Chapter 411 Skeletal Diseases Influencing Pulmonary Function
Pulmonary function is influenced by the structure of the chest wall (Chapter 365). Chest wall abnormalities can lead to restrictive or obstructive pulmonary disease, impaired respiratory muscle strength, and decreased ventilatory performance in response to physical stress. The congenital chest wall deformities include pectus excavatum, pectus carinatum, sternal clefts, Poland syndrome, and skeletal and cartilage dysplasias. Vertebral anomalies such as kyphoscoliosis can alter pulmonary function in children and adolescents.
411.1 Pectus Excavatum (Funnel Chest)
Epidemiology
Pectus excavatum occurs in 1 : 400 births with a 9 : 1 male preponderance and accounts for >90% of congenital chest wall anomalies. There is a positive family history in one third of cases.
Etiology
Midline narrowing of the thoracic cavity is usually an isolated skeletal abnormality. The cause is unknown. Pectus excavatum can occur in isolation or it may be associated with a connective tissue disorder (Marfan [Chapter 693] or Ehlers-Danlos syndrome [Chapter 651]). It may be acquired secondarily to chronic lung disease, neuromuscular disease, or trauma.
Clinical Manifestations
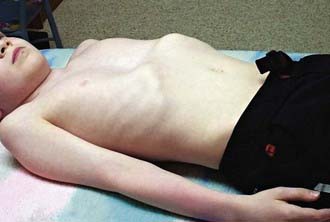
The deformity is present at or shortly after birth but is usually not associated with any symptoms at that time. In time, decreased exercise tolerance, fatigue, chest pain, palpitations, recurrent respiratory infections, wheezing, stridor, and cough may be present. Because of the cosmetic nature of this deformity, children may experience significant psychologic stress. Physical examination may reveal sternal depression, protracted shoulders, kyphoscoliosis, inferior rib flares, rib cage rigidity, forward head tilt, scapular winging, and loss of vertebral contours (Fig. 411-1). Patients exhibit paroxysmal sternal motion and a shift of point of maximal impulse to the left. Innocent systolic murmurs may be heard.
Laboratory Findings
Lateral chest radiograms demonstrate the sternal depression. Use of the Haller index on chest CT (maximal internal transverse diameter of the chest divided by the minimal anteroposterior diameter at the same level) in comparison with age- and gender-appropriate normative values for determining the extent of depression of the chest wall anomaly has become useful in determining the extent of the anatomic abnormality. An electrocardiogram may show a right-axis deviation or Wolff-Parkinson-White syndrome (Chapter 429); an echocardiogram may demonstrate mitral valve prolapse (Chapter 422.3) and ventricular compression. Results of static pulmonary function tests may be normal but commonly show an obstructive defect in the lower airways and, less commonly, a restrictive defect due to abnormal chest wall mechanics. Exercise testing may demonstrate either normal tolerance or limitations from underlying cardiopulmonary dysfunction that appear associated with the severity of the defect. Ventilatory limitations are commonly seen in younger children and adolescents, whereas cardiac limitations secondary to stroke volume impairments are more commonly seen in older adolescents and young adults.
Treatment
Treatment is based on the severity of the deformity and the extent of physiologic compromise. Therapeutic options include careful observation, use of physical therapy to address musculoskeletal compromise, and corrective surgery. For patients with significant physiologic compromise, surgical correction may improve the cosmetic deformity and may help minimize or even improve the cardiopulmonary compromise. The 2 main surgical interventions are the Ravitch and Nuss procedures. Although superiority of 1 approach has not been established, there is now >20 years of successful experience with the minimally invasive Nuss procedure. For teenagers with exercise limitations, surgical repair may result in improved exercise tolerance. Normalization of lung perfusion scans and maximal voluntary ventilation have also been observed after surgery. Ongoing treatment to address the secondary musculoskeletal findings is commonly employed before and after the operation.
Brigato RR, Campos JR, Jatene FB, et al. Pectus excavatum: evaluation of Nuss technique by objective methods. Interact Cardiovasc Thorac Surg. 2008;7:1084-1088.
Borowitz D, Cerny F, Zallen G, et al. Pulmonary function and exercise response in patients with pectus excavatum after Nuss repair. J Pediatr Surg. 2003;38:544-547.
Daunt SW, Cohen JH, Miller SF. Age-related normal ranges for the Haller index in children. Pediatr Radiol. 2004;34:326-330.
Haller JAJr, Loughlin GM. Cardiorespiratory function is significantly improved following corrective surgery for severe pectus excavatum: proposed treatment guidelines. J Cardiovasc Surg. 2000;41:125-130.
Koumbourlis AC, Stolar CJ. Lung growth and function in children and adolescents with idiopathic pectus excavatum. Pediatr Pulmonol. 2004;38:339-343.
Malek MH, Fonkalsrud EW, Cooper CB. Ventilatory and cardiovascular responses to exercise in patients with pectus excavatum. Chest. 2003;124:870-882.
Nuss D, Kelly REJr. Minimally invasive surgical correction of chest wall deformities in children (Nuss procedure). Adv Pediatr. 2008;55:395-410.
Ohno K, Morotomi Y, Nakahira M, et al. Indications for surgical repair of funnel chest based on indices of chest wall deformity and psychologic state. Surg Today. 2003;33:662-665.
Protopapas AD, Athanasiou T. Peri-operative data on the Nuss procedure in children with pectus excavatum: independent survey of the first 20 years’ data. J Cardiothorac Surg. 2008 Jul 4;3:40.
Rowland T, Moriarty K, Banever G. Effect of pectus excavatum deformity on cardiorespiratory fitness in adolescent boys. Arch Pediatr Adolesc Med. 2005;159:1069-1073.
Shamberger RC, Welch KJ, Sanders SP. Mitral valve prolapse associated with pectus excavatum. J Pediatr. 1987;111:404-407.
Zhao L, Feinberg MS, Gaides M, et al. Why is exercise capacity reduced in subjects with pectus excavatum? J Pediatr. 2000;136:163-167.
411.2 Pectus Carinatum and Sternal Clefts
Pectus Carinatum
Etiology/Epidemiology
Pectus carinatum is a sternal deformity accounting for 5-15% of congenital chest wall anomalies. Anterior displacements of the mid and lower sternum and adjacent costal cartilages are the most common types. They are most commonly associated with protrusion of the upper sternum; depression of the lower sternum occurs in only 15% of patients. Asymmetry of the sternum is common, and localized depression of the lower anterolateral chest is also often observed. Males are affected 4 times more often than females. There is a high familial occurrence and a common association of mild to moderate scoliosis. Mitral valve disease and coarctation of the aorta are associated with this anomaly. Three types of anatomic deformity occur (upper, lower, and lateral pectus carinatum), with corresponding physiologic changes and treatment algorithms.
Clinical Manifestations
In early childhood, symptoms appear minimal. School-aged children and adolescents, however, commonly complain of dyspnea with mild exertion, decreased endurance with exercise, and exercise-induced wheezing. The incidence of increased respiratory infections and use of asthma medication is higher than in nonaffected subjects. On physical examination, a marked increase in the anteroposterior chest diameter is seen, with resultant reduction in chest excursion and expansion. The increased residual volume results in tachypnea and diaphragmatic respirations. Chest radiographs show an increased anteroposterior diameter of the chest wall, emphysematous-appearing lungs, and a narrow cardiac shadow. The pectus severity score (width of chest divided by distance between sternum and spine; analogous to the Haller index) is reduced.
Sternal Clefts
Sternal clefts are rare congenital malformations that result from the failure of the fusion of the sternum. Partial sternal clefts are more common and may involve the superior sternum in association with other lesions, such as vascular dysplasias and supraumbilical raphe, or the inferior sternal clefts, which are often associated with other midline defects (pentalogy of Cantrell). Complete sternal clefts with complete failure of sternal fusion are rare. These disorders may also occur in isolation. The paradoxic movement of thoracic organs with respiration may alter pulmonary mechanics. Rarely, respiratory infections and even significant compromise result. Surgery is required early in life, before fixation and immobility occur.
Abramson H, D’Agostino J, Wuscovi S. A 5-year experience with a minimally invasive technique for pectus carinatum repair. J Pediatr Surg. 2009;44:118-123.
Coelho Mde S, Guimarães Pde S. Pectus carinatum. J Bras Pneumol. 2007;33:463-474.
Fonkalsrud EW, Anselmo DM. Less extensive techniques for repair of pectus carinatum: the undertreated chest deformity. J Am Coll Surg. 2004;198:898-905.
Goretsky MJ, Kelly RE, Croitoru D, et al. Chest wall anomalies: pectus excavatum and pectus carinatum. Adolesc Med Clin. 2004;15:455-471.
Ohye RG, Rutherford JA, Bove EL. Congenital sternal clefts. Pediatr Cardiol. 2002;23:472-473.
Waters P, Welch K, Micheli LJ, et al. Scoliosis in children with pectus excavatum and pectus carinatum. J Pediatr Orthop. 1989;9:551-556.
Williams AM, Crabbe DC. Pectus deformities of the anterior chest wall. Paediatr Respir Rev. 2003;4:237-242.
411.3 Asphyxiating Thoracic Dystrophy (Thoracic-Pelvic-Phalangeal Dystrophy)
Pathogenesis
A multisystem autosomal recessive disorder, asphyxiating thoracic dystrophy results in a constricted and narrow rib cage. Also known as Jeune syndrome, the disorder is associated with a characteristic skeletal abnormalities as well as variable involvement of other systems, including renal, hepatic, neurologic, pancreatic, and retinal abnormalities. Several genetic loci have been identified in affected individuals.
Clinical Manifestations
Most patients with this disorder die shortly after birth from respiratory failure, although less aggressive forms have been reported in older children. For those who survive the neonatal period, progressive respiratory failure often ensues, owing to impaired lung growth, recurrent pneumonia, and atelectasis originating from the rigid chest wall.
Diagnosis
Physical examination reveals a narrowed thorax that, at birth, is much smaller than the head circumference. The ribs are horizontal, and the child has short extremities. Chest radiographs demonstrate a bell-shaped chest cage with short, horizontal, flaring ribs and high clavicles.
Treatment
No specific treatment exists, although thoracoplasty to enlarge the chest wall and long-term mechanical ventilation have been tried. Rib-expanding procedures have resulted in improved survival.
Prognosis
For some children with asphyxiating thoracic dystrophy, improvement in the bony abnormalities occurs with age. However, children <1 yr old often succumb to respiratory infection and failure. Progressive renal disease often occurs with older children. Use of vaccines for influenza and other respiratory pathogens is warranted, as is aggressive use of antibiotics for respiratory infections.
Davis JT, Long FR, Adler BH, et al. Lateral thoracic expansion for Jeune syndrome: evidence of rib healing and new bone formation. Ann Thorac Surg. 2004;77:445-448.
Kajantic E, Anderson S, Kaitila I. Familial asphyxiating thoracic dysplasia: clinical variability and impact of improved neonatal intensive care. J Pediatr. 2001;139:130-133.
Phillips JD, van Aalst JA. Jeune’s syndrome (asphyxiating thoracic dystrophy): congenital and acquired. Semin Pediatr Surg. 2008;17:167-172.
Sharoni E, Erez E, Chorev G, et al. Chest reconstruction in asphyxiating thoracic dystrophy. J Pediatr Surg. 1998;33:1578-1581.
Tahernia AC, Stamps P. “Jeune syndrome” (asphyxiating thoracic dystrophy): report of a case, a review of the literature, and an editor’s commentary. Clin Pediatr. 1977;16:903-908.
Wiebicke W, Pasterkamp H. Long-term continuous positive pressure in a child with asphyxiating thoracic dystrophy. Pediatr Pulmonol. 1988;4:54-58.
411.4 Achondroplasia
Achondroplasia is the most common condition characterized by disproportionate short stature. This condition is inherited as an autosomal dominant disorder that results in disordered growth (Chapter 687). Much has been learned about this disorder, including its genetic origins and how to minimize its serious complications.
Clinical Manifestations
Restrictive pulmonary disease, affecting <5% of children with achondroplasia who are younger than 3 yr, is more likely at high elevation. Recurrent infections, cor pulmonale, and dyspnea are commonly associated. There is an increased risk of obstructive sleep apnea, although most patients are not affected. Hypoxemia during sleep is a common feature. Onset of restrictive lung disease can begin at a very young age. On examination, the breathing pattern is rapid and shallow, with associated abdominal breathing. The anteroposterior diameter of the thorax is reduced. Special growth curves for chest circumference of patients with achondroplasia from birth to 7 yr are available. Three distinct phenotypes exist: phenotypic group 1 patients possess relative adenotonsillar hypertrophy, group 2 patients have muscular upper airway obstruction and progressive hydrocephalus, and group 3 patients have upper airway obstruction without hydrocephalus. Kyphoscoliosis may develop during infancy.
Diagnosis
Pulmonary function tests reveal a reduced vital capacity that is more pronounced in males. The lungs are small but functionally normal. Chest radiographs demonstrate the decreased anteroposterior diameter along with anterior cupping of the ribs. The degree of foramen magnum involvement correlates with the extent of respiratory dysfunction.
Treatment
Treatment of sleep apnea, if present, is supportive (Chapter 17). Physiotherapy and bracing may minimize the complications of both kyphosis and severe lordosis. Aggressive treatment of respiratory infections and scoliosis is warranted.
Prognosis
The life span is normal for most children with this condition, except for the phenotypic groups with hydrocephalus or with severe cervical or lumbar spinal compression.
Boulet S, Althuser M, Nugues F, Schaal JP, et al. Prenatal diagnosis of achondroplasia: new specific signs. Prenat Diagn. 2009;29:697-702.
Hunter AG, Reid CS, Pauli RM, et al. Standard curves of chest circumference in achondroplasia and the relationship of chest circumference to respiratory problems. Am J Med Genet. 1996;62:91-97.
Mogayzel PJJr, Carroll JL, Loughlin GM, et al. Sleep-disordered breathing in children with achondroplasia. J Pediatr. 1998;132:667-671.
Stokes DC, Phillips JA, Leonard CO, et al. Respiratory complications of achondroplasia. J Pediatr. 1983;102:534-541.
Stokes DC, Wohl ME, Wise RA, et al. The lungs and airways in achondroplasia: do little people have little lungs? Chest. 1990;98:145-152.
Tasker RC, Dundas I, Laverty A, et al. Distinct patterns of respiratory difficulty in young children with achondroplasia: a clinical, sleep, and lung function study. Arch Dis Child. 1998;79:99-108.
Trotter TL, Hall JG, American Academy of Pediatrics Committee on Genetics. Health supervision for children with achondroplasia. Pediatrics. 2005;116:771-783.
411.5 Kyphoscoliosis: Adolescent Idiopathic Scoliosis and Congenital Scoliosis
Pathogenesis
Adolescent idiopathic scoliosis (AIS) is characterized by lateral bending of the spine (Chapter 671). It commonly affects children during their teen years as well as during periods of rapid growth. The cause is unknown. Congenital scoliosis is uncommon, affecting girls more than boys, and is apparent in the 1st year of life (Chapter 671.2).
Clinical Manifestations
The pulmonary manifestations of scoliosis may include chest wall restriction, leading to a reduction in total lung capacity. The angle of scoliosis deformity has been correlated with the degree of lung impairment only for patients with thoracic curves. Vital capacity, forced expiratory volume in 1 sec (FEV1), work capacity, diffusion capacity, chest wall compliance, and PaO2 decrease as the severity of thoracic curve increases. These findings can be seen in even mild to moderate AIS (Cobb angle <30 degrees) but do not occur in other, nonthoracic curves. Reduction in peripheral muscle function has been associated with AIS through either intrinsic mechanisms or deconditioning. Severe impairment can lead to cor pulmonale or respiratory failure and can occur before age 20 yr. Children with severe scoliosis, especially boys, may have abnormalities of breathing during sleep, and the resultant periods of hypoxemia may contribute to the eventual development of pulmonary hypertension.
Diagnosis
Physical examination and an upright, posteroanterior radiograph with subsequent measurement of the angle of curvature (Cobb technique) remain the gold standard for assessment. Curves >10 degrees define the presence of scoliosis.
Treatment
Depending on the extent of the curve and the degree of skeletal maturation, treatment options include reassurance, observation, bracing, and surgery (spinal fusion). Influenza vaccine should be administered, given the extent of pulmonary compromise that may coexist. Because vital capacity is a strong predictor for the development of respiratory failure in untreated AIS, surgical goals are to diminish the scoliotic curve, maintain the correction, and prevent deterioration in pulmonary function. Abnormalities of vital capacity and total lung capacity, exercise intolerance, and the rate of change of these variables over time should be taken into consideration for the timing of surgical correction. Preoperative assessment of lung function may assist in predicting postsurgical pulmonary difficulties. Many patients undergoing surgical correction may be managed postoperatively without mechanical ventilation. Even patients with mild scoliosis may have pulmonary compromise immediately after spinal fusion, secondary to pain and a body cast that may restrict breathing and interfere with coughing. Children with a preoperative FEV1 <40% predicted are at risk for requiring prolonged postoperative mechanical ventilation. Rib-expanding procedures have been successful in severe cases of congenital scoliosis.
Chen S, Huang T, Lee Y, Hsu RW. Pulmonary function after thoracoplasty in adolescent idiopathic scoliosis. Clin Orthop Related Res. 2002;399:152-161.
Hedequist D, Emans J. Congenital scoliosis. J Am Acad Orthop Surg. 2004;12:266-275.
Kearon C, Viviani GR, Killian KJ. Factors influencing work capacity in adolescent idiopathic thoracic scoliosis. Am Rev Respir Dis. 1993;148:295-303.
Leech JA, Ernst P, Rogala EJ, et al. Cardiorespiratory status in relation to mild deformity in adolescent idiopathic scoliosis. J Pediatr. 1985;106:143-149.
McNicholas WT. Impact of sleep in respiratory failure. Eur Respir J. 1997;10:920-933.
Mezon BL, West P, Israels J, et al. Sleep breathing abnormalities in kyphoscoliosis. Am Rev Respir Dis. 1980;122:617-621.
Yuan N, Skaggs DL, Dorey F, et al. Preoperative predictors of prolonged postoperative mechanical ventilation in children following scoliosis repair. Pediatr Pulmonol. 2005;40:414-419.
411.6 Congenital Rib Anomalies
Clinical Manifestations
Isolated defects of the highest and lowest ribs have minimal clinical pulmonary consequences. Missing midthoracic ribs are associated with the absence of the pectoralis muscle, and lung function can become compromised. Associated kyphoscoliosis and hemivertebrae may accompany this defect. If the rib defect is small, no significant sequelae ensue. When the 2nd to 5th ribs are absent anteriorly, lung herniation and significant abnormal respiration ensue. The lung is soft and nontender and may be easily reducible on examination. Complicating sequelae include severe lung restriction (secondary to scoliosis), cor pulmonale, and congestive heart failure. Symptoms are often minimal but can cause dyspnea. Respiratory distress is rare in infancy.
Diagnosis
Chest radiographs demonstrate the deformation and absence of ribs with secondary scoliosis. Most rib abnormalities are discovered as incidental findings on a chest film.
Treatment
If symptoms are severe enough to cause clinical compromise or significant lung herniation, then homologous rib grafting can be performed. Rib-expanding procedures are also of great value. Adolescent girls with congenital rib anomalies may require cosmetic breast surgery.
Bronsther B, Coryllos E, Epstein B, et al. Lung hernias in children. J Pediatr Surg. 1968;3:544-550.
Campbell RMJr, Smith MD, Mayes TC, et al. The characteristics of thoracic insufficiency syndrome associated with fused ribs and congenital scoliosis. J Bone Joint Surg Am. 2003;85-A:399-408.
Mehta MH, Patel RV, Mehta LV, et al. Congenital absence of ribs. Indian Pediatr. 1992;29:1149-1152.
Rickham PP. Lung hernia secondary to congenital absence of ribs. Arch Dis Child. 1959;34:14-17.