Chapter 19 Neuro-ophthalmology
Neuroimaging
Computed tomography
Physics
Computed tomography (CT) uses X-ray beams to obtain tissue density values from which detailed cross-sectional images are formed by a computer. Tissue density is represented by a grey scale, white being maximum density (e.g. bone) and black being minimum density (e.g. air). Advanced CT scanners are able to acquire thinner slices leading to improved spatial resolution, together with faster examination times, without a proportionate increase in radiation dose. Images are acquired in an axial form and can be viewed in any plane using computer reconstruction. This multiplanar information can be an advantage over MR with regard to anatomical detail. CT is widely available, easy to perform, relatively inexpensive and quick but unlike MR it exposes the patient to ionizing radiation.
Contrast enhancement
Iodinated contrast material improves sensitivity and specificity but is contraindicated in patients allergic to iodine and in those with renal failure. Contrast is not indicated in acute haemorrhage, bony injury or localization of foreign bodies because it may mask visualization of these high density structures.
Indications
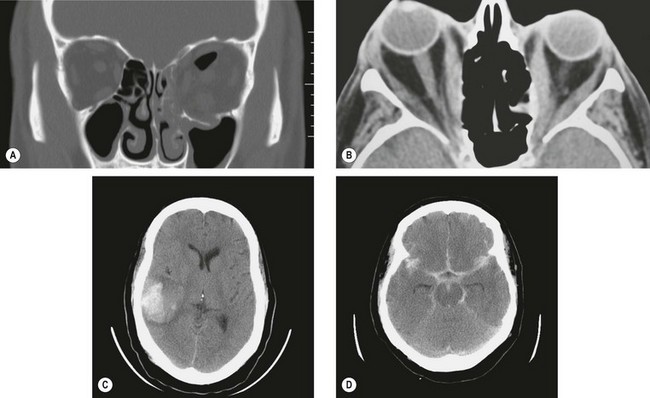
Fig. 19.1 CT scans. (A) Coronal image shows blow-out fractures of the left orbital floor and medial wall with orbital emphysema; (B) axial image shows bilateral enlargement of extraocular muscles and right proptosis; (C) axial image shows an acute parenchymal haematoma in the right temporal lobe; (D) axial image shows extensive subarachnoid blood in the basilar cisterns, and the Sylvian and interhemispheric fissures
(Courtesy of N Sibtain – figs A, C and D; A Pearson – fig. B)
Magnetic resonance imaging
Physics
Magnetic resonance imaging (MR) depends on the rearrangement of positively-charged hydrogen nuclei (protons) when a tissue is exposed to a short electromagnetic pulse. When the pulse subsides, the nuclei return to their normal position re-radiating some of the absorbed energy. Sensitive receivers pick up this electromagnetic echo. Unlike CT, it does not subject the patient to ionizing radiation. The signals are analyzed and displayed as a cross-sectional image which may be: (a) axial, (b) coronal or (c) sagittal.
Weighting
Weighting refers to two methods of measuring the relaxation times of the excited protons after the magnetic field has been switched off. Various body tissues have different relaxation times so that a given tissue may be T1- or T2-weighted, (i.e. best visualized on that particular type of image). In practice, both types of scans are usually performed. It is easy to tell the difference between CT and MR because bone appears white on CT but is not clearly demonstrated on MR.
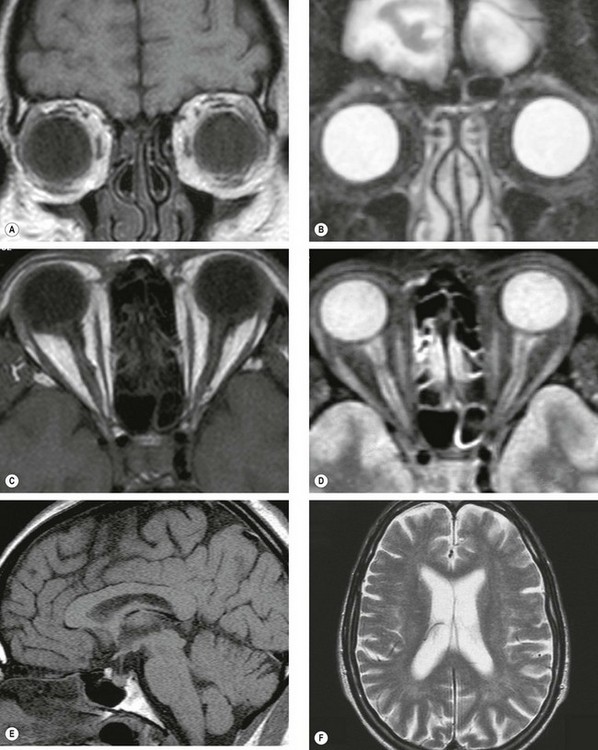
Fig. 19.2 MR scans. (A) T1-weighted coronal image through the globe in which vitreous is hypointense (dark) and orbital fat is hyperintense (bright); (B) T2-weighted scan in which vitreous is hyperintense and orbital fat is hypointense; (C) T1-weighted axial image through the globes and optic nerves in which vitreous and CSF around the optic nerves are hypointense and orbital fat is hyperintense; (D) T2-weighted axial image in which vitreous and CSF are hyperintense; (E) T1-weighted midline sagittal image through the brain in which the CSF in the third ventricle is hypointense; (F) T2-weighted axial image through the brain in which the CSF in the lateral ventricles is hyperintense
Contrast enhancement
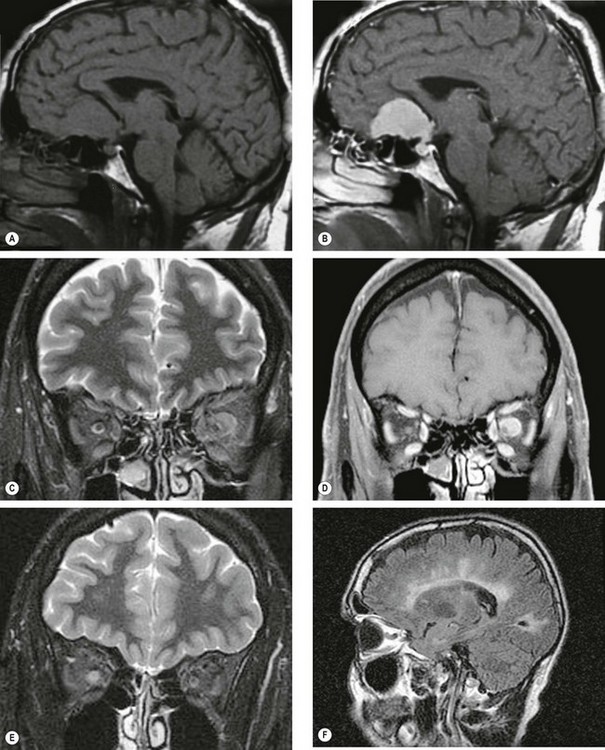
Fig. 19.3 Enhancement techniques. (A) Pre-contrast sagittal T1-weighted image of a meningioma; (B) post-contrast image shows enhancement of the tumour; (C) coronal STIR image shows an intermediate signal intensity mass surrounding the left optic nerve consistent with an optic nerve sheath meningioma; (D) T1-weighted fat saturated coronal image of the same patient shows avid homogeneous enhancement of the meningioma; (E) coronal STIR image of right retrobulbar neuritis shows a high signal within the optic nerve with enlargement of the optic nerve sheath complex; (F) sagittal FLAIR image shows multiple periventricular plaques of demyelination
(Courtesy of D Thomas – figs A and B; N Sibtain – figs C–F)
Limitations
Neuro-ophthalmic indications
MR is the technique of choice for lesions of the intracranial pathways.
Angiography
Magnetic resonance angiography
Magnetic resonance angiography (MRA) is a non-invasive method of imaging the intra- and extracranial carotids and vertebrobasilar circulations (Fig. 19.4A) to demonstrate stenosis, dissection, occlusion, arteriovenous malformations and aneurysms. This technique uses the motion sensitivity of MR to visualize blood flow within vessels and does not require contrast. However, because it relies on flow, thrombosed aneurysms may be missed and turbulent flow may lead to difficulties in interpretation. Furthermore MRA is unreliable in detecting very small aneurysms.
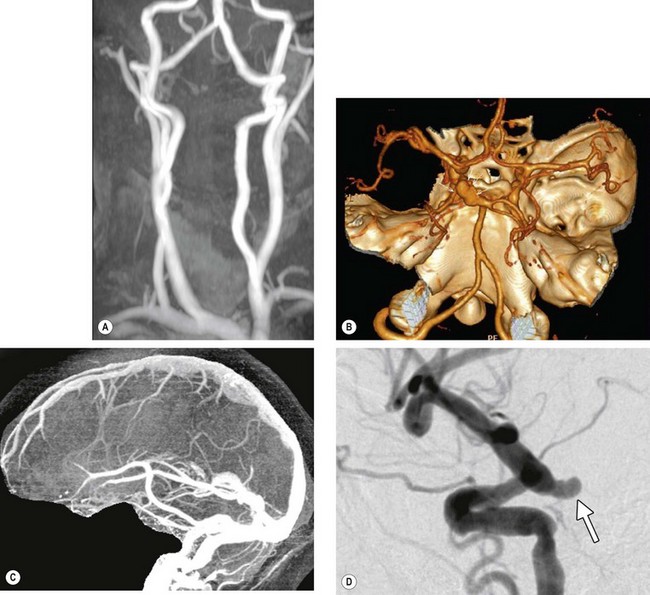
Fig. 19.4 Cerebral angiography. (A) Normal MRA of the external carotid and vertebral circulation; (B) CT angiogram shows a left posterior communicating aneurysm (arrows); (C) normal CT venogram; (D) conventional catheter angiogram with subtraction shows an aneurysm arising from the internal carotid artery at its junction with the posterior communicating artery (arrow)
(Courtesy of N Sibtain – figs A–C; JD Trobe, from Neuro-ophthalmology, in Rapid Diagnosis in Ophthalmology, Mosby 2008)
Computed tomographic angiography
Computed tomographic angiography (CTA) is emerging as the method of choice in the investigation of intracranial aneurysms (Fig. 19.4B). It enables acquisition of extremely thin slice images of the brain following intravenous contrast. Images of the vessels can be reconstructed in three dimensions and viewed from any direction. The investigation is safe and quick and without the 1% risk of stroke associated with conventional catheter angiography.
Computed tomographic venography
Computed tomographic venography (CTV) may be useful when MRA is contraindicated or there are difficulties in distinguishing slow flow from thrombus on MR. The technique is similar to CTA, except that images are acquired in the venous phase of contrast enhancement (Fig. 19.4C). However, it is not as sensitive as MRA in detecting associated parenchymal changes and this usually limits its use to equivocal cases.
Conventional catheter angiography
Conventional intra-arterial catheter angiography is usually performed under local anaesthetic. A catheter is passed via the femoral artery into the internal carotid and vertebral arteries in the neck under fluoroscopic guidance. Following contrast injection images are taken in rapid succession. Digital subtraction results in images of the contrast-filled vessels without any background structure such as bone (Fig. 19.4D). Until recently, this technique was the first-line investigation in the diagnosis of intracranial aneurysms but is now mainly reserved for cases where CTA is equivocal or negative.
Optic nerve
Anatomy
General structure
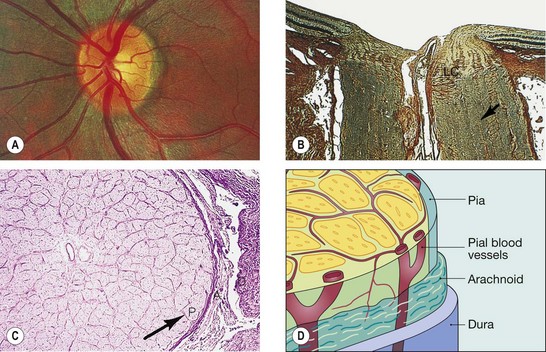
Fig. 19.5 Structure of the optic nerve. (A) Clinical appearance; (B) longitudinal section, LC = lamina cribrosa; arrow points to a fibrous septum; (C) transverse section, P = pia, A = arachnoid, D = dura; (D) surrounding sheaths and pial blood vessels
(Courtesy of Wilmer Institute – figs A, B and C)
Anatomical subdivisions
The optic nerve is approximately 50 mm long from globe to chiasm. It can be subdivided into four segments:
Visual evoked potential
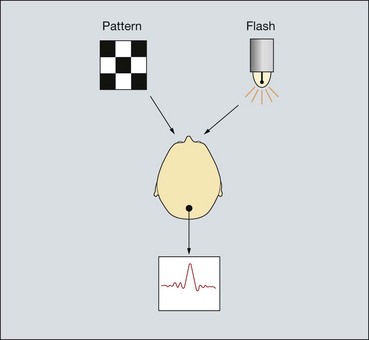
Signs of optic nerve dysfunction
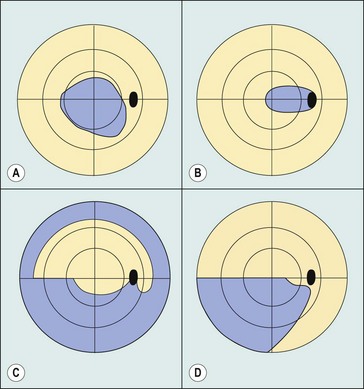
Table 19.1 Focal visual field defects in optic neuropathies
Optic atrophy
Primary optic atrophy
Primary optic atrophy occurs without antecedent swelling of the optic nerve head. It may be caused by lesions affecting the visual pathways from the retrolaminar portion of the optic nerve to the lateral geniculate body. Lesions anterior to the optic chiasm result in unilateral optic atrophy, whereas those involving the chiasm and optic tract will cause bilateral changes.
Secondary optic atrophy
Secondary optic atrophy is preceded by long-standing swelling of the optic nerve head.
Consecutive optic atrophy
Consecutive optic atrophy is caused by diseases of the inner retina or its blood supply. The cause is usually obvious on fundus examination such as retinitis pigmentosa, old vasculitis (Fig. 19.7C), retinal necrosis and excessive retinal photocoagulation.
Classification of optic neuritis
Optic neuritis is an inflammatory, infective or demyelinating process affecting the optic nerve. It can be classified both ophthalmoscopically and aetiologically as follows.
Ophthalmoscopical classification
Demyelinating optic neuritis
Overview
Demyelination is a pathological process by which normally myelinated nerve fibres lose their insulating myelin layer. The myelin is phagocytosed by microglia and macrophages, subsequent to which astrocytes lay down fibrous tissue in plaques. Demyelinating disease disrupts nervous conduction within the white matter tracts of the brain, brainstem and spinal cord. Diseases which may cause ocular problems are the following:
Multiple sclerosis
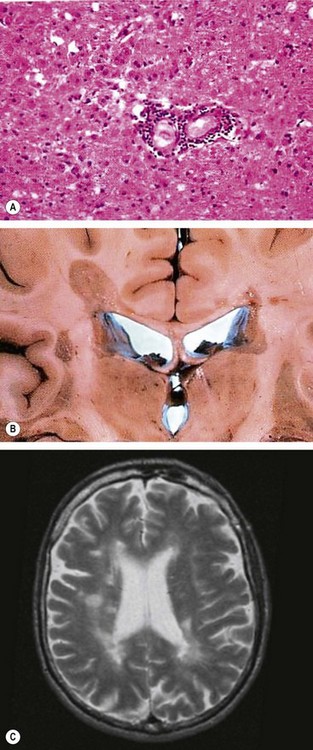
Multiple sclerosis (MS) is an remitting idiopathic demyelinating disease involving white matter within the CNS (Fig. 19.9A and B).
Association between optic neuritis and multiple sclerosis
Although some patients with optic neuritis have no clinically demonstrable associated systemic disease, the following close association exists between optic neuritis and MS.
Clinical features of demyelinating optic neuritis
Treatment of demyelinating optic neuritis
Parainfectious optic neuritis
Optic neuritis may be associated with various viral infections such as measles, mumps, chickenpox, rubella, whooping cough and glandular fever. It may also occur following immunization. Children are affected much more frequently than adults.
Infectious optic neuritis
Non-infectious optic neuritis
Sarcoid
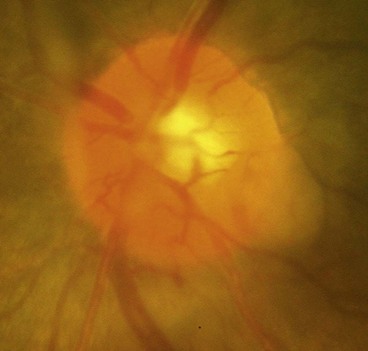
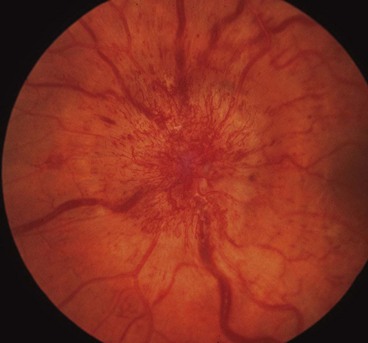
Optic neuritis affects 1–5% of patients with neurosarcoid. It may occasionally be the presenting feature of sarcoidosis but usually develops during the course of established systemic disease. The optic nerve head may exhibit a lumpy appearance suggestive of granulomatous infiltration and there may be associated vitritis (Fig. 19.11). The response to steroid therapy is often rapid although vision may decline if treatment is tapered or stopped prematurely, and some patients require long-term low-dose therapy. Methotrexate may also be used as an adjunct to steroids or as monotherapy in steroid-intolerant patients.
Autoimmune
Autoimmune optic nerve involvement may be in the form of retrobulbar neuritis or anterior ischaemic optic neuropathy (see below). Some patients may also experience slowly progressive visual loss suggestive of optic nerve compression. Treatment is with systemic steroids and other immunosuppressants.
Neuroretinitis
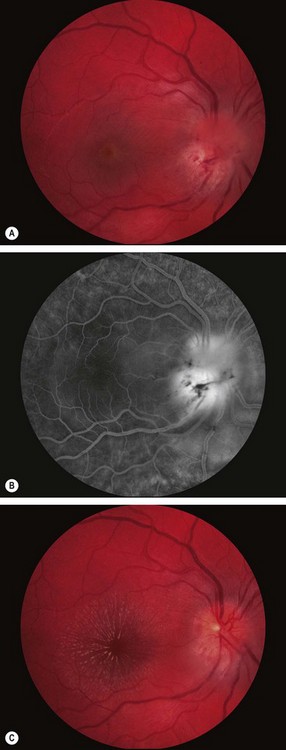
Non-arteritic anterior ischaemic optic neuropathy
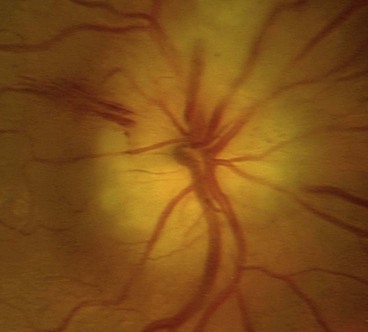
Arteritic anterior ischaemic optic neuropathy
Arteritic anterior ischaemic optic neuropathy (AAION) is caused by giant cell arteritis.
Diagnosis of giant cell arteritis
Giant cell arteritis (GCA) is a granulomatous necrotizing arteritis (Fig. 19.14A) with a predilection for large and medium-size arteries, particularly the superficial temporal, ophthalmic, posterior ciliary and proximal vertebral. The severity and extent of involvement are associated with the quantity of elastic tissue in the media and adventitia. Intracranial arteries, which possess little elastic tissue, are usually spared.
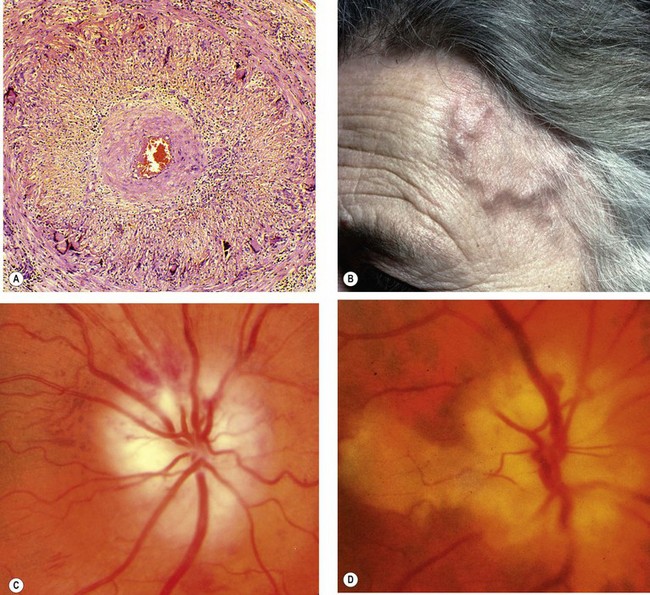
Fig. 19.14 Giant cell arteritis. (A) Histology shows transmural granulomatous inflammation, disruption of the internal elastic lamina, proliferation of the intima and gross narrowing of the lumen; (B) the superficial temporal artery is pulseless, nodular and thickened; (C) ischaemic optic neuropathy; (D) ischaemic optic neuropathy and cilioretinal artery occlusion
(Courtesy of J Harry and G Misson, from Clinical Ophthalmic Pathology, Butterworth-Heinemann 2002 – fig. A; S Farley, T Cole and S Rimmer – fig. C; SS Hayreh – figs C and D)
Treatment of giant cell arteritis
Treatment involves systemic steroids, the duration of which is governed by symptoms and the level of the ESR or CRP. Symptoms may, however, recur without a corresponding rise in ESR or CRP and vice versa. Most patients need treatment for 1–2 years, although some may require indefinite maintenance therapy. CRP may play an important role in monitoring disease activity, as the level seems to fall more rapidly than the ESR in response to treatment.
Ophthalmic manifestations of giant cell arteritis
Arteritic anterior ischaemic optic neuropathy
AAION affects 30–50% of untreated patients of which one-third develop involvement of the fellow eye, usually within 1 week of the first. Posterior ischaemic optic neuropathy is much less common (see below).
Other manifestations
Posterior ischaemic optic neuropathy
Posterior ischemic optic neuropathy (PION) is much less common than the anterior variety. It is caused by ischaemia of the retrolaminar portion of the optic nerve which is supplied by the surrounding pial capillary plexus; only a small number of capillaries actually penetrate the nerve and extend to its central portion among the pial septae. The diagnosis of PION is made after other causes of retrobulbar optic neuropathy, such as compression or inflammation, have been excluded. PION occurs in the following three settings.
Diabetic papillopathy
Diabetic papillopathy is an uncommon condition that may occur in both type 1 and type 2 diabetics. The underlying pathogenesis is unclear but it may be the result of small-vessel disease.
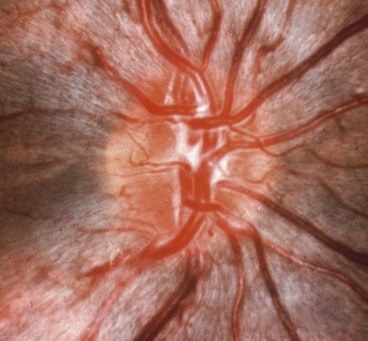
Leber hereditary optic neuropathy
Leber hereditary optic neuropathy (LHON) is a rare disease associated with maternally-inherited mitochondrial DNA mutations, most notably 11778. The condition typically affects males between the ages of 15 and 35 years, although in atypical cases the condition may affect females and present at any age between 10 and 60 years. The diagnosis of LHON should therefore be considered in any patient with bilateral optic neuropathy, irrespective of age. Unaffected carriers show thickening of temporal retinal fibres on OCT.
Hereditary optic atrophy
The hereditary optic atrophies (neuropathies) are a very rare heterogeneous group of disorders that are primarily characterized by bilateral optic atrophy.
Kjer-type optic atrophy
Behr syndrome
Nutritional optic neuropathy
Nutritional optic neuropathy (tobacco-alcohol amblyopia) typically affects heavy drinkers and smokers who are deficient in protein and the B vitamins. Most patients have neglected their diet, obtaining their calories from alcohol instead. Some of those affected also have defective vitamin B12 absorption and may develop pernicious anaemia.
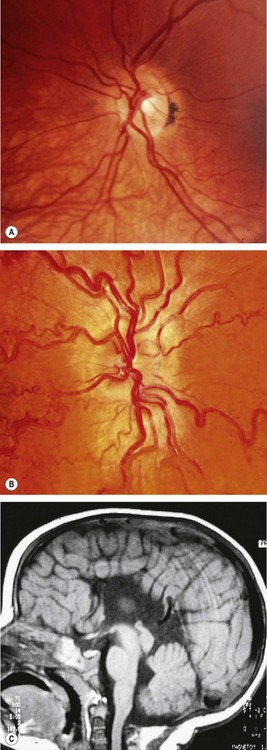
Papilloedema
Pathogenesis
Papilloedema is swelling of the optic nerve head secondary to raised intracranial pressure. It is nearly always bilateral, although it may be asymmetrical. All other causes of disc oedema in the absence of raised intracranial pressure are referred to as ‘disc swelling’ and are usually associated with persistent visual impairment. All patients with papilloedema should be suspected of having an intracranial mass unless proved otherwise. However, not all patients with raised intracranial pressure will necessarily develop papilloedema. Tumours of the cerebral hemispheres tend to produce papilloedema later than those in the posterior fossa. Patients with a history of previous papilloedema may develop a substantial increase in intracranial pressure but fail to re-develop papilloedema because of glial scarring of the optic nerve head.
Cerebrospinal fluid and causes of raised intracranial pressure
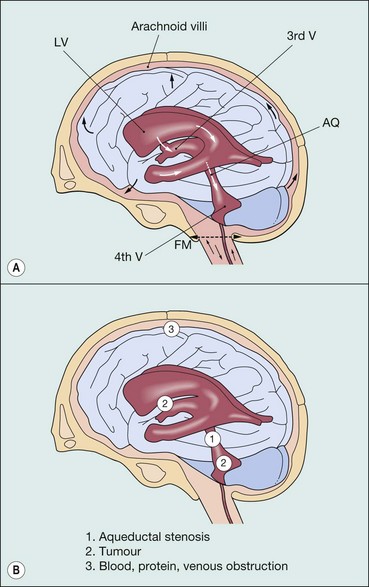
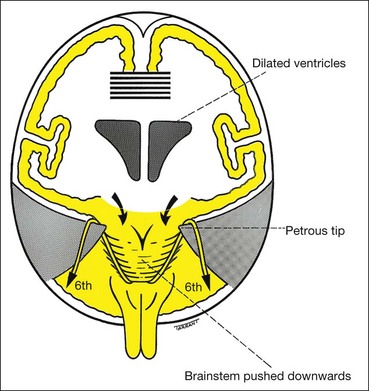
Diagnosis of raised intracranial pressure
Stages of papilloedema
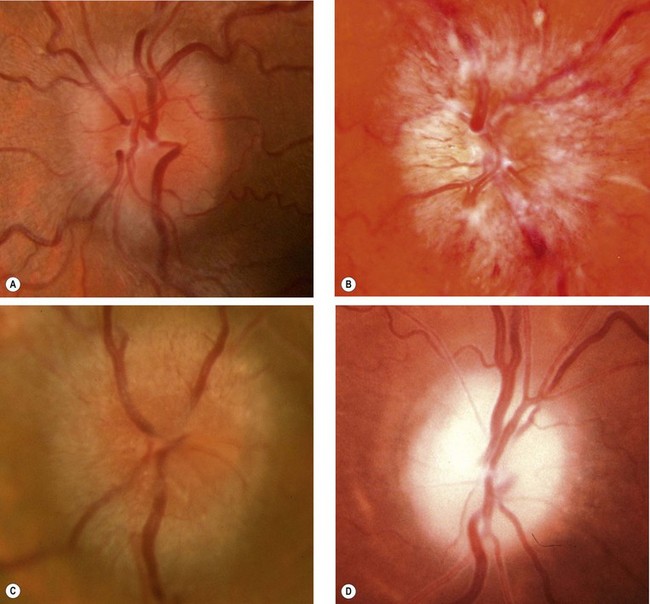
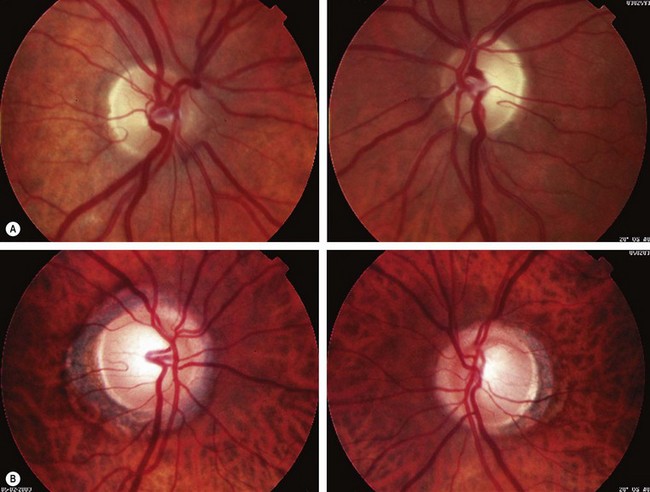
Fig. 19.20 Papilloedema. (A) Early; (B) established; (C) chronic; (D) atrophic
(Courtesy of S Farley, T Cole and S Rimmer – fig. B)
Table 19.2 Causes of optic disc elevation
Congenital optic disc anomalies
Tilted disc
A tilted optic disc is a common, usually bilateral, anomaly caused by an oblique entry of the optic nerve into the globe. This results in pseudorotation of the superior pole of the disc, angulation of the optic cup axis and elevation of the neuroretinal rim.
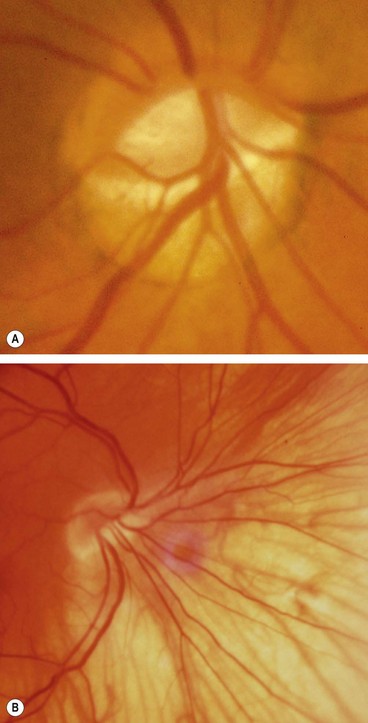
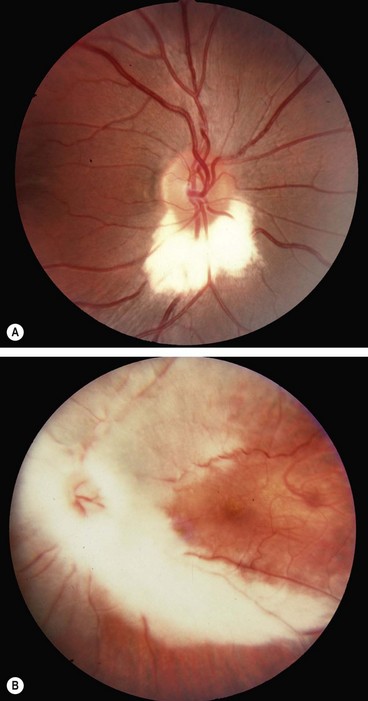
Optic disc pit
Optic disc drusen
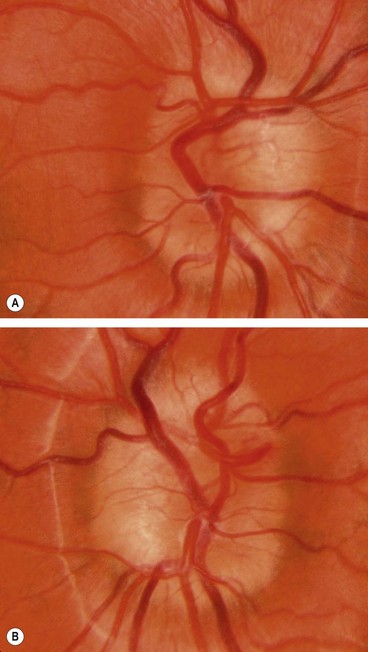
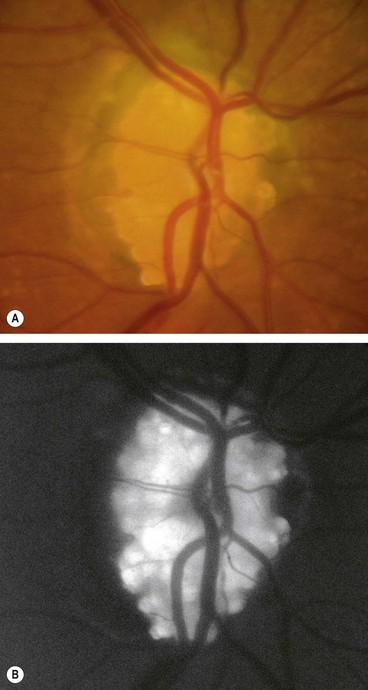
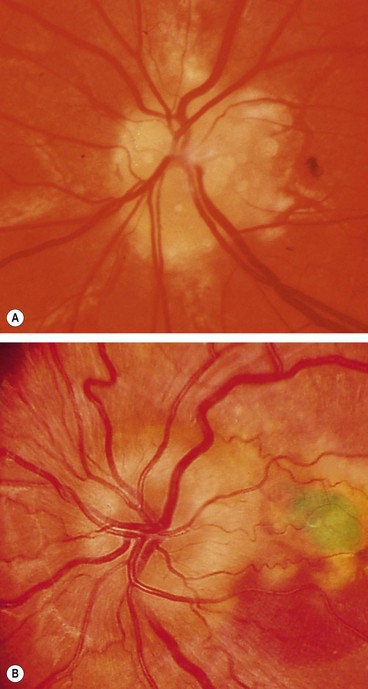
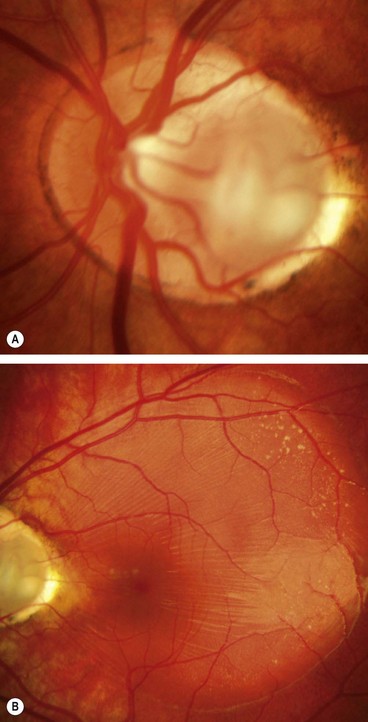
Fig. 19.25 (A) Disc drusen associated with angioid streaks; (B) drusen with secondary choroidal neovascularization
(Courtesy of J Donald M Gass, from Stereoscopic Atlas of Macular Diseases, Mosby 1997 – fig. B)
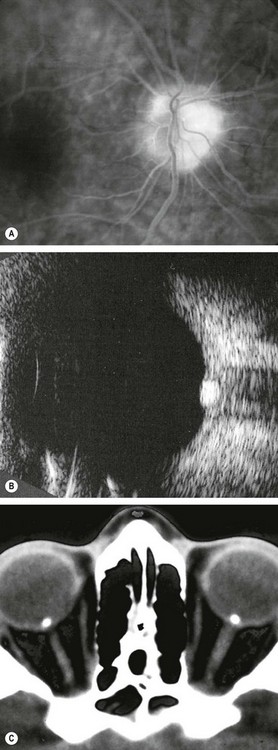
Fig. 19.26 Imaging of disc drusen. (A) FA late phase shows hyperfluorescence without leakage; (B) B-scan shows high acoustic reflectivity; (C) axial CT shows bilateral drusen
(Courtesy of P Gili – figs A and B; JD Trobe, from Neuro-ophthalmology, in Rapid Diagnosis in Ophthalmology, Mosby 2008 – fig. C)
Optic disc coloboma
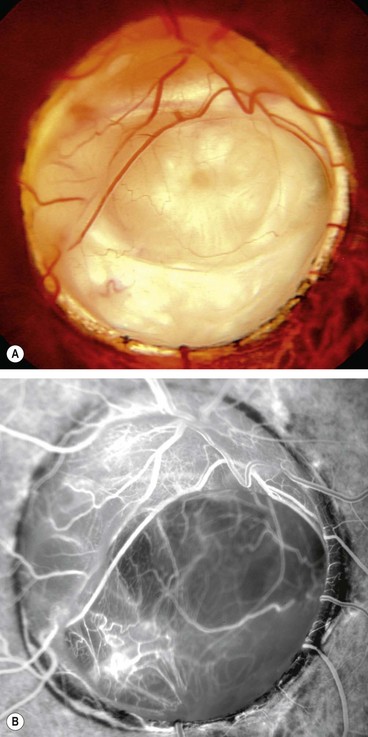
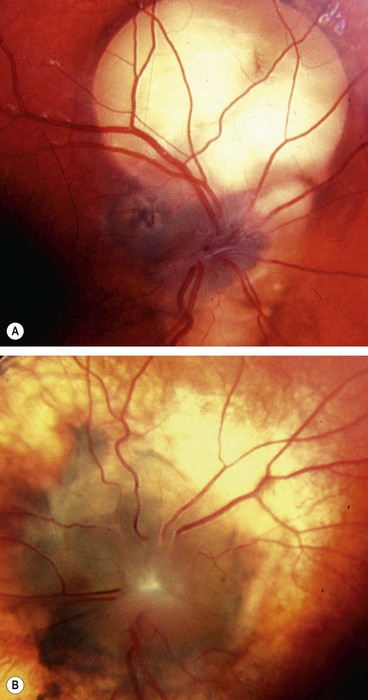
Morning glory anomaly
Morning glory anomaly is a very rare, usually unilateral sporadic condition that has a spectrum of severity. Bilateral cases, which are rarer still, may be hereditary.
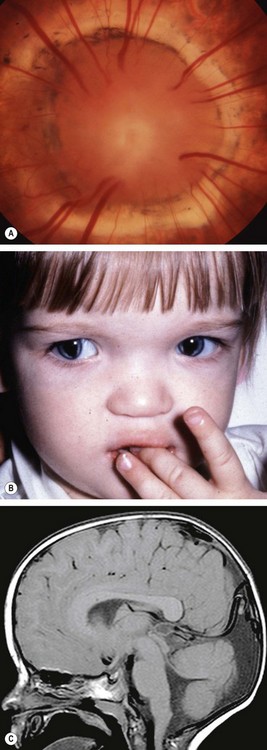
Optic nerve hypoplasia
The hypoplastic optic nerve, unilateral or bilateral, is characterized by a diminished number of nerve fibres. It may occur as an isolated anomaly in an otherwise normal eye, in a grossly malformed eye or in association with a heterogeneous group of disorders most commonly involving the midline structures of the brain.
Myelinated nerve fibres
In normal eyes, optic nerve myelination stops at the cribriform plate. In eyes with myelinated nerve fibres the ganglion cells retain a myelin sheath.
Aicardi syndrome
Miscellaneous anomalies
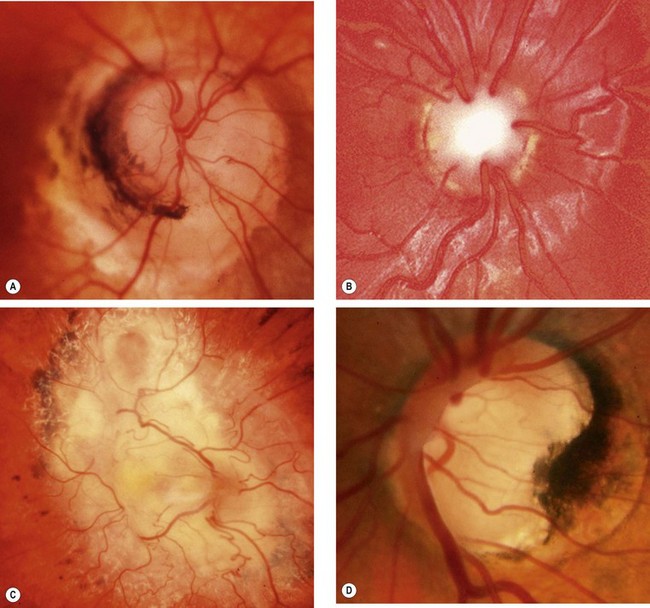
Pupillary reactions
Anatomy
Light reflex
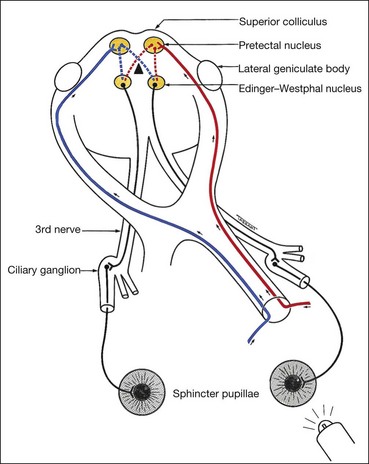
The light reflex is mediated by the retinal photoreceptors and subserved by four neurones (Fig. 19.33).
Near reflex
The near reflex, a synkinesis rather than a true reflex, is activated when gaze is changed from a distant to a near target (see also Ch. 18). It comprises accommodation, convergence and miosis. Vision is not a prerequisite and there is no clinical condition in which the light reflex is present but the near response absent. Although the final pathways for the near and light reflexes are identical (i.e. 3rd nerve, ciliary ganglion, short ciliary nerves), the centre for the near reflex is ill-defined. There are probably two supranuclear influences: the frontal and occipital lobes. The midbrain centre for the near reflex is probably located more ventrally than the pretectal nucleus and this may explain why compressive lesions such as pinealomas, preferentially involving the dorsal internuncial neurones involved in the light reflex, spare the near reflex fibres until later.
Afferent pupillary defect
Absolute afferent pupillary defect
An absolute afferent pupillary defect (amaurotic pupil) is caused by a complete optic nerve lesion and is characterized by the following:
Relative afferent pupillary defect
A relative pupillary defect (Marcus Gunn pupil) is caused by an incomplete optic nerve lesion or severe retinal disease, but never by a dense cataract. The clinical features are those of an amaurotic pupil but more subtle. Thus the pupils respond weakly to stimulation of the diseased eye and briskly to that of the normal eye. The difference between the pupillary reactions of the two eyes is highlighted by the ‘swinging flashlight test’ in which a light source is alternatively switched from one eye to the other and back, thus stimulating each eye in rapid succession. A right relative defect is characterized by the following (Fig. 19.34A-D):
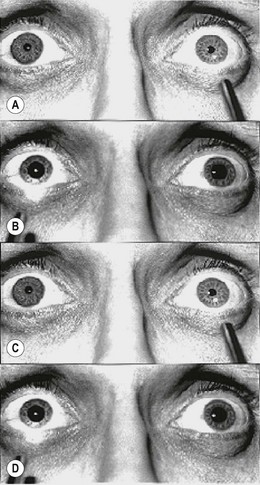
Fig. 19.34 ‘Swinging flashlight test’ in a right afferent pupillary defect
(Courtesy of ES Rosen, P Eustace, HS Thompson and WJK Cumming, from Neuro-ophthalmology, Mosby 1998)
This paradoxical dilatation of the pupils in response to light occurs because the dilatation produced by withdrawing the light from the normal eye outweighs the constriction produced by stimulating the abnormal eye.
It should be emphasized that in afferent (sensory) lesions, the pupils are equal in size. Anisocoria (inequality of pupillary size) implies disease of the efferent (motor) nerve, iris or muscles of the pupil.
Oculosympathetic palsy (Horner syndrome)
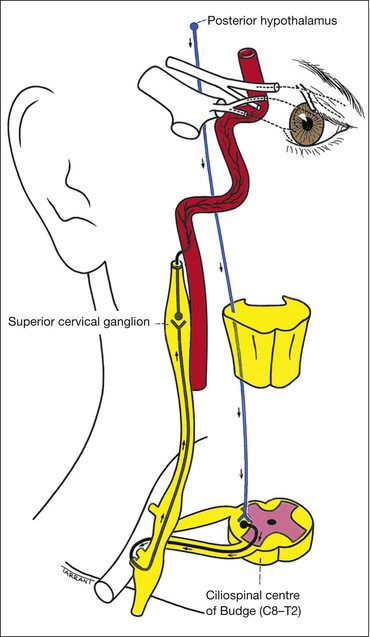
Anatomy
The sympathetic supply involves three neurones (Fig. 19.35):
Causes
The causes of Horner syndrome are shown in Table 19.3.
Table 19.3 Causes of Horner syndrome
Signs
The vast majority of cases are unilateral. Causes of bilateral involvement include cervical spine injuries and as part of systemic autonomic diabetic neuropathy.
Pharmacological tests
Cocaine confirms the diagnosis. Hydroxyamphetamine (Paredrine) may be used to differentiate a preganglionic from a postganglionic lesion. Adrenaline may also be used to assess denervation supersensitivity.
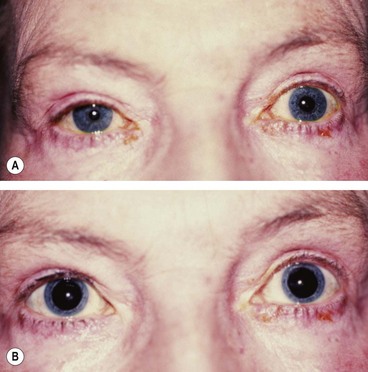
Adie pupil
Adie (tonic) pupil is caused by denervation of the postganglionic supply to the sphincter pupillae and the ciliary muscle, which may follow a viral illness. It typically affects young adults and presents as a unilateral condition in 80% of cases although involvement of the second eye typically develops within months or years.
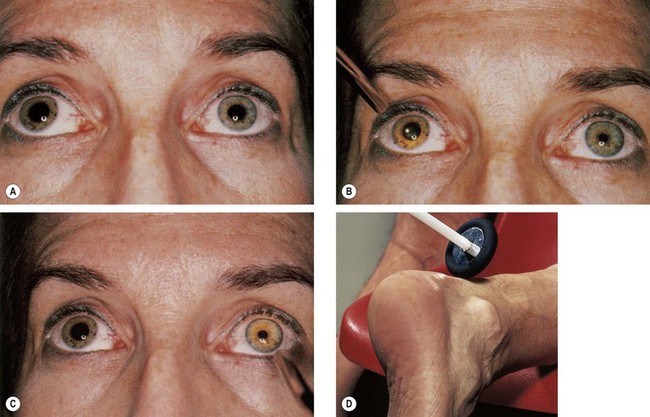
Fig. 19.38 Right Adie pupil. (A) Right pupil is large and regular; (B) direct light reflex is absent or sluggish; (C) consensual light reflex is similar; (D) diminished deep tendon reflex
(Courtesy of DM Albert and FA Jakobiec, from Principles and Practice of Ophthalmology, Saunders 1994 – figs A, B and C; MA Mir, from Atlas of Clinical Diagnosis, Saunders 2003 – fig. D)
Other abnormal reactions
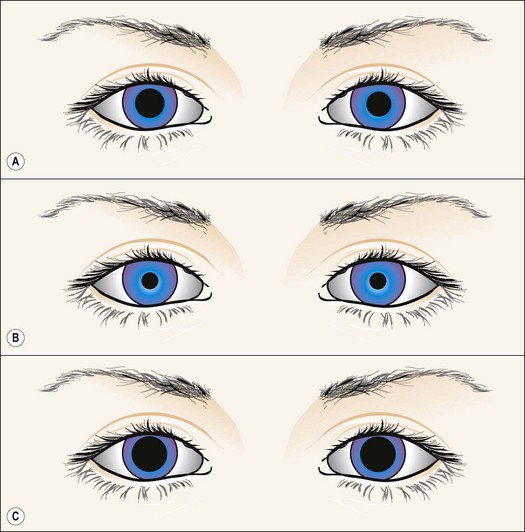
Fig. 19.39 Right physiological anisocoria
From Kanski JJ, Signs in Ophthalmology: Causes and Differential Diagnosis. Mosby 2010
Fig. 19.40 Right pharmacological mydriasis
From Kanski JJ, Signs in Ophthalmology: Causes and Differential Diagnosis. Mosby 2010
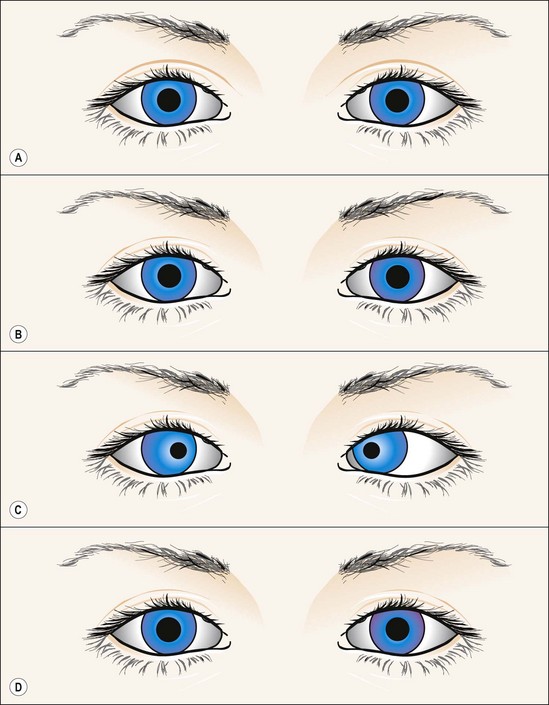
Fig. 19.41 Argyll Robertson pupils
From Kanski JJ, Signs in Ophthalmology: Causes and Differential Diagnosis. Mosby 2010
The pupils do not dilate well in the dark but atropine or cocaine induces mydriasis unless extensive iris atrophy is present.
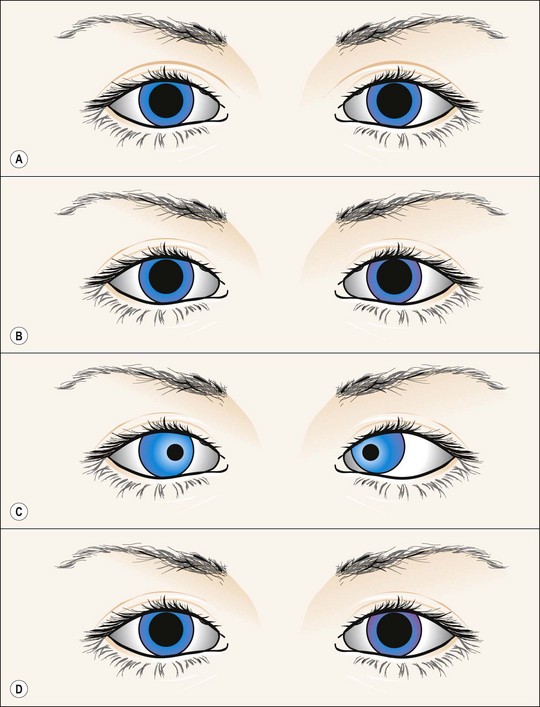
Fig. 19.42 Tectal (dorsal midbrain) pupils
From Kanski JJ, Signs in Ophthalmology: Causes and Differential Diagnosis. Mosby 2010
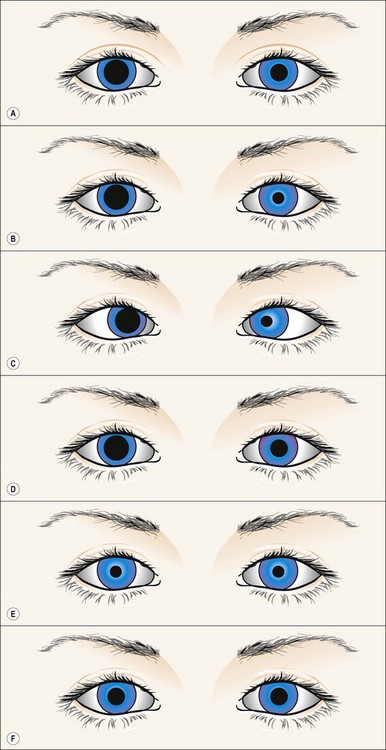
Fig. 19.43 Right episodic mydriasis
From Kanski JJ, Signs in Ophthalmology: Causes and Differential Diagnosis. Mosby 2010
Light-near dissociation
In this condition the light reflex is absent or sluggish but the near response is normal. The causes are shown in Table 19.4.
Table 19.4 Causes of light-near dissociation
Chiasm
Anatomy
Pituitary gland
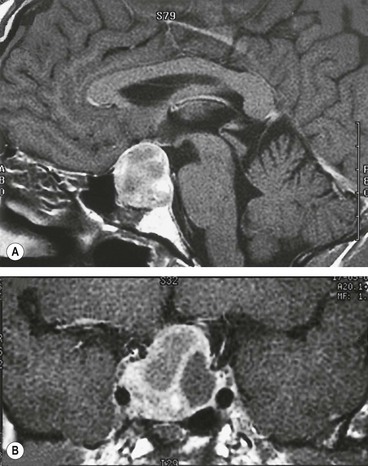
The sella turcica (Turkish saddle) is a deep saddle-shaped depression in the superior surface of the body of the sphenoid bone in which the pituitary gland lies (Fig. 19.44). The roof of the sella is formed by a fold of dura mater which stretches from the anterior to the posterior clinoids (diaphragma sellae). The optic nerves and chiasm lie above the diaphragma sellae; a visual field defect in a patient with a pituitary tumour therefore indicates suprasellar extension. Tumours less than 10 mm in diameter (microadenomas) often remain intrasellar, whereas those larger than 10 mm (macroadenomas) tend to manifest extrasellar extension. Posteriorly the chiasm is continuous with the optic tracts and forms the anterior wall of the 3rd ventricle.
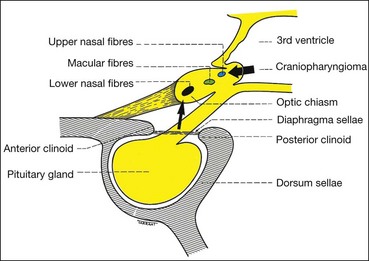
Chiasmal neural pathways
Optic nerve fibres passing through the chiasm are arranged as follows:
Anatomical variants
The following anatomical variations in the location of the chiasm may have important clinical significance (Fig. 19.45):
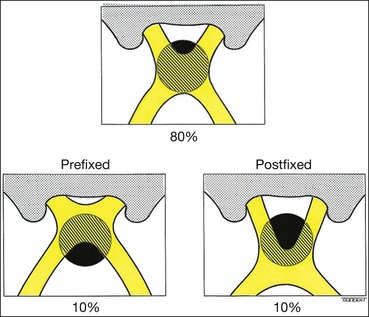
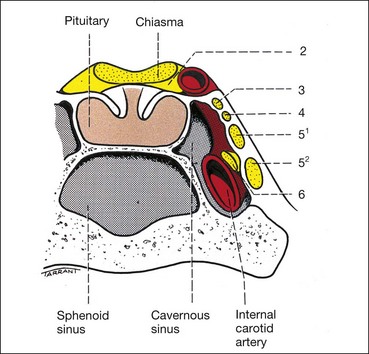
Parachiasmal vascular structures
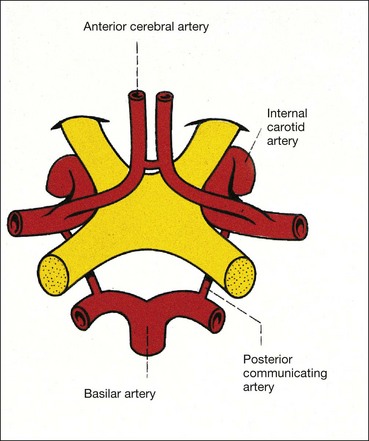
Physiology
Pituitary hormones
The lobules of the anterior part of the pituitary gland are composed of six cell types (Fig. 19.47). Five of these secrete hormones and the sixth (follicular cell) has no secretory function. The hormones secreted by the anterior pituitary gland are follicle stimulating hormone (FSH), luteinizing hormone (LH), adrenocorticotrophic hormone (ACTH), thyroid stimulating hormone (TSH), and beta-lipotrophin (C-terminal part of the ACTH precursor molecule). The posterior pituitary releases antidiuretic hormone (ADH) and oxytocin.
The anterior pituitary is itself under the control of the various inhibiting and releasing factors which are synthesized in the hypothalamus and which pass to the anterior pituitary through the hypothalamo-hypophyseal portal system.
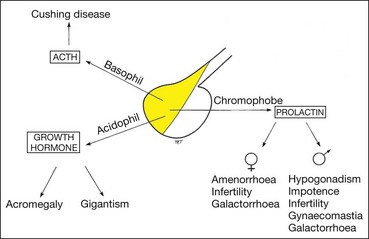
Although pituitary adenomas are classified as basophil, acidophil and chromophobe, tumours of mixed-cell type are common; any of the six cell types may proliferate to produce an adenoma.
Causes of hypopituitarism
The clinical features are dictated by both the pattern of hormone deficiency and the stage of growth and development of the patient at the time. Usually gonadotrophin secretion is impaired first, followed by that of growth hormone; deficiencies in other hormones occur later. Table 19.5 shows the causes of chiasmal disease.
Table 19.5 Causes of chiasmal disease
Pituitary adenomas
Basophil adenoma
Basophil tumours secrete ACTH and cause Cushing disease (Cushing syndrome refers to the clinical picture due to any cause of increased blood cortisol).
Acidophil adenoma
Acidophil tumours cause gigantism in children and acromegaly in adults. Acromegaly is caused by excessive growth hormone (GH) occurring during adult life, after epiphyseal closure and is almost invariably due to a secreting pituitary acidophil adenoma (hypersecretion of growth hormone in childhood, prior to epiphyseal closure, results in gigantism).
Chromophobe adenoma
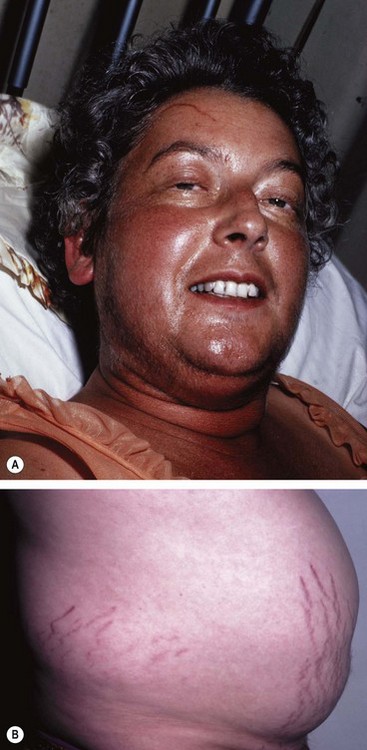
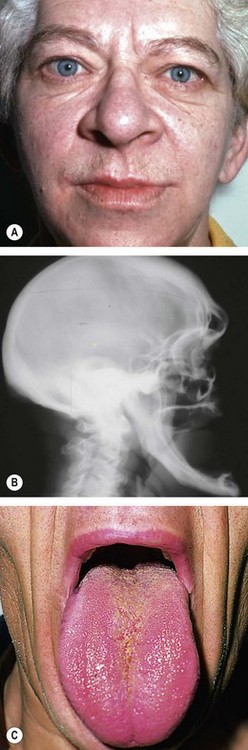
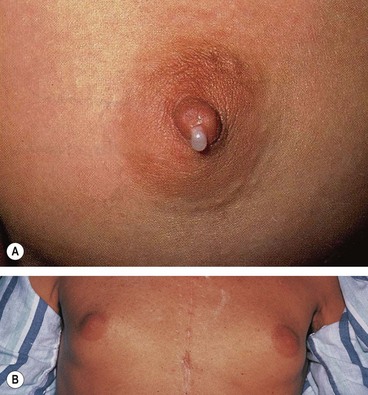
Chromophobe adenomas may secrete prolactin and are referred to as prolactinomas. In women excessive levels of prolactin lead to the infertility-amenorrhoea-galactorrhoea syndrome (Fig. 19.50A), and in men they cause hypogonadism, impotence, sterility, decreased libido, and occasionally gynaecomastia (Fig. 19.50B) and galactorrhoea. Some chromophobe adenomas are non-secreting. Chromophobe adenoma is the most common primary intracranial tumour to produce neuro-ophthalmological features. Although generally detected by endocrinologists, non-secreting tumours may first present to ophthalmologists.
Special investigations of pituitary adenomas
Treatment of pituitary adenomas
Not all tumours require treatment; observation may be appropriate for incidentally discovered and clinically silent tumours.
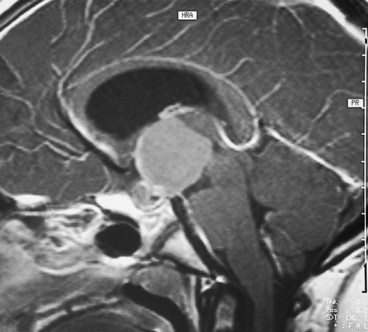
Craniopharyngioma
Craniopharyngioma is a slow-growing tumour arising from vestigial remnants of the Rathke pouch along the pituitary stalk.
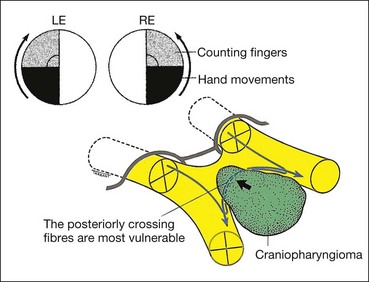
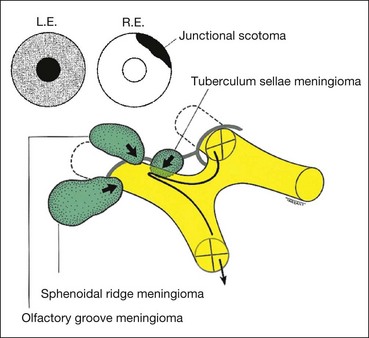
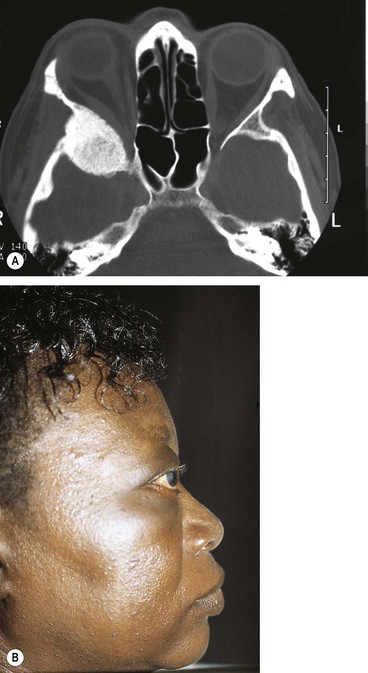
Meningioma
Intracranial meningiomas typically affect middle-aged women. Visual field defects and clinical signs depend on the location of the tumour (Fig. 19.55).
Retrochiasmal pathways
Optic tract
Overview
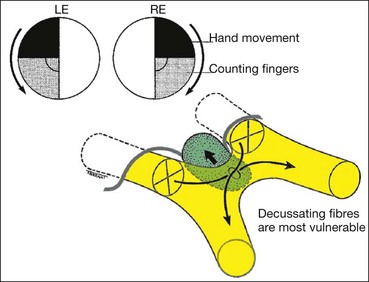
Retrochiasmal pathology results in binocular visual field defects involving contralateral visual space. Both eyes therefore manifest partial or total visual hemifield loss opposite the side of a retrochiasmal lesion. Such a ‘hemianopia’ involving the same side of visual space in both eyes is homonymous, in contradistinction to that seen in chiasmal compression, which produces heteronymous (bitemporal) hemianopia, in which opposite sides of the visual field are affected in each eye.
Incongruity
A homonymous hemianopia may be incomplete or complete. In the context of incomplete hemianopia, congruity refers to how closely the extent and pattern of field loss in one eye matches that of the other. Almost identical field defects in either eye are therefore highly congruous, while mismatching right and left visual field defects are incongruous. Hemianopia secondary to pathology in the anterior retrochiasmal visual pathways is characteristically incongruous, while that due to pathology further back (i.e. the posterior optic radiations) manifests a higher degree of congruity.
Clinical features
Optic radiations
Anatomy
The optic radiations extend from the lateral geniculate body to the striate cortex, which is located on the medial aspect of the occipital lobe, above and below the calcarine fissure (Fig. 19.57). As the radiations pass posteriorly, fibres from corresponding retinal elements lie progressively closer together. For this reason, incomplete hemianopia caused by lesions of the posterior radiations are more congruous than those involving the anterior radiations. Because these fibres are third order neurones that originate in the lateral geniculate body, lesions of the optic radiations do not produce optic atrophy. The optic radiations and visual cortex have a dual blood supply from the middle and posterior cerebral arteries.
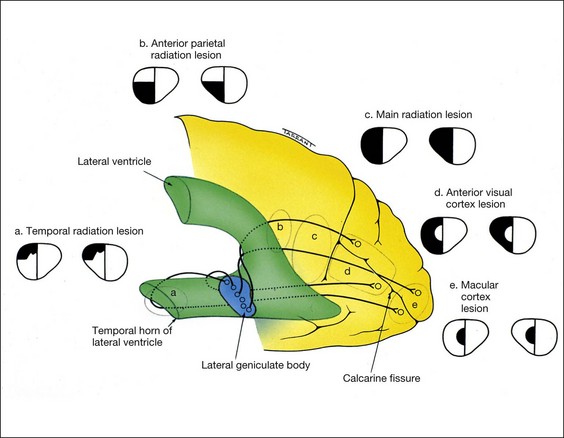
Temporal radiations
Anterior parietal radiations
Main radiations
Striate cortex
Clinical features
Ocular motor nerves
3rd nerve
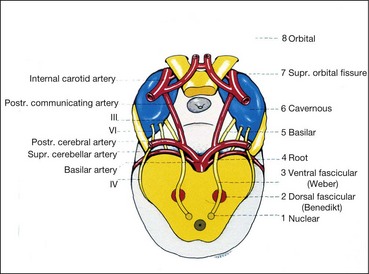
Nuclear complex
The nuclear complex of the 3rd (oculomotor) nerve is situated in the midbrain at the level of the superior colliculus, ventral to the Sylvian aqueduct (Fig. 19.58). It is composed of the following paired and unpaired subnuclei.
Fasciculus
The fasciculus consists of efferent fibres which pass from the 3rd nerve nucleus through the red nucleus and the medial aspect of the cerebral peduncle. They then emerge from the midbrain and pass into the interpeduncular space. The causes of nuclear and fascicular lesions are similar, except that demyelination may affect the fasciculus.
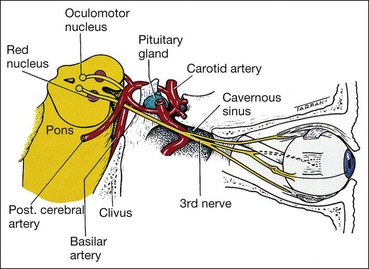
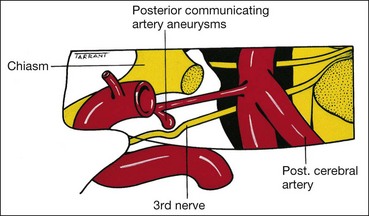
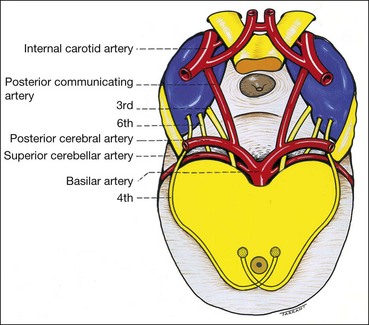
Basilar
The basilar part starts as a series of ‘rootlets’ which leave the midbrain on the medial aspect of the cerebral peduncle, before coalescing to form the main trunk. The nerve then passes between the posterior cerebral and superior cerebellar arteries, running lateral to and parallel with the posterior communicating artery (Fig. 19.59). As the nerve traverses the base of the skull along its subarachnoid course unaccompanied by any other cranial nerve, isolated 3rd nerve palsies are commonly basilar. The following two are important causes:
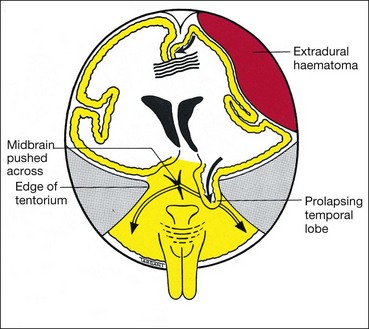
Intracavernous
The 3rd nerve then enters the cavernous sinus by piercing the dura just lateral to the posterior clinoid process. Within the cavernous sinus, the 3rd nerve runs in the lateral wall above the 4th nerve (Fig. 19.62). In the anterior part of the cavernous sinus, the nerve divides into superior and inferior branches which enter the orbit through the superior orbital fissure within the annulus of Zinn. The following are important causes of intracavernous 3rd nerve palsies:
Intraorbital
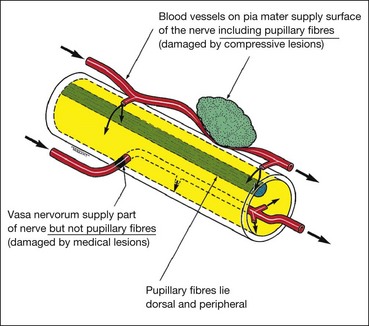
Pupillomotor fibres
Between the brainstem and the cavernous sinus, the pupillomotor parasympathetic fibres are located superficially in the superomedial part of the 3rd nerve (Fig. 19.63). They derive their blood supply from the pial blood vessels, whereas the main trunk of the 3rd nerve is supplied by the vasa nervorum. Involvement or otherwise of the pupil is of great importance because it frequently differentiates a ‘surgical’ from a ‘medical’ lesion. Pupillary involvement, like other features of 3rd nerve palsy, may be complete or partial, and may demonstrate features of recovery. Mild mydriasis and non-reactivity may therefore be clinically significant.
These principles are, however, not infallible because pupillary involvement may be seen in some diabetic-associated 3rd nerve palsies, while pupillary sparing does not invariably exclude aneurysm or some other compressive lesion. Pupillary involvement may develop a few days after onset of diplopia as an aneurysm expands. Sometimes pupillary involvement may be the only sign of 3rd nerve palsy (basal meningitis, uncal herniation).
Signs
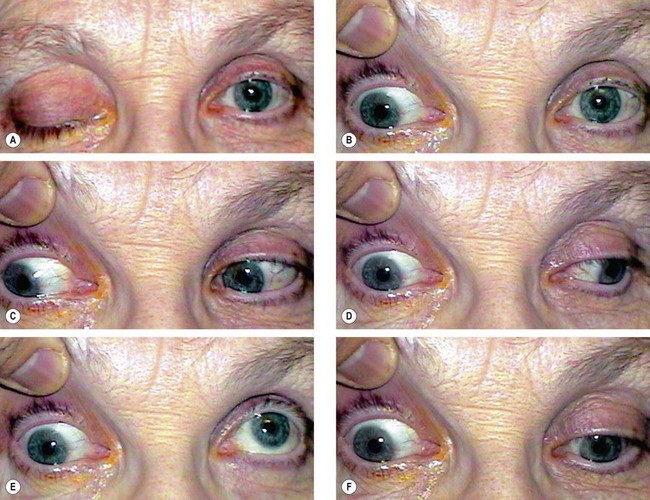
Right 3rd nerve palsy is characterized by the following (Fig. 19.64A–F):
Aberrant regeneration
Aberrant regeneration may follow acute traumatic and compressive, but not vascular, 3rd nerve palsies. This is because the endoneural nerve sheaths, which may be breached in traumatic and compressive lesions, remain intact in vascular pathology. Bizarre defects in ocular motility such as elevation of the upper eyelid on attempted adduction or depression (the pseudo-Graefe phenomenon), are caused by misdirection of regenerating axons which reinnervate the wrong extraocular muscle. The pupil may also be involved.
Causes of isolated 3rd nerve palsy
Treatment
4th nerve
Anatomy
Signs
Acute onset of vertical diplopia in the absence of ptosis, combined with a characteristic head posture, strongly suggests 4th nerve disease. The features of nuclear, fascicular and peripheral 4th nerve palsies are clinically identical, except that nuclear palsies produce contralateral superior oblique weakness.
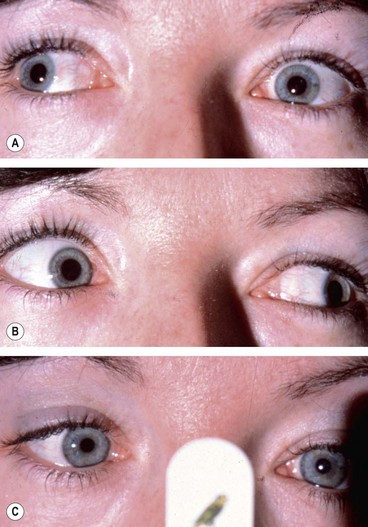
Left 4th nerve palsy is characterized by the following (Fig. 19.66A–F):
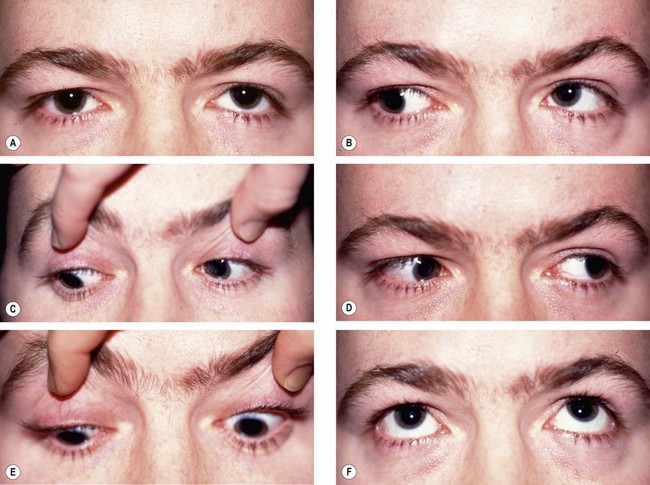
Fig. 19.66 Left 4th nerve palsy. (A) Left hypertropia (left-over-right) in the primary position; (B) Increase in left hypertropia on right gaze due to left inferior oblique overaction; (C) limitation of left depression on adduction; left limitation of depression in adduction; (D) normal left abduction; (E) normal left depression; (F) normal left elevation
Abnormal head posture (Fig. 19.67) avoids diplopia which is vertical, torsional and worse on looking down.

Bilateral involvement
Bilateral involvement should always be suspected until proved otherwise. It is characterized by the following:
Special tests
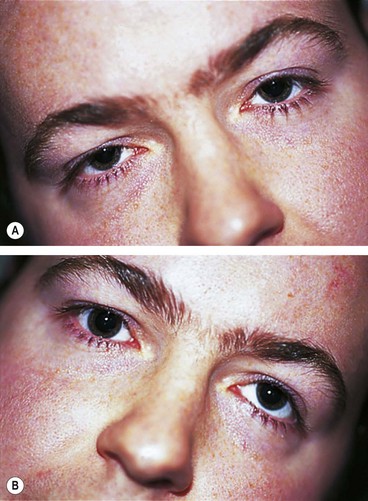
Causes of isolated 4th nerve palsy
6th nerve
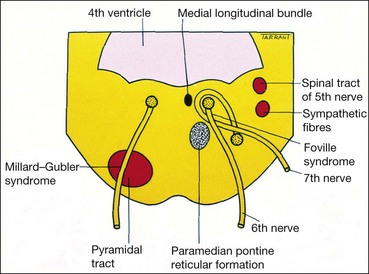
Nucleus
The nucleus of the 6th (abducens) nerve lies at the mid-level of the pons, ventral to the floor of the 4th ventricle, where it is closely related to the horizontal gaze centre. The fasciculus of the 7th nerve curves around the abducent nucleus and produces an elevation in the floor of the 4th ventricle (facial colliculus; Fig. 19.69). Isolated 6th nerve palsy is therefore never nuclear in origin. A lesion in and around the 6th nerve nucleus causes the following signs:
Fasciculus
The fasciculus passes ventrally to leave the brainstem at the pontomedullary junction, just lateral to the pyramidal prominence.
Basilar
The basilar part enters the prepontine basilar cistern. It then passes upwards close to the base of the skull and is crossed by the anterior inferior cerebellar artery (Fig. 19.70). It pierces the dura below the posterior clinoids and angles forwards over the tip of the petrous bone, passing through or around the inferior petrosal sinus, through the Dorello canal (under the petroclinoid ligament), to enter the cavernous sinus. The following are important causes of damage to the basilar part of the nerve:
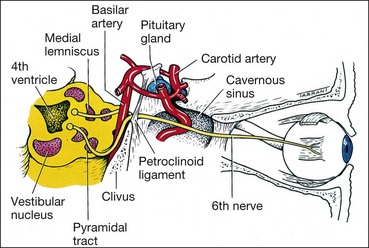
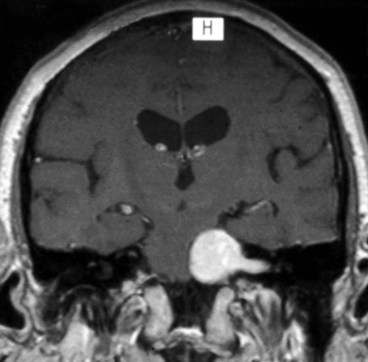
Intracavernous and intraorbital
Signs
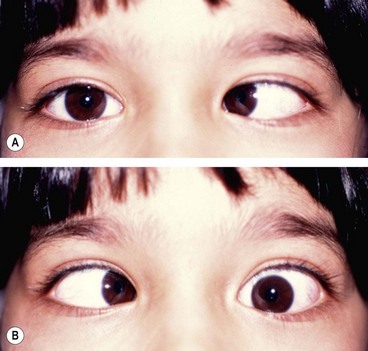
Fig. 19.72 Acute left 6th nerve palsy. (A) Left esotropia in the primary position; (B) marked limitation of left abduction
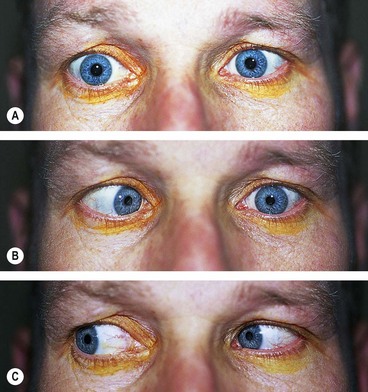
Fig. 19.73 Long-standing left 6th nerve palsy. (A) Slight left esotropia in the primary position; (B) limitation of left abduction; (C) normal left adduction
Compensatory face turn is into the field of action of the paralyzed muscle (i.e. to the left) to minimize diplopia, so that the eyes do not need to look towards the field of action of the paralyzed muscle (i.e. to the left).
Differential diagnosis
The following conditions may mimic 6th nerve palsy.
Supranuclear disorders of ocular motility
Conjugate eye movements
Conjugate eye movements or ‘versions’ are binocular movements in which the eyes move synchronously and symmetrically in the same direction. The three main types are: (a) saccadic, (b) smooth pursuit and (c) non-optical reflex. Saccadic and pursuit movements are controlled at both cerebral and brainstem levels. Supranuclear disturbances produce gaze palsies, characterized by absence of diplopia and normal vestibulo-ocular reflexes (e.g. oculocephalic movements and caloric stimulation).
Saccadic movements
Smooth pursuit movements
Horizontal gaze palsy
Anatomy
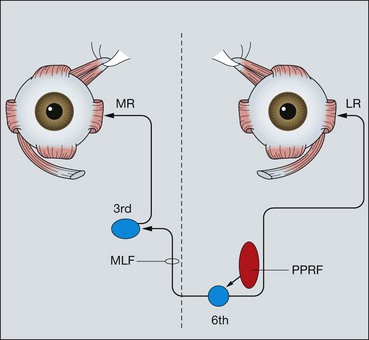
Fig. 19.74 Anatomical pathways for horizontal eye movements (PPRF = paramedian pontine reticular formation; MLF = medial longitudinal fasciculus; MR = medial rectus; LR = lateral rectus)
Table 19.6 Causes of internuclear ophthalmoplegia
Signs
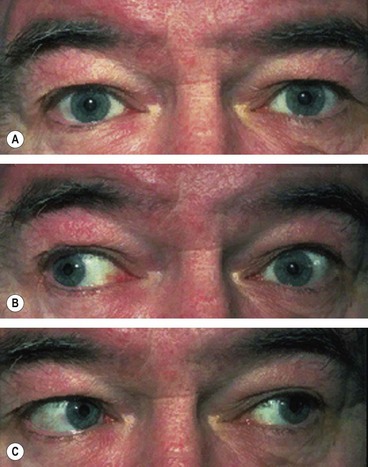
Fig. 19.75 Left internuclear ophthalmoplegia. (A) Straight in the primary position; (B) limitation of left adduction on right gaze; (C) normal left abduction on left gaze
Convergence is intact if the lesion is discrete; this may help to differentiate INO from myasthenia. Vertical nystagmus occurs on attempted upgaze.
Vertical gaze palsy
Anatomy
Vertical eye movements are generated from the vertical gaze centre (rostral interstitial nucleus of the MLF) which lies in the midbrain just dorsal to the red nucleus. From the vertical gaze centre, impulses pass to the subnuclei of the eye muscles controlling vertical gaze in both eyes. Cells mediating upward and downward eye movements are intermingled in the vertical gaze centre, although selective paralysis of upgaze and downgaze may occur in spite of this.
Parinaud (dorsal midbrain) syndrome
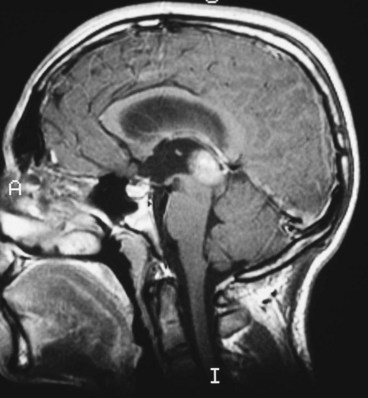
Nystagmus
Introduction
Physiological principles
Classification
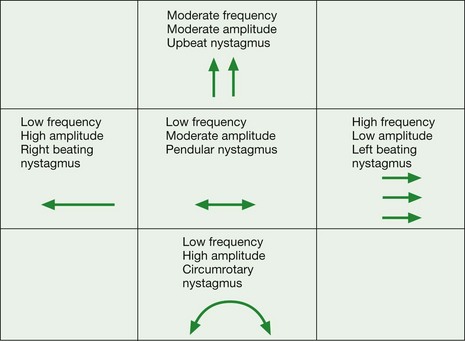
The characteristics of any form of nystagmus can be documented using the scheme shown in Figure 19.79.
Physiological nystagmus
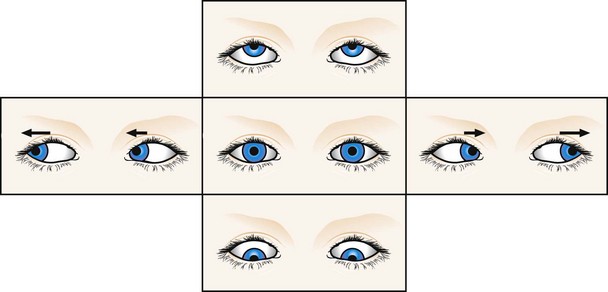
Vestibular nystagmus
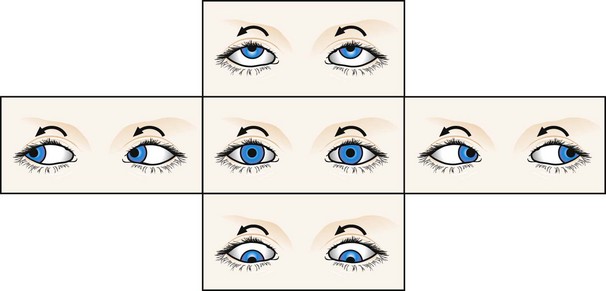
Motor imbalance nystagmus
Motor imbalance nystagmus is the result of primary defects in the efferent mechanisms.
Primary congenital nystagmus
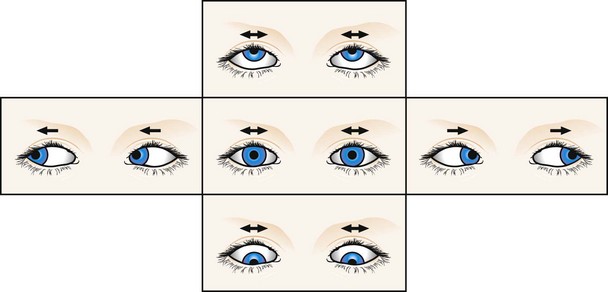
Fig. 19.82 Primary congenital nystagmus
From Kanski JJ, Signs in Ophthalmology: Causes and Differential Diagnosis. Mosby 2010
Adults with congenital forms of nystagmus do not notice oscillopsia but it is noticed by adults with acquired nystagmus.
Spasmas nutans
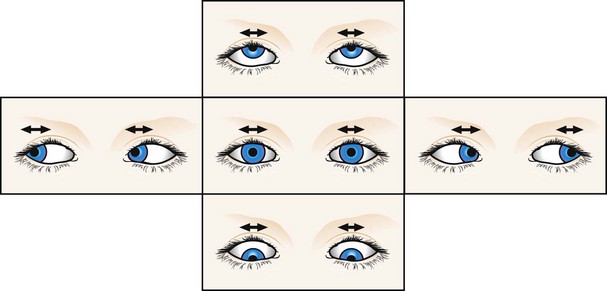
Latent nystagmus
Latent nystagmus is associated with infantile esotropia and dissociated vertical deviation (see Ch. 18). It is characterized by the following:
Periodic alternating nystagmus
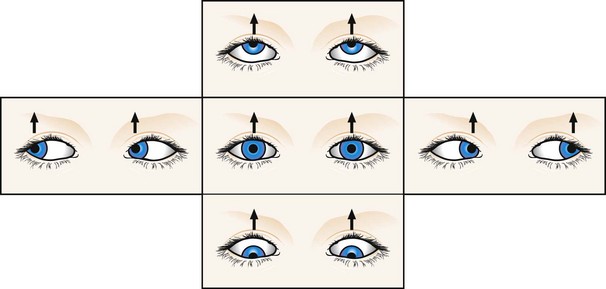
Convergence-retraction nystagmus
Convergence-retraction nystagmus is the result of co-contraction of the extraocular muscles, particularly the medial recti.
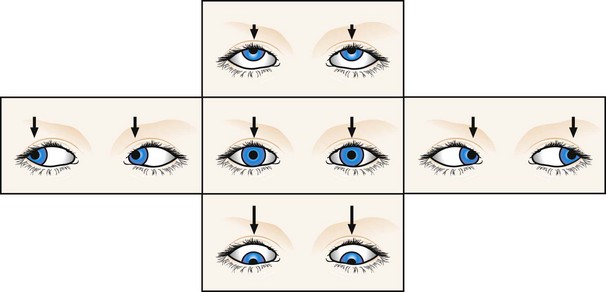
Downbeat nystagmus
See-saw nystagmus
Sensory deprivation nystagmus
Sensory deprivation (ocular) nystagmus is caused by defective vision. Horizontal and pendular, it can often be dampened by convergence. The severity depends on the degree of visual loss. An abnormal head posture may be adopted to decrease the amplitude of the nystagmus. It is caused by severe impairment of central vision in early life (e.g. congenital cataract, macular hypoplasia). In general, children who sustain bilateral loss of central vision before the age of 2 years develop nystagmus.
Surgery for nystagmus
Surgery for nystagmus may be considered if there is an abnormal head posture with a null position or for congenital motor/sensory nystagmus without a null point.
Carotid stenosis
Carotid stenosis involves atheromatous narrowing, often associated with ulceration, at the bifurcation of the common carotid artery. The irregularity of the vessel wall may act as a source of cerebral and retinal emboli composed of platelets and fibrin (white emboli – Fig. 19.86A) or tiny fragments of atheromatous material (Hollenhorst plaques – Fig. 19.86B).
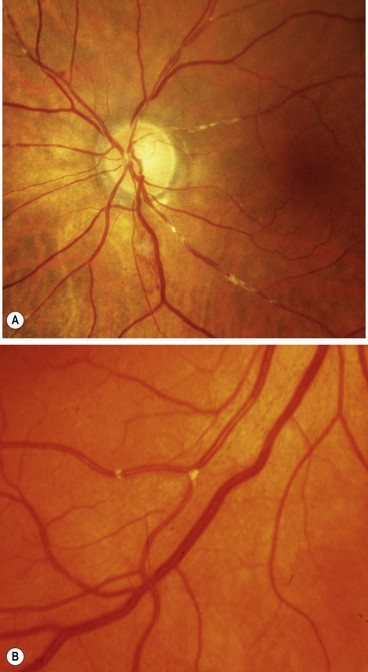
Diagnosis
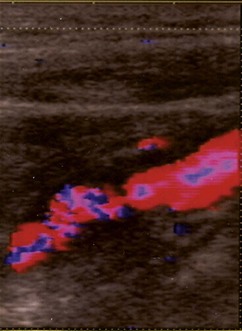
Treatment
Treatment is aimed at preventing stroke and permanent visual impairment by the following:
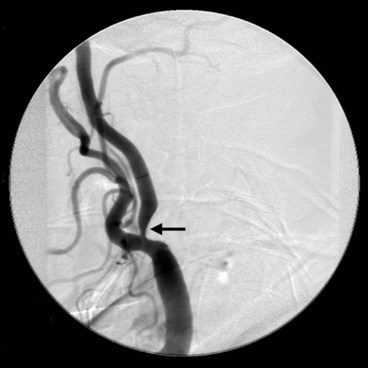
Intracranial aneurysms
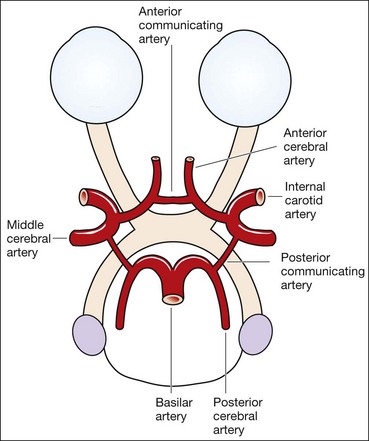
Anatomy
The arterial supply to the brain comes from the internal carotid and vertebral arteries.
Neurological considerations
Intracranial aneurysms are saccular arterial outpouchings that most commonly develop at the branching points of the major arteries coursing through the subarachnoid space at the base of the brain. Eighty-five percent arise from the anterior half of the circle of Willis. Their prevalence ranges from 1–6% among adults in large autopsy series. Aneurysms are multiple (usually two or three) in about 25% of cases. The majority remain asymptomatic during life although occasionally they may cause the following life-threatening complications:
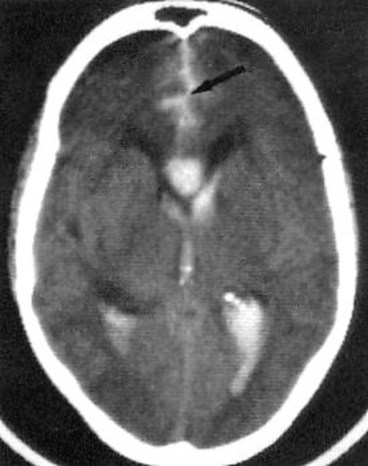
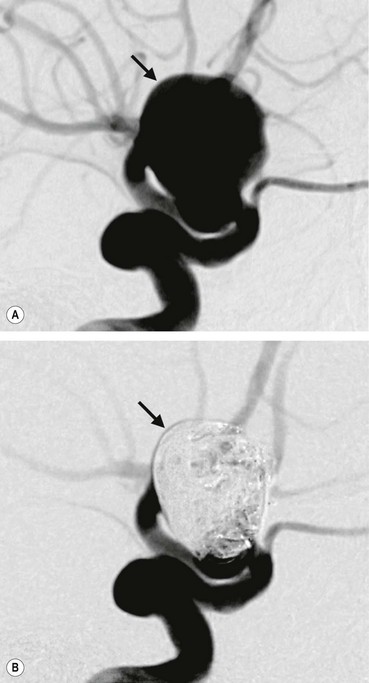
Neuro-ophthalmic considerations
Ocular motor nerve palsies
Visual loss
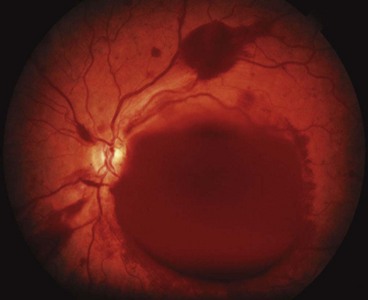
Terson syndrome
Ocular myopathies
Myasthenia gravis
Myasthenia gravis is an autoimmune disease in which antibodies mediate damage and destruction of acetylcholine receptors in striated muscle. The resultant impairment of neuromuscular conduction causes weakness and fatigability of skeletal musculature, but not of cardiac and involuntary muscles. The disease affects females twice as commonly as males. Myasthenia may be (a) ocular, (b) bulbar or (c) generalized.
Systemic myasthenia
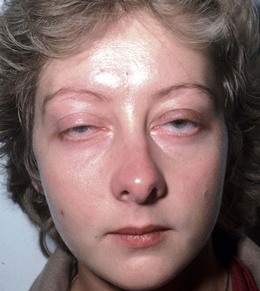
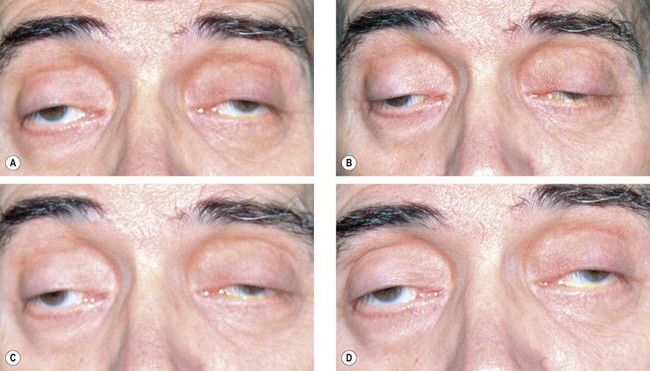
Ocular myasthenia
Ocular involvement occurs in 90% of cases and is the presenting feature in 60%. Two-thirds of patients have both ptosis and diplopia. Less than 10% of patients have ptosis alone and less than 30% have diplopia alone.
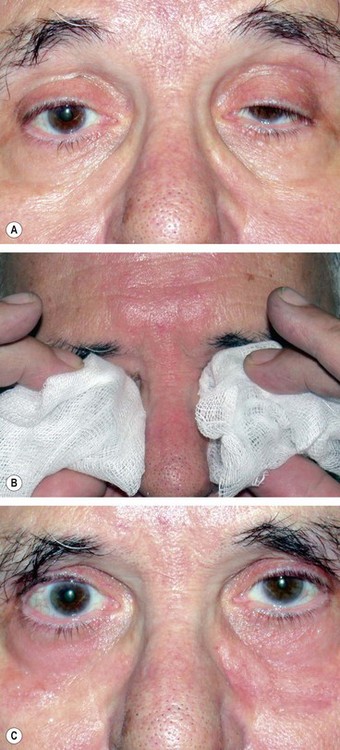
Edrophonium test
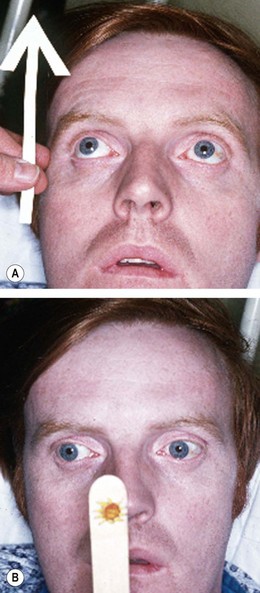
Edrophonium is a short-acting anticholinesterase agent which increases the amount of acetylcholine available at the neuromuscular junction. In myasthenia this results in transient improvement of symptoms and signs. The estimated sensitivity is 85% in ocular and 95% in systemic myasthenia. Potential but uncommon complications include bradycardia, loss of consciousness and even death. The test should therefore never be performed without an assistant, and a resuscitation trolley should also be close at hand in case of sudden cardiorespiratory arrest. The test is performed as follows (Fig. 19.95):
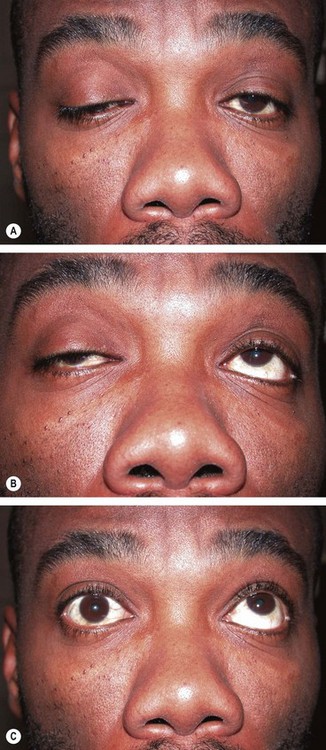
Myotonic dystrophy
Myotonic dystrophy (dystrophia myotonica) is characterized by delayed muscular relaxation after cessation of voluntary effort (myotonia). There are two forms: the classic form, dystrophia myotonica 1 (DM1), is caused by a mutation in the dystrophia myotonica protein kinase gene DMPK on chromosome 19q13. DM2 (proximal muscle myopathy, PROMM) involves the gene ZNF9 on 3q; DM2 has fewer systemic features (although cataract is common) and a better long-term prognosis. The following refers to DM1:
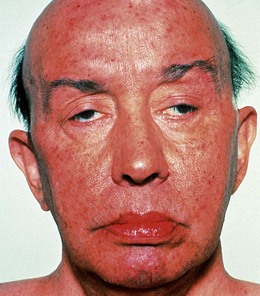
Chronic progressive external ophthalmoplegia
Chronic progressive external ophthalmoplegia (CPEO) refers to a group of disorders characterized by ptosis and slowly progressive bilateral ocular immobility. The condition may occur in isolation or in association with the Kearns–Sayre syndrome or oculopharyngeal dystrophy.
Signs
Kearns–Sayre syndrome
Eaton–Lambert myasthenic syndrome
Neurofibromatosis
Neurofibromatosis type 1
Neurofibromatosis is a disorder that primarily affects cell growth of neural tissues. Inheritance is AD with irregular penetrance and variable expressivity. The mutation rate is high. The two main types are: (a) type 1 (NF1) and (b) type 2 (NF2). Both may show segmental involvement in which the features are confined to one or more body segments. NF1 (von Recklinghausen disease) is the most common phacomatosis affecting 1 : 4000 individuals. Inheritance is AD with irregular penetrance and variable expressivity with the gene locus on 17q11, though about 50% have new mutations.
Diagnostic criteria
Two or more of the following must be present:
Systemic features
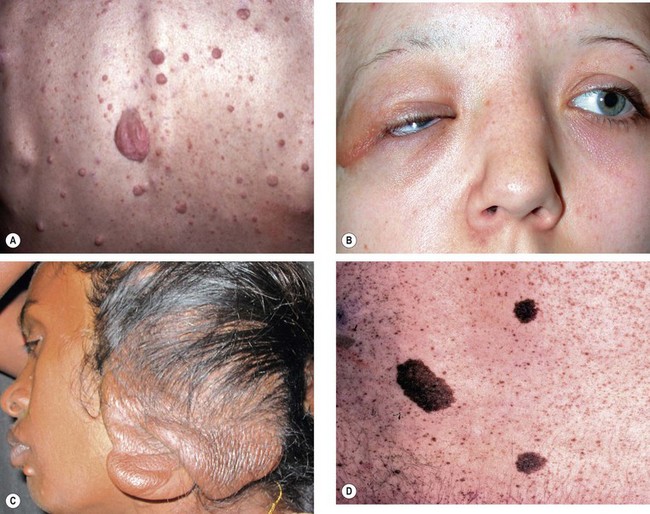
Ophthalmic features
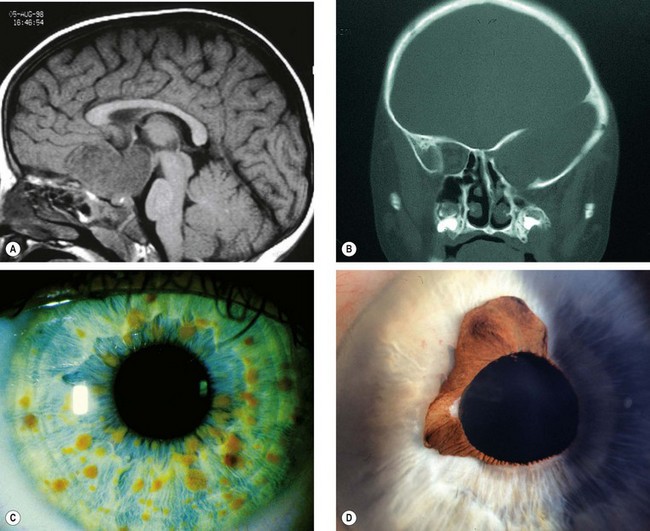
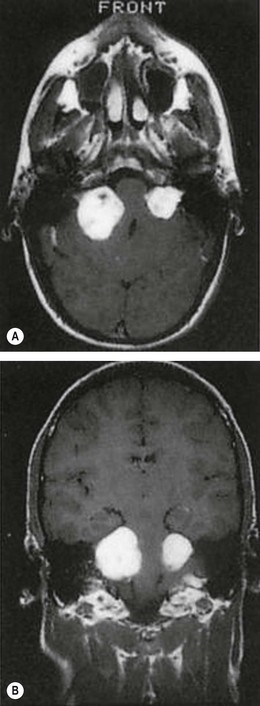
Fig. 19.101 Ocular features of NF1. (A) Sagittal T1-weighted MR image shows optic nerve glioma invading the hypothalamus; (B) coronal CT image shows absence of the greater wing of the left sphenoid bone; (C) Lisch nodules; (D) congenital ectropion uveae
(Courtesy of D Armstrong – fig. A; K Nischal – fig. B)
Neurofibromatosis type 2
Neurofibromatosis 2 (NF2) is less common than NF1. Inheritance is AD with the gene locus on 22q12.
Diagnostic criteria
Ophthalmic features
The following ocular lesions are often the first signs of the disease and may therefore assist in pre-symptomatic diagnosis:
Migraine
Clinical features
Migraine is often a familial disorder, more prevalent in females, characterized by recurrent attacks of headache widely variable in intensity, duration and frequency. The headache is commonly unilateral, associated with nausea and vomiting and may be preceded by, or associated with, neurological and mood disturbances. However, all these characteristics are not necessarily present during each attack or in every patient. The main types of migraine are discussed below.
Common migraine
Common migraine (migraine without aura) is characterized by headache with autonomic nervous system dysfunction (e.g. pallor and nausea), but without stereotypical neurological or ophthalmic features as in classical migraine (see later).
Classical migraine
Classical migraine (migraine with aura) is less common but better recognized.

Cluster headache
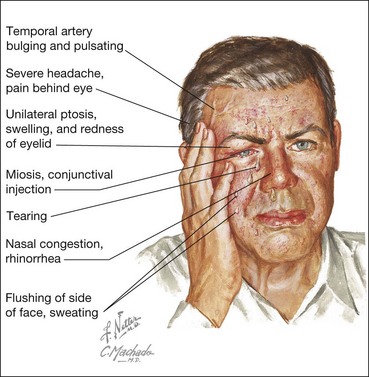
Cluster headache (migrainous neuralgia) is a migraine variant which typically affects men during the 4th–5th decades. It is of particular interest to ophthalmologists because it is associated with ocular features and may initially be misdiagnosed as a local ocular problem. The condition is characterized by a stereotyped headache accompanied by various autonomic phenomena occurring almost every day for a period of some weeks (Fig. 19.104).
Other types of migraine
Treatment
Differential diagnosis
Visual phenomena
The visual phenomena of migraine are typically binocular, zig-zag, scintillating and migrate within the visual field. This is often associated with a scotoma and/or homonymous visual loss. A patient may often report loss of vision only in the eye ipsilateral to the hemianopic symptoms. The following conditions should be considered in the differential diagnosis:
Neuralgias
The following conditions should be considered in the differential diagnosis of ocular or periocular pain in the absence of apparent physical disease:
Facial spasm
Essential blepharospasm
Clinical features
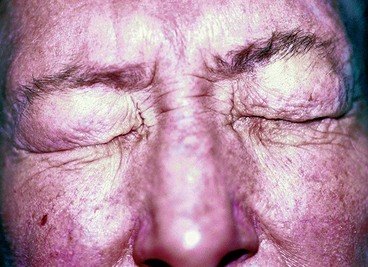
Essential blepharospasm is an uncommon but distressing idiopathic disorder which often presents in the 6th decade and affects women more commonly than men by a 3 : 1 ratio. It is characterized by progressive bilateral involuntary spasm of the orbicularis oculi and upper facial muscles. In severe cases blepharospasm is disabling because it may temporarily render the patient functionally blind (Fig. 19.105). Spasms may be precipitated by reading, driving, stress or bright light, and alleviated by talking, walking and relaxation. It does not occur during sleep.
Treatment
Prior to commencing treatment, it is important to exclude reflex blepharospasm, most commonly due to ocular surface disease such as filamentary keratitis, as well as extrapyramidal disease such as parkinsonism.
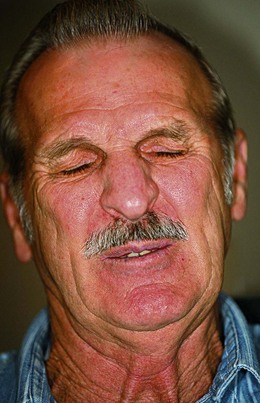
Hemifacial spasm
Hemifacial spasm is a unilateral condition that presents in the 5th–6th decades of life. It is characterized by brief spasm of the orbicularis oculi which later spreads along the distribution of the facial nerve (Fig. 19.107). The condition may be idiopathic or the result of irritation at any region from the facial nucleus to the peripheral nerve. Neuroimaging should be performed to exclude a compressive aetiology. Facial hyperkinesia may occur several months or years after a Bell palsy. Treatment is similar to that of essential blepharospasm.
