active attention to manifestations of the syndrome and institution of appropriate treatment measures are challenging and sometimes paradoxical nursing responsibilities (e.g. administering heparin to a bleeding patient). Table 30-7 and NCP 30-2 provide assessments and interventions appropriate for the patient with DIC. Early detection of bleeding, both occult and overt, must be a primary goal. The patient is assessed for signs of external bleeding (e.g. petechiae, oozing at IV or injection sites) and signs of internal bleeding (e.g. increased heart rate, changes in mental status, increasing abdominal girth, pain). Any sites of bleeding are carefully monitored for continued bleeding. Tissue damage should be minimised and the patient protected from additional foci of bleeding.
An additional nursing responsibility is to administer blood products and medications correctly. Blood product transfusion is discussed on page 797.
Leucopenia refers to a decrease in the total WBC count (granulocytes, monocytes and lymphocytes). Granulocytopenia is a deficiency of granulocytes, which include neutrophils, eosinophils and basophils. The neutrophilic granulocytes, which play a major role in phagocytosing pathogenic microbes, are closely monitored in clinical practice as an indicator of a patient’s risk of infection. A reduction in neutrophils is termed neutropenia. (Some clinicians use the terms granulocytopenia and neutropenia interchangeably because the largest constituency of granulocytes is the neutrophils.) The absolute neutrophil count is determined by multiplying the total WBC count by the percentage of neutrophils. Neutropenia is defined as a neutrophil count of less than 1–1.5 × 109/L. Severe neutropenia is defined as a neutrophil count of less than 0.5 × 109/L. Normally, neutrophils range from 2.0 × 109/L to 7.5 × 109/L. However, in considering the clinical significance of neutropenia it is important to know the rapidity of the decrease in the neutrophil count (gradual or rapid), the degree of neutropenia and the duration of the episode of neutropenia. The faster the drop and the longer the duration, the greater the likelihood of developing life-threatening infection, sepsis and death. Other factors and comorbid conditions, such as being older than 60, the presence of an existing minor infection, being in the hospital and having diabetes can increase the risk of a serious infection.42
Neutropenia is a clinical consequence that occurs with a variety of conditions or diseases (Box 30-10). It can also be an expected effect, a side effect or an unintentional effect of taking certain drugs. The most common cause of neutropenia is iatrogenic, resulting from widespread use of chemotherapeutic and immunosuppressive therapy in the treatment of malignancies and autoimmune diseases.
BOX 30-10 Causes of neutropenia
• Antitumour antibiotics (daunorubicin, doxorubicin)
• Alkylating agents (nitrogen mustards, busulfan)
• Antimetabolites (methotrexate, 6-mercaptopurine)
• Anti-inflammatory drugs (phenylbutazone)
• Psychotropics and antidepressants (clozapine, imipramine)
• Miscellaneous (gold, penicillamine)
• Antimicrobial agents (zidovudine, trimethoprim–sulfamethoxazole)
The patient with neutropenia is predisposed to infection with non-pathogenic organisms that constitute normal body flora, as well as opportunistic pathogens. When the WBC count is depressed or immature WBCs are present, normal phagocytic mechanisms are impaired. Also, because of the diminished phagocytic response, the classic signs of inflammation—redness, heat and swelling—may not occur. WBCs are the major component of pus. Therefore, in the patient with neutropenia, pus formation (e.g. as a visible skin lesion or as pulmonary infiltrates on a chest X-ray) is also absent. Because neutropenia masks some of the signs and symptoms of infection, the presence of even a low-grade fever is of great significance.
When fever occurs in a neutropenic patient, it is assumed to be caused by infection and requires immediate attention. The immunocompromised, neutropenic patient has little or no ability to fight infection. Thus minor infections can lead rapidly to sepsis. The mucous membranes of the throat and mouth, the skin, the perianal area and the pulmonary system are common entry points for pathogenic organisms in susceptible hosts. Clinical manifestations related to infection at these sites include complaints of sore throat and dysphagia, the appearance of ulcerative lesions of the pharyngeal and buccal mucosa, diarrhoea, rectal tenderness, vaginal itching or discharge, shortness of breath and non-productive cough. Any minor complaint of pain or any other symptom by the patient should be taken seriously and reported to the doctor immediately. These seemingly minor complaints can progress to fever, chills, sepsis and septic shock if not recognised and treated in the early stages.
Systemic infections caused by bacterial, fungal and viral organisms are common in patients with neutropenia. The patient’s own flora (normally non-pathogenic) contributes significantly to life-threatening infections, such as pneumonia. Organisms that are known to be common sources of infection include Gram-positive Staphylococcus aureus and aerobic Gram-negative organisms. Fungi that are involved include Candida (usually C. albicans) and Aspergillus. Viral infections caused by reactivation of herpes simplex and zoster are common following prolonged periods of neutropenia.
The primary diagnostic tests for assessing neutropenia are the peripheral WBC count and bone marrow aspiration and biopsy (see Box 30-11). A total WBC count of less than 4 × 109/L reflects leucopenia. However, only a differential count can confirm the presence of neutropenia (neutrophil count 1–1.5 × 109/L). If the differential WBC count reflects an absolute neutropenia of 0.5–1.0 × 109/L, the patient is at moderate risk of a bacterial infection. An absolute neutropenia of less than 0.5 × 109/L places the patient at severe risk.
MULTIDISCIPLINARY CARE
History and physical examination
WBC count with differential count
Reticulocyte and platelet count
Bone marrow aspiration or biopsy
Cultures of nose, throat, sputum, urine, stool, obvious lesions, blood (as indicated)
Identification and removal of cause of neutropenia (if possible)
Identification of site of infection (if present) and causative organism
Haematopoietic growth factors (G-CSF, GM-CSF)
Strict hand-washing and patient hygiene
Single patient room, positive pressure or high-efficiency particulate air (HEPA) filtration, depending on risk
Community isolation and home precautions if outpatient
G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte–macrophage colony-stimulating factor; Hb, haemoglobin; Hct, haematocrit; WBC, white blood cell.
A peripheral blood smear is used to assess for immature forms of WBCs (e.g. bands). The haematocrit level, reticulocyte count and platelet count are done to evaluate bone marrow function. A review of the patient’s recent past and current drug history should also be done. If the cause of neutropenia is unknown, bone marrow aspirations and biopsies are done to examine cellularity and cell morphology. Additional studies may be done as indicated to assess spleen and liver function.
NURSING AND COLLABORATIVE MANAGEMENT: NEUTROPENIAThe factors involved in the nursing and multidisciplinary care of neutropenia include: (1) determining the cause of the neutropenia; (2) identifying the offending organisms if an infection has developed; (3) instituting prophylactic, empiric or therapeutic antibiotic therapy; (4) administering haematopoietic growth factors (e.g. granulocyte colony-stimulating factor [G-CSF] and granulocyte–macrophage colony-stimulating factor [GM-CSF]); and (5) instituting protective environmental practices, such as strict hand-washing, visitor restrictions and a private room if hospitalised. A positive pressure room or high-efficiency particulate air (HEPA) filtration may be used for patients undergoing HSCT (see Box 30-11).
Occasionally, the cause of the neutropenia can be easily treated (e.g. nutritional deficiencies). However, neutropenia can also be a side effect that must be tolerated as a necessary step in therapy (e.g. chemotherapy, radiation therapy). In some situations the neutropenia resolves when the primary disease is treated (e.g. tuberculosis).
The nurse needs to monitor the neutropenic patient for signs and symptoms of infection (e.g. any fever ≥38°C) and early septic shock. Early identification of a potentially infective organism depends on acquiring cultures from various sites. Serial blood cultures (at least two) or one from a peripheral site and one from a venous access device should be done promptly and antibiotics started immediately. Additionally, cultures of sputum, throat, lesions, wounds, urine and faeces are essential in the surveillance of the patient. It may also be necessary to do a tracheal aspiration, bronchoscopy with bronchial brushings or lung biopsy to diagnose the cause of pneumonic infiltrates. However, invasive diagnostic studies are often contraindicated due to the concern of introducing infection and the fact that patients are also often thrombocytopenic. Despite these many tests, the causative organism is identified in only approximately half of neutropenic patients.
When a febrile episode occurs in a neutropenic patient, antibiotic therapy must be initiated immediately (within 1 hour) even before the determination of a specific causative organism by culture. Administration of broad-spectrum antibiotics is usually by the IV route because of the rapidly lethal effects of infection. However, some oral antibiotics are highly effective and routinely used for prophylaxis against infection in some neutropenic patients. The use of a third- or fourth-generation cephalosporin (e.g. cefepime, ceftazidime), a carbapenem (e.g. imipenem/cilastatin) or a combination of an aminoglycoside plus an antipseudomonal offers broad-spectrum coverage for initial management.43 Regardless of the combination, the nurse must initiate therapy promptly and observe the patient for side effects of antimicrobial agents. Side effects common to aminoglycosides include nephrotoxicity and ototoxicity; side effects common to cephalosporins include rashes, fever and pruritus.
The duration of the neutropenia affects the infection risk of the patient. The longer the neutropenia, the greater the risk of a fungal infection. Antifungal therapy is initiated whenever a culture is positive, or in patients who do not become afebrile with broad-spectrum antibiotic coverage.
G-CSF (filgrastim and pegfilgrastim) can be used to treat a neutropenic patient. G-CSF stimulates the production and function of neutrophils. GM-CSF stimulates the production and function of neutrophils and monocytes. These agents can be given IV or SC. The nurse can teach the patient and family how to administer the SC medication. These factors are especially beneficial in enhancing granulocyte recovery after chemotherapy and shorten the period of vulnerability to fatal infections. Keratinocyte growth factor may also be used to reduce the duration and severity of mucositis, which may contribute to infection. (See Ch 14.)
An important consideration is the determination of the best means to protect the patient whose own defences against infection are compromised. To accomplish this goal, the following issues must be kept in mind: (1) the patient’s normal flora are the most common source of microbial colonisation and infection; (2) transmission of organisms from humans most commonly occurs by direct contact with the hands; (3) air, food, water and equipment provide additional opportunities for transmission of infection; and (4) healthcare providers with transmittable illnesses and other patients with infections can be sources of infection under certain conditions.
Hand-washing is the single most important preventative measure in minimising the risk of infection in the neutropenic patient. Strict hand-washing by all persons coming in contact with the compromised patient is the major method to prevent transmission of harmful pathogens. The National Institute for Health and Clinical Excellence advocates hand-washing before, during and after care.44 This seemingly routine technique has a significant effect in reducing infection. It must be emphasised and enforced, despite its seeming simplicity.
Separating immunocompromised patients from those who are infected or who have conditions that increase the probability of transmitting infections (e.g. poor hygiene caused by lack of understanding or cognitive dysfunction) is strongly recommended. Often, patients can be managed on an outpatient basis if the patient and family can astutely monitor for fevers and other signs of infection and then report promptly to a nearby hospital (see Box 30-12). If the patient is hospitalised, private rooms should be used. HEPA filtration is an air-handling method with a high-flow filtering system that can reduce or eliminate the number of aerosolised pathogens in the environment. Although it is expensive to install, it is often used for patients with severe prolonged neutropenia (e.g. bone marrow transplant patients). Care routines in a HEPA environment are essentially the same as care in any other private room. Additional neutropenic precaution guidelines may be employed, such as the avoidance of tap water, fresh fruits and vegetables, and prophylactic antibiotics and antifungals. The nursing measures presented in NCP 30-3 are important in the treatment of the patient with neutropenia.
PATIENT & FAMILY TEACHING GUIDE
This instruction sheet explains precautions you should take to protect yourself when your neutrophil count is low. Please make sure to ask your healthcare provider about specific precautions you should take that relate to your infection risk factors.
• WASH YOUR HANDS frequently and make sure those around you wash their hands frequently, particularly if they help with your care. An antibacterial hand gel may also be used.
• Notify your nurse or healthcare provider if you have any of the following:
• If you are at home, take your temperature as directed and follow instructions on what to do if you have a fever.
• Avoid crowds and people with colds, flu or infections.
• Avoid raw foods, such as sushi, caesar salad dressing (may have raw eggs), blue cheese and fruits that cannot be peeled or vegetables that cannot be well cleaned. Ask your healthcare provider about specific dietary guidelines for you.
• Bathe or shower daily. A moisturiser may be used to prevent skin from drying and cracking.
• Do not perform gardening or clean-up after pets. Feeding and petting your dog or cat is fine as long as you wash your hands well after handling.
Quality-of-life issues for patients should not be overlooked. Potential patient experiences of fatigue, malaise, a decrease in functioning, social isolation and depression, as well as family support, require appropriate interventions.
The value of effective nursing care in reducing the development of infection or limiting its extent cannot be overemphasised. Regular assessment and early detection of infectious sources are key roles for the nurse in reducing morbidity and mortality rates from infection.
Gerontological considerations: thrombocytopenia and neutropenia
About 55–60% of cancers are currently diagnosed in individuals aged over 65 years. This proportion is expected to further increase in the next two decades. Age-related changes of bone marrow function are rather subtle and probably not significant for the haematopoietic function of normal older individuals. These changes may, however, become clinically evident under conditions of severe haematopoietic stress, such as the administration of repeated courses of chemotherapy and/or radiation therapy and the resultant sequelae of myelosupression.45 The use of supportive therapies, such as haemapoietic growth factors, increases the likelihood that older individuals will be treated with standard and even aggressive therapies, leading to neutropenia and thrombocytopenia. The nurse also needs to be aware that older individuals may have signs and symptoms different from those in younger individuals. For example, the older adult may present with delirium as compared to cough as a clinical manifestation of pneumonia. (See Ch 5.)
Myelodysplastic syndrome (MDS) is a group of related haematological disorders characterised by a change in the quantity and quality of bone marrow elements. Peripheral blood cytopenias in combination with a hypercellular bone marrow exhibiting dysplastic changes are the hallmark of MDS. The risk of MDS increases with age as the disease most commonly affects people between the ages of 58 and 75.
The aetiology of MDS is unknown. Its manifestations result from neoplastic transformation of the pluripotent haematopoietic stem cells within the bone marrow. Occasionally one type of MDS transforms into another. In some cases, MDS will progress to acute myelogenous leukaemia.
MDS is referred to as a clonal disorder because some bone marrow stem cells continue to function normally whereas others (a specific clone) do not. The abnormal clone of the stem cells is usually found in the bone marrow but eventually may be found in the circulation. In contrast to acute myelogenous leukaemia, in which the leukaemic cells show little normal maturation, the clonal cells in MDS always display some degree of maturity. Disease progression is slower than in acute myelogenous leukaemia. However, eventually the abnormal cells replace the bone marrow. Typically, life-threatening anaemia, thrombocytopenia and neutropenia occur during the advanced stage of MDS.
MDS commonly manifests as infection and bleeding caused by inadequate numbers of ineffective functioning circulating granulocytes or platelets. MDS is often discovered in the elderly as a result of testing for the symptoms of anaemia, thrombocytopenia or neutropenia. It may also be diagnosed incidentally from a routine FBC.
Bone marrow biopsy with aspirate analysis is essential for both the diagnosis and classification of the specific types of myelodysplasia. In MDS the bone marrow is normocellular, hypocellular or hypercellular and the patient has peripheral cytopenia. Laboratory data and bone marrow studies will help rule out other causes of the dysplasia, such as non-malignant disorders, vitamin B12 and folate deficiencies, and infectious causes. MDS is staged according to clinical and laboratory findings. The relationship between the number of circulating blast cells and the number of blast cells in the bone marrow serves as the main indicator of prognosis in this disease. The findings of the biopsy also help in deciding how aggressive the treatment will be.
NURSING AND COLLABORATIVE MANAGEMENT: MYELODYSPLASTIC SYNDROMESupportive treatment of MDS is based on the premise that the aggressiveness of treatment should match the aggressiveness of the disease. Supportive treatment consists of haematological monitoring (serial bone marrow and peripheral blood examinations), antibiotic therapy or transfusions with blood products. Side effects and toxicities from supportive treatment include anaemia, thrombocytopenia and blood transfusion reactions. The overall goal is to improve haematopoiesis and ensure age-related quality of life.
Low-risk patients can often be treated with EPO and G-CSF. Only about one-third of high-risk patients are treated with intensive chemotherapy and/or HSCT. Treatments for MDS include cytarabine with or without antitumour antibiotics (anthracyclines), ATG, cyclosporin and thalidomide. High-dose chemotherapy and allogeneic HSCT have been used in an attempt to treat bone marrow dysfunction of MDS and restore it with normal haematopoiesis. However, because of the aggressiveness of this treatment, it is generally recommended for patients less than 55–60 years old.
Nursing care of the patient with MDS is similar to that of the patient with manifestations of anaemia (see NCP 30-1), thrombocytopenia (see NCP 30-2) and neutropenia (see NCP 30-3).
Leukaemia is the general term used to describe a group of malignant disorders affecting the blood and blood-forming tissues of the bone marrow, lymph system and spleen. Leukaemia occurs in all age groups. It results in an accumulation of dysfunctional cells because of a loss of regulation in cell division. It follows a progressive course that is eventually fatal if untreated. In Australia, it was projected that in 2010, 3138 Australians would be diagnosed with leukaemia.46 Leukaemia is the number one childhood cancer in Australia, but although it is often thought of as a disease of children, the number of adults affected is approximately 10 times that of children.46
Regardless of the specific type of leukaemia, there is generally no single causative agent in the development of leukaemia. Most leukaemias result from a combination of factors, including genetic and environmental influences. Chromosomal changes, first recognised in chronic myelogenous leukaemia, have led to discoveries of how normal genes, once transformed, can result in abnormal genes (oncogenes) capable of causing many types of cancer, including leukaemia (see Ch 15). Chemical agents (e.g. benzene), chemotherapeutic agents (e.g. alkylating agents), viruses, radiation and immunological deficiencies have all been associated with the development of leukaemia in susceptible hosts. There is an increased incidence of leukaemia in radiologists, people who have lived near nuclear bomb test sites or nuclear reactor accidents (e.g. Chernobyl), survivors of the bombings of Nagasaki and Hiroshima, and people previously treated with radiation therapy or chemotherapy. Although ribonucleic acid (RNA) retroviruses cause a number of leukaemias in animals, a viral cause for a human leukaemia has been established only for some patients with adult T-cell leukaemia. This form of leukaemia is endemic in south-western Japan and parts of the Caribbean and central Africa, and is caused by the human T-cell leukaemia virus type 1 (HTLV-1).
Classification of leukaemia can be made according to whether it is acute or chronic and the type of WBC involved. The terms acute and chronic refer to cell maturity and the nature of disease onset. Acute leukaemia is characterised by the clonal proliferation of immature haematopoietic cells. The leukaemia develops following malignant transformation of a single type of immature haematopoietic cell, followed by cellular replication and expansion of that malignant clone. Chronic leukaemia involves more mature forms of the WBC and the disease onset is more gradual.
Leukaemia can also be classified by identifying the type of leucocyte involved—that is, whether it is of myelogenous or lymphocytic origin. By combining the acute and chronic categories with the cell type involved, specific types of leukaemia can be identified. Four major types of leukaemia are acute myelogenous leukaemia (AML), acute lymphocytic leukaemia (ALL), chronic myelogenous (granulocytic) leukaemia (CML) and chronic lymphocytic leukaemia (CLL). Other defining features of these leukaemic subtypes are presented in Table 30-12.
TABLE 30-12 Types of leukaemia
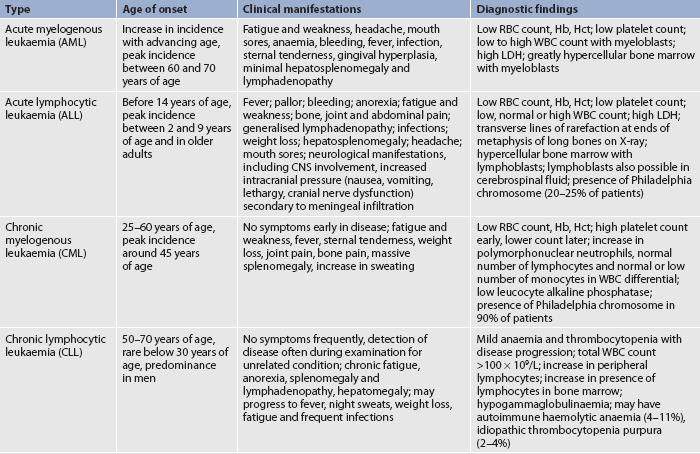
CNS, central nervous system; Hb, haemoglobin; Hct, haematocrit; LDH, lactic dehydrogenase; RBC, red blood cell; WBC, white blood cell.
AML represents only 25% of all leukaemias but makes up about 85% of acute leukaemias in adults. Its onset is often abrupt and dramatic. A patient may have serious infections and abnormal bleeding from the onset of the disease (see Fig 30-13).
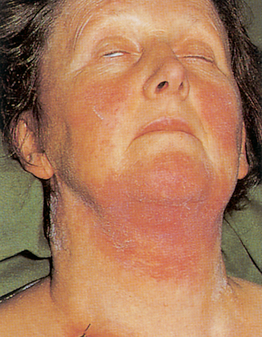
Figure 30-13 Complications of acute leukaemia. Spreading cellulitis of the neck and chin in this woman with acute myelogenous leukaemia results from streptococcal and candidal infection. She was at risk because of previous chemotherapy and prolonged neutropenia.
AML is characterised by uncontrolled proliferation of myeloblasts, the precursors of granulocytes. There is hyperplasia of the bone marrow. The clinical manifestations are usually related to replacement of normal haematopoietic cells in the marrow by leukaemic myeloblasts and, to a lesser extent, to infiltration of other organs and tissue (see Table 30-12).
ALL is the most common type of leukaemia in children and accounts for about 15% of acute leukaemias in adults. Five years after diagnosis, about half of adults with leukaemia will have survived, while 80% of children (0–14 years) will survive.46
In ALL, immature lymphocytes proliferate in the bone marrow; most are of B-cell origin. Fever is present in the majority of patients at the time of diagnosis. Signs and symptoms may appear abruptly, with bleeding or fever, or they may be insidious, with progressive weakness, fatigue, bone and/or joint pain and bleeding tendencies.
CNS manifestations are especially common in ALL and represent a serious problem. Leukaemic meningitis caused by arachnoids infiltration occurs in many patients with ALL.
CML is caused by excessive development of mature neoplastic granulocytes in the bone marrow. The excess neoplastic granulocytes move into the peripheral blood in massive numbers and ultimately infiltrate the liver and spleen. These cells contain a distinctive cytogenetic abnormality, the Philadelphia chromosome, which serves as a disease marker and results from translocation of genetic material between chromosomes 9 and 22.
The natural history of CML is a chronic stable phase, followed by the development of a more acute, aggressive phase referred to as the blastic phase. The chronic phase of CML can last for several years and can usually be well controlled with treatment. Even with treatment, the chronic phase of the disease will eventually progress to the accelerated phase, ending in the blastic phase. Once CML transforms to the acute or blastic phase, it is often refractory to therapy and the patient may live for only a few months.
CLL is the most common leukaemia in adults. It is characterised by the production and accumulation of functionally inactive but long-lived, small, mature-appearing lymphocytes. The type of lymphocyte involved is usually the B cell. The lymphocytes infiltrate the bone marrow, spleen and liver. Lymph node enlargement (lymphadenopathy) is present throughout the body, and there is an increased incidence of infection due to T-cell deficiencies or hypogammaglobulinaemia. B-cell CLL is considered to be identical to the mature B-cell small lymphocytic lymphoma, a type of non-Hodgkin’s lymphoma, and 5–8% of these patients will experience a transformation to a diffuse, large B-cell non-Hodgkin’s lymphoma, called Richter’s syndrome.47
Complications from early-stage CLL are rare but may develop as the disease advances. Pressure on nerves from enlarged lymph nodes causes pain and even paralysis. Mediastinal node enlargement leads to pulmonary symptoms. Because CLL is usually a disease of older adults, treatment decisions must be made by considering the progression of the disease and the side effects of treatment. Many individuals in the early stages of CLL require no treatment. Others may be followed closely and receive treatment only when the disease progresses; approximately one-third will require immediate intervention at the time of diagnosis.47
Occasionally, the subtype of leukaemia cannot be identified. The malignant leukaemic cells may have lymphoid, myeloid or mixed characteristics. Frequently these patients do not respond to treatment and have a poor prognosis. Other rare types include hairy cell leukaemia and biphenotypic (both abnormal myeloid and lymphoid clones) leukaemia.
The clinical manifestations of leukaemia are varied (see Table 30-12). Essentially they relate to problems caused by bone marrow failure and the formation of leukaemic infiltrates. Bone marrow failure results from: (1) bone marrow overcrowding by abnormal cells; and (2) inadequate production of normal marrow elements. The patient is predisposed to anaemia, thrombocytopenia and decreased number and function of WBCs.
As leukaemia progresses, fewer normal blood cells are produced. The abnormal WBCs continue to accumulate because they do not go through the normal cell life cycle to death (apoptosis). The leukaemic cells infiltrate the patient’s organs, leading to problems such as splenomegaly, hepatomegaly, lymphadenopathy, bone pain, meningeal irritation and oral lesions. Solid masses resulting from collections of leukaemic cells, called chloromas, can also occur.
Peripheral blood evaluation and bone marrow examination are the primary methods of diagnosing and classifying the subtypes of leukaemia. Morphological, histochemical, immunological and cytogenetic methods are all used to identify cell subtypes and the stage of development of leukaemic cell populations. This is important because different subtypes have different natural histories, prognoses and chemotherapeutic regimens. Other studies, such as lumbar puncture and computed tomography (CT) scan, can determine the presence of leukaemic cells outside the blood and bone marrow.
The malignant cells in most patients with AML and ALL have chromosomal abnormalities. In some cases, specific cytogenetic abnormalities are associated with distinct subsets of the disease. In addition to establishing the type of AML or ALL, specific cytogenetic abnormalities have diagnostic, prognostic and therapeutic importance. For example, in CML, the finding of the Philadelphia chromosome has a good prognostic significance, but not so in ALL.48
Once a diagnosis of leukaemia has been made, multidisciplinary care is focused on the initial goal of attaining remission. Age and chromosome analysis often help form the basis of important treatment decisions. Because cytotoxic chemotherapy is the mainstay of the treatment, the nurse must understand the principles of cancer chemotherapy, including cellular kinetics, the use of multiple drugs rather than single agents and the cell cycle. (See the section on chemotherapy in Ch 15.)
In some cases, such as non-symptomatic patients with CLL, watchful waiting with active supportive care may be appropriate. Attaining remission or disease control is a realistic option for the majority of patients, and in some cases cure is a realistic goal. In complete remission there is no evidence of overt disease on physical examination, and the bone marrow and peripheral blood appear normal. A lesser state of control is known as partial remission. Partial remission is characterised by a lack of symptoms and a normal peripheral blood smear, but there is still evidence of disease in the bone marrow. Minimal residual disease is defined as tumour cells that cannot be detected by morphological examination, but can be identified by molecular testing. Molecular remission indicates that all molecular studies are negative for residual leukaemia. The patient’s prognosis is directly related to the ability to maintain a remission. The patient’s prognosis becomes more unfavourable with each relapse. Each time there is a relapse, the succeeding remission may be more difficult to achieve and shorter in duration.
Sometimes patients present with such a high WBC count (e.g. >100 × 109/L) that initial emergent treatment may employ the use of leucapheresis and hydroxyurea. The purpose of these treatments is to reduce the WBC count and the risk of leukaemia-induced thrombosis.
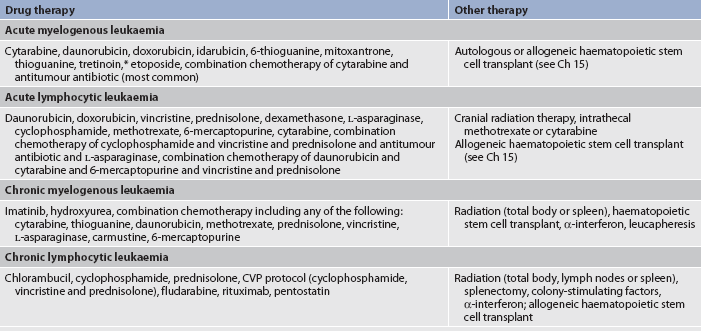
The chemotherapeutic treatment of acute leukaemia is often divided into stages. The first stage, induction therapy, is the attempt to induce or bring about a remission. Induction is aggressive treatment that seeks to destroy leukaemic cells in the tissues, peripheral blood and bone marrow in order to eventually restore normal haematopoiesis upon bone marrow recovery. During induction therapy a patient may become critically ill because the bone marrow is severely depressed by the chemotherapeutic agents. Throughout the induction phase, nursing interventions focus on neutropenia, thrombocytopenia and anaemia, as well as providing psychosocial support to the patient and family. Common chemotherapy agents for induction of AML include cytarabine and antitumour antibiotics (anthracyclines), such as daunorubicin, doxorubicin or idarubicin hydrochloride. After one course of induction therapy, approximately 70% of newly diagnosed patients achieve complete remission. There is one subtype of AML, called promyelocytic leukaemia (M3), in which tretinoin is used along with chemotherapy to induce a remission.49 It is generally assumed that leukaemia cells persist undetected after induction therapy. This could lead to relapse within a few months if no further therapy is administered.
Terms used to describe post-induction or post-remission stages of chemotherapy include intensification, consolidation and maintenance. Intensification therapy, or high-dose therapy, may be given immediately after induction therapy for several months. This therapy may use the same drugs as those used in induction but at higher dosages. Other drugs that target the cell in a different way than those administered during induction may also be added.
Consolidation therapy is started after a remission is achieved. It may consist of one or two additional courses of the same drugs given during induction or it may involve high-dose therapy (intensive consolidation). The purpose of consolidation therapy is to eliminate remaining leukaemic cells that may not be clinically or pathologically evident.
Maintenance therapy is treatment with lower doses of the same drugs used in induction or other drugs given every 3–4 weeks for a prolonged period of time. Like intensification and consolidation therapy, the goal is to keep the body free of leukaemic cells. Each leukaemia requires different maintenance therapy. In AML, maintenance therapy is rarely effective and therefore rarely administered.
In addition to chemotherapy, corticosteroids and radiation therapy may have a role in the complex therapeutic plans for the patient with leukaemia. Total body radiation may be used to prepare the patient for bone marrow transplantation or it may be restricted to certain areas (fields), such as the liver and spleen or other organs affected by infiltrates. In ALL, prophylactic intrathecal methotrexate or cytarabine is given to decrease the chance of CNS involvement, which is common in this particular type of leukaemia. When CNS leukaemia does occur, cranial radiation may be given. Biological therapy may be indicated for specific leukaemias. (Biological therapy is discussed in Ch 15.)
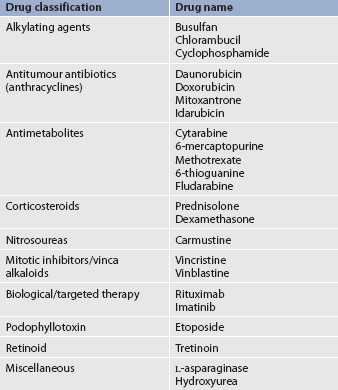
The therapeutic agents used to treat leukaemia vary. Table 30-13 lists various agents used to treat leukaemia. Table 30-14 gives examples of treatment regimens used in various types of leukaemia.
Combination therapy is the mainstay of treatment for leukaemia. The three purposes for using multiple drugs are to: (1) decrease drug resistance; (2) minimise the drug toxicity to the patient by using multiple drugs with varying toxicities; and (3) interrupt cell growth at multiple points in the cell cycle.
Acronyms made from the letters of the drugs used in combination chemotherapy are used to identify the regimen. For example, COAP stands for cyclophosphamide, oncovin, arabinoside and prednisolone. This combination of drugs is used to treat ALL.
Newer therapeutic drugs are aimed at affecting small molecules that promote the growth and differentiation of leukaemia cells. Imatinib represents a new class of drugs that specifically target an abnormal cell. (Targeted therapy is discussed in Ch 15.) Imatinib targets an abnormal version of a normal cell protein (the Bcr-Abl protein) that is present in nearly all patients with CML. This abnormal protein is probably the cause of the disease. The Bcr-Abl gene is located on the Philadelphia chromosome. Thus this drug kills only cancer cells, leaving healthy cells alone.
The use of specific targeted therapy in the form of monoclonal antibodies is a new treatment modality in haematopoietic malignancies (see Table 15-14). Rituximab binds to the B-cell antigen (CD 20) and has been used with CLL.
HSCT is another type of therapy used for patients with different forms of leukaemia. The goal of HSCT is to totally eliminate leukaemic cells from the body using combinations of chemotherapy with or without total body irradiation. This treatment also eradicates the patient’s haematopoietic stem cells, which are then replaced with those of an HLA-matched sibling or volunteer donor (allogeneic), an identical twin (syngeneic) or the patient’s own (autologous) stem cells that were removed (harvested) before the intensive therapy. (HSCT is discussed in Ch 15.)
The primary complications of patients with allogeneic HSCT are graft-versus-host disease (GVHD), relapse of leukaemia (especially ALL) and infection (especially interstitial pneumonia). GVHD is discussed in Chapter 13. Because HSCT has serious associated risks, the patient must weigh the significant risks of treatment-related death or treatment failure (relapse) with the hope of cure.
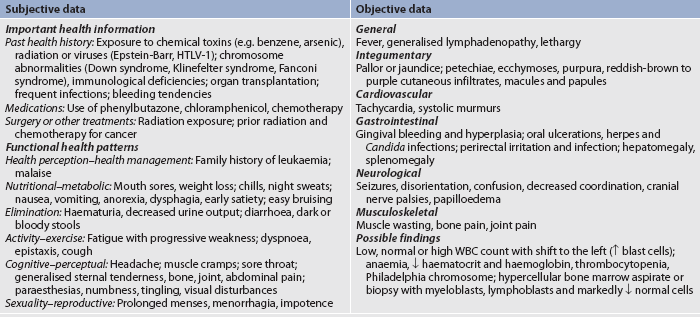
NURSING MANAGEMENT: LEUKAEMIA Nursing assessmentSubjective and objective data that should be obtained from the patient with leukaemia are presented in Table 30-15.
Nursing diagnosesNursing diagnoses for the patient with leukaemia include those appropriate for anaemia, thrombocytopenia and neutropenia (see NCPs 30-1, 30-2 and 30-3).
PlanningThe overall goals are that the patient with leukaemia will: (1) understand and cooperate with the treatment plan; (2) experience minimal side effects and complications associated with both the disease and its treatment; and (3) feel hopeful and supported during the periods of treatment, relapse or remission.
Nursing implementation Acute interventionThe nursing role during acute phases of leukaemia is extremely challenging because the patient has many physical and psychosocial needs. As with other forms of cancer, the diagnosis of leukaemia can evoke great fear and be equated with death. It may be viewed as a hopeless, horrible disease with many painful and undesirable consequences. It is important that the nurse has an understanding of the patient’s type of leukaemia, the prognosis, the treatment plan and goals. By doing this, the nurse can help the patient realise that although the future may be uncertain, they can have a meaningful quality of life while in remission or with disease control and that, in some cases, there is reasonable hope for cure.
The family also needs help in adjusting to the stress of this abrupt onset of serious illness (e.g. dependence, withdrawal, changes in role responsibilities, alterations in body image) and the losses imposed by the sick role. The diagnosis of leukaemia often brings with it the need to make difficult decisions at a time of profound stress for the patient and family.
As the majority of patients are older than 65 years, they may have comorbid conditions that affect treatment decisions. In addition, although older adults have often learned to live with periods of life hardships, disappointments and loss, healthcare providers need to assess all patients for potential symptoms of depression and provide appropriate care and referrals.
The nurse is an important advocate in helping the patient and family to understand the complexities of treatment decisions and manage the side effects and toxicities. A patient empowered by knowledge of the disease and treatment can have a more positive outlook and improved quality of life. The patient may require periods of intense hospitalisation or may need to temporarily relocate to an appropriate treatment centre. These situations can lead the patient to feel deserted and isolated at a time when support is most needed. The nurse can help reverse feelings of abandonment, isolation and loneliness by balancing the demanding technical needs with a humane, caring approach. Nurses face special challenges when meeting the intense psychosocial needs of a patient with leukaemia while continuing to offer the complex physical care that is required. The needs of the patient with leukaemia are best met by a multidisciplinary team (e.g. mental health and oncology clinical nurse specialists, case managers, dieticians, chaplains and social workers).
From a physical care perspective, the nurse is challenged to make astute assessments and plan care to help the patient manage the severe side effects of chemotherapy. The life-threatening results of bone marrow suppression (neutropenia, thrombocytopenia and anaemia) require aggressive nursing interventions (see NCPs 30-1, 30-2 and 30-3). Additional complications of chemotherapy may affect the patient’s GI tract, nutritional status, skin and mucosa, cardiopulmonary status, liver, kidneys and neurological system. (Nursing interventions related to chemotherapy are discussed in Ch 15.)
The nurse needs to ensure that the patient’s doctor has discussed all treatment options with the patient prior to the commencement of treatment and that this is documented in the patient’s medical record. The nurse must be knowledgeable about all drugs being administered. This includes the mechanism of action, purpose, routes of administration, usual doses, potential side effects, safe-handling considerations and toxic effects of the drugs. In addition, the nurse must know how to assess laboratory data reflecting the effects of the drugs. Patient survival and comfort during aggressive chemotherapy are significantly affected by the quality of nursing care.
Ambulatory and home careOngoing care for the patient with leukaemia is necessary to monitor for signs and symptoms of disease control or relapse. For a patient requiring long-term or maintenance chemotherapy, the fatigue of long-term chronic disease management can become arduous and discouraging. Therefore, the patient and significant other must be taught to understand the importance of continued diligence in disease management and the need for follow-up care. They must also be taught about the drugs, self-care measures and when to seek medical attention.
The goals of rehabilitation for long-term survivors of childhood and adult leukaemia are to manage the physical, psychological, social and spiritual consequences and delayed effects from the disease and its treatment. (Delayed effects are discussed in Ch 15.) The patient may need assistance to re-establish the various relationships that are a part of their life. Friends and family may not know how to interact with the patient. The patient and family must learn to regain attitudes of health and life while facing the real fear of relapse of the disease. Involving the patient in survivor networks, support groups or services offered through health organisations may help the patient to adapt to living after a life-threatening illness. Exploring resources in the community (e.g. the Leukaemia Foundation) may reduce the financial burden and feelings of dependence. Spiritual support may give the patient inner strength and peace. Studies examining spirituality and nursing care have highlighted the importance of nurses meeting patients’ spiritual needs.50,51
The patient will need support in adapting to any physical limitations or changes imposed by the illness. Vigilant follow-up care by providers who are aware of the unique needs of a cancer survivor is of the utmost importance for early recognition and treatment of long-term or delayed physical, psychological and social effects. The nurse may involve other healthcare providers in meeting the patient’s needs. However, often these needs will require the initiation of a referral or consultation. For example, physiotherapists may be asked to develop an exercise program to prevent post-treatment deficits caused by drug-induced peripheral neuropathy. Most patients should receive the pneumococcal vaccine at diagnosis and every 5 years as well as an annual influenza vaccine. These needs may also include other concerns, such as growth and development concerns for childhood survivors, and vocational retraining and reproductive concerns for a patient of childbearing age. The long-term recovery following treatment for leukaemia affects the quality of the patient’s life.
Lymphomas are malignant neoplasms originating in the bone marrow and lymphatic structures that result in the proliferation of lymphocytes. Lymphomas are the fifth most common type of cancer in Australian women and the sixth most common in Australian men.46
There are two major types of lymphoma: Hodgkin’s lymphoma and non-Hodgkin’s lymphomas. A comparison between them is presented in Table 30-16. Clinical practice guidelines on caring for the patient with lymphoma are available from the Cancer Council Australia (see Resources on p 805).52
Hodgkin’s lymphoma, also called Hodgkin’s disease, makes up about 12% of all lymphomas. It is a malignant condition characterised by proliferation of abnormal giant, multinucleated cells, called Reed-Sternberg cells, which are located in lymph nodes. The disease has a bimodal age-specific incidence, occurring most frequently in persons 15–35 years of age and above 50 years of age. It is twice as prevalent in men as in women and has a 5-year survival rate after diagnosis of 85%.53
Although the cause of Hodgkin’s lymphoma remains unknown, several key factors are thought to play a role in its development. The main interacting factors include infection with Epstein-Barr virus (EBV), genetic predisposition and exposure to occupational toxins. The incidence of Hodgkin’s lymphoma is increased among HIV-infected patients.9
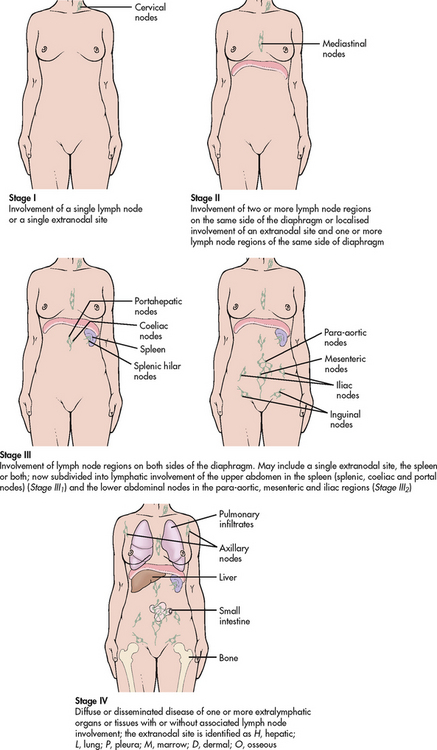
Normally, the lymph nodes are composed of connective tissues that surround a fine mesh of reticular fibres and cells. In Hodgkin’s lymphoma the normal structure of lymph nodes is destroyed by hyperplasia of monocytes and macrophages. The main diagnostic feature of Hodgkin’s lymphoma is the presence of Reed-Sternberg cells in lymph node biopsy specimens. The disease is believed to arise in a single location (it originates in lymph nodes in 90% of patients) and then spread along adjacent lymphatics. However, in recurrent disease, it may be more diffuse, and not necessarily contiguous. It eventually infiltrates other organs, especially the lungs, spleen and liver. In approximately two-thirds of patients the cervical lymph nodes are the first to be affected. When the disease begins above the diaphragm, it remains confined to lymph nodes for a variable period of time. Disease originating below the diaphragm frequently spreads to extra-lymphoid sites such as the liver.
The onset of symptoms in Hodgkin’s lymphoma is usually insidious. The initial development is most often enlargement of cervical, auxiliary or inguinal lymph nodes (see Fig 30-14). A mediastinal node mass is the second most common presenting location. This lymphadenopathy affects discrete nodes that remain movable and non-tender. The enlarged nodes are not painful unless they exert pressure on adjacent nerves.
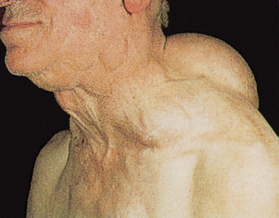
Figure 30-14 Hodgkin’s lymphoma (stage IIA). This patient has enlargement of the cervical lymph nodes.
The patient may notice weight loss, fatigue, weakness, fever, chills, tachycardia or night sweats. A group of initial findings including fever, night sweats and weight loss (termed B symptoms) correlates with a worse prognosis. After the ingestion of even small amounts of alcohol, individuals with Hodgkin’s lymphoma may complain of a rapid onset of pain at the site of disease. The cause of alcohol-induced pain is unknown.54 Generalised pruritus without skin lesions may develop. Cough, dyspnoea, stridor and dysphagia may all reflect mediastinal node involvement.
In more advanced disease there may be hepatomegaly and splenomegaly. Anaemia results from increased destruction and decreased production of erythrocytes. Other physical signs vary depending on where the disease is located. For example, intrathoracic involvement may lead to superior vena cava syndrome, enlarged retroperitoneal nodes may cause palpable abdominal masses or interfere with renal function, jaundice may occur from liver involvement, and spinal cord compression leading to paraplegia may occur with extradural involvement. Bone pain occurs as a result of bone involvement.
Peripheral blood analysis, excisional lymph node biopsy, bone marrow examination and radiological evaluation are important means of evaluating Hodgkin’s lymphoma. Peripheral blood analysis often reveals a microcytic hypochromic anaemia, neutrophilic leucocytosis (15–28 × 109/L), which may be associated with lymphopenia and an increased platelet count. Leucopenia and thrombocytopenia may develop, but they are usually a consequence of treatment, advanced disease or superimposed hypersplenism. Other blood studies may show hypoferraemia caused by excessive iron uptake by the liver and spleen, elevated leucocyte alkaline phosphatase from liver and bone involvement, hypercalcaemia from bone involvement and hypoalbuminaemia from liver involvement.
Excisional lymph node biopsy offers a definitive means of diagnosis. The removed peripheral lymph node is examined for the presence of the diagnostic Reed-Sternberg cells and to identify the subtype (the most common is nodular sclerosing). Bone marrow biopsy is performed as an important aspect of staging. Reed-Sternberg cells may also be found in the bone marrow.
Radiological evaluation can help define all sites and determine the clinical stage of the disease. CT or magnetic resonance imaging (MRI) scans are used as initial staging tools. Positron emission tomography (PET), with or without CT scans, is used to assess response to therapy and to differentiate residual tumour from fibrotic masses after treatment. These scans may show increased uptake (by PET) and masses (by CT) such as mediastinal lymphadenopathy, renal displacement caused by retroperitoneal node enlargement, abdominal lymph node enlargement, and liver, spleen, bone and brain infiltration.55
NURSING AND COLLABORATIVE MANAGEMENT: HODGKIN’S LYMPHOMAUsing all of the information from the various diagnostic studies, a clinical stage of disease is determined (see Fig 30-15). The final staging is based on the clinical stage (the extent of the disease) as well as the presence of B symptoms. Treatment depends on the nature and extent of the disease. The nomenclature used in staging involves an A or B classification, depending on whether symptoms are present when the disease is found, and a Roman numeral (I–IV) that reflects the location and extent of the disease. Additional features, such as an elevated erythrocyte sedimentation rate, age over 50 years, the presence of a large mediastinal mass and a low serum albumin, haemoglobin and lymphocyte count, may move an early stage (I–II) to an unfavourable prognosis, warranting more aggressive therapy.56
Once the stage of Hodgkin’s lymphoma is established, management focuses on selecting a treatment plan. The standard for chemotherapy is the ABVD regimen: doxorubicin, bleomycin, vinblastine and dacarbazine. Patients with favourable early-stage disease will receive two to four cycles of chemotherapy. Patients with early-stage disease but unfavourable prognostic features (e.g. the presence of B symptoms) or intermediate stage disease will be treated with four to six cycles of chemotherapy. Advanced-stage Hodgkin’s lymphoma is treated more aggressively using six to potentially eight cycles of chemotherapy. Other chemotherapy regimens include MOPP alternating with ABVD. MOPP consists of mechlorethamine, vincristine (oncovin), procarbazine and prednisolone. The role of radiation as a supplement to chemotherapy varies depending on the sites of disease and the presence of resistant disease after chemotherapy. The nurse needs to ensure that the doctor has discussed the risks and benefits associated with treatment or no treatment and the outcomes of alternative treatments with the patient prior to commencement of treatment and that consent is documented in the patient’s medical record.
Intensive chemotherapy, with or without the use of autologous or allogeneic HSCT and haematopoietic growth factors, is the treatment of choice for advanced, refractory or relapsed Hodgkin’s lymphoma (stages IIIB and IV). HSCT has allowed patients to receive higher, potentially curative doses of chemotherapy while reducing life-threatening leucopenia (see Ch 15). Combination chemotherapy works well because, as in leukaemia, drugs are used that have an additive antitumour effect without increasing side effects. As with leukaemia, therapy must be aggressive. Therefore, potentially life-threatening problems are addressed in an attempt to achieve a remission.
Maintenance chemotherapy does not contribute to increased survival once a complete remission is achieved. Occasionally, single drugs may be administered palliatively to patients who cannot tolerate intensive combination therapy. A serious consequence of the treatment for Hodgkin’s lymphoma is the later development of secondary malignancies (see Ch 15) as well as potential long-term toxicities from the treatment, such as endocrine, cardiac and pulmonary dysfunction.56 The estimated risk of a secondary cancer is between 2% and 6% and generally occurs within the first 10 years after treatment.54 The most common secondary malignancies are acute myelogenous leukaemia, non-Hodgkin’s lymphoma and solid tumours.
The nursing care for Hodgkin’s lymphoma is largely based on managing problems related to the disease (e.g. pain due to tumour), pancytopenia and other side effects of therapy. Because the survival of patients with Hodgkin’s lymphoma depends on their response to treatment, supporting the patient through the consequences of treatment is extremely important.
The patient undergoing radiation therapy has special nursing needs. The skin in the radiation field requires attention. Also, the nurse must understand the concepts related to administration of radiation therapy (see Ch 15).
Psychosocial considerations are just as important as they are with leukaemia. However, the prognosis for Hodgkin’s lymphoma is better than that for many forms of cancer or leukaemia. Attention must be given to the physical, psychological, social and spiritual consequences of the patient’s disease. Fertility issues may be of particular concern because this disease is frequently seen in adolescents and young adults. The nurse must help ensure that these issues have been addressed soon after diagnosis. Evaluation of patients for long-term effects of therapy is important because delayed consequences of disease and treatment may not be apparent for many years. (Secondary malignancies and delayed effects are discussed in Ch 15.)
Non-Hodgkin’s lymphomas (NHL) are a heterogeneous group of malignant neoplasms of primarily B- or T-cell origin affecting all ages. B-cell lymphomas constitute about 90% of all NHL. They are classified according to different cellular and lymph node characteristics. A variety of clinical presentations and courses are recognised, from indolent (slowly developing) to rapidly progressive disease. NHL is the most commonly occurring haematological cancer, and 89% of people diagnosed in Australia with lymphoma will have NHL.53 Australia’s incidence of NHL is similar to that of New Zealand and Canada but higher than in the UK and lower than in the US.
As with Hodgkin’s lymphoma, the cause of NHL is usually unknown. However, as the population has aged and the incidence of HIV infection has increased, the incidence of NHL has increased by 2–3% per year for at least the past 30 years. It is also more common in individuals who have used immunosuppressive medications (e.g. to prevent rejection following an organ transplant or to treat autoimmune disorders) or who have received chemotherapy or radiation therapy. EBV is associated with Burkitt’s lymphoma, but not all individuals with EBV get NHL.
There is no hallmark feature in NHL that parallels the Reed-Sternberg cells of Hodgkin’s lymphoma. However, all types of NHL involve lymphocytes arrested in various stages of development. For example, lymphoblastic lymphoma and lymphoblastic leukaemia result from malignant proliferation of small naive B lymphocytes, the latter having a majority of disease within the bone marrow (as compared to the lymph nodes).57 Diffuse large B-cell lymphoma, the most common aggressive lymphoma in adults, is a neoplasm that originates in the lymph nodes.58 Burkitt’s lymphoma is a highly aggressive disease thought to originate from B-cell blast cells in the lymph nodes.
NHL can originate outside the lymph nodes, the method of spread can be unpredictable and the majority of patients have widely disseminated disease at the time of diagnosis (see Fig 30-16). The primary clinical manifestation is painless lymph node enlargement. The lymphadenopathy can be present or absent in indolent disease. Because the disease is usually disseminated when it is diagnosed, other symptoms will be present depending on where the disease has spread (e.g. hepatomegaly with liver involvement, neurological symptoms with CNS disease). NHL can also present in non-specific ways, such as an airway obstruction, hyperuricaemia and renal failure from tumour lysis syndrome, pericardial tamponade and GI complaints.
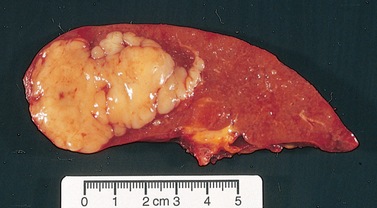
Figure 30-16 Non-Hodgkin’s lymphoma involving the spleen. The presence of an isolated mass is typical.
Patients with high-grade lymphomas may have lymphadenopathy and constitutional (B) symptoms, such as fever, night sweats and weight loss. The peripheral blood is usually normal, but some lymphomas manifest in a ‘leukaemic’ phase.
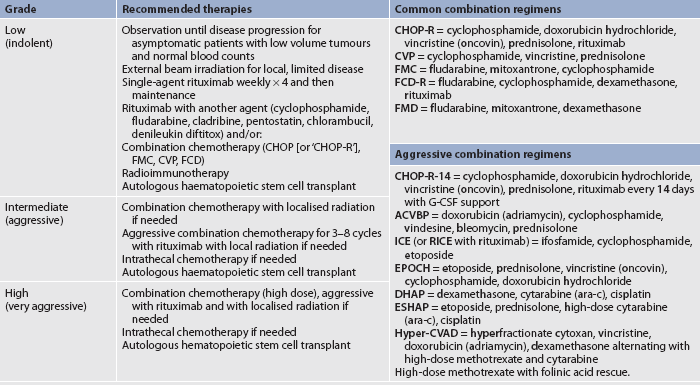
Diagnostic studies used for NHL resemble those used for Hodgkin’s lymphoma. However, because NHL is more often in extranodal sites, more diagnostic studies may be done, such as an MRI to rule out CNS or bone marrow infiltration, and a barium enema or CT scan to visualise suspected GI involvement. Clinical staging, as described for Hodgkin’s lymphoma, is used to help guide therapy (see Fig 30-15), but establishing the precise histological subtype is extremely important. Lymph node biopsy establishes the cell type and pattern. NHL can be classified based on morphological, genetic and immunophenotypic findings as well as clinical presentation.55 A useful system for classifying NHL is the International Working Formulation (IWF), which simply divides each subtype of lymphoma into low grade (indolent), intermediate grade (aggressive) and high grade (very aggressive; see Box 30-13). Additional factors, known as the International Prognostic Index (IPI), may be considered to help select the appropriate treatment for these patients. Factors considered are advanced disease, number of extranodal sites, age over 60 years, high serum LDH and performance status. Immunological, cytogenetic and molecular studies are also useful for making therapeutic decisions and assessing prognosis. The prognosis for NHL is generally not as good as that for Hodgkin’s lymphoma.
BOX 30-13 Non-Hodgkin’s lymphoma classification*
Small lymphocytic, plasmacytoid
Follicular, predominantly large cell
Diffuse, mixed, small and large cell
*Partial listing.
NURSING AND COLLABORATIVE MANAGEMENT: NON-HODGKIN’S LYMPHOMATreatment for NHL involves chemotherapy and sometimes radiation therapy (see Table 30-17). Paradoxically, more aggressive lymphomas are more responsive to treatment and are more likely to be cured. In contrast, indolent lymphomas have a naturally long course but are difficult to treat effectively.
Patients with low-grade (indolent) lymphoma have a median overall survival of 9 years. However, most patients relapse several times and cure is very unlikely. A previous approach was that patients who were asymptomatic were followed with watchful waiting to assess the progress of the disease. However, some initial therapies can be well tolerated and have been shown to reduce the time to progression of the disease. Therapy for these patients is indicated if the patient has local symptoms from progressive, bulky or painful disease or a compromise of normal organ function. An option for these patients is rituximab every week and then every 2 months as maintenance therapy. Rituximab, a genetically engineered monoclonal antibody against the CD20 antigen on the surface of normal and malignant B lymphocytes, binds to the cells, causing lysis and cell death. Once the disease is symptomatic, rituximab with chemotherapy, such as cyclophosphamide with or without prednisolone or even the CHOP-R regimen (cyclophosphamide, doxorubicin hydrochloride, vincristine [oncovin], prednisolone and rituximab), may be used. Complete remissions are uncommon, but the majority of patients will respond with improvement in adenopathy and symptoms. Numerous chemotherapy combinations have been used to try to overcome the resistant nature of this disease.
Intermediate-grade (aggressive) and high-grade (very aggressive) lymphomas may be treated similarly, depending on the extent of the disease and the patient’s prognostic factors. Diffuse large B-cell NHL is the most common high-grade lymphoma and the most common subtype. The most common standard chemo-therapeutic regimen is CHOP-R. Other combination therapies may be used for very aggressive or refractory disease. These include RICE, EPOCH, DHAP and ESHAP (see Table 30-17). Dose-dense (intensified) treatment with CHOP-R-14 every 2 weeks (versus every 3–4) with haematopoietic growth factor support may be used in patients with aggressive disease. High-dose chemotherapy with autologous HSCT has shown a better outcome than conventional chemotherapy in the treatment of patients with relapsed, aggressive NHL who are still responding to salvage chemotherapy. The role of allogeneic transplant in NHL is uncertain but it is thought that it may have some benefit in certain subtypes with aggressive or refractory lymphoma.59
Since NHL represents a large variety of neoplasms, some subtypes may be treated differently from the general standards described above. For example, cutaneous T-cell lymphoma may be treated with topical corticosteroids or topical chemotherapy for limited-stage disease. For more diffuse disease, treatment may include phototherapy or α-interferon.
Other therapies for NHL include the monoclonal antibodies. These antibodies are linked to a radioactive isotope (yttrium-90 and iodine I-130, respectively; see Table 15-14). The monoclonal antibody targets the CD20 antigen, which is on the surface of mature B cells and B-cell tumours. This allows the delivery of radiation directly to the malignant cells. Currently, these therapies are primarily used for patients with indolent lymphomas, particularly those with chemotherapy-refractory disease. Side effects of this type of treatment include pancytopenia. The nurse needs to be aware of the necessary precautions in caring for these patients, both to educate patients about safety issues and to minimise the risk of radiation exposure to staff and others. The nurse also needs to make sure that the doctor has discussed the risks and benefits associated with treatment and/or no treatment and the outcome of alternative treatments with the patient prior to treatment commencing and that consent is documented in the patient’s medical record.
The nursing care for NHL is similar to that for Hodgkin’s lymphoma. Nursing care is largely based on managing problems related to the disease (e.g. pain due to the tumour, spinal cord compression, tumour lysis syndrome), pancytopenia and other side effects of therapy. However, because NHL can be more extensive and involve specific organs (e.g. CNS, spleen, liver, gastrointestinal tract, bone marrow), it is important that the nurse has an understanding of the subtype and extent of the disease. For example, a patient with known involvement of the colon may complain of acute abdominal pain. The patient most likely would have abdominal guarding and an enlarged and tympanic abdomen. This could indicate a bowel perforation and be considered a medical emergency. A patient with Burkitt’s NHL starting chemotherapy would be at high risk for tumour lysis syndrome and would need to have frequent laboratory parameters drawn and monitored, as well as being on strict intake and output. For oncological problems, refer to Chapter 15. Because most of these patients receive therapy that is potentially myelosuppressive, NCPs 30-1, 30-2 and 30-3 apply to these patients as well.
The patient undergoing radiation therapy has special nursing needs. The skin in the radiation field requires attention. In addition, the nurse needs to understand the concepts related to administration of and safety issues regarding radiation therapy (see Ch 15).
Psychosocial considerations are very important. Helping the patient and family understand the disease, treatment and expected and potential untoward side effects is paramount in enlisting their help to ensure the patient’s wellbeing and safety. Fertility issues may be of concern in young patients. As in Hodgkin’s lymphoma, evaluation of patients with NHL for long-term effects of therapy is important because delayed consequences of disease and treatment may not be apparent for many years. (Secondary malignancies and delayed effects are discussed in Ch 15.)
Multiple myeloma, or plasma cell myeloma, is a condition in which neoplastic plasma cells infiltrate the bone marrow and destroy bone. It is a relatively rare disease, representing 1% of all cancers in Australia.60 The disease is more common after 60 years of age and is uncommon in people under 40 years of age.60 Multiple myeloma occurs in African Americans more commonly than in whites.61 A patient usually lives for approximately 2 years after diagnosis if untreated. However, many patients are now living 7 years or more after diagnosis due to the variety of treatments that can be provided throughout the course of the disease.62
The cause of multiple myeloma is unknown. Exposure to radiation, organic chemicals (such as benzene), herbicides and insecticides may play a role. Genetic factors and viral infection may also influence the risk of developing multiple myeloma.
The disease process involves excessive production of plasma cells. Plasma cells are activated B cells, which produce immunoglobulins (antibodies) that normally serve to protect the body. However, in multiple myeloma the malignant plasma cells infiltrate the bone marrow and produce abnormal and excessive amounts of immunoglobulin (usually IgG [most common], IgA, IgD or IgE). This abnormal immunoglobulin is termed a myeloma protein, or M protein. Furthermore, plasma cell production of excessive and abnormal amounts of cytokines (interleukins [ILs]; IL-4, IL-5 and IL-6) also plays an important role in the pathological process of bone destruction. As myeloma protein increases, normal plasma cells are reduced, which further compromises the body’s normal immune response. In some patients, excessive production and secretion of free light-chain proteins (called Bence Jones proteins) from the myeloma cell are also seen and can be detected in the urine. Proliferation of malignant plasma cells and the overproduction of immunoglobulin and proteins result in the end-organ effects of myeloma to the bone marrow, bone and kidneys and possibly the spleen, lymph nodes, liver and even heart muscle.
Multiple myeloma develops slowly and insidiously. The patient often does not manifest symptoms until the disease is advanced, at which time skeletal pain is the major manifestation. Pain in the pelvis, spine and ribs is particularly common. The pain is triggered by movement. Diffuse osteoporosis develops as the myeloma protein destroys bone. Osteolytic lesions are seen in the skull, vertebrae and ribs (see Fig 30-17). Vertebral destruction can lead to collapse of vertebrae with ensuing compression of the spinal cord. Loss of bone integrity can lead to the development of pathological fractures; about 30% of patients present with pathological fractures.
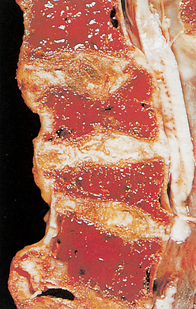
Figure 30-17 Multiple myeloma. This segment of the lower thoracic spine has been sectioned to show the extensive replacement of the bone and marrow with red gelatinous tissue.
Bony degeneration also causes calcium to be lost from bones, eventually causing hypercalcaemia. Hypercalcaemia may cause renal, GI or neurological manifestations, such as polyuria, anorexia, confusion and ultimately seizures, coma and cardiac problems. High protein levels caused by the presence of the myeloma protein can result in renal failure, from renal tubular obstruction by the myeloma protein and interstitial nephritis. The patient may also display manifestations of anaemia, thrombocytopenia and neutropenia, all of which are related to the replacement of normal bone marrow with plasma cells.
Evaluating multiple myeloma involves laboratory, radiological and bone marrow examination. The presence of a monoclonal (M) antibody protein can be found in the blood and urine. Pancytopenia, hypercalcaemia, the presence of Bence Jones protein in the urine and an elevated serum creatinine are possible findings.
X-rays show distinct lytic areas of bone erosions, generalised thinning of the bones and/or fractures, especially in the vertebrae, ribs, pelvis and bones of the thigh and upper arms. Bone marrow analysis shows significantly increased numbers of plasma cells in the bone marrow. The simplest measure of prognosis in multiple myeloma is based on blood levels of two markers: β2-microglobulin and albumin. In general, higher levels of β2-microglobulin and lower levels of albumin are associated with a poorer prognosis.
Multidisciplinary care involves managing both the disease and its symptoms. The current treatment options include watchful waiting (for early multiple myeloma), corticosteroids, chemotherapy, biological therapy and HSCT. Multiple myeloma is seldom cured, but treatment can relieve symptoms, produce remission and prolong life.
Ambulation and adequate hydration are used to treat hypercalcaemia, dehydration and potential renal damage. Weight-bearing helps the bones reabsorb some calcium, and fluids dilute calcium and prevent protein precipitates from causing renal tubular obstruction. Control of pain and prevention of pathological fractures are other goals of management. Analgesics, orthopaedic supports and localised radiation help reduce the skeletal pain. Bisphosphonates, such as pamidronate, zoledronic acid and etidronate, inhibit bone breakdown and are used for the treatment of skeletal pain and hypercalcaemia. They inhibit bone resorption without inhibiting bone formation and mineralisation. They are given monthly by IV infusion. Radiation therapy is another important component of treatment, primarily because of its effect on localised lesions. Surgical procedures, such as vertebroplasty, may be done to support degenerative vertebrae.
Chemotherapy is usually the first treatment recommended for multiple myeloma. It is used to reduce the number of plasma cells. The agents most frequently used are the alkylating drugs, including melphalan, cyclophosphamide, vincristine and doxorubicin. Corticosteroids (prednisolone, dexamethasone) may be added because they exert an antitumour effect in some patients. A common regimen is VAD—vincristine, doxorubicin and dexamethasone—given once a month over several months prior to an autologous HSCT. Because of improved response rates and survival rates, high-dose chemotherapy, such as melphalan, followed by autologous HSCT has evolved as the standard of care in eligible patients.61
Other medications may also be used to keep the myeloma under control for some time. Bortezomib is a new targeted therapy known as a proteasome inhibitor (see Table 15-14). Proteasomes are present in all cells and regulate cell growth. Normal cells are capable of recovery from proteasome inhibition, but cancer cells undergo death when proteasomes are inhibited. Bortezomib is indicated for patients whose disease has relapsed after two prior treatments and who have demonstrated resistance to their last treatment. α-interferon therapy has been used following chemotherapy. It works by altering the receptors for IL-6, which is a growth factor for myeloma cells.
Drugs may be used to treat complications of multiple myeloma. For example, allopurinol may be given to reduce hyperuricaemia, and IV frusemide promotes renal excretion of calcium. Calcitonin can be used to decrease the risk of fractures and reduce bone pain.
NURSING MANAGEMENT: MULTIPLE MYELOMAA major focus of care relates to the bone involvement and sequelae from bone breakdown. Maintaining adequate hydration is a primary nursing consideration to minimise problems from hypercalcaemia. Fluids are administered to attain a urinary output of 1.5–2 L per day. This may require an intake of 3–4 L per day. In addition, weight-bearing helps bones reabsorb some of the circulating calcium, and corticosteroids may augment the excretion of calcium. Once chemotherapy is initiated, the uric acid levels may rise because of the increased cell destruction. Hyperuricaemia is treated by ensuring adequate hydration and using allopurinol to prevent any renal damage. Because of the myeloma proteins, the patient is at additional risk of renal dysfunction. The nurse needs to monitor electrolytes and fluid balance. The nurse also needs to ensure that the doctor has informed the patient of the risks and benefits associated with treatment, alternative treatments and no treatment prior to commencing therapy and that consent is documented in the patient’s medical record.
Due to the potential for pathological fractures, the nurse must be careful when moving and ambulating the patient. A slight twist or strain in the wrong area (e.g. a weak area in the patient’s bones) may be sufficient to cause a fracture. In addition, the development of peripheral neuropathy is common with several therapies for multiple myeloma and can contribute to discomfort, the inability to perform basic activities of daily living and the risk of injury from falling.
Pain management requires innovative and knowledgeable nursing interventions. Analgesics, such as non-steroidal anti-inflammatory drugs, codeine or a codeine/paracetamol combination, may be more effective than opioids alone in diminishing bone pain. Braces, especially for the spine, may also help control pain. As in any pain management situation, the nurse is responsible for assessing the patient and for implementing necessary measures to alleviate the pain. The pain team or the pain nurse consultant should be involved in the patient care. (Pain management is discussed in Ch 8.) Patients may also be at risk for deep vein thrombosis related to chemotherapy and immobility and should have appropriate preventative measures employed.63
Assessment and prompt treatment of infection is important in the care of patients with multiple myeloma: 50–70% of patients will die as a result of bacterial infections. These recurrent infections may be due to a decrease in the production of normal immunoglobins, the ineffectiveness of the overproduced and abnormal immunoglobins and/or the neutropenia resulting from the bone marrow infiltration or side effects of treatment. Nursing care plans for anaemia (see NCP 30-1), thrombocytopenia (see NCP 30-2) and neutropenia (see NCP 30-3) apply to the patient with multiple myeloma.
The patient’s psychosocial needs require sensitive, skilled management. It is important to help the patient and significant others to adapt to changes fostered by chronic sickness, deal with reality and adjust to the losses related to the disease process. The symptoms of multiple myeloma remit and exacerbate. Consequently, acute care is needed at various times during the course of the illness. The final, acute phase is unresponsive to treatment and usually short in duration. The way in which patients and families deal with confronting death may be affected by the manner in which they have learned to accept and live with the chronic nature of the disease. Guidelines on caring for patients with multiple myeloma have been developed.64
The spleen can be affected by many illnesses, most of which can cause some degree of splenomegaly (enlarged spleen; see Box 30-14). The term hypersplenism refers to the occurrence of splenomegaly and peripheral cytopenias (anaemia, leucopenia and thrombocytopenia). The degree of splenic enlargement varies with the disease. For example, massive splenic enlargement occurs with chronic myelogenous leukaemia and thalassaemia major. Mild splenic enlargement occurs with heart failure and systemic lupus erythematosus. The normal spleen contains 20–40 mL of blood and does not usually serve as a reservoir for blood volume or erythrocytes.65
BOX 30-14 Causes of splenomegaly
Bacterial infections: endocarditis
Mycobacterial infections: tuberculosis
Spirochetes: syphilis, Lyme disease
Viral infections: hepatitis, human immunodeficiency virus, cytomegalovirus, mononucleosis
Parasitic infections: malaria, trypanosomiasis, schistosomiasis, leishmaniasis, toxoplasmosis, echinococcosis
Rickettsial infections: Rocky Mountain spotted fever, typhoid fever, kala-azar
When the spleen enlarges, its normal filtering and sequestering capacity increases. Consequently, there is often a reduction in the number of circulating blood cells. In addition, there are unusual findings in the peripheral smear, such as pitted or pocked erythrocytes or Howell-Jolly bodies (clusters of DNA in the RBCs where there should be none). These findings assist with diagnosing a malfunctioning spleen. A slight-to- moderate enlargement of the spleen is usually asymptomatic and found during a routine examination of the abdomen. Even massive splenomegaly can be well tolerated but the patient may complain of abdominal discomfort and early satiety. In addition to physical examination, other techniques to assess the size of the spleen include a technetium-98–sulfur colloid liver–spleen scan, CT scan, MRI and ultrasound scan. Occasionally laparoscopy or open laparotomy and splenectomy are indicated in the evaluation or treatment of splenomegaly. Splenectomy can have a dramatic effect in increasing peripheral RBC, WBC and platelet counts. Another major indication for splenectomy is splenic rupture. The spleen may rupture from trauma, inadvertent tearing during other surgical procedures, and diseases such as mononucleosis, malaria and lymphoid neoplasms.
Nursing responsibilities for the patient with spleen disorders vary depending on the nature of the problem. Splenomegaly may be painful and may require analgesic administration; care in moving, turning and positioning; and evaluation of lung expansion because spleen enlargement may impair diaphragmatic excursion. If anaemia, thrombocytopenia or leucopenia develops from splenic enlargement, nursing measures must be instituted to support the patient and prevent life-threatening complications. If splenectomy is performed, the nurse must provide the meticulous care warranted after any surgery. In addition, there must be special observation for haemorrhage, which could lead to shock, fever and abdominal distension.66
After splenectomy, immunological deficiencies may develop. IgM levels are reduced, and IgG and IgA values remain within normal limits. Postsplenectomy patients have a lifelong risk of infection, especially from encapsulated organisms such as pneumococcus. This risk is reduced by immunisation with pneumococcal vaccine.
Blood component therapy is frequently used in managing haematological diseases. Many therapeutic and surgical procedures depend on blood product support. However, blood component therapy only temporarily supports the patient until the underlying problem is resolved. Because transfusions are not free from hazards, they should be used only if necessary. The research regarding RBC substitutes is ongoing. To reduce the use of blood products, antifibrinolytic agents and other products may be used in some situations. Nurses must be careful to avoid developing a complacent attitude about this common but potentially dangerous therapy. The nurse also needs to make sure that the doctor has discussed the risks, benefits and alternatives with the patient and that consent is documented in the patient’s medical record. An informative resource for safety related to blood transfusions is the BloodSafe eLearning program,67 and clinical practice guidelines for transfusions are available from the Australian and New Zealand Society of Blood Transfusion68 and the National Health and Medical Research Council69 (see Resources on p 805).
Traditionally, the term blood transfusion meant the administration of whole blood. Blood transfusion now has a broader meaning because of the ability to administer specific components of blood such as platelets, packed RBCs or plasma. Usually, a specific component is ordered, although whole blood may be used rarely with massive haemorrhage or for an exchange transfusion (see Table 30-18). (See Ch 15 for discussion of haematopoietic stem cell transplants.)
TABLE 30-18 Blood products*
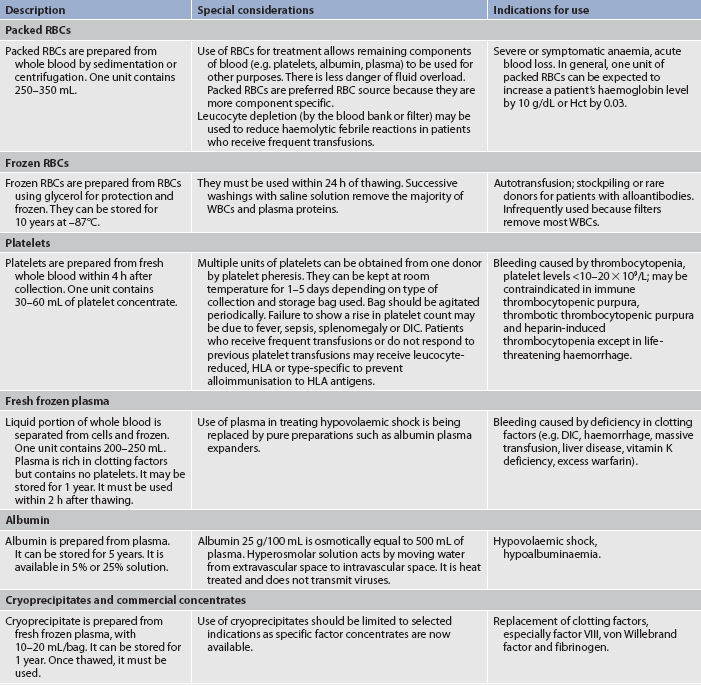
DIC, disseminated intravascular coagulation; HLA, human leucocyte antigen; RBCs, red blood cells; WBCs, white blood cells.
* Component therapy has replaced the use of whole blood, which accounts for less than 10% of all transfusions. Granulocyte transfusions are not included here because they are rarely used.
CLINICAL PRACTICE
An older woman is transferred from a nursing home because of gastrointestinal bleeding from an unknown cause. Some of her family members tell the nurse that she is a Jehovah’s Witness and must not receive blood products. If she does not have exploratory surgery and transfusions, the surgeon believes that she will die.
• Competent adults have the right to make healthcare decisions based on their religious beliefs, including the right to refuse treatment.
• Healthcare professionals should make every effort to incorporate the patient’s values and beliefs into the treatment plan. When the extent of the patient’s beliefs is not clear, members of the healthcare team should consult other available resources, such as family, friends or church officials, in an attempt to ascertain the patient’s commitment to their faith.
• Jehovah’s Witnesses believe that if they receive blood or blood products there are eternal consequences.
• When a clear determination of the patient’s beliefs cannot be made, the patient is unable to communicate their wishes or there are no advance directives, a decision by the healthcare team to perform the lifesaving surgery and transfusion would be acceptable.
Blood components can be administered safely through a 23-gauge needle, but this usually impedes flow and could cause haemolysis, particularly if using a pressured infusion device. If a small gauge is to be used, it is best to have the unit split (the blood bank issues half of the unit at a time) or diluted with normal saline during administration to ensure that it does not run over the maximum time of 4 hours that blood can be kept out of refrigeration.67 Therefore, a larger gauge of at least 19 is preferred, running the unit into a free-flowing IV line. Larger needles (e.g. 18 or 16 gauge) may be preferred if rapid transfusions are given. Smaller needles can be used for platelets, albumin and clotting factor replacement. Most blood product administration tubing is of a ‘Y type’ with a microaggregate filter (which filters out particulate), with one arm of the Y for the isotonic saline solution and the other for the blood product. Dextrose solutions or Hartmann’s solution should not be used because they induce RBC haemolysis. No other additives (including medications) should be given via the same tubing as the blood unless the tubing is cleared with saline solution.67
When the blood or blood components have been obtained from the blood bank, positive identification of the donor blood and recipient must be made. Improper product-to-patient identification causes the majority of haemolytic transfusion reactions, thus placing a great responsibility on nursing personnel to carry out the identification procedure appropriately.67 The nurse must follow the policy and procedures at the place of employment. The blood bank is responsible for typing and cross-matching the donor’s blood with the recipient’s blood; the result of the compatibility testing should be noted on the product bag or tag, if pertinent.
The nurse should ensure that the patient understands the procedure and the signs and symptoms to report, and that they agree with the treatment plan. The patient’s vital signs should be taken prior to beginning the transfusion to provide a baseline measure; if the patient has abnormal vital signs, such as an elevated fever, the doctor should clarify when the blood component may be administered. The blood should be administered as soon as it is brought to the patient. It should not be refrigerated in the nursing unit. If the blood is not used within 30 minutes, it should be returned to the blood bank. During the first 15 minutes or 50 mL of blood infusion, the nurse should remain with the patient—if there are any untoward reactions, they are most likely to occur at this time. The rate of infusion during this period should be no more than 2 mL/min. Packed RBCs should not be infused quickly unless in emergency. Rapid infusion of cold blood may cause the patient to become chilled. If rapid replacement of large amounts of blood is necessary, a blood-warming device may be used. Other blood components, such as fresh frozen plasma and platelets, may be infused over 15–30 minutes.
After the first 15 minutes, vital signs are usually retaken, and the rate of infusion is governed by the clinical condition of the patient and the product being infused. Most patients not in danger of fluid overload can tolerate the infusion of 1 unit of packed RBCs over 2 hours. The transfusion should not take more than 4 hours to administer due to the increased risk of bacterial growth in the product once it is out of refrigeration. Blood unrefrigerated for 4 hours or longer should not be infused and should be returned to the blood bank.
A blood transfusion reaction is an adverse reaction to blood transfusion therapy that can range in severity from mild symptoms to a life-threatening condition. Because complications of transfusion therapy may be significant, judicious evaluation of the patient is required. Blood transfusion reactions can be broadly classified as acute or delayed and examples of these are provided in Tables 30-19 and 30-20. The Australian Red Cross Blood Service and the New Zealand Blood Service provide information about adverse events from transfusions (see Resources on p 805).70,71
TABLE 30-19 Acute transfusion reactions
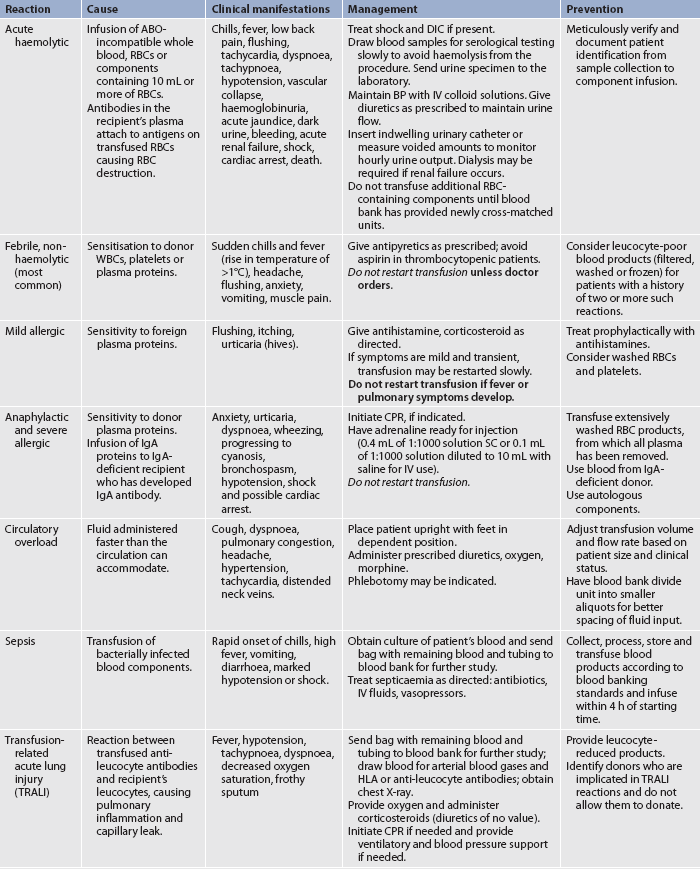
BP, blood pressure; CPR, cardiopulmonary resuscitation; DIC, disseminated intravascular coagulation; IgA, immunoglobulin A; IV, intravenous; RBC, red blood cell; SC, subcutaneous; WBC, white blood cell.
TABLE 30-20 Delayed transfusion reactions

ALT, alanine aminotransferase; AST, aspartate aminotransferase; HCV, hepatitis C virus; HTLV-1, human T-cell leukaemia virus, type 1; RBCs, red blood cells.
* New cases of transfusion-related hepatitis B and C are not common.
If any acute transfusion reaction occurs, the nurse should: (1) stop the transfusion immediately; (2) check and monitor vital signs; (3) maintain a patent IV line; (4) check the right pack has been given to the right patient; and (5) notify the blood bank or transfusion service and doctor immediately.70 Nurses need to follow their health organisation’s policies and may also be required to: (1) recheck identifying tags and numbers; (2) treat symptoms as per the doctor’s order; (3) save the blood bag and tubing and send them to the blood bank for examination; (4) complete an incident report and a transfusion reaction report; (5) collect required blood and urine specimens at intervals stipulated by hospital policy to evaluate for haemolysis; and (6) document the details in the patient chart. The blood bank and laboratory are responsible for identifying the type of reaction.
The most common cause of haemolytic reactions is transfusion of ABO-incompatible blood (see Table 30-19). This is an example of a type II cytotoxic hypersensitivity reaction (see Ch 13). Severe haemolytic reactions are rare. Mislabelling specimens and administering blood to the wrong individual cause most acute haemolytic reactions. This points to the importance of using proper patient identifiers when drawing blood samples and when administering medications and blood products.
When an acute haemolytic reaction occurs, antibodies in the recipient’s serum react with antigens on the donor’s RBCs. This results in agglutination of cells, which can obstruct capillaries and block blood flow. Haemolysis of the RBCs releases free haemoglobin into the plasma. The haemoglobin is filtered by the kidneys and may be found in the urine (haemoglobinuria). Haemoglobin may obstruct the renal tubules, leading to acute renal failure, DIC and death (see Ch 46).
The clinical manifestations of an acute haemolytic reaction may be mild or severe and usually develop within the first 15 minutes of transfusion. Free haemoglobin in blood and urine specimens obtained at the onset of the reaction will provide evidence of an acute haemolytic reaction. Delayed transfusion reactions occur 2–14 days after the administration of blood.70 (The clinical manifestations and nursing management for the patient with a haemolytic reaction are presented in Table 30-19.)
Febrile reactions are most commonly caused by leucocyte incompatibility. Many individuals who receive five or more transfusions develop circulating antibodies to the small amount of WBCs in the blood product. Febrile reactions can often be prevented by using additional filters in the tubing to leucocyte-deplete RBCs and platelets. Leucocyte-poor blood products (filtered, washed or frozen) can also be used to prevent febrile reactions.
Allergic reactions result from the recipient’s sensitivity to plasma proteins in the donor’s blood. These reactions are more common in an individual with a history of allergies. Antihistamines may be used to prevent allergic reactions. Adrenaline or corticosteroids may be used to treat a severe reaction.
An individual with cardiac or renal insufficiency is at risk of developing circulatory overload. This is especially true if a large quantity of blood is infused over a short period of time, particularly in an elderly patient. Packed RBCs can be split by the blood bank, allowing half of a unit to be given over a time frame of up to 4 hours. A fluid balance assessment, including baseline auscultation of the patient’s lungs, is performed. Complaints of shortness of breath and the presence of adventitious breath sounds may indicate fluid overload in any patient.
Blood products can become infected from improper handling and storage. Bacterial contamination of blood products can result in bacteraemia, sepsis or septic shock.
Transfusion-related acute lung injury (TRALI) is characterised by the sudden development of non-cardiogenic pulmonary oedema (acute lung injury). It usually occurs within 2–6 hours after transfusion of blood products, but may occur as late as 48 hours after transfusion. With the reduction in administrative errors, the advent of leucocyte-reduced products, more effective screening and the prevention of the transmission of infectious agents, TRALI has surpassed haemolytic reactions as the leading cause of transfusion-related mortality. It is thought to be due to an antibody-mediated reaction between the recipient’s leucocytes and antileucocyte antibodies from donors who were sensitised during pregnancy or by previous transfusions. This causes pulmonary capillary inflammation and increased permeability, leading to respiratory distress and potentially death.70
An acute complication of transfusing large volumes of blood products is termed massive blood transfusion reaction. This reaction can occur when replacement of RBCs or blood exceeds the total blood volume within 24 hours. In this situation, an imbalance of normal blood elements results because clotting factors, albumin and platelets are not found in RBC transfusions.
Additional problems such as hypothermia, citrate toxicity, hypocalcaemia and hyperkalaemia may occur when massive blood transfusions are given. Hypothermia and cardiac dysrhythmias can result from rapid infusion of large quantities of cold blood. Blood-warming equipment prevents this problem. Citrate toxicity and hypocalcaemia can occur from the use of large quantities of blood products, because citrate is part of the storage solution; calcium binds to the citrate. Citrate toxicity is likely to develop when blood is transfused at a rate of 1 unit in 10 minutes (or 8–10 units of RBCs within a few hours). Manifestations such as muscle tremors and ECG changes may be observed with hypocalcaemia but can be prevented or reversed by the infusion of 10% calcium gluconate (10 mL with every litre of citrated blood). Hyperkalaemia results when potassium leaks from RBCs in stored blood. Mild-to-severe signs and symptoms can occur, including nausea, muscle weakness, diarrhoea, paraesthesias, flaccid paralysis of the cardiac or respiratory muscles, and cardiac arrest. Electrolyte monitoring is an important aspect of care of the patient receiving massive transfusions of blood products.70
Delayed transfusion reactions include delayed haemolytic reactions (discussed previously), infection, iron overload and graft-versus-host disease (see Table 30-20).70
Infectious agents transmitted by blood transfusion include hepatitis B and C viruses, HIV, human herpes virus type 6 (HSV-6), EBV, HTLV-1, cytomegalovirus (CMV) and malaria. Hepatitis is still the most common viral infection transmitted, although its incidence has been decreasing. Hepatitis B virus can be detected in the blood by the presence of hepatitis B surface antigen (HBsAg). A test for hepatitis C antibodies in donor blood is used to exclude the use of any donated blood testing positive for hepatitis C. Therefore, the risk of transmission of hepatitis C has been reduced. Leucocyte-reduced blood products drastically reduce the risk of blood transfusion-associated viral infections, including CMV.
In the past, HIV was transmitted by contaminated blood and blood products. This posed a serious problem for those who received infected transfusions. Patients with haemophilia who received antihaemophilic factors, which had been prepared from the pooled plasma of a large number of donors of whom some were infected, have a high rate of HIV infection from transfusion sources. Presently, the use of recombinant antihaemophilic factors (see Table 30-9), donor education, donor screening and HIV antibody testing has greatly reduced the transmission of HIV by blood transfusion or factor replacement therapy.
Autotranfusion, or autologous transfusion, consists of removing whole blood from a person and transfusing that blood back into the same person. The problems of incompatibility, allergic reactions and transmission of disease can thereby be avoided. Methods of autotransfusion include the following:
• Autologous donation or elective phlebotomy (preoperative autologous deposit; PAD).70 The individual donates blood before a planned surgical procedure. The blood can be frozen and stored for up to 10 years. Usually the blood is stored without being frozen and is given to the person within a few weeks of donation. This technique is especially beneficial to the patient with a rare blood type or for any patient who might be expected to require limited blood product support during a major surgical procedure (e.g. elective orthopaedic surgery).
• Autotransfusion. This newer method for replacing blood volume involves safely and aseptically collecting, filtering and returning the patient’s own blood lost during a major surgical procedure or from a traumatic injury. This system was originally developed in response to patients’ concerns about the safety of blood from blood products. However, today it provides an important way to safely replace volume and stabilise bleeding patients. Collection devices are most often used during surgeries. Some systems allow blood to be automatically and continuously reinfused; others require collection for a period of time (usually no longer than 4 hours) and then the blood is reinfused.
Drainage after the first 24 hours or drainage that is suspected to contain pathogens should not be reinfused. Anticoagulants may or may not be added before reinfusion. Development of clots after blood is filtered through the collection system can sometimes prevent reinfusion of the blood. Sometimes blood that has been collected has become depleted of its normal coagulation factors; therefore monitoring coagulation studies in the patient receiving an autotransfusion is important.72
CASE STUDY
John, a 35-year-old Caucasian man, went to the emergency department because of severe bruising caused by a fall while hiking.
1. What components of the laboratory test results suggest acute leukaemia?
2. How is acute myelogenous leukaemia treated?
3. What is the prognosis for this patient?
4. What are the main priorities for patient teaching for a newly diagnosed young adult with leukaemia?
5. What are the life-threatening problems that can occur as a result of this disease and treatment? How can the nurse anticipate and assess for these problems?
6. Based on the assessment data presented, write one or more nursing diagnoses. Are there any collaborative problems?
1. In a severely anaemic patient, the nurse would expect to find:
2. When obtaining assessment data from a patient with a microcytic, hypochromic anaemia, the nurse would question the patient about:
3. A nursing intervention for a patient with severe anaemia of chronic kidney disease includes:
4. The nursing management of a patient in sickle cell crisis includes:
5. A complication of the hyperviscosity of polycythaemia is:
6. When providing care for a patient with thrombocytopenia, the nurse instructs the patient to:
7. The nurse would anticipate that a patient with von Willebrand’s disease undergoing surgery would be treated with administration of vWF and:
8. Disseminated intravascular coagulation is a disorder in which:
9. Appropriate nursing actions when caring for a hospitalised patient with severe neutropenia include:
10. Because myelodysplastic syndrome arises from the pluripotent haematopoietic stem cell in the bone marrow, laboratory results the nurse would expect to find include:
11. A most common type of leukaemia in older adults is:
12. Multiple drugs are often used in combinations to treat leukaemia and lymphoma because:
13. The nurse is aware that a major difference between Hodgkin’s lymphoma and non-Hodgkin’s lymphomas is that:
14. A patient with multiple myeloma becomes confused and lethargic. The nurse would expect that these clinical manifestations may be explained by diagnostic results that indicate:
15. When reviewing the patient’s haematological laboratory values after a splenectomy, the nurse would expect to find:
16. Complications of transfusions that can be decreased by the use of leucocyte-reduction filters for red blood cells and platelets are:
1 Marks PW, Glader B. Approach to anemia in the adult and child. In Hoffman R, Benz EJ, Shattil SJ, et al, eds.: Hematology: basic principles and practice, 5th edn., Philadelphia: Elsevier, 2009.
2 Coyer SM, Lash AA. Pathophysiology of anemia and nursing care implications. Medsurg Nurs. 2008;17:77.
3 Beers MH. The Merck manual of geriatrics, 3rd edn. New Jersey: Merck & Co, 2008.
4 Patel KV. Epidemiology of anemia in older adults. Semin Hematol. 2008;45:210.
5 Dietitians Association of Australia. Anaemia. Available at www.daa.asn.au/index.asp?pageID=2145834335. accessed 3 April 2011.
6 Brittenham G. Disorders of iron metabolism: iron deficency and overload. In Hoffman R, Benz EJ, Shattil SJ, et al, eds.: Hematology: basic principles and practice, 5th edn., Philadelphia: Elsevier, 2009.
7 Department of Health and Ageing, National Health and Medical Research Council. Dietary guidelines for all Australians: a guide to healthy eating. Available at www.nhmrc.gov.au/publications/synopses/dietsyn.htm, 2003. accessed 3 April 2011.
8 Auerbach M, Goodnough LT, Picard D, Maniatis A. The role of intravenous iron in anemia management and transfusion avoidance. Transfusion. 2008;48:988.
9 Howard MR, Hamilton PJ. Haematology: an illustrated colour text, 3rd edn. Edinburgh: Churchill Livingstone, 2008.
10 Kwiatkowski JL. Oral iron chelators. Pediatr Clin North Am. 2008;55:461.
11 Lucarelli G, Gaziev J. Advances in the allogeneic transplantation for thalassemia. Blood Rev. 2008;22:53.
12 Giardina PJ, Forget BG. Thalassemia syndromes. In Hoffman R, Benz EJ, Shattil SJ, et al, eds.: Hematology: basic principles and practice, 5th edn., Philadelphia: Elsevier, 2009.
13 Antony AC. Megaloblastic anemias. In Hoffman R, Benz EJ, Shattil SJ, et al, eds.: Hematology: basic principles and practice, 5th edn., Philadelphia: Elsevier, 2009.
14 Adamson JW. The anemia of inflammation/malignancy: mechanisms and management. Hematology Am Soc Hematol Educ Program. 2008:159–165.
15 Gardner LB, Benz EJ. Anemia of chronic diseases. In Hoffman R, Benz EJ, Shattil SJ, et al, eds.: Hematology: basic principles and practice, 5th edn., Philadelphia: Elsevier, 2009.
16 Young NS, Scheinberg P, Calado RE. Aplastic anemia. Curr Opin Hematol. 2008;15:162.
17 Dokal I, Vulliamy T. Inherited aplastic anaemias/bone marrow failure syndromes. Blood Rev. 2008;22:141.
18 Afable MG, Lyon DE. Severe fatigue: could it be aplastic anemia? Clin J Oncol Nurs. 2008;12:569.
19 Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008;359:2254.
20 Geller AK, O’Connor MK. The sickle cell crisis: a dilemma in pain relief. Mayo Clin Proc. 2008;83:320.
21 Thomas VJ, Cohn T. Communication skills and cultural awareness courses for healthcare professionals who care for patients with sickle cell disease. J Adv Nurs. 2006;53(4):480–488.
22 Duffin C. Sickle cell disease. Emerg Nurse. 2008;16:10–13.
23 Hagar W, Vichinsky E. Advances in clinical research in sickle cell disease. Br J Haematol. 2008;141:346.
24 Benjamin L. Pain management in sickle cell disease: palliative care begins at birth? Hematol. 2008:466–474.
25 Heeney MM, Ware RE. Hydroxyurea for children with sickle cell disease. Pediatr Clin North Am. 2008;55:483.
26 Lanzkron S, Strouse JJ, Wilson R, et al. Systematic review: hydroxyurea for the treatment of adults with sickle cell disease. Ann Intern Med. 2008;148:939.
27 Downes TM. Gene replacement therapy for sickle cell disease and other blood disorders. Hematology Am Soc Hematol Educ Program. 2008:193–196.
28 Fowler C. Hereditary hemochromatosis: pathophysiology, diagnosis, and management. Crit Care Nurs Clin North Am. 2008;20:191.
29 Matthews AL, Grimes SJ, Wiesner GL, et al. Clinical consult: iron overload—hereditary hemochromatosis. Prim Care. 2004;31(3):767–770.
30 Tefferi A. Essential thrombocythemia, polycythemia vera, and myelofibrosis: current management and the prospect of targeted therapy. Am J Hematol. 2008;83:491.
31 Finazzi G, Barbui T. Evidence and expertise in the management of polycythemia vera and essential thrombocythemia. Leukemia. 2008;22:1494.
32 Stasi R, Provan D. Helicobacter pylori and chronic ITP. Hematology Am Soc Hematol Educ Program. 2008:206–211.
33 Kremer Hovinga JA, Meyer SC. Current management of thrombotic thrombocytopenic purpura. Curr Opin Hematol. 2008;15:445.
34 Baldwin ZK, Spitzer AL, Ng VL, Harken AH, et al. Contemporary standards for the diagnosis and treatment of heparin-induced thrombocytopenia (HIT). Surgery. 2008;143:305.
35 Dolan JP, Sheppard BC, DeLoughery TG. Splenectomy for immune thrombocytopenic purpura: surgery for the 21st century. Am J Hematol. 2008;83:93.
36 Kobos R, Bussel JB. Overview of thrombopoietic agents in the treatment of thrombocytopenia. Clin Lymphoma Myeloma. 2008;8:33.
37 Haemophilia Foundation Australia. Haemophilia fact sheet. Available at www.haemophilia.org.au/bleedingdisorders/cid/2/parent/0/pid/2/t/bleedingdisorders/title/haemophilia. accessed 5 April 2011.
38 Haemophilia Foundation of New Zealand. What is haemphilia? Available at www.haemophilia.org.nz/page.php?onClickbd=bd&page_id=16. accessed 5 April 2011.
39 Friedman KD, Rodgers GM. Inherited coagulation disorders. In Greer JP, et al, eds.: Wintrobe’s clinical hematology, 12th edn., Philadelphia: Lippincott Williams & Wilkins, 2009.
40 Ragni MV, Kessler CM, Lozier JN. Clinical aspects and therapy for hemophilia. In Hoffman R, Benz EJ, Shattil SJ, et al, eds.: Hematology: basic principles and practice, 5th edn., Philadelphia: Elsevier, 2009.
41 Dalainas I. Pathogenesis, diagnosis, and management of disseminated intravascular coagulation: a literature review. Eur Rev Med Pharmacol Sci. 2008;12:19.
42 Latto C. An overview of sepsis. Dimens Crit Care Nurs. 2008;27:195.
43 Baden LR, Segal BH, et al. Prevention and treatment of cancer-related infections. NCCN Clinical Practice Guidelines in Oncology: Evidence-based Information for Clinical Practice. Available at www.nccn.org, 2008. accessed 7 April 2011.
44 National Institute for Health and Clinical Excellence. Infection control: prevention of healthcare-associated infections in primary and community care. Available at http://guidance.nice.org.uk/CG2, 2003. accessed 7 April 2011.
45 Gazit R, Weissman IL, Rossi DJ. Hematopoietic stem cells and the aging hematopoietic system. Semin Hematol. 2008;45:218.
46 Leukaemia Foundation. About diseases—leukaemias. Available at www.leukaemia.org.au/web/aboutdiseases/factsheets.php. accessed 7 April 2011.
47 Elphee EE. Caring for patients with chronic lymphocytic leukemia. Clin J Oncol Nurs. 2008;12:417.
48 Pui CH, Robison LL, Cook AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030.
49 Estey E. New drugs in acute myeloid leukemia. Semin Oncol. 2008;35:439.
50 Lundmark M. Attitudes to spiritual care among nursing staff in a Swedish oncology clinic. J Clin Nurs. 2006;15(7):863–874.
51 Holloway M. Death the great leveler? Towards a transcultural spirituality of dying and bereavement. J Clin Nurs. 2006;15(7):833–839.
52 Cancer Council Australia. Clinical practice guidelines for the diagnosis and management of lymphoma. Available at www.cancer.org.au/Healthprofessionals/clinicalguidelines/Lymphoma.htm, 2005. accessed 7 April 2011.
53 Leukaemia Foundation. About diseases—lyphoma. Available at www.leukaemia.org.au/fileadmin/dl-docs/factsheets2/FactSheet_LF_Lymphomas.pdf. accessed 7 April 2011.
54 Diehl V, Klimm B, Re D. Hodgkin lymphoma: clinical manifestations, staging, and therapy. In Hoffman R, Benz EJ, Shattil SJ, et al, eds.: Hematology: basic principles and practice, 5th edn., Philadelphia: Elsevier, 2009.
55 Matasar MJ, Zelenetz AD. Overview of lymphoma diagnosis and management. Radiol Clin North Am. 2008;46:175.
56 Evens AM, Hutchings M, Diehl V. Treatment of Hodgkin lymphoma: the past, present, and future. Nat Clin Pract Oncol. 2008;5:543.
57 Peinert S, Seymour JF. Indolent lymphomas other than follicular and marginal zone lymphomas. Hematol Oncol Clin North Am. 2008;22:903.
58 Friedberg JW, Fisher RI. Diffuse large B-cell lymphoma. Hematol Oncol Clin North Am. 2008;22:941.
59 Peggs KS, Mackinnon S, Linch DC. The role of allogeneic transplantation in non-Hodgkin’s lymphoma. Br J Haematol. 2005;128:153–168.
60 Leukaemia Foundation. About diseases—myeloma. Available at www.leukaemia.org.au/web/aboutdiseases/myeloma_index.php. accessed 8 April 2011.
61 Kyle RA, Rajkumar SV. Multiple myeloma. Blood. 2008;111:2962.
62 Tricot G. Multiple myeloma. In Hoffman R, Benz EJ, Shattil SJ, et al, eds.: Hematology: basic principles and practice, 5th edn., Philadelphia: Elsevier, 2009.
63 Rome S, Doss D, Miller K, et al. Thromboembolic events associated with novel therapies in patients with multiple myeloma. Clin J Oncol Nurs. 2008;12(3 suppl):S21.
64 Smith A, Wisloff F, Samson D. Guidelines on the diagnosis and management of multiple myeloma 2005. Br J Haematol. 2005;132:410–451.
65 Shurin SB. The spleen and its disorders. In Hoffman R, Benz EJ, Shattil SJ, et al, eds.: Hematology: basic principles and practice, 5th edn., Philadelphia: Elsevier, 2009.
66 Cadili A, de Gara C. Complications of splenectomy. Am J Med. 2008;121:371.
67 BloodSafe eLearning Australia. BloodSafe eLearning program. Available at www.bloodsafelearning.org.au. accessed 8 April 2011.
68 Australian and New Zealand Society of Blood Transfusion. Clinical practice guidlines. Available at www.anzsbt.org.au. accessed 8 April 2011.
69 National Health and Medical Research Council. Clinical practice guidelines on the use of blood components. Available at www.nhmrc.gov.au/_files_nhmrc/file/publications/synopses/cp78.pdf. accessed 8 April 2011.
70 Australian Red Cross Blood Service. Available at www.transfusion.com.au accessed 8 April 2011.
71 New Zealand Blood Service. Transfusion medicine. Available at www.nzblood.co.nz/?t=10. accessed 8 April 2011.
72 Lemaire R. Strategies for blood management in orthopaedic and trauma surgery. J Bone Joint Surg. 2008;90B:1128.
American Cancer Society. www.cancer.org
American Sickle Cell Anemia Association. www.ascaa.org
Australian and New Zealand Society of Blood Transfusion. www.anzsbt.org.au
Australian Institute of Health & Welfare. www.aihw.gov.au
Australian Red Cross Blood Service. www.transfusion.com.au
Australian Society for HICV Medicine. www.ashm.org.au
BloodSafe eLearning program. www.bloodsafelearning.org.au
British Liver Trust. www.britishlivertrust.org.uk
Cancer Council Australia. www.cancer.org.au
Cancer Society of New Zealand. www.cancernz.org.nz
Haemochromatosis Society Australia. www.haemochromatosis.org.au
Haemophilia Foundation Australia. www.haemophilia.org.au
International Myeloma Foundation. www.myeloma.org.uk
Leukaemia Foundation. www.leukaemia.org.au
National Health and Medical Research Council. www.nhmrc.gov.au
New Zealand Blood Service. www.nzblood.co.nz
Peter MacCallum Cancer Centre. www.petermac.org
Thalassaemia Australia. www.thalassaemia.org.au
The Royal Children’s Hospital, Melbourne. www.rch.org.au