Production of sterile products
Introduction
The production of sterile medicinal products has special requirements. These products must be produced in conditions that ensure that they are pure. They must also be free from viable organisms and pyrogens with limited, or ideally no, particulate contamination. It is thus important that only carefully regulated and tested procedures are used to manufacture sterile products.
Owing to their special manufacturing requirements, sterile medicines are prepared in special facilities known as clean rooms. These rooms are designed to reduce the risk of microbial and particulate contamination at all stages of the manufacturing process.
The clean area used to produce sterile products is commonly designed as a suite of clean rooms. With this system, the operators enter the clean rooms by way of a changing room. Within this area, the operators put on clean room clothing before entering into the clean rooms. The changing room has a lower standard of environmental quality. A clean room with a lower environmental standard is also used to prepare solutions. These solutions are then sterilized by filtration before being transferred into the filling room. The clean room used to fill and seal the product containers is the highest quality of clean room. This will reduce the risk of product contamination.
Sterile products that are marketed in the European Union must be produced in conditions which conform with the conditions given in the revised Annex 1 of Good Manufacturing Practices (Volume IV) of The Rules Governing Medicinal Products in the European Union. This guidance on the procedures for manufacturing sterile products describes the cleanliness of the clean room environment and recommends how pharmaceutical clean rooms should be built and used.
Sterile product production
Production of sterile products should be carried out in a clean environment with a limit for the environmental quality of particulate and microbial contamination. This limit for contamination is necessary to reduce the risk of product contamination. In addition, however, the temperature, humidity and air pressure of the environment should be regulated to suit the clean room processes and the comfort of the operators.
Clean areas for the production of sterile products are classified into grades A, B, C and D. These grades are categorized by the particulate quality of the environmental air when the clean area is operating in both a ‘manned’ and ‘unmanned’ state. In addition, these areas are graded by the microbial monitoring of the environmental air, surfaces and operators when the area is functioning. The standards are shown in Tables 40.1 and 40.2.
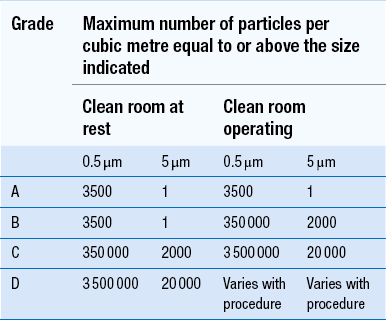
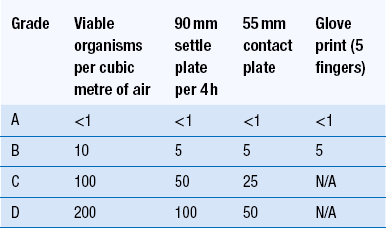
There are two common procedures used to manufacture sterile products. The first method involves the preparation of products that will be terminally sterilized. The second method involves the aseptic filling of containers that are not exposed to terminal sterilization. Aseptic filling requires a higher environmental quality for the preparation of solutions and the filling of containers. The qualities of the clean rooms used for these production procedures are detailed in Tables 40.3 and 40.4.
Table 40.3
Conditions for preparing terminally sterilized products
| Procedure | Required standard before terminal sterilization |
| Preparation of solutions for filtration and sterilization | Grade C is used for products which support microbial growth |
| Grade D acceptable if solutions subsequently filtered | |
| Filling small and large volume parenterals | Grade C. For products with a high risk of contamination such as wide-necked containers, a Grade A laminar airflow workstation with Grade C background |
| Preparation and filling of ointments, creams, suspensions and emulsions | Grade C |
Table 40.4
Conditions for the production of aseptically prepared products
| Procedure | Required standard |
| Handling of sterile starting materials | Grade A with Grade B background or Grade C if solution filtered later in production process |
| Preparation of production solutions | Grade A with Grade B background or Grade C if sterile during filtered production |
| Filling of aseptically prepared products such as small and large volume parenterals | Grade A with Grade B background |
| Preparation and filling of ointments, creams, suspensions and emulsions | Grade A with Grade B background |
Premises
The sterile production unit must be separated from the general manufacturing area within the hospital pharmacy or factory. This sterile production unit must not be accessible to unauthorized personnel.
The unit is designed to allow each stage of production to be segregated. It should also ensure a safe and organized workflow and reduce the need for personnel to move around the clean rooms. The unit is built and the equipment positioned to protect the product from contamination. The layout must allow efficient cleaning of the area and avoid the build up of dust. Premises are also arranged to decrease the risk of mix up or contamination of one product or material by another.
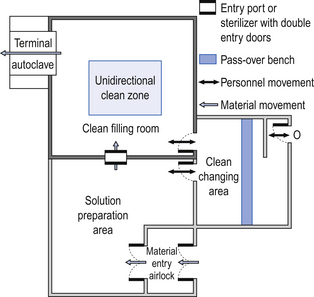
The filling room is typically serviced from an adjacent preparation room. This allows supporting personnel to assemble and prepare materials. Staff within the filling room area then use these materials. Figure 40.1 shows the layout of rooms for the production of terminally sterilized medicines such as small or large volume injections.
Design and construction
Access to clean and aseptic filling areas is limited to authorized personnel. Operators enter clean rooms by way of changing rooms. Within the changing room the operators can don and remove their clean room garments.
A low physical barrier, commonly known as a pass-over (or cross-over) bench, extends across the changing room. It forms a physical barrier that separates the different areas for changing by the operators.
Special precautions are needed to avoid contamination of clean and aseptic filling areas when materials are passed through airlocks or hatchways. Thus, sterilizers and entry ports are fitted with double-sided doors. The doors are interlocked to prevent both doors being opened simultaneously.
Surfacing materials
All clean room surfaces, including the floors, walls and ceilings, should be smooth, impervious and unbroken. This will decrease the release and build-up of contaminating particles and organisms. The surfaces are made of materials that allow the use of cleaning agents and disinfectants. The ceilings are sealed to prevent the entry of contaminants from the space above them. Uncleanable recesses within the clean room should be avoided. This will reduce the collection of contaminating particles. Thus, the junction between the wall and the floor is commonly coved. The presence of shelves, ledges, cupboards and equipment is minimized. Windows should be non-opening and sealed. This will prevent the ingress of contaminants.
Services
Piped liquids and gases should be filtered before entering the clean room. This will ensure that the liquid or gas at the work position will be as clean as the clean room air. The pipes and ducts must be positioned for easy cleaning. All other fittings such as fuse boxes and switch panels should be positioned outside the clean rooms.
Sinks and drains must be excluded from areas where aseptic procedures are performed in clean room areas. They should be avoided in the whole unit wherever possible. In areas where sinks and drains are installed they must be designed, positioned and maintained to decrease the risk of microbial contamination. They are thus often fitted with easily cleanable traps. The traps may contain electrically heated devices for disinfection.
There should be a limited number of entry doors for personnel and ports for materials. Entry doors should be self-closing and allow the easy movement of personnel.
Airlock doors, wall ports, through-the-wall autoclaves and dry heat sterilizers should be fitted with interlocked doors. This will prevent both doors being opened simultaneously. An alarm system should be fitted to all the doors to prevent the opening of more than one door.
Lights in clean rooms are fitted flush with the ceiling to reduce the collection of dust and avoid disturbing the airflow pattern within the room. Similarly, equipment should be positioned in clean rooms to avoid the distribution and the collection of particles and microbial contaminants.
Environmental control
Potential sources of particles and microbial contaminants occurring within the clean room are:
Each of these possible sources can be minimized as described below.
Air supply
The air supply to a Grade A, B or C clean room must be filtered to ensure the removal of particulate and microbial contamination. This is carried out by filtering the air with high-efficiency particulate air (HEPA) filters. The HEPA filter should be positioned at the inlet to the clean room or close to it. A prefilter may be fitted upstream of the HEPA filter. This will prolong the life of the final filter. A fan is required to pump the air through the filter.
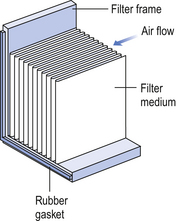
The HEPA filters use pleated fibreglass paper as the filter medium. Parallel pleats of this filter material increase the surface area of the filter and increase the airflow through the filter. This structure allows the filter to retain a compact volume. Aluminium foil is used to form spacers in the traditional type of HEPA filter. Spacers are not used in the more modern ‘mini-pleat’ type of filter design. These mini-pleat filters are now widely used. They have a shallower depth in construction than the traditional HEPA filter. Within the structure of the filter, the filter material is sealed to an aluminium frame (Fig. 40.2). At least one side of the filter is protected with a coated mild steel mesh. HEPA filters exhibit:
HEPA filters remove larger particles from the air by inertial impaction, the medium-sized particles by direct interception and the small particles by Brownian diffusion. The HEPA filters are least efficient at removing particles of about 0.3 μm. However, the efficiency of removing particles is affected by the air velocity and the filter packing. Larger and smaller particles will be removed more efficiently.
With a new HEPA filter fitted in a clean room, the air exits from the filter face at a rate of about 0.45 m/s and has a 99.997% efficiency at removing 0.3 μm particles. The pressure difference across the depth of a new filter is about 130 Pascal (Pa). At the end of the effective life of the filter the pressure drop across the filter will increase to about 490 Pa. To retain the operating efficiency of the filter, the fan forcing air through the filter must be able to maintain this pressure difference. Sensors are fitted upstream and downstream of the filters to indicate the pressure differential across the filter. An automatic alarm system should be fitted to indicate failure in the air supply or filter blockage.
The HEPA filters for clean room use must conform with the British Standard 5295 (1989) aerosol test. The filters may have faulty seals and can be damaged during delivery or installation. It is thus important that they are tested in situ before use.
The filter material possesses a uniform resistance and is constructed with a large number of parallel pleats. This results in the air downstream of the filter face flowing uniformly with a unidirectional configuration.
The number of air changes in clean rooms is affected by:
In practice 25–35 air changes per hour are common. The airflow pattern within the clean room must be carefully regulated to avoid generating particles from the clean room floor and from the operators. Various options for ventilating clean rooms may be categorized by the airflow pattern within the room. These are:
Unidirectional airflow systems
Air enters the room through a complete wall or ceiling of high-efficiency filters. This air will sweep contamination in a single direction to the exhaust system on the opposing wall or floor (Fig. 40.3). In the interests of economy, the exhaust grill may be fitted low down on the wall. The velocity of the air is about 0.3 m/s in downflow air from ceiling filters and 0.45 m/s in crossflow air. These are highly efficient airflow systems. However, one major disadvantage of these rooms for pharmaceutical use is that they are expensive to construct. They also use much more conditioned air than rooms with non-unidirectional airflow. This greatly increases their operating costs. Owing to these factors, unidirectional airflow clean rooms are seldom used for pharmaceutical purposes.
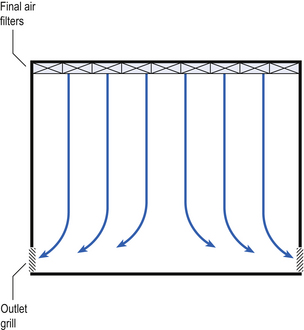
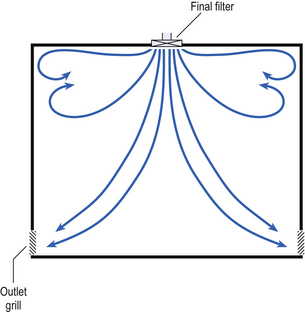
Non-unidirectional airflow systems
Air enters the clean rooms through filters and diffusers that are usually located in the ceiling. It exits through outlet ducts positioned low down on the wall or in the floor at sites remote from the air inlet (Fig. 40.4). With the use of this system, the filtered inlet air mixes with and dilutes the contaminated air within the room. As the clean room air has been previously heated and cleaned, it can be recirculated to save energy, a little fresh air being introduced with each air change cycle.
Various designs of diffuser are used with this ventilation system. These affect the air movement and the cleanliness of the rooms. The perforated plate diffuser produces a jet flow of air directly beneath it. This jet of air will carry contamination at its edges. However, it does produce high-quality air directly under the diffuser. It is thus important that production procedures are located directly below the diffuser. By contrast, the air released from the bladed diffuser will mix with the clean room air. This diffuser thus produces a reasonably constant quality of air throughout the room.
Combination systems
In many pharmaceutical clean rooms, it is common to find that the background area is ventilated by a non-unidirectional airflow system. Meanwhile, the critical areas are supplied with high-quality air from unidirectional airflow units.
The combination airflow system is often selected for pharmaceutical clean room applications as it:
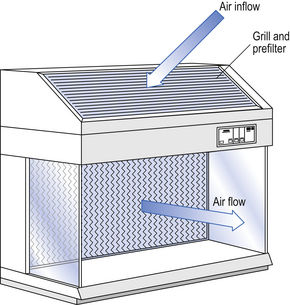
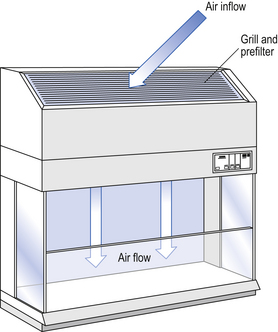
Several types of unidirectional flow workstations or benches are used in this combination-type room. Various vertical unidirectional airflow systems are used in combination clean rooms. With one system, the critical area is surrounded by a plastic curtain with vertical unidirectional downflow air ‘washing’ over the manufacturing process and exiting under the plastic curtains into the general clean room area (Fig. 40.5). An alternative system is often used with the small-scale combination-type clean room in hospital pharmacies. With this system, a horizontal airflow cabinet (Fig. 40.6) is used as the workstation. With these cabinets, a fan forces air through a HEPA filter located at the rear wall of the workstation. The air that exits from the filter first washes over the critical work area before washing over the arms and upper body areas of the operator. Contamination arising from the operator is thus kept downstream of the critical procedures. Grade A environmental conditions are achieved at the critical work area. A similar workstation known as a vertical laminar airflow cabinet (Fig. 40.7) could also be used in the combination room. This cabinet passes air vertically downwards from the ceiling of the cabinet over the critical working area. It produces a Grade A environmental quality. The air exits from the front of the workstation.
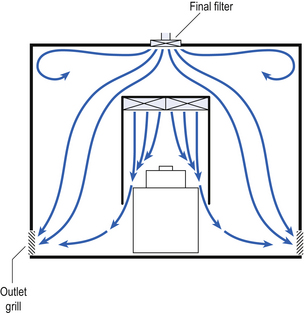
Figure 40.5 Airflow patterns in a mixed-flow clean room with non-unidirectional airflow background environment and unidirectional airflow protection for a critical area.
In recent times, there has been a trend towards protecting the critical procedures within combination clean rooms by using isolator cabinets. The isolator cabinet gives a localized high-quality environment. Isolators give protection from potential contamination in clean rooms as they are positively pressurized with air supplied through HEPA filters. The operator works outside the confines of the isolator using glove ports to perform procedures within the enclosed chamber. The gloved hands of clean room operators can transfer microbial contamination into critical working areas within the clean room. To indicate that the required clean room standards have been achieved (Table 40.2), the fingertips of a gloved hand are depressed onto the surface of a suitable solid growth medium. This medium is incubated to show any contamination.
There is also a need to avoid contaminated external air passing into the clean room environment. Thus, the clean room air pressure must exceed that of the surrounding areas. The pressure differential between different standards of clean room should be 10–15 Pa. This level should be comparatively easy to monitor and will decrease the unregulated outflow of air. Adjusting grills known as pressure stabilizers located in the walls of rooms regulate the outflow of clean room air and the room pressure. The air moves from an area of high pressure to an area of lower pressure. To maintain the room pressure, it is important that the rooms are airtight. However, a small quantity of air will exit from the rooms by way of door spaces.
Temperature and humidity control
The temperature and the humidity are adjusted to suit the procedures being carried out within the clean room and maintain the comfort of the operators. A target temperature of about 20°C with a relative humidity of about 35–45% is usually preferred.
Personnel
The clean room environment is supplied with high-quality air at positive pressure. The main source of contamination in these areas arises from skin scales that are released by the clean room operators.
To limit clean room contamination by personnel, there is a need to:
 Restrict the number of operators working in the clean room
Restrict the number of operators working in the clean room
Restrict operator conversations
Instruct operators to move slowly
Minimize general movement throughout the room
Avoid operators interrupting the airflow between the inlet filter and the work area.
The clean room operator is constantly shedding dead skin scales from the body surface. Not all of these skin particles are contaminated with bacteria. Males shed more particles that are contaminated with bacteria than females. In addition, individual males and females show variable rates of bacterial dispersal. This dispersal from the individual is affected by:
Body movements of personnel will increase the number of contaminated particles released from the skin surface. Each individual releases more than 106 skin scales/min during normal walking movements. There is a need to contain the dispersion of skin particles from the operators in clean rooms and protect both the environment and the product. Containment of particles is achieved by the operators wearing clean room clothing. This clothing is made from synthetic fabrics that filter out particulate and microbial contamination from the operators without the fabrics releasing contamination. However, this clothing is not absolute and particles can pass through the garments. Operators wearing clean room undergarments reduce this effect. The outer garments are close fitting at the neck, wrists and ankles, but these sites still provide an exit route for particulate matter.
Clean garments should be used for each work session and must provide operator comfort. Disposable single-use garments are available, although most production units employ reusable garments. The clothing is specially laundered in an area with similar standards to those used in the clean room. Garments are laundered by a wet-wash process using particle-free solutions. This is followed by an antibacterial rinse and hot air drying and then the garments are packaged in sealed bags to avoid particulate contamination. This cleaning process fulfils the needs of most pharmaceutical clean room applications, which are a balance between cost and acceptability. For a higher level of sterility assurance the garments are gamma irradiated using 60Co, each garment receiving an approved dose of 25 kGy. This treatment is expensive and decreases the life of the garments. The donning of clean room clothing without contaminating the outer surface of the garments is a rather difficult procedure that is performed in the changing room.
Changing room
Entry of personnel into clean rooms should be through a changing room fitted with interlocking doors. These doors act as an airlock to prevent the influx of external air. This access route is intended for the entry of personnel only. The changing room is subdivided into three areas. Movement through these areas must comply with a strict protocol. They are often colour coded as black, grey and white, black representing the dirtiest area while white represents the cleanest area.
The black area is where jewellery, cosmetics, factory or hospital protective garments and shoes are removed. Long hair may be contained and a mobcap donned to contain the hair completely. The pass-over bench forms a physical separation between the black and the grey areas in the changing room. The operator sits on the pass-over bench, swings his/her legs over the bench and fits clean room covers over the feet before they are placed on the floor of the grey area.
The operator then stands up in the grey area. Wrappings on the various garments are opened to avoid contacting the outer surface of the packaging following the hand-washing procedure. Then the operator washes hands and forearms using an antiseptic solution. Special attention is paid to cleaning the fingernails. The hands are then dried using an automatic air-blow drier as towels shed particles when used for drying hands.
Clothing garments are donned in sequence from head to foot. Throughout this procedure, care must be taken to avoid the hands contacting the outer surface of the clean room clothing. First, the head and shoulder hood is fitted, ensuring that all the hair is contained within the head cover. A face mask is fitted to prevent the shedding of droplets. The one-piece coverall (or alternatively two-piece trouser suit) is put on. Care must be taken to avoid these garments contacting the floor surface. The shoulder cover of the head and shoulder hood is tucked into the coverall. Then the zip is closed and the studs fastened. Overboots are then fitted over the clean room shoes. The overboots are kept in position with ties that are suitably fastened for operator comfort. For entry into aseptic filling rooms an antiseptic cream is applied to the hands. The clean room powder-free gloves are then donned. Care is needed to avoid contacting the outer surface of the gloves. The cuffs of the coverall are secured within the gloves and the gloved hands disinfected. The operator now enters into the white clean room area and begins work. During the work procedures, the gloved hands of the operators are regularly disinfected. Key features of the clothing are given in Table 40.5.
Table 40.5
| Clean room grade | Description of clothing |
| A/B | Head cover and face mask |
| Single- or two-piece trouser suit | |
| Overboots and sterile powder-free rubber or plastic gloves | |
| C | Hair (and beard) cover |
| Single- or two-piece trouser suit | |
| Clean room shoes or overshoes | |
| D | Hair (and beard) cover |
| Protective suit | |
| Appropriate shoes or overshoes |
Cleaning
A strict cleaning and disinfection policy is essential to minimize particulate and microbial contamination in the clean room. Operators release microbial and particulate contamination within the clean room. These contaminants are mostly deposited onto horizontal surfaces. However, other areas of the clean room can become contaminated due to direct contact with the operators’ clothing. It is thus essential that a strict cleaning and disinfection policy is implemented within the clean room to minimize both the particulate and the microbial contamination.
There are two main methods of cleaning. Vacuuming is effective at removing gross particulate contamination of particles greater than 100 μm. However, vacuuming is not very effective at removing smaller particles. Small particles are removed by wet wiping. It is important that the wet wipe is sterile and must not generate particulate contamination. The use of wet wipes involves the use of cleaning agents that will remove particulate contamination and have an antibacterial effect.
The ideal cleaning agent should be:
Anionic or cationic surfactants are used as cleaning agents within the clean room. The disinfectants of choice for clean room use are generally quaternary ammonium compounds, phenols, alcohols and polymeric biguanides. The disinfectant solutions should be freshly prepared before use. Different types of disinfectants should be used in rotation to prevent the development of resistant microbial strains. Most surfactants or detergents will dissipate surface static electricity but the most effective and widely used antistatic agents used in clean rooms are cationic surfactants.
Trained personnel regularly clean critical production areas of clean rooms. A less stringent cleaning protocol is required in the general clean room areas. This applies to the walls and floors where contamination cannot directly contaminate the product. As part of the cleaning protocol, regular microbiological monitoring should be carried out to determine the effectiveness of the disinfection procedures.
Isolators
Commercial manufacturers are using isolators increasingly for the aseptic filling of products, with combination isolators being used. Isolators are also used for sterility testing of products. Robots have been used in isolators for repetitive processes such as sterility testing but they are expensive. Isolators are used in hospital pharmacy departments as an alternative to clean rooms for the small-scale aseptic processing of sterile products. Aseptic procedures performed in the best isolators cannot reach the same levels of sterility assurance achieved by terminal heat sterilization (see Table 40.6). However, when suitably operated they can produce a sterility assurance level better than the conventional clean room. Isolators are often selected for aseptic manipulations of sterile products as they are:
Isolators are composed of a chamber that controls the environment surrounding the work procedure (Fig. 40.8). The inlet and exhaust air passes through HEPA filters. The airflow pattern within the isolator chamber may be either unidirectional, non-unidirectional or a combination of both. Vertical unidirectional airflow has the advantage of rapidly purging particles from the isolator chamber. This is an advantage for aseptic processes. The air within the isolator chamber should be frequently changed to maintain the aseptic chamber environment. Particle and microbial contamination of the environment within the isolator chamber must conform with the Grade A standard as detailed in Tables 40.1 and 40.2.
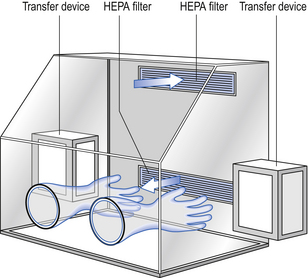
The operator remains outside the isolator chamber environment. To perform manual manipulations within the chamber, the operator inserts his hands and arms into the chamber. Entry occurs by way of a glove port using either a one-piece full-arm-length glove or a glove and sleeve system. With the glove and sleeve system, the easily changeable glove is attached to a sleeve that is attached to the wall of the chamber through an airtight seal. Using either of these glove systems, the operator is able to perform aseptic manipulations in comfort up to a distance of about 0.5 m within the chamber. The glove system avoids contamination arising from the operator and maintains the integrity of the isolator chamber environment. As cytotoxic materials can diffuse through the gloves it is important that they are changed regularly. To perform the work procedure within the chamber, materials must be introduced and prepared products removed without compromising the chamber environment. This transfer procedure is a critical factor in the operation of the isolator and is carried out using a transfer system. The transfer system separates the external environment from the controlled isolator environment. It restricts airflow between these areas while allowing the transfer of materials between them. The transfer system is fitted with an interlocked double door entry system. This will provide an airlock that avoids both doors being opened to the external environment simultaneously. A filtered air inlet and exhaust is fitted to the transfer system. However, a risk of microbial contamination during the transfer does exist. The isolator must be positioned in a suitable background environment of at least a Grade D classification. This is typically achieved by positioning the isolator in a dedicated room that is only used for the isolator and its related activities.
Isolators are divided into positive and negative pressure isolators.
Positive pressure isolator
This isolator operates under positive pressure and protects the product from contamination arising from an external source and from the aseptic process itself. It is used for the aseptic preparation of pharmaceutical products and can be used as a sterility test chamber.
Negative pressure isolator
This isolator will protect the product from contamination arising from an external source and from the aseptic manipulation. In addition, however, this isolator should protect the operator from hazardous materials such as cytotoxic preparations or radiopharmaceuticals in the isolator chamber. This type of isolator operates under negative pressure. The exhaust air is ducted to the outside through at least one HEPA filter and through an adsorption material such as activated carbon. Rigid negative pressure isolators should be used for radiopharmaceutical manipulations. In this situation, the isolator is frequently used with a lead-free vision panel and a lead glass protector around the product. Alternatively, isolators are available with lead acrylic glass windows.
The chambers of isolators are gas sterilized. The ideal sterilant for use in the isolator chamber should have the following properties:
The sterilants in most general use for pharmaceutical applications in isolators do not comply with all of these ideal properties. Those used are peracetic acid vapour and hydrogen peroxide vapour. To reduce the risk of chemical contamination of the sterile product, the sterilant contact time should be carefully regulated. The sterilant must be flushed from the isolator before beginning the aseptic manipulations.
Currently marketed isolators are constructed with either a flexible canopy or a rigid containment medium. The rigid type of isolator is often preferred, owing to the reduced risk of the chamber being punctured. This occurs more readily with the flexible canopy design. Rigid isolators are often constructed from a stainless steel frame with a moulded acrylic window. A further isolator known as a half-suit isolator is currently in use. This is a flexible canopy isolator that is made from material such as nylon-lined polyvinyl chloride. It is designed using a half-suit sealed to a wall of the chamber. This system allows the torso of the operator to be introduced into the suit that is located within the chamber of the isolator. To improve visibility, a transparent helmet is sealed to the neck of the suit that is ventilated by a pressurized air supply. This provides operator comfort over prolonged work sessions. The advantage of the half-suit isolator is that the operator can easily access a large area of the chamber and manoeuvre heavier and larger materials. The half-suit isolator is used as dedicated production equipment for the aseptic compounding of products such as total parenteral nutrition (TPN) fluids.
During a 2-week period in September 1992, eight children died from infection after receiving contaminated TPN fluids at four different hospitals in South Africa. These fluids had been prepared in flexible film isolators. The investigation of this incident revealed that the production equipment was suitable for its purpose but inadequate procedures had allowed contamination and subsequent growth of pathogenic bacteria in the TPN fluids. It should therefore be carefully noted that the use of isolators requires trained staff and good manufacturing practices to maintain product quality.
Isolator tests
Isolators must be frequently tested to ensure that they operate as a sealed chamber and conform with the required level of air quality and surface contamination. They are thus subjected to both physical and microbial tests.
Integrity tests. These tests will detect leaks that compromise the integrity of the isolator chamber. The procedure is routinely carried out by sealing the chamber and recording changes in the chamber pressure over time
Glove inspection. The glove and sleeve are visually inspected and leak tested for pin holes
HEPA filter test. The integrity of the HEPA filter should be tested with an aerosol generator and a detector
Air-borne particle count. This is carried out in the isolator chamber and the transfer device using a particle counter.
Microbial tests use microbial growth media suitable for the growth of potential contaminants. The tests include:
Active air sampling. This test determines the number of organisms in the air of the isolator chamber. The procedure uses impact and agar impingement samplers
Settle plates. Settle plates containing growth media are exposed in the chamber for 2–4 h. Particles and organisms settle by gravity onto the agar surface. The plates are then incubated
Surface tests. Surfaces are sampled using direct contact plates that are then incubated. Following sampling, it is important to remove materials deposited onto the sampled surfaces during the test. Alternatively, surfaces are sampled using sterile moistened swabs. The swabs are then streaked onto solid growth media and incubated. Soluble swabs may be dissolved in sterile diluent and the viable count determined
Finger dabs. The fingertips of the gloved hand are pressed onto the surface of solid growth medium. The medium is then incubated
Broth fill test. This test challenges both the manipulative procedure of the operator and the facilities. The test simulates routine aseptic procedures by using nutrient medium in place of a product to produce broth-filled units. These units are incubated to indicate microbial contamination.
Environmental monitoring
Following construction of a clean room, it must be tested to ensure that it is providing the required quality of environment. These verification tests are rigorously performed and are similar to the tests that are used to monitor the clean room. The monitoring tests ensure that the clean room continues to provide satisfactory operation.
To ensure that the pharmaceutical clean room is providing the required environmental standards, the following are determined.
Air quality
The air supplied to the clean room must not contribute to particulate or microbial contamination within the room. The HEPA filters for the inlet air must be tested to ensure that neither the filter fabric nor the filter seals are leaking. This is done by introducing a smoke with a known particle size upstream of the filter. The clean room surface of the filter is then scanned for smoke penetration using a photometer or a particle counter.
Air movement
Adequate ventilation throughout the clean room can be determined by air movement tests. These are carried out at the time of clean room validation. Air movement within the clean room is determined by measuring the decay profile of smoke particles released into the clean room. Smoke particle release is also used to ensure that a clean area within a unidirectional workstation is not being contaminated with air from the clean room environment.
The outflow of air from a clean room with a higher standard of cleanliness to an area with a lower standard is indicated by the pressure differential between the rooms. This is determined using a manometer or magnehelic gauge.
Air velocity
The velocity of the air at several points in a clean room area of critical importance should be determined. This is done both at validation of the clean room and at timed intervals. The procedure involves the use of an anemometer.
Air-borne particulate and microbial contamination
The particle count and the microbial bioburden of the clean room provide the basis for the air classification system for grading a clean room, as detailed in Table 40.1. The points for sampling and the number of samples taken at each position are determined by the size and the grade of the clean room. Air-borne particles are normally sized and counted by optical particle counters.
Microbial monitoring
There should be very few viable organisms present in the clean room air. However, operators within the clean room disperse large numbers of skin particles. Many of these particles are contaminated with bacteria. The dispersal of contaminated particles by the clean room operator is greatly decreased by the wearing of occlusive clothing together with appropriate air ventilation. Sampling for microbial contamination is necessary when people are present in the clean room during production. Monitoring of the microbial contamination during production will ensure that both the use of clean room clothing by the operators and the air ventilation system are producing the required environmental standards. Air sampling is carried out by volumetric sampling or by the use of settle plates. With volumetric sampling, a measured volume of air is drawn from the environment and contaminants are impinged onto a suitable microbial growth medium. The medium is then incubated and the colonies of microbial growth counted. Settle plates rely on bacteria-carrying particles being deposited onto the exposed solid surface of sterile microbial growth media contained in a 90 or 140 mm diameter Petri dish. When positioning the plates, care is needed to avoid accidental contamination. Owing to the small number of microbial contaminants in the clean room, the settle plates are preferably exposed for about 4 h.
The surfaces of the clean room should also be tested for microbial contamination, notably in areas that may be contacted by the clothing of the operators. This is achieved by using contact plates or by using sterile moistened swabs. The contact plates allow a sterile agar surface to be pressed onto the clean room surface. These plates are then incubated to reveal microbial growth. Swabbing procedures are carried out as previously detailed in isolator tests.
Aseptic preparation
Parenteral products such as injections, infusions and eye products must be sterile for administration to the patient (see Chs 41 and 42). The preferred method of manufacturing parenteral medicines is to place the product in its final container and then seal this package. The product is then protected from further contamination and is terminally sterilized. At worst, this achieves the risk of one product in a million being contaminated following terminal sterilization by dry or moist heat or by irradiation. Some products cannot withstand this sterilization process. An alternative approach known as aseptic preparation must then be used to prepare these medicines. This procedure is carried out in industry with selected products but is extensively used in hospital pharmacy where products are specially compounded to meet the specific needs of patients (see Chs 44 and 46).
As shown in Table 40.6, aseptic preparation of parenteral products provides the lowest level of assurance of sterility of all the methods currently used to produce these formulations. In the pharmaceutical industry, pre-sterilized medicines are aseptically filled into sterile containers. The filling process must avoid recontamination of the sterile medicine and its container during this process. A sterility assurance level of 10−6 is achieved, but to achieve this requires highly sophisticated industrial production procedures. In hospital pharmacy, pre-sterilized product components are aseptically compounded using sterile apparatus and then aseptically added to appropriate packaging for subsequent patient administration. It is critical that the sterile product components and the packaging are not recontaminated with organisms or particulate matter during these aseptic procedures. In order to achieve this, the sterile product components and the sterile package must be manipulated in a high-quality environment. The aseptic preparation and filling of products is performed in a localized Grade A zone that is achieved by a laminar airflow cabinet with a Grade B background. The Grade A environment within an isolator cabinet is also suitable for the compounding of aseptic preparations. It is important that this quality environment is continuous throughout the aseptic preparation process. Great reliance is not only placed on the facility and equipment used to produce the product, but also on the ability of the trained operators to avoid product contamination. It achieves a sterility assurance level of about one in a thousand. The manufacture of aseptic products also needs a stringent quality assurance system to ensure production of a quality product that is fit for its intended purpose. The quality assurance system should have documented, validated and audited procedures with in-process monitoring and standard operating procedures defining each step of the production process.
There is a need for awareness of the potential risk of infection that can occur during the aseptic preparation of pharmacy products. This has been shown by the tragic outcome of the supply of contaminated parenteral nutrition fluids to children at the Royal Manchester Children’s Hospital in 1994. These fluids were aseptically compounded in an isolator. Microorganisms were unknowingly transferred from a sink into the isolator chamber on components used to prepare the feeds. The contaminating organisms grew in fluid remaining in assembled tubing used to prepare the feeds in the isolator. Reuse of this tubing resulted in contamination of the feeds that infected the patients. During these events, it was shown that the equipment was not faulty, only the manner in which it had been used. This demonstrates the importance of adequately disinfecting the components being transferred into the isolator and for a total quality system for the manufacture of aseptic products.
In order to aseptically prepare a parenteral medicine, it is critical that validated procedures are stringently followed. This must go hand in hand with the other components of the quality assurance system for the preparation of aseptically prepared products of quality that are right first time and every time.
Testing for sterility
Sterility testing is the final method of assuring sterility of the manufactured product. The test is required in most countries for assuring the sterility of aseptically prepared sterile products. Aseptic manufacturing units in hospital pharmacy often perform the test retrospectively following patient administration. A few commercial manufacturers are exempt from performing the sterility test on products that have been terminally sterilized and prepared using highly developed quality assurance procedures incorporating validated and controlled sterilization procedures. This has been referred to as parametric release.
Sterility testing attempts to indicate the presence or absence of viable microorganisms in containers selected from a batch of product. A decision is made as to the sterility of the entire batch from the results obtained by testing the sample. The test has both technical and numerical limitations and thus only provides a partial indication to the state of sterility of each product within a manufactured batch. The numerical limitation arises as only 10% of a batch of parenteral product is sampled, but the probability of accidental contamination in an aseptically manufactured batch can be as high as one in a thousand (10−3), while the probability of contamination of a terminally sterilized batch is at worst only one in a million (10−6).
The details of the test for sterility are provided in the British Pharmacopoeia (BP 2012) and this test conforms with the standards of the European Pharmacopoeia (EP 2013). These are also very similar to the test in the Japanese Pharmacopoeia (2012) and United States Pharmacopoeia 36 (USP 2013).
Key Points
Particulate and microbial contamination of sterile products is minimized by preparation in a clean environment
Quality of clean areas is graded A, B, C, D in decreasing stringency for particulate and microbial content
Premises must allow segregation of stages of production and protect products from contamination by all possible means of design and operation
Access to clean areas is restricted and special clothing must be worn
Environmental control, particularly of the air supply to the room, is required to ensure a minimal contamination hazard
HEPA filters have a 99.997% efficiency at removing 0.3 μm particles, the size at which their efficiency is lowest
Airflow may be designed as unidirectional, non-unidirectional or as a combination system
In addition to general air quality, localized areas of higher quality can be produced either by airflow design in enclosed areas or by isolator cabinets
The main source of contamination in clean rooms is the skin scales from operators
Clean room clothing, made from synthetic fabrics, is designed to minimize release of operator contaminants
Changing areas are designed and used to minimize the entry of contamination on personnel
During cleaning, vacuuming and wet wiping are used to remove large and small particles, respectively
Isolators give protection to both the product and the operator at relatively low cost
Type II isolators protect the operator from hazardous materials in addition to providing the Type I facilities of protection of the product from contamination
Isolator interiors are sterilized using a gas sterilant
Isolator integrity is tested using physical and microbial tests
A range of environmental tests is used in clean rooms to monitor air quality, movement and velocity, air-borne particles and microbial contamination
Aseptic preparation is involved with repackaging sterile products for patient use without terminal sterilization
Aseptic preparation is performed in laminar airflow cabinets in clean rooms or in isolator cabinets to avoid product contamination
A stringent quality assurance system is required for aseptic production to ensure a quality product is prepared
The test for sterility has numerical limitations due to the sample size – cannot guarantee to detect small levels of product contamination