CHAPTER 19 Endodontic Pharmacology
Even in the 21st century, the terms root canal and pain often are considered synonymous. Every clinician who provides endodontic therapy has had to deal with this misperception, and the skill of the clinician is often judged primarily by their success or failure of pain control. Achieving successful endodontic pain control in a predictable and efficient manner requires a working knowledge of the underlying biology of the trigeminal pain system and the mechanisms by which available drugs and therapies provide relief. This chapter presents insights into basic science discoveries regarding acute pain and reviews the best evidence available for successful treatment of pain of pulpal and periradicular origins. It will also include a brief overview of the field of pain pharmacogenomics, which promises the possibility of predicting the effects of an analgesic drug depending on the genetic makeup of the patient. A truly inclusive review of the topic of pain control in endodontics would also include the diagnosis of odontogenic pain (see Chapters 1 and 2), the diagnosis of nonodontogenic pain (see Chapter 3), a discussion of the mediators of pulpal and periradicular inflammation (see Chapters 12 to 14), a review of the pharmacology of local anesthetics (see Chapter 20), and behavioral modification (see Chapter 26).
The Trigeminal Pain System
When activated by a stimulus sufficient to cause tissue damage or release of inflammatory mediators, nerve endings in the pulp and periradicular tissues begin to send bursts of messages to the central nervous system (CNS) that may eventually be perceived as pain. The anatomic pathway for this transmission of information has been fairly well established, and it is tempting to view the perception of pain of orofacial origin as a simple graded response to the intensity of the stimulus. However, researchers have come to realize that the pain system is a complex, multilevel system that begins with the detection of tissue-damaging stimuli in the periphery, the processing of that input at the level of the medullary spinal cord, and the perception of what is felt as pain in higher brain regions such as the cerebral cortex (Fig. 19-1). After a noxious stimulus is detected in the periphery, there is ample opportunity for a great deal of endogenous and possibly exogenous modification of the message prior to its ultimate perception. The clinician deals with all three levels of the pain system in diagnosing and treating odontalgia, and a practitioner with a basic understanding of each level will be able to recognize therapeutic opportunities and apply effective pain control methods.
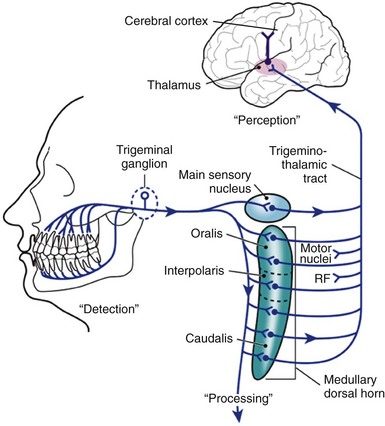
FIG. 19-1 Schematic diagram of the pathway of transmission of nociceptive information from the orofacial region. The trigeminal pain system is a complex multilevel system that begins with the DETECTION of tissue damaging stimuli in the periphery, the PROCESSING of that input at the level of the medullary spinal cord, and the final PERCEPTION of what is felt as pain in the cerebral cortex. There is growing appreciation for the concept that once a noxious stimulus is detected in the periphery, there is the opportunity for a great deal of modification of the message prior to ultimate perception.
Detection: The First Step in Pain Perception
Various types of peripheral neurons are found in the trigeminal system, including large-diameter, heavily myelinated Aα, Aβ and Aγ fibers associated with motor, proprioception, touch, pressure, and muscle spindle stretch functions. But it is the smaller, less myelinated Aδ and yet smaller and unmyelinated C fibers that conduct information likely to be perceived as pain. These two classes of pain-sensing nerve fibers, or nociceptors, are both found in the tooth pulp, but there are three to eight times more unmyelinated C fibers than Aδ fibers.22,23,113,227 It should be noted that this classification system is based purely upon the size and myelination of the neurons and does not necessarily indicate function. For example, another class of pulpal C fibers are the postganglionic sympathetic efferents found in association with blood vessels, where they regulate pulpal blood flow2,127,191,192 and may also influence the activity of peripheral nociceptors (for reviews, see Perl189 or Hargreaves91). Because most pulpal sensory fibers are nociceptive, their terminal branches are free nerve endings, and physiologic stimulation by any modality (temperature, hyperosmotic fluids) results in the perception of pure pain which can be very difficult for patients to localize. Under experimental conditions, electrical stimulation can result in a prepain sensation that is also difficult to localize. Once inflammation has extended to the periodontal ligament, which is well endowed with Aβ discriminative touch receptors, localization of pain is more predictable with light mechanical stimuli such as the percussion test.
In the normal uninflamed pulp and periradicular tissues, a noxious stimulus causes depolarization of nociceptors sufficient to generate action potentials by means of the opening of voltage-gated sodium channels (Nav). Of the nine known voltage-gated sodium channel subtypes (Nav1.1 through Nav1.9), five are believed to exist in the trigeminal system. As will be discussed in Chapter 21, they likely play an important role in the altered sensitivity of pulpal and periradicular nociceptors during inflammation, where even non-noxious stimuli can result in the perception of pain (for review see Henry and Hargreaves101).
After generation of an action potential, not only is information sent to the CNS, but in an antidromic fashion (i.e., in the reverse direction of the impulse), proinflammatory neuropeptides such as substance P (SP), calcitonin gene-related peptide (CGRP), neurokinins, and the classic neurotransmitter, glutamate, are released from afferent terminals in the inflamed pulp and periradicular tissues.
Peripheral Sensitization
Following repeated noxious stimuli, both A and polymodal C fiber nociceptors undergo a process of sensitization manifested by three obvious changes in response patterns. First, firing thresholds may decrease, so that previously non-noxious stimuli may trigger discharges, contributing to the sensation of pain (allodynia). Second, after-discharges may occur, so that noxious stimuli may produce an even greater increase in the perceived intensity of pain (hyperalgesia). And third, firing may occur spontaneously, contributing to the development of spontaneous pain. These changes are often seen in endodontic pain patients and may be explained in part by the effects of chemical mediators released into inflamed pulp and periradicular tissues. Such mediators include substances produced from damaged tissues, agents of vascular origin, and peptides released from the nerve fibers themselves (Table 19-1). Other mechanisms of peripheral sensitization are listed in Box 19-1.
TABLE 19-1 Effect of Inflammatory Mediators on Nociceptive Afferent Fibers
| Mediator | Effect on Nociceptors | Effect on Human Volunteers |
|---|---|---|
| Potassium115 | Activate | ++ |
| Protons143,212 | Activate | ++ |
| Serotonin15,115 | Activate | ++ |
| Bradykinin15,115,137 | Activate | +++ |
| Histamine115 | Activate | + |
| Tumor necrosis factor-α | Activate | ? |
| Prostaglandins16 | Sensitize | ± |
| Leukotrienes16,145 | Sensitize | ± |
| Nerve growth factor139,190 | Sensitize | ++ |
| Substance P86 | Sensitize | ± |
| Interleukin 162 | Sensitize (?) | ? |
+, Positive; ++, very positive; +++, extremely positive; ±, equivalent; ?, unknown.
Modified from Fields H: Pain, New York, 1987, McGraw-Hill.
Inflammatory Mediators
Among the best characterized of the inflammatory mediators are the prostaglandins (PGs), which are derived from arachidonic acid via the action of the cyclooxygenase (COX) enzyme systems. The human COX enzyme is known to exist in at least two forms, COX-1 and COX-2. COX-1 is constitutively expressed and produces PGs that are involved in basic housekeeping functions such as cytoprotection in the stomach, regulation of blood flow in the kidneys, and the formation of thromboxane A2. The formation of thromboxane A2 can ultimately lead to platelet aggregation; therefore, inhibition of thromboxane A2 should decrease platelet aggregation. COX-2 is inducible, synthesized in inflamed tissues (including dental pulp),170 and is important in the production of the proinflammatory PGs as well as the vasodilating prostacyclin (PGI2). Although they do not produce pain if applied alone, PGs are known to sensitize peripheral nociceptors, which increases the algogenic (pain-producing) properties of serotonin and bradykinin.50,80 The exact mechanism by which PGs increase neuronal excitability is not clear, but there is a growing body of evidence to suggest that they activate PG receptors EP2 and EP3 in the trigeminal system184 and exert their effects by regulating the activity of certain ion channels,234 including voltage-gated sodium channels (for review see England56). For example, application of prostaglandin E2 (PGE2) to isolated dorsal root ganglion neuronal somata more than doubles the responsiveness of certain sodium channels found predominantly on nociceptors—channels thought to be relatively resistant to lidocaine.8,79 When administered to rats before an inflammatory insult, ibuprofen, a nonselective COX inhibitor, has been shown to block the increased expression of Nav1.7 and Nav1.8.84,85 Therefore, if the concentrations of PGs in inflamed pulp and periradicular tissues can be decreased with nonsteroidal antiinflammatory drugs (NSAIDs) or corticosteroids, postoperative pain may be relieved and, in addition, more profound local anesthesia may be achieved in patients with hyperalgesia of pulpal origin.95,106,160 It is interesting to note that sensory neurons themselves are a source of PGs; during inflammation, the levels of PGE2 appear to increase in dorsal root ganglia and spinal cord, suggesting that NSAIDs also have a central site of action (discussed later in this chapter).148,239
Bradykinin (BK) is a proinflammatory mediator derived from circulating plasma proteins and also causes direct activation of nociceptive neurons, resulting in pain. Increased levels of BK have been demonstrated in the inflamed dental pulp,135 and the presence of growth factors associated with inflammation (e.g., nerve growth factor) have been reported to cause an increase in the expression of mRNA encoding B1 and B2 receptors in primary cultures of rat trigeminal ganglia,218 as well as other receptors such as TRPV1 and TRPA1.41,112 The transient receptor potential subtype V1 (TRPV1) is the “capsaicin receptor”; it plays a key role in mediating inflammatory pain. TRPA1 is expressed on capsaicin-sensitive neurons41 and interacts with TRPV1.200 Bradykinin likely increases the excitability of nociceptive neurons through its action on TRPV1 and TRPA1 (for review, see Tominaga et al.217).
Cytokines are a diverse group of regulatory proteins synthesized and secreted by a variety of cell types, such as leukocytes, neurons, and glia. In particular, tumor necrosis factor α (TNF-α), and the interleukins IL-1β, IL-6, and IL-8 are thought to play a role in the neuroplastic changes that occur in nociceptors innervating inflamed tissues, leading to hyperalgesia.130 Application of TNF-α rapidly sensitizes TRPV1,124 contributing to activation of the capsaicin-sensitive class of nociceptors. All of the above are thought to exist in the inflamed pulp (for review see Fouad64) and are thought to act at least in part by causing increased release of prostanoids.214
Changes in Neuronal Phenotypes
Sensory neurons innervating inflamed peripheral tissues undergo striking phenotypic changes, including changes in transduction mechanisms and plasticity of nociceptor organization. In the dental pulp, it is likely that C fibers predominate in the response to inflammation, because the quality of the pain described by patients with pulpal inflammatory disease (e.g., dull, aching, throbbing sensations) is similar to that reported by patients experiencing experimental C fiber activation.5,169,174 As described previously, certain combinations of inflammatory mediators causes both activation and sensitization of these fibers. Interestingly, when pulpal and periradicular tissues undergo inflammatory changes, C fibers innervating those tissues are found to contain increased amounts of proinflammatory neuropeptides such as SP and CGRP.19,24,26 It has also been shown that C fiber terminals in inflamed pulpal tissues undergo a process called sprouting, whereby those terminals closest to the inflamed tissues actually grow to form additional terminal endings,22-24 possibly leading to increased participation in detecting and modulating tissue responses to injury.
Another consideration in the neural response to inflammation is the possibility of a change in the distribution and activity of voltage-gated sodium channels. In particular, mice lacking the gene for Nav1.7 show reduced painlike behaviors when treated with a variety of proinflammatory agents.175 Also implicated in the altered firing characteristics of nociceptors innervating inflamed tissues are the sodium channels that are resistant to tetrodotoxin (TTX), the biotoxin found in the tetraodon pufferfish. The two main TTX-R sodium channels are Nav1.8 and Nav1.9, and both have been shown to be increased two- to fourfold in inflamed dental pulp collected from patients with a diagnosis of irreversible pulpitis.235,237 When exposed to PGE2, neurons isolated from dorsal root ganglia cells have been shown to increase TTX-resistant sodium channel currents within minutes,79 indicating an increased activation of existing channels, rather than de novo protein synthesis. Interestingly, these sodium channels are relatively resistant to lidocaine,199 and this may explain the difficulty in achieving profound anesthesia in inflamed tissues (see also Chapter 20).95
Signs and Symptoms of Pulpalgia and Acute Periradicular Periodontitis
Again, the hallmarks of pain perception due to acute inflammation are allodynia, hyperalgesia, and spontaneous pain. The peripheral mechanisms for these symptoms include a decrease in firing threshold, an increase in responsiveness to noxious stimuli, and development of spontaneous discharges of nociceptors. All three of these characteristics can be seen in patients experiencing inflammatory pain of pulpal origin (Table 19-2). From this perspective, the histology of the pulp would not be expected to correlate with the patient’s symptoms. Simply put, inflammatory pain is the result of molecules interacting with nociceptors, an event that cannot be detected by conventional histologic methods.
TABLE 19-2 Signs of Hyperalgesia and Allodynia and Endodontic Diagnostic Tests
| Signs of Hyperalgesia | Related Diagnostic Tests or Symptoms |
|---|---|
| Spontaneous pain | Spontaneous pain |
| Reduced pain threshold | Percussion test, palpation test, throbbing pain |
| Increased response to painful stimuli | Increased response to pulp test (EPT or thermal test) |
EPT, Electric pulp test.
From Hargreaves KM, Swift JQ, Roszkowski MT, et al: Pharmacology of peripheral neuropeptide and inflammatory mediator release, Oral Surg Oral Med Oral Pathol 78:503–510, 1994.
Thermal allodynia is the term that best describes a patient whose chief complaint is “I have pain when I drink cold beverages.” Mechanical allodynia is involved when the chief complaint is “It now hurts when I bite on this tooth.” These previously non-noxious stimuli now cause the perception of pain. Hyperalgesia is manifested in endodontic pain patients when noxious stimuli (e.g., refrigerant sprays or carbon dioxide snow used in the cold test) produce much more pain than they would in teeth with normal pulp tissues. Spontaneous pain involves episodes of pain that seem to be unprovoked. All these changes can be partly explained by sensitization of peripheral nerve endings in the pulp and periradicular tissues.
Analgesic Drugs With the Potential to Act in the Periphery
Pain control can be effected by endogenous mechanisms in the periphery. For example, immune cells in inflamed peripheral tissues may release opioids that act on receptors found on sensory neurons.213 Several drug classes (discussed later in this chapter) also have the potential for reducing the production of agents that serve to cause pain directly or sensitize peripheral nociceptors. These drugs include NSAIDs,104,105 corticosteroids,69 and opiates.47
Processing: The Second Step in Pain Perception
The Medullary Dorsal Horn
After activation of peripheral nociceptors, nerve impulses in the form of action potentials convey information about the intensity (encoded by firing frequency), quality (encoded by type of neuron activated), and temporal features (encoded by onset, duration, and offset of depolarization) of the peripheral stimuli to the CNS. In the trigeminal pain system, these action potentials arrive at the trigeminal spinal tract nuclear complex located in the medulla.94,141,204 Three distinct subnuclei can be found in this complex. Named for their anatomic position, they are the subnuclei oralis, interpolaris, and caudalis (see Fig. 19-1). Although the more rostral subnuclei (oralis and interpolaris) receive some nociceptive input from oral tissues,39 most such input is received at the level of the subnucleus caudalis.57,146,206 Because of its organizational similarity to the dorsal horn of the spinal cord (which receives nociceptive input from the somatosensory system), the subnucleus caudalis has been termed the medullary dorsal horn.
Components of the Medullary Dorsal Horn
The medullary dorsal horn relays information to higher centers in the brain and serves as the site of much potential processing of the signals from primary afferent sensory nerve fibers. Output from this region can be increased (hyperalgesia), decreased (analgesia), or misinterpreted (referred pain). Understanding the functional components involved in such processing not only helps explain some of these clinical phenomena but also allows evaluation of potential therapeutic modalities currently under investigation. Functional components include the central terminals of primary nociceptors (Aδ and C fiber afferents), the second-order projecting neurons, interneurons, the terminals of descending neurons, and glial152 cells (for review see Hargreaves91).
Primary afferent fibers (whose cell bodies are located in the trigeminal ganglion) transmit signals to projection neurons via the release of transmitters such as the excitatory amino acid, glutamate, and the neuropeptide, substance P. Receptors for these neurotransmitters are found on postsynaptic membranes and include the NMDA and AMPA classes of glutamate receptors and the neurokinin 1 (NK1) class of substance P receptors. Antagonists to these receptors have been shown to reduce hyperalgesia in animal studies.29 In a human clinical trial using an oral surgery model, the AMPA/kainate antagonist, LY293558, was shown to be antihyperalgesic.75 NK1 antagonists have shown promising results in animal studies, but in general, they have displayed limited analgesic efficacy in humans.103
The cell bodies of the second-order (projection) neurons in the trigeminal pain system are found in the medullary dorsal horn; their processes cross the midline and project rostrally to the thalamus via the trigeminothalamic tract (Fig. 19-2). From the thalamus, third-order neurons relay information to the cerebral cortex via a thalamocortical tract. Once signals have reached the cortex, the input may be perceived as pain. Evidence exists that referred pain is caused by convergence of afferent input from different areas onto the same projection neurons.
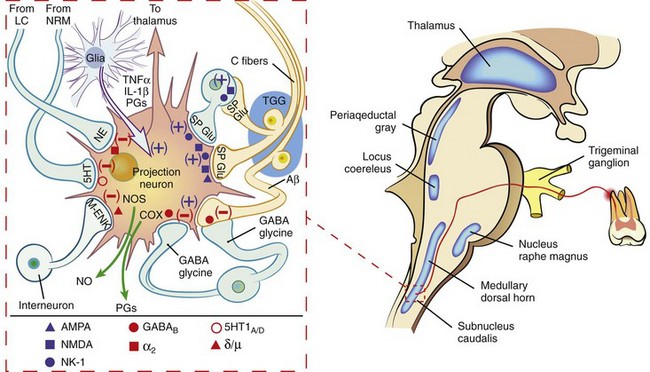
FIG. 19-2 Schematic diagram of the perception and modulation of orofacial pain. Activation of primary afferent fibers (in this example from an inflamed maxillary molar) leads to the entry of a nociceptive signal that is conveyed across a synapse in the subnucleus caudalis of the trigeminal spinal nucleus. The second-order neuron projects to the thalamus; the information is then relayed to the cortex. A great deal of processing of nociceptive input can occur at the level of the medullary dorsal horn (MDH). The inset depicts a typical wide dynamic range (WDR) projection neuron and its relationship with other components of the MDH. Primary afferent fibers release the excitatory amino acid, glutamate—which binds and activates either AMPA or NMDA receptors—and substance P—which activates NK-1 receptors on the WDR neuron or excitatory interneurons. Descending fibers from the locus coeruleus (LN) and nucleus raphe magnus (NRM) secrete serotonin (5HT) and norepinephrine (NE), respectively, which inhibit transmission. Release of γ amino butyric acid (GABA), the amino acid, glycine, and endogenous opioid peptides such as metenkephalin (M-ENK) also inhibit transmission of nociceptive information. Projection neurons may have autocrine and/or paracrine effects by the synthesis and release of prostaglandins (PGs) and nitric oxide (NO) via the action of cyclooxygenase (COX) and nitric oxide synthase (NOS), respectively. Glial cells can modulate nociceptive processing by the release of cytokines such as tumor necrosis factor alpha (TNF-α), and interleukin 1 beta (IL-1β). The + sign indicates an excitatory action, whereas the − sign denotes an inhibitory action.
Approximately 50% of subnucleus caudalis neurons are estimated to receive convergence of sensory input from cutaneous and deep structures.206 In one study of a cat, a single nucleus caudalis neuron received input from sensory neurons innervating the cornea, the skin overlying the maxilla, a maxillary premolar tooth, and a mandibular canine and premolar tooth on one side.208 Subnuclei oralis and interpolaris also receive converging input from orofacial and muscle afferents.207 This would explain the clinical observation of patients who perceive pain in a particular tooth that actually originates from either a different tooth or structure (see also Chapter 3). In such cases, anesthetizing the tooth suspected by the patient would afford no relief. However, if an anesthetic is delivered selectively to the suspected primary source of pain, the patient’s discomfort should be greatly diminished.180 Likewise, if the source of a perceived toothache were located in a muscle of mastication, palpation of that muscle should aggravate the pain.247
In the medullary dorsal horn, local circuit interneurons have the potential to affect transmission of nociceptive input from primary afferents to projection neurons. Depending on the transmitter released, these neurons have the ability to enhance or diminish the signal. Typically, excitatory interneurons release glutamate and/or substance P, whereas inhibitory interneurons release the amino acid, glycine, and/or gamma amino butyric acid (GABA).141,205
The terminals of neurons that descend from brain structures such as the locus coeruleus and nucleus raphe magnus tend to inhibit nociceptive transmission at the level of the medullary dorsal horn.13 These terminals release a variety of neuroeffective agents, including the endogenous opioid peptides (EOP). The EOPs, similar in three-dimensional structure to many of the exogenous opiates from which their name derives, are released in response to nociceptive input and act to suppress the pain system. The EOPs likely are partly responsible for the placebo effect seen in pain control studies, because this effect can be reversed by administration of the opioid antagonist, naloxone.92,138
The final component of the medullary dorsal horn complex to be considered is the glial cell population. Historically considered to be solely supportive in function, they are now recognized to play an important role in the pain processing system.193,236 Following nociceptive input from primary afferents, glia release cytokines such as TNF-α and IL-1, as well as certain PGs that may facilitate the activity of projection neurons. Glial modulating agents have been shown to be effective in experimental models of neuropathic pain,216 and NSAIDs potentially could exert part of their analgesic mechanism by acting at this level.
Central Sensitization
Central sensitization can be defined as an increased responsiveness of central nociceptive neurons to peripheral stimulation that occurs in addition to peripheral sensitization of the primary afferent nociceptors. Central sensitization is thought to be a major cause of hyperalgesia and allodynia.245 Recent clinical trials implicate central sensitization in patients reporting pain due to irreversible pulpitis. In one survey of nearly 1000 patients, 57% of patients with irreversible pulpitis reported mechanical allodynia (pain due to percussion).182 This appears to be due at least in part to central sensitization, since both the ipsilateral (pulpitis) tooth and a contralateral (normal) tooth demonstrated mechanical allodynia to a force transducer.125 Thus, central sensitization contributes to endodontic pain, and the clinical application of bite force transducers may provide a novel method for diagnosing pain mechanisms.125,126
Different studies shed light on the molecular mechanisms involved in central sensitization (for review, see Cousins and Power36 and Hargreaves91), but the process is generally initiated by a barrage of nociceptive impulses from peripheral C fibers. The level and duration of pain prior to endodontic intervention has been cited in several studies as a predictor of postoperative endodontic pain,108,219,233 and this may be due to such a prolonged and intense input from C nociceptors. Any reduction of such a barrage should limit the occurrence of central sensitization and the development of pain of longer duration after tissue injury (including surgical and nonsurgical endodontic procedures). The use of long-acting local anesthetics following tonsillectomies and third molar extractions has shown to provide pain relief far beyond the duration of the peripheral tissue anesthesia.83,111
A reduction in the chemical mediators of inflammation at the level of the medullary dorsal horn also should serve to reduce sensitization of second-order neurons. Decreasing the synthesis of proinflammatory PGs, cytokines, nitric oxide, and/or the use of drugs that block the receptors of such agents probably will become accepted pharmacotherapy in the future. For example, application of an inflammatory agent to the tooth pulp of rat maxillary molars results in an increased receptive field of Aβ touch receptors on the face. This can be blocked by pretreatment with a glutamate NMDA receptor antagonist, indicating that such centrally acting drugs may offer highly efficacious means of treating odontogenic pain.29 A similar investigation implicated nitric oxide synthesis at the level of the subnucleus caudalis in the development of tactile hypersensitivity following dental injury.252 Reduction in nitric oxide synthase levels may also provide protection from central sensitization.149,157
Perception: Thalamus to Cortex
The final anatomic step in the trigeminal pain pathway relies on neurons that leave the thalamus and extend to the cerebral cortex (see Fig. 19-1). The patient actually perceives a stimulus as painful at the cortical level. It is interesting to note (but likely of no surprise to the experienced clinician) that a disproportionately large portion of the sensory cortex in humans is devoted to input from orofacial regions.186
It is becoming increasingly obvious that higher-order (i.e., cortical) perceptual processes have a profound effect on the ultimate state of pain the patient experiences (for review, see Yaksh250). Memories of previous pain experiences provide a framework by which similar new experiences are judged and serve to shape the patient’s response to a given stimulus. In the field of dentistry, the anxiety level of the patient at the time of treatment has been shown to affect not only the patient’s response to pain experienced during treatment52,241 but also the tendency of the patient to recall the experience as painful or unpleasant even 18 months posttreatment.72 The clinician should do everything possible to control a patient’s anxiety level prior to endodontic treatment (see also Chapter 26). One simple pretreatment method is to provide patients with positive written information regarding the control of pain during their endodontic treatment. In a placebo-controlled clinical trial, 437 endodontic patients were given one of five informative paragraphs to read prior to treatment. One of the paragraphs contained positive information about pain during treatment. Patients completed questionnaires following treatment that evaluated their dental anxiety and dental fear. Subjects given positive information were shown to be less fearful of pain during endodontic therapy.228 Along with a positive and caring attitude, pharmacologic intervention may help reduce anxiety. Nitrous oxide has been shown to be effective in a dental setting44 but may interfere with radiography procedures during endodontic therapy. In a placebo-controlled clinical trial in patients undergoing the extraction of impacted third molars, 0.25 mg of oral triazolam (a benzodiazepine) provided comparable anxiolysis to intravenous diazepam titrated to a typical clinical endpoint.116 Of course, the patient so medicated must be provided transportation to and from the dental office, and the potential drug-drug interactions with other centrally acting agents such as opioids, barbiturates, and alcohol must be considered. One interaction that should be considered is the capacity of grapefruit juice to prolong the half-life of triazolam.142 It has been shown that furanocoumarins in grapefruit juice inhibit cytochrome P450 3A4,183 which is the enzyme responsible for the metabolism of triazolam in the liver. Patients should be told not to take oral triazolam with grapefruit juice.
Predictors of Postoperative Endodontic Pain
Although current endodontic treatment can be virtually pain free during the procedure itself, certain patients may still experience some pain after the appointment. Early studies investigating postoperative endodontic pain have reported an incidence of moderate to severe pain in the range of 15% to 25%.30,97,179 In a prospective clinical study, 57% of patients reported no pain after débridement and shaping of the root canal system, although 21% had slight pain, 15% had moderate pain, and 7% had severe pain.74 Although some patients may experience moderate to severe pain after endodontic treatment, very few experience what is now commonly referred to as a flare-up or a postoperative problem requiring an unscheduled visit with unplanned treatment intervention to manage the patient’s symptoms.231 Patients with a flare-up usually describe severe pain, swelling, or the sensation of pressure within their mandible or maxilla within 1 or 2 days of treatment. The incidence of flare-up varies across studies and ranges from about 2% to 20% of patients, with the higher prevalence generally reported in older studies using classical cleaning and shaping techniques.12,165,224,225
Numerous studies have evaluated factors related to postoperative endodontic pain and flare-up to better predict when these conditions are more likely to occur. Table 19-3 summarizes the results of clinical endodontic studies evaluating more than 12,000 patients for predictors of postoperative pain. Differences in experimental design prevent direct comparison of studies, but interestingly, the presence of preoperative pain or mechanical allodynia (reduced mechanical pain threshold or percussion sensitivity) was a positive predictor of postoperative pain in more than 15 studies involving more than 6600 patients.
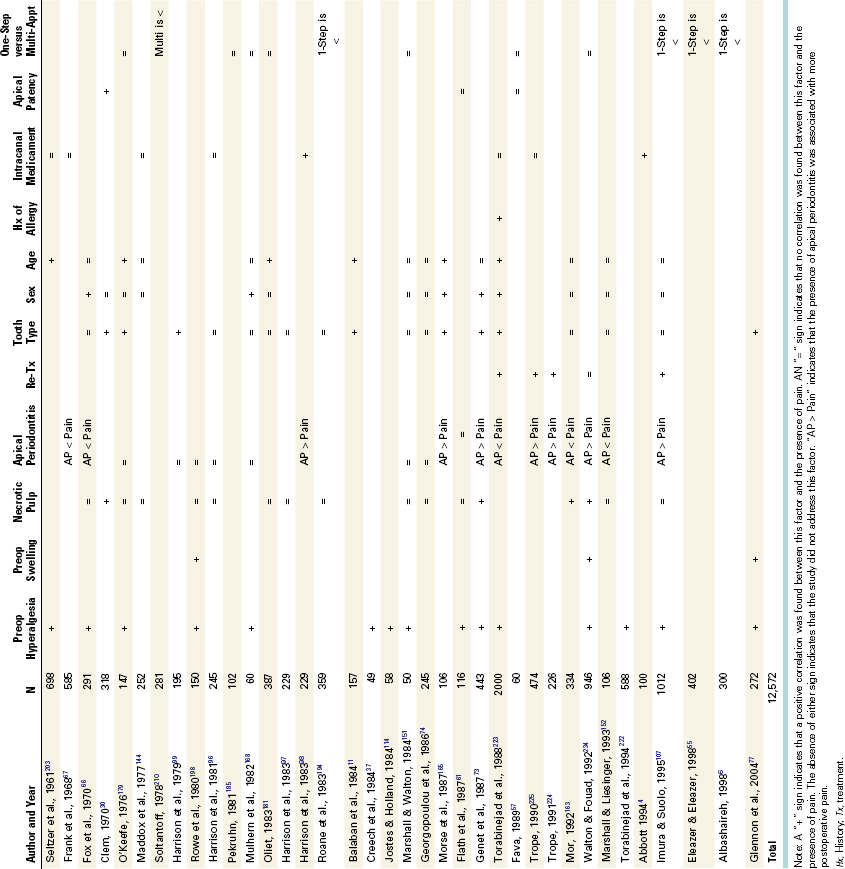
Other factors were more variable in their predictive value of postoperative pain. In a retrospective study, the dental records of 1000 patients who had received nonsurgical root canal treatment and experienced no flare-ups (i.e., unscheduled return visits) were compared with the records of 1000 patients who experienced flare-ups after the cleansing and shaping of their necrotic root canals.220 The results showed that factors such as presence of preoperative pain, tooth type, sex, age, history of allergy, and retreatment, were significantly predictive for the incidence of flare-up, although intracanal medicaments, systemic disease, and establishment of patency of the apical foramen had no significant relation to the incidence of flare-ups.220 Specifically, the highest incidences of flare-ups were associated with mandibular teeth, retreatment procedures, females over the age of 40, and patients with a history of allergies.220
In another retrospective study, the flare-up rate was evaluated in patients treated in multiple visits by undergraduate dental students.163 The incidence in this study was 4.2%, and a positive correlation was found between flare-ups and teeth with necrotic pulp. No correlation was found between the occurrence of a flare-up and the presence or absence of a periradicular radiolucency.
In a prospective study of one-visit nonsurgical root canal treatments, the overall flare-up rate was only 1.8%.224 However, this same study determined that one-visit endodontic retreatment cases involving teeth with apical periodontitis had almost a 10-fold higher incidence of flare-ups (13.6%), with the recommendation that retreatment of teeth with apical periodontitis should not be completed in one visit. Another prospective clinical study reported an overall flare-up rate of 3.2% of 946 endodontic cases.233 Patients with severe preoperative pain had a flare-up rate of 19%, although the presence of localized or diffuse swelling related to an incidence of 15%. In this study, pulpal status predicted the flare-up rate, with necrotic teeth having a significantly greater incidence of flare-ups compared with vital teeth (6.5% versus 1.3%). Periradicular status also predicted flare-up rates, with differences noted between chronic apical periodontitis (3.4%), acute apical periodontitis (4.8%), and acute apical abscess (13.1%). No significant difference was seen between single and multiple visits. Finally, no significant increase in flare-ups was seen for teeth undergoing retreatment of a failed root canal, although a subset analysis of retreatment with or without apical periodontitis was not performed.
A recent systematic review of clinical trials evaluating the effectiveness of single versus multiple visits for endodontic therapy concluded that while there was no difference in ultimate outcome of the treatment, those patients treated in one visit were more likely to take analgesic drugs.132
In summary, the most consistent factor that predicts postendodontic pain is the presence of preoperative pain or mechanical allodynia. Although no single factor completely predicts the occurrence and magnitude of postoperative pain, an astute clinician should interpret the presence of preoperative pain or mechanical allodynia as a warning sign in selecting the pain control regimen for each patient.
Nonnarcotic Analgesics
Management of endodontic pain is multifactorial and directed at reducing the peripheral and central components of hyperalgesia (see Box 19-1) through combined endodontic procedures and pharmacotherapy. A major class of drugs for managing endodontic pain is nonnarcotic analgesics, which include both NSAIDs and acetaminophen. NSAIDs have been shown to be very effective in managing pain of inflammatory origin and, by virtue of their binding to plasma proteins, actually exhibit increased delivery to inflamed tissue via the extravasation of plasma proteins.20,48,89,93 Although these drugs classically are thought to produce analgesia through peripheral mechanisms, the CNS now is believed to be an additional site of action.147,215 Some researchers have proposed that a splice variant of COX1—COX3—is expressed predominantly in the CNS and is the major site of action of acetaminophen.28,128,202 However, recent studies indicate that the antipyretic and analgesic effects of acetaminophen do not involve inhibition of COX3102 but are more likely exerted through effects of an active metabolite on CNS cannabinoid receptors.7
Numerous NSAIDs are available for management of pain and inflammation (Table 19-4). Unfortunately, comparatively few studies (particularly for endodontic pain) directly compare one NSAID to one another for analgesia and side-effect liability, particularly regarding endodontic pain. The lack of comprehensive comparative studies in endodontic models means that only general recommendations can be made, and clinicians are encouraged to be familiar with several of these drugs. Ibuprofen generally is considered the prototype of contemporary NSAIDs and has a well-documented efficacy and safety profile.45 Other NSAIDs may offer certain advantages over ibuprofen. For example, etodolac (i.e., Lodine) has minimal gastrointestinal (GI) irritation,9 and ketoprofen (i.e., Orudis) has been shown in some studies to be somewhat more analgesic than ibuprofen.34 The advantages of NSAIDs include their well-established analgesic efficacy for inflammatory pain. Many of the NSAIDs listed in Table 19-4 have been shown to be more effective than traditional acetaminophen and opioid combinations such as Tylenol with codeine no. 3.33,45,226
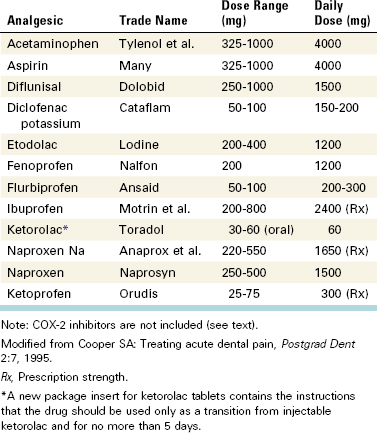
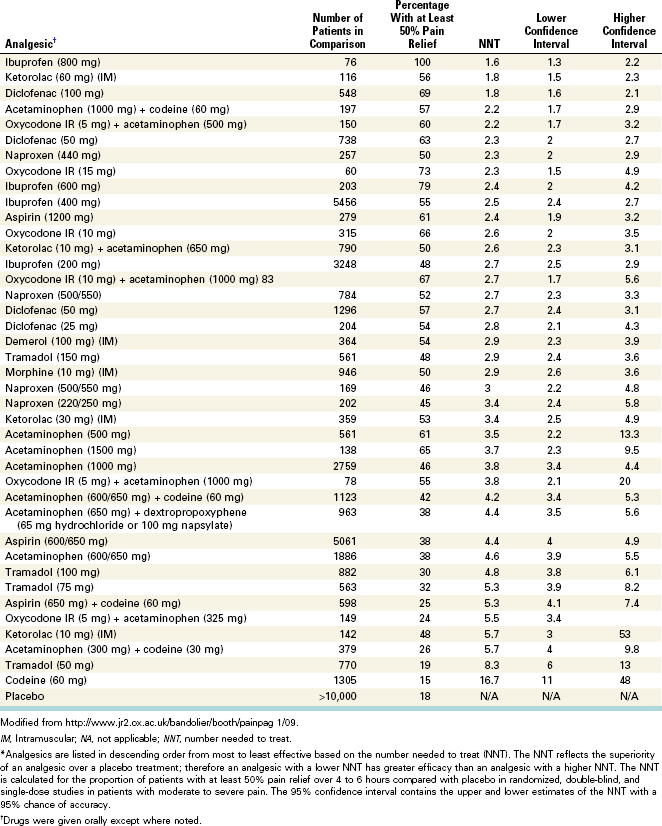
A 2002 paper represents the first systematic review comparing all endodontic pain studies evaluating oral NSAIDs.105 This study also provides a framework for other investigators interested in conducting systematic reviews for endodontic research. The authors concluded that NSAIDs combined with other drugs (e.g., flurbiprofen with tramadol49) or pretreatment and posttreatment application of NSAIDs provides effective pain control. Although relatively new to endodontic research, systematic reviews of analgesic drugs in inflammatory pain models have been conducted for several years. Table 19-5 lists the results of a large ongoing systematic review of the relative efficacy of analgesics in inflammatory pain conditions. Importantly, the data are generated based on postoperative patients having moderate to severe pain, and the number needed to treat (NNT) is based on the relative superiority of the analgesic over placebo for producing a 50% relief in pain. These data therefore constitute important, clinically relevant information for clinicians who want to compare the relative efficacy of posttreatment analgesics. Of course, other issues such as the potential adverse effects of drugs and the patient’s medical history must be considered when developing a postendodontic pain treatment plan.
The introduction of selective inhibitors of COX-2 offered the potential for both analgesic and antiinflammatory benefits and reduced GI irritation.43,123 Oral surgery pain studies evaluating COX-2 inhibitors have indicated that rofecoxib (i.e., Vioxx) has significant analgesic efficacy in this model.53 In one study, a 50-mg dose of rofecoxib produced analgesia equivalent to 400 mg of ibuprofen, with the two drugs displaying similar times for onset of analgesia.53 Moreover, COX-2 levels are increased in inflamed human dental pulp,171 and a COX-2 inhibitor (rofecoxib) is analgesic in patients with endodontic pain. Concern has been raised that the COX-2 inhibitors may also display at least some GI irritation in patients with preexisting GI disease.230
Another major concern centers on the recognized prothrombic adverse effects of the COX-2 inhibitors. This debate originally started when patients randomized to 50 mg/day of rofecoxib in the VIGOR study had a fivefold increase in thromboembolic cardiovascular (CV) events compared to 1000 mg/day of naproxen.17 The debate continued until the demonstration of an increased risk for prothrombic events following long-term administration of rofecoxib, which led to the withdrawal of this drug from the market in 2004.59 Two meta-analyses have examined the CV safety of traditional NSAIDs and COX-2 inhibitors. Kearney et al. conducted a meta-analysis of 138 randomized trials, and McGettigan and Henry conducted a meta-analysis of 23 controlled observational studies.118,155 Kearney et al. estimated a relative risk for CV events associated with COX-2 to be 1.42 (95% CI, 1.64 to 2.91). Naproxen was found to have no significant adverse effects on the CV system in both meta-analyses. Diclofenac (Voltaren) is a relatively COX-2-selective drug and seems to have a similar degree of COX-2 selectivity as celecoxib. Diclofenac was associated with increased CV events. In the randomized trial analysis, there was an increase in CV risk with high-dose ibuprofen. Based on the available data, the FDA has requested that manufacturers of all prescription products containing nonselective NSAIDs revise their product labeling to include (1) a boxed warning regarding the potential serious adverse CV events and the serious, potentially life-threatening GI adverse events associated with the use of this class of drugs; (2) a contraindication for use in patients who have recently undergone coronary artery bypass surgery; and (3) a medication guide for patients, regarding the potential for CV and GI adverse events associated with the use of this class of drugs. The available data do not suggest an increased risk of serious CV events for the short-term, low-dose use of NSAIDs available over the counter, but the U.S. Food and Drug Administration (FDA) has requested changes to the label to better inform consumers regarding the safe use of these products. Given this situation and reasonable alternative NSAIDs, we recommend not considering COX-2 inhibitors for treating routine endodontic pain patients.
Limitations and Drug Interactions
Clinicians should educate themselves not only on the efficacy of the nonnarcotic analgesics but also on their limitations and interactions with other drugs.25 For example, NSAIDs exhibit an analgesic ceiling that limits the maximal level of analgesia and induces side effects, including those affecting the GI system (3% to 11% incidence) and the CNS (1% to 9% incidence of dizziness and headache). NSAIDs are contraindicated in patients with ulcers and aspirin hypersensitivity.31-33,68,248 They also are associated with severe GI complications, and the risk of adverse effects increases with increasing lifetime-accumulated dose of these drugs.46,240
The NSAIDs have been reported to interact with a number of other drugs (Table 19-6). Acetaminophen and opioid combination drugs represent alternatives for those patients unable to take NSAIDs.32 Further information is available on the pharmacology and adverse effects of this important class of drugs.25,31,46,68,248 Other resources are also available for evaluation of drug interactions, including Internet drug search engines such as rxlist.com, Epocrates.com, and Endodontics.UTHSCSA.edu.
TABLE 19-6 Summary of Selected NSAID Drug Interactions
| Drug | Possible Effect |
|---|---|
| Anticoagulants | Increase in prothrombin time or bleeding with anticoagulants (e.g., coumarins) |
| ACE inhibitors | Reduced antihypertensive effectiveness of captopril (especially indomethacin) |
| Beta-blockers | Reduced antihypertensive effects of beta-blockers (e.g., propranolol, atenolol, pindolol) |
| Cyclosporine | Increased risk of nephrotoxicity |
| Digoxin | Increase in serum digoxin levels (especially ibuprofen, indomethacin) |
| Dipyridamole | Increased water retention (especially indomethacin) |
| Hydantoins | Increased serum levels of phenytoin |
| Lithium | Increased serum levels of lithium |
| Loop diuretics | Reduced effectiveness of loop diuretics (e.g., furosemide, bumetanide) |
| Methotrexate | Increased risk of toxicity (e.g., stomatitis, bone marrow suppression) |
| Penillamine | Increased bioavailability (especially indomethacin) |
| Sympathomimetics | Increased blood pressure (especially indomethacin with phenylpropanolamine) |
| Thiazide diuretics | Reduced antihypertensive effectiveness |
Data from Drug facts and comparisons, ed 54, St Louis, 2000, Facts and Comparisons; Gage T, Pickett F: Mosby’s dental drug reference, ed 5, St Louis, 2000, Mosby; Wynn R, Meiller T, Crossley H: Drug information handbook for dentistry, Hudson, Ohio, 2000, Lexi-Comp.
Acetaminophen
Acetaminophen is one of the most commonly used drugs. It is also one of the most common drugs found in combination products for the relief of pain and symptoms of cold or flu. It is considered safe when taken at normal doses, but in higher doses, acetaminophen causes liver toxicity and has become the most common cause of acute liver failure.134 Most acetaminophen is conjugated in the liver to form inactive metabolites. A small portion is metabolized by the cytochrome P450 system to form N-acetyl-p-benzoquinone imine (NAPQI), which is very toxic but is generally detoxified by glutathione and converted into nontoxic compounds. Large doses of acetaminophen saturate the main route of metabolism, causing more acetaminophen to be converted to NAPQI. Liver injury occurs once glutathione becomes depleted and NAPQI is allowed to accumulate. Healthy adults should not take more than 4 g (4000 mg) of acetaminophen in a 24-hour period. However, the FDA convened an Advisory Panel meeting in June 2009, with discussions on limiting the maximal daily dose of acetaminophen and possibly removing certain medications from the market.58 Although no action was taken by late 2009, the clinician is encouraged to continue to monitor the FDA website for further information (www.FDA.gov). Opioid pain medications may contain up to 650 to 750 mg of acetaminophen per tablet. Care must be taken when prescribing such combinations to keep the maximum acetaminophen dose below 4000 mg. In an effort to address this, manufacturers have made high-dose opioid/low-dose acetaminophen combinations, such as hydrocodone 10 mg and acetaminophen 325 mg.
Opioid Analgesics
Opioids are potent analgesics and are often used in dentistry in combination with acetaminophen, aspirin, or ibuprofen. Most clinically available opioids activate mu opioid receptors located at several important sites in the brain (see Fig. 19-2). Activation of this receptor inhibits the transmission of nociceptive signals from the trigeminal nucleus to higher brain regions, and recent studies indicate that opioids also activate peripheral opioid receptors located in dental pulp.60 Intraligamentary injection of morphine has been shown to significantly reduce pain in endodontic patients and other inflammatory pain states.47,88 Gender-dependent differences appear to exist in responsiveness to at least the kappa opioid agonists; women have a significantly greater analgesic response to pentazocine compared to men.71
Although opioids are effective as analgesics for moderate to severe pain, their use is generally limited by their adverse side effects, which can include nausea, emesis, dizziness, drowsiness, and the potential for respiratory depression and constipation. Chronic use is associated with tolerance and dependence. Because dosage is limited by side effects, these medications are almost always used in combination drugs to manage dental pain. A combination formulation is preferred because it permits a lower dose of the opioid, thereby reducing side effects (Table 19-7).
TABLE 19-7 Selected Opioid Combination Analgesic Drugs
| Formulation | Trade Name* | Possible Rx |
|---|---|---|
| APAP 300 mg and codeine 30 mg | Tylenol with codeine no. 3 | 2 tabs q4h |
| APAP 500 mg and hydrocodone 5 mg | Vicodin, Lortab 5/500 | 1-2 tabs q6h |
| APAP 325 mg and oxycodone 5 mg | Percocet | 1 tab q6h |
| APAP 500 mg and oxycodone 5 mg | Tylox | 1 tab q6h |
| ASA 325 mg and codeine 30 mg | Empirin with codeine no. 3 | 2 tabs q4h |
| ASA 325 mg and oxycodone 5 mg | Percodan | 1 tab q6h |
APAP, Acetaminophen; ASA, aspirin; Rx, prescription.
* Several generics are available for most formulations.
Codeine is often considered the prototype opioid for orally available combination drugs. Most studies have found that the 60-mg dose of codeine (the amount in two tablets of Tylenol with codeine no. 3) produces significantly more analgesia than placebo, although it often produces less analgesia than either aspirin 650 mg or acetaminophen 600 mg.32,33,89 In general, patients taking only 30 mg of codeine report about as much analgesia as those taking a placebo.14,226 Table 19-8 provides comparable doses of other opioids equivalent to 60 mg of codeine.
TABLE 19-8 Analgesic Doses of Representative Opioids
| Opioid | Dose Equivalent to Codeine 60 mg |
|---|---|
| Codeine | 60 mg |
| Oxycodone | 5-6 mg |
| Hydrocodone | 10 mg |
| Dihydrocodeine | 60 mg |
| Propoxyphene HCl | 102 mg |
| Propoxyphene-N | 146 mg |
| Meperidine | 90 mg |
| Tramadol | 50 mg |
HCl, Hydrochloride; N, napsylate.
Modified from Troullos E, Freeman R, Dionne RA: The scientific basis for analgesic use in dentistry. Anesth Prog 33:123, 1986.
Corticosteroids
Posttreatment pain or flare-up after endodontic treatment can be attributed to inflammation, infection, or both in the periradicular tissues. Establishing patency and subsequently débriding and shaping the root canal system may irritate the periradicular tissues and inadvertently introduce bacteria, bacterial products, necrotic pulp tissue, or caustic irrigating solution through apical foramina.
In response to this irritation, inflammatory mediators (e.g., prostaglandins, leukotrienes, bradykinin, platelet-activating factor, substance P, etc.) are released into the tissues surrounding the apical area of the tooth. As a result, pain fibers are directly stimulated or sensitized, and an increase in vascular dilation and permeability results in edema and increased interstitial tissue pressure.
Glucocorticosteroids are known to reduce the acute inflammatory response by suppressing vasodilation, migration of polymorphonuclear (PMN) leukocytes, and phagocytosis and by inhibiting formation of arachidonic acid from neutrophil and macrophage cell membrane phospholipids, thus blocking the COX and lipoxygenase pathways and respective synthesis of PGs and leukotrienes. It is not surprising that a number of investigations have evaluated the efficacy of corticosteroids (administered via either intracanal or systemic routes) in the prevention or control of postoperative endodontic pain or flare-ups.150
Intracanal Administration
Several studies have evaluated intracanal administration of steroids. In 50 consecutive patients requiring nonsurgical root canal treatment of vital teeth, one investigator alternately placed a dexamethasone solution or saline placebo as intracanal medicaments after the root canals had been cleaned and shaped.167 Pretreatment pain ratings were collected, and at 24, 48, and 72 hours after treatment. Results indicated a significant reduction in pain at 24 hours but no significant difference at 48 and 72 hours. In a similar double-blind clinical trial, intracanal placement of a 2.5% steroid solution or saline placebo on completion of instrumentation resulted in a significant reduction of the incidence of postoperative pain in teeth in which the pulp was vital.27 When the pulp was necrotic, however, there was no significant difference between the steroid and placebo in reducing postoperative discomfort.
Another study found no significant difference in the flare-up rate when either formocresol, Ledermix (a corticosteroid antibiotic paste), or calcium hydroxide was placed as an intracanal medicament in strict sequence, irrespective of the presence or absence of symptoms or radiographic signs of apical periodontitis.225 However, a large-scale clinical trial of 223 patients reported significantly less posttreatment pain in patients after intracanal administration of Ledermix compared with either calcium hydroxide or no intracanal dressing.54 Intracanal steroids appear to have significant effects for reducing postoperative pain.195
Systemic Administration
Some studies have evaluated the systemic route of administration of corticosteroids on posttreatment pain or flare-ups. In one double-blind, randomized, placebo-controlled study, dexamethasone (4 mg/ml) or saline was injected intramuscularly at the conclusion of a single-visit endodontic appointment or at the first visit of a multivisit procedure.151 The results indicated that the steroid significantly reduced the incidence and severity of pain at 4 hours when compared with the placebo. Pain was reduced at 24 hours, but it was not statistically significant, and no difference in incidence or severity was seen at 48 hours.
In a similar study, 106 patients with irreversible pulpitis and acute periradicular periodontitis were given an intraoral intramuscular injection of dexamethasone at different doses, either on completion of a single-visit endodontic treatment or after the first visit of a multivisit procedure.140 Systemic administration of dexamethasone was shown to significantly reduce the severity of pain at 4 and 8 hours, with an optimum dose between 0.07 and 0.09 mg/kg. However, no significant reduction in the severity of pain was noted at 24, 48, and 72 hours, and no overall effect was seen on the incidence of pain.
Another study compared the effect an intraligamentary injection of methylprednisolone, mepivacaine, or placebo in preventing posttreatment endodontic pain.117 The results showed that methylprednisolone significantly reduced postoperative pain within a 24-hour follow-up period.
In a double-blind placebo controlled study, patients with irreversible pulpitis were given 4 mg of dexamethasone or placebo by means of a supraperiosteal injection at the apex of the treated tooth following pulpectomy.156 This is an injection technique that most clinicians would be familiar with (as opposed to intramuscular injection). Posttreatment pain was significantly reduced in the steroid group during the first 24 hours. There was no difference at 48 hours.
Yet another study evaluated the effect of intraosseous injection of methylprednisolone or placebo to patients with irreversible pulpitis. Highly significant pain reduction in the steroid group was maintained for 7 days after a single injection.70
Animal studies have histologically evaluated the antiinflammatory effects of corticosteroids on inflamed periradicular tissues. In one study, after an acute inflammatory reaction was induced in the molar teeth of rats by overextending endodontic instruments, sterile saline or dexamethasone was infiltrated supraperiosteally into the buccal vestibule adjacent to the treated teeth. Dexamethasone significantly reduced the number of neutrophils present and thus had an antiinflammatory effect on the periradicular tissues of the treated teeth.178
Other studies of systemic administration have evaluated the efficacy of oral administration of corticosteroids on the incidence and severity of posttreatment endodontic pain. In a double-blind controlled clinical trial, 50 patients randomly received either 0.75 mg dexamethasone or a placebo tablet orally after initial endodontic treatment.129 Oral dexamethasone significantly reduced posttreatment pain after 8 and 24 hours compared with placebo. A follow-up study evaluated the effect a larger oral dose of dexamethasone (i.e., 12 mg given every 4 hours) on the severity of posttreatment endodontic pain.76 Results showed that dexamethasone was effective in reducing posttreatment endodontic pain up to 8 hours after the treatment was completed; no affect on the severity of pain was seen at 24 and 48 hours after treatment.
Collectively, these studies on systemic steroid administration indicate that corticosteroids reduce the severity of posttreatment endodontic pain compared with placebo treatment. However, given the relative safety/efficacy relationship between steroids and NSAIDs, most investigators choose an NSAID as the drug of first choice for postoperative pain control.
Antibiotics
Because bacteria are involved in endodontic cases with apical periodontitis, the incidence of a posttreatment infection or flare-up is a concern to clinicians providing endodontic treatment. Prescribing an antibiotic to prevent such an occurrence might make sense, but use of antibiotics is controversial for several reasons.63 First, overprescribing antibiotics, especially when these drugs are not indicated, has led to increased bacterial resistance and patient sensitization. Second, antibiotics have been mistakenly prescribed for patients with severe pain who have a vital tooth (i.e., when bacteria are unlikely to be a causative factor in periradicular pain).251 Third, even when bacteria are likely to be present, data from controlled clinical trials provide little or no support for the hypothesis that antibiotics reduce pain.119
A series of clinical studies evaluated the efficacy of prophylactically administered systemic antibiotics for preventing posttreatment endodontic flare-ups. Working on the premise that the incidence of infectious flare-ups after endodontic treatment is 15%, Morse et al. randomly prescribed a prophylactic dose of either penicillin or erythromycin to patients after endodontic treatment of teeth with a diagnosis of necrotic pulp and chronic periradicular periodontitis (no placebo was used).166 The results showed that the overall incidence of flare-ups was 2.2%, with no difference between penicillin or erythromycin. Similar results were obtained in a study in which dental students (instead of private practitioners) provided the endodontic treatment.3 Outcomes found a 2.6% incidence of flare-up, with no statistically significant differences between penicillin or erythromycin. However, neither the original nor the follow-up study was a randomized, placebo-controlled clinical trial. This observation appears to be highly significant for clinical recommendations, since in general, randomized controlled studies fail to detect any analgesic benefit to antibiotics, whereas open-label or historical control studies often report profound effects.63
To determine whether the timing of administration of an antibiotic altered the occurrence of flare-ups and pain unassociated with flare-ups, researchers analyzed components of two separate prospective studies of patients undergoing endodontic treatment for teeth with necrotic pulps and chronic periradicular periodontitis. In the first study, prophylactic penicillin was provided; in the second study, patients were instructed to take penicillin (or erythromycin if they were allergic to penicillin) at the first sign of swelling.165,166 The authors concluded that prophylactic administration of antibiotics is preferable to having the patient take antibiotics at the first sign of an infection.
Another study of similar design compared the incidence of flare-up when a cephalosporin or erythromycin was given prophylactically.164 When the data from previous studies were pooled and retrospectively compared, the authors concluded that prophylactically administered antibiotics, including cephalosporins, significantly reduced the incidence of flare-ups in endodontic cases involving necrotic pulp and chronic periradicular periodontitis. However, these studies have been questioned because of the lack of concurrent placebo-treated groups and the use of historical controls.
In a multicenter, two-part clinical study, 588 consecutive patients received one of nine medications or placebos and were monitored for 72 hours after treatment.221,222 The results showed that ibuprofen, ketoprofen, erythromycin, penicillin, and penicillin plus methylprednisolone significantly reduced the severity of pain within the first 48 hours after treatment.221 The second part of the study then evaluated the incidence of posttreatment pain after obturation of the same teeth in the first phase of the study.222 Although only 411 of the original 588 patients participated in this phase, they were randomly given the same medications or placebo after completion of the obturation appointment. The incidence of posttreatment pain was lower after obturation (5.83%) than after cleaning and shaping (21.76%) of the root canal system, and no significant difference was found in the effectiveness of the various medications and placebo in controlling posttreatment pain after obturation.
Walton and Chiappinelli232 were concerned that previous studies were uncontrolled, retrospective, or carried out on different patient groups at different times and with different treatment modalities. They conducted a randomized, prospective, double-blind clinical trial to test the hypothesis that an antibiotic (e.g., penicillin) can prevent a posttreatment endodontic flare-up.232 Eighty patients with teeth that had necrotic pulp and chronic periradicular periodontitis were randomly divided into three groups. In the first two groups, either penicillin or a placebo was administered 1 hour before and 6 hours after the individual appointments on a double-blind basis. Upon completion of the individual appointments, which included débridement, shaping, and possibly obturation of the root canal system, the patients completed questionnaires at 4, 8, 12, 24, and 48 hours. No significant difference was found among the three groups in the incidence of flare-ups, pain, or swelling. The authors concluded that the use of prophylactic penicillin offers no benefit for postoperative pain or flare-up, and prophylactic administration of penicillin should not be used routinely in patients undergoing endodontic treatment of necrotic teeth and chronic periradicular periodontitis.
In another randomized, prospective, placebo-controlled clinical study, a group of investigators examined whether supplemental penicillin reduced symptoms or shortened the course of recovery of emergency patients diagnosed with necrotic pulp and acute apical abscess.65 Patients were randomly given penicillin, a placebo, or no medication. Using a visual analog scale, the subjects then evaluated their postoperative pain and swelling for up to 72 hours. No significant difference was found among the three groups. Recovery occurred as a result of endodontic treatment alone.
It is well recognized that antibiotics may be indicated for the management of infections of endodontic origin. However, a review of the available literature indicates that prophylactic use is contraindicated in immunocompetent patients with no systemic signs of infection and with swelling localized to the vestibule. Controlled clinical studies indicate that antibiotics offer little or no benefit for pain reduction under these circumstances, but they may be indicated for immunocompromised patients and for patients with systemic signs and symptoms of an infection or an infection that has spread into the fascial spaces of the head and neck.
Pain-Management Strategies
When managing pain in an individual patient, the skilled clinician must customize the treatment plan, balancing the general principles of endodontics, mechanisms of hyperalgesia, and pain-management strategies with the particular factors of the individual patient (e.g., medical history, concurrent medications).93,120,121,197,209
Effective management of endodontic pain starts with the “three Ds”: diagnosis, definitive dental treatment, and drugs (Box 19-2). Comprehensive reviews on diagnosis and definitive dental treatment (e.g., incision and drainage, pulpectomy) are provided elsewhere in this text. As described earlier in this chapter, the management of endodontic pain should focus on removal of peripheral mechanisms of hyperalgesia and allodynia (see Box 19-1). This generally requires treatment that removes and reduces causative factors (e.g., bacterial and immunologic factors). For example, both pulpotomy and pulpectomy have been associated with substantial reduction in patient reports of pain compared to pretreatment pain levels.49,100,154,187 However, pharmacotherapy often is required to reduce continued nociceptor input (e.g., NSAIDs, local anesthetics) and suppress central hyperalgesia (e.g., NSAIDs, opioids).
Pretreatment
Treatment with an NSAID before a procedure has been shown to have a significant benefit in many studies45,109 but not all of them.177 The rationale for pretreatment is to block the development of hyperalgesia by reducing the input from peripheral nociceptors. For patients who cannot take NSAIDs, pretreatment with acetaminophen also has been shown to reduce postoperative pain.162 Patients can be pretreated 30 minutes before the procedure with either an NSAID (e.g., ibuprofen 400 mg or flurbiprofen 100 mg) or with acetaminophen 1000 mg.49,109,162
Long-Acting Local Anesthetics
A second pharmacologic approach for pain management is the use of long-acting local anesthetics such as bupivacaine and ropivacaine (see Chapter 20 for an extensive review of this topic). Clinical trials indicate that long-acting local anesthetics not only provide anesthesia during the procedure but also significantly delay the onset of posttreatment pain when compared with local anesthetics that contain lidocaine.38,45,81,82,111 Administration of long-acting local anesthetics by block injection has been shown to reduce posttreatment pain for 2 to 7 days after the oral procedure,81,82,111 because an afferent barrage of nociceptors can induce central hyperalgesia.242-244 The analgesic benefit of long-acting local anesthetics is more prominent with block injections than infiltration injections, but the clinician should also be aware of adverse effects attributed to these agents.10,161
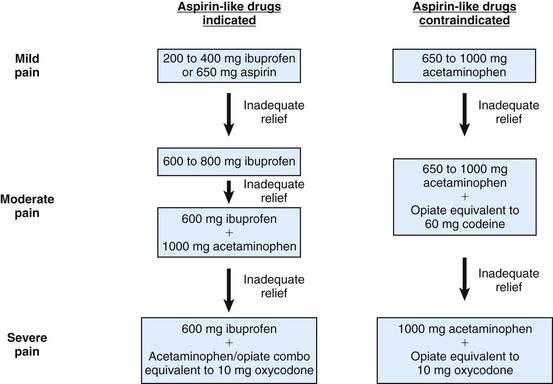
Flexible Plan
A third pharmacologic approach is the use of a flexible plan for prescribing analgesics (Fig. 19-3).* A flexible prescription plan serves to minimize both postoperative pain and side effects. Given these goals, the clinician’s strategy is twofold: (1) to achieve a maximally effective dose of the nonnarcotic analgesic (either an NSAID or acetaminophen for patients who cannot take NSAIDs), and (2) in the rare cases in which the patient still has moderate to severe pain, to consider adding drugs that increase the NSAID’s analgesia. Because of its predictive value, the presence of preoperative pain or mechanical allodynia may be an indicator that such NSAID combinations should be considered.
Studies have shown that treatment combining an NSAID with acetaminophen 1000 mg alone (i.e., no opioid) produces nearly twice the analgesic response as just the NSAID.18,35,158 Administration of ibuprofen 600 mg with acetaminophen 1000 mg produced significant relief of posttreatment endodontic pain, compared to ibuprofen alone or placebo (Fig. 19-4). Studies have also shown that concurrent administration of an NSAID and an acetaminophen-opioid combination drug provided significantly greater analgesia than an NSAID alone.18,211 The concurrent administration of acetaminophen and NSAIDs appears to be well tolerated, with no detectable increase in side effects or alterations in pharmacokinetics.18,133,211,246
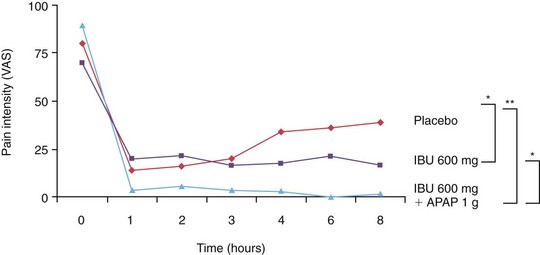
FIG. 19-4 Comparison of ibuprofen 600 mg with acetaminophen 1000 mg to ibuprofen alone or to placebo treatment in postendodontic pain patients.
(From Menhinick KA, Gutmann JL, Regan JD, et al: The efficacy of pain control following nonsurgical root canal treatment using ibuprofen or a combination of ibuprofen and acetaminophen in a randomized, double-blind, placebo-controlled study. Int Endod J 37:531–541, 2004. Reprinted with permission.)
In cases of moderate to severe pain, an NSAID may need to be administered with an opioid. Two general methods are used to combine NSAIDs and opioids to achieve the analgesic benefits of both. The first involves an alternating regimen of an NSAID followed by an acetaminophen-opioid combination.1,33 Aspirin and opioid combinations are not used in this alternating schedule because of the potential for NSAID/aspirin interactions. The second method involves administration of a single drug consisting of an NSAID-opioid combination. For example, a tablet of Vicoprofen contains both ibuprofen (200 mg) and hydrocodone (7.5 mg). Postoperative pain studies have shown that this combination was about 80% more effective for analgesia than ibuprofen (200 mg) alone, with about the same incidence of side effects.238 Other opioids can also be added to an NSAID for increased analgesia. Ibuprofen 400 mg with a 10-mg oxycodone tablet produces significantly greater analgesia than ibuprofen alone.42 A study on posttreatment endodontic pain demonstrated short-term benefits of the combination of flurbiprofen and tramadol.49 Other NSAID and opioid combinations have also been evaluated.46 However, the results of clinical trials on the use of NSAIDs alone and combined with acetaminophen (see Fig. 19-4) suggest that opioid combinations may be required only rarely.
Not all patients require concurrent use of NSAIDs with acetaminophen-opioid combinations or combinations of an NSAID and opioid. The basic premise of a flexible prescription plan is that the analgesic prescribed be matched to the patient’s need. The major advantage of a flexible plan is that the clinician is prepared for those rare cases when additional pharmacotherapy is indicated to increase the efficacy of pain control. As mentioned, preoperative hyperalgesia may be an indicator for more comprehensive pharmacotherapy. When considering the combination of various analgesics, the clinician must make sure to use dosing regimens that do not exceed any of the drugs’ maximum daily dosage.
Future Directions
Current use of analgesics has been driven by clinical trials to arrive at doses that obtain the desired level of pain relief with acceptable side effect profiles. After administration, drugs are absorbed and distributed to their site of action where they interact with functional targets. They then undergo metabolism and eventual excretion. All of the steps along the way are influenced by a variety of environmental and genetic factors. The ability to predict how a patient’s genome may affect the efficacy of a given analgesic drug is being discovered in the field of pain pharmacogenomics (for review, see Rollason et al.196).
One example relevant to pain control in dentistry is the variable efficacy of codeine in a specific population. Many analgesic drugs are metabolized by the cytochrome P450 (CYP) family of hepatic enzymes, and the genes that encode for their biosynthesis have been identified. Codeine is a prodrug that is demethylated to produce morphine, which is responsible for its analgesic action. This demethylation is catalyzed by the enzyme, cytochrome P402D6. It has been estimated that 6% to 7% of the Caucasian population has a nonfunctional CYP2D6 mutant allele, making them poor metabolizers of codeine to morpine.41 These patients may be aware that codeine is ineffective in their systems from past experience and may request a different (usually more potent) form of narcotic. The clinician may suspect drug-seeking behavior, when in fact there is a biochemical reason for their request. Given the current state of clinical DNA analysis, it is not incomprehensible to envision a time when rapid (chairside) genomic evaluation will lead to specific recommendations for analgesic prescription.
Summary
The information and recommendations provided in this chapter were selected to aid the clinician in the management of acute endodontic pain. Clinical judgment must also take into account other sources of information—patient history, concurrent medications, the nature of the pain, and the overall treatment plan—when determining the best means of alleviating patient pain. An effective approach to managing endodontic pain demands integration of general principles of pain mechanisms and management with thorough, individualized clinical assessment.
1. American Association of Endodontists. Post-Endodontic Pain Control. Chicago: The Association; 1995.
2. Aars H, Gazelius B, Edwall L, Olgart L. Effects of autonomic reflexes on tooth pulp blood flow in man. Acta Physiol Scand. 1992;146:423.
3. Abbott A, et al. A prospective randomized trial on efficacy of antibiotic prophylaxis in asymptomatic teeth with pulpal necrosis and associated periapical pathosis. Oral Surg. 1988;66:722.
4. Abbott P. Factors associated with continuing pain in endodontics. Aust Dent J. 1994;39:157.
5. Ahlquist ML, Franzen OG. Inflammation and dental pain in man. Endod Dent Traumatol. 1994;10:201.
6. Albashaireh Z, Alnegrish A. Postobturation pain after single- and multiple-visit endodontic therapy. A prospective study. J Dent. 1998;26:227.
7. Anderson B. Paracetamol (Acetaminophen): mechanisms of action. Paediatr Anaesth. 2008;18:915.
8. Arbuckle JB, Docherty RJ. Expression of tetrodotoxin-resistant sodium channels in capsaicin-sensitive dorsal root ganglion neurons of adult rats. Neurosci Lett. 1995;185:70.
9. Arnold J, Salom I, Berger A. Comparison of gastrointestinal microbleeding associated with use of etodolac, ibuprofen, indomethacin, and naproxen in normal subjects. Curr Ther Res. 1985;37:730.
10. Bacsik C, Swift J, Hargreaves K. Toxic systemic reactions of bupivacaine and etidocaine: review of the literature, Oral Surg. Oral Med Oral Pathol. 1995;79:18.
11. Balaban F, Skidmore A, Griffin J. Acute exacerbations following initial treatment of necrotic pulps. J Endod. 1984;10:78.
12. Barnett F, Tronstad L. The incidence of flare-ups following endodontic treatment. J Dent Res. 1989;68:1253.
13. Basbaum AI, Fields HL. Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Ann Rev Neurosci. 1984;7:309.
14. Beaver W. Mild analgesics. A review of their clinical pharmacology. Am J Med Sci. 1966;251:576.
15. Beck P, Handwerker H. Bradykinin and serotonin effects on various types of cutaneous nerve fibers. Pflugers Arch. 1974;347:209.
16. Bisgaard H, Kristensen J. Leukotriene B4 produces hyperalgesia in humans. Prostaglandins. 1985;30:791.
17. Bombardier C, Laine L, Reicin A, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343:1520.
18. Breivik E, Barkvoll P, Skovlund E. Combining diclofenac with acetaminophen or acetaminophen-codeine after oral surgery: a randomized, double-blind, single oral dose study. Clin Pharmacol Ther. 1999;66:625.
19. Buck S, Reese K, Hargreaves KM. Pulpal exposure alters neuropeptide levels in inflamed dental pulp and trigeminal ganglia: evaluation of axonal transport. J Endod. 1999;25:718.
20. Bunczak-Reeh M, Hargreaves K. Effect of inflammation on delivery of drugs to dental pulp. J Endod. 1998;24:822.
21. Byers M, et al. Effects of injury and inflammation on pulpal and periapical nerves. J Endod. 1990;16:78.
22. Byers MR. Dynamic plasticity of dental sensory nerve structure and cytochemistry. Arch Oral Biol. 1994;39(Suppl):13S.
23. Byers MR, Narhi MV. Dental injury models: experimental tools for understanding neuroinflammatory interactions and polymodal nociceptor functions. Crit Rev Oral Biol Med. 1999;10:4.
24. Byers MR, Taylor PE, Khayat BG, Kimberly CL. Effects of injury and inflammation on pulpal and periapical nerves. J Endod. 1990;16:78.
25. Byrne B. Drug interactions: a review and update. Endod Topics. 2004;4:9.
26. Caviedes-Bucheli J, Munoz HR, Azuero-Holguin MM, Ulate E. Neuropeptides in dental pulp: the silent protagonists. J Endod. 2008;34:773.
27. Chance K, Lin L, Shovlin F, Skribner J. Clinical trial of intracanal corticosteroid in root canal therapy. J Endod. 1987;13:466.
28. Chandrasekharan NV, Dai H, Roos KL, et al COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression [see comment] Proc Natl Acad Sci U S A 99 2002 13926
29. Chiang CY, Park SJ, Kwan CL, Hu JW, Sessle BJ. NMDA receptor mechanisms contribute to neuroplasticity induced in caudalis nociceptive neurons by tooth pulp stimulation. J Neurophysiol. 1998;80:2621.
30. Clem W. Post-treatment endodontic pain. J Am Dent Assoc. 1970;81:1166.
31. Comparisons DFa. Drug Facts and Comparisons. St Louis: Facts and Comparisons Inc; 2000.
32. Cooper S. New peripherally acting oral analgesics. Ann Rev Pharmacol Toxicol. 1983;23:617.
33. Cooper S. Treating acute dental pain. Postgrad Dent. 1995;2:7.
34. Cooper S, Berrie R, Cohn P. The analgesic efficacy of ketoprofen compared to ibuprofen and placebo. Adv Ther. 1988;5:43.
35. Cooper SA. The relative efficacy of ibuprofen in dental pain. Compend Contin Educ Dent. 1986;7:580.
36. Cousins M, Power I. Acute and postoperative pain. In: Wall P, Melzack R, editors. Textbook of pain. Edinburgh: Churchill Livingstone; 2002:456.
37. Creech J, Walton R, Kaltenbach R. Effect of occlusal relief on endodontic pain. J Am Dent Assoc. 1984;109:64.
38. Crout R, Koraido G, Moore P. A clinical trial of long-acting local anesthetics for periodontal surgery. Anesth Prog. 1990;37:194.
39. Dallel R, Clavelou P, Woda A. Effects of tractotomy on nociceptive reactions induced by tooth pulp stimulation in the rat. Exp Neurol. 1989;106:78.
40. Desmeules J, Gascon MP, Dayer P, Magistris M. Impact of environmental and genetic factors on codeine analgesia. Eur J Clin Pharmacol. 1991;41:23.
41. Diogenes A, Akopian AN, Hargreaves KM. NGF upregulates TRPA1: implications for orofacial pain. J Dent Res. 2007;86:550-555.
42. Dionne R. Additive analgesic effects of oxycodone and ibuprofen in the oral surgery model. J Oral Maxillofac Surg. 1999;57:673.
43. Dionne R. COX-2 inhibitors: better than ibuprofen for dental pain? Compendium. 1999;20:518.
44. Dionne R. Oral sedation. Compend Contin Educ Dent. 1998;19:868.
45. Dionne R. Suppression of dental pain by the preoperative administration of flurbiprofen. Am J Med Sci. 1986;80:41.
46. Dionne RA, Berthold C. Therapeutic uses of non-steroidal anti-inflammatory drugs in dentistry. Crit Rev Oral Biol Med. 2000;12:315.
47. Dionne RA, Lepinski AM, Gordon SM, Jaber L, Brahim JS, Hargreaves KM. Analgesic effects of peripherally administered opioids in clinical models of acute and chronic inflammation. Clin Pharmacol Ther. 2001;70:66.
48. Dionne RA. Additive analgesic effects of oxycodone and ibuprofen in the oral surgery model. J Oral Maxillofac Surg. 1999;57:673.
49. Doroshak A, Bowles W, Hargreaves K. Evaluation of the combination of flurbiprofen and tramadol for management of endodontic pain. J Endod. 1999;25:660.
50. Dray A. Inflammatory mediators of pain. Br J Anaesth. 1995;75:125.
51. Dubner R, Bennett GJ. Spinal and trigeminal mechanisms of nociception. Ann Rev Neurosci. 1983;6:381.
52. Dworkin SF. Anxiety and performance in the dental environment: an experimental investigation. J Am Soc Psychosom Dent Med. 1967;14:88.
53. Ehrich E, et al. Characterization of rofecoxib as a cyclooxygenase inhibitor and demonstration of analgesia in the dental pain model. Clin Pharmacol Ther. 1999;65:336.
54. Ehrmann EH, Messer HH, Adams GG. The relationship of intracanal medicaments to postoperative pain in endodontics. Int Endod J. 2003;36:868.
55. Eleazer P, Eleazer K. Flare-up rate in pulpally necrotic molars in one-visit versus two-visit endodontic treatment. J Endod. 1998;24:614.
56. England S. Molecular basis of peripheral hyperalgesia. In: Wood J, editor. Molecular basis of pain induction. ed 1. New York: Wiley-Liss; 2000:261.
57. Fava L. A comparison of one versus two appointment endodontic therapy in teeth with non-vital pulps. Int Endod J. 1989;22:179.
58. FDA. Joint Meeting of the Drug Safety and Risk Management Advisory Committee with the Anesthetic and Life Support Drugs Advisory Committee and the Nonprescription Drugs Advisory Committee. http://www.fda.gov/AdvisoryCommittees/Calendar/ucm143083.htm. accessed 8/25/09
59. FDA. MedWatch: Rofecoxib. http://www.fda.gov/medwatch/SAFETY/2004/safety04.htm#vioxx, 2004.
60. Fehrenbacher J, Sun XX, Locke E, Henry M, Hargreaves KM. Capsaicin-evoked iCGRP release from human dental pulp: a model system for the study of peripheral neuropeptide secretion in normal healthy tissue. Pain. 2009;144:253.
61. Flath R, et al. Pain suppression after pulpectomy with preoperative flurbiprofen. J Endod. 1987;13:339.
62. Follenfant R, et al. Inhibition by neuropeptides of interleukin-1ß-induced, prostaglandin-independent hyperalgesia. Br J Pharmacol. 1989;98:41.
63. Fouad A. Are antibiotics effective for endodontic pain? An evidence-based review. Endod Topics. 2002;3:52.
64. Fouad A. Molecular mediators of pulpal inflammation. In: Hargreaves KM, Goodis HE, editors. Seltzer and Bender’s dental pulp. Chicago: Quintessence Publishing Co, Inc; 2002:247.
65. Fouad A, Rivera E, Walton R. Penicillin as a supplement in resolving the localized acute apical abscess. Oral Surg Oral Med Oral Pathol. 1996;81:590.
66. Fox J, et al. Incidence of pain following one-visit endodontic treatment. Oral Surg Oral Med Oral Pathol. 1970;30:123.
67. Frank A, et al. The intracanal use of sulfathiazole in endodontics to reduce pain. J Am Dent Assoc. 1968;77:102.
68. Gage T, Pickett F. Mosby’s dental drug reference, ed 4. St Louis: Mosby; 2000.
69. Gallatin E, Reader A, Nist R, Beck M. Pain reduction in untreated irreversible pulpitis using an intraosseous injection of Depo-Medrol. J Endod. 2000;26:633.
70. Geborek P, Mansson B, Wollheim FA, Moritz U. Intraarticular corticosteroid injection into rheumatoid arthritis knees improves extensor muscles strength. Rheum Int. 1990;9(6):265-270.
71. Gear R, et al. Kappa-opioids produce significantly greater analgesia in women than in men. Nat Med. 1996;2:1248.
72. Gedney JJ, Logan H, Baron RS. Predictors of short-term and long-term memory of sensory and affective dimensions of pain. J Pain. 2003;4:47.
73. Genet JM, Hart AA, Wesselink, et al. Preoperative and operative factors associated with pain after the first endodontic visit. Int Endod J. 1987;20:53.
74. Georgopoulou M, Anastassiadis P, Sykaras S. Pain after chemomechanical preparation. Int Endod J. 1986;19:309.
75. Gilron I, Max MB, Lee G, et al. Effects of the 2-amino-3-hydroxy-5-methyl-4-isoxazole-proprionic acid/kainate antagonist LY293558 on spontaneous and evoked postoperative pain. Clin Pharmacol Ther. 2000;68:320.
76. Glassman G, Krasner P, Morse DR, et al. A prospective randomized double-blind trial on efficacy of dexamethasone for endodontic interappointment pain in teeth with asymptomatic inflamed pulps. Oral Surg Oral Med Oral Pathol. 1989;67:96.
77. Glennon JP, Ng Y-L, Setchell DJ, Gulabivala K. Prevalence of and factors affecting postpreparation pain in patients undergoing two-visit root canal treatment. Int Endod J. 2004;37:29.
78. Gold M. Tetrodotoxin-resistant Na currents and inflammatory hyperalgesia. Proc Natl Acad Sci U S A. 1999;96:7645.
79. Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci U S A. 1996;93:1108.
80. Goodis H, Bowles W, Hargreaves K. Prostaglandin E2 enhances bradykinin-evoked iCGRP release in bovine dental pulp. J Dent Res. 2000;79:1604.
81. Gordon SM. Blockade of peripheral neuronal barrage reduces postoperative pain. Pain. 1997;306:264.
82. Gordon SM, Brahim JS, Dubner R, et al. Attenuation of pain in a randomized trial by suppression of peripheral nociceptive activity in the immediate postoperative period. Anesth Analg. 2002;95:1351.
83. Gordon SM, Dionne RA, Brahim J, et al. Blockade of peripheral neuronal barrage reduces postoperative pain. Pain. 1997;70:209.
84. Gould HJ3rd, Gould TN, Paul D, et al. Development of inflammatory hypersensitivity and augmentation of sodium channels in rat dorsal root ganglia. Brain Res. 1999;824:296.
85. Gould HJ, England JD, Soignier RD, et al. Ibuprofen blocks changes in Nav1.7 and 1.8 sodium channels associated with complete Freund’s adjuvant-induced inflammation in rat. J Pain. 2004;5:270.
86. Hagermark O, Hokfelt T, Pernow B. Flare and itch produced by substance P in human skin. J Invest Dermatol. 1979;71:233.
87. Hargreaves K. Neurochemical factors in injury and inflammation in orofacial tissues. In: Lund JP, Lavigne GJ, Dubner R, Sessle BJ, editors. Orofacial pain: basic sciences to clinical management. Chicago: Quintessence Publishers, 2000.
88. Hargreaves K, Joris J. The peripheral analgesic effects of opioids. J Am Pain Soc. 1993;2:51.
89. Hargreaves K, Troullos E, Dionne R. Pharmacologic rationale for the treatment of acute pain. Dent Clin North Am. 1987;31:675.
90. Hargreaves KM, Swift JQ, Roszkowski MT, et al. Pharmacology of peripheral neuropeptide and inflammatory mediator release. Oral Surg Oral Med Oral Pathol. 1994;78:503.
91. Hargreaves KM. Pain mechanisms of the pulpodentin complex. In: Hargreaves KM, Goodis HE, editors. Seltzer and Bender’s dental pulp. Chicago: Quintessence Publishing Company, Inc; 2002:181.
92. Hargreaves KM, Dionne RA, Mueller GP, Goldstein DS, Dubner R. Naloxone, fentanyl, and diazepam modify plasma beta-endorphin levels during surgery. Clin Pharmacol Ther. 1986;40:165.
93. Hargreaves KM, Keiser K. New advances in the management of endodontic pain emergencies. J Calif Dental Assoc. 2004;32:469.
94. Hargreaves KM, Milam SB. Mechanisms of pain and analgesia. In: Dionne R, Phero J, editors. Management of pain and anxiety in dental practice. New York: Elsevier; 2001:18.
95. Hargreaves KM, Keiser K. Local anesthetic failure in endodontics: mechanisms and management. Endod Topics. 2002;1:26.
96. Harrison J, Baumgartner C, Zielke D. Analysis of interappointment pain associated with the combined use of endodontic irrigants and medicaments. J Endod. 1981;7:272.
97. Harrison J, Baumgartner J, Svec T. Incidence of pain associated with clinical factors during and after root canal therapy. I. Interappointment pain. J Endod. 1983;9:384.
98. Harrison J, Baumgartner J, Svec T. Incidence of pain associated with clinical factors during and after root canal therapy. II. Postobturation pain. J Endod. 1983;9:434.
99. Harrison J, Bellizzi R, Osetek E. The clinical toxicity of endodontic medicaments. J Endod. 1979;5:42.
100. Hasselgren G, Reit C. Emergency pulpotomy: pain relieving effect with and without the use of sedative dressings. J Endod. 1989;15:254.
101. Henry MA, Hargreaves KM. Peripheral mechanisms of odontogenic pain. Dent Clin North Am. 2007;51:19.
102. Hersh EV, Lally ET, Moore PA. Update on cyclooxygenase inhibitors: has a third COX isoform entered the fray? Curr Med Res Opin. 2005;21:1217.
103. Hill R NK1 (substance P) receptor antagonists–why are they not analgesic in humans? [see comment] Trends Pharmacol Sci 21 2000 244
104. Hofacker A, Coste O, Nguyen HV, Marian C, Scholich K, Geisslinger G. Downregulation of cytosolic prostaglandin E2 synthase results in decreased nociceptive behavior in rats. J Neurosci. 2005;25(39):9005-9009.
105. Holstein A, Hargreaves KM, Niederman R. Evaluation of NSAIDs for treating post-endodontic pain. Endod Topics. 2002;3:3.
106. Ianiro SR, Jeansonne BG, McNeal SF, Eleazer PD. The effect of preoperative acetaminophen or a combination of acetaminophen and ibuprofen on the success of inferior alveolar nerve block for teeth with irreversible pulpitis. J Endod. 2007;33:11.
107. Imura N, Zuolo M. Factors associated with endodontic flare-ups: a prospective study. Int Endod J. 1995;28:261.
109. Jackson D, Moore P, Hargreaves K. Preoperative nonsteroidal anti-inflammatory medication for the prevention of postoperative dental pain. J Am Dent Assoc. 1989;119:641.
110. Janig W, Kollman W. The involvement of the sympathetic nervous system in pain. Fortschr Arzneimittelforsch. 1984;34:1066.
111. Jebeles JA, Reilly JS, Gutierrez JF, et al. Tonsillectomy and adenoidectomy pain reduction by local bupivacaine infiltration in children. IntJ Ped Otorhinolaryngology. 1993;25:149.
112. Jeske NA, Diogenes A, Ruparel NB, et al. A-kinase anchoring protein mediates TRPV1 thermal hyperalgesia through PKA phosphorylation of TRPV1. Pain. 2008;138(3):604-616.
113. Johnsen DC, Harshbarger J, Rymer HD. Quantitative assessment of neural development in human premolars. Anat Rec. 1983;205:421.
114. Jostes J, Holland G. The effect of occlusal reduction after canal preparation on patient comfort. J Endod. 1984;10:34.
115. Juan H, Lembeck F. Action of peptides and other analgesic agents on paravascular pain receptors of the isolated perfused rabbit ear. Naunyn Schmiedebergs Arch Pharmacol. 1974;283:151.
116. Kaufman E, Hargreaves KM, Dionne RA. Comparison of oral triazolam and nitrous oxide with placebo and intravenous diazepam for outpatient premedication. Oral Surg Oral Med Oral Pathol. 1993;75:156.
117. Kaufman E, et al. Intraligamentary injection of slow-release methylprednisolone for the prevention of pain after endodontic treatment. Oral Surg Oral Med Oral Pathol. 1994;77:651.
118. Kearney PM, Baigent C, Godwin J, et al Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials [See comment.] BMJ 332 2006 1302
119. Keenan JV, Farman AG, Fedorowicz Z, Newton JT. A Cochrane systematic review finds no evidence to support the use of antibiotics for pain relief in irreversible pulpitis. J Endod. 2006;32:87.
120. Keiser K. Strategies for managing the endodontic pain patient. Texas Dent J. 2003;120:250.
121. Keiser K, Hargreaves K. Building effective strategies for the management of endodontic pain. Endod Topics. 2002;3:93.
122. Keiser K, Hargreaves KM. Strategies for managing the endodontic pain patient. J Tenn Dent Assoc. 2003;83:24.
123. Khan AA, Dionne RA. The COX-2 inhibitors: new analgesic and anti-inflammatory drugs. Dent Clin North Am. 2002;46:679.
124. Khan A, Diogenes A, Jeske N, Henry M, Akopian A, Hargreaves KM. Tumor necrosis factor alpha enhances the sensitivity of trigeminal ganglion neurons to capsaicin. Neuroscience. 2008;155:503-509.
125. Khan AA, Owatz CB, Schindler WG, et al. Measurement of mechanical allodynia and local anesthetic efficacy in patients with irreversible pulpitis and acute periradicular periodontitis. J Endod. 2007;33:796.
126. Khan AA, McCreary B, Owatz CB, et al. The development of a diagnostic instrument for the measurement of mechanical allodynia. J Endod. 2007;33:663.
127. Kim S. Regulation of pulpal blood flow. J Dent Res. 1985;64(Spec No):590.
128. Kis B, Snipes A, Bari F, Busija DW. Regional distribution of cyclooxygenase-3 mRNA in the rat central nervous system. Brain Res Mol Brain Res. 2004;126:78.
129. Krasner P, Jackson E. Management of posttreatment endodontic pain with oral dexamethasone: a double-blind study. Oral Surg Oral Med Oral Pathol. 1986;62:187.
130. Kress M, Sommer C. Neuroimmunology and pain: peripheral effects of proinflammatory cytokines. In: Brune K, Handwerker H, editors. Hyperalgesia: molecular mechanisms and clinical implications. Seattle: IASP Press; 2003:57.
131. Kumazawa T, Mizumura K. Thin-fiber receptors responding to mechanical, chemical and thermal stimulation in the skeletal muscle of the dog. Am J Physiol. 1977;273:179.
132. Figini GL, Gorni F, Gagliani M: Single versus multiple visits for endodontic treatment of permanent teeth. Cochrane Database Syst Rev 4:CD005296, 2007.
133. Lanza F, et al. Effect of acetaminophen on human gastric mucosal injury caused by ibuprofen. Gut. 1986;27:440.
134. Larson AM, Polson J, Fontana RJ, et al Acute liver failure study G: acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study [See comment.] Hepatology 42 2005 1364
135. Lepinski AM, Hargreaves KM, Goodis HE, Bowles WR. Bradykinin levels in dental pulp by microdialysis. J Endod. 2000;26:744.
136. Levine J, Moskowitz M, Basbaum A. The contribution of neurogenic inflammation in experimental arthritis. J Immunol. 1985;135:843.
137. Levine J, Taiwo Y. Inflammatory pain. In: IWPaM R, editor. Textbook of pain. Edinburgh: Churchill-Livingston, 1994.
138. Levine JD, Gordon NC, Fields HL. The mechanism of placebo analgesia. Lancet. 1978;2:654.
139. Lewin G, Rueff A, Mendell L. Peripheral and central mechanisms of NGF-induced hyperalgesia. Eur J Neurosci. 1994;6:1903.
140. Liesinger A, Marshall F, Marshall J. Effect of variable doses of dexamethasone on posttreatment endodontic pain. J Endod. 1993;19:35.
141. Light A. The initial processing of pain and its descending control: spinal and trigeminal systems. Basel: Karger; 1992.
142. Lilja JJ, Kivisto KT, Backman JT, Neuvonen PJ. Effect of grapefruit juice dose on grapefruit juice-triazolam interaction: repeated consumption prolongs triazolam half-life. Eur J Clin Pharmacol. 2000;56:411.
143. Lindahl O. Pain-a chemical explanation. Acta Rheumatol Scand. 1962;8:161.
144. Maddox D, Walton R, Davis C. Influence of posttreatment endodontic pain related to medicaments and other factors. J Endod. 1977;3:447.
145. Madison S, et al. Sensitizing effects of leukotriene B4 on intradental primary afferents. Pain. 1992;49:99.
146. Maixner W, Dubner R, Kenshalo DRJr, Bushnell MC, Oliveras JL. Responses of monkey medullary dorsal horn neurons during the detection of noxious heat stimuli. J Neurophysiol. 1989;62:437.
147. Malmberg A, Yaksh T. Antinociceptive actions of spinal nonsteroidal anti-inflammatory agents on the formalin test in rats. J Pharmacol Exp Ther. 1992;263:136.
148. Malmberg AB, Yaksh TL. Cyclooxygenase inhibition and the spinal release of prostaglandin E2 and amino acids evoked by paw formalin injection: a microdialysis study in unanesthetized rats. J Neurosci. 1995;15:2768.
149. Malmberg AB, Yaksh TL. Spinal nitric oxide synthesis inhibition blocks NMDA-induced thermal hyperalgesia and produces antinociception in the formalin test in rats. Pain. 1993;54:291.
150. Marshall G. Consideration of steroids for endodontic pain. Endod Topics. 2002;3:41.
151. Marshall J, Walton R. The effect of intramuscular injection of steroid on posttreatment endodontic pain. J Endod. 1984;10:584.
152. Marriott D, Wilkin GP, Coote PR, Wood JN. Eicosanoid synthesis by spinal cord astrocytes is evoked by substance P; possible implications for nociception and pain. Advances in Prostaglandin, Thromboxane, & Leukotriene Res. 1991;21B:739-741.
153. Martin H, et al. Leukotriene and prostaglandin sensitization of cutaneous high-threshold C- and A-delta mechanoreceptors in the hairy skin of rat hindlimbs. Neuroscience. 1987;22:651.
154. McDougal RA, Delano EO, Caplan D, Sigurdsson A, Trope M. Success of an alternative for interim management of irreversible pulpitis. J Am Dent Assoc. 2004;135:1707.
155. McGettigan P, Henry D Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2 [See comment.] JAMA 296 2006 1633
156. Mehrvarzfar P, Shababi B, Sayyad R, Fallahdoost A, Kheradpir K. Effect of supraperiosteal injection of dexamethasone on postoperative pain. Aust Endod J. 2008;34:25.
157. Meller ST, Dykstra C, Gebhart GF. Production of endogenous nitric oxide and activation of soluble guanylate cyclase are required for N-methyl-D-aspartate-produced facilitation of the nociceptive tail-flick reflex. Eur J Pharmacol. 1992;214:93.
158. Menhinick K, Gutmann J, Regan J, et al. The efficacy of pain control following nonsurgical root canal treatment using ibuprofen or a combination of ibuprofen and acetaminophen in a randomized, double-blind, placebo-controlled study. Int Endod J. 2003;37:531.
159. Meyer R, Campbell J. Myelinated nociceptive afferents account for the hyperalgesia that follows a burn to the hand. Science. 1981;213:1527.
160. Modaresi J, Dianat O, Mozayeni MA. The efficacy comparison of ibuprofen, acetaminophen-codeine, and placebo premedication therapy on the depth of anesthesia during treatment of inflamed teeth. Oral Surg Oral Med Oral Pathol. 2006;102:399.
161. Moore P. Long-acting local anesthetics: a review of clinical efficacy in dentistry. Compendium. 1990;11:24.
162. Moore P, et al. Analgesic regimens for third molar surgery: pharmacologic and behavioral considerations. J Am Dent Assoc. 1986;113:739.
163. Mor C, Rotstein I, Friedman S. Incidence of interappointment emergency associated with endodontic therapy. J Endod. 1992;18:509.
164. Morse D, et al. A comparison of erythromycin and cefadroxil in the prevention of flare-ups from asymptomatic teeth with pulpal necrosis and associated periapical pathosis. Oral Surg Oral Med Oral Pathol. 1990;69:619.
165. Morse D, et al. Infectious flare-ups and serious sequelae following endodontic treatment: a prospective randomized trial on efficacy of antibiotic prophylaxis in cases of asymptomatic pulpal-periapical lesions. Oral Surg Oral Med Oral Pathol. 1987;64:96.
166. Morse D, et al. Prophylactic penicillin versus erythromycin taken at the first sign of swelling in cases of asymptomatic pulpal-periapical lesions: a comparative analysis. Oral Surg Oral Med Oral Pathol. 1988;65:228.
167. Moskow A, et al. Intracanal use of a corticosteroid solution as an endodontic anodyne. Oral Surg Oral Med Oral Pathol. 1984;58:600.
168. Mulhern J, et al. Incidence of postoperative pain after one-appointment endodontic treatment of asymptomatic pulpal necrosis in single-rooted teeth. J Endod. 1982;8:370.
169. Mumford JM, Bowsher D. Pain and protopathic sensibility. A review with particular reference to the teeth. Pain. 1976;2:223.
170. Nakanishi T, Shimizu H, Hosokawa Y, Matsuo T, Hafez AA. An immunohistological study on cyclooxygenase-2 in human dental pulp. J Endod. 2001;27:385.
171. Nakanishi T, Shimuzu H, Matsuo T. Immunohistochemical analysis of cyclooxygenase-2 in human dental pulp. J Dent Res. 1999;78:142. abstract
172. Narhi M. The characteristics of intradental sensory units and their responses to stimulation. J Dent Res. 1985;64:564.
173. Narhi M, et al. Role of intradental A and C type nerve fibers in dental pain mechanisms. Proc Finn Dent Soc. 1992;88:507.
174. Narhi MV, Hirvonen T. The response of dog intradental nerves to hypertonic solutions of CaCl2 and NaCl, and other stimuli, applied to exposed dentine. Arch Oral Biol. 1987;32(11):781-786.
175. Nassar M, Stirling L, Forlani G, et al. Nociceptor-specific gene deletion reveals a major role for NAv 1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A. 2004;101:12706.
176. Neumann S, et al. Inflammatory pain hypersensitivity mediated by phenotype switch in myelinated primary sensory neurons. Nature. 1996;384:360.
177. Niv D. Intraoperative treatment of postoperative pain. In: IC J, editor. Pain 1996—an updated review. Seattle: IASP Press, 1996.
178. Nobuhara WK, Carnes DL, Gilles JA. Anti-inflammatory effects of dexamethasone on periapical tissues following endodontic overinstrumentation. J Endod. 1993;19:501.
179. O’Keefe E. Pain in endodontic therapy: preliminary study. J Endod. 1976;2:315.
180. Okeson JP. Assessment of orofacial pain disorders. In: Okeson JP, editor. Orofacial pain. Guidelines for assessment, diagnosis, and management. Chicago: Quintessence Publishing Company, Inc; 1996:35.
181. Oliet S. Single-visit endodontics: a clinical study. J Endod. 1983;9:147.
182. Owatz CB, Khan AA, Schindler WG, et al. The incidence of mechanical allodynia in patients with irreversible pulpitis. J Endod. 2007;33:552.
183. Paine MF, Widmer WW, Hart HL, et al A furanocoumarin-free grapefruit juice establishes furanocoumarins as the mediators of the grapefruit juice-felodipine interaction [Erratum appears in Am J Clin Nutr. 2006 Jul;84(1):264.] Am J Clin Nutr 83 2006 1097
184. Patwardhan A, Cerka K, Vela J, Hargreaves KM. Trigeminal nociceptors express prostaglandin receptor subtypes EP2 and EP3. J Dent Res. 2008;87:262.
185. Pekruhn R. Single-visit endodontic therapy: a preliminary clinical study. J Am Dent Assoc. 1981;103:875.
186. Penfield W, Rasmussen G. The cerebral cortex of man. New York: MacMillan; 1950.
187. Penniston S, Hargreaves K. Evaluation of periapical injection of ketorolac for management of endodontic pain. J Endod. 1996;22:55.
188. Perl E. Causalgia, pathological pain and adrenergic receptors. Proc Natl Acad Sci U S A. 1999;96:7664.
189. Perl ER Ideas about pain, a historical view [Review] [140 refs] Nature Revi Neuroscie 8 1 2007 71-80
190. Petty Bea. The effect of systemically administered recombinant human nerve growth factor in healthy human subjects. Ann Neurol. 1994;36:244.
191. Pohto P. Sympathetic adrenergic innervation of permanent teeth in the monkey (Macaca irus). Acta Odontol Scand. 1972;30:117.
192. Pohto P, Antila R. Demonstration of adrenergic nerve fibres in human dental pulp by histochemical fluorescence method. Acta Odontol Scand. 1968;26:137.
193. Pomonis JD, Rogers SD, Peters CM, Ghilardi JR, Mantyh PW. Expression and localization of endothelin receptors: implications for the involvement of peripheral glia in nociception. J Neurosci. 2001;21:999.
194. Roane J, Dryden J, Grimes E. Incidence of postoperative pain after single- and multiple-visit endodontic procedures. Oral Surg Oral Med Oral Pathol. 1983;55:68.
195. Rogers MJ, Johnson BR, Remeikis NA, BeGole EA. Comparison of effect of intracanal use of ketorolac tromethamine and dexamethasone with oral ibuprofen on post treatment endodontic pain. J Endod. 1999;25:381.
196. Rollason V, Samer C, Piguet V, Dayer P, Desmeules J. Pharmacogenetics of analgesics: toward the individualization of prescription. Pharmacogenomics. 2008;9:905.
197. Rosenberg P. Clinical strategies for managing endodontic pain. Endod Topics. 2002;3:78.
198. Rowe N, et al. Control of pain resulting from endodontic therapy: a double-blind, placebo-controlled study. Oral Surg Oral Med Oral Pathol. 1980;50:257.
199. Roy ML, Narahashi T. Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J Neurosci. 1992;12:2104.
200. Ruparel NB, Patwardhan AM, Akopian A, Hargreaves KM. Homologous and heterologous desensitization of capsaicin and mustard oil responses utilize different cellular pathways in nociceptors. Pain. 2008;135:271-279.
201. Schaible H, Schmidt R. Discharge characteristics of receptors with fine afferents from normal and inflamed joints: influence of analgesics and prostaglandins. Agents Actions. 1986;19:99.
202. Schwab JM, Schluesener HJ, Meyermann R, Serhan CN. COX-3 the enzyme and the concept: steps towards highly specialized pathways and precision therapeutics? Prostaglandins Leukot Essent Fatty Acids. 2003;69:339.
203. Seltzer S, Bender IB, Ehrenreich J. Incidence and duration of pain following endodontic therapy. Relationship to treatment with sulfonamides and to other factors. Oral Surg Oral Med Oral Path. 1961;14:74.
204. Sessle B. Recent developments in pain research: central mechanisms of orofacial pain and its control. J Endodon. 1986;12:435.
205. Sessle BJ. Acute and chronic craniofacial pain: brainstem mechanisms of nociceptive transmission and neuroplasticity, and their clinical correlates. Crit Rev Oral Biol Med. 2000;11:57.
206. Sessle BJ. Recent developments in pain research: central mechanisms of orofacial pain and its control. J Endod. 1986;12:435.
207. Sessle BJ, Greenwood LF. Inputs to trigeminal brain stem neurones from facial, oral, tooth pulp and pharyngolaryngeal tissues: I. Responses to innocuous and noxious stimuli. Brain Res. 1976;117:211.
208. Sessle BJ, Hu JW, Amano N, Zhong G. Convergence of cutaneous, tooth pulp, visceral, neck and muscle afferents onto nociceptive and non-nociceptive neurones in trigeminal subnucleus caudalis (medullary dorsal horn) and its implications for referred pain. Pain. 1986;27:219.
209. Siqueira J, Barnett F. Interappointment pain: mechanisms, diagnosis, and treatment. Endod Topics. 2004;3:93.
210. Soltanoff W. A comparative study of the single-visit and the multiple-visit endodontic procedure. J Endod. 1978;4:278.
211. Stambaugh J, Drew J. The combination of ibuprofen and oxycodone/acetaminophen in the management of chronic cancer pain. Clin Pharmacol Ther. 1988;44:665.
212. Steen Kea. Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin in vitro. J Neurosci. 1992;21:86.
213. Stein C. The control of pain in peripheral tissues by opioids. N Engl J Med. 1995;332:1685.
214. Sundqvist G, Lerner UH. Bradykinin and thrombin synergistically potentiate interleukin 1 and tumour necrosis factor induced prostanoid biosynthesis in human dental pulp fibroblasts. Cytokine. 1996;8:168.
215. Svensson CI, Yaksh TL. The spinal phospholipase-cyclooxygenase-prostanoid cascade in nociceptive processing. Ann Rev Pharmacol Toxicol. 2002;42:553.
216. Sweitzer SM, Schubert P, DeLeo JA. Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J Pharmacol Exp Ther. 2001;297:1210.
217. Tominaga M, Numazaki M, Iida T, et al Molecular mechanisms of TRPV1-mediated thermal hypersensitivity vol 30 2003 IASP Press Seattle p 37
218. Tonioli M, Patel T, Diogenes A, et al. Effect of neurotrophic factors on bradykinin expression in rat trigeminal sensory neurons determined by real-time polymerase chain reaction. J Endod. 2004;30:263.
219. Torabinejad M, Cymerman JJ, Frankson M, et al. Effectiveness of various medications on postoperative pain following complete instrumentation. J Endod. 1994;20:345.
220. Torabinejad M, Kettering JD, McGraw JC, et al. Factors associated with endodontic interappointment emergencies of teeth with necrotic pulps. J Endod. 1988;14:261.
222. Torabinejad M, et al. Effectiveness of various medications on postoperative pain following root canal obturation. J Endod. 1994;20:427.
223. Torabinejad M, Walton RE. Managing endodontic emergencies. J Am Dent Assoc. 1991;122(5):99. 101, 103
224. Trope M. Flare-up rate of single-visit endodontics. Int Endod J. 1991;24:24.
225. Trope M. Relationship of intracanal medicaments to endodontic flare-ups. Endod Dent Traumatol. 1990;6:226.
226. Troullos E, Freeman R, Dionne R. The scientific basis for analgesic use in dentistry. Anesth Prog. 1986;33:123.
227. Trowbridge HO. Review of dental pain–histology and physiology. J Endod. 1986;12:445.
228. van Wijk AJ, Hoogstraten J. Reducing fear of pain associated with endodontic therapy. Int Endod J. 2006;39:384.
229. Wagner R, Myers R. Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. Neuroreport. 1996;7:2897.
230. Wallace J. Selective COX-2 inhibitors: is the water becoming muddy? Trends Pharmacol Sci. 1999;20:4.
231. Walton R. Interappointment flare-ups: incidence, related factors, prevention, and management. Endod Topics. 2002;3:67.
232. Walton R, Chiappinelli J. Prophylactic penicillin: effect on posttreatment symptoms following root canal treatment of asymptomatic periapical pathosis. J Endod. 1993;19:466.
233. Walton R, Fouad A. Endodontic interappointment flare-ups: a prospective study of incidence and related factors. J Endod. 1992;18:172.
234. Wang C, Li GW, Huang LY. Prostaglandin E2 potentiation of P2X3 receptor mediated currents in dorsal root ganglion neurons. Molecular Pain. 2007;3:22.
235. Warren C, Mok L, Gordon S, Fouad AF, Gold M. Quantification of neural protein in extirpated tooth pulp. J Endod. 2008;34:7.
236. Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450.
237. Wells J, Bingham V, Rowland K, Hatton J. Expression of Nav1.9 channels in human dental pulp and trigeminal ganglion. J Endod. 2007;33:1172.
238. Wideman G, et al. Analgesic efficacy of a combination of hydrocodone with ibuprofen in postoperative pain. Clin Pharmacol Ther. 1999;65:66.
239. Willingale HL, Gardiner NJ, McLymont N, Giblett S, Grubb BD. Prostanoids synthesized by cyclo-oxygenase isoforms in rat spinal cord and their contribution to the development of neuronal hyperexcitability. Br J Pharmacol. 1997;122:1593.
240. Wolf M, Lichtenstein D, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. New Engl J Med. 1999;340:1888.
241. Wong M, Lytle WR. A comparison of anxiety levels associated with root canal therapy and oral surgery treatment. J Endod. 1991;17:461.
242. Woolf C. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686.
243. Woolf C. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci U S A. 1999;96:7723.
244. Woolf C. Windup and central sensitization are not equivalent. Pain. 1996;66:105.
245. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10(9):895-926.
246. Wright C, et al. Ibuprofen and acetaminophen kinetics when taken concurrently. Clin Pharmacol Ther. 1983;34:707.
247. Wright EF Referred craniofacial pain patterns in patients with temporomandibular disorder [See comment][erratum appears in J Am Dent Assoc 2000 Nov;131(11):1553.] J Am Dent Assoc 131 2000 1307
248. Wynn R, Meiller T, Crossley H. Drug information handbook for dentistry. Hudson, Ohio: Lexi-Comp Inc; 2000.
249. Xiao W-Hea. TNF-alpha applied to the sciatic nerve trunk elicits background firing in nociceptive primary afferent fibers. In: Abstracts 8th world congress on pain. Seattle: IASP Press; 1996.
250. Yaksh TL. Central pharmacology of nociceptive transmission. In: Wall P, Melzack R, editors. Textbook of pain. Edinburgh: Churchill Livingstone; 2002:285.
251. Yingling NM, Byrne BE, Hartwell GR. Antibiotic use by members of the American Association of Endodontists in the year 2000: report of a national survey. J Endod. 2002;28:396.
252. Yonehara N, Amano K, Kamisaki Y. Involvement of the NMDA-nitric oxide pathway in the development of hypersensitivity to tactile stimulation in dental injured rats. Jpn J Pharmacol. 2002;90:145.