21 The heart
Overview
In this chapter, we review briefly the physiology of cardiac function in terms of electrophysiology, of contraction, of oxygen consumption and coronary blood flow, of autonomic control and as a source of peptide hormones. This provides a basis for understanding effects of drugs on the heart and their place in treating cardiac disease. The main drugs considered are drugs that act directly on the heart, namely antidysrhythmic drugs and drugs that increase the force of contraction of the heart (especially digoxin); antianginal drugs are also covered in this chapter. The commonest forms of heart disease are caused by atheroma in the coronary arteries, and thrombosis on ruptured atheromatous plaques; drugs to treat and prevent these are considered in Chapters 23 and 24. Heart failure is mainly treated by drugs that work indirectly on the heart via actions on vascular smooth muscle, discussed in Chapter 22, by diuretics (Ch. 28) and β-adrenoceptor antagonists (Ch. 14).
Introduction
In this chapter, we consider effects of drugs on the heart under three main headings:
The effects of drugs on these aspects of cardiac function are not, of course, independent of each other. For example, if a drug affects the electrical properties of the myocardial cell membrane, it is likely to influence both cardiac rhythm and myocardial contraction. Similarly, a drug that affects contraction will inevitably alter metabolism and blood flow as well. Nevertheless, from a therapeutic point of view, these three classes of effect represent distinct clinical objectives in relation to the treatment, respectively, of cardiac dysrhythmias, cardiac failure and coronary insufficiency (as occurs during angina pectoris or myocardial infarction).
Physiology of Cardiac Function
Cardiac Rate and Rhythm
The chambers of the heart normally contract in a coordinated manner, pumping blood efficiently by a route determined by the valves. Coordination of contraction is achieved by a specialised conducting system. Physiological sinus rhythm is characterised by impulses arising in the sinoatrial (SA) node and conducted in sequence through the atria, the atrioventricular (AV) node, bundle of His, Purkinje fibres and ventricles. Cardiac cells owe their electrical excitability to voltage-sensitive plasma membrane channels selective for various ions, including Na+, K+ and Ca2+, the structure and function of which are described in Chapter 4. Electrophysiological features of cardiac muscle that distinguish it from other excitable tissues include:
Thus several of the special features of cardiac rhythm relate to Ca2+ currents. The heart contains intracellular calcium channels (i.e. ryanodine receptors and inositol trisphosphate-activated calcium channels described in Ch. 4, which are important in myocardial contraction) and voltage-dependent calcium channels in the plasma membrane, which are important in controlling cardiac rate and rhythm. The main type of voltage-dependent calcium channel in adult working myocardium is the L-type channel, which is also important in vascular smooth muscle; L-type channels are important in specialised conducting regions as well as in working myocardium.
The action potential of an idealised cardiac muscle cell is shown in Figure 21.1A and is divided into five phases: 0 (fast depolarisation), 1 (partial repolarisation), 2 (plateau), 3 (repolarisation) and 4 (pacemaker).
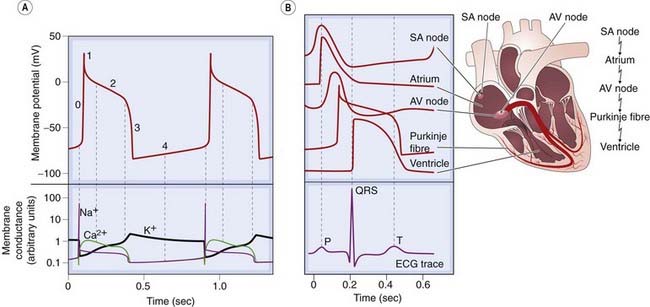
Fig. 21.1 The cardiac action potential.
[A] Phases of the action potential: 0, rapid depolarisation; 1, partial repolarisation; 2, plateau; 3, repolarisation; 4, pacemaker depolarisation. The lower panel shows the accompanying changes in membrane conductance for Na+, K+ and Ca2+. [B] Conduction of the impulse through the heart, with the corresponding electrocardiogram (ECG) trace. Note that the longest delay occurs at the atrioventricular (AV) node, where the action potential has a characteristically slow waveform. SA, sinoatrial.
(Adapted from: [A] Noble D 1975 The initiation of the heartbeat. Oxford University Press, Oxford.)
 Ionic mechanisms underlying these phases can be summarised as follows.
Ionic mechanisms underlying these phases can be summarised as follows.
Phase 0, rapid depolarisation, occurs when the membrane potential reaches a critical firing threshold (about −60 mV), at which the inward current of Na+ flowing through the voltage-dependent sodium channels becomes large enough to produce a regenerative (‘all-or-nothing’) depolarisation. This mechanism is the same as that responsible for action potential generation in neurons (see Ch. 4). Activation of sodium channels by membrane depolarisation is transient, and if the membrane remains depolarised for more than a few milliseconds, they close again (inactivation). They are therefore closed during the plateau of the action potential and remain unavailable for the initiation of another action potential until the membrane repolarises.
Phase 1, partial repolarisation, occurs as the Na+ current is inactivated. There may also be a transient voltage-sensitive outward current.
Phase 2, the plateau, results from an inward Ca2+ current. Calcium channels show a pattern of voltage-sensitive activation and inactivation qualitatively similar to sodium channels, but with a much slower time course. The plateau is assisted by a special property of the cardiac muscle membrane known as inward-going rectification, which means that the K+ conductance falls to a low level when the membrane is depolarised. Because of this, there is little tendency for outward K+ current to restore the resting membrane potential during the plateau, so a relatively small inward Ca2+ current suffices to maintain the plateau.
Phase 3, repolarisation, occurs as the Ca2+ current inactivates and a delayed outwardly rectifying K+ current (analogous to, but much slower than, the K+ current that causes repolarisation in nerve fibres; Ch. 4) activates, causing outward K+ current. This is augmented by another K+ current, which is activated by high intracellular Ca2+ concentrations, [Ca2+]i during the plateau, and sometimes also by other K+ currents, including one through channels activated by acetylcholine (see below) and another that is activated by arachidonic acid, which is liberated under pathological conditions such as myocardial infarction.
Phase 4, the pacemaker potential, is a gradual depolarisation during diastole. Pacemaker activity is normally found only in nodal and conducting tissue. The pacemaker potential is caused by a combination of increasing inward currents and declining outward currents during diastole. It is usually most rapid in cells of the SA node, which therefore acts as pacemaker for the whole heart. Cells in the SA node have a greater background conductance to Na+ than do atrial or ventricular myocytes, leading to a greater background inward current. In addition, inactivation of voltage-dependent calcium channels wears off during diastole, resulting in increasing inward Ca2+ current during late diastole. Activation of T-type calcium channels during late diastole contributes to pacemaker activity in the SA node. The negative membrane potential early in diastole activates a cation channel that is permeable to Na+ and K+, giving rise to another inward current, called If.1 An inhibitor of this current, ivabradine, slows the heart and is used therapeutically (see below).
Several voltage- and time-dependent outward currents play a part as well: delayed rectifier K+ current (IK), which is activated during the action potential, is turned off by the negative membrane potential early in diastole. Current from the electrogenic Na+/K+ pump also contributes to the outward current during the pacemaker potential.
Figure 21.1B shows the action potential configuration in different parts of the heart. Phase 0 is absent in the nodal regions, where the conduction velocity is correspondingly slow (∼5 cm/s) compared with other regions such as the Purkinje fibres (conduction velocity ∼200 cm/s), which propagate the action potential rapidly to the ventricles. Regions that lack a fast inward current have a much longer refractory period than fast-conducting regions. This is because recovery of the slow inward current following its inactivation during the action potential takes a considerable time (a few hundred milliseconds), and the refractory period outlasts the action potential. With fast-conducting fibres, inactivation of the Na+ current recovers rapidly, and the cell becomes excitable again almost as soon as it is repolarised.
The orderly pattern of sinus rhythm can be disrupted either by heart disease or by the action of drugs or circulating hormones, and an important therapeutic use of drugs is to restore a normal cardiac rhythm where it has become disturbed. The commonest cause of cardiac dysrhythmia is ischaemic heart disease, and many deaths following myocardial infarction result from ventricular fibrillation (‘fibrillation’ is a state where heart chambers stop contracting in a coordinated way because of chaotic electrical activity; instead, the affected heart chambers ‘fibrillate’—rapid uncoordinated contractions within ventricles or atria that are visible to the naked eye but do not support output from the affected chambers) rather than directly from contractile failure.
Disturbances of Cardiac Rhythm
Clinically, dysrhythmias are classified according to:
They may cause palpitations (awareness of the heartbeat) or symptoms from cerebral hypoperfusion (faintness or loss of consciousness). Their diagnosis depends on the surface electrocardiogram (ECG), and details are beyond the scope of this book—see Braunwald & Opie (2001). The commonest types of tachyarrhythmia are atrial fibrillation, where the heartbeat is completely irregular, and supraventricular tachycardia (SVT), where the heartbeat is rapid but regular. Occasional ectopic beats (ventricular as well as supraventricular) are common. Sustained ventricular tachyarrhythmias are much less common but much more serious; they include ventricular tachycardia, and ventricular fibrillation where the electrical activity in the ventricles is completely chaotic and cardiac output ceases. Bradyarrhythmias include various kinds of heart block (e.g. at the AV or SA node) and complete cessation of electrical activity (‘asystolic arrest’). It is often unclear which of the various mechanisms discussed below are responsible. These cellular mechanisms nevertheless provide a useful starting point for understanding how antidysrhythmic drugs work. Four basic phenomena underlie disturbances of cardiac rhythm:
The main cause of delayed after-depolarisation is abnormally raised [Ca2+]i, which triggers inward current and hence a train of abnormal action potentials (Fig. 21.2). After-depolarisation is the result of a net inward current, known as the transient inward current. A rise in [Ca2+]i activates Na+/Ca2+ exchange. This transfers one Ca2+ ion out of the cell in exchange for entry of three Na+ ions, resulting in a net influx of one positive charge and hence membrane depolarisation. Additionally, raised [Ca2+]i opens non-selective cation channels in the plasma membrane, causing depolarisation analogous to the endplate potential at the neuromuscular junction (Ch. 13). Consequently, hypercalcaemia (which increases the entry of Ca2+) promotes after-depolarisation. Hypokalaemia also influences repolarisation,via an effect on the gating of cardiac delayed rectifier potassium channels. Many drugs, including ones whose principal effects are on other organs, delay cardiac repolarisation by binding to potassium or other cardiac channels or by influencing electrolyte concentrations (see Roden, 2004). Delayed repolarisation increases Ca2+ entry during the prolonged action potential, leading to after-depolarisation. Prolongation of the QT interval, which carries a risk of causing dangerous ventricular dysrhythmias, is a concern in drug development (see section below, Class III drugs, and see Ch. 57).
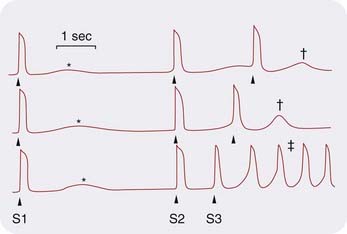
Fig. 21.2 After-depolarisation in cardiac muscle recorded from a dog coronary sinus in the presence of noradrenaline (norepinephrine).
The first stimulus (S1) causes an action potential followed by a small after-depolarisation. As the interval S2–S3 is decreased, the after-depolarisation gets larger (†) until it triggers an indefinite train of action potentials (‡).
(Adapted from Wit A L, Cranefield P F 1977 Circ Res 41: 435.)
Normally, a cardiac action potential dies out after it has activated the ventricles because it is surrounded by refractory tissue, which it has just traversed. Re-entry (Fig. 21.3) describes a situation in which the impulse re-excites regions of the myocardium after the refractory period has subsided, causing continuous circulation of action potentials. It can result from anatomical anomalies or, more commonly, from myocardial damage. Re-entry underlies many types of dysrhythmia, the pattern depending on the site of the re-entrant circuit, which may be in the atria, ventricles or nodal tissue. A simple ring of tissue can give rise to a re-entrant rhythm if a transient or unidirectional conduction block is present. Normally, an impulse originating at any point in the ring will propagate in both directions and die out when the two impulses meet, but if a damaged area causes either a transient block (so that one impulse is blocked but the second can get through; Fig. 21.3) or a unidirectional block, continuous circulation of the impulse can occur. This is known as circus movement and was demonstrated experimentally on rings of jellyfish tissue many years ago.
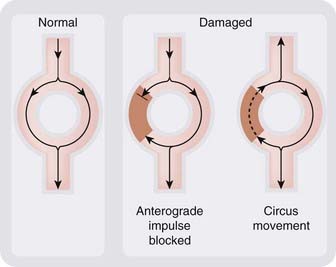
Fig. 21.3 Generation of a re-entrant rhythm by a damaged area of myocardium.
The damaged area (brown) conducts in one direction only. This disturbs the normal pattern of conduction and permits continuous circulation of the impulse to occur.
Although the physiological pacemaker resides in the SA node, other cardiac tissues can take on pacemaker activity. This provides a safety mechanism in the event of failure of the SA node but can also trigger tachyarrhythmias. Ectopic pacemaker activity is encouraged by sympathetic activity and by partial depolarisation, which may occur during ischaemia. Catecholamines, acting on β1 adrenoceptors (see below), increase the rate of depolarisation during phase 4 and can cause normally quiescent parts of the heart to take on a spontaneous rhythm. Several tachyarrhythmias (e.g. paroxysmal atrial fibrillation) can be triggered by circumstances associated with increased sympathetic activity. Pain (e.g. during myocardial infarction) increases sympathetic discharge and releases adrenaline (epinephrine) from the adrenal gland. Partial depolarisation resulting from ischaemic damage also causes abnormal pacemaker activity.
Heart block results from fibrosis of, or ischaemic damage to, the conducting system (often in the AV node). In complete heart block, the atria and ventricles beat independently of one another, the ventricles beating at a slow rate determined by whatever pacemaker picks up distal to the block. Sporadic complete failure of AV conduction causes sudden periods of unconsciousness (Stokes–Adams attacks) and is treated by implanting an artificial pacemaker.
Cardiac Contraction
Cardiac output is the product of heart rate and mean left ventricular stroke volume (i.e. the volume of blood ejected from the ventricle with each heartbeat). Heart rate is controlled by the autonomic nervous system (Chs 13 and 14, and see below). Stroke volume is determined by a combination of factors, including some intrinsic to the heart itself and other haemodynamic factors extrinsic to the heart. Intrinsic factors regulate myocardial contractility via [Ca2+]i and ATP, and are sensitive to various drugs and pathological processes. Extrinsic circulatory factors include the elasticity and contractile state of arteries and veins, and the volume and viscosity of the blood, which together determine cardiac load (preload and afterload). Drugs that influence these circulatory factors are of paramount importance in treating patients with heart failure. They are covered in Chapter 22.
Myocardial Contractility and Viability
The contractile machinery of myocardial striated muscle is basically the same as that of voluntary striated muscle (Ch. 4). It involves binding of Ca2+ to troponin C; this changes the conformation of the troponin complex, permitting cross-bridging of myosin to actin and initiating contraction. Levosimendan (a drug used to treat acute decompensated heart failure; Ch. 22), increases the force of contraction of the heart by binding troponin C and sensitising it to the action of Ca2+.
Many effects of drugs on cardiac contractility can be explained in terms of actions on [Ca2+]i, via effects on calcium channels in plasma membrane or sarcoplasmic reticulum, or on the Na+/K+ pump, which indirectly influences the Na+/Ca2+ pump (see below). Other factors that affect the force of contraction are the availability of oxygen and a source of metabolic energy such as free fatty acids. Myocardial stunning—contractile dysfunction that persists after ischaemia and reperfusion despite restoration of blood flow and absence of cardiac necrosis—is incompletely understood but can be clinically important. Its converse is known as ischaemic preconditioning; this refers to an improved ability to withstand ischaemia following previous ischaemic episodes. This potentially beneficial state could be clinically important. There is some evidence that it is mediated by adenosine (see Ch. 2), which accumulates as ATP is depleted. Exogenous adenosine affords protection similar to that caused by ischaemic preconditioning, and blockade of adenosine receptors prevents the protective effect of preconditioning (see Gross & Auchampach, 2007). There is considerable interest in developing strategies to minimise harmful effects of ischaemia while maximising preconditioning.
Ventricular Function Curves and Heart Failure
The force of contraction of the heart is determined partly by its intrinsic contractility (which, as described above, depends on [Ca2+]i and availability of ATP), and partly by extrinsic haemodynamic factors that affect end-diastolic volume and hence the resting length of the muscle fibres. The end-diastolic volume is determined by the end-diastolic pressure, and its effect on stroke work is expressed in the Frank–Starling law of the heart, which reflects an inherent property of the contractile system. The Frank–Starling law can be represented as a ventricular function curve (Fig. 21.4). The area enclosed by the pressure–volume curve during the cardiac cycle provides a measure of ventricular stroke work. It is approximated by the product of stroke volume and mean arterial pressure. As Starling showed, factors extrinsic to the heart affect its performance in various ways, two patterns of response to increased load being particularly important.
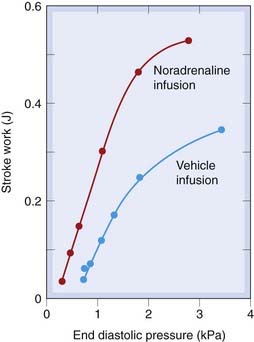
Fig. 21.4 Ventricular function curves in the dog. Infusion of physiological saline increases blood volume and hence end-diastolic pressure.
This increases stroke work (‘extrinsic’ control) by increasing the force of contraction of the heart. This relationship is called the Starling curve. Noradrenaline has a direct action on the heart (‘intrinsic’ control), increasing the slope of the Starling curve.
(Redrawn from Sarnoff S J et al. 1960 Circ Res 8: 1108.)
Normal ventricular filling pressure is only a few centimetres of water, on the steep part of the ventricular function curve, so a large increase in stroke work can be achieved with only a small increase in filling pressure. The Starling mechanism plays little part in controlling cardiac output in healthy subjects (e.g. during exercise), because changes in contractility, mainly as a result of changes in sympathetic nervous activity, achieve the necessary regulation without any increase in ventricular filling pressure (Fig. 21.4). In contrast, the denervated heart in patients who have received a heart transplant relies on the Starling mechanism to increase cardiac output during exercise.
In heart failure, the cardiac output is insufficient to meet the needs of the body, initially only when these are increased during exercise but ultimately, as disease progresses, also at rest. It has many causes, most commonly ischaemic heart disease. In patients with heart failure (see Ch. 22), the heart may be unable to deliver as much blood as the tissues require, even when its contractility is increased by sympathetic activity. Under these conditions, the basal (i.e. at rest) ventricular function curve is greatly depressed, and there is insufficient reserve, in the sense of extra contractility that can be achieved by sympathetic activity, to enable cardiac output to be maintained during exercise without a large increase in central venous pressure (Fig. 21.4). Oedema of peripheral tissues (causing swelling of the legs) and the lungs (causing breathlessness) is an important consequence of cardiac failure. It is caused by the increased venous pressure, and retention of Na+ (see Ch. 22).
Myocardial contraction 
Myocardial Oxygen Consumption and Coronary Blood Flow
Relative to its large metabolic needs, the heart is one of the most poorly perfused tissues in the body. Coronary flow is, under normal circumstances, closely related to myocardial oxygen consumption, and both change over a nearly 10-fold range between conditions of rest and maximal exercise.
Physiological Factors
The main physiological factors that regulate coronary flow are:
Physical factors
During systole, the pressure exerted by the myocardium on vessels that pass through it equals or exceeds the perfusion pressure, so coronary flow occurs only during diastole. Diastole is shortened more than systole during tachycardia, reducing the period available for myocardial perfusion. During diastole, the effective perfusion pressure is equal to the difference between the aortic and ventricular pressures (Fig. 21.5). If diastolic aortic pressure falls or diastolic ventricular pressure increases, perfusion pressure falls and so (unless other control mechanisms can compensate) does coronary blood flow. Stenosis of the aortic valve reduces aortic pressure but increases left ventricular pressure upstream of the narrowed valve, and often causes ischaemic chest pain (angina) even in the absence of coronary artery disease.
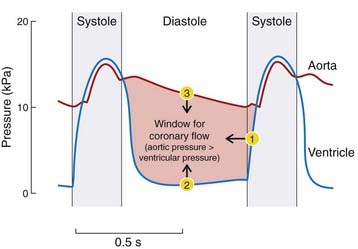
Vascular control by metabolites/mediators
Vascular control by metabolites is the most important mechanism by which coronary flow is regulated. A reduction in arterial partial pressure of oxygen (PO2) causes marked vasodilatation of coronary vessels in situ but has little effect on isolated strips of coronary artery. This suggests that it is a change in the pattern of metabolites produced by the myocardial cells, rather than the change in PO2 per se, that controls the state of the coronary vessels, a popular candidate for the dilator metabolite being adenosine (see Ch. 16).
Neural and humoral control
Coronary vessels have a dense sympathetic innervation, but sympathetic nerves (like circulating catecholamines) exert only a small direct effect on the coronary circulation. Large coronary vessels possess α adrenoceptors that mediate vasoconstriction, whereas smaller vessels have β2 adrenoceptors that have a dilator effect. Coronary vessels are also innervated by purinergic, peptidergic and nitrergic nerves, and basal coronary blood flow in patients with angiographically normal coronary arteries is reduced by about one-third by selective inhibition of nNOS (Seddon et al., 2009). Coronary vascular responses to altered mechanical and metabolic activity during exercise or pathological events overshadow neural and endocrine effects.
Autonomic Control of the Heart
The sympathetic and parasympathetic systems (see Chs 12–14) each exert a tonic effect on the heart at rest. They influence each of the aspects of cardiac function that have been discussed above, namely rate and rhythm, myocardial contraction, and myocardial metabolism and blood flow.
Sympathetic System
The main effects of sympathetic activity on the heart are:
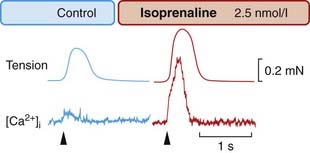
Fig. 21.6 The calcium transient in frog cardiac muscle.
A group of cells was injected with the phosphorescent Ca2+ indicator aequorin, which allows [Ca2+]i to be monitored optically. Isoprenaline causes a large increase in the tension and in the [Ca2+]i transient caused by an electrical stimulus ( ).
).
(From Allen D G, Blinks J R 1978 Nature 273: 509.)
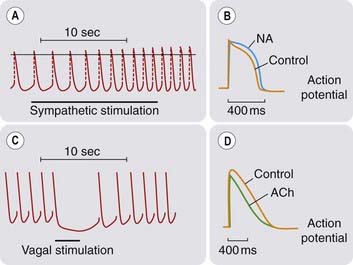
Fig. 21.7 Autonomic regulation of the heartbeat.
[A] and [B] Effects of sympathetic stimulation and noradrenaline (NA). [C] and [D] Effects of parasympathetic stimulation and acetylcholine (ACh). Sympathetic stimulation [A] increases the slope of the pacemaker potential and increases heart rate, whereas parasympathetic stimulation [C] abolishes the pacemaker potential, hyperpolarises the membrane and temporarily stops the heart (frog sinus venosus). NA [B] prolongs the action potential, while ACh [D] shortens it (frog atrium).
(From: [A] and [C] Hutter O F, Trautwein W 1956 J Gen Physiol 39: 715; [B] Reuter H 1974 J Physiol 242: 429; [D] Giles W R, Noble S J 1976 J Physiol 261: 103.)
These effects all result from activation of β1-adrenoceptors. The β1 effects of catecholamines on the heart, although complex, probably all occur through increased intracellular cAMP (see Ch. 3). cAMP activates protein kinase A, which phosphorylates sites on the α1 subunits of calcium channels. This increases the probability that the channels will open, increasing inward Ca2+ current and hence force of cardiac contraction (Fig. 21.6). Activation of β1-adrenoceptors also increases the Ca2+ sensitivity of the contractile machinery, possibly by phosphorylating troponin C; furthermore, it facilitates Ca2+ capture by the sarcoplasmic reticulum, thereby increasing the amount of Ca2+ available for release by the action potential. The net result of catecholamine action is to elevate and steepen the ventricular function curve (Fig. 21.4). The increase in heart rate results from an increased slope of the pacemaker potential (Figs 21.1 and 21.7A). Increased Ca2+ entry also increases automaticity because of the effect of [Ca2+]i on the transient inward current, which can result in a train of action potentials following a single stimulus (Fig. 21.2).
Activation of β1-adrenoceptors repolarises damaged or hypoxic myocardium by stimulating the Na+/K+ pump. This can restore function if asystole has occurred following myocardial infarction, and adrenaline is one of the most important drugs used during cardiac arrest.
The reduction of cardiac efficiency by catecholamines is important because it means that the oxygen requirement of the myocardium increases. This limits the use of β agonists such as adrenaline and dobutamine for circulatory shock (Ch. 22). Myocardial infarction activates the sympathetic nervous system (see Fig. 21.8), which has the undesirable effect of increasing the oxygen needs of the damaged myocardium.
Parasympathetic System
Parasympathetic activity produces effects that are, in general, opposite to those of sympathetic activation. However, in contrast to sympathetic activity, the parasympathetic nervous system has little effect on contractility, its main effects being on rate and rhythm, namely:
These effects result from activation of muscarinic (M2) acetylcholine receptors, which are abundant in nodal and atrial tissue but sparse in the ventricles. These receptors are negatively coupled to adenylyl cyclase and thus reduce cAMP formation, acting to inhibit the opening of L-type Ca2+ channels and reduce the slow Ca2+ current, in opposition to β1-adrenoceptors. M2 receptors also open a potassium channel (called KACh). The resulting increase in K+ permeability produces a hyperpolarising current that opposes the inward pacemaker current, slowing the heart and reducing automaticity (see Fig. 21.7C). Vagal activity is often increased during myocardial infarction, both in association with vagal afferent stimulation and as a side effect of opioids used to control the pain, and parasympathetic effects are important in predisposing to acute dysrhythmias.
Vagal stimulation decreases the force of contraction of the atria associated with marked shortening of the action potential (Fig. 21.7D). Increased K+ permeability and reduced Ca2+ current both contribute to conduction block at the AV node, where propagation depends on the Ca2+ current. Shortening the atrial action potential reduces the refractory period, which can lead to re-entrant arrhythmias. Coronary vessels lack cholinergic innervation; consequently, the parasympathetic nervous system has little effect on coronary artery tone (see Ch. 13).2
Autonomic control of the heart
Cardiac Natriuretic Peptides
Cardiac natriuretic peptides are an important family of mediators (see Potter et al., 2009, for a review). Atrial cells contain secretory granules, and store and release atrial natriuretic peptide (ANP). This has powerful effects on the kidney and vascular system. Release of ANP occurs during volume overload in response to stretching of the atria, and intravenous saline infusion is sufficient to stimulate its release. B-natriuretic peptide (BNP) is released from ventricular muscle and opposes ventricular fibrosis; its plasma concentration is increased in patients with heart failure and is used as an aid to diagnosis. C-natriuretic peptide (CNP) is stored in endothelium and in addition to vascular actions influences development of long bones.
The main effects of natriuretic peptides are to increase Na+ and water excretion by the kidney; relax vascular smooth muscle (except efferent arterioles of renal glomeruli; see below); increase vascular permeability; and inhibit the release and/or actions of several hormones and mediators, including aldosterone, angiotensin II, endothelin and antidiuretic hormone. They exert their effects by combining with membrane receptors (natriuretic peptide receptors, NPRs, which exist in at least two subtypes, designated A and B).3
Both NPR-A and NPR-B incorporate a catalytic guanylyl cyclase moiety (see Ch. 3), and, when activated, increase intracellular cGMP. Organic nitrates (see later) and endothelium-derived nitric oxide (Ch. 20) act similarly, though they interact with soluble rather than membrane-bound guanylyl cyclase. Renal glomerular afferent arterioles are dilated by ANP but efferent arterioles are constricted, so filtration pressure is increased, leading to increased glomerular filtration and enhanced Na+ excretion. Elsewhere, natriuretic peptides cause vasorelaxation and reduce blood pressure. Their therapeutic potential, which remains controversial (see Richards, 2009, for a recent editorial commentary), is considered in Chapter 22.
Ischaemic Heart Disease
Atheromatous deposits are ubiquitous in the coronary arteries of adults living in developed countries. They are asymptomatic for most of the natural history of the disease (see Ch. 23), but can progress insidiously, culminating in acute myocardial infarction and its complications, including dysrhythmia and heart failure. Details of ischemic heart disease are beyond the scope of this book, and excellent accounts (e.g. Braunwald, 2005) are available for those seeking pathological and clinical information. Here, we merely set the scene for understanding the place of drugs that affect cardiac function in treating this most common form of heart disease.
Important consequences of coronary atherosclerosis include:
Angina
Angina occurs when the oxygen supply to the myocardium is insufficient for its needs. The pain has a characteristic distribution in the chest, arm and neck, and is brought on by exertion, cold or excitement. A similar type of pain occurs in skeletal muscle when it is made to contract while its blood supply is interrupted, and Lewis showed many years ago that chemical factors released by ischaemic muscle are responsible. Possible candidates include K+, H+ and adenosine (Ch. 16), all of which sensitise or stimulate nociceptors (see Ch. 41). It is possible that the same mediator that causes coronary vasodilatation is responsible, at higher concentration, for initiating pain.
Three kinds of angina are recognised clinically: stable, unstable and variant.
Stable angina
This is predictable chest pain on exertion. It is produced by an increased demand on the heart and is caused by a fixed narrowing of the coronary vessels, almost always by atheroma. Symptomatic therapy is directed at reducing cardiac work with organic nitrates, β-adrenoceptor antagonists and/or calcium antagonists (as described below), together with treatment of the underlying atheromatous disease, usually including a statin (Ch. 23), and prophylaxis against thrombosis with an antiplatelet drug, usually aspirin (Ch. 24).
Unstable angina
This is characterised by pain that occurs with less and less exertion, culminating in pain at rest. The pathology is similar to that involved in myocardial infarction, namely platelet–fibrin thrombus associated with a ruptured atheromatous plaque, but without complete occlusion of the vessel. Treatment is as for myocardial infarction without ST-segment elevation on the cardiogram (NSTEMI). Antiplatelet drugs (aspirin and/or an ADP antagonist such as clopidogrel or prasugrel) reduce the risk of myocardial infarction in this setting, and heparin and platelet glycoprotein receptor antagonists add to this benefit (Ch. 24) at the cost of increased risk of haemorrhage, and organic nitrates relieve ischaemic pain.
Myocardial Infarction
Myocardial infarction occurs when a coronary artery has been blocked by thrombus. This may be fatal and is a common cause of death, usually as a result of mechanical failure of the ventricle or from dysrhythmia. Cardiac myocytes rely on aerobic metabolism. If the supply of oxygen remains below a critical value, a sequence of events leading to cell death (by necrosis or apoptosis) ensues (see Ch. 5 for a fuller account of apoptosis), detected clinically by an elevation of circulating troponin (the gold-standard biochemical marker of myocardial injury). The sequences leading from vascular occlusion to cell death via the two pathways are illustrated in Figure 21.8. The relative importance of necrosis and apoptosis in myocardial cell death in clinically distinct settings is unknown, but it has been suggested that apoptosis may be an adaptive process in hypoperfused regions, sacrificing some jeopardised myocytes but thereby avoiding the disturbance of membrane function and risk of dysrhythmia inherent in necrosis. Consequently, it is currently unknown if pharmacological approaches to promote or inhibit this pathway could be clinically beneficial.
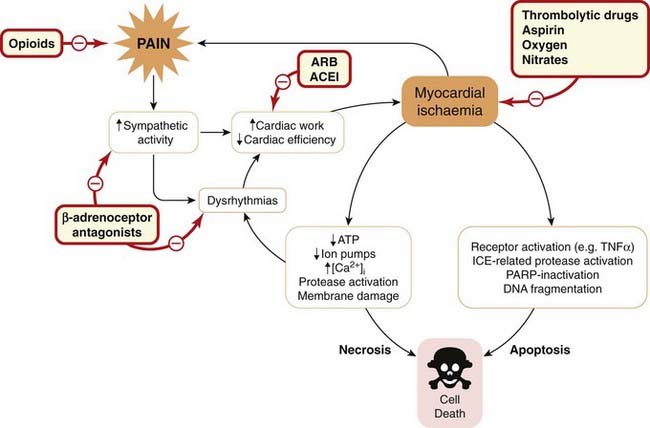
Fig. 21.8 Effects of myocardial ischaemia.
This leads to cell death by one of two pathways: necrosis or apoptosis. ACEI, angiotensin-converting enzyme inhibitor; ARB, angiotensin AT1 receptor antagonist; ICE, interleukin-1-converting enzyme; PARP, poly-[ADP-ribose]-polymerase; TNF-α, tumour necrosis factor-α.
Prevention of irreversible ischaemic damage following an episode of coronary thrombosis is an important therapeutic aim. Opening the occluded artery is key, and it is important that this is achieved promptly, irrespective of the means by which it is done. If logistically possible, angioplasty (performed using a catheter with an inflatable balloon near its tip, with a glycoprotein IIb/IIIa antagonist—see Chapter 24—to prevent reocclusion) is somewhat more effective than thrombolytic drugs. The main therapeutic drugs (see Fig. 21.8) include drugs to improve cardiac function by maintaining oxygenation and reducing cardiac work as well as treating pain and preventing further thrombosis. They are used in combination, and include:
β-Adrenoceptor antagonists reduce cardiac work and thereby the metabolic needs of the heart, and are used as soon as the patient is stable. ACEIs and ARBs also reduce cardiac work and improve survival as does opening the coronary artery (with angioplasty or thrombolytic drug) and antiplatelet treatment.
Drugs that Affect Cardiac Function
Drugs that have a major action on the heart can be divided into three groups.
Antidysrhythmic Drugs
A classification of antidysrhythmic drugs based on their electrophysiological effects was proposed by Vaughan Williams in 1970. It provides a good starting point for discussing mechanisms, although many useful drugs do not fit neatly into this classification (Table 21.1). Furthermore, emergency treatment of serious dysrhythmias is usually by physical means (e.g. pacing or electrical cardioversion by applying a direct current shock to the chest or via an implanted device) rather than drugs.
Table 21.1 Antidysrhythmic drugs unclassified in the Vaughan Williams system
| Drug | Use |
|---|---|
| Atropine | Sinus bradycardia |
| Adrenaline (epinephrine) | Cardiac arrest |
| Isoprenaline | Heart block |
| Digoxin | Rapid atrial fibrillation |
| Adenosine | Supraventricular tachycardia |
| Calcium chloride | Ventricular tachycardia due to hyperkalaemia |
| Magnesium chloride | Ventricular fibrillation, digoxin toxicity |
There are four classes (see Table 21.2).
Table 21.2 Summary of antidysrhythmic drugs (Vaughan Williams classification)
| Class | Example(s) | Mechanism |
|---|---|---|
| Ia | Disopyramide | Sodium channel block (intermediate dissociation) |
| Ib | Lidocaine | Sodium channel block (fast dissociation) |
| Ic | Flecainide | Sodium channel block (slow dissociation) |
| II | Propranolol | β-Adrenoceptor antagonism |
| III | Amiodarone, sotalol | Potassium channel block |
| IV | Verapamil | Calcium channel block |
The phase of the action potential on which each of these classes of drug have their main effect is shown in Figure 21.9.
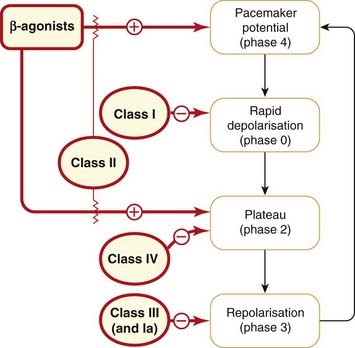
Fig. 21.9 Effects of antidysrhythmic drugs on the different phases (as defined in Fig. 21.1) of the cardiac action potential.
Mechanisms of Action
Class I drugs
Class I drugs block sodium channels, just as local anaesthetics do, by binding to sites on the α subunit (see Chs 4 and 42). Because this inhibits action potential propagation in many excitable cells, it has been referred to as ‘membrane-stabilising’ activity, a phrase best avoided now that the ionic mechanism is understood. The characteristic effect on the action potential is to reduce the maximum rate of depolarisation during phase 0.
The reason for further subdivision of these drugs into classes Ia, Ib and Ic is that the earliest examples, quinidine and procainamide (class Ia), have different effects from many of the more recently developed drugs, even though all share the same basic mechanism of action. A partial explanation for these functional differences comes from electrophysiological studies of the characteristics of the sodium channel block produced by different class I drugs.
The central concept is of use-dependent channel block. It is this characteristic that enables all class I drugs to block the high-frequency excitation of the myocardium that occurs in tachyarrhythmias, without preventing the heart from beating at normal frequencies. Sodium channels exist in three distinct functional states: resting, open and refractory (see Ch. 4). Channels switch rapidly from resting to open in response to depolarisation; this is known as activation. Maintained depolarisation, as in ischaemic muscle, causes channels to change more slowly from open to refractory (inactivation), and the membrane must then be repolarised for a time to restore the channel to the resting state before it can be activated again. Class I drugs bind to channels most strongly when they are in either the open or the refractory state, less strongly to channels in the resting state. Their action therefore shows the property of ‘use dependence’ (i.e. the more frequently the channels are activated, the greater the degree of block produced).
Class Ib drugs, for example lidocaine, associate and dissociate rapidly within the timeframe of the normal heartbeat. The drug binds to open channels during phase 0 of the action potential (affecting the rate of rise very little, but leaving many of the channels blocked by the time the action potential reaches its peak). Dissociation occurs in time for the next action potential, provided the cardiac rhythm is normal. A premature beat, however, will be aborted because the channels are still blocked. Furthermore, class Ib drugs bind selectively to refractory channels and thus block preferentially when the cells are depolarised, for example in ischaemia.
Class Ic drugs, such as flecainide and encainide, associate and dissociate much more slowly, thus reaching a steady-state level of block that does not vary appreciably during the cardiac cycle. They markedly inhibit conduction through the His–Purkinje system.
Class Ia, the oldest group (e.g. quinidine, procainamide, disopyramide), lies midway in its properties between Ib and Ic but, in addition, prolongs repolarisation, albeit less markedly than class III drugs (see below).
Class II drugs
Class II drugs comprise the β-adrenoceptor antagonists (e.g. metoprolol).
Adrenaline can cause dysrhythmias by its effects on the pacemaker potential and on the slow inward Ca2+ current (see above). Ventricular dysrhythmias following myocardial infarction are partly the result of increased sympathetic activity (see Fig. 21.8), providing a rationale for using β-adrenoceptor antagonists in this setting. AV conduction depends critically on sympathetic activity; β-adrenoceptor antagonists increase the refractory period of the AV node and can therefore prevent recurrent attacks of supraventricular tachycardia (SVT). The β-adrenoceptor antagonists are also used to prevent paroxysmal attacks of atrial fibrillation when these occur in the setting of sympathetic activation.
Class III drugs
The class III category was originally based on the unusual behaviour of a single drug, amiodarone (see below), although others with similar properties (e.g. sotalol) have since been described. Both amiodarone and sotalol have more than one mechanism of antidysrhythmic action. The special feature that defines them as class III drugs is that they substantially prolong the cardiac action potential. The mechanism of this effect is not fully understood, but it involves blocking some of the potassium channels involved in cardiac repolarisation, including the outward (delayed) rectifier. Action potential prolongation increases the refractory period, accounting for powerful and varied antidysrhythmic activity, for example by interrupting re-entrant tachycardias and suppressing ectopic activity. However, all drugs that prolong the cardiac action potential (detected clinically as prolonged QT interval on the ECG; see above) can paradoxically also have proarrhythmic effects, notably a polymorphic form of ventricular tachycardia called (somewhat whimsically) torsade de pointes (because the appearance of the ECG trace is said to be reminiscent of this ballet sequence). This occurs particularly in patients taking other drugs that can prolong QT, including several antipsychotic drugs; those with disturbances of electrolytes involved in repolarisation (e.g. hypokalaemia, hypercalcaemia); or individuals with hereditary prolonged QT (Ward–Romano syndrome).4 The mechanism of the dysrhythmia is not fully understood; possibilities include increased dispersion of repolarisation (i.e. lack of spatial homogeneity) and increased Ca2+ entry during the prolonged action potential, leading to increased after-depolarisation.
Class IV drugs
Class IV agents act by blocking voltage-sensitive calcium channels. Class IV drugs in therapeutic use as antidysrhythmic drugs (e.g. verapamil) act on L-type channels. Class IV drugs slow conduction in the SA and AV nodes where action potential propagation depends on slow inward Ca2+ current, slowing the heart and terminating SVT by causing partial AV block. They shorten the plateau of the action potential and reduce the force of contraction. Reduced Ca2+ entry reduces after-depolarisation and thus suppresses premature ectopic beats. In contrast, L-type calcium channel blockers that act mainly on vascular smooth muscle (e.g. nifedipine) indirectly increase sympathetic tone via their hypotensive effect and so may actually provoke tachyarrhythmias.
Details of Individual Drugs
Quinidine, procainamide and disopyramide (class Ia)
Quinidine and procainamide are pharmacologically similar. They are now mainly of historical interest. Disopyramide resembles quinidine, including in its marked atropine-like effects, which result in blurred vision, dry mouth, constipation and urinary retention. It has more negative inotropic action than quinidine but is less likely to cause hypersensitivity reactions.
Lidocaine (class Ib)
Lidocaine, also well known as a local anaesthetic (see Ch. 42), is given by intravenous infusion to treat and prevent ventricular dysrhythmias in the immediate aftermath of myocardial infarction. It is almost completely extracted from the portal circulation by hepatic first-pass metabolism (Ch. 9), and so cannot usefully be administered orally. Its plasma half-life is normally about 2 h, but its elimination is slowed if hepatic blood flow is reduced, for example by reduced cardiac output following myocardial infarction or by drugs that reduce cardiac contractility (e.g. β-adrenoceptor antagonists). Dosage must be reduced accordingly to prevent accumulation and toxicity. Indeed, its clearance has been used to estimate hepatic blood flow, analogous to the use of para-aminohippurate clearance to measure renal blood flow.
The adverse effects of lidocaine are mainly due to its actions on the central nervous system and include drowsiness, disorientation and convulsions. Because of its relatively short half-life, the plasma concentration can be adjusted fairly rapidly by varying the infusion rate.
Phenytoin (an antiepileptic drug, see Ch. 44), acts similarly, but is no longer used in treating dysrhythmias.
Flecainide and encainide (class Ic)
Flecainide and encainide suppress ventricular ectopic beats. They are long acting and reduce the frequency of ventricular ectopic beats when administered orally. However, in clinical trials, they increase the incidence of sudden death associated with ventricular fibrillation after myocardial infarction, so they are no longer used in this setting. This counterintuitive result had a profound impact on the way clinicians and drug regulators view the use of seemingly reasonable intermediate end points (in this case, reduction of frequency of ventricular ectopic beats) as evidence of efficacy in clinical trials. Currently, the main use of flecainide is in prophylaxis against paroxysmal atrial fibrillation.
β-Adrenoceptor antagonists (class II)
The most important β-adrenoceptor antagonists are described in Chapter 14. Their clinical use for rhythm disorders is shown in the clinical box. Propranolol, like several other drugs of this type, has some class I action in addition to blocking β-adrenoceptors. This may contribute to its antidysrhythmic effects, although probably not very much, because an isomer with little β antagonist activity has little antidysrhythmic activity, despite similar activity as a class I agent.
Adverse effects are described in Chapter 14, the most important being worsening bronchospasm in patients with asthma, a negative inotropic effect, bradycardia and fatigue. It was hoped that the use of β1-selective drugs (e.g. metoprolol, atenolol) would reduce the risk of bronchospasm, but their selectivity is insufficient to achieve this goal in clinical practice, although the once-a-day convenience of several such drugs has led to their widespread use in patients without lung disease.
Class III
Amiodarone is highly effective at suppressing dysrhythmias (see the clinical box). Like other drugs that interfere with cardiac repolarisation, it is important to monitor plasma electrolyte concentrations (especially of K+) during its use to avoid precipitating torsades de pointes. Unfortunately it has several peculiarities that complicate its use. It is extensively bound in tissues, has a long elimination half-life (10–100 days) and accumulates in the body during repeated dosing. For this reason, a loading dose is used, and for life-threatening dysrhythmias this is given intravenously via a central vein (it causes phlebitis if given into a peripheral vessel). Adverse effects are numerous and important; they include photosensitive skin rashes and a slate-grey/bluish discoloration of the skin; thyroid abnormalities (hypo- and hyper-, connected with its high iodine content); pulmonary fibrosis, which is late in onset but may be irreversible; corneal deposits; and neurological and gastrointestinal disturbances, including hepatitis. Dronedarone is a related benzofuran with somewhat different effects on individual ion channels. It lacks iodine and was designed to be less lipophilic than amiodarone in hopes of reducing thyroid and pulmonary toxicities. Its elimination t1/2 is shorter than that of amiodarone and while it increased mortality in patients with severe heart failure (Køber et al., 2008), it improved survival in high-risk patients with atrial fibrillation (Hohnloser et al., 2009) and has recently been approved by the Food and Drug Administration for this indication.
Sotalol is a non-selective β-adrenoceptor antagonist, this activity residing in the L isomer. Unlike other β antagonists, it prolongs the cardiac action potential and the QT interval by delaying the slow outward K+ current. This class III activity is present in both L and D isomers. Racemic sotalol (the form prescribed) appears to be somewhat less effective than amiodarone in preventing chronic life-threatening ventricular tachyarrhythmias. It shares the ability of amiodarone to cause torsades de pointes but lacks its other adverse effects; it is valuable in patients in whom β-adrenoceptor antagonists are not contraindicated. As with amiodarone, close monitoring of plasma K+ is important during its use because of the risk of proarryhthmia.
Verapamil and diltiazem (class IV)
Verapamil is given by mouth. (Intravenous preparations are available but are dangerous and almost never needed.) It has a plasma half-life of 6–8 h and is subject to quite extensive first-pass metabolism, which is more marked for the isomer that is responsible for its cardiac effects. A slow-release preparation is available for once-daily use, but it is less effective when used for prevention of dysrhythmia than the regular preparation because the bioavailability of the cardioactive isomer is reduced through the presentation of a steady low concentration to the drug-metabolising enzymes in the liver. If verapamil is added to digoxin in patients with poorly controlled atrial fibrillation, the dose of digoxin should be reduced and plasma digoxin concentration checked after a few days, because verapamil both displaces digoxin from tissue-binding sites and reduces its renal elimination, hence predisposing to digoxin accumulation and toxicity (see Ch. 56).
Verapamil is contraindicated in patients with Wolff–Parkinson–White syndrome (a pre-excitation syndrome caused by a rapidly conducting pathway between atria and ventricles anatomically distinct from the physiological conducting pathway that predisposes to re-entrant tachycardia), and is ineffective and dangerous in ventricular dysrhythmias. Adverse effects of verapamil and diltiazem are described below in the section on calcium channel antagonists.
Diltiazem is similar to verapamil but has relatively more effect on smooth muscle while producing less bradycardia (said to be ‘rate neutral’).
Adenosine (unclassified in the Vaughan Williams classification)
Adenosine is produced endogenously and is an important chemical mediator (Ch. 16) with effects on breathing, cardiac and smooth muscle, vagal afferent nerves and on platelets, in addition to the effects on cardiac conducting tissue that underlie its therapeutic use. The A1 receptor is responsible for its effect on the AV node. These receptors are linked to the same cardiac potassium channel (KACh) that is activated by acetylcholine, and adenosine hyperpolarises cardiac conducting tissue and slows the rate of rise of the pacemaker potential accordingly. It is administered intravenously to terminate SVT if this rhythm persists despite manoeuvres such as carotid artery massage designed to increase vagal tone. It has largely replaced verapamil for this purpose, because it is safer owing to its effect being short lived. This is a consequence of its pharmacokinetics: it is taken up via a specific nucleoside transporter by red blood cells and is metabolised by enzymes on the lumenal surface of vascular endothelium. Consequently, the effects of a bolus dose of adenosine last only 20–30 s. Once SVT has terminated, the patient usually remains in sinus rhythm, even though adenosine is no longer present in plasma. Its short-lived unwanted effects include chest pain, shortness of breath, dizziness and nausea. Theophylline and other xanthine alkaloids block adenosine receptors and inhibit the actions of intravenous adenosine, whereas dipyridamole (a vasodilator and antiplatelet drug; see below and Ch. 24) blocks the nucleoside uptake mechanism, potentiating adenosine and prolonging its adverse effects. Both these interactions are clinically important.
Drugs that Increase Myocardial Contraction
Cardiac Glycosides
Cardiac glycosides come from foxgloves (Digitalis spp.) and related plants. Withering (1775) wrote on the use of the foxglove: ‘it has a power over the motion of the heart to a degree yet unobserved in any other medicine …’ Foxgloves contain several cardiac glycosides with similar actions. Their basic chemical structure consists of three components: a sugar moiety, a steroid and a lactone ring, The lactone is essential for activity, the other parts of the molecule mainly determining potency and pharmacokinetic properties. Therapeutically the most important cardiac glycoside is digoxin.
Endogenous cardiotonic steroids (CTSs), also called digitalis-like factors, have been mooted for nearly half a century. There is evidence in mammals of an endogenous digitalis-like factor closely similar to ouabain, a short-acting cardiac glycoside (see Schoner & Scheiner-Bobis, 2007). CTSs were first considered important in the regulation of renal sodium transport and arterial pressure, but they have now been implicated in the regulation of cell growth, differentiation, apoptosis, fibrosis, the modulation of immunity and of carbohydrate metabolism, and the control of various central nervous functions (Bagrov et al., 2009).
Actions and adverse effects
The main actions of glycosides are on the heart, but some of their adverse effects are extracardiac, including nausea, vomiting, diarrhoea and confusion. The cardiac effects are:
Adverse effects are common and can be severe. One of the main drawbacks of glycosides in clinical use is the narrow margin between effectiveness and toxicity.
Mechanism
The mechanism whereby cardiac glycosides increase the force of cardiac contraction (positive inotropic effect) is inhibition of the Na+/K+ pump in the cardiac myocytes. Cardiac glycosides bind to a site on the extracellular aspect of the α subunit of the Na+-K+-ATPase (which is an αβ heterodimer), and are useful experimental tools for studying this important transport system. The molecular mechanism underlying increased vagal tone (negative chronotropic effect) is unknown, but could also be due to inhibition of the Na+/K+ pump.
Rate and rhythm
Cardiac glycosides slow AV conduction by increasing vagal outflow. Their beneficial effect in established rapid atrial fibrillation results partly from this. If ventricular rate is excessively rapid, the time available for diastolic filling is inadequate. Increasing the refractory period of the AV node reduces ventricular rate. The atrial dysrhythmia persists, but the pumping efficiency of the heart improves owing to improved ventricular filling. SVT can be terminated by cardiac glycosides, which slow AV conduction, although other drugs are usually employed for this indication (see below).
Larger doses of glycosides disturb sinus rhythm. This can occur at plasma concentrations of digoxin within, or only slightly above, the therapeutic range. Slowing of AV conduction can progress to AV block. Glycosides can also cause ectopic beats. Because Na+/K+ exchange is electrogenic, inhibition of the pump by glycosides causes depolarisation, predisposing to disturbances of cardiac rhythm. Furthermore, the increased [Ca2+]i causes increased after-depolarisation, leading first to coupled beats (bigeminy), in which a normal ventricular beat is followed by an ectopic beat; this is followed by ventricular tachycardia and eventually by ventricular fibrillation.
Force of contraction
Glycosides cause a large increase in twitch tension in isolated preparations of cardiac muscle. Unlike catecholamines, they do not accelerate relaxation (compare Fig. 21.6 with Fig. 21.10). Increased tension is caused by an increased [Ca2+]i transient (Fig. 21.10). The action potential is only slightly affected and the slow inward current little changed, so the increased [Ca2+]i transient probably reflects a greater release of Ca2+ from intracellular stores. The most likely mechanism is as follows (see also Ch. 4):
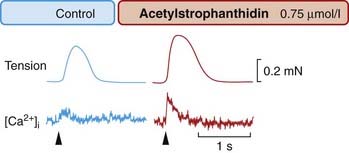
Fig. 21.10 Effect of a cardiac glycoside (acetylstrophanthidin) on the Ca2+ transient and tension produced by frog cardiac muscle.
The effect was recorded as in Figure 21.6
(From Allen D G, Blinks J R 1978 Nature 273: 509.)
The effect of extracellular potassium
Effects of cardiac glycosides are increased if plasma [K+] decreases, because of reduced competition at the K+-binding site on the Na+-K+-ATPase. This is clinically important, because diuretics (Ch. 28) are often used to treat heart failure, and most such drugs decrease plasma [K+], thereby increasing the risk of glycoside-induced dysrhythmia.
Pharmacokinetic aspects
Digoxin is administered by mouth or, in urgent situations, intravenously. It is a polar molecule; elimination is mainly by renal excretion and involves P-glycoprotein (Ch. 8), leading to clinically significant interactions with other drugs used to treat heart failure, such as spironolactone, and with antidysrhythmic drugs such as verapamil and amiodarone. Elimination half-time is approximately 36 h in patients with normal renal function, but considerably longer in elderly patients and those with overt renal failure, in whom reduced doses are indicated. A loading dose is used in urgent situations. The therapeutic range of plasma concentrations, below which digoxin is unlikely to be effective and above which the risk of toxicity increases substantially, is fairly well defined (1–2.6 nmol/l). Determination of plasma digoxin concentration is useful when lack of efficacy or toxicity is suspected.
Clinical uses of cardiac glycosides (e.g. digoxin) 
Other Drugs that Increase Myocardial Contraction
Certain β1-adrenoceptor agonists, for example dobutamine, are used to treat acute but potentially reversible heart failure (e.g. following cardiac surgery or in some cases of cardiogenic or septic shock) on the basis of their positive inotropic action. Dobutamine, for reasons that are not well understood, produces less tachycardia than other β1 agonists. It is administered intravenously. Glucagon also increases myocardial contractility by increasing synthesis of cAMP, and has been used in patients with acute cardiac dysfunction owing to overdosage of β-adrenoceptor antagonists.
Inhibitors of the heart-specific subtype (type III) of phosphodiesterase, the enzyme responsible for the intracellular degradation of cAMP, increase myocardial contractility. Consequently, like β-adrenoceptor agonists, they increase intracellular cAMP but cause dysrhythmias for the same reason. Compounds in this group include amrinone and milrinone. They improve haemodynamic indices in patients with heart failure but paradoxically worsen survival, presumably because of dysrhythmias. This dichotomy has had a sobering effect on clinicians and drug regulatory authorities.
Antianginal Drugs
The mechanism of anginal pain is discussed above. Angina is managed by using drugs that improve perfusion of the myocardium or reduce its metabolic demand, or both. Two of the main groups of drugs, organic nitrates and calcium antagonists, are vasodilators and produce both these effects. The third group, β-adrenoceptor antagonists, slow the heart and hence reduce metabolic demand. Organic nitrates and calcium antagonists are described below. The β-adrenoceptor antagonists are covered in Ch. 14, and their antidysrhythmic actions are described above. Ivabradine slows the heart by inhibiting the sinus node If current (see above), and is an alternative to β-adrenoceptor antagonists in patients in whom these are not tolerated or are contraindicated.
Organic Nitrates
The ability of organic nitrates (see also Chs 20 and 22) to relieve angina was discovered by Lauder Brunton, a distinguished British physician, in 1867. He had found that angina could be partly relieved by bleeding, and also knew that amyl nitrite, which had been synthesised 10 years earlier, caused flushing and tachycardia, with a fall in blood pressure, when its vapour was inhaled. He thought that the effect of bleeding resulted from hypotension, and found that amyl nitrite inhalation worked much better. Amyl nitrite has now been replaced by glyceryl trinitrate (GTN).5 Several related organic nitrates, of which the most important is isosorbide mononitrate, have a prolonged action.
Actions
Organic nitrates relax smooth muscle (especially vascular smooth muscle, but also other types including oesophageal and biliary smooth muscle). They relax veins, with a consequent reduction in central venous pressure (reduced preload). In healthy subjects, this reduces stroke volume; venous pooling occurs on standing and can cause postural hypotension and dizziness. Therapeutic doses have less effect on small resistance arteries than on veins, but there is a marked effect on larger muscular arteries. This reduces pulse wave reflection from arterial branches (as appreciated in the 19th century by Murrell but neglected for many years thereafter), and consequently reduces central (aortic) pressure and cardiac afterload (see Ch. 22 for the role of these factors in determining cardiac work). The direct dilator effect on coronary arteries opposes coronary artery spasm in variant angina. With larger doses, resistance arteries and arterioles dilate, and arterial pressure falls. Nevertheless, coronary flow is increased as a result of coronary vasodilatation. Myocardial oxygen consumption is reduced because of the reductions in both cardiac preload and afterload. This, together with the increased coronary blood flow, causes a large increase in the oxygen content of coronary sinus blood. Studies in experimental animals have shown that glyceryl trinitrate diverts blood from normal to ischaemic areas of myocardium. The mechanism involves dilatation of collateral vessels that bypass narrowed coronary artery segments (Fig. 21.11).
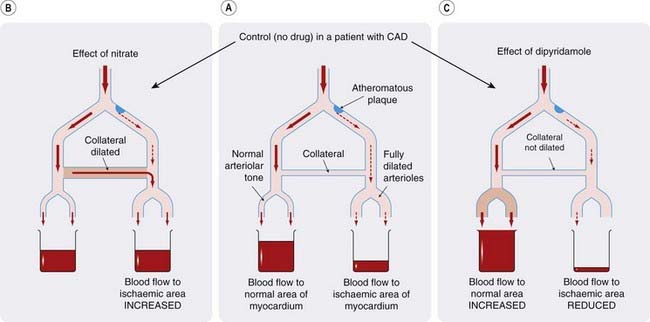
Fig. 21.11 Comparison of the effects of organic nitrates and an arteriolar vasodilator (dipyridamole) on the coronary circulation.
[A] Control. [B] Nitrates dilate the collateral vessel, thus allowing more blood through to the underperfused region (mostly by diversion from the adequately perfused area). [C] Dipyridamole dilates arterioles, increasing flow through the normal area at the expense of the ischaemic area (in which the arterioles are anyway fully dilated). CAD, coronary artery disease.
It is interesting to compare this effect with that of other vasodilators, notably dipyridamole, which dilate arterioles but not collaterals. Dipyridamole is at least as effective as nitrates in increasing coronary flow in normal subjects but actually worsens angina. This is probably because arterioles in an ischaemic region are fully dilated by the ischaemia, and drug-induced dilatation of the arterioles in normal areas has the effect of diverting blood away from the ischaemic areas (Fig. 21.11), producing what is termed a vascular steal. This effect is exploited in a pharmacological ‘stress’ test for coronary arterial disease, in which dipyridamole is administered intravenously to patients in whom this diagnosis is suspected but who cannot exercise, while monitoring myocardial perfusion and the ECG.
In summary, the antianginal action of nitrates involves:
In addition to its effects on smooth muscle, nitric oxide increases the rate of relaxation of cardiac muscle (dubbed a ‘lusiotropic’ action). It is probable that organic nitrates mimic this action, which could be important in patients with impaired diastolic function, a common accompaniment of hypertension and of heart failure.
Mechanism of action
Organic nitrates are metabolised with release of nitric oxide. At concentrations achieved during therapeutic use, this involves an enzymic step and possibly a reaction with tissue sulfhydryl (–SH) groups. Nitric oxide activates soluble guanylyl cyclase (see Ch. 20), increasing formation of cGMP, which activates protein kinase G (Ch. 4) and leads to a cascade of effects in smooth muscle culminating in dephosphorylation of myosin light chains, sequestration of intracellular Ca2+ and consequent relaxation.
Tolerance and unwanted effects
Repeated administration of nitrates to smooth muscle preparations in vitro results in diminished relaxation, possibly partly because of depletion of free –SH groups, although attempts to prevent tolerance by agents that restore tissue –SH groups have not been clinically useful. Tolerance to the antianginal effect of nitrates does not occur to a clinically important extent with ordinary formulations of short-acting drugs (e.g. glyceryl trinitrate), but does occur with longer acting drugs (e.g. isosorbide mononitrate) or when glyceryl trinitrate is administered by prolonged intravenous infusion or by frequent application of slow-release transdermal patches (see below).
The main adverse effects of nitrates are a direct consequence of their main pharmacological actions, and include postural hypotension and headache. This was the cause of ‘Monday morning sickness’ among workers in explosives factories. Tolerance to these effects develops quite quickly but wears off after a brief nitrate-free interval (which is why the symptoms appeared on Mondays and not later in the week). Formation of methaemoglobin, an oxidation product of haemoglobin that is ineffective as an oxygen carrier, seldom occurs when nitrates are used clinically but is induced deliberately with amyl nitrite in the treatment of cyanide poisoning, because methaemoglobin binds and inactivates cyanide ions.
Pharmacokinetic and pharmaceutical aspects
Glyceryl trinitrate is rapidly inactivated by hepatic metabolism. It is well absorbed from the mouth and is taken as a tablet under the tongue or as a sublingual spray, producing its effects within a few minutes. If swallowed, it is ineffective because of first-pass metabolism. Given sublingually, the trinitrate is converted to di- and mononitrates. Its effective duration of action is approximately 30 min. It is appreciably absorbed through the skin, and a more sustained effect can be achieved by applying it as a transdermal patch. Once a bottle of the tablets has been opened, its shelf-life is quite short because the volatile active substance evaporates; spray preparations avoid this problem.
Isosorbide mononitrate is longer acting than glyceryl trinitrate because it is absorbed and metabolised more slowly but has similar pharmacological actions. It is swallowed rather than taken sublingually, and is taken twice a day for prophylaxis (usually in the morning and at lunch, to allow a nitrate-free period during the night, when patients are not exerting themselves, to avoid tolerance). It is also available in slow-release form for once-daily use in the morning.
Organic nitrates
Clinical uses of organic nitrates
Potassium Channel Activators
Nicorandil combines activation of the potassium KATP channel (see Ch. 4) with nitrovasodilator (nitric oxide donor) actions. It is both an arterial and a venous dilator, and causes the expected unwanted effects of headache, flushing and dizziness. It is used for patients who remain symptomatic despite optimal management with other drugs, often while they await surgery or angioplasty.
β-Adrenoceptor Antagonists
β-Adrenoceptor antagonists (see Ch. 14) are important in prophylaxis of angina, and in treating patients with unstable angina. They work for these indications by reducing cardiac oxygen consumption. In addition, they reduce the risk of death following myocardial infarction, probably via their antidysrhythmic action. Any effects on coronary vessel diameter are of minor importance, although these drugs are avoided in variant angina because of the theoretical risk that they will increase coronary spasm. Their very diverse clinical uses are summarised in the clinical box.
Calcium Antagonists
The term ‘calcium antagonist’ is used for drugs that block cellular entry of Ca2+ through calcium channels rather than its intracellular actions (Ch. 4). Some authors use the term ‘Ca2+ entry blockers’ to make this distinction clearer. Therapeutically important calcium antagonists act on L-type channels. L-type calcium antagonists comprise three chemically distinct classes: phenylalkylamines (e.g. verapamil), dihydropyridines (e.g. nifedipine, amlodipine) and benzothiazepines (e.g. diltiazem).
Mechanism of action: types of calcium channel
The properties of voltage-gated calcium channels have been studied by voltage clamp and patch clamp techniques (see Ch. 3). Drugs of each of the three chemical classes mentioned above all bind the α1 subunit of the L-type calcium channel but at distinct sites. These interact allosterically with each other and with the gating machinery of the channel to prevent its opening (see below), thus reducing Ca2+ entry. Many calcium antagonists show properties of use dependence (i.e. they block more effectively in cells in which the calcium channels are most active; see the discussion of class I antidysrhythmic drugs above). For the same reason, they also show voltage-dependent blocking actions, blocking more strongly when the membrane is depolarised, causing calcium channel opening and inactivation.
Dihydropyridines affect calcium channel function in a complex way, not simply by physical plugging of the pore. This became clear when some dihydropyridines, exemplified by BAY K 8644, were found to bind to the same site but to do the opposite; that is, to promote the opening of voltage-gated calcium channels. Thus BAY K 8644 increases the force of cardiac contraction, and constricts blood vessels; it is competitively antagonised by nifedipine. Calcium channels can exist in one of three distinct states, termed ‘modes’ (Fig. 21.12). When a channel is in mode 0, it does not open in response to depolarisation; in mode 1, depolarisation produces a low opening probability, and each opening is brief. In mode 2, depolarisation produces a very high opening probability, and single openings are prolonged. Under normal conditions, about 70% of the channels at any one moment exist in mode 1, with only 1% or less in mode 2; each channel switches randomly and quite slowly between the three modes. Dihydropyridine antagonists bind selectively to channels in mode 0, thus favouring this non-opening state, whereas agonists bind selectively to channels in mode 2 (Fig. 21.12). This type of two-directional modulation resembles the phenomenon seen with the GABA/benzodiazepine interaction (Ch. 43), and invites speculation about possible endogenous dihydropyridine-like mediator(s) with a regulatory effect on Ca2+ entry.
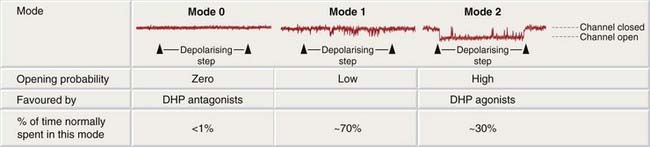
Fig. 21.12 Mode behaviour of calcium channels.
The traces are patch clamp recordings (see Ch. 3) of the opening of single calcium channels (downward deflections) in a patch of membrane from a cardiac muscle cell. A depolarising step is imposed close to the start of each trace, causing an increase in the opening probability of the channel. When the channel is in mode 1 (centre), this causes a few brief openings to occur; in mode 2 (right), the channel stays open for most of the time during the depolarising step; in mode 0 (left), it fails to open at all. Under normal conditions, the channel spends most of its time in modes 1 and 0, and only rarely enters mode 2. DHP, dihydropyridine.
(Redrawn from Hess et al. 1984 Nature 311: 538–544.)
Mibefradil blocks T- as well as L-type channels at therapeutic concentrations, but was withdrawn from therapeutic use because it caused adverse drug interactions by interfering with drug metabolism. Ethosuximide (a carbonic anhydrase inhibitor used to treat absence seizures, Ch. 44) also blocks T channels in thalamic and reticular neurones.
Pharmacological effects
The main effects of calcium antagonists, as used therapeutically, are on cardiac and smooth muscle. Verapamil preferentially affects the heart, whereas most of the dihydropyridines (e.g. nifedipine) exert a greater effect on smooth muscle than on the heart. Diltiazem is intermediate in its actions.
Cardiac actions
The antidysrhythmic effects of verapamil and diltiazem have been discussed above. Calcium antagonists can cause AV block and cardiac slowing by their actions on conducting tissues, but this is offset by a reflex increase in sympathetic activity secondary to their vasodilator action. For example, nifedipine typically causes reflex tachycardia; diltiazem causes little or no change in heart rate and verapamil slows the heart rate. Calcium antagonists also have a negative inotropic effect, from their inhibition of Ca2+ entry during the action potential plateau. Verapamil has the most marked negative inotropic action, and is contraindicated in heart failure, whereas amlodipine does not worsen cardiovascular mortality in patients with severe but stable chronic heart failure.
Vascular smooth muscle
Calcium antagonists cause generalised arterial/arteriolar dilatation, thereby reducing blood pressure, but do not much affect the veins. They affect all vascular beds, although regional effects vary considerably between different drugs. They cause coronary vasodilatation and are used in patients with coronary artery spasm (variant angina). Other types of smooth muscle (e.g. biliary tract, urinary tract and uterus) are also relaxed by calcium antagonists, but these effects are less important therapeutically than their actions on vascular smooth muscle, although they do cause adverse effects (see below).
Protection of ischaemic tissues
There are theoretical reasons (see Fig. 21.8) why calcium antagonists might exert a cytoprotective effect in ischaemic tissues and thus be of use in treating heart attack and stroke (see Ch. 39). However, randomised clinical trials have been disappointing, with little or no evidence of beneficial (or harmful) effects of calcium antagonists on cardiovascular morbidity or mortality in patient groups other than patients with hypertension, in whom calcium antagonists have beneficial effects comparable with those of other drugs that lower blood pressure to similar extents (see Ch. 22). Nimodipine is partly selective for cerebral vasculature and is sometimes used to reduce cerebral vasospasm following subarachnoid haemorrhage.
Pharmacokinetics
Calcium antagonists in clinical use are all well absorbed from the gastrointestinal tract, and are given by mouth except for some special indications, such as following subarachnoid haemorrhage, for which intravenous preparations are available. They are extensively metabolised. Pharmacokinetic differences between different drugs and different pharmaceutical preparations are clinically important, because they determine the dose interval and also the intensity of some of the unwanted effects, such as headache and flushing (see below). Amlodipine has a long elimination half-life and is given once daily, whereas nifedipine, diltiazem and verapamil have shorter elimination half-lives and are either given more frequently or are formulated in various slow-release preparations to permit once-daily dosing.
Unwanted effects
Most of the unwanted effects of calcium antagonists are extensions of their main pharmacological actions. Short-acting dihydropyridines cause flushing and headache because of their vasodilator action, and in chronic use, dihydropyridines often cause ankle swelling related to arteriolar dilatation and increased permeability of postcapillary venules. Verapamil can cause constipation, probably because of effects on calcium channels in gastrointestinal nerves or smooth muscle. Effects on cardiac rhythm (e.g. heart block) and force of contraction (e.g. worsening heart failure) are discussed above.
Apart from these predictable effects, calcium channel antagonists, as a class, appear rather free from idiosyncratic adverse effects.
Calcium antagonists
Clinical uses of calcium antagonists
References and Further Reading
Opie L.H., Gersh B.J. Drugs for the heart, seventh ed. Philadelphia: Saunders/ Elsevier; 2009.
Vaughan Williams E.M. Classification of antiarrhythmic actions. In: Vaughan Williams E.M., editor. Antiarrhythmic drugs. Handbook of experimental pharmacology, vol. 89. Berlin: Springer-Verlag; 1989. (For a different approach, see Circulation 1994, 84, 1848)
Physiological and pathophysiological aspects
Bagrov A.Y., Shapiro J.I., Fedorova O.V. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev.. 2009;61:9-38. (Reviews physiological interactions between CTS and other regulatory systems that may be important in the pathophysiology of essential hypertension, pre-eclampsia, end-stage renal disease, congestive heart failure and diabetes)
Gross G.J., Auchampach J.A. Reperfusion injury: does it exist? J. Mol. Cell Cardiol.. 2007;42:12-18. (Mounting evidence supports the concept of reperfusion injury, based on work conducted with adenosine and opioid receptor ligands, and the discovery of ‘postconditioning’ [POC] and of the reperfusion injury salvage kinase [RISK] signalling pathway)
Potter L.R., Yoder A.R., Flora D.R., et al. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb. Exp. Pharmacol.. 2009;191:341-366. (Reviews the history, structure, function and clinical applications of natriuretic peptides and their receptors)
Richards A.M. Therapeutic potential of infused cardiac natriuretic peptides in myocardial infarction. Heart. 2009;95:1299-1300. (Recent editorial on controversial issue)
Rockman H.A., Koch W.J., Lefkowitz R.J. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206-212.
Schoner W., Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am. J .Physiol. Cell Physiol.. 2007;293:C509-C536. (Review: touches on anticancer potential also)
Seddon M., Melikian N., Dworakowski R., et al. Effects of neuronal nitric oxide synthase on human coronary artery diameter and blood flow in vivo. Circulation. 2009;119:2656-2662. (Local nNOS-derived NO regulates basal blood flow in the human coronary vascular bed, whereas substance P-stimulated vasodilatation is eNOS mediated)
Welsh M.J., Hoshi T. Molecular cardiology—ion channels lose the rhythm. Nature. 1995;376:640-641. (Commentary on Ward–Romano syndrome)
COMMIT Collaborative Group. Early intravenous then oral metoprolol in 45 852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1622-1632. (Early β blockade reduced ventricular fibrillation and reinfarction, benefits that were offset by increased cardiogenic shock in patients with signs of heart failure; see accompanying comment by Sabatine, M.S. pp. 1587–1589 in the same issue)
Fox K., Ford I., Steg P.G., et al. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:807-816. (Ivabradine does not improve cardiac outcomes in all patients with stable coronary artery disease and left-ventricular systolic dysfunction, but improved outcome in patients with heart rates > 70 bpm. See also adjacent paper: Fox, K., et al., 2008. Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet 372, 817–821)
Hohnloser S.H., Crijns H.J., van Eickels M., et al. Effect of dronedarone on cardiovascular events in atrial fibrillation. N. Engl. J. Med.. 2009;360:668-678. (4628 patients with atrial fibrillation who had additional risk factors for death; those randomised to dronedarone had fewer hospitalisations due to cardiovascular events and longer survival)
ISIS-4 Collaborative Group. ISIS-4: a randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58 050 patients with suspected acute myocardial infarction. Lancet. 1995;345:669-685. (Impressive trial: disappointing results! Magnesium was ineffective; oral nitrate did not reduce 1-month mortality)
Køber L., Torp-Pedersen C., McMurray J.J.V., et al. Increased mortality after dronedarone therapy for severe heart failure. N. Engl. J. Med.. 2008;358:2678-2687.
Kochegarov A.A. Pharmacological modulators of voltage-gated calcium channels and their therapeutical application. Cell Calcium. 2003;33:145-162.
Lee L., Horowitz J., Frenneaux M. Metabolic manipulation in ischaemic heart disease, a novel approach to treatment. Eur. Heart. J.. 2004;25:634-641. (Reviews four metabolic antianginal drugs: perhexiline, trimetazidine, ranolazine and etomoxir)
Marzilli M. Cardioprotective effects of trimetazidine: a review. Curr. Med. Res. Opin.. 2003;19:661-672. (Reviews metabolic effects of this 3-ketoacyl-CoA thiolase inhibitor and its lack of haemodynamic effects in stable angina pectoris)
Rahimtoola S.H. Digitalis therapy for patients in clinical heart failure. Circulation. 2004;109:2942-2946. (Review)
Roden D.M. Drug therapy: drug-induced prolongation of the QT interval N. Engl. J. Med. 350:2004:1013-1022 (Adverse effect of great concern in drug development; see also Ch. 57)
Ruskin J.N. The cardiac arrhythmia suppression trial (CAST). N. Engl. J. Med.. 1989;321:386-388. (Enormously influential trial showing increased mortality with active treatment despite suppression of dysrhythmia)
1‘f’ for ‘funny’, because it is unusual for cation channels to be activated by hyperpolarisation; electrophysiologists have a peculiar sense of humour!
2The Creator has, however, thoughtfully provided coronary endothelium with muscarinic receptors linked to nitric oxide synthesis (see Ch. 20), presumably for the delectation of vascular pharmacologists.
3The nomenclature of natriuretic peptides and their receptors is peculiarly obtuse. The peptides are named ‘A’ for atrial, ‘B’ for brain—despite being present mainly in cardiac ventricle—and ‘C’ for A, B, C …; NPRs are named NPR-A, which preferentially binds ANP; NPR-B, which binds C natriuretic peptide preferentially; and NPR-C for ‘clearance’ receptor, because until recently clearance via cellular uptake and degradation by lysosomal enzymes was the only definite known function of this binding site.
4A 3-year-old girl began to have blackouts, which decreased in frequency with age. Her ECG showed a prolonged QT interval. When 18 years of age, she lost consciousness running for a bus. When she was 19, she became quite emotional as a participant in a live television audience and died suddenly. The molecular basis of this rare inherited disorder is now known. It is caused by a mutation in either the gene coding for a particular potassium channel—called HERG—or another gene, SCN5A, which codes for the sodium channel and disruption of which results in a loss of inactivation of the Na+ current (see Welsh & Hoshi, 1995, for a commentary).
5Nobel discovered how to stabilise GTN with kieselguhr, enabling him to exploit its explosive properties in dynamite, the manufacture of which earned him the fortune with which he endowed the eponymous prizes.