CHAPTER 14 Pathobiology of the Periapex
Periradicular tissues consist of cementum, periodontal ligament, and alveolar bone. Cementum is a mineralized, avascular connective tissue and consists of three different types. Acellular afibrillar cementum covers the teeth at and along the cemento-enamel junction. Acellular extrinsic fiber cementum is confined to the coronal half of the root. Cellular intrinsic fiber cementum is present on the apical half of the root where no acellular extrinsic fiber cementum has been laid down.171 Many growth factors, such as insulin-like growth factor-1 (IGF-1), fibroblast growth factors (FGFs), epidermal growth factor (EGF), bone morphogenetic proteins (BMPs), transforming growth factor-β (TGF-β), and platelet-derived growth factor (PDGF) are contained in the cementum matrix.41,81,147 These growth factors may be released under certain conditions, since they have been shown to be associated with cementoblast proliferation, migration, and differentiation during cementum wound healing.81
The periodontal ligament is a soft, specialized connective tissue that connects the cementum to the alveolar bone. Periodontal ligament contains heterogeneous cell populations128 and extracellular matrix (ECM). The cells of the periodontal ligament include osteoblasts, osteoclasts, fibroblasts, epithelial cell rests of Malassez, macrophages, cementoblasts, and undifferentiated mesenchymal cells (stem cells).171 Fibroblasts, osteoblasts, and epithelial cells are differentiated cells that have retained the ability to undergo cell division and proliferation upon stimulation by appropriate signals. Multipotent stem cells of the periodontal ligament are capable of differentiating into cementoblast-like cells and periodontal ligament cells as well as osteoblasts.102,161,209 The ECM of the periodontal ligament consists of collagen fibers, fibronectin, elastin, other noncollagenous proteins, and proteoglycans. The ECM serves as stratum for cell adhesion and promotes cell spreading and cytoskeletal organization. The collagen fibers (Sharpey’s fiber) of the periodontal ligament connect the tooth with the alveolar bone. The periodontal ligament is highly vascularized and innervated. The tissue apical to the dentinocemental junction should be considered part of the periodontal ligament because cementum is not a normal component of the pulp tissue.
Epithelial cell rests of Malassez (ERM), the remnants of Hertwig’s epithelial root sheath that disintegrates after tooth development, are present in the periodontal ligament near the root surface in all teeth after root formation.187 They are nests of epithelial cells connected as a network and surrounded by a basal lamina.171 ERM are quiescent in normal periodontal ligament171 but can be stimulated to proliferate in apical periodontitis.139 They are believed to be the cellular source that when properly stimulated can form radicular cysts in certain apical periodontitis lesions.139,171,178
Alveolar bone or alveolar process is that part of bone of the jaws housing the sockets for the teeth. It consists of outer cortical plate, a central spongy or cancellous bone, and bone lining the sockets.171 Bone matrix contains IGFs, TGF-β, BMPs, FGF, and PDGF.36,222 These growth factors are essential for osteoblast proliferation, migration, and differentiation during bone wound healing.145
The response of the periradicular tissues to various injuries is similar to that of other connective tissues elsewhere in the body. The response is manifested as an immunoinflammatory reaction. Although microbial infection of the pulp in the root canals is the primary cause of apical periodontitis,108,158 the pathologic changes of the periapical tissues in apical periodontitis are usually not directly caused by microbes themselves, but rather by their toxins, noxious metabolic byproducts, and disintegrated pulp tissue in the root canal system. These irritants are capable of inducing both innate and adaptive immune responses; they can activate either nonantigenic pathways or serve as antigens to activate adaptive responses. The host’s immunoinflammatory responses are quite diverse and can involve changes in microvasculature, transmigration of blood-borne cells and plasma proteins out of the blood circulation into the tissue space, and activation of sensory nerves. In addition, endothelial cells, mast cells, platelets, fibroblasts, neutrophils, macrophages, dendritic cells, innate and adaptive immune cells, immunoglobulins, inflammatory mediators, proinflammatory cytokines, chemokines, and neuropeptides are also involved in the immunoinflammatory response. Apical periodontitis can be protective or destructive, depending on the dynamic interaction between microbial insult and the host’s defenses in the periapical tissues. Unfortunately, the bacterial biofilm formed in the root canal system with necrotic pulp is protected from host’s defenses and antibiotic therapy because of a lack of blood circulation in the root canal system. Consequently, any attempt of wounded periradicular tissues to repair/regenerate is futile, since bacterial toxins and noxious metabolic byproducts in the root canal system continuously egress into the periapical area and irritate the periapical tissues. Emerging lines of evidence suggest that under most conditions, bacterial biofilms in the complex root canal system can be greatly reduced, but not eliminated, by conventional endodontic procedures such as mechanical instrumentation, antiseptic irrigation, and intracanal medication. If microbes in the root canal system are effectively eliminated or entombed within the root canal filling material, and the root canal system is adequately sealed and protected from coronal microleakage, then periradicular tissues have the ability to restore their original structures by means of a repair/regeneration process. Nevertheless, the presence of posttreatment apical periodontitis may be due to persistent microbial biofilms,70 and this recognition has spurred considerable research into treating biofilms (see Chapters 9 and 15).
Apical Periodontitis
Prevalence
Epidemiologic study of apical periodontitis documents that the prevalence of apical periodontitis varies among patients aged 20 to 30 (33% prevalence of apical periodontitis), 30 to 40 (40%), 40 to 50 (48%), 50 to 60 (57%), and older than 60 years of age (62%).181 Most studies on the prevalence of apical periodontitis are from European and Scandinavian countries.64,106,212 According to a survey by the American Dental Association in 1990, an estimated 14 million root canal treatments were performed in the United States alone.8 Apical periodontitis is a very prevalent problem.65
Etiology
The etiology, pathogenesis, and histopathology of apical periodontitis are very similar to that of marginal periodontitis (see also Chapter 15). Both diseases are caused by bacterial infection and involve pathologic changes of alveolar bone, periodontal ligament, and cementum. Marginal periodontitis affects coronal periodontal tissues, whereas apical periodontitis affects apical periodontal tissues. Bone loss is one of characteristic features in both diseases: crestal bone is lost in marginal periodontitis, and apical bone undergoes resorption in apical periodontitis.
Apical periodontitis can be caused by both exogenous and endogenous factors. Exogenous factors include microbes and their toxins and noxious metabolic byproducts, chemical agents, mechanical irritation, foreign bodies, and trauma. Endogenous factors include the host’s metabolic products, such as urate and cholesterol crystals, as well as cytokines or other inflammatory mediators that activate osteoclasts. These irritants can activate nonantigenic pathways or antigenic pathways to induce innate and adaptive immunoinflammatory responses, respectively.
In the root canal system, infection of the pulp tissue caused by caries or other pathways is the primary cause of apical periodontitis.108,158,232 The classic study of Kakehashi et al.108 demonstrated that pulp necrosis and periradicular inflammation developed in conventional rats when the pulps of teeth were exposed to oral microorganisms. However, in germ-free laboratory rats, no pulp necrosis and periradicular inflammation occurred even when the pulps of teeth were exposed to the oral environment and packed with sterile food debris. A similar response occurs in humans. Using bacterial culturing, it has been demonstrated that traumatized human teeth with intact crowns and necrotic pulps without bacterial contamination did not show radiographic evidence of periapical bone destruction. In contrast, if bacteria were isolated from traumatized teeth with intact crowns and necrotic pulps, radiographic evidence of periradicular bone destruction was observed.232 These important findings have been replicated in nonhuman primate experiments. When the pulps of intact vital teeth were intentionally devitalized under aseptic conditions and left in the root canals with bacteria-tight, sealed coronal restoration for 6 months to 1 year, no periradicular inflammatory reaction was observed.137,158 Taken together, there is considerable evidence that bacteria constitute a major etiologic factor in the development of apical periodontitis.
Bacterial toxins (e.g., lipopolysaccharide [LPS], lipoteichoic acid [LTA]) and noxious metabolic byproducts that egress from the root canal system into the periapical tissues are capable of inducing a periapical immunoinflammatory reaction.47,58,203,280 These substances can activate the innate immune system via receptors that recognize the stereotypic pathogen-associated molecular patterns (PAMPs) that are found in the structure of these toxins. Different classes of microbes express different molecular patterns that are recognized by different pattern recognition receptors (PRRs) or toll-like receptors (TLRs) on host cells, such as phagocytes, dendritic cells, and B lymphocytes.1,154 PRRs or TLRs are encoded in the germline. In mammalian species, there are at least 10 TLRs, and each appears to have a distinct function in innate immune recognition.154 For example, LPS can stimulate sensory nerve fibers to release calcitonin gene–related peptide (CGRP) and substance P (SP)55,96 to cause vasodilation and increased vascular permeability. LPS and lipoproteins can also activate TLRs on dendritic cells to stimulate T lymphocyte differentiation.4 The common shared structural features of various toxins (i.e., PAMPS) are recognized by certain subtypes of TLRs. Since the TLRs are synthesized before an infection, they are classified as part of the innate immune system.
Apical periodontitis can be caused either by entry into the periapical tissues of bacterial toxins, enzymes, and noxious metabolic byproducts or by direct invasion of the periapical tissues by microbes originating from the root canal system. It is important to differentiate between apical inflammation and apical infection. Apical inflammation is the periapical tissue reaction to irritants emerging from the root canal system that manifests as vasodilation, increased vascular permeability and exudation. In contrast, apical infection is due to the physical presence of pathogenic microorganisms in the periapical tissues that subsequently produce tissue damage. There can be infection without inflammation, for instance, in a severely immunocompromised patient. There can also be inflammation without infection, such as in a myocardial infarct, cerebral infarct, physical, or chemical injury.148 In diseases caused by infection, bacteria are usually present in the involved tissues or organs,265 such as acute necrotizing gingivitis, marginal periodontitis, actinomycosis, tuberculosis, and bacterial bronchitis. Although apical periodontitis is primarily an infectious disease, bacteria are usually not present in the periapical tissues but in the root canal system,123,162,267 except in certain cases of apical periodontitis associated with abscess formation,179,262,274 a draining sinus tract,83,181,272 or extraradicular endodontic infection.231,255 One major current hypothesis is that apical periodontitis is triggered by entry into the periapical tissues of bacterial toxins, enzymes, and noxious metabolic byproducts.240 The mere presence of bacteria (colonization) in some apical periodontitis lesions does not necessarily denote a periradicular infection. Periapical infection is related to both virulence and the number and perhaps combinations of microorganisms in the periapical tissues.235 Bacteria may be temporarily present in the inflamed periradicular tissues only to be killed by the host’s defense mechanisms when the focus of infection in the root canal system is effectively eliminated by mechanical instrumentation, antiseptic irrigation, and intracanal medication. For instance, the majority of apical periodontitis lesions with abscess formation or draining sinus tracts heal satisfactorily after nonsurgical root canal treatment without the need for systemic antimicrobial therapy.181
Primary root canal infection in untreated root canals is a polymicrobial mix with approximately equal proportions of gram-positive and gram-negative species, dominated by obligate anaerobes (see Chapter 15).233,235 In root-filled teeth with apical periodontitis, gram-positive microorganisms, with a relatively equal distribution of facultative and anaerobic species, appear to dominate other microorganisms.156,236 A high prevalence of E. faecalis is frequently observed in filled root canals associated with persistent apical periodontitis.* These issues are described in greater detail in Chapter 15.
Physical (overinstrumentation, overfilling) and chemical (irrigants, intracanal medication, root canal filling materials) insults,205 as well as traumatic injury10 to the periapical tissues, can also cause apical periodontitis, depending on the severity of injury and cytotoxicity of the chemicals. Foreign bodies, such as root canal filling materials, have been shown to cause persistent periapical inflammation.91,118,166,183,282 However, the possibility of bacterial contamination in foreign body–induced apical periodontitis lesions was not carefully ruled out in many studies, so it is possible that the foreign bodies served as carriers for the microorganisms. In addition, foreign bodies have the odd property of favoring infection because they cause granulocytes to develop phagocytic defect, loss of ammunition.283 Although most root canal filling materials are not inert and are capable of inducing certain degrees of inflammation, in general they are biocompatible and well tolerated by periapical tissues.91
Infection: A Conflict Between Host and Parasites
Every infection is a race between the capabilities of the microorganism to multiply, spread, and cause disease and the ability of the host to control and finally eliminate the microorganisms and resulting infection.265 The host has physical barriers—surface epithelium, enamel, and dentin—as well as innate and adaptive immune defenses to prevent pulpal and periapical infection. Nevertheless, parasites also possess weapons, leading to inhibition of phagocytosis, inhibited lysosomal function, reduced killing by phagocytes, inactivation of complement system and immunoglobulins, and specific mechanisms that permit invasion of the host’s physical barriers.265 Infection of a tissue can manifest different histopathologic features as a result of specific host-parasite interactions that occur. Many infections are asymptomatic in more than 90% of individuals.265 For instance, pulp necrosis and chronic apical periodontitis caused by root canal infection are usually asymptomatic, and patients are often surprised to find out that this infection has been present for a sufficient time to lead to destruction of periapical bone. Therefore, there is no simple correlation between infection and clinical symptoms of apical periodontitis, except in cases of symptomatic apical periodontitis and acute apical abscess.
Pathogenesis
When pulps are infected/inflamed, many innate and adaptive immune cells release elevated amounts of various inflammatory mediators, including cytokines, chemokines, and neuropeptides. As the pulpal inflammation spreads, the inflammatory mediators begin to alter the physiology of the periapical tissues. Clinically, the observable changes on radiographic examination are widening of the periodontal ligament space or development of apical osteolytic lesions due to bone resorption. The loss of bone is mainly caused by activated osteoclasts. Many cytokines, such as interleukin (IL)-1, IL-11, IL-17, and tumor necrosis factor α (TNF-α), are found to have the ability to induce osteoclast differentiation and activation.21 The inflammation-induced bone resorption in the periapical tissues is accompanied by recruitment of immune cells that essentially build a defensive line against the spread of microbial invasion from the root canal.152 The pathogenesis of apical periodontitis involves innate and adaptive immune responses as well as sensory nerve response in the periapical tissues. Immune cells present in human periradicular lesions consist of lymphocytes, macrophages, plasma cells, neutrophils, and natural killer (NK) cells with the former two types as the majority.135,151 The characteristic features of innate and adaptive immunity are summarized in Table 14-1.
TABLE 14-1 Features of Innate and Adaptive Immunity
| Property | Innate | Adaptive |
|---|---|---|
| Recognition | Structures shared by groups of related microbes (conserved molecular patterns) | Antigens of microbes and of nonmicrobial antigens (details of molecular structure) |
| Diversity | Limited | Very large |
| Memory | None | Yes |
| Receptors | Encoded in the genome | Encoded in gene segments (somatic recombination) |
| Blood proteins | Complement | Antibodies |
| Cells | Macrophages | Lymphocytes |
| Neutrophils | ||
| NK cells | ||
| Action time | Immediate activation of effectors | Delayed activation of effectors |
| Response | Costimulatory molecules, Cytokines | |
| (IL-1, IL-6) | ||
| IL-2 | Clonal expansion | |
| Chemokines (IL-8) | Effector cytokines (IL-4, IFNr) |
IFN, Interferon; IL, interleukin; NK, natural killer.
Data from Janeway CA, Medzhitov R: Innate immune recognition. Annu Rev Immunol 20:197, 2002; and Abbas AK, Lichtman AH, Pober JS: Cellular and molecular immunology, ed 5, Philadelphia, Saunders, 2003.
Innate Immune Response
Specificity of Innate Immune Response
In recent years, the concept of the nonspecific nature of innate immunity has changed since identification of a network of germline-encoded receptors, the pattern-recognition receptors (PRRs) mentioned earlier, that recognize specific molecular motifs of microorganisms.5,153 PRRs can be expressed on the cell surface (macrophages, dendritic cells, neutrophils, NK cells, B cells), in intracellular compartments, or secreted into the blood and tissue fluids.104 There are numerous microbial constitutive and conserved products, the pathogen-associated molecular patterns (PAMPs), also noted earlier. Importantly, the PRRs of the innate immune system recognize PAMPs.5,153
The specificity of innate immunity is due to the recognition of PAMPs of microorganisms by PRRs, also called toll-like receptors (TLRs), of the host’s cells. Activation of PRRs triggers numerous host responses, including opsonization, activation of complement and coagulation cascades, phagocytosis, activation of proinflammatory signaling pathways, and induction of apoptosis.104 For example, TLR4/CD14 is the receptor for the gram-negative bacterial LPS. TLR4-mutated C3H/HeJ mice (LPS hyporesponsive) have reduced response to gram-negative bacteria and are highly susceptible to infection by Salmonella typhimurium or Neisseria meningitidis.42 Importantly, there is a reduced expression of IL-1 and IL-12 and decreased periradicular bone destruction in TLR-4 deficient mice when teeth are subjected to pulpal exposures and infection with a mixture of four anaerobic pathogens: Prevotella intermedia, Fusobacterium nucleatum, Streptococcus intermedius (G+), and Peptostreptococcus micros (G+).94 In addition, LPS is shown to be capable of inducing pain via direct activation of TLR4/CD14 expressed on nociceptive sensory neurons.264 Thus, the TLR4 PRR receptor is importantly involved in odontogenic infections.
Components such as lipoteichoic acid (LTA) of gram-positive bacterial cell walls can also stimulate innate immunity in a way similar to LPS. TLR2 plays a major role in detecting gram-positive bacteria and is involved in the recognition of a variety of microbial components, including LTA, lipoproteins, and peptidoglycan. The importance of TLR2 in the host defense against gram-positive bacteria has been demonstrated using TLR2-deficient (TLR2−/−) mice, which were found to be highly susceptible to challenge with Staphylococcus aureus or Streptococcus pneumoniae.60,241 LTA also stimulates leukocytes to release inflammatory mediators, including TNF-a, IL-1b, IL-6, IL-8, and prostaglandin (PG) E2, which are known to play a role in various phases of the inflammatory response. All these inflammatory mediators have been detected in periapical samples, and each has a well-known tissue-damaging effect by activating various host responses.
The innate immune response to bacterial infection induces expression of proinflammatory cytokines, chemokines, and costimulators, which are essential for activation and influence of the nature of adaptive immune response.4,154 The innate immune system is capable of recognizing nonself and self-antigens, while the adaptive immune system does not; thus, many autoimmune diseases are disorders of adaptive immunity.154
Nonspecific Innate Immune Response
The primary nonspecific innate immune defense mechanism in apical periodontitis is phagocytosis of microbes by specialized phagocytes such as polymorphonuclear leukocytes (PMNs) and macrophages. Tissue inflammation leads to the recruitment of PMNs from the blood circulation into the periradicular tissue. Activated PMNs exhibit an abrupt increase in oxygen consumption, the well-known respiratory burst, resulting in the release of oxygen radicals, a family of extremely destructive, short-lived substances that destroy nearby microorganisms and host cells.12 Phagocytosed microbes or foreign particles are exposed to a very toxic environment containing specific and azurophil granules and oxygen-derived free radicals and are eventually degraded.173 PMNs also possess an extracellular killing mechanism via neutrophil extracellular traps (NETs), which are extracellular structures composed of chromatin with specific proteins from the neutrophilic granules. Upon activation (e.g., by IL-8, LPS, bacteria, fungi, activated platelets), neutrophils start a cellular program called apoptosis that leads to their death and the formation of NETs, which have antimicrobial activities.27,71 Besides their role in the innate immunity as professional phagocytes, macrophages also serve as antigen-presenting cells by expressing MHC class II molecules that interact with antigen-specific clones of T-helper lymphocytes. Circulating monocytes are the precursors of both tissue macrophages and many dendritic cell subsets.1,208 The details of the immunologic activities of neutrophils and macrophages in periapical pathosis are described in Chapters 12 and 13.
Adaptive/Specific Immune Response
The specificity of adaptive immunity is regulated at genetic levels in B and T lymphocytes through a complex process leading to the generation of molecules that recognize and bind to foreign or self-antigens. These molecules are specific receptors on T cells (T-cell antigen receptors or TCRs) and on B cells (B-cell antigen receptor or BCRs; also termed immunoglobulins). TCRs on T cells interact with antigens that are presented by MHC molecules along with other accessory molecules, whereas BCRs on B cells interact with antigens directly. BCRs may be secreted in the blood circulation or in the tissues as antibodies. The variable region of both TCR and BCR proteins are rearranged at the genomic level via genetic recombination of the V(D)J segments. The estimated total diversity after this recombination for TCR is approximately 1018 and for BCR is approximately 1014 that generates the repertoire of different individual T and B cell clones.1,105 Each T or B cell clone generated in the bone marrow carries a specific TCR and BCR. They undergo a positive and negative selection process through which most clones are deleted via apoptosis because they bind to self-antigens. This initial “negative screening” process serves to reduce the potential for autoimmune disorders. Only those that do not interact with self-antigens are released into the lymphatic system and blood circulation. The naïve T cells circulate back and forth between the lymphatic system and blood circulation until they encounter foreign antigens presented by antigen-presenting cells. About 97% of T cells undergo apoptosis, and only a small percentage of these cells are exported to the periphery as mature T cells.1
The interaction between TCR and the antigen peptide/MHC complex and costimulators activate T cells, leading to the synthesis of T-cell growth factor, IL-2, and its receptor that causes T-cell clonal expansion/proliferation. Some of these T cells differentiate into armed, effector T cells and other become memory cells. There are a number of T-cell subpopulations, categorized by their functions: (1) T helper cells (TH), (2) T regulatory cells (Treg), (3) T suppressor cells (TS), and (4) T cytotoxic (cytolytic) (TC) cells.1,50,261 Some of them can be distinguished by their cell surface markers, cytokine profiles, or transcriptional factors. See also Chapter 13 for additional details.
Upon antigen stimulation, naïve CD4 T cells proliferate and differentiate into TH0, which are subsequently committed to develop into TH1 or TH2 cells. Monocytoid DC (DC1) induces TH1-type responses; plasmacytoid DC (DC2) selectively induces TH2 responses. Each subset of TH cells has distinct functions and cytokine profiles. TH1 cells mainly produce IL-2 and interferon (IFN)-γ, which activate macrophages and induce B cells to produce opsonizing antibody. TH2 cells produce IL-4, -5, -10, and -13, which activate B cells to make neutralizing antibody. Overall, TH1 and TH2 have mutually inhibitory effects.1 The development of CD4 TH cells involves the encounter of antigen presented by antigen-presenting cells (APCs) in association with class II MHC. All cells express MHC class I, but only certain cells express class II MHC. These class II MHC–expressing cells comprise the body’s population of APCs and consist of (1) dendritic cells, (2) macrophages, (3) B cells, (4) vascular endothelial cells, and (5) epithelial cells. The former three are considered “professional” APCs, since they are dedicated to this function. The latter two APCs are quiescent under normal conditions but can be induced to express class II MHC when exposed to elevated concentrations of IFN-γ.1,105
Dendritic cells and macrophages phagocytose foreign antigens, while B cells utilize the membrane-bound immunoglobulin to bind and internalize antigens. Other APCs endocytose foreign antigens into the cytoplasm for antigen processing. The processed antigens are partially degraded into small peptides. Many of them are 10 to 30 amino acids long and capable of binding onto the newly synthesized class II MHC molecules before the antigen/class II MHC complex is transported to the surface of cells and presented to TCRs of CD4+ T cells.
While controversial, evidence has suggested that CD8+ TS and CD8+ CTLs represent distinct subpopulations of CD8+ T cells. TS are MHC class I–restricted CD8+/CD28− TS cells, which act on antigen-presenting cells (APC) by a contact-dependent manner, rendering them tolerogenic to TH cells. They inhibit the proliferation of TH cells.44,285 T cytotoxic cells (CD8+TC), also known as cytolytic T lymphocytes (CTLs), are a subset of T cells that kill target cells expressing MHC-associated peptide antigens. The majority of TC express CD8 and recognize antigens degraded in the cytosol and expressed on the cell surface in association with class I MHC molecules of the target cells. Functional TC acquire specific membrane-bound cytoplasmic granules, including a membrane pore-forming protein called perforin or cytolysin and enzymes called granzymes.1
The role of B cells in adaptive immunity is mainly the production of antibodies that constitute the host humoral immune response. The V(D)J gene recombination occurs in both heavy and light (κ and λ) chains. A recombinase system, which is an enzymatic complex consisting of several enzymes—including recombination activating gene 1 and 2 (RAG-1, RAG-2), terminal deoxynucleotidyl transferase (TdT), DNA ligase IV, Ku proteins, and XRCC4—is essential for successful recombination. This recombinase system is also used for TCR recombination. Mature IgM/IgD coexpressing B cells undergo isotype switching via a process called switch recombination after encountering antigen. The rearranged V(D)J gene segment recombines with a downstream C region gene (γ, ε, or α), and the intervening DNA sequence is deleted. This gives rise to other classes of immunoglobulins (IgG, IgE, IgA) besides IgM. In addition to isotype switching, activated B cells also undergo somatic mutation in the V region gene, leading to affinity maturation of antibodies and alternative splicing of VDJ RNA to membrane or secreted immunoglobulin mRNA. A large quantity of antibody is secreted when B cells terminally differentiate into plasma cells.1,105 The ability of antigens to selectively stimulate the differentiation of plasma cells supports the clinical finding that plasma cells isolated from periapical lesions secrete antibodies specific for the particular bacteria found in the adjacent root canal system.
The immune response and the role of lymphocyte subpopulations in apical periodontitis lesions were investigated by employing lymphocyte-deficient rodents as study models. T-cell deficiency appears to accelerate bone loss at the early phase of apical periodontitis lesions (2 weeks) but does not affect the overall course of lesion development.242,266 Using RAG-2 SCID mice (both T- and B-cell-deficient), it was found that approximately a third of the RAG-2 mice developed endodontic abscesses, while no immunocompetent controls had abscesses.244 In another study, specific RAG 2 knockout (k/o) mice were used to determine which immune element was important for the defense mechanism in endodontic infection. The results demonstrated that B cells, but not T cells, played a pivotal role in preventing dissemination of endodontic infection.95 Therefore, both T and B cells mediate the observed immune responses in apical periodontitis lesions.152
Neurogenic Inflammation
Certain primary afferent nerve fibers, upon stimulation by various irritants, release neuropeptides, which cause vasodilation, protein extravasation, and recruitment/regulation of immune cells such as macrophages, neutrophils, mast cells, and lymphocytes. This phenomenon is termed neurogenic inflammation. Pivotal neuropeptides in the induction of neurogenic inflammation are CGRP, for vasodilation, and SP, for the induction of protein extravasation. Neuropeptides and their receptors are widely distributed throughout the body. During inflammation, there is a sprouting of afferent fibers89 and local increases in inflammatory mediators that trigger neuropeptide release, leading to neurogenic inflammation.23,38 Besides the cardinal functions of the key neuropeptides that cause the first sign of inflammation—vasodilation and increased vascular permeability—the role of these neuropeptides in the process of inflammation is now known to be far more complex. In the development of chronic apical periodontitis lesions, neuropeptides are also involved in immune regulation, bone resorption, and wound healing. At sufficient concentrations, SP increases the secretion of IL-1, TNF-α, and IL-6 from macrophages and stimulates T-lymphocyte proliferation and enhances antigen-induced IFN-γ production by T cells.225 Certain neuropeptides, such as SP, upregulate immune and inflammatory responses, whereas other neuropeptides, such as vasoactive intestinal peptide (VIP) and neuropeptide Y (NPY), inhibit inflammatory responses. Synergistic interactions among neuropeptides, such as CGRP and other inflammatory mediators, eicosanoids, and bradykinin, suggests a complex interplay between these molecules in the immune response.77,189,225
In chronic apical periodontitis lesions, specific receptors for SP and CGRP are expressed in certain immune cells, including macrophages and lymphocytes. Both CGRP and VIP may play a role in inhibiting bone resorption by suppressing osteoclastic functions. The level of VIP in apical periodontitis lesions is inversely related to lesion size. Osteoclast cell culture studies have demonstrated that the presence of greater concentrations of VIP leads to a decrease in their ability to form lacunae of osseous resorption, causing rapid cytoplasmatic contraction and reduced cell mobility. VIP exerts an effect on macrophages to block the production of TNF-α, IL-6 and IL-12, suggesting that VIP could have a role in controlling growth of apical periodontitis lesions.38
The major innate and adaptive immune responses and neurogenic inflammation in the pathogenesis of apical periodontitis caused by root canal infection is illustrated in Fig. 14-1.
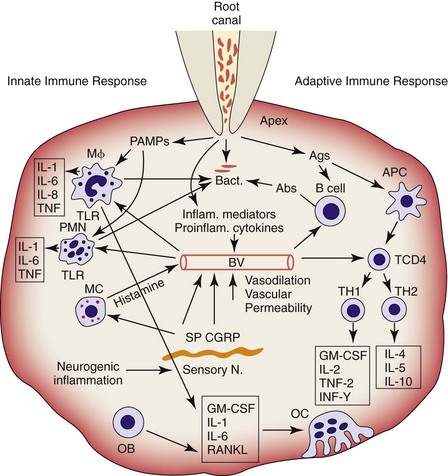
FIG. 14-1 Major innate and adaptive immune responses and neurogenic inflammation in the pathogenesis of apical periodontitis. APC, Antigen presenting cell; GM-CSF, granulocyte/monocyte colony-stimulating factor; MC, mast cell; Mφ, macrophage; OB, osteoblast; OC, osteoclast; PAMPs, pathogen-associated molecular patterns; PMN, polymorphonuclear leukocyte; RANKL, receptor activator of nuclear factor κB ligand; TKR, toll-like receptor.
(Courtesy Dr. Lin.)
Diagnosis
Correlation Between Clinical and Histologic Findings
Clinical diagnosis of inflammatory periapical disease is mainly based on clinical signs and/or symptoms, duration of disease, pulp tests, percussion, palpation, and radiographic findings. In contrast, a histologic diagnosis is a morphologic and biologic description of cells and extracellular matrix of diseased tissues. The clinical diagnosis represents a provisional diagnosis based upon signs, symptoms, and testing results, whereas the histologic diagnosis is a definitive diagnosis of tissue disease.
Similar to pulpitis,206 apical periodontitis is not always symptomatic or painful. Although many inflammatory mediators (histamine, bradykinin, prostaglandins) and proinflammatory cytokines (IL-1, IL-6, TNF, nerve growth factor [NGF]), as well as neuropeptides (SP, CGRP), are capable of sensitizing and activating nociceptive sensory nerve fibers,89,90 other mediators such as endogenous opioids and somatostatin released by inflammatory cells during inflammation are able to inhibit firing of sensory nerve fibers.93,211 Activation of nociceptive sensory nerve fibers may also be related to concentrations of inflammatory mediators. The complexity of these findings supports the clinical observation that there is no good correlation between clinical symptoms and histopathologic findings of apical periodontitis.24,143,213 For example, many teeth with apical periodontitis are free of symptoms.
Correlation Between Radiographic and Histologic Findings
Radiography is designed to detect pathologic changes at tissue not cellular levels. Even using very sensitive imaging systems such as ultrasound, cone-beam computed tomography, and other technologies, it is impossible to detect the presence of inflammatory cells or other subtle changes in the periapical tissues. Using conventional radiographic and histologic methods in the same cadavers, evidence of inflammation was often observed in the periapical tissues of endodontically treated teeth with normal radiographic features.15,32,78 This finding is supported by the fact that lesions localized in the cancellous bone may not be visible radiographically unless they involve cortical bone.17,19,98 In addition, radiographic findings are unable to predict asymptomatic apical periodontitis (granuloma) from asymptomatic apical periodontitis with cyst formation (radicular cyst).125,238 Accordingly, radiographic findings and histopathologic features of apical periodontitis have a poor correlation based on available case series studies.
The absence of clinical symptoms and negative periapical radiographic findings of endodontically involved teeth does not necessarily indicate absence of apical periodontitis. By the same token, clinical success of endodontically involved teeth (i.e., absence of signs and symptoms and negative periapical radiographic findings) after nonsurgical root canal therapy does not necessarily imply complete histologic healing of periapical lesions. Thus, currently available diagnostic methods used in endodontics, such as percussion, palpation, and pulp tests (cold, heat, electric), are not sensitive enough to provide histologic diagnosis of an inflammatory periapical disease. In fact, all endodontic tests are basically used to examine the functions of nociceptive sensory nerves and not the pathologic changes of the pulp and/or periapical tissues. Until we have more advanced and sophisticated clinical diagnostic technologies, we will continue to face the problem of clinical diagnosis of inflammatory periapical disease. Nevertheless, the treatment of various types of apical periodontitis lesions is basically the same: nonsurgical root canal therapy. Interestingly, this treatment does provide a consistently good clinical outcome, as measured by either clinical signs of success or survival of the treated tooth. Future research should focus on developing testing methods that provide greater insight into the status of the periapical tissue.
Histopathology
The study of the histopathology of diseased tissues and organs has gone through an interesting evolution. It began at macroscopic, light microscopic, and then electron microscopic observations of diseased tissues and organs. Nowadays, histopathology is focused on cellular and molecular biology of diseased tissues and organs. Biochemical disorders occur inside the cells before light and electron microscopic observable morphologic changes of cell injury. Cell death (protein denaturation) occurs before observable morphologic changes of cell necrosis. Gene transformation or mutation occurs before observable morphologic changes of neoplastic cells.148
Based on etiology, clinical signs and symptoms, and duration of the disease process, the World Health Organization (WHO) classifies disease of periapical tissues into many categories.278 There are also many classifications of inflammatory periradicular disease in several endodontic textbooks99,181,253 and by the American Board of Endodontists, depending on clinical manifestations and histologic appearances. In addition, the American Association of Endodontists held a consensus conference on diagnostic terms in the fall of 2008. Traditionally, there has been a lack of consensus on clinical diagnostic terminology of pulpal and periapical disease in the endodontic community because of the paucity of studies with high levels of evidence. Since the focus of this chapter is on pathobiology of the periapex, inflammatory periapical disease will be classified based on histologic appearances and cell biology of the injured periapical tissues. To avoid confusion between histologic and clinical diagnosis, readers are encouraged to read the chapters related to clinical examination, radiographic interpretation, and clinical diagnosis of inflammatory periapical disease (see Chapters 1, 2, and 4).
The histopathologic analysis of a diseased tissue or organ only shows structural changes of cells and extracellular matrix at the time the tissue or organ is removed. Therefore, it does not represent the complete kinetics or spectrum of disease development. Histologic classification of apical periodontitis is based on types of cells participating in immunoinflammatory responses in the periapical tissues. In general, inflammation can be divided into acute and chronic responses, depending on types of cells present at the site of injured tissue.121,148,220,270 Acute immunoinflammatory response is characterized by participation of neutrophilic leukocytes and chronic immunoinflammatory response by participation of macrophages and lymphocytes. However, many factors, such as severity of tissue injury, specific etiology, host’s resistance, and particular tissue involved, can modify the course and morphologic variations as well as cell biology of both acute and chronic immunoinflammatory responses.121,148 Acute and chronic immunoinflammatory responses are in fact not rigid phases of a single programmed event but two overlapping responses with partially different triggering mechanisms and programs.148
Symptomatic Apical Periodontitis
It is a general belief that the development of apical periodontitis follows total pulp necrosis. This belief is based on (1) the pulpal strangulation theory due to a generalized increase in pulpal interstitial pressure inside the uncompromised pulp space during pulpal inflammation that causes collapse of venules and cessation of blood flow89; and (2) animal and human studies that concluded that uncontaminated necrotic pulps which are intentionally devitalized or accidentally traumatized are generally incapable of inducing periapical inflammation unless they are infected.137,158,232 However, if the vital pulps become infected due to caries or other pathways, periapical inflammation can develop even when inflamed, but vital tissue is still present in the apical portion of the root canal. Most of our information related to the histopathology of apical periodontitis comes from analysis of longstanding, chronic human lesions caused by caries or from time-course studies of development of apical periodontitis induced by artificial root canal infection in animals. In these instances, the moment of transition from pulpitis to apical periodontitis was not captured. In fact, apical periodontitis has been demonstrated to be a direct extension of apical pulpitis into the periapical tissues before total pulp necrosis caused by root canal infection (Fig. 14-2).* For example, Kovacevic et al.119 studied the transition from pulpitis to apical periodontitis in dogs’ teeth by artificial exposure of pulps to the oral cavity and observed that pulpitis was coupled with an acute apical periodontitis. Similarly, Kimberly and Byers115 demonstrated that periapical changes, including sprouting of nerve fibers, appeared 3 to 5 weeks following establishment of irreversible pulpitis subsequent to pulp exposure lesions in animals. Yamasaki et al.279 and Stashenko et al.226 also showed that periapical inflammatory infiltrates, increased osteoclast numbers, and bone destruction were apparent well in advance of total pulpal necrosis, with vital pulp tissue still present in the apical portion of the root canal. The biologic basis for these observations appears to hinge on the apical development of pulpal infection/inflammation leading to the diffusion of many inflammatory mediators, proinflammatory cytokines, chemokines, and bacterial toxins into the periapical area140 prior to total pulpal necrosis.
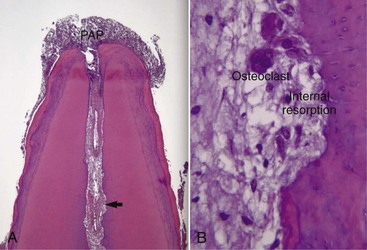
FIG. 14-2 A, Inflammation of the pulp tissue in the apical root canal extends into the periapical tissues in a mature tooth (H & E, ×100). B, Arrow in (A). High magnification of the pulp tissue in the apical root canal in A. The pulp tissue is vital and infiltrated with chronic inflammatory cells. Note resorption of canal wall and multinucleated clast cells (H & E, ×200).
(Courtesy Dr. Domenico Ricucci, Rome, Italy.)
The development of acute apical periodontitis largely reflects the innate immune system and is the first line of active defense against irritants from the root canal. Acute apical periodontitis is an immediate defense reaction to irritants and does not require exquisite specificity and memory. Characteristic features of acute apical periodontitis are similar to the typical acute inflammatory reaction and consists of vasodilatation, increased vascular permeability, and transmigration of leukocytes from the blood vessels into perivascular tissue space. The beneficial actions of acute inflammation are (1) infiltration of leukocytes to the injured tissue to phagocytose and kill microbial agents; (2) accumulation and activation of humoral factors such as immunoglobulins, complement factors, and plasma proteins in the injured tissue to recruit more neutrophils and macrophages; and (3) neutralization or degradation of bacterial toxins and their harmful metabolic byproducts.121,148
Cell Biology
The immunoinflammatory response is a dynamic interaction between host defense mechanisms and microbial insults. The interlacing activation and control pathways of cellular and humoral components involved in the immunoinflammatory response are complex. The cells involved—neutrophils, monocytes/macrophages, platelets, mast cells, T lymphocytes, B lymphocytes, NK cells, dendritic cells, endothelial cells, fibroblasts, eosinophils, and basophils—each have numerous functions that are activated and modulated by a multiplicity of biochemical messengers.1,121,148 Cell activation means that the cell acquires the ability to perform one or more new functions or to perform normal functions at a higher rate;1 it often results in transcription of new genes and synthesis of new proteins.1
Mast Cells
Histamine stored in the cytoplasmic granules of mast cells is the mediator that first appears in acute inflammation to induce vasodilation and increased vascular permeability; mast cells are the designated triggers of acute inflammation. They are widely distributed in perivascular tissue spaces and originate in the bone marrow from precursor cells. Mature mast cells contain numerous cytoplasmic granules, which are the source of vasoactive mediators. The preformed histamine is released by mast cell degranulation and can be triggered by (1) physical stimuli such as cold, heat, and mechanical trauma; (2) binding of IgE-specific antigen to mast cells, membrane-bound IgE antibodies; (3) binding of complement components (C3a and C5a) to their complementary receptors on mast cells; and (4) stimulation by neuropeptide (SP) and cytokines (IL-1, IL-8).1,26,121,148 In addition, activated mast cells secret cytokines (TNF-α, IL-1, IL-3, IL-4, IL-5, IL-6, IL-13), prostaglandins, and leukotrienes to enhance inflammatory defense mechanisms.1,33,114
Endothelial Cells
Endothelial cells are important players in the immunoinflammatory response. Without participation of endothelial cells, the host is unable to deliver its cellular and humoral defense components from the circulating blood to the site of tissue injury. Inflammatory mediators, complement components, proinflammatory cytokines, nitric oxide, neuropeptides, and bacterial toxins can all affect endothelial cells, resulting in vasodilation and increased vascular permeability.1,121,148 IL-1 and TNF released by activated macrophages and NK cells can stimulate endothelial cells to express intercellular adhesion molecules (ICAMs), such as ICAM-1, ICAM-2, ICAM-3, vascular cell adhesion molecule (VCAM), and platelet endothelial cell adhesion molecule (PECAM), which enhance leukocyte adhesion to endothelial cells and transmigration through the blood vessels.1,121,132,148 IL-1, TNF, and LPS also can activate endothelial cells to synthesize chemokine (IL-8), a potent chemotactic mediator for neutrophils.1
Polymorphonuclear Neutrophilic Leukocytes
Polymorphonuclear neutrophilic leukocytes (PMNs) are the principal effector cells in acute apical periodontitis.3,111,180 They are derived from bone marrow stem cells. Neutrophils have a lobulated nucleus and contain primary or azurophil (elastase and myeloperoxidase) and secondary or specific (lysozyme and other protease) granules in their cytoplasm.114,121,148 Neutrophils are only present in the blood circulation. They are the first leukocytes to transmigrate through the blood vessels into perivascular tissue space and then are directed toward the wound or irritants, peaking at 24 to 48 hours. The transmigration of neutrophilic leukocytes from the blood vessels into perivascular space involves complex cellular and molecular biology. Following vasodilation and increased vascular permeability, leukocyte margination, rolling, capture, and activation in the blood vessel and then transmigration through the blood vessel are mediated by an intricate interaction of cell adhesion molecules expressed on leukocytes (L-selectin, integrins) and on endothelial cells (P- and E-selectins, ICAM, VCAM, PECAM-1). Neutrophil rolling is mediated by interaction between leukocyte selectin ligands and P-selectins on endothelial cells. Leukocyte sticking is mediated by interaction between leukocyte integrins and ICAMs and VCAMs on endothelial cells. Leukocyte transmigration is mediated by interaction between PECAM-1 on both leukocytes and endothelial cells.148 Chemokines (IL-8) increase the affinity of leukocyte integrins for their ligands on endothelial cells.1 Once transmigrating through the junction between endothelial cells and basement membrane into the perivascular tissue space, neutrophilic leukocytes are directed toward stimuli by chemotactic factors or chemotaxins, such as bacterial products (fLMP), C3a, C5a, leukotriene B4 (LTB4), platelet-activating factor (PAF), fibrinopeptides, dead cells, and chemokines (IL-8) by receptor-mediated mechanisms.1,121,148,220
Neutrophils can be activated by bacteria and pathogen-associated molecular patterns (also known as the TLRs, described earlier). They can also be stimulated by IL-1, TNF, and chemokines produced by activated macrophages and NK cells to enhance their phagocytic activity of infectious agents and synthesis of defensins, which are broad-spectrum antibiotics.1 Neutrophilic leukocytes are terminally differentiated cells and short lived—within hours to a few days. Most neutrophilic leukocytes in acute inflammatory response die as a result of apoptosis or programmed cell death. The apoptotic neutrophils are phagocytosed by macrophages.82,121,148 However, some neutrophilic leukocytes die after a furious battle against microbial infection and release intracellular proteolytic lysosomal enzymes, oxygen-derived active metabolites, nitric oxide, proinflammatory cytokines, eicosanoids, and matrix metalloproteinases into tissue to intensify inflammation and tissue damage.121,148 The release of lysosomal enzymes by neutrophils and macrophages can also occur by lysosomal suicide due to rupture of phagolysosome in the cytosol, regurgitation during phagocytosis of irritants, or frustrated phagocytosis of indigestible foreign bodies.121,148 The main effector functions of neutrophilic leukocytes are phagocytosis, killing of microbes, and release of inflammatory mediators (including proinflammatory cytokines) to recruit more leukocytes to prevent spread of infection.
Macrophages
Macrophages make their appearance as a second wave in acute apical periodontitis within 48 to 96 hours. Macrophages are blood-borne but have a counterpart in the connective tissue.148 Blood monocytes transmigrate through the blood vessel into the tissues and become macrophages. Mature macrophages have an irregular-shaped nucleus and contain abundant lysosomes and many phagocytic vesicles (phagosomes). Monocytes use mechanisms similar to neutrophils to adhere to endothelial cells in high endothelial venule–expressing adhesion molecules for mononuclear leukocytes (ICAM for macrophages and VCAM for lymphocytes); they then transmigrate through the blood vessel and are directed toward the site of tissue injury by chemotactic factors.148
Macrophages can be activated by bacteria, PAMPs, and interferon-r (INF-r) and produce numerous products: lysosomal enzymes, coagulation factors, bioactive lipids, reactive oxygen species, chemokines, cytokines/growth factors, and angiogenesis factors.121,148,220 Macrophages are the most dynamic and versatile leukocytes. The main functions of macrophages are numerous. They include phagocytosis of microbes and foreign bodies, production of inflammatory mediators, initiation of the immune response, cleanup operation of necrotic cells and tissue, induction of systemic effects (fever, acute phase reaction, cachexia), synthesis of molecules or cytokines affecting cell and vascular growth in wound healing, as well as antibacterial and antiviral defenses.148 Tissue macrophages are not terminally differentiated cells and are able to undergo mitosis. Their lifespan is from several weeks to months.
Activated neutrophilic leukocytes and macrophages are very capable of phagocytosing and killing pathogens. They recognize microbes through TLRs and receptors for the Fc fragment of immunoglobulin IgG as well as complement component C3b on their cell surface. C3b opsonization and antibody coating of microbes enhance recognition and phagocytosis of pathogens by activated neutrophilic leukocytes and macrophages.1,121,148 Activated neutrophilic leukocytes are effective in phagocytosing and killing extracellular microbes, whereas macrophages activated by IFN-r produced by NK cells and TH1 cells are more effective in phagocytosing and killing intracellular microbes.1 Killing of phagocytosed microbes by neutrophilic leukocytes and macrophages is mediated through oxygen-dependent and oxygen-independent mechanisms. Oxygen-dependent mechanisms are more effective in killing all kinds of bacteria than oxygen-independent mechanisms.1,121,220 The effector molecules of an oxygen-dependent system are hydrogen peroxide, superoxide anion, hydroxyl radical, singlet oxygen, and hypochlorite. An oxygen-independent system is also important in killing microbes and is dependent on lysozymes, defensins, and lactoferrin contained in phagocyte granules.1,121,148,220 Some antimicrobial peptides and other oxygen-independent mechanisms possessed by phagocytes are specialized for killing of certain groups of microbes.1,220
Microbes phagocytosed by phagocytes are enclosed in membrane-bound phagosomes in the cytosol. The phagosomes fuse with lysosomes to form phagolysosomes. Irritants such as microbes, foreign protein antigens, and dead cells inside the phagolysosome are destroyed or degraded by proteolytic enzymes stored in lysosomes.1 There are also granules that are fused with the nascent phagosome and release their contents into the phagosome. Some of these granules have direct antimicrobial action, such as defensins and the bactericidal permeability-increasing protein, azurocidin. Others are proteases, such as elastase and cathepsins, lactoferrin, and peroxidase myeloperoxidase. Lysosomal enzymes, reactive oxygen intermediates, and nitric oxide released by neutrophils and macrophages indiscriminately kill not only bacteria but also tissue cells. Much of periapical tissue damage that occurs during acute inflammation can be attributed to release of proteolytic lysosomal enzymes and matrix metalloproteinases from disintegrated neutrophilic leukocytes and macrophages rather than to bacteria and their toxins.1,148
Platelets
Platelets normally circulate in the blood but also play an important role in inflammation. They are small cytoplasmic fragments derived from the megakaryocyte.114 Platelets are essential for blood clotting, hemostasis, and fibrinolysis. Platelets produce vasoactive amines (PAF, serotonin), chemokines, and growth factors (PGDF, FGF, TGF) during inflammation.148
Natural Killer Cells
NK cells may also be players in acute apical periodontitis. They are a subset of lymphocytes found in blood and lymphoid tissues. NK cells are derived from the bone marrow stem cells but lack the specific T-cell receptor for antigen recognition.1 NK cells also possess TLRs for microbial constitutive and conserved products. The effector functions of NK cells are to lyse virus-infected cells without expressing class 1 MHC molecules and to secret IFN-r to activate macrophages. Antibody-coated cells, cells infected by viruses, some intracellular bacteria, and IL-2 released by activated macrophages can activate NK cells. Viruses have been isolated in apical periodontitis lesions.196,221 NK cells kill target cells by antibody-dependent cell-mediated cytotoxicity because NK cells express receptor for the Fc fragment of IgG.1 NK cells provide a link between the innate and adaptive immune systems.
Inflammatory Mediators
Numerous biochemical mediators are involved in the acute innate immunoinflammatory response. They are mainly derived from plasma and cells. The main biologic functions of inflammatory mediators are to cause vasodilation and increased vascular permeability and recruit inflammatory cells, mainly neutrophilic leukocytes and macrophages from blood circulation to the site of tissue injury. Some mediators can also cause tissue damage. The major mediators involved in vascular changes, cell recruitment, and tissue damage in the acute immunoinflammatory response are listed in Table 14-2.
TABLE 14-2 Major Mediators in Inflammation
| Mediator | Source | Effector Cells and Tissues |
|---|---|---|
| VASODILATOR | ||
| Histamine | Mast cells | Endothelial cells |
| Platelets | ||
| Prostaglandins | Leukocytes | Endothelial cells |
| Mast cells | ||
| Nitric oxide | Endothelial cells | Vascular smooth muscle |
| Macrophages | ||
| CGRP | Sensory nerve | Endothelial cells |
| Fibrin degradation products | Plasma | Endothelial cells |
| INCREASED VASCULAR PERMEABILITY | ||
| Bradykinin | Plasma | Endothelial cells |
| Leukotrienes C4, D4, E4 | Leukocytes | Endothelial cells |
| Mast cells | ||
| PAF | Leukocytes | Endothelial cells |
| Mast cells | ||
| Endothelial cells | ||
| C3a, C5a | Plasma | Endothelial cells |
| Fibrinopeptides | Plasma | Endothelial cells |
| Substance P | Sensory nerve | Endothelial cells |
| INCREASED EXPRESSION OF ENDOTHELIAL ADHESION MOLECULES (SELECTIN, ICAM, VCAM, PEAM) | ||
| TNF | Activated | Endothelial cells |
| macrophages | ||
| NK cells | ||
| IL-1 | Activated | Endothelial cells |
| macrophages | ||
| NK cells | ||
| Chemokines | Macrophages | Endothelial cells |
| Neutrophils | ||
| Endothelial cells | ||
| Fibroblasts | ||
| LEUKOCYTE ACTIVATION AND CHEMOTAXIS | ||
| C3a, C5a | Plasma | Leukocytes |
| Leukotriene B4 | Mast cells | Leukocytes |
| Leukocytes | ||
| Chemokines (IL-8) | Macrophages | Leukocytes |
| Neutrophils | ||
| Endothelial cells | ||
| Fibroblasts | ||
| Fibrinopeptides | Plasma | Leukocytes |
| Bacterial products (fMLP) | Bacteria | Leukocytes |
| TNF | Activated | Leukocytes |
| macrophages | ||
| Dead cells | ||
| OPSONINS | ||
| C3b, C5b, immunoglobulins | Plasma | Microbes |
| TISSUE DAMAGE | ||
| Lysosomal enzymes | Neutrophils | Cells and tissues |
| Macrophages | ||
| Free oxygen radicals | Activated | Cells and tissues |
| leukocytes | ||
| Nitric oxide | Macrophages | Cells and tissues |
| Bacterial products (LPS) | Bacteria | Cells and tissue |
CGRP, Calcitonin gene–related peptide; fMLP, formyl-methionyl-leucyl-phenylalanine; IL-1, interleukin-1; PAF, platelet activation factor; NK cell, natural killer cell; SP, substance P; TNF, tumor necrosis factor.
Data from Kumar V, Abbas AK, Fausto N, et al: Robbins and Cotran pathologic basis of disease, ed 8, Philadelphia, Saunders, 2010; Slauson DO, Cooper BJ: Mechanisms of disease, ed 3, St Louis, Mosby, 2002; Majno G, Joris I: Cell, tissues, and disease, ed 2, Oxford, Oxford University Press, 2004; Abbas AK, Lichtman, Pober JS: Cellular and molecular immunology, ed 5, Philadelphia, Saunders, 2003.
All listed inflammatory mediators have been shown to be present in apical periodontitis.249,250 Bradykinin is the product of kinin-system activation; fibrinopeptides, the products of blood-clotting system activation; and fibrin degradation products, the products of fibrinolytic-system activation. Kinin, fibrinolytic, and clotting systems are initiated by activated Hageman factor. Prostaglandins and leukotrienes are the products of arachidonic acid metabolism. Activated phospholipase A2 splits cell membrane phospholipid molecules into arachidonic acid and platelet activating factor. Arachidonic acid molecules can be processed along two pathways: the cyclooxygenase pathway, which leads to production of prostaglandins and thromboxanes, and the lipoxygenase pathway, which produces leukotrienes.121,148
Complement components, such as C3a, C3b, C5a, C5b, and C5-C9, are products of the complement cascade, which can be activated by two pathways. The classic pathway is initiated by activation of C1 by multimolecular aggregates of IgG or IgM antibody complexed with specific antigen. The alternate pathway is activated by microbial cell components (lipopolysaccharide, teichoic acid) and plasmin. C3a and C5a are anaphylatoxins which stimulate mast cells and basophils to release histamine. They also cause phagocytes to release lysosomal enzymes. C3b is an opsonin and can coat bacteria to enhance phagocytosis by phagocytes. In addition, C3b can also bind to antibody bound to antigen or microbes. C5a is a strong chemotaxin for neutrophils. C5b-C9 is a membrane attack complex and able to cause cell lysis if activated on the host cell and bacterial cell membrane.121,148
Besides inflammatory mediators, the immunoinflammatory response is also dependent upon the timing and extent of various cytokine and chemokine secretions. IL-1, TNF, IL-6, IL-12, and IFN-γ are present in the acute inflammatory response.1,84,148 IL-6 is produced by activated mononuclear phagocytes, endothelial cells, and fibroblasts in response to microbial infection and other cytokines, such as IL-1 and TNF. IL-6 stimulates the synthesis of acute-phase proteins by hepatocytes.1 The major source of IL-12 is activated mononuclear phagocytes and dendritic cells. IL-12 provides a link between innate and adaptive immune responses.1 IFN-r is produced by activated NK cells, TH1 cells, and cytotoxic T cells and can activate macrophages and enhance their microbial killing ability. IL-1β and TNF are associated with apical bone resorption during chronic apical periodontitis.224
Chemokines are a large family of structurally homologous cytokines that stimulate leukocyte movement and regulate the transmigration of leukocytes from the blood vessel to tissue space.1 They are produced by leukocytes, endothelial cells, and fibroblasts. The secretion of chemokines is induced by microbial infection, TNF, and IL-1.1,84,214 Different types of leukocytes express different chemokine receptors.1
Neuropeptides are released via axon reflexes of afferent sensory neurons in response to various stimuli. Neuropeptides, as mediators of the immunoinflammatory process, are described in detail in Neurogenic Inflammation in this chapter. Inflammatory mediators such as histamine, kinins, prostaglandins, and proinflammatory cytokines are capable of sensitizing and activating sensory nerves to release neuropeptides.26,89,90,191,202
Histopathology
The acute immunoinflammatory response is practically immutable in all vascularized living tissues, largely due to the programmed actions of the innate immune system. Initially, blood vessels are engorged by a local infiltration of inflammatory cells, mainly activated neutrophilic leukocytes and some macrophages in the apical periodontal ligament of the infected/inflamed root canal.3,180 In addition, sprouting of sensory nerve fibers has been shown early in the inflamed periapical tissues.35,115 Several studies have also demonstrated the presence of inflamed vital pulp tissue with intact nerve fibers in the apical portion of the root canal in association with apical periodontitis.140,192,193,226,279 This explains the clinical observation that a patient may experience some pain if an instrument is introduced into the canal short of the apex in some teeth with apical periodontitis lesions.
In primary acute apical periodontitis, apical bone destruction is usually not observed radiographically because the duration of acute response is short, and activated neutrophilic leukocytes and macrophages are not able to resorb bone. Only osteoclasts are capable of resorbing bone, and they have to differentiate from the monocyte/macrophage cell lineage in the blood circulation. However, in a rat model experiment, Stashenko et al.226 showed histologically that periapical inflammatory cell infiltration increases osteoclast numbers, and that bone destruction was apparent well in advance of total pulpal necrosis.
Bacteria are usually not present in acute apical periodontitis lesions.
Clinical Features
As discussed earlier, there is no correlation between clinical and radiographic findings and histologic appearance of inflammatory periapical disease. Teeth with acute apical periodontitis are usually symptomatic and painful to bite and percussion, which results from mechanical allodynia and hyperalgesia.113 Pain is induced by sensitization and activation of nociceptive sensory nerve fibers by inflammatory mediators, proinflammatory cytokines, nerve growth factor, and pressure.113 Sprouting of sensory nerve fibers in inflamed periapical tissues could also increase receptive field size in teeth with apical periodontitis.34,35 Radiographic examination usually does not show periapical bone destruction of involved tooth in acute apical periodontitis, although occasional slight widening of the apical periodontal ligament space and loss of the apical lamina dura of the involved tooth may be present.
Outcomes
The fundamental purpose of the acute immunoinflammatory response is to restore the structural and functional integrity of damaged tissue by eliminating irritants as soon as possible.270 Tissue damage can also occur in acute inflammation by release of lysosomal enzymes, toxic oxygen radicals, and nitric acid from disintegrated neutrophilic leukocytes and macrophages into tissue.148 Depending on the dynamic interaction between host defenses and microbial insults, acute apical periodontitis can result in (1) restitution of normal periapical tissues if irritants are immediately eliminated by root canal therapy; (2) abscess formation if massive invasion of periapical tissues by highly pyogenic bacteria occurs; (3) organization by scarring if extensive destruction of periapical tissues results; or (4) progression to chronic apical inflammation if irritants continue to persist.
Abscess is a focal localized collection of purulent exudate in a tissue or an organ.121 It is a morphologic variation of acute and chronic inflammation.121,148,270 The development of an abscess in apical periodontitis lesions is probably caused by invasion of a combination of specific pyogenic bacteria in the inflamed periapical tissues.29,179,234,262 Neutrophilic leukocytes are the predominant cells in acute apical periodontitis with abscess formation. Abscess begins as a furious battle between highly virulent pathogens and an army of neutrophilic leukocytes. The pathogens produce massive toxins to kill neutrophils. As neutrophils attack the pathogens, they secrete lysosomal enzymes that digest not only the dead cells but also some live ones. Many neutrophils die fighting against pathogens. The resulting purulent fluid is poorly oxygenated and has a low pH. The bactericidal ability of leukocytes appears to be impaired because of deprivation of oxygen and interference of respiratory burst. The purpose of respiratory burst is to generate bactericidal agents.12,148 However, the phagocytic activity of leukocytes is not impaired in aerobic conditions.148
Histologically, apical abscess formation is characterized by local collection of suppurative or purulent exudate composed of dead and live neutrophilic leukocytes, disintegrated tissue cells, degraded extracellular matrix, and lysosomal enzymes released by dead neutrophilic leukocytes. It also contains dead and live bacteria and bacterial toxins released by dead bacteria in the inflamed periapical tissues. Abscess formation also involves destruction of periodontal ligament and sometimes periapical bone—especially in chronic apical periodontitis with abscess formation—and a surrounding layer of viable neutrophilic leukocytes and a band of fibrovascular granulation tissue. Both layers are thought to serve as protective barriers to prevent the spread of infection. Epithelial cell proliferation is scanty in acute apical periodontitis with abscess formation (Fig. 14-3).39,193,205
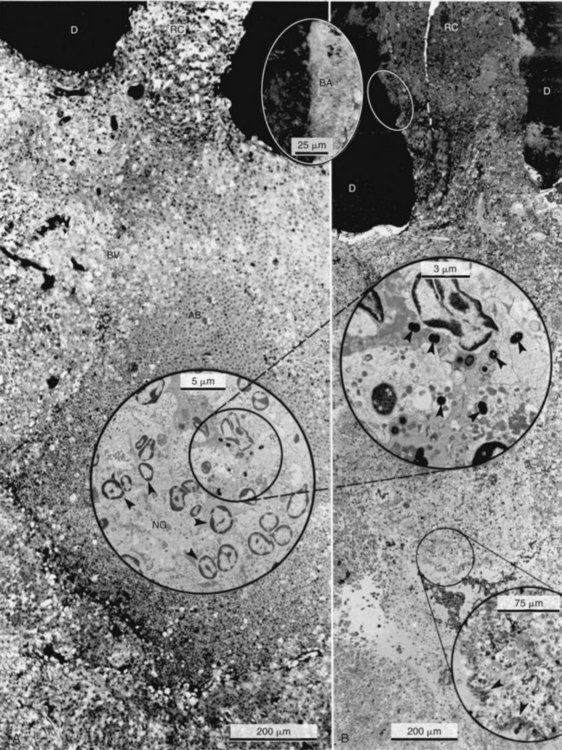
FIG. 14-3 Structure of a secondary periapical abscess. A, Axial section of an abscessed apical periodontitis. The microabscess (AB) contains a focus of neutrophils (NG inset in A). Note the phagocytosed bacteria in one of the neutrophils (further magnified in large inset in B). A secondary abscess forms when bacteria (BA in oval inset) from the apical root canal (RC) advance into the chronic apical periodontitis lesion (B). Note the tissue necrosis immediately in front of the apical foramen and the bacterial front in the body of the lesion (arrowhead in lower inset). BV, Blood vessels; D, dentin. (A ×130; B ×100; oval inset, ×400; inset in A, ×2680; upper inset in B, ×4900; lower inset in B, ×250.)
(From Nair PNR: Periodontol 2000 13:121, 1997.)
Clinically, teeth with acute apical abscess formation usually have symptoms such as pain to biting and percussion. The periapical area of the involved tooth may be very tender to palpation. Intraoral or extraoral swelling is often present. Because of a sudden outpouring of suppurative exudate in the periapical area, tissue pressure increases such that mechanical stimuli are capable of activating terminals of nociceptive neurons in the inflamed periapical tissues. The severe pain of an acute apical abscess can be due to activation of nociceptors by inflammatory mediators and sensitization to mechanical stimuli due to increased interstitial pressures.207 If the periapical purulent exudate can be evacuated through the root canal (or through incision and drainage when indicated) during root canal therapy, the patient usually experiences an immediate relief of acute pain. Radiographically, the involved teeth may show slight widening of the apical periodontal ligament space to loss of apical lamina dura. In chronic apical abscess, the involved teeth may be symptomatic or asymptomatic. If an intraoral or extraoral draining sinus tract is present, swelling is usually absent. Radiographic bone destruction is obvious in teeth with chronic apical abscess formation.
In most cases, if the source of infection in the root canal is eliminated by root canal therapy, the abscess will heal by reabsorption of the pus in teeth with an acute apical abscess. Phagocytes will kill all bacteria in the abscess. The continued influx of leukocytes stops because the chemotactic stimuli have been removed, and the existing neutrophilic leukocytes die of apoptosis. Finally, macrophages move in to clean up necrotic neutrophilic leukocytes and disintegrated tissue cells. In chronic apical abscess formations, wound healing will take place mainly by means of regeneration and to some degree by tissue repair. However, if bacterial virulence and numbers of pathogens overwhelm the host’s defenses, the abscess may break through the cortical bone, periosteum, and oral mucosa or facial skin to develop an intraoral or extraoral draining sinus tract. Sometimes, the uncontrolled abscess may spread along the fascial planes of the head and neck to develop serious cellulitis (see also Chapter 15).126,205
Asymptomatic Apical Periodontitis: Apical Granuloma, Chronic Apical Periodontitis
If pathogens in the root canal are not eliminated, the symptomatic apical periodontitis may progress to become an asymptomatic apical periodontitis. Asymptomatic apical periodontitis is characterized by the persistence of inflammatory stimuli, adaptation of the host’s response to stimuli, presence of adaptive immune responses, and initiation of the repair process.121,148,220,270 Chronic inflammation is good news and bad news. The good news is that the host’s defenses are able to maintain an active defense against the invading microorganisms and toxins; the bad news is that the host response is inadequate to eliminate these factors.148
Asymptomatic apical periodontitis is a form of adaptive immune response that requires exquisite specificity and memory. The adaptive immune response enhances bacterial killing compared with the innate immune response. Traditionally, asymptomatic chronic apical periodontitis and periapical granuloma are terms used interchangeably. A granuloma is a focal area of granulomatous inflammation, which is a histologic term for a chronic inflammatory reaction.121,148,258 Granulomatous inflammation is characterized by the presence of activated macrophages with modified epithelioid cells in diseases such as tuberculosis, leprosy, syphilis, cryptococcosis, sarcoidosis, rheumatic fever, and foreign body granuloma.92,121,148,275 The presence of poorly digestible irritants (nonantigenic or antigenic), T cell–mediated immunity to irritants, or both appears to be necessary for granuloma formation.1,121,148 A granuloma is relatively avascular,121,148 whereas a chronic apical periodontitis is very vascular. Histologically, some but not all chronic apical periodontitis lesions may show some features of granulomatous inflammation,121,148 so the terms apical granuloma and asymptomatic chronic apical periodontitis should not be used interchangeably. A granuloma is best considered a histologic term used to describe a specific form of chronic inflammation such as foreign body granuloma or immune granuloma.121,148
The foreign body reaction is a specific subtype of chronic inflammation.121,148,270 Foreign materials such as root canal filling materials, paper points, cotton fibers, and surgical sutures can trigger a foreign body giant cell granuloma.118,166,282 If activated macrophages are unable to engulf large indigestible foreign particles, they can fuse to form giant cells on the surface of the particles and continuously release lysosomal enzymes, inflammatory mediators, and proinflammatory cytokines as a result of frustrated phagocytosis. Giant cells appear to be at least as active metabolically as a regular macrophage.148 In addition, foreign bodies can favor infection in several ways, since they can be a source of bacterial biofilm45,176 and they lower the infectious dose of bacteria to induce infection. For example, if a small, sterile plastic cage is implanted under the skin of a guinea pig, as few as 100 Staphylococcus aureus are sufficient to infect the tissue, whereas even 108 bacteria (i.e., a million-fold increase in dose) fails to produce an abscess in normal guinea pig skin.284 Finally, foreign bodies can make the infection hard to treat because bacteria in biofilms can switch on appropriate genes and cover themselves with a thick layer of biopolymer that is resistant to both host defense mechanisms and antimicrobial agents.107,148
Cell Biology
Macrophages and Lymphocytes
Macrophages and lymphocytes are the primary players in asymptomatic apical periodontitis.* Lymphocytes are blood-borne and have counterparts in the connective tissue.148 Macrophages play a dual role in host defenses. In the innate immune response, activated macrophages phagocytose microbes, dead cells, and foreign bodies and produce inflammatory mediators and proinflammatory cytokines to enhance the host defense against stimuli. In the adaptive immune response, activated macrophages function as APCs. They phagocytose and present the processed foreign antigens in association with MHC to T cells. Thus, activated macrophages are effector cells of adaptive immune response.
Lymphocytes are the only cells in the body capable of specifically recognizing and distinguishing different antigenic determinants; they are responsible for the two defining characteristics of the adaptive immune response, specificity and memory. The functions of lymphocytes were described earlier in the section on the adaptive immune response.
Dendritic Cell
Dendritic cells play a vital role in asymptomatic apical periodontitis. They have been shown to be present in apical periodontitis lesions in rats.109,180 Dendritic cells are accessory immune cells derived from bone marrow stem cells and may be related to the mononuclear phagocyte lineage.1 They function as antigen-presenting cells for naïve T lymphocytes and are important for the initiation of adaptive immune responses to protein antigen.1 Activated dendritic cells produce IL-12, which is a key inducer of cell-mediated immunity.
Osteoclasts
Periapical bone destruction is a hallmark of asymptomatic apical periodontitis. During the chronic stage of apical periodontitis, both osteoclast and osteoblast activity are decreased,268 so the periapical osteolytic lesion remains stationary. Based on magnified radiographic and automated image analysis, periapical bone destruction was observed at 7 days after the pulps of experimental teeth were exposed to oral microorganisms in animal studies. A period of rapid bone destruction took place between 10 and 20 days, with slower bone resorption thereafter.268 The stationary phase of bone resorbing activity was correlated to asymptomatic apical periodontitis.268 Increased expression of bone resorptive cytokines such as IL-1, IL-6, and TNF was related to the period of active bone resorption.269 TH cells appear to outnumber TS cells during the active stage of periapical bone destruction in induced rat periapical lesions, but TS cells dominate TH cells during the stationary stage of bone destruction.227
Bone resorption is caused by osteoclasts. The formation of osteoclasts involves differentiation of the osteoclast precursor from the monocyte-macrophage cell lineage in bone marrow. Several cytokines and growth factors, such as granulocyte/macrophage colony-stimulating factor (GM-CSF), RANKL (receptor activator of nuclear factor κB ligand), osteoprotegerin (OPG), IL-1, IL-6, TNF, as well as prostaglandins, bradykinin, kallidin, and thrombin, have been shown to mediate osteoclast differentiation.59,131,159,186,243 Parathyroid hormone is capable of stimulating osteoblasts to synthesize GM-CSF and RANKL. Bone stromal cells and T cells also produce RANKL. Osteoclast progenitor cells express receptor activator of nuclear factor κB (RANK). OPG, a decoy receptor for RANKL secreted by osteoblasts, negatively regulates the differentiation of osteoclasts by absorbing RANKL and reducing its ability to activate the RANK pathway.114
RANKL activates the RANK pathway on osteoclast progenitor cells, resulting in differentiation of these cells along the osteoclast lineage. Proinflammatory cytokines, IL-1, TNF, and IL-6 also mediate the differentiation of osteoclast progenitor cells to osteoclasts. The differentiation of the mononuclear osteoclast progenitor cells terminates with fusion to latent multinucleated osteoclasts, which are finally activated to become bone-resorbing osteoclasts. The mature osteoclasts then attach to mineralized bone surface after osteoblasts have primed the unmineralized bone surface and released chemotactic factor to attract osteoclasts.169 Osteoclasts attach to bone by the vitronectin receptor (integrin superfamily), expressed preferentially in the sealing zone. Vitronectin has binding sites for the arginine-glycine-aspartic amino acid (RDG) sequences present in many extracellular matrix proteins, including osteopontin, bone sialoprotein, and fibronectin, on the surface of the exposed mineralized bone.75 When bound to bone extracellular matrix, osteoclasts develop a ruffled border. Inside the ruffled border, osteoclasts use ATP to drive H+ pumps, leading to the acidification of the extracellular compartment. They subsequently secrete proteolytic lysosomal enzymes and carbonic anhydrase to degrade both the mineralized and unmineralized components of bone.13,14,25,243 (See Fig. 14-5 for the mechanism of bone resorption by osteoclasts in apical periodontitis.)
The resorption of root cementum and/or dentin in apical periodontitis lesions is less well understood than resorption of bone. Bone is constantly remolding (resorption and deposition) through physiologic and functional processes and thus is much easier to study. In contrast, cementum and dentin are more stable. The cells responsible for dental hard-tissue resorption are called odontoclasts.197 It has been shown in ultrastructure and gene expression studies that odontoclasts and osteoclasts are similar.198,199 Therefore, it is believed that the cellular mechanisms of bone, cementum, and dentin resorption are similar.198 Nevertheless, little is known about how the precursors of the odontoclasts appear and what causes odontoclast differentiation and activation to resorb dentin and cementum. Bone resorption by osteoclasts in apical periodontitis is illustrated in Fig. 14-4.
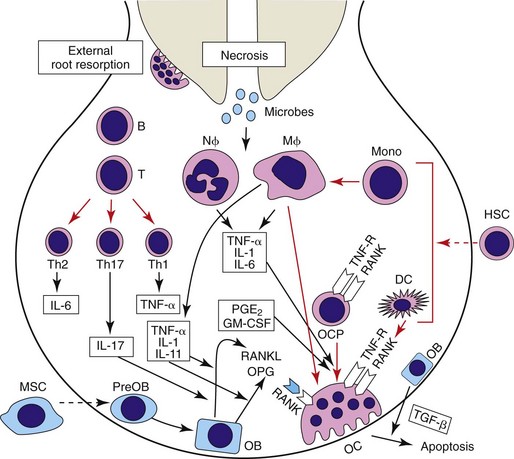
FIG. 14-4 Bone resorption by osteoclasts in apical periodontitis. DC, Dendritic cell; HSC, hematopoietic stem cell; Mφ, macrophage; MSC, mesenchymal stromal cell; Nφ, neutrophil; OB, osteoblast; OC, osteoclast; OCP, osteoclast precursor; OPG, osteoprotegerin; RANK, receptor activator of nuclear factor κB; RANKL, receptor activator of nuclear factor κB ligand.
(Courtesy Dr. Huang.)
Epithelial Cell Rests of Malassez
Proliferation of epithelial cell rests of Malassez (ERM) is evident in approximately 52% of inflammatory periapical lesions collected from extracted teeth.164 It is a form of pathologic (inflammatory) hyperplasia. This type of hyperplasia is caused by stimulation of growth factors and cytokines produced during the immunoinflammatory response. Hyperplasia is a self-limiting process and is reversible when the causative stimulus is eliminated.121,148,220 During periapical inflammation, innate and adaptive immune cells and stromal cells (e.g., activated macrophages, neutrophilic leukocytes, NK cells, T cells, fibroblasts) in the periapical tissues produce many inflammatory mediators (e.g., prostaglandin, histamine), proinflammatory cytokines (e.g., IL-1, IL-6, TNF), and growth factors (e.g., PDGF, epidermal growth factor [EGF], keratinocyte growth factor [KGF], FGF). These inflammatory mediators,31 proinflammatory cytokines,80,155,201 and growth factors* are capable of stimulating epithelial cell rests to proliferate (Fig. 14-5). The extent of cellular proliferation appears to be related to the degree of inflammatory cell infiltration.192,193
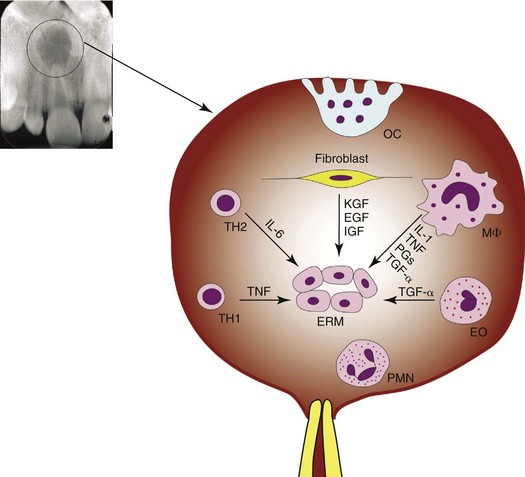
FIG. 14-5 Schematic illustration of the major mechanisms that activate proliferation of epithelial cell rests in apical periodontitis.
(From Lin LM, Huang GT-J, Rosenberg P: Proliferation of epithelial cell rests, formation of apical cysts, and regression of apical cysts after periapical wound healing. J Endod 33:908, 2007.)
Fibroblasts
Fibroblasts are important cells in chronic inflammation and wound healing. They are derived from undifferentiated mesenchymal cells and exist in all connective tissues. They synthesize and secret proteoglycans, glycoproteins, and precursor molecules of various types of collagens and elastin.114 In chronic inflammation, fibroblast migration and proliferation are triggered by multiple growth factors (TGF-β, PDGF, EGF, FGF) and the fibrogenic cytokines, IL-1 and TGF-α, produced by activated platelets, macrophages, endothelial cells, and inflammatory cells.121,148 In turn, activated fibroblasts produce an array of cytokines such as IL-1, IL-6, and granulocyte/macrophage colony-stimulating factor, which influence leukocyte development. Fibroblasts stimulated by inflammation also produce matrix metalloproteinase to degrade the proteins that comprise the extracellular matrix.148,258
Fibrovascular granulation tissue is also a prominent feature of chronic apical periodontitis as a repair process. Migration and proliferation of endothelial cells from preexisting capillaries and venules into the site of chronic inflammation is called neovascularization. It is mediated by angiogenesis factors, such as vascular endothelial growth factor (VEGF) and TGF-β, produced by activated macrophages, platelets, and endothelium.148,220 Neovascularization supplies oxygen and nutrients for the support of metabolically active macrophages and fibroblasts during wound healing.
Inflammatory Mediators
Many inflammatory mediators present in symptomatic apical periodontitis are also expressed in asymptomatic apical periodontitis. In addition, several different cytokines and growth factors are secreted by cells such as activated macrophages, lymphocytes, dendritic cells, and fibroblasts, in the chronic/adaptive immunoinflammatory response described earlier.
Histopathology
Macrophages and lymphocytes are predominant cells in asymptomatic apical periodontitis lesions (Fig. 14-6). Occasionally, foamy macrophages and giant cells are seen, especially associated with cholesterol crystal deposits, which are products of disintegrated cell membranes. Cholesterol crystals are present in approximately 18% to 44% of all apical periodontitis lesions.30,210,257 Bone resorption is a hallmark of asymptomatic apical periodontitis. Multinucleated osteoclasts may sometimes be seen in resorptive Howship’s lacunae. Proliferation of epithelial cell rests is often present in asymptomatic apical periodontitis lesions. ERM proliferate in three dimensions and form irregular strands or islands of epithelium, which are often infiltrated by varying degrees of inflammatory cells (see Fig. 14-6, B). A key feature of asymptomatic apical periodontitis is a proliferation of fibrovascular granulation tissue that is an attempt to prevent further spread of infection/inflammation and to repair wounded periapical tissues.
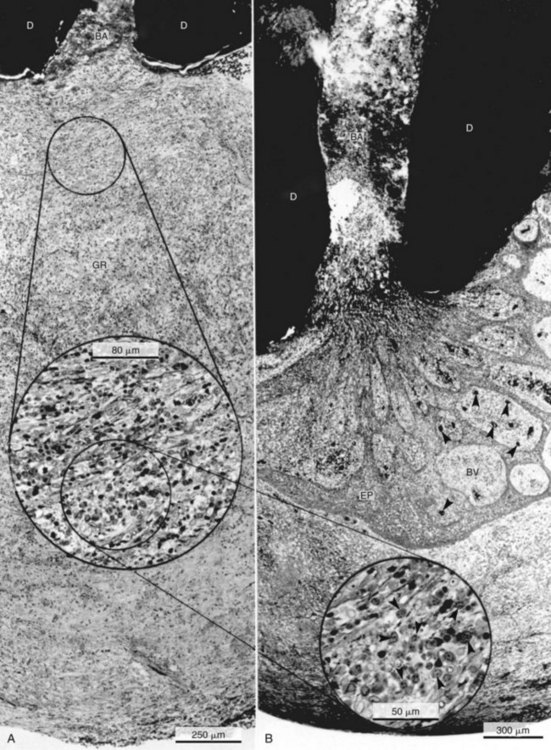
FIG. 14-6 Chronic asymptomatic apical periodontitis without (A) and with (B) epithelium (EP). The root canal contains bacteria (BA) in A and B. The lesion in A has no acute inflammatory cells, even at the mouth of the root canal with visible bacteria at the apical foramen (BA). Note the collagen-rich maturing granulation tissue (GR) infiltrated with plasma cells and lymphocytes (inset in A and B). BV, Blood vessels; D, dentin. (A ×80; B ×60; inset in A, ×250; inset in B, ×400.)
(From Nair PNR: Periodontol 2000 13:121, 1997.)
Little information is available concerning cemental changes in apical periodontitis. Using a scanning electron microscope, Simon et al.216 observed that projections, depressions, and fibers of cementum were haphazardly arranged in root canal infection under scanning electron microscopy. There was an increase of mineralized projections, cementum lacunae, and surface resorptions and a decrease in fibers. All these changes may provide a more favorable condition for attachment of bacterial biofilm on the apical external root surface in extraradicular infection.
It is a general belief that asymptomatic apical periodontitis lesions are usually devoid of innervation, so local anesthesia may not be necessary if teeth with asymptomatic apical periodontitis require root canal therapy. However, light and transmission electron microscopic studies have demonstrated that teeth with chronic apical periodontitis were well innervated (Fig. 14-7).141,150 This explains why instruments accidentally introduced into inflamed periapical tissues without local anesthesia can cause the patient pain.
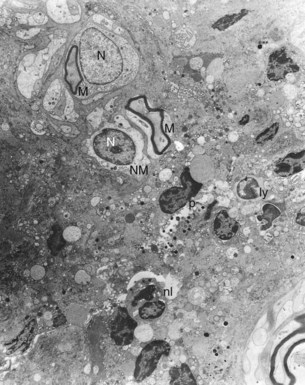
FIG. 14-7 Presence of intact myelinated and nonmyelinated nerve fibers in chronic apical periodontitis lesion. ly, Lymphocyte; M, myelinated nerve fiber; N, nucleus of Schwann cell; nl, neutrophilic leukocyte; NM, nonmyelinated nerve fiber; p, plasma cell (EM, ×1600).
(From Lin L, Langeland K: Innervation of inflammatory periapical lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 51:535, 1981.)
External root resorption that involves cementum or both cementum and dentin usually occurs in asymptomatic apical periodontitis lesions.127,263 Fortunately, cementum and dentin appear to be less readily resorbed than bone by inflammatory processes.85 However, the radiographic presence of root “blunting” should be interpreted by the astute clinician as evidence for resorption of dentin and cementum, and the working length of the instrumentation and obturation phases of nonsurgical endodontic treatment should be adjusted by a corresponding amount.
Using light and transmission electron microscopy and microbiologic culturing, bacteria have been shown to be present in many asymptomatic apical periodontitis lesions.2,101,162,255,271 Inadvertent contamination is a major concern when culturing bacteria from these lesions. Under meticulous transmission electron microscopic examination, Nair162 was unable to observe bacteria in most asymptomatic apical periodontitis lesions. If bacteria are present in the inflamed periapical tissues, they are usually found inside the phagocytes (Fig. 14-8).140 Importantly, the mere presence of bacteria in the inflamed periapical tissues (colonization) does not necessarily imply periapical infection. Bacteria have to be able to establish an infectious process, such as survival and tissue destruction, in the inflamed periapical tissues to be considered etiologic factors for the resulting infection. Most apical periodontitis lesions are not infected; nonsurgical root canal therapy of teeth with apical periodontitis can achieve a high success rate, provided root canal infection is controlled.
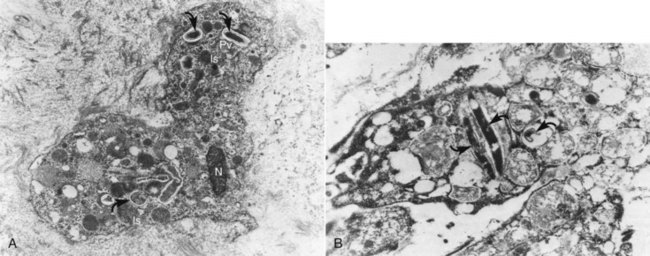
FIG. 14-8 Bacteria phagocytosed by phagocytes in chronic apical periodontitis lesion. A, Bacteria (arrows) in phagosome of a neutrophil. ls, Lysosome; N, part of nucleus; pv, phagocytic vacuole (EM, ×6300). B, Part of the cell structure of a macrophage. Note longitudinally and cross-cut bacteria cells with unit membrane (arrows) in phagocytic vacuoles (EM, ×10,000).
(From Lin L, Langeland K: Light and electron microscopic study of teeth with carious pulp exposures. Oral Surg Oral Med Oral Pathol 51:292, 1981.)
Clinical Features
Involved teeth are usually asymptomatic and show an ill-defined or well-defined periapical radiolucent lesion. The chronic apical abscess and associated sinus tract are usually asymptomatic. Occasionally, the sinus tract from an apical abscess may drain along the root surface and opens into the gingival sulcus. This leads to the development of a deep, narrow pseudopocket that often mimics a periodontal pocket or a vertical root fracture (see Chapter 18 for details).
Outcomes
Asymptomatic apical periodontitis may result in (1) regeneration or repair of the periapical tissues after root canal therapy; (2) severe periapical tissue destruction; (3) acute exacerbation; (4) development of an abscess with an intraoral or extraoral draining sinus tract; or (5) development of a serious cellulitis.
Asymptomatic Apical Periodontitis with Cyst Formation: Radicular Cyst, Chronic Apical Periodontitis with Cyst Formation
The radicular cyst is unique because no cysts in the body have similar pathogenesis. It is believed that the radicular cyst is likely formed by inflammatory proliferation of epithelial cell rests in the inflamed periodontal ligament.139,168,178,245 The radicular cyst is a pathologic cavity completely lined by nonkeratinized stratified squamous epithelium of variable thickness in a three-dimensional structure in an apical periodontitis lesion. A radicular cyst can be a “pocket cyst” (i.e., attached to the apical foramen) or a “true cyst” (no attachment to the root structure), but it cannot form by itself. Therefore, a radicular cyst should not be considered a separate disease entity from asymptomatic apical periodontitis. A radicular cyst (pocket or true) is classified as an inflammatory and not a neoplastic lesion in the WHO Histological Typing of Odontogenic Tumors, Jaw Cysts and Allied Lesions.120 The prevalence of radicular cysts in apical periodontitis lesions from extracted teeth ranges from 15% to 20%.164
Cell Biology
In addition to the presence of all chronic inflammatory cells, the epithelial cells are the most prominent cell type found in asymptomatic apical periodontitis with cyst formation. ERM can be considered unipotent or restricted-potential stem cells. They can be stimulated to divide symmetrically and asymmetrically into basal cells and suprabasal squamous cells of lining epithelium of radicular cysts.139 Many theories about apical cyst formation have been proposed. The nutritional deficiency theory assumes that when islands of epithelium keep growing, the central cells of the epithelial island will move farther away from their source of nutritional supply and undergo necrosis and liquefaction degeneration. The products accumulated attract neutrophils and granulocytes into the necrotic area. Microcavities then coalesce to form a cyst cavity lined by stratified squamous epithelium.245 The abscess theory postulates that when an abscess is formed in connective tissue, epithelial cells proliferate and line the abscess cavity because of their inherent tendency to cover exposed connective-tissue surface.168,178 The theory of merging of epithelial strands proposes that proliferating epithelial strands merge in all directions to form a three-dimensional spherelike structure composed of fibrovascular connective tissue, with varying degrees of inflammatory cells trapped, that gradually degenerates due to lack of blood supply. This leads to formation of a cyst cavity.139 Regardless of how a radicular cyst (pocket or true) is formed, it is likely caused by inflammatory proliferation (hyperplasia) of epithelial cell rests in these lesions.
It has been speculated that radicular cyst expansion is caused by increased osmotic pressure in the cyst cavity,248 but this hypothesis overlooks the cellular aspects of cyst growth and the biochemistry of bone destruction.66,155 Radicular cyst expansion is likely caused by degradation of fibrous connective tissue capsule by matrix metalloproteinases, which are produced by activated neutrophils, fibroblasts, and macrophages246 and bone resorption. The ERM have also been shown to be capable of secreting a bone resorbing factor.22 Most inflammatory mediators and proinflammatory cytokines that stimulate proliferation of epithelial cell rests also mediate bone resorption in apical periodontitis lesions.
Inflammatory Mediators
Mediators are basically similar to those present in chronic apical periodontitis.
Histopathology
Two types of cysts have been described in chronic apical periodontitis lesions. The pocket cyst’s lumen opens into the root canal of the involved tooth (Fig. 14-9).164,215 The true cyst is completely enclosed by a lining epithelium, and its lumen has no communication with the root canal of the involved tooth (Fig. 14-10).164,215 Apical cysts are lined by hyperplastic, nonkeratinized stratified squamous epithelium of variable thickness, which is separated from the fibrovascular connective tissue capsule by a basement membrane. Both the lining epithelium and the connective tissue capsule are usually infiltrated with inflammatory cells, indicating that inflammatory cells are attracted to these tissues by chemotactic irritants, either in the root canal system and/or in the periapical tissues. In nonproliferating epithelium, there is less inflammatory cell infiltration.39,205 Occasionally, mucous cell metaplasia or ciliated respiratory-type cells are present in cystic epithelial lining.174 The epithelial cells of lining epithelium do not show any characteristic features of neoplastic changes, such as pleomorphism, lack of polarity, nuclear enlargement, large nucleoli, abnormal nuclear/cytoplasmic ratio, hyperchromatism, or abnormal mitosis. Unlike odontogenic keratocysts and calcifying odontogenic cysts, the basal cells of radicular cystic lining epithelium are incapable of proliferation by themselves without stimulation of growth factors or cytokines released by innate and adaptive immune cells during periapical inflammation. The lumen of cysts may contain inflammatory exudates, cholesterol crystals, clear fluid, or bacterial colonies (Fig. 14-11; Fig. 14-12).164,193
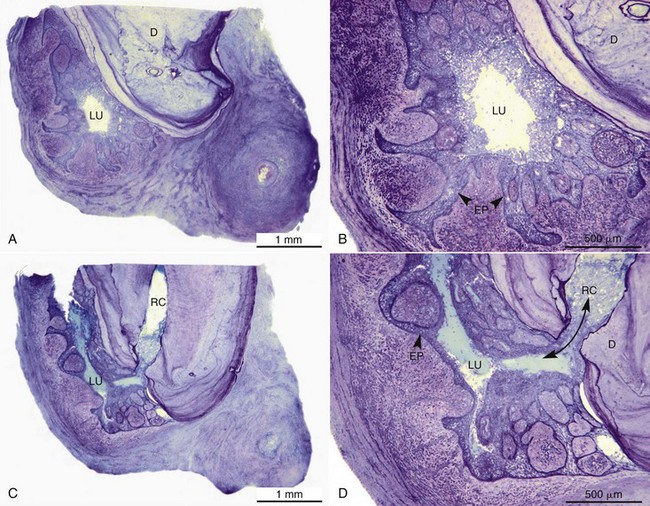
FIG. 14-9 A, A well-developed pocket cyst in apical periodontitis lesion. B, Note the saclike epithelial lumen. C-D, Sequential axial serial section passing through the apical foramen in the root canal plane (RC) shows the continuity of the lumen with the root canal. D, Dentin (A, C, ×16; B, D, ×40).
(From Nair PNR: Endod Topics 6:96, 2003.)
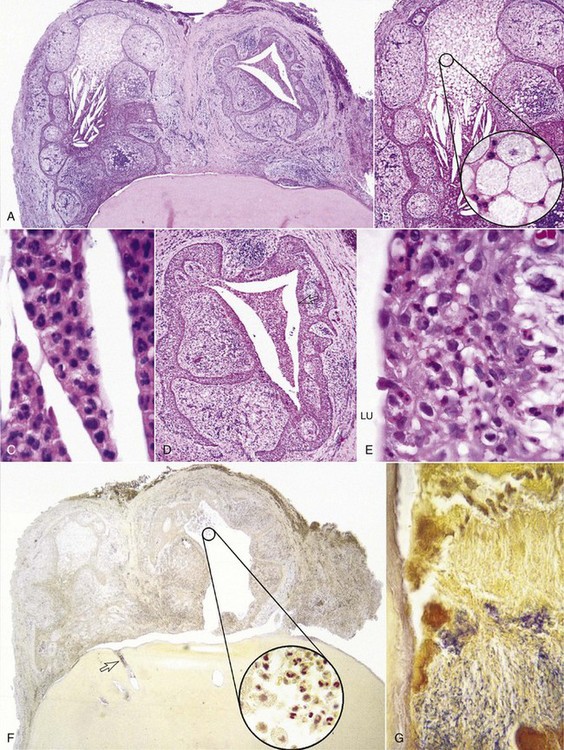
FIG. 14-10 A, Two distinct cysts lined by epithelium in apical periodontitis lesion. There is no evidence of communication with the foramen in serial sections (H & E, ×25). B, Cyst cavity on the left side in A. Accumulation of foamy macrophages (H & E, ×50; inset ×1000). C, Lower part of cavity in B. Dense infiltration of neutrophilic leukocytes (H & E, ×1000). D, Cyst cavity on the right side in A. Necrotic tissue debris in the lumen; infiltration of inflammatory cells in the epithelium (H & E, ×50). E, Cystic wall in D. Cyst’s lining epithelium is infiltrated with acute and chronic inflammatory cells. Lu, Lumen (H & E, ×1000). F, Foamy macrophages, neutrophilic leukocytes, and necrotic tissue inside the cyst (inset), but no bacteria (Brown & Brenn, ×25, inset ×1000). G, Area indicated by an open arrow in F. Bacterial colonies in the apical foramen (Brown & Brenn, ×1000).
(From Ricucci D, Pascon EA, Pitt Ford TR, Langeland K: Epithelium and bacteria in periapical lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 101:241, 2006.)
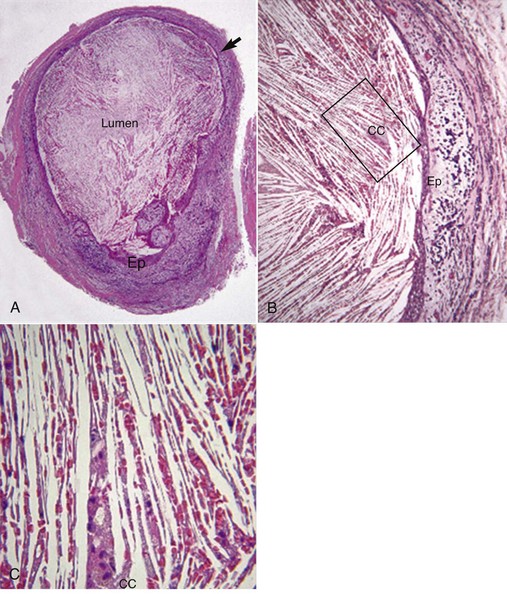
FIG. 14-11 A, A radicular cyst in which the lumen is completely filled with cholesterol crystals (H & E, ×25). B, High magnification of cholesterol crystals in A (H & E, ×100). C, High magnification of rectangular area in B. Multinucleated giant cells associated with cholesterol crystals (H & E, ×400).
(Courtesy Dr. Ricucci, Rome, Italy.)
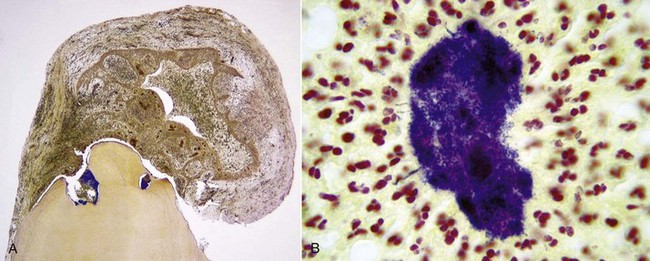
FIG. 14-12 A, An apical true cyst in apical periodontitis lesion. Bacterial colonies are seen in the foraminal areas (Brown & Brenn, ×25). B, Bacterial colonies in the cyst lumen, surrounded by inflammatory cells (Brown & Brenn, ×400).
(From Ricucci D, Bergenholtz G: Histologic features of apical periodontitis in human biopsies. Endod Topics 8:68, 2004, Fig. 6.)
Clinical Features
The involved tooth is usually asymptomatic. Periapical osteolytic lesions of endodontically involved teeth may sometimes show a well-demarcated radiolucency surrounded by a rim of radiopaque border.
Outcomes
There is no direct evidence demonstrating whether asymptomatic apical periodontitis with cyst formation (pocket and true) can or cannot regress after nonsurgical root canal therapy. Unfortunately, apical cysts cannot be diagnosed clinically and can only be diagnosed after surgical biopsy or extraction of teeth with apical periodontitis. Clinical outcome studies have shown that after proper nonsurgical root canal therapy, teeth with apical periodontitis heal in 78% of cases, regardless of whether preobturation bacterial cultures are negative or positive.219 Therefore, it is speculated that some cysts, especially pocket cysts, may heal after nonsurgical root canal therapy.163 Apical true cysts are less likely to heal after nonsurgical root canal therapy because of their self-sustaining nature; surgical intervention is necessary.163 Nonetheless, similar to the periapical pocket cyst, a periapical true cyst is also formed within an apical periodontitis lesion and is not a neoplastic lesion. Any disease caused by inflammation/infection should be able to heal if the causative irritant/irritants are removed, unless the irritant/irritants are neoplasm-inducing agents or a carcinogen.
Asymptomatic Apical Periodontitis with Reactive Bone Formation: Condensing Osteitis or Chronic Focal Sclerosing Osteomyelitis
Asymptomatic apical periodontitis with reactive bone formation is analogous to chronic osteomyelitis with proliferative periostitis. The etiology and pathogenesis of these two diseases are not well understood. It is generally believed that both lesions are caused by a long-term, low-grade inflammation/infection or a high resistance of local tissue to inflammation/infection.174,188 Instead of bone resorption, the inflammation induces reactive bone formation of alveolar trabecular or spongy bone around the periapex of endodontically involved teeth.
Cell Biology
As described previously, during the chronic stage of apical periodontitis, both osteoclast and osteoblast activity is decreased.268 However, in asymptomatic apical periodontitis with reactive bone formation, osteoblasts appear to be stimulated to produce more bone. It is not clear what factor/factors stimulate osteoblasts to produce more bone mass. It could be due to increased expression of growth factors/cytokines, such as TGF-β, BMP, PDGF, and transcription factor Cbfa1 (core-binding factor family).114
Histopathology
There is an excessive apposition of bone mass without bone resorption in the apical area. As the bone marrow spaces become smaller and obliterated, the bone resembles compact bone which is infiltrated by a small number of lymphocytes. The compact bone has few lacunae, and many of them are empty of osteocytes. It has many prominent resting and reversal lines, similar to an idiopathic osteosclerosis or a Pagetoid appearance.39,174,188
Clinical Features
The lesion is usually observed in young patients, and the mandibular first molar is most commonly involved. The teeth often have gross carious lesions and can be vital or nonvital. They are usually asymptomatic. Radiographically, the lesion may have a well-defined or ill-defined radiopaque mass associated with the apex of an endodontically involved tooth. The lamina dura around the root apex is usually intact.
Outcomes
Most apical periodontitis lesions with reactive bone formation demonstrate healing after nonsurgical root canal therapy. The excessive compact bone in most cases will be remodeled to normal appearance.63
Periapical Lesions of Nonendodontic Origin
There are many periapical lesions that are not of endodontic origin and should be considered in the differential diagnosis of apical periodontitis. These lesions include (but are not limited to) trauma,9 foreign bodies,118,166,282 host metabolic byproducts,167 advanced periodontal disease,124 fibro-osseous lesions, and benign and malignant tumors.174 A complete description of these lesions is beyond the scope of this chapter; the interested reader is encouraged to seek the cited references.
Extraradicular Endodontic Infection
Extraradicular endodontic infection implies that bacteria have established an infectious process outside the root canal system in the periapical area.162,217,231,255 Extraradicular endodontic infection can only be diagnosed after surgical biopsy or microbiologic sampling or molecular detection of therapy-resistant periapical lesions during apical surgery. The method of sample collection should be carefully reviewed, since contamination is a problem in bacteriologic study of apical periodontitis lesions during apical surgery.
Extraradicular endodontic infection can be a part of intraradicular infection or an independent entity without an intraradicular component. To be considered an independent extraradicular infection, the presence of pathogens in the root canal system must be completely ruled out, because intraradicular bacteria pose more potential problems than extraradicular bacteria in periapical inflammation. Bacteria in the complex root canal system are well protected from root canal procedures (i.e., mechanical instrumentation, antiseptic irrigation, intracanal medication), host defenses, and antimicrobial therapy because of lack of blood circulation and areas resistant to access (e.g., isthmi; see Chapter 7). In our opinion, the independent extraradicular infection probably does not occur often and is usually associated with apical actinomycosis.88,165,237 Bacteria have been observed as biofilm on the apical external root surface.217,254 In addition, viruses have also been isolated from apical periodontitis lesions.133,196,221 If microbiologic root canal cultures are not completely reliable (because bacteria may remain in the complex root canal system),200 then interpretation of independent extraradicular infections as endodontic treatment failures should be carefully evaluated.
Although virulence and number of pathogens, as well as the host’s defenses, determine whether extraradicular infection will develop or not, the development of extraradicular infection is still not fully understood. What are the predisposing factors to extraradicular infection? How are bacteria able to evade the host’s powerful defenses in order to colonize and establish an infectious process in the periapical tissues? Are all extraradicular infections resistant to nonsurgical root canal therapy? For instance, are bacteria present in apical periodontitis with abscess formation?29,83,179,234,262 Importantly, most periapical abscesses heal after proper nonsurgical root canal therapy. Similarly, most apical periodontitis lesions with draining sinus tracts also heal after proper nonsurgical root canal therapy, because the primary source of infection in the root canal system is removed. The preponderance of evidence does not support the consistent presence of an independent periapical infection in endodontic infections.
Apical Periodontitis and Systemic Diseases
Information concerning apical periodontitis and systemic diseases is limited. Systemic conditions affecting the severity of inflammatory periapical lesions have been documented and include diabetes, smoking, hypertension, cytokine gene polymorphisms, and nonspecific disorders of the immune system. In general, diabetic patients are prone to infections due to generalized vascular disorders caused by accumulation of atheromatous plaques in the vascular walls, particularly of capillaries. Atherosclerotic vessels cause impaired leukotactic response and failure in delivering humoral and cellular components of the immune system.18 Clinical and radiographic studies have shown that there is a greater prevalence of periapical lesions in diabetics than in nondiabetics.18 Type 2 diabetes mellitus is significantly associated with an increased prevalence of apical periodontitis.204 Other findings demonstrated that type 2 diabetes is associated with increased risk of a poor response by the periradicular tissues to odontogenic pathogens.28 In rodent models, diabetic conditions enhance the development of periradicular lesions and increase mortality.67,103,116 Mice deficient in P/E-selectin are shown to be more prone to infection-mediated early-onset periodontal disease.175
Genetic polymorphisms may also play a role in the individual’s response to various diseases. Haplotype analyses indicated that a variant of the IL-1β gene was likely to be most important for early-onset periodontitis risk.56 In a Chinese male population, it was reported that the polymorphisms of IL1A+4845 and IL1B−511 might play an important role in determining generalized aggressive periodontitis susceptibility. A possible combined effect of the polymorphism of IL1B−511 and smoking on this type of periodontitis susceptibility was suggested.134 In a study on patients from Chile, the heterozygous inheritance of the IL1B+3954 gene was found significantly higher in periodontitis cases than in control patients. The prevalence of a positive genotype (at least one allele of the two variants present at each locus) is significantly higher in cases (26%) than in controls (10%) and significantly associated with periodontitis, irrespective of smoking status and periodontitis severity.146 Those displaying high-producer IL6 genotype, intermediate and high-producer IL1B genotypes, and low-producer TNFA genotype tend to exhibit symptomatic dental abscesses.54
Apical periodontitis was once considered a focus of infection, and the microorganisms from the focus could disseminate through blood circulation to remote parts of the body, where secondary disease occurred.97 Even though bacteremias do occur during root canal treatment of teeth with apical periodontitis when instrumentation is carried through the apical foramen into the periapical tissues, the incidence and magnitude of bacteremias is not clinically significant for a healthy person and in fact appears to be lower than that observed by flossing.16,51,52 Microorganisms in the blood circulation are rapidly eliminated by the host’s humoral and cellular defense components within minutes.16,52 However, bacteremia may pose a potential danger to immunocompromised patients or patients with congenital heart valve disease.51 No correlation between apical periodontitis and coronal heart disease and rheumatoid arthritis has been observed.37,160 There is insufficient evidence to support that apical periodontitis could serve as a focus of infection and cause significant systemic diseases.62,182
Wound Healing of Apical Periodontitis
Periapical Wound Healing After Nonsurgical Root Canal Therapy
Understanding of wound healing is as important as knowing the pathogenesis of disease, because satisfactory wound healing is the ultimate goal of treatment. If we are able to understand the mechanisms of periapical wound healing, we can design treatment approaches that maximize favorable conditions for wound healing to occur, such as effective disinfection of the root canal system in nonsurgical root canal therapy, control of periapical inflammation by medication, or incorporation of growth factors in bone grafts in surgical endodontic therapy (see also Chapters 9 and 21).
Interestingly, healing begins as soon as inflammation starts. When irritants (microbial and nonmicrobial) in the canal systems or in the periapical tissues are eliminated by nonsurgical or surgical endodontic therapy, inflammatory mediators are no longer produced in the periapical tissues because of reduction of immunoinflammatory cells. Inflammatory mediators already present are inactivated by the body’s control mechanisms to prevent an inflammatory reaction from going unchecked. This process precedes wound healing. Although a great deal of information is known about what turns on the inflammation, relatively little is known about what turns off the inflammatory system after elimination of irritants. Examples of host antiinflammatory control mechanisms are (1) enzyme destruction of inflammatory activators; (2) natural inhibitors of inflammatory mediators (e.g., opioids, somatostatin, glucocorticoids, etc.); (3) relative balance between intracellular levels of cyclic AMP and cyclic GMP; (4) the antiphlogistic role of histamine; (5) inhibitors of the complement system258; and (6) antiinflammatory cytokines, such as IL-4, IL-10, IL-13 and TGF-β.86,172 In addition, the major cellular inducer of inflammation, neutrophilic leukocytes, undergo apoptosis,82 and the major cellular player of wound healing, macrophages, secrete antiinflammatory molecules.172
The process of wound healing is tightly regulated by cell-to-cell and cell-to–extracellular matrix interactions, as well as the temporal and spatial expression of a variety of cytokines, growth factors, and neuropeptides and apoptosis (Table 14-3).26,79,82,228,273 All these cellular and humoral factors operate together in either an antagonistic or synergistic manner and are precisely orchestrated during wound healing. This results in a highly organized response permitting regeneration of the original tissue architecture. Wound healing appears to be a programmed event. Much information concerning the pathogenesis of apical periodontitis has been gained from animal studies.69,108,251 Although studies of wound healing of teeth with apical periodontitis after nonsurgical root canal therapy and endodontic surgery are available in a few animal experiments and a human study,11,129,130 no studies of wound healing of apical periodontitis lesions with cyst formation after nonsurgical root canal therapy exist in the literature.
TABLE 14-3 Important Growth Factors/Cytokines in Wound Healing
| Cytokines | Major Source | Target Cells and Major Effects |
|---|---|---|
| EGF | Macrophages, platelets, fibroblasts | Epithelial cells, fibroblasts, endothelial cells |
| FGF | Macrophages, endothelial cells | Endothelial cells (angiogenesis), mesenchymal cells |
| TGF-α | Macrophages, platelets, keratinocytes | Angiogenesis Fibroblasts |
| TGF-β | Macrophages, platelets | Similar to EGF |
| PDGF | Macrophages, platelets, endothelial cells | Chemoattractant for macrophages, fibroblasts |
| VEGF | Macrophages, epithelial cells | Angiogenesis |
| IGF | Fibroblasts, epithelial cells | Granulation tissue formation, reepithelialization |
| CSF | Multiple cells | Macrophages, granulation tissue formation |
| SP, CGRP | Sensory nerve | Endothelial cells, fibroblasts, keratinocytes |
CGRP, Calcitonin gene–related peptide; CSF, colony-stimulating factor; EGF, epidermal growth factor; FGF, fibroblast growth factor; IGF, insulin-like growth factor; PDGF, platelet-derived growth factor; TGF, transforming growth factor; VEGF, vascular endothelial growth factor.
Modified from Majno G, Joris I: Cell, tissues, and disease, ed 2, Oxford, Oxford University Press, 2004; Slauson DO, Cooper BJ: Mechanisms of disease, ed 3, St Louis, Mosby, 2002; Werner S, Grose R: Regulation of wound healing by growth factors and cytokines. Physiol Rev 83:835, 2003.
Wound healing of apical periodontitis lesions after proper nonsurgical root canal therapy follows the general principle of wound healing of connective tissues elsewhere in the body, with the formation of fibrovascular granulation tissue, removal of necrotic tissue and dead bacteria by activated macrophages, and finally repair and/or regeneration of the wounded tissue. Healing of apical periodontitis lesions is largely accomplished by regeneration and to some degree by fibrosis. Local tissue resident cells involved in periapical wound healing are osteoblasts and bone marrow stromal cells in alveolar bone and multipotent stem cells in periodontal ligament.209 During periapical wound healing, many unwanted hyperplastic cells (e.g., endothelial cells, fibroblasts, epithelial cells) are deleted by apoptosis,53,79,82 and the extracellular matrix is remodeled by metalloproteinases. Pathologic processes such as extensive fibrosis do not occur, and the damaged periapical tissues can be ideally restored to their original structure by the process of regeneration.
The temporal and spatial relationship between alveolar bone, cementum, and periodontal ligament during periapical wound healing after nonsurgical root canal therapy cannot be clearly delineated. Nevertheless, wound healing appears to recapitulate the initial histogenesis of damaged tissues or organs in many instances. The process of periapical wound healing after nonsurgical root canal therapy may be similar to wound healing following guided tissue regeneration in periodontal therapy: regeneration of new periodontal ligament, new cementum, and new alveolar bone.61,256 Both nonsurgical root canal therapy and guided tissue regeneration therapy are able to remove irritants and provide a favorable environment conducive to regeneration of periodontal tissues damaged by apical periodontitis and marginal periodontitis, respectively.
During periapical wound healing, the cells of viable periodontal ligament from adjacent root surfaces proliferate to cover the root surfaces in which the periodontal ligament was damaged by apical periodontitis and removed by macrophages. Proteins derived from Hertwig’s epithelial root sheath (i.e., enamel matrix proteins) are required for cementoblast differentiation from ectomesenchymal stem cells in the dental follicle during tooth development.171 The cells of Hertwig’s epithelial root sheath are absent in mature teeth.147,223
Nevertheless, the extracellular matrix and growth factors of cementum (i.e., IGF-1, FGFs, EGF, BMP, TGF-β, PDGF) sequestered after cemental resorption in mature teeth are capable of inducing proliferation, migration, attachment, and differentiation of multipotent stem cells in the periodontal ligament into cementoblast-like cells and produce cementoid tissue on the root surface denuded of periodontal ligament.81,209 This is similar to reparative dentin formation by pulp stem cells in direct pulp capping procedures where growth factors such as TGF-β are released from dentin matrix binding sites.170,195,260 Root resorption that involves cementum or both cementum and dentin can only be repaired by cementoid tissue, because multipotent stem cells of the periodontal ligament are incapable of differentiating into odontoblasts that produce dentin.209
Bone has a remarkable capacity for regeneration in response to injury. During periapical wound healing, the osteoblasts or mesenchymal cells lining the surfaces of endosteum—stimulated by TGF-β, BMPs, IGFs, PDGF, VEGF, and cytokines released by stromal cells, osteoblasts, platelets, and bone matrix after bone resorption—can undergo proliferation and differentiation into osteoblasts and produce bone matrix.6,145 When one of the cortical bone plates (buccal or lingual/palatal) is destroyed, mesenchymal cells in the inner layer of periosteum beneath the oral mucosa—stimulated by TGF-β, BMPs, IGFs, PDGF, and VEGF—are also capable of proliferation and differentiation into osteoblasts and can produce bone matrix.114,145 If both buccal and lingual/palatal cortical bone plates are destroyed by large apical periodontitis lesions, it is possible that the lesion may be repaired with fibrous scar tissue because of extensive destruction of the periosteum beneath the oral mucosa.11 Accordingly, a guided tissue regeneration procedure using membrane barriers and bone grafts is recommended to prevent ingrowth of fibroblasts from periosteum or submucosa into the bony defect and to enhance periapical wound healing if periapical surgery is necessary.48,49 The cellular and molecular mechanisms of excess scar tissue formation in periapical wound healing is not completely understood. Growth factors/cytokines may play an important role in regulating fibroblast gene expression and excess scar tissue formation.259
The newly regenerated periodontal ligament will finally undergo remodeling into mature periodontal ligament, with one group of collagen fibers (Sharpey’s fibers) inserted into the newly formed cementum and another group of collagen fibers inserted into the newly formed alveolar bone. Thereby, regeneration of damaged periapical tissues, cementum, periodontal ligament, and alveolar bone is completed.
Periapical Wound Healing After Surgical Endodontic Therapy
The mechanism of periapical wound healing after nonsurgical and surgical endodontic therapy is quite similar, but the kinetics of healing of the periapical wound after endodontic surgery is much faster than nonsurgical endodontic therapy.122 In surgical endodontic therapy, a surgeon performs removal of irritants, such as necrotic cells, tissue debris, and bacteria in the periapical lesions, which is called surgical débridement. In contrast, in nonsurgical endodontic therapy, activated macrophages perform bacterial killing and cleanup of periapical lesions, which is called biologic débridement.138 Surgical débridement is very effective and of course quite rapid, whereas biologic débridement takes time. However, endodontic surgery is more invasive. In addition, proper case selection is more important in endodontic surgery than in nonsurgical endodontic therapy. The distinctive difference between nonsurgical and surgical endodontic therapy is that the goal of nonsurgical endodontic therapy is to remove primary microbial etiology from the root canal system, and the goal of surgical endodontic therapy is often to seal microbial etiology within the root canal system by root-end filling in most cases (see also Chapter 21).
Can Radicular Cysts in Apical Periodontitis Lesions Regress After Nonsurgical Endodontic Therapy?
Based on histology and cell biology, no studies have ever shown that apical true cysts are different from apical pocket cysts. It was suggested that pocket cysts in apical periodontitis lesions might regress after nonsurgical root canal therapy by the mechanism of apoptosis or programmed cell death, based on molecular cell biology.139 In contrast, apical true cysts may be less likely to heal after nonsurgical root canal therapy because of their self-sustaining nature.163 Histologically, inflammatory cell infiltration is always present in the lining epithelium and/or fibrous connective tissue capsule of apical true cysts.164,192,193 This indicates there is continued presentation of irritants (such as bacteria)—present in the root canal system, the periapical tissues, or the lumen of cysts—to attract inflammatory cells to the cystic lining epithelium and/or fibrous connective tissue capsule.164,192,193 It is not known if epithelial cells of apical true cysts alone are capable of acting as autocrine cells and secreting growth factors to sustain their own survival. It is important to realize that the apical true cyst is completely different from the odontogenic keratocyst, which is self-sustaining because it is a neoplastic lesion. Biologically, it is very unlikely that the hyperplastic epithelial cells of inflammatory apical true cysts would suddenly transform into cells that behave like self-sustaining neoplasms. Any disease caused by infection should be able to regress after removal of its causative irritant(s), unless the irritants themselves are neoplasm-inducing agents or carcinogens, such as some viruses and human malignant tumors.148,265 From pathogenesis, histology, and molecular cell biology, apical true cysts are similar to pocket cysts. Accordingly, apical true cysts, similar to pocket cysts, may be able to regress after nonsurgical root canal therapy by the mechanism of apoptosis if root canal infection is effectively under control.139 This prediction is consistent with the high level of healing observed after nonsurgical root canal treatment.
In apical periodontitis lesions with cyst formation, the cysts have to regress before the periapical tissues can be restored to their original architecture. It is not known what matrix serves as a scaffold for endothelial cells, fibroblasts, and osteoblasts to migrate into the lumen of regressing cysts after nonsurgical root canal therapy. Complete regression of radicular cysts after nonsurgical root canal therapy could be due to any of several possible scenarios. Regression of radicular cysts and regeneration of bone may occur concurrently; or during regression of radicular cysts, part of the cystic lining epithelium could disintegrate due to apoptosis of local epithelial cells. Together with degradation of the basal lamina by matrix metalloproteinases, this could allow a fibrous connective tissue capsule to grow into the lumen of radicular cysts. Eventually, the cystic lining epithelium will completely regress or become remnants of epithelial cell rests remaining in the periodontal ligament.
Taken together, our knowledge of cyst mechanisms and healing offer tantalizing clinical implications and clearly support the conclusion that more studies are required to understand the complex mechanisms of regression of inflammatory apical cysts, both pocket and true.
Factors Influencing Periapical Wound Healing
Local and systemic factors may affect periapical wound healing. Infection will complicate and prevent wound healing, foreign bodies can impair wound healing,166,282 and nutrition can also affect wound healing.276 Diabetes was reported to reduce the likelihood of healing of apical periodontitis lesions after nonsurgical root canal therapy.68 Impaired nonspecific immune response and disorders of the vascular system appeared to have a significant influence on the success rate of nonsurgical root canal therapy on teeth with apical periodontitis.149 However, immunocompromised patients, such as HIV patients, responded as well as counterpart patients after nonsurgical endodontic therapy.43,76,185 Although smoking has not been shown to be associated with increased incidence of apical periodontitis and prognosis of nonsurgical root canal therapy,20,57 smoking may increase complications of periapical surgery, such as pain and swelling.74
Patients receiving radiotherapy of jaws and bisphosphonate therapy have a risk of developing osteonecrosis of jawbones,190,194,277 so nonsurgical root canal therapy is recommended for these patients.110,136 However, for endodontic surgery, the guidelines of the American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws are highly recommended.7 For asymptomatic patients who have taken an oral bisphosphonate for less then 3 years and have no clinical risk factors, endodontic surgery is not contraindicated.7 Nevertheless, the patient’s physician should be consulted. Any kind of surgical procedures should be avoided for patients receiving intravenous bisphosphonate.7 To prevent the complication of osteonecrosis, if patients are going to receive radiation of jaws or bisphosphonate therapy, nonsurgical or surgical endodontic therapy should be completed before initiation of radiation or bisphosphonate therapy.7
1. Abbas AK, Lichtman AH, Pober JS. Cellular and molecular immunology. Philadelphia: Saunders; 2007.
2. Abous-Rass M, Bogen G. Microorganisms in closed periapical lesions. Int Endod J. 1998;31:39.
3. Akamine A, Hashiguchi I, Toriya Y, Maeda K. Immunohistochemical examination of the localization of macrophages and plasma cells in induced rat’s periapical lesions. Endod Dent Traumatol. 1994;10:121.
4. Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nature Immunol. 2001;2:675.
5. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783.
6. Al-Aql ZS, Alagl AS, Graves DT, Gerstenfeld LC, Einborn TA. Molecular mechanisms controlling bone formation during fracture healing and distraction osteogenesis. J Dent Res. 2008;87:107.
7. American Association of Oral and Maxillofacial Surgeons: Position paper on bisphosphonate-related osteonecrosis of the jaws. Rosemont, IL: AAOMS Board of Trustees, Sept, 2006.
8. American Dental Association. 1990 survey of dental services rendered. Chicago: American Dental Association; 1994.
9. Andreasen FM. Transient apical breakdown and its relation to color and sensitivity changes after luxation injuries to teeth. Endod Dent Traumatol. 1986;2:9.
10. Andreasen JO, Andreasen FM. Textbook and color atlas of traumatic injuries to the teeth, ed 3. Copenhagen: Munksgaard; 1994.
11. Andreasen JO, Rud J. Modes of healing histologically after endodontic surgery in 70 cases. Int Oral Surg. 1972;1:148.
12. Babior BM. The respiratory burst of phagocytes. J Clin Invest. 1984;73:599.
13. Bartkiewicz M, Hernando N, Reddy SV, Roodman DG, Baron R. Characterization of the osteoclast vacuolar H+-ATPase B-subunit. Gene. 1995;160:157.
14. Baron R. Molecular mechanisms of bone resorption by the osteoclasts. Anat Rec. 1989;224:317.
15. Barthel CR, Zimmer S, Trope M. Relationship of radiologic and histologic signs of inflammation in human root-filled teeth. J Endod. 2004;30:75.
16. Baumgartner JC, Heggers JP, Harrison JW. The incidence of bacteremias related to endodontic procedures. I. Non-surgical endodontics. J Endod. 1976;2:135.
17. Bender IB. Factors influencing the radiographic appearance of bone lesions. J Endod. 1982;8:161.
18. Bender IB, Bender AB. Diabetes mellitus and the dental pulp. J Endod. 2003;29:383.
19. Bender IB, Seltzer S. Roentgenographic and direct observation of experimental lesions in bone: 1. J Am Dent Assoc. 1961;62:152.
20. Bergstrom J, Babcan J, Eliasson S. Tobacco smoking and dental periapical condition. Eur J Oral Sci. 2004;112:115.
21. Bezerra MC, Carvalho JF, Prokopowitsch AS, Pereira RM. RANK, RANKL and osteoprotegerin in arthritic bone loss. Braz J Med Biol Res. 2005;38:161.
22. Birek C, Heersche JN, Jez D, Brunette DM. Secretion of a bone resorbing factor by epithelial cells cultured from porcine rests of Malassez. J Periodontal Res. 1983;18:75.
23. Birklein F, Schmelz M. Neuropeptides, neurogenic inflammation and complex regional pain syndrome (CRPS). Neurosci Lett. 2008;437:199.
24. Block RM, Bushell A, Rodrigues H, Langeland K. A histopathologic, histobacteriologic, and radiographic study of periapical endodontic surgical specimens. Oral Surg Oral Med Oral Pathol. 1976;42:656.
25. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337.
26. Brian SD. Sensory peptides: their role in inflammation and wound healing. Immunopharmacology. 1997;37:133.
27. Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. 2007;5:577.
28. Britto LR, Katz J, Guelmann M, Heft M. Periradicular radiographic assessment in diabetic and control individuals. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96:449.
29. Brook I, Fraizier EH, Gher ME. Aerobic and anaerobic microbiology of periapical abscess. Oral Microbiol Immunol. 1991;6:123.
30. Browne RM. The origin of cholesterol in odontogenic cysts in man. Arch Oral Biol. 1971;16:107.
31. Brunette DM. Cholera toxin and dibutyl cyclic-AMP stimulate the growth of epithelial cells derived from epithelial cell rests from porcine periodontal ligament. Arch Oral Biol. 1984;29:303.
32. Brynolf I. A histological and roentgenological study of the periapical region of human upper incisors. Odontol Rev. 1967;18(Suppl 11):1,.
33. Burd PR, Rogers HW, Gordon JR, et al. Interleukin 3-dependent and –independent mast cells stimulated with IgE and antigen express multiple cytokines. J Exp Med. 1989;170:245.
34. Byers MR, Narhi MVO. Dental injury models. Experimental tools for understanding neuroinflammatory nociceptor functions. Crit Rev Oral Biol. 1999;10:4.
35. Byers MR, Taylor PE, Khayat BG, Kimberly CL. Effects of injury and inflammation on the pulp and periapical nerves. J Endod. 1990;16:78.
36. Canalis E, McCarthy TL, Centrella M. Growth factors and cytokines in bone cell metabolism. Annu Rev Med. 1991;42:17.
37. Caplan DJ, Chasen JB, Krall EA, et al. Lesions of endodontic origin and risk of coronary heart disease. J Dent Res. 2006;85:996.
38. Caviedes-Bucheli J, Muñoz HR, Azuero-Holguín MM, Ulate E. Neuropeptides in dental pulp: the silent protagonists. J Endodon. 2008;34:773.
39. Cawson RA, Everson JW. Oral pathology and diagnosis. Philadelphia: Saunders; 1987.
40. Chedid M, Rubin JS, Csaky KG, Aaronson SA. Regulation of keratinocyte growth factor gene expression by interleukin 1. J Biol Chem. 1994;269:10753.
41. Cochran DL, Wozney JM. Biological mediators for periodontal regeneration. Periodontol 2000. 1999;19:40.
42. Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol. 2004;5:975.
43. Cooper H. Root canal treatment in patients with HIV infection. Int Endod J. 1993;26:369.
44. Cortesini R, LeMaoult J, Ciubotariu R, Cortesini NS. CD8+CD28- T suppressor cells and the induction of antigen-specific, antigen-presenting cell-mediated suppression of TH reactivity. Immunol Rev. 2001;182:201.
45. Costerton JW, Geesey GG, Chen KJ. How bacteria stick. Sci Am. 1978;238:86.
46. Cymerman JJ, Cymerman DH, Walters I, Nevins AJ. Human T lymphocyte subpopulations in chronic periapical lesions. J Endod. 1984;10:9.
47. Dahlen G, Magnusson BC, Moller A. Histological and histochemical study of the influence of lipopolysaccharide extracted from Fusobacterium nucleatum on the periapical tissues in the monkey Macaca fascicularis. Arch Oral Biol. 1981;26:591.
48. Dahlin C, Gottlow J, Linde A, Nyman S. Healing of maxillary and mandibular bone defects using a membrane technique. An experimental study in monkeys. Scand J Plast Reconstr Hand Surg. 1990;24:13.
49. Dahlin C, Linde A, Gottlow J, Nyman S. Healing of bone defects by guided tissue regeneration. Plast Reconstr Surg. 1988;81:672.
50. Damoiseaux J. Regulatory T cells: back to the future. Neth J Med. 2006;64:4.
51. Debelian GJ, Olsen I, Transtad L. Systemic diseases caused by oral microorganisms. Endod Dent Traumatol. 1994;10:5.
52. Debelian GJ, Olsen I, Transtad L. Bacteremia in conjunction with endodontic therapy. Endod Dent Traumatol. 1995;11:142.
53. Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56.
54. de Sa AR, Moreira PR, Xavier GM, et al. Association of CD14, IL1B, IL6, IL10 and TNFA functional gene polymorphisms with symptomatic dental abscesses. Int Endod J. 2007;40:563.
55. Dickerson C, Undem B, Bullock B, Winchurch RA. Neuropeptides regulation of proinflammatory cytokine responses. J Leukocyte Biol. 1998;63:602.
56. Diehl SR, Wang Y, Brooks CN, et al. Linkage disequilibrium of interleukin-1 genetic polymorphisms with early-onset periodontitis. J Periodontol. 1999;70:418.
57. Duncan HF, Pitt Ford TR. The potential association between smoking and endodontic disease. Int Endo J. 2006;39:843.
58. Dwyer TG, Torabinejad M. Radiographic and histologic evaluation of the effect of endotoxin on the periapical tissues of the cat. J Endod. 1980;7:31.
59. Dziak R. Biochemical and molecular mediators of bone metabolism. J Periodontol. 1993;64:407.
60. Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, Landmann R. Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis. 2002;186:798.
61. Egelberg J. Regeneration and repair of periodontal tissues. J Periodontal Res. 1987;22:233.
62. Ehrmann EH. Focal infection. The endodontic point of view. Oral Surg Oral Med Oral Pathol. 1977;44:628.
63. Eliasson S, Halvarson C, Ljunheimer C. Periapical condensing osteitis and endodontic treatment. Oral Surg Oral Med Oral Pathol. 1984;57:195.
64. Eriksen HM, Bjertness E. Prevalence of apical periodontitis and results of endodontic treatment in middle-aged adults in Norway. Dent Traumatol. 1991;7:1.
65. Figdor D. Apical periodontitis: a very prevalent problem. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:65.
66. Formigli L, Orlandini SZ, Tonelli P, Gianelli M, Martini M, Brandi M. Osteolytic processes in human radicular cysts: morphological and biochemical results. J Oral Pathol Med. 1995;24:216.
67. Fouad AF, Barry J, Russo J, Radolf J, Zhu Q. Periapical lesion progression with controlled microbial inoculation in a type I diabetic mouse model. J Endod. 2002;28:8.
68. Fouad AF, Burleson J. The effect of diabetes mellitus on endodontic treatment outcome: data from an electronic patient record. J Am Dent Assoc. 2003;134:43.
69. Fouad AF, Walton RE, Rittman BR. Induced periapical lesions in ferret canines: histological and radiographic evaluation. Dent Traumatol. 1992;8:56.
70. Friedman S. Considerations and concepts of case selection in the management of post-treatment endodontic disease (treatment failure). Endod Topics. 2002;1:54.
71. Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231.
72. Gao Z, Falitz CM, Mackenzie IC. Expression of keratinocyte growth factor in periapical lesions. J Dent Res. 1996;75:1658.
73. Gao Z, Mackenzie IC, Rittman BR, Korszun AK, Williams DM, Cruchley AT. Immunocytochemical examination of immune cells in periapical granulomas and odontogenic cysts. J Oral Pathol. 1988;17:84.
74. Garcia B, Penerrocha M, Marti E, Gay-Escodad C, von Arx T. Pain and swelling after periapical surgery related to oral hygiene and smoking. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;104:271.
75. Giachelli CM, Steitz S. Osteopontin: a versatile regulator of inflammation and biomineralization. Matrix Biol. 2000;19:615.
76. Glick M, Abel SN, Muzyka BC, DeLorenzo M. Dental complications after treating patients with AIDS. J Am Dent Assoc. 1994;125:296.
77. Gonzalez-Rey E, Chorny A, Delgado M. Regulation of immune tolerance by anti-inflammatory neuropeptides. Nat Rev Immunol. 2007;7:52.
78. Green TL, Walton RE, Taylor JK, Merrell P. Radiographic and histologic findings of root canal treated teeth in cadaver. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:707.
79. Greenhalgh DG. The role of apoptosis in wound healing. Int J Biochem Cell Biol. 1998;30:1019.
80. Grossman RM, Kreuger J, Yourish D, et al. Interleukin-6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocyte. Proc Nat Acad Sci U S A. 1989;86:6367.
81. Grzesik WJ, Narayanan AS. Cementunm and periodontal wound healing and regeneration. Crit Rev Oral Biol Med. 2002;13:474.
82. Haanen C, Vermes IV. Apoptosis and inflammation. Mediators Inflamm. 1995;4:5.
83. Haapasalo M, Ranta K, Ranta H. Mixed anaerobic periapical infection with sinus tract. Endod Dent Traumatol. 1987;3:83.
84. Hahn C-L, Liewehr FR. Innate immune responses of the dental pulp to caries. J Endod. 2007;33:643.
85. Hammarstrom L, Lindskog S. General morphological aspects of resorption of teeth and alveolar bone. Int Endod J. 1985;18:93.
86. Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13:413.
87. Hancock HHIII, Sigurdsson A, Trope M, Moiseiwitsch J. Bacteria isolated after unsuccessful endodontic treatment in a north American population. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001;91:579.
88. Happonen RP. Periapical actinomycosis: a follow-up study of 16 surgically treated cases. Endod Dent Traumatol. 1986;2:205.
89. Hargreaves KM, Goodis HE. Dental Pulp. Chicago: Quintessence; 2002.
90. Hargreaves KM, Swift JQ, Roszkowski MT, et al. Pharmacology of peripheral neuropeptide and inflammatory mediator release. Oral Surg Oral Med Oral Pathol. 1994;78:503.
91. Hauman CHJ, Love RM. Biocompatibility of dental materials used in contemporary endodontic therapy: a review. Part 2. Root-canal filling materials. Int Endod J. 2003;36:147.
92. Hirsh BC, Johnson WC. Concepts of granulomatous inflammation. Int J Dermatol. 1984;23:90.
93. Hirvonen T, Hippi P, Narhi M. The effect of an opioid antagonist and somatostatin antagonist on the nerve function in normal and inflamed dental pulps. J Dent Res. 1998;77:1329.
94. Hou L, Sasaki H, Stashenko P. Toll-like receptor 4-deficient mice have reduced bone destruction following mixed anaerobic infection. Infect Immun. 2000;68:4681.
95. Hou L, Sasakj H, Stashenko P. B-Cell deficiency predisposes mice to disseminating anaerobic infections: protection by passive antibody transfer. Infect Immun. 2000;68:5645.
96. Hou L, Wang X. PKC and PKA, but not PKG mediate LPS-induced CGRP release and (Ca2+)I elevation in DRG neurons of neonatal rats. J Neurosci Res. 2001;66:592.
97. Hunter W. Oral sepsis as a cause of disease. Br Med J. 1900;2:215.
98. Huumonen S, Orstavik D. Radiological aspects of apical periodontitis. Endod Topics. 2002;1:3.
99. Ingle J, Bakland L, Baugartner C. Ingle’s endodontics, ed 6. Hamilton: BC Decker; 2008.
100. Irwin CR, Schor SL, Ferguson NW. Expression of EGF-receptor on epithelial and stromal cells of normal and inflamed gingiva. J Periodont Res. 1991;26:388.
101. Iwu C, MacFarlane TW, MacKenzie D, Stenhouse D. The microbiology of periapical granulomas. Oral Surg Oral Med Oral Pathol. 1990;69:502.
102. Isaka J, Ohazama A, Kobayashi M, et al. Participation of periodontal ligament cells with regeneration of alveolar bone. J Periodontol. 2001;72:314.
103. Iwama A, Nishigaki N, Nakamura K, et al. The effect of high sugar intake on the development of periradicular lesions in rats with type 2 diabetes. J Dent Res. 2003;82:322.
104. Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197.
105. Janeway CA, Travers P, Walport M, Shlomchik MJ. Immunobiology: the immune system in health and disease, 6th ed. New York: Garland Science; 2005.
106. Jimenez-Pinzon A, Segura-Egea JJ, Poyato-Ferrera M, Velasco-Ortega E, Rios-Santos V. Prevalence of apical periodontitis and frequency of root-filled teeth in an adult Spanish population. Int Endod J. 2004;37:167.
107. Johnson GM, Lee DA, Regelmann WE, Gray ED, Peters G, Quie PG. Interference with granulocyte function by staphylococcus epidermis slime. Infect Immun. 1986;54:13.
108. Kakehashi S, Stanley H, Fitzgerald R. The effect of surgical exposures of dental pulps in germ-free and conventional laboratory rats. Oral Surg Oral Med Oral Pathol. 1965;20:340.
109. Kaneko T, Okiji T, Kan L, Tagagi M, Suda H. Ultrastructural analysis of MHC class II molecule-expressing cells in experimentally induced periapical lesions in the rat. J Endod. 2001;27:337.
110. Katz H. Endodontic implications of bisphosphonate-associated osteonecrosis of the jaws: a report of three cases. J Endod. 2005;31:831.
111. Kawashima N, Okiji T, Kosaka T, Suda H. Kinetics of macrophages and lymphoid cells during the development of experimentally induced periapical lesions in rat molars. J Endod. 1996;22:311.
112. Kayaoglu G, Orstavik D. Virulence factors of Enterococcus faecalis: Relationship to endodontic disease. Crit Rev Oral Biol Med. 2004;15:308.
113. Khan AA, Hargreaves KM. Dental pain. In: Trup JC, Sommer C, Hugger A, editors. The puzzle of orofacial pain. Basel: Karger, 2007.
114. Kierszenbaum AL. Histology and cell biology. St. Louis: Mosby; 2002.
115. Kimberly CL, Byers MR. Inflammation of rat molar pulp and periodontium causes increased calcitonin gene-related peptide and axonal sprouting. Ant Rec. 1988;222:289.
116. Kohsaka T, Kumazawa M, Yamasaki M, Nakamura H. Periapical lesions in rats with streptozotocin-induced diabetes. J Endod. 1996;22:418.
117. Kopp W, Schwarting R. Differentiation of T lymphocyte subpopulations, macrophages, and HLA-DR-restricted cells of apical granulation tissue. J Endod. 1989;15:72.
118. Koppang HS, Koppang R, Solheim T, Aarnes H, Stolen SO. Cellulose fibers from endodontic paper points as an etiological factor in postendodontic periapical granulomas and cysts. J Endod. 1989;15:369.
119. Kovacevic M, Tamarut T, Jonjic N, Braut A, Kovacevic M. The transition from pulpitis to periapical periodontitis in dog’s teeth. Aust Endod J. 2008;34:12.
120. Kramer IR, Pindberg JJ, Shear M. WHO histological typing of odontogenic tumors, ed 2. Geneva: Springer Verlag; 1992.
121. Kumar V, Abbas AK, Fausto N, et al, editors. Robbins and Cotran pathologic basis of disease, ed 8, Philadelphia: Saunders, 2010.
122. Kvist T, Reit C. Results of endodontic retreatment: a randomized clinical study comparing surgical and nonsurgical procedures. J Endod. 1999;25:814.
123. Langeland K, Block RM, Grossman LI. A histopathological and histobacteriologic study of 35 periapical endodontic surgical specimens. J Endod. 1977;3:8.
124. Langeland K, Rodrigues H, Dowden W. Periodontal disease, bacteria and pulpal histopathology. Oral Surg Oral Med Oral Pathol. 1974;37:257.
125. Lalonde ER. A new rationale for the management of periapical granulomas and cysts. An evaluation of histopathological and radiographic findings. J Am Dent Assoc. 1970;80:1056.
126. Laskin DM. Anatomic considerations in diagnosis and treatment of odontogenic infections. J Am Dent Assoc. 1964;69:308.
127. Laux M, Abbott PV, Pajarola G, Nair PNR. Apical inflammatory root resorption: a correlative radiographic and histological assessment. Int Endod J. 2000;33:483.
128. Lekic P, Rojas J, Birek C, Tenenbaum H, McCulloch CA. Phenotypic comparison of periodontal ligament cells in vivo and in vitro. J Periodontal Res. 2001;36:71.
129. Leonardo MR, Silva LAB, Utrilla LS, Assed S, Ether SS. Calcium hydroxide root canal sealers—histologic evaluation of apical and periapical repair after endodontic treatment. J Endod. 1997;23:232.
130. Leonardo MR, Hemandez MEFT, Silva LAB, Tanomaru-Filho M. Effect of a calcium hydroxide-based root canal dressing on periapical repair in dogs: a histological study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;102:680.
131. Lerner UH. Inflammation-induced bone remodeling in periodontal disease and the influence of post-menopausal osteoporosis. J Dent Res. 2006;85:596.
132. Ley K. Molecular mechanisms of leukocyte recruitment in the inflammatory process. Cardiovasc Res. 1996;32:733.
133. Li H, Chen V, Chen Y, Baumgartner JC, Machida CA. Herpesviruses in endodontic pathosis: association of Epstein-Barr virus with irreversible pulpitis and apical periodontitis. J Endod. 2009;35:23-29.
134. Li QY, Zhao HS, Meng HX, et al. Association analysis between interleukin-1 family polymorphisms and generalized aggressive periodontitis in a Chinese population. J Periodontol. 2004;75:1627.
135. Liapatas S, Nakou M, Rontogianni D. Inflammatory infiltrate of chronic periradicular lesions: an immunohistochemical study. Int Endod J. 2003;36:464.
136. Lilly JP, Cox D, Arcuri M, Krell KV. An evaluation of root canal treatment in patients who have received irradiation to the mandible and maxilla. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;86:224.
137. Lin LM, Di Fiore PM, Lin JL, Rosenberg PA. Histological study of periradicular tissue responses to uninfected and infected devitalized pulps in dogs. J Endod. 2006;32:34.
138. Lin LM, Gaengler P, Langeland K. Periapical curettage. Int Endod J. 1996;29:220.
139. Lin LM, Huang G. T-J, Rosenberg P. Proliferation of epithelial cell rests, formation of apical cysts, and regression of apical cysts after periapical wound healing. J Endod. 2007;33:908.
140. Lin L, Langeland K. Light and electron microscopic study of teeth with carious pulp exposures. Oral Surg Oral Med Oral Pathol. 1981;51:292.
141. Lin L, Langeland K. Innervation of inflammatory periapical lesions. Oral Surg Oral Med Oral Pathol. 1981;51:535.
142. Lin L, Shovlin F, Skribner J, Langeland K. Pulp biopsies from the teeth associated with periapical radiolucency. J Endod. 1984;10:436.
143. Lin LM, Pascon EA, Skribner J, Gangler P, Langeland K. Clinical, radiographic, and histological study of endodontic treatment failures. Oral Surg Oral Med Oral Pathol. 1991;11:603.
144. Lin LM, Wang SL, Wu-Wang C, Chang KM, Leung C. Detection of epidermal growth factor receptor in inflammatory periapical lesions. Int Endod J. 1996;29:179.
145. Linkhart TA, Mohan S, Baylink DJ. Growth factors for bone growth and repair. Bone. 1996;19:1S.
146. Lopez NJ, Jara L, Valenzuela CY. Association of interleukin-1 polymorphisms with periodontal disease. J Periodontol. 2005;76:234.
147. MacNeil RL, Somerman MJ. Development and regeneration of the periodontium: parallels and contrasts. Periodontol. 1999;19:8.
148. Majno G, Joris I. Cell, tissues, and disease, ed 2. Oxford: Oxford University Press; 2004.
149. Marending M, Peters OA, Zehnder M. Factors affecting the outcome of orthograde root canal therapy in a general dentistry hospital practice. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:119.
150. Martinelli C, Rulli MA. The innervation of chronic inflammatory human periapical lesions. Arch Oral Biol. 1967;12:593-600.
151. Marton IJ, Kiss C. Characterization of inflammatory cell infiltrate in dental periapical lesions. Int Endod J. 1993;26:131.
152. Marton IJ, Kiss C. Protective and destructive immune reactions in apical periodontitis. Oral Microbiol Immunol. 2000;15:139.
153. Medzhitove R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135.
154. Medzhitov R, Janeway CJr. Innate immunity. New Engl J Med. 2000;343:338.
155. Meghji S, Qureshi W, Henderson B, Harris M. The role of endotoxin and cytokines in the pathogenesis of odontogenic cysts. Arch Oral Biol. 1996;41:523.
156. Molander A, Reit C, Dahlen G, Kvist T. Microbiological status of root-filled teeth with apical periodontitis. Int Endod J. 1998;31:1.
157. Moldauer I, Velez I, Kuttler S. Upregulation of basic fibroblast growth factor in human periapical lesions. J Endod. 2006;32:408.
158. Moller AJR, Fabricius L, Dahlen G, Ohman AE, Heyden G. Influence on periapical tissues of indigenous oral bacteria and necrotic pulp tissue in monkeys. Scand J Dent Res. 1981;89:29.
159. Mundy GR. Inflammatory mediators and the destruction of bone. J Periodont Res. 1991;26:213.
160. Murray CA, Saunders WP. Root canal treatment and general health: a review of the literature. Int Endod J. 2000;33:1.
161. Nagatomo K, Komaki M, Sekiya Y, et al. Stem cell properties of human periodontal ligament cells. J Periodontal Res. 2006;41:303.
162. Nair PNR. Apical periodontitis: a dynamic encounter between root canal infection and host response. Periodontol 2000. 1997;13:121.
163. Nair PNR. New perspectives on radicular cysts: do they heal. Int Endod J. 1998;31:155.
164. Nair PNR, Pajarola G, Schroeder HE. Types and incidence of human periapical lesions obtained with extracted teeth. Oral Surg Oral Med Oral Pathol. 1996;81:93.
165. Nair PNR, Schroeder HE. Periapical actinomycosis. J Endod. 1984;10:567.
166. Nair PNR, Sjogren U, Krey G, Sundqvist G. Therapy-resistant foreign body giant cell granuloma at the periapex of a root-filled human tooth. J Endod. 1990;26:225.
167. Nair PNR, Sjogren U, Sundqvist G. Cholesterol crystals as an etiological factor in non-resolving chronic inflammation: an experimental study in guinea pigs. Eur J Oral Sci. 1998;106:644.
168. Nair PNR, Sundqvist G, Sjogren U. Experimental evidence supports the abscess theory of development of radicular cysts. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106:294.
169. Nakamura I, Takahashi N, Sasaki T, Jimi E, Kurokawa T, Suda T. Chemical and physical properties of extracellular matrix are required for the actin ring formation in osteoclasts. J Bone Miner Res. 1996;11:1873.
170. Nakashima M. Induction of dentin formation on canine amputated pulp by recombinant human bone morphogenic proteins (BMP)-2 and -4. J Dent Res. 1994;73:1515.
171. Nanci A. Ten Cate’s oral histology: development, structure, and function, ed 7. St. Louis: Mosby; 2008.
172. Nathan C. Points of control in inflammation. Nature. 2002;420:846.
173. Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88.
174. Neville BW, Damm DD, Allen CM, Bouquot JE. Oral and maxillofacial pathology, ed 3. St. Louis: Saunders; 2009.
175. Niederman R, Westernoff T, Lee C, et al. Infection-mediated early-onset periodontal disease in P/E-selectin-deficient mice. J Clin Periodontol. 2001;28:569.
176. Noirti Y, Ehara A, Kawahara T, Takemura N, Ebisu S. Participation of bacterial biofilms in refractory and chronic periapical periodontitis. J Endod. 2002;28:679.
177. Nordlund L, Hormia M, Saxen L, Thesleff I. Immunohistochemical localization of epidermal growth factor receptors in human gingival epithelia. J Periodont Res. 1991;26:333.
178. Oehlers FAC. Periapical lesions and residual dental cysts. Br J Oral Surg. 1970;8:103.
179. Oguntebi B, Slee AM, Tanzer JM, Langeland K. Predominant microflora associated with human dental periradicular abscesses. J Clin Microbiol. 1982;15:964.
180. Okiji T, Kawashima N, Kosaka T, Kobayashi C, Suda H. Distribution of la antigen-expressing nonlymphoid cells in various stages of induced periapical lesions in rat molars. J Endod. 1994;20:27.
181. Orstavik D, Pitt Ford TR. Essential endodontology: prevention and treatment of apical periodontitis, ed 2. Philadelphia: Wiley-Blackwell; 2008.
182. Pallasch TJ, Wahl MJ. Focal infection: new age or ancient history? Endod Topics. 2003;4:32.
183. Pascon EA, Leonardo MR, Safavi K, Langeland K. Tissue reaction to endodontic materials: methods, criteria, assessment, and observations. Oral Surg Oral Med Oral Pathol. 1991;2:222.
184. Peciuliene V, Reynaud AH, Balciuniene I, Haapasalo M. Isolation of yeasts and enteric bacteria in root-filled teeth with chronic apical periodontitis. Int Endod. 2001;34:429.
185. Quesnell BT, Alves M, Hawkinson RW, Johnson BR, Wenkus CS, BeGole EA. The effect of human immunodeficiency virus on endodontic treatment outcome. J Endod. 2005;31:633.
186. Reddy SV, Roodman GD. Control of osteoclast differentiation. Crit Rev Eukaryotic Gene Expression. 1998;8:1.
187. Reeve CM, Wentz FM. The prevalence, morphology, and distribution of epithelial cell rests in the human periodontal ligament. Oral Surg. 1962;15:785.
188. Regezi JA, Scubba JJ, Jordan RCK. Oral pathology: clinical pathologic correlations, ed 5. St. Louis: Saunders; 2008.
189. Reinke E, Fabry Z. Breaking or making immunological privilege in the central nervous system: the regulation of immunity by neuropeptides. Immunol Lett. 2006;104:102.
190. Reuther T, Schuster T, Mende U, Kubler A. Osteoradionecrosis of the jaws as a side effect of radiotherapy of head and neck tumor patients—a report of a thirty year retrospective review. Int J Oral Maxillofacial Surg. 2003;32:289.
191. Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther. 2002;302:839.
192. Ricucci D, Bergenholtz G. Histologic features of apical periodontitis in human biopsies. Endod Topics. 2004;8:68.
193. Ricucci D, Pascon EA, Pitt Ford TR, Langeland K. Epithelium and bacteria in periapical lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:241.
194. Ruggiero SL, Drew SJ. Osteonecrosis of the jaws and bisphosphonate therapy. J Dent Res. 2007;86:1013.
195. Rutherford RB, Spangberg L, Tucker M, Ruger D, Charette M. The time-course of the induction of reparative dentin formation in monkeys by recombinant human osteogenic protein-1. Arch Oral Biol. 1994;39:833.
196. Sabeti M, Simon JH, Howzari H, Slot J. Cytomegalovirus and Epstein-Barr virus active infection in periapical lesions of teeth with intact crowns. J Endod. 2003;29:321.
197. Sahara N, Toyoki A, Ashizawa Y, Deguchi T, Suzuki K. Cytodifferentiation of the odontoclast prior to the shedding of human deciduous teeth: an ultrastructural and cytochemical study. Anat Rec. 1996;244:33.
198. Sasaki T. Differentiation and functions of osteoclasts and odontoclasts in mineralized tissue resorption. Microscopy Res Tech. 2003;61:483.
199. Sasaki T, Sasaki T, Motegi N, et al. Dentin resorption mediated by odontoblasts in physiological root resorption of human deciduous teeth. Am J Anat. 1988;183:303.
200. Sathorn C, Parashos P, Messer HH. How useful is root canal culturing in predicting treatment outcome? J Endod. 2007;33:220.
201. Saunder DN. Interleukin-1 in dermatological disease. In: Bomford R, Henderson B, editors. Interleukin-1, inflammation and Disease. North Holland: Elsevier, 1989.
202. Schaffer M, Beiter T, Becker HD, Hunt TK. Neuropeptides. Mediators of inflammation and tissue repair? Arch Surg. 1998;133:1107.
203. Schoenfeld SE, Greening AB, Glick DH, Frank AL, Simon JH, Herless SM. Endotoxic activity in periapical lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1982;53:82.
204. Segura-Egea JJ, Jimenez A, Rios-Santos JV, Velasco-Ortega E, Cisneros-Cabello R, Poyato-Ferrera M. High prevalence of apical periodontitis amongst type 2 diabetic patients. Int Endod J. 2005;38:564.
205. Seltzer S. Endodontology, ed 2. Philadelphia: Lea & Fibiger; 1988.
206. Seltzer S, Bender IB, Zionitz M. The dynamics of pulp inflammation: correlation between diagnostic data and actual histological finding in the pulp. Part 1 and Part 2. Oral Surg Oral Med Oral Pathol. 1963;16:846.
207. Seltzer S, Naidorf IJ. Flare-ups in endodontics: 1. Etiological factors. J Endod. 1985;11:472.
208. Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Ann Rev Immunol. 2008;26:421.
209. Seo B-M, Miura M, Gronthos S, et al. Investigation of multipotent postnatal stem cells from human periodontal ligament. Lancet. 2004;364:149.
210. Shear M. The histogenesis of dental cysts. Dent Pract. 1963;13:238.
211. Shimono M, Suda H, Maeda T, editors. Dentin/pulp complex. New York: Quintessence, 1996.
212. Sidaravicius B, Aleksejuniene J, Eriksen HM. Endodontic treatment and prevalence of apical periodontitis in an adult population of Vilnius, Lithuania. Dent Traumatol. 2000;15:210.
213. Sigurdsson A. Pulpal disgnosis. Endod Topics. 2003;5:12.
214. Silva TA, Garlet GP, Fukada SY, Silva JS, Cunha FQ. Chemokines in oral inflammatory diseases: apical periodontitis and periodontal disease. J Dent Res. 2007;86:306.
215. Simon JHS. Incidence of periapical cysts in relation to root canal. J Endod. 1980;6:116.
216. Simon JHS, Yonemoto GS, Bakland LK. Comparison of cellular cementum in normal and diseased teeth—a scanning electron microscope study. J Endod. 1981;7:370.
217. Siqueira JF, Lopes HP. Bacteria on the apical root surfaces of untreated teeth with periradicular lesions: a scanning electron microscopy study. Int Endod J. 2001;34:216.
218. Siqueira JFJr, Rocas IN. Polymerase chair reaction-based analysis of microorganisms associated with failed endodontic treatment. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:85.
219. Sjogren U, Hagglund B, Sundqvist G, Wing K. Factors affecting the long-term results of endodontic treatment. J Endod. 1990;16:498.
220. Slauson DO, Cooper BJ. Mechanisms of disease, ed 3. St. Louis: Mosby; 2002.
221. Slots J, Nowzari H, Sabeti M. Cytomegalovirus infection in symptomatic periapical pathosis. Int Endod J. 2004;37:519.
222. Solheim E. Growth factors in bone. Int Orthop. 1998;22:410.
223. Sonoyama W, Seo B-M, Yamaza T, Shi S. Human Hertwig’s epithelial root sheath cells play crucial roles in cementum formation. J Dent Res. 2007;86:594.
224. Stashenko F. The role of immune cytokines in the pathogenesis of periapical lesions. Endod Dent Traumatol. 1990;6:89.
225. Stashenko P, Teles R, D’Souza R. Periapical inflammatory responses and their modulation. Crit Rev Oral Biol Med. 1998;9:498.
226. Stashenko P, Wang CY, Riley E, Wu Y, Ostroff G, Niederman R. Reduction of infection-stimulated bone periapical bone resorption by the biological response modifier PGG glucan. J Dent Res. 1995;74:323.
227. Stashenko P, Yu SM. T helper and T suppressor cell reversal during the development of induced rat periapical lesions. J Dent Res. 1989;68:830.
228. Steed DL. The role of growth factors in wound healing. Surg Clin North Am. 1997;77:575.
229. Stern MH, Dreizen S, Mackler BF, Selbst AG, Levy BM. Quantitative analysis of cellular composition of human periapical granulomas. J Endod. 1981;7:117.
230. Stern MH, Dreizen S, Mackler BF, Selbst AG, Levy BM. Isolation and characterization of inflammatory cells from the human periapical granuloma. J Dent Res. 1982;61:1408.
231. Sunde P, Olsen I, Debelian G, Transtad L. Microbiota of periapical lesions refractory to endodontic therapy. J Endod. 2002;28:304.
232. Sundqvist G. Bacteriological studies of necrotic dental pulps. Sweden: Dissertation, Umea; 1976.
233. Sundqvist G. Taxonomy, ecology, and pathogenicity of the root canal flora. Oral Surg Oral Med Oral Pathol. 1994;78:522.
234. Sundqvist G, Eckerbom MI, Larsson AP, Sjogren UT. Capacity of anaerobic bacteria from necrotic pulp to induce purulent infection. Infect Immun. 1979;25:685.
235. Sundqvist G, Figdor D. Life as an endodontic pathogen. Endod Topics. 2003;6:3.
236. Sundqvist G, Figdor D, Persson S, Sjogren U. Microbiological analysis of teeth with failed endodontic treatment and the outcome of conservative re-treatment. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85:86.
237. Sundqvist G, Reuterving CO. Isolation of Actinomyces israelii from periapical lesion. J Endod. 1980;6:60.
238. Syrjanen S, Tammisalo E, Lilja R, Syrjanen K. Radiographical interpretation of the periapical cysts and granulomas. Dentomaxillofac Radiol. 1982;11:89.
239. Tagger M, Massler M. Periapical tissue reactions after pulp exposure in rat molars. Oral Surg Oral Med Oral Pathol. 1975;39:304.
240. Takahashi K. Microbiological, pathological, inflammatory, immunological and molecular biological aspects of periradicular disease. Int Endod J. 1998;31:311.
241. Takeuchi O, Hoshino K, Akira S. Cutting Edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392.
242. Tani N, Kuchiba K, Osada T, Watanabe Y, Umemoto T. Effect of T-cell deficiency on the formation of periapical lesions in mice: histological comparison between periapical lesion formation in BALB/c and BALB/c nu/nu mice. J Endod. 1995;21:195.
243. Teitelbaum SL, Tondravi MM, Ross FP. Osteoclasts, macrophages, and the molecular mechanisms of bone resorption. J Leukocyte Biol. 1997:381.
244. Teles R, Wang CY, Stashenko P. Increased susceptibility of RAG-2 SCID mice to dissemination of endodontic infections. Infect Immun. 1997;65:3781.
245. Ten Cate AR. The epithelial cell rests of Malassez and the genesis of the dental cyst. Oral Surg Oral Med Oral Pathol. 1972;34:56.
246. Teronen O, Salo T, Laitinen J, et al. Characterization of interstitial collagenases in jaw cyst wall. Eur J Oral Sci. 1995;103:141.
247. Thesleff I. Epithelial cell rests of Malassez bind epidermal growth factor intensely. J Periodont Res. 1987;22:419.
248. Toller PA. The osmolality of fluids from cysts of the jaws. Br Dent J. 1970;129:275.
249. Torabinejad M. Mediators of acute and chronic periradicular lesions. Oral Surg Oral Med Oral Pathol. 1994;78:511.
250. Torabinejad M, Eby WC, Naidorf IJ. Inflammatory and immunological aspects of the pathogenesis of human periapical lesions. J Endod. 1985;11:479.
251. Torabinejad M, Kiger RD. Experimentally induced alterations in periapical tissues of the cat. J Dent Res. 1980;59:87.
252. Torabinejad M, Kettering J. Identification and relative concentration of B and T lymphocytes in human chronic periapical lesions. J Endod. 1985;11:122.
253. Torabinejad M, Walton RE. Endodontics: principles and practice, ed 4. St. Louis: Saunders; 2009.
254. Tronstad L, Barnett F, Cervone F. Periapical bacterial plaque in teeth refractory to endodontic treatment. Dent Traumatol. 1990;6:73.
255. Tronstad L, Barnett F, Riso K, Slots J. Extraradicular endodontic infection. Dent Traumatol. 1987;3:86.
256. Trombelli L, Lee MB, Promsudthi A, Guglielmoni PG, Wilesjo UME. Periodontal repair in dogs: histologic observations of guided tissue regeneration with a prostaglandin E1 analog/methacrylate composite. Clin Periodontol. 1999;26:381.
257. Trott JR, Chebeb F, Galindo Y. Factors related to cholesterol formation in cysts and granulomas. J Cand Dent Assoc. 1973;38:76.
258. Trowbridge H, Emling RC. Inflammation. A review of the process, ed 5. Chicago: Quintessence Publishing Co; 1997.
259. Tuan T-L, Nichter LS. The molecular basis of keloid and hypertrophic scar formation. Mol Med Today. 1998;4:19.
260. Tzaifas D, Alvanou A, Papadimitrious S, Gasic J, Komnenou A. Effects of recombinant basic fibroblast growth factor, insulin-like growth factor-II and transforming growth factor-β 1 on dog dental pulp cells in vivo. Arch Oral Biol. 1998;43:431.
261. van Oosterhout AJ, Bloksma N. Regulatory T-lymphocytes in asthma. Eur Respir J. 2005;26:918.
262. van Winkelhoff AJ, Carless AW, de Graaff J. Bacteroides endodontalis and other black-pigmented Bacteroides species in odontogenic abscesses. Infect Immun. 1985;49:494.
263. Vier FV, Figueiredo JA. Prevalence of different periapical lesions associated human teeth and their correlation with the presence and extension of apical external root resorption. Int Endod J. 2002;35:710.
264. Wadachi R, Hargreaves KW. Trigeminal nociceptors express TLR-4 and CD14: a mechanism for pain due to infection. J Dent Res. 2006;85:49.
265. Wakelin D, Roitt I, Mims C, et al, editors. Mims’ medical microbiology, ed 4, Philadelphia: Mosby, 2008.
266. Wallstrom JB, Torabinejad M, Kettering J, McMillan P. Role of T cells in the pathogenesis of periapical lesions. A preliminary report. Oral Surg Oral Med Oral Pathol. 1993;76:213.
267. Walton RE, Ardjmand K. Histological evaluation of the presence of bacteria in induced periapical lesions in monkeys. J Endod. 1992;18:216.
268. Wang CY, Stashenko P. Kinetics of bone-resorbing activity in developing periapical lesions. J Dent Res. 1991;70:1362.
269. Wang CY, Tani-Ishii N, Stashenko P. Bone resorptive cytokine gene expression in developing rat periapical lesions. Oral Microbiol Immunol. 1997;12:65.
270. Warren JR, Scarpelli DG, Reddy JK, Kanwar YS. Essentials of general pathology. New York: Macmillan Publishing Co; 1987.
271. Wayman BE, Murata SM, Almeida RJ, Fowler CB. A bacteriological and histological evaluation of 58 periradicular lesions. J Endod. 1992;18:152.
272. Weiger R, Manncke B, Werner H, Lost C. Microbial flora of sinus tracts and root canals of non-vital teeth. Endod Dent Traumatol. 1995;11:15.
273. Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83:835.
274. Williams BL, McCann GF, Schoenknecht FD. Bacteriology of dental abscesses of endodontic origin. J Clin Microbiol. 1983;18:770.
275. Williams GT, Williams WJ. Granulomatous inflammation—a review. J Clin Pathol. 1983;36:723.
276. Williams JZ, Barbul A. Nutrition and wound healing. Surg Clin North Am. 2003;83:571.
277. Woo S-B, Hellstein JW, Kalmar JR. Systematic review: bisphosphonates and osteonecrosis of the jaws. Ann Int Med. 2006;144:753.
278. World Health Organization. Application of the international classification of disease to dentistry and stomatology, ed 3. Geneva: WHO; 1995.
279. Yamasaki M, Kumazawa M, Kohsaka T, Nakamura H, Kameyama Y. Pulp and periapical tissue reactions after experimental pulpal exposure in rats. J Endod. 1994;20:13.
280. Yamasaki M, Nakane A, Kumazawa M, Hasioka K, Horiba N, Nakamura H. Endotoxin and gram-negative bacteria in the rat periapical lesion. J Endod. 1992;18:501.
281. Yu SM, Stashenko P. Phenotypic characterization of inflammatory cells in induced rat periapical lesions. J Endod. 1987;13:535.
282. Yusulf H. The significance of the presence of foreign material periapically as a cause of failure of root treatment. Oral Surg Oral Med Oral Pathol. 1982;54:566.
283. Zimmerli W, Lew PD, Walvogel FA. Pathogenesis of foreign body infection. Evidence for a local granulocyte defect. J Clin Invest. 1984;73:1191.
284. Zimmerli W, Waldvogel FA, Vaudaux P, Nydegger UE. Pathogenesis of foreign body infection: description and characteristics of an animal model. J Infect Dis. 1982;146:487.
285. Zimring JC, Kapp JA. Identification and characterization of CD8+ suppressor T cells. Immunol Res. 2004;29:303.