33 The thyroid
Overview
Diseases of the thyroid gland are prevalent, and in this chapter we deal with drug therapy used to mitigate these disorders. We set the scene by briefly outlining the structure, regulation and physiology of the thyroid, and highlight the most common abnormalities of thyroid function. We then go on to consider the drugs that replace the thyroid hormones when these are deficient or cease to function adequately, and the drugs that decrease thyroid function when this is excessive.
Synthesis, Storage and Secretion of Thyroid Hormones
The thyroid gland secretes three main hormones: thyroxine (T4), tri-iodothyronine (T3) and calcitonin. T4 and T3 are critically important for normal growth and development and for controlling energy metabolism. Calcitonin is involved in the control of plasma [Ca2+] and is used to treat osteoporosis and other metabolic bone diseases. It is dealt with in Chapter 35. The term thyroid hormone will be used here solely to refer to T4 and T3.
The functional unit of the thyroid is the follicle or acinus. Each follicle consists of a single layer of epithelial cells around a cavity, the follicle lumen, which is filled with a thick colloid containing thyroglobulin. Thyroglobulin is a large glycoprotein, each molecule of which contains about 115 tyrosine residues. It is synthesised, glycosylated and then secreted into the lumen of the follicle, where iodination of the tyrosine residues occurs. Surrounding the follicles is a dense capillary network, and the rate of blood flow through the gland is very high in comparison with other tissues. The main steps in the synthesis, storage and secretion of thyroid hormone (Fig. 33.1) are:
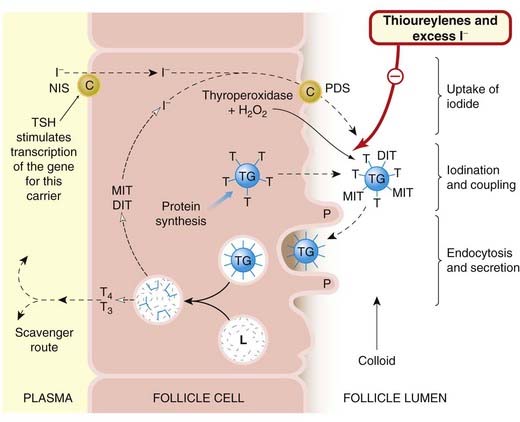
Fig. 33.1 Diagram of thyroid hormone synthesis and secretion, with the sites of action of drugs used in the treatment of thyroid disorders.
Iodide in the blood is transported by the carriers NIS and pendrin (PDS) through the follicular cell and into the colloid-rich lumen, where it is incorporated into thyroglobulin under the influence of the thyroperoxidase enzyme (see text for details). The hormones are produced by processing of the endocytosed thyroglobulin and exported into the blood. DIT, di-iodotyrosine; L, lysosome; MIT, monoiodotyrosine; P, pseudopod; T, tyrosine; T3, tri-iodothyronine; T4, thyroxine; TG, thyroglobulin; TSH, thyroid-stimulating hormone (thyrotrophin).
Uptake of Plasma Iodide by the Follicle Cells
Iodide uptake is an energy-dependent process occurring against a gradient, which is normally about 25:1. Iodide is captured from the blood and moved to the lumen by two transporters: the Na+/I− symporter (NIS) located at the basolateral surface of the thyrocytes (the energy being provided by Na+-K+-ATPase), and pendrin1 (PDS), an I−/Cl− porter in the apical membranes (Nilsson, 2001; Yoshida et al., 2004). Uptake is very rapid: labelled iodide (125I) is found in the lumen within 40 s of intravenous injection. Numerous mutations have been discovered in the NIS and PDS genes, and these contribute to thyroid disease in some patients.
Oxidation of Iodide and Iodination of Tyrosine Residues
The oxidation of iodide and its incorporation into thyroglobulin (termed the organification of iodide) is catalysed by thyroperoxidase, an enzyme situated at the inner surface of the cell at the interface with the colloid. The reaction requires the presence of hydrogen peroxide (H2O2) as an oxidising agent. Iodination occurs after the tyrosine has been incorporated into thyroglobulin. The process is shown in Figure 33.2.
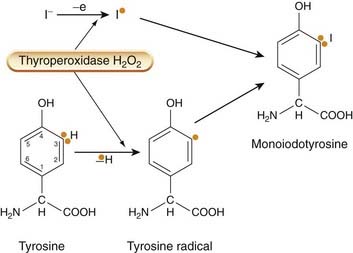
Fig. 33.2 Iodination of tyrosyl residues by the thyroperoxidase–H2O2 complex.
This probably involves two sites on the enzyme, one of which removes an electron from iodide to give the free radical I•; another removes an electron from tyrosine to give the tyrosyl radical (shown by orange dot). Monoiodotyrosine results from the addition of the two radicals.
Tyrosine residues are iodinated first at position 3 on the ring, forming monoiodotyrosine (MIT) and then, in some molecules, on position 5 as well, forming di-iodotyrosine (DIT). While still incorporated into thyroglobulin, these molecules are then coupled in pairs, either MIT with DIT to form T3, or two DIT molecules to form T4. The mechanism for coupling is believed to involve a peroxidase system similar to that involved in iodination. About one-fifth of the tyrosine residues in thyroglobulin are iodinated in this way.
The iodinated thyroglobulin of the thyroid forms a large store of thyroid hormone within the gland, with a relatively slow turnover. This is in contrast to some other endocrine secretions (e.g. the hormones of the adrenal cortex), which are not stored but synthesised and released as required.
Secretion of Thyroid Hormone
The thyroglobulin molecule is taken up into the follicle cell by endocytosis (Fig. 33.1). The endocytotic vesicles then fuse with lysosomes, and proteolytic enzymes act on thyroglobulin, releasing T4 and T3 to be secreted into the plasma. The surplus MIT and DIT, which are released at the same time, are scavenged by the cell, where the iodide is removed enzymatically and reused.
Regulation of Thyroid Function
Thyrotrophin-releasing hormone (TRH), released from the hypothalamus in response to various stimuli, releases thyroid-stimulating hormone (TSH; thyrotrophin) from the anterior pituitary (Fig. 33.3), as does the synthetic tripeptide protirelin (pyroglutamyl-histidyl-proline amide), which is used in this way for diagnostic purposes. TSH acts on receptors on the membrane of thyroid follicle cells through a mechanism that involves cAMP and phosphatidylinositol 3-kinase. It has a trophic action on thyroid cells and controls all aspects of thyroid hormone synthesis, including:
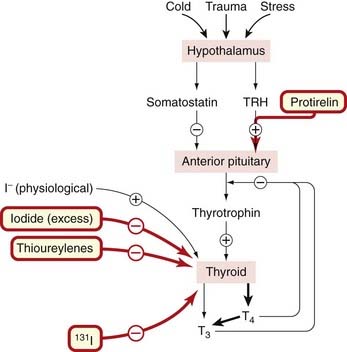
Fig. 33.3 Regulation of thyroid hormone secretion.
Iodide (I−) is essential for thyroid hormone synthesis, but excess of endogenous or exogenous iodide (30 times the daily requirement of iodine) actually inhibits the increased thyroid hormone production, which occurs in thyrotoxicosis. Protirelin as well as recombinant thyrotrophin-releasing hormone (TRH) is sometimes used to stimulate the system for diagnostic purposes, as is the administration of 131I (see text for details). T3, tri-iodothyronine; T4, thyroxine.
The production of TSH is also regulated by a negative feedback effect of thyroid hormones on the anterior pituitary gland, T3 being more active than T4 in this respect. The peptide somatostatin also reduces basal TSH release. The control of the secretion of TSH thus depends on a balance between the actions of T3/T4 and TRH (and probably also somatostatin) on the pituitary, although even high concentrations of thyroid hormone do not totally inhibit TSH secretion.
The other main factor influencing thyroid function is the plasma iodide concentration. About 100 nmol of T4 is synthesised daily, necessitating uptake by the gland of approximately 500 nmol of iodide each day (equivalent to about 70 µg of iodine). A reduced iodine intake, with reduced plasma iodide concentration, will result in a decrease of hormone production and an increase in TSH secretion. An increased plasma iodide has the opposite effect, although this may be modified by other factors (see below). The overall feedback mechanism responds to changes of iodide slowly over fairly long periods of days or weeks, because there is a large reserve capacity for the binding and uptake of iodide in the thyroid. The size and vascularity of the thyroid are reduced by an increase in plasma iodide and this is exploited therapeutically in preparing hyperthyroid patients for surgery to the gland (see below). Diets deficient in iodine eventually result in a continuous excessive compensatory secretion of TSH, and eventually in an increase in vascularity and (sometimes gross) hypertrophy of the gland.2
Actions of the Thyroid Hormones
The physiological actions of the thyroid hormones fall into two categories: those affecting metabolism and those affecting growth and development.
Effects on Metabolism
The thyroid hormones produce a general increase in the metabolism of carbohydrates, fats and proteins, and regulate these processes in most tissues, T3 being three to five times more active than T4 in this respect (Fig. 33.4). Although the thyroid hormones directly control the activity of some of the enzymes of carbohydrate metabolism, most effects are brought about in conjunction with other hormones, such as insulin, glucagon, the glucocorticoids and the catecholamines. There is an increase in oxygen consumption and heat production, which is manifested as an increase in the measured basal metabolic rate. This reflects the action of these hormones on tissues such as heart, kidney, liver and muscle, although not on others, such as the gonads, brain or spleen. The calorigenic action is important as part of the response to a cold environment. Administration of thyroid hormone results in augmented cardiac rate and output, and increased tendency to dysrhythmias such as atrial fibrillation.
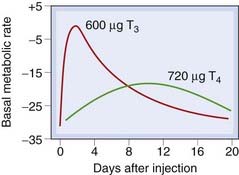
Fig. 33.4 The effect of equimolar doses of tri-iodothyronine (T3) and thyroxine (T4) on basal metabolic rate (BMR) in a hypothyroid subject.
Note that this figure is meant only to illustrate overall differences in effect; thyroxine is not given clinically in a single bolus dose as here, but in regular daily doses so that the effect builds up to a plateau. The apparent differences in potency really represent differences in kinetics, reflecting the prehormone role of T4.
(From Blackburn C M et al. 1954 J Clin Invest 33: 819.)
Effects on Growth and Development
The thyroid hormones have a critical effect on growth, partly by a direct action on cells, and also indirectly by influencing growth hormone production and potentiating its effects on its target tissues. The hormones are important for a normal response to parathormone and calcitonin as well as for skeletal development; they are also essential for normal growth and maturation of the central nervous system.
Mechanism of Action
While there is some evidence for non-genomic actions (see Bassett et al., 2003; Lazar, 2003), these hormones act mainly through a specific nuclear receptor, TR (Ch. 3 and Fig. 3.17). Two distinct genes, TRα and TRβ, code for several receptor isoforms that have distinct functions. T4 may be regarded as a prohormone, because when it enters the cell, it is converted to T3, which then binds with high affinity to a member of the TR family. This interaction is likely to take place in the nucleus, where TR isoforms generally act as a constitutive repressor of target genes. When T3 is bound, the receptors change conformation, the co-repressor complex is released and a co-activator complex is recruited, which then activates transcription, resulting in generation of mRNA and protein synthesis.
Transport and Metabolism of Thyroid Hormones
Both thyroid hormones are transported in the blood bound mainly to thyroxine-binding globulin (TBG). Plasma concentrations of these hormones can be measured by radioimmunoassay, and are approximately 1 × 10−7 mol/l (T4) and 2 × 10−9 mol/l for T3. Both are eventually metabolised in their target tissues by deiodination, deamination, decarboxylation and conjugation with glucuronic and sulfuric acids. The liver is a major site of metabolism, and the free and conjugated forms are excreted partly in the bile and partly in the urine. The metabolic clearance of T3 is 20 times faster than that of T4 (plasma half-life about 6 days). The long half-life of T4 is a consequence of its strong binding to TBG. Abnormalities in the metabolism of these hormones may occur naturally or be induced by drugs or heavy metals, and this may give rise to a variety of (uncommon) clinical conditions such as the ‘low T3 syndrome’.
Abnormalities of Thyroid Function
Thyroid disorders are among the most common endocrine diseases, and subclinical thyroid disease is particularly prevalent in the middle-aged and elderly. They are accompanied by many extrathyroidal symptoms, particularly in the heart and skin. One (rare) cause of organ dysfunction is thyroid cancer. Many other thyroid disorders have an autoimmune basis and like other autoimmune diseases are more common in women than men. The ultimate reason for this is not clear, although it may be linked to polymorphisms in the PDS, tumour necrosis factor (TNF)-α or other genes. Regardless of causation, thyroid dysfunction is often associated with enlargement of the gland, known as goitre.
Hyperthyroidism (Thyrotoxicosis)
In thyrotoxicosis, there is excessive activity of the thyroid hormones, resulting in a high metabolic rate, an increase in skin temperature and sweating, and a marked sensitivity to heat. Nervousness, tremor, tachycardia, heat sensitivity and increased appetite associated with loss of weight occur. There are several types of hyperthyroidism, but only two are common: diffuse toxic goitre (also called Graves’ disease3 or exophthalmic goitre) and toxic nodular goitre.
Diffuse toxic goitre is an organ-specific autoimmune disease caused by autoantibodies to the TSH receptor which actually stimulate it, increasing thyroxine secretion. Constitutively active mutations of the TRH receptor may also be involved. As is indicated by the name, patients with exophthalmic goitre have protrusion of the eyeballs. The pathogenesis of this condition is not fully understood, but it is thought to be caused by the presence of TSH receptor-like proteins in orbital tissues. There is also an enhanced sensitivity to catecholamines. Toxic nodular goitre is caused by a benign neoplasm or adenoma, and may develop in patients with long-standing simple goitre (see below). This condition does not usually have concomitant exophthalmos. The antidysrhythmic drug amiodarone (Ch. 21) is rich in iodine and can cause either hyperthyroidism or hypothyroidism. Some other iodine-containing drugs, such as iopanoic acid and its congeners, which are used as imaging agents used to visualise the gall bladder, may also interfere with thyroid function.
Simple, Non-Toxic Goitre
A dietary deficiency of iodine, if prolonged, causes a rise in plasma TRH and eventually an increase in the size of the gland. This condition is known as simple or non-toxic goitre. Another cause is ingestion of goitrogens (e.g. from cassava root). The enlarged thyroid usually manages to produce normal amounts of thyroid hormone, although if the iodine deficiency is very severe, hypothyroidism may supervene.
Hypothyroidism
A decreased activity of the thyroid results in hypothyroidism, and in severe cases myxoedema. Once again, this disease is immunological in origin, and the manifestations include low metabolic rate, slow speech, deep hoarse voice, lethargy, bradycardia, sensitivity to cold and mental impairment. Patients also develop a characteristic thickening of the skin (caused by the subcutaneous deposition of glycosaminoglycans), which gives myxoedema its name. Hashimoto’s thyroiditis, a chronic autoimmune disease in which there is an immune reaction against thyroglobulin or some other component of thyroid tissue, can lead to hypothyroidism and myxoedema. Genetic factors play an important role. Therapy of thyroid tumours with radioiodine (see below) is another cause of hypothyroidism.
Thyroid deficiency during development, which is the most prevalent endocrine disorder in the newborn (1 in 3000–4000 births) causes congenital hypothyroidism,4 characterised by gross retardation of growth and mental deficiency. Pendred’s syndrome, an autosomal recessive disorder caused by mutations in the PDS transporter gene, may cause goitre as well as deafness and other symptoms (see Hadj Kacem et al., 2003).
The thyroid 
Drugs Used in Diseases of the Thyroid
Hyperthyroidism
Hyperthyroidism may be treated pharmacologically or surgically. In general, surgery is used only when there are mechanical problems resulting from compression of the trachea, and it is usual to remove only part of the organ. Although the condition of hyperthyroidism can be controlled with antithyroid drugs, these drugs do not alter the underlying autoimmune mechanisms or improve the exophthalmos associated with Graves’ disease.
Radioiodine
Radioiodine is a first-line treatment for hyperthyroidism (particularly in the USA). The isotope used is 131I (usually as the sodium salt), and the dose generally 5–15 millicuries. Given orally, it is taken up and processed by the thyroid in the same way as the stable form of iodide, eventually becoming incorporated into thyroglobulin. The isotope emits both β and γ radiation. The γ rays pass through the tissue without causing damage, but the β particles have a very short range; they are absorbed by the tissue and exert a powerful cytotoxic action that is restricted to the cells of the thyroid follicles, resulting in significant destruction of the tissue. 131I has a half-life of 8 days, so by 2 months its radioactivity has effectively disappeared. It is given as one single dose, but its cytotoxic effect on the gland is delayed for 1–2 months and does not reach its maximum for a further 2 months.
Hypothyroidism will eventually occur after treatment with radioiodine, particularly in patients with Graves’ disease, but is easily managed by replacement therapy with T4. Radioiodine is best avoided in children and also in pregnant patients because of potential damage to the fetus. There is theoretically an increased risk of thyroid cancer but this has not been seen following the therapeutic treatment.
The uptake of 131I and other isotopes of iodine is also used diagnostically as a test of thyroid function. A tracer dose of the isotope is given orally or intravenously, and the amount accumulated by the thyroid is measured by a γ-scintillation counter placed over the gland. Another use for this drug is the treatment of thyroid cancer.
Thioureylenes
The thioureylene group of drugs comprises carbimazole, methimazole and propylthiouracil. Chemically, they are related to thiourea, and the thiocarbamide (S–C–N) group is essential for antithyroid activity.
Mechanism of action
Thioureylenes decrease the output of thyroid hormones from the gland, and cause a gradual reduction in the signs and symptoms of thyrotoxicosis, the basal metabolic rate and pulse rate returning to normal over a period of 3–4 weeks. Their mode of action is not completely understood, but there is evidence that they inhibit the iodination of tyrosyl residues in thyroglobulin (see Figs 33.1 and 33.2). It is thought that they inhibit the thyroperoxidase-catalysed oxidation reactions by acting as substrates for the postulated peroxidase–iodinium complex, thus competitively inhibiting the interaction with tyrosine. Propylthiouracil has the additional effect of reducing the deiodination of T4 to T3 in peripheral tissues.
Pharmacokinetic aspects
Thioureylenes are given orally. Carbimazole is rapidly converted to its active metabilite methimazole, which is distributed throughout the body water and has a plasma half-life of 6–15 h. An average dose of carbimazole produces more than 90% inhibition of thyroid incorporation of iodine within 12 h. The clinical response to this and other antithyroid drugs, however, may take several weeks (Fig. 33.5). This is not only because T4 has a long half-life, but also because the thyroid may have large stores of hormone, which need to be depleted before the drug’s action can be fully manifest. Propylthiouracil is thought to act somewhat more rapidly because of its additional effect as an inhibitor of the peripheral conversion of T4 to T3.
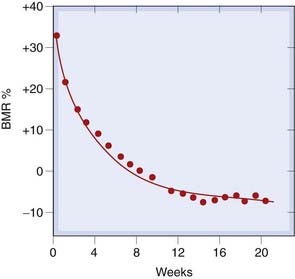
Fig. 33.5 Time course of fall of basal metabolic rate (BMR) during treatment with an antithyroid drug, carbimazole.
The curve is exponential, corresponding to a daily decrease in BMR of 3.4%.
(From Furth E O et al. 1963 J Clin Endocrinol Metab 23: 1130.)
Both methimazole and propylthiouracil cross the placenta and also appear in the milk, but this effect is less pronounced with propylthiouracil, because it is more strongly bound to plasma protein. After degradation, the metabolites are excreted in the urine, propylthiouracil being excreted more rapidly than methimazole. The thioureylenes may be concentrated in the thyroid.
Unwanted effects
The most dangerous unwanted effect of thioureyline drugs is neutropenia and agranulocytosis (see Ch. 24). This is relatively rare, having an incidence of 0.1–1.2%, and is reversible on cessation of treatment. Patients must be warned to report symptoms (especially sore throat) immediately and have a blood count. Rashes are more common (2–25%), and other symptoms, such as headaches, nausea, jaundice and pain in the joints, can also occur with the thiourelyenes.
Iodine/Iodide
Iodine is converted in vivo to iodide (I−), which temporarily inhibits the release of thyroid hormones. When high doses of iodine are given to thyrotoxic patients, the symptoms subside within 1–2 days. There is inhibition of the secretion of thyroid hormones and, over a period of 10–14 days, a marked reduction in vascularity of the gland, which becomes smaller and firmer. Iodine is often given orally in a solution with potassium iodide (‘Lugol’s iodine’). With continuous administration, its effect reaches maximum within 10–15 days and then decreases. The mechanism of action is not entirely clear; it may inhibit iodination of thyroglobulin, possibly by reducing the H2O2 generation that is necessary for this process.
The main uses of iodine/iodide are for the preparation of hyperthyroid subjects for surgical resection of the gland, and as part of the treatment of severe thyrotoxic crisis (thyroid storm). Allergic reactions can occur; these include angio-oedema, rashes and drug fever. Lacrimation, conjunctivitis, pain in the salivary glands and a cold-like syndrome are dose-related adverse effects connected to the concentration of iodide by transport mechanisms in tears and saliva.
Other Drugs Used
The β-adrenoceptor antagonists, for example propranolol (Ch. 14), are not antithyroid agents as such, but they are useful for decreasing many of the signs and symptoms of hyperthyroidism—the tachycardia, dysrhythmias, tremor and agitation. They are used during the preparation of thyrotoxic patients for surgery, as well as in most hyperthyroid patients during the initial treatment period while the thioureylenes or radioiodine take effect, or as part of the treatment of acute hyperthyroid crisis. Eye drops containing guanethidine, a noradrenergic-blocking agent (Ch. 14), are used to ameliorate the exophthalmos of hyperthyroidism (which is not relieved by antithyroid drugs); it acts by relaxing the sympathetically innervated smooth muscle that causes eyelid retraction. Glucocorticoids (e.g. prednisolone or hydrocortisone) or surgical decompression may be needed to mitigate severe exophthalmia in Graves’ disease. Some other drugs (e.g. cholecystographic agents or antiepileptic drugs) as well as ‘endocrine disruptors’5 may interfere with the normal production of thyroid hormones.
Hypothyroidism
There are no drugs that specifically augment the synthesis or release of thyroid hormones. The only effective treatment for hypothyroidism, unless it is caused by iodine deficiency (which is treated with iodide; see above), is to administer the thyroid hormones themselves as replacement therapy. Thyroxine (official name: levothyroxine) and tri-iodothyronine (official name: liothyronine) are synthetic compounds, identical to the natural hormones, and are given orally. Thyroxine as the sodium salt in doses of 50–100 µg/day is the usual first-line drug of choice. Liothyronine has a faster onset but a shorter duration of action, and is generally reserved for acute emergencies such as the rare condition of myxoedema coma, where these properties are an advantage.
Unwanted effects may occur with overdose, and in addition to the signs and symptoms of hyperthyroidism there is a risk of precipitating angina pectoris, cardiac dysrhythmias or even cardiac failure. The effects of less severe overdose are more insidious; the patient feels well but bone resorption is increased, leading to osteoporosis (Ch. 35).
The use of drugs acting on the thyroid is summarised in the clinical box.
Drugs in thyroid disease
Drugs for hyperthyroidism

References and Further Reading
Bassett J.H.D., Harvey C.B., Williams G.R. Mechanisms of thyroid hormone receptor-specific nuclear and extra nuclear actions. Mol. Cell Endocrinol.. 2003;213:1-11. (An excellent and comprehensive review dealing with the actions of thyroid hormones through the nuclear receptor mechanism as well as other actions through G-protein-coupled receptors and other pathways)
Braga M., Cooper D.S. Clinical review 129. Oral cholecystographic agents and the thyroid. J. Clin. Endocrinol. Metab.. 2001;86:1853-1860. (Discusses the deleterious effect of imaging agents on thyroid function)
Hadj Kacem H., Rebai A., Kaffel N., et al. PDS is a new susceptibility gene to autoimmune thyroid diseases: association and linkage study. J. Clin. Endocrinol. Metab.. 2003;88:2274-2280. (Interesting article on the PDS transporter protein and its contribution to disease susceptibility)
Kahaly G.J., Dillmann W.H. Thyroid hormone action in the heart. Endocr. Rev.. 2005;26:704-728. (A very interesting review focusing on the cardiac actions of thyroid hormones; much historical detail)
Kelly G.S. Peripheral metabolism of thyroid hormones: a review. Altern. Med. Rev.. 2000;5:306-333. (This review focuses on the role of peripheral metabolism in thyroid hormone action)
Lazar M.A. Thyroid hormone action: a binding contract. J. Clin. Invest. 2003;112:497-499. (A short and accessible article dealing with the main nuclear receptor-mediated effects of thyroid hormones as well as some other potential mechanisms of action)
Lazarus J.H. Hyperthyroidism. Lancet. 1997;349:339-343. (A ‘seminar’ covering aetiology, clinical features, pathophysiology, diagnosis and treatment)
Lindsay R.S. Hypothyroidism. Lancet. 1997;349:413-417. (A ‘seminar’ emphasising the management of hypothyroidism)
Mastorakos G., Karoutsou E.I., Mizamtsidi M., Creatsas G. The menace of endocrine disruptors on thyroid hormone physiology and their impact on intrauterine development. Endocrine.. 2007;3:219-237. (A review of endocrine disrupters and their effects on the thyroid. Not mainstream reading but an interesting topic)
Nilsson M. Iodide handling by the thyroid epithelial cell. Exp. Clin. Endocrinol. Diabetes. 2001;109:13-17. (Useful and readable review of iodide handling by the thyroid gland)
Paschke R., Ludgate M. The thyrotropin receptor and its diseases. N. Engl. J. Med.. 1997;337:1675-1679. (Reviews aspects of TSH biology and disease)
Roberts C.G., Ladenson P.W. Hypothyroidism. Lancet. 2004;363:793-803. (Authoritative and accessible review dealing with this thyroid pathology)
Schmutzler C., Kohrle J. Implications of the molecular characterization of the sodium–iodide symporter (NIS). Exp. Clin. Endocrinol. Diabetes. 1998;106:S1-S10. (Discusses the diagnostic and therapeutic implications of the information now available as a result of the cloning of NIS)
Surks M.I., Ortiz E., Daniels G.H., et al. Subclinical thyroid disease: scientific review and guidelines for diagnosis and management. JAMA. 2004;291:228-238. (Discusses and reviews the treatment of subclinical thyroid disease in detail; primarily of interest to clinical students)
Yen P.M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev.. 2001;81:1097-1142. (Comprehensive review of thyroid hormone–receptor interaction and the effects of thyroid hormone on target tissues)
Yoshida A., Hisatome I., Taniguchi S., et al. Mechanism of iodide/chloride exchange by pendrin. Endocrinology. 2004;145:4301-4308. (An original research article that takes an electrophysiological approach to understanding of the PDS transporter and its relationship to Pendred’s syndrome)
Zhang J., Lazar M. The mechanism of action of thyroid hormones. Annu. Rev. Physiol.. 2000;62:439-466. (Detailed review of the molecular aspects of thyroid hormone–receptor interaction)
1So called because it is implicated in the pathophysiology of Pendred’s syndrome, named after the eponymous English physician who first described this form of familial goitre.
2’Derbyshire neck’ was the name given to this condition in a part of the UK where sources of dietary iodine were once scarce.
3After a Dublin physician who connected ‘violent and long continued palpitations in females’ with enlargement of the thyroid gland. The young ladies’ complaints of fluttering hearts and lumps in their throats had previously been attributed to hysteria.
4An older term for this condition, cretinism, has been dropped.
5These are man-made chemicals such as pesticides or herbicides (e.g. polychlorinated biphenyls) that linger in the environment and are ingested in foodstuffs. The endocrine system is particularly sensitive to these, especially during development.