24 Haemostasis and thrombosis
Overview
This chapter summarises the main features of blood coagulation, platelet function and fibrinolysis. These processes underlie haemostasis and thrombosis, and provide a basis for understanding haemorrhagic disorders (e.g. haemophilia) and thrombotic diseases both of arteries (e.g. thrombotic stroke, myocardial infarction) and of veins (e.g. deep vein thrombosis). Drugs that act on the coagulation cascade, on platelets and on fibrinolysis are considered. Anticoagulants, antiplatelet drugs and fibrinolytic drugs are especially important clinically because of the prevalence of thrombotic disease, and are emphasised for this reason.
Introduction
Haemostasis is the arrest of blood loss from damaged blood vessels and is essential to life. A wound causes vasoconstriction, accompanied by:
Platelet activation leads to the formation of a haemostatic plug, which stops the bleeding and is subsequently reinforced by fibrin. The relative importance of each process depends on the type of vessel (arterial, venous or capillary) that has been injured.
Thrombosis is the pathological formation of a ‘haemostatic’ plug within the vasculature in the absence of bleeding (‘haemostasis in the wrong place’). Over a century ago, Rudolph Virchow defined three predisposing factors—‘Virchow’s triad’: injury to the vessel wall—for example, when an atheromatous plaque ruptures or becomes eroded; altered blood flow—for example, in the left atrial appendage of the heart during atrial fibrillation, or in the veins of the legs while sitting awkwardly on a long journey; and abnormal coagulability of the blood—as occurs, for example, in the later stages of pregnancy or during treatment with certain oral contraceptives (see Ch. 34). Increased coagulability of the blood can be inherited and is referred to as thrombophilia. A thrombus, which forms in vivo, should be distinguished from a clot, which forms in static blood in vitro. Clots are amorphous, consisting of a diffuse fibrin meshwork in which red and white blood cells are trapped indiscriminately. By contrast, arterial and venous thrombi each have a distinct structure.
An arterial thrombus (see Fig. 24.1) is composed of so-called white thrombus consisting mainly of platelets in a fibrin mesh. It is usually associated with atherosclerosis and can interrupt blood flow, causing ischaemia or death (infarction) of tissue downstream. Venous thrombus is composed of ‘red thrombus’ and consists of a small white head and a large jelly-like red tail, similar in composition to a blood clot, which streams away in the flow. Thrombus can break away from its attachment and float through the circulation, forming an embolus; venous emboli usually lodge in the lungs, while thrombus that embolises from the left heart or a carotid artery usually lodges in an artery in the brain or other organs, causing death, stroke or other disaster.
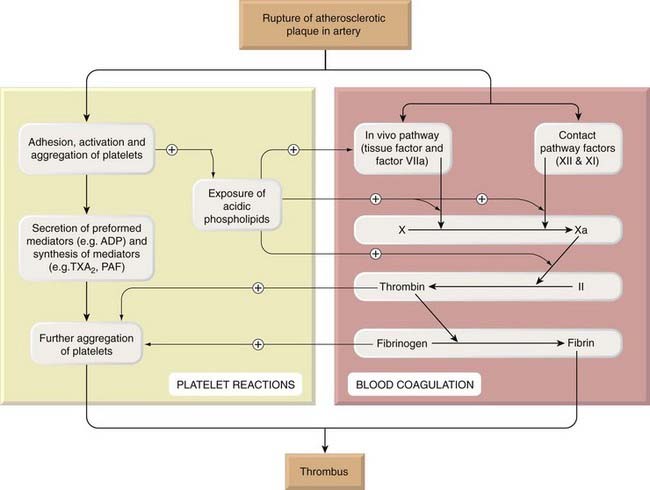
Fig. 24.1 The main events in the formation of an arterial thrombus.
Exposure of acidic phospholipids during platelet activation provides a surface on which factors IXa and VIIa interact with factor X; factor Xa then interacts with factor II, as illustrated in more detail in Figure 24.4. Activation of factor XII also initiates the fibrinolytic pathway, which is shown in Figure 24.10. (A similar series of events occurs when there is vascular damage, leading to haemostasis.) PAF, platelet-activating factor; TXA2, thromboxane A2.
Drug therapy to promote haemostasis (e.g. antifibrinolytic and haemostatic drugs; see below) is indicated when this essential process is defective (e.g. coagulation factors in haemophilia or following excessive anticoagulant therapy), or when it proves difficult to staunch haemorrhage following surgery or for menorrhagia. Drug therapy to treat or prevent thrombosis or thromboembolism is extensively used because such diseases are common as well as serious. Drugs affect haemostasis and thrombosis in three distinct ways, by influencing:
Blood Coagulation
Coagulation Cascade
Blood coagulation means the conversion of liquid blood to a gel or clot. The main event is the conversion by thrombin of soluble fibrinogen to insoluble strands of fibrin, the last step in a complex enzyme cascade. The components (called factors) are present in blood as inactive precursors (zymogens) of proteolytic enzymes and co-factors. They are activated by proteolysis, the active forms being designated by the suffix ‘a’. Factors XIIa, XIa, Xa, IXa and thrombin (IIa) are all serine proteases. Activation of a small amount of one factor catalyses the formation of larger amounts of the next factor, which catalyses the formation of still larger amounts of the next, and so on; consequently, the cascade provides a mechanism of amplification.1 As might be expected, this accelerating enzyme cascade has to be controlled by inhibitors, because otherwise all the blood in the body would solidify within minutes of the initiation of haemostasis. One of the most important inhibitors is antithrombin III, which neutralises all the serine proteases in the cascade. Vascular endothelium also actively limits thrombus extension (see below).
Two pathways of fibrin formation were described traditionally (termed ‘intrinsic’—because all the components are present in the blood—and ‘extrinsic’—because some components come from outside the blood). The intrinsic or ‘contact’ pathway is activated when shed blood comes into contact with an artificial surface such as glass, but physiologically the system functions as a single in vivo pathway (Fig. 24.2). Tissue damage exposes blood to tissue factor, initiating the process and leading to production of a small amount of thrombin. This acts through several positive feedbacks (on Va, VIIIa and on platelets) that amplify and propagate the process with production of more thrombin.
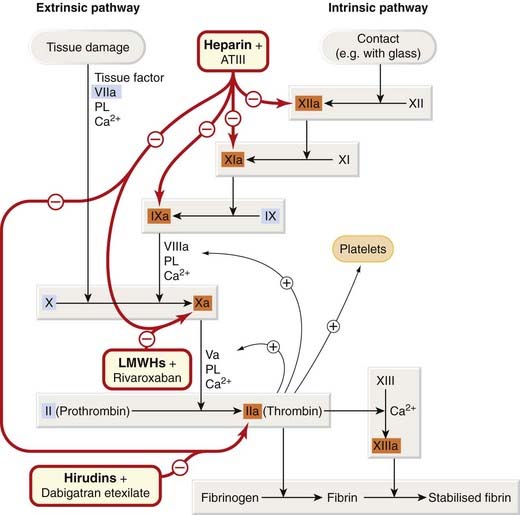
Fig. 24.2 The coagulation cascade: sites of action of anticoagulant drugs.
Oral anticoagulants interfere with post-translational γ-carboxylation of factors II, VII, IX and X (shown in blue boxes); see Figure 24.4. Heparins activate antithrombin III. ATIII, antithrombin III; LMWHs, low-molecular-weight heparins; PL, negatively charged phospholipid supplied by activated platelets.
 The in vivo pathway is initiated by ‘tissue factor’. This is the cellular receptor for factor VII, which, in the presence of Ca2+, undergoes an active site transition. This results in rapid autocatalytic activation of factor VII to VIIa. The tissue factor–VIIa complex activates factors IX and X. Acidic phospholipids function as surface catalysts. They are provided during platelet activation, which exposes acidic phospholipids (especially phosphatidylserine), and these activate various clotting factors, closely juxtaposing them in functional complexes. Platelets also contribute by secreting coagulation factors, including factor Va and fibrinogen. Coagulation is sustained by further generation of factor Xa by IXa–VIIIa–Ca2+–phospholipid complex. This is needed because the tissue factor–VIIa complex is rapidly inactivated in plasma by tissue factor pathway inhibitor and by antithrombin III. Factor Xa, in the presence of Ca2+, phospholipid and factor Va, activates prothrombin to thrombin, the main enzyme of the cascade. The contact (intrinsic) pathway commences when factor XII (Hageman factor) adheres to a negatively charged surface and converges with the in vivo pathway at the stage of factor X activation (see Fig. 24.2). The proximal part of this pathway is not crucial for blood coagulation in vivo.2 The two pathways are not entirely separate even before they converge, and various positive feedbacks promote coagulation.
The in vivo pathway is initiated by ‘tissue factor’. This is the cellular receptor for factor VII, which, in the presence of Ca2+, undergoes an active site transition. This results in rapid autocatalytic activation of factor VII to VIIa. The tissue factor–VIIa complex activates factors IX and X. Acidic phospholipids function as surface catalysts. They are provided during platelet activation, which exposes acidic phospholipids (especially phosphatidylserine), and these activate various clotting factors, closely juxtaposing them in functional complexes. Platelets also contribute by secreting coagulation factors, including factor Va and fibrinogen. Coagulation is sustained by further generation of factor Xa by IXa–VIIIa–Ca2+–phospholipid complex. This is needed because the tissue factor–VIIa complex is rapidly inactivated in plasma by tissue factor pathway inhibitor and by antithrombin III. Factor Xa, in the presence of Ca2+, phospholipid and factor Va, activates prothrombin to thrombin, the main enzyme of the cascade. The contact (intrinsic) pathway commences when factor XII (Hageman factor) adheres to a negatively charged surface and converges with the in vivo pathway at the stage of factor X activation (see Fig. 24.2). The proximal part of this pathway is not crucial for blood coagulation in vivo.2 The two pathways are not entirely separate even before they converge, and various positive feedbacks promote coagulation.
Haemostasis and thrombosis 
The Role of Thrombin
Thrombin (factor IIa) cleaves fibrinogen, producing fragments that polymerise to form fibrin. It also activates factor XIII, a fibrinoligase, which strengthens fibrin-to-fibrin links, thereby stabilising the coagulum. In addition to coagulation, thrombin also causes platelet aggregation, stimulates cell proliferation and modulates smooth muscle contraction. Paradoxically, it can inhibit as well as promote coagulation (see below). Effects of thrombin on platelets and smooth muscle are initiated by interaction with specific protease-activated receptors (PARs; see Ch. 3), which belong to the superfamily of G-protein-coupled receptors. PARs initiate cellular responses that contribute not only to haemostasis and thrombosis, but also to inflammation and perhaps angiogenesis. The signal transduction mechanism is unusual: receptor activation requires proteolysis by thrombin of the extracellular N-terminal domain of the receptor, revealing a new N-terminal sequence that acts as a ‘tethered agonist’ (see Fig. 3.7).
Vascular Endothelium in Haemostasis and Thrombosis
Vascular endothelium, the container of the circulating blood, can change focally from a non-thrombogenic to a thrombogenic structure in response to different demands. Normally, it provides a non-thrombogenic surface by virtue of surface heparan sulfate, a glycosaminoglycan related to heparin, which is, like heparin, a co-factor for antithrombin III. Endothelium thus plays an essential role in preventing intravascular platelet activation and coagulation. However, it also plays an active part in haemostasis, synthesising and storing several key haemostatic components; von Willebrand factor,3 tissue factor and plasminogen activator inhibitor (PAI)-1 are particularly important. PAI-1 is secreted in response to angiotensin IV, receptors for which are present on endothelial cells, providing a link between the renin–angiotensin system (see Ch. 22) and thrombosis. These prothrombotic factors are involved, respectively, in platelet adhesion and in coagulation and clot stabilisation. However, the endothelium is also implicated in thrombus limitation. Thus it generates prostaglandin (PG) I2 (prostacyclin; Ch. 17) and nitric oxide (NO; Ch. 20); converts the platelet agonist ADP to adenosine, which inhibits platelet function (Ch. 16); synthesises tissue plasminogen activator (tPA; see below); and expresses thrombomodulin, a receptor for thrombin. After combination with thrombomodulin, thrombin activates protein C, a vitamin K-dependent anticoagulant. Activated protein C, helped by its co-factor protein S, inactivates factors Va and VIIa. This is known to be physiologically important, because a naturally occurring mutation of the gene coding for factor V (factor V Leiden), which confers resistance to activated protein C, results in the commonest recognised form of inherited thrombophilia. A synthetic form of activated protein C, drotrecogin alpha (activated), is licensed for the treatment of severe septic shock with multiple organ failure (Ch. 22).
Endotoxin and cytokines, including tumour necrosis factor, tilt the balance of prothrombotic and antithrombotic endothelial functions towards thrombosis by causing loss of heparan (see above) and expression of tissue factor, and impair endothelial NO function. If other mechanisms limiting coagulation are also faulty or become exhausted, disseminated intravascular coagulation can result. This is a serious complication of sepsis and of certain malignancies, and the main treatment is to correct the underlying disease.
Blood coagulation (fibrin formation)
The clotting system consists of a cascade of proteolytic enzymes and cofactors.
Drugs that Act on the Coagulation Cascade
Drugs are used to modify the cascade either when there is a defect in coagulation or when there is unwanted coagulation.
Coagulation Defects
Genetically determined deficiencies of clotting factors are not common. Examples are classic haemophilia, caused by lack of factor VIII, and an even rarer form of haemophilia (haemophilia B or Christmas disease) caused by lack of factor IX (also called Christmas factor). Missing factors can be supplied by giving fresh plasma or concentrated preparations of, respectively, factor VIII or factor IX.
Acquired clotting defects are more common than hereditary ones. The causes include liver disease, vitamin K deficiency (universal in neonates) and excessive oral anticoagulant therapy, each of which may require treatment with vitamin K.
Vitamin K
Vitamin K (for Koagulation in German) is a fat-soluble vitamin (Fig. 24.3) occurring naturally in plants (vitamin K1) and as a series of bacterial menaquinones (vitamin K2) formed in the gut (see Shearer & Newman, 2008, for a review). It is essential for the formation of clotting factors II, VII, IX and X. These are all glycoproteins with several γ-carboxyglutamic acid (Gla) residues. The interaction of factors Xa and prothrombin (factor II) with Ca2+ and phospholipid is shown in Figure 24.4. γ-Carboxylation occurs after the synthesis of the amino acid chain, and the carboxylase enzyme requires reduced vitamin K as a co-factor (Fig. 24.5). Binding does not occur in the absence of γ-carboxylation. Similar considerations apply to the proteolytic activation of factor X by IXa and by VIIa (see Fig. 24.2).
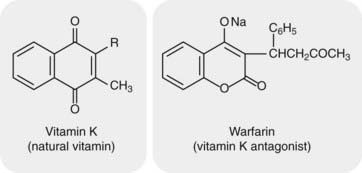
Fig. 24.3 Vitamin K and warfarin.
Warfarin, a vitamin K antagonist, is an oral anticoagulant. It competes with vitamin K (note the similarity in their structures) for the reductase enzyme (VKORC1) that activates vitamin K and is the site of its action (see Fig. 24.5).
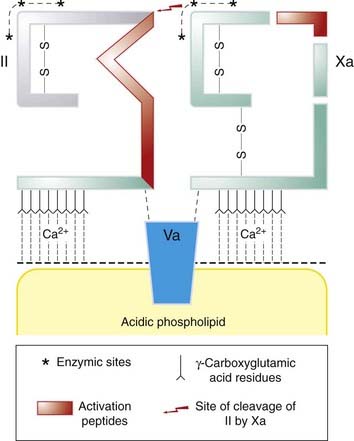
Fig. 24.4 Activation of prothrombin (factor II) by factor Xa.
The complex of factor Va with a negatively charged phospholipid surface (supplied by aggregating platelets) forms a binding site for factor Xa and prothrombin (II), which have peptide chains (shown schematically) that are similar to one another. Platelets thus serve as a localising focus. Calcium ions are essential for binding. Xa activates prothrombin, liberating thrombin (shown in grey).
(Modified from Jackson C M 1978 Br J Haematol 39: 1.)
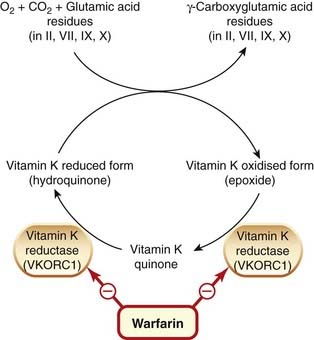
Fig. 24.5 Mechanism of vitamin K and of warfarin.
After the peptide chains in clotting factors II, VII, IX and X have been synthesised, reduced vitamin K (the hydroquinone) acts as a co-factor in the conversion of glutamic acid to γ-carboxyglutamic acid. During this reaction, the reduced form of vitamin K is converted to the epoxide, which in turn is reduced to the quinone and then the hydroquinone by vitamin K epoxide reductase component 1 (VKORC1), the site of action of warfarin.
There are several other vitamin K-dependent Gla proteins, including proteins C and S (see above) and osteocalcin in bone: the effect of the vitamin on osteoporosis is under investigation.
Administration and pharmacokinetic aspects
Natural vitamin K1 (phytomenadione) may be given orally or by injection. If given by mouth, it requires bile salts for absorption, and this occurs by a saturable energy-requiring process in the proximal small intestine. A synthetic preparation, menadiol sodium phosphate, is also available. It is water soluble and does not require bile salts for its absorption. This synthetic compound takes longer to act than phytomenadione. There is very little storage of vitamin K in the body. It is metabolised to more polar substances that are excreted in the urine and the bile.
Clinical uses of vitamin K are summarised in the clinical box.
Thrombosis
Thrombotic and thromboembolic disease is common and has severe consequences, including myocardial infarction, stroke, deep vein thrombosis and pulmonary embolus. The main drugs used for platelet-rich ‘white’ thrombi are the antiplatelet drugs and fibrinolytic drugs, which are considered below. The main drugs used to prevent or treat ‘red’ thrombus are:
Heparins and direct thrombin inhibitors act immediately, whereas warfarin and other vitamin K antagonists take several days to exert their effect. Consequently, if warfarin is used to treat patients with venous thrombosis, an agent that acts immediately is also administered until the effect of warfarin has become established.
Heparin (Including Low-Molecular-Weight Heparins)
Heparin was discovered in 1916 by a second-year medical student at Johns Hopkins Hospital. He was attempting to extract thromboplastic (i.e. coagulant) substances from various tissues during a vacation project, but found instead a powerful anticoagulant activity.4 This was named heparin, because it was first extracted from liver.
Heparin is not a single substance but a family of sulfated glycosaminoglycans (mucopolysaccharides). It is present together with histamine in the granules of mast cells. Commercial preparations are extracted from beef lung or hog intestine and, because preparations differ in potency, assayed biologically against an agreed international standard: doses are specified in units of activity rather than of mass.
Heparin fragments (e.g. enoxaparin, dalteparin) or a synthetic pentasaccharide (fondaparinux), referred to as low-molecular-weight heparins (LMWHs), are often used in place of unfractionated heparin, which is reserved for special situations such as patients with renal failure in whom LMWHs are contraindicated (see below).
Mechanism of action
Heparin inhibits coagulation, both in vivo and in vitro, by activating antithrombin III (see above). Antithrombin III inhibits thrombin and other serine proteases by binding to the active serine site. Heparin modifies this interaction by binding, via a unique pentasaccharide sequence, to antithrombin III, changing its conformation and increasing its affinity for serine proteases.
Thrombin is considerably more sensitive to the inhibitory effect of the heparin–antithrombin III complex than is factor Xa. To inhibit thrombin, it is necessary for heparin to bind to the enzyme as well as to antithrombin III; to inhibit factor Xa, it is necessary only for heparin to bind to antithrombin III (Fig. 24.6). Antithrombin III deficiency is very rare but can cause thrombophilia and resistance to heparin therapy.
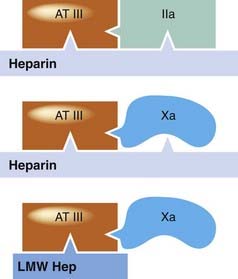
The schematic shows interactions of heparins, antithrombin III (AT III) and clotting factors. To increase the inactivation of thrombin (IIa) by AT III, heparin needs to interact with both substances (top), but to speed up its effect on factor Xa it need only interact with AT III (middle). Low-molecular-weight heparins (LMW Hep) increase the action of AT III on factor Xa (bottom), but cannot increase the action of AT III on thrombin because they cannot bind both simultaneously.
(Modified from Hirsh J, Levine M 1992 Blood 79: 1–17.)
The LMWHs increase the action of antithrombin III on factor Xa but not its action on thrombin, because the molecules are too small to bind to both enzyme and inhibitor, essential for inhibition of thrombin but not for that of factor Xa (Fig. 24.6).
Administration and pharmacokinetic aspects
Heparin is not absorbed from the gut because of its charge and high molecular weight, and it is therefore given intravenously or subcutaneously (intramuscular injections would cause haematomas).
After intravenous injection of a bolus dose, there is a phase of rapid elimination followed by a more gradual disappearance owing both to saturable processes (involving binding to sites on endothelial cells and macrophages) and to slower non-saturable processes including renal excretion. As a result, once the dose exceeds the saturating concentration, a greater proportion is dealt with by these slower processes, and the apparent half-life increases with increasing dose (saturation kinetics; see Ch. 10).
Heparin acts immediately following intravenous administration, but the onset is delayed by up to 60 min when it is given subcutaneously. The elimination half-life is approximately 40–90 min. In urgent situations, it is therefore usual to start treatment with a bolus intravenous dose, followed by a constant-rate infusion. The activated partial thromboplastin time (APTT), or some other in vitro clotting test, is measured and the dose of heparin adjusted to achieve a value within a target range (e.g. 1.5–2.5 times control).
Low-molecular-weight heparins are given subcutaneously. They have a longer elimination half-life than unfractionated heparin, and this is independent of dose (first-order kinetics), so the effects are more predictable and dosing less frequent (once or twice a day). LMWHs do not prolong the APTT. Unlike unfractionated heparin, the effect of a standard dose is sufficiently predictable that monitoring is not required routinely. LMWHs are eliminated mainly by renal excretion, and unfractionated heparin is preferred in renal failure, but with this exception LMWHs are at least as safe and effective as unfractionated heparin and are more convenient to use, because patients can be taught to inject themselves at home and there is generally no need for blood tests and dose adjustment.
Unwanted effects
Haemorrhage. The main hazard is haemorrhage, which is treated by stopping therapy and, if necessary, giving protamine sulfate. This heparin antagonist is a strongly basic protein that forms an inactive complex with heparin; it is given intravenously. The dose is estimated from the dose of heparin that has been administered recently, and it is important not to give too much, as this can itself cause bleeding. If necessary, an in vitro neutralisation test is performed on a sample of blood from the patient to provide a more precise indication of the required dose.
Thrombosis. This is an uncommon but serious adverse effect of heparin and, as with warfarin necrosis (see below), may be misattributed to the natural history of the disease for which heparin is being administered.
Paradoxically, it is associated with heparin-induced thrombocytopenia (HIT). A transitory early decrease in platelet numbers is not uncommon after initiating heparin treatment, and is not clinically important. More serious thrombocytopenia occurring 2–14 days after the start of therapy is uncommon and is referred to as type II HIT. This is caused by IgM or IgG antibodies against complexes of heparin and platelet factor 4. Circulating immune complexes bind to Fc receptors (see Ch. 6) on circulating platelets, thereby activating them and releasing more platelet factor 4 and causing thrombocytopenia. Antibody also binds to platelet factor 4 complexed with glycosaminoglycans on the surface of endothelial cells, leading to immune injury of the vessel wall, thrombosis and disseminated intravascular coagulation. LMWHs are less liable than standard heparin to activate platelets to release platelet factor 4, and they bind less avidly to platelet factor 4. LMWHs are less likely than unfractionated heparin to cause thrombocytopenia and thrombosis by this mechanism. HIT is usually treated with either danaparoid or with a direct thrombin inhibitor (see below). Danaparoid is a low-molecular-weight heparinoid consisting of a mixture of heparan, dermatan and chondroitin sulfates, with well-established antithrombotic activity.
Osteoporosis with spontaneous fractures has been reported with long-term (6 months or more) treatment with heparin (usually during pregnancy, when warfarin is contraindicated or problematic—see below). Its explanation is unknown.
Hypoaldosteronism (with consequent hyperkalaemia) is uncommon, but increases with prolonged treatment. It is recommended to check plasma K+ concentration if treatment is to be continued for > 7 days.
Hypersensitivity reactions are rare with heparin but more common with protamine. (Protamine sensitivity also occurs in patients treated with protamine zinc insulin; Ch. 30. Protamine is extracted from fish roe, and sensitivity to protamine occurs in some people with fish allergy.)
Direct Thrombin Inhibitors and Related Drugs
Hirudins are direct thrombin inhibitors derived from the anticoagulant present in saliva from the medicinal leech. Unlike the heparins they do not depend on activation of antithrombin. Lepirudin is used clinically. It is a polypeptide related to hirudin that binds irreversibly both to the fibrin-binding and catalytic sites on thrombin and is used for thromboembolic disease in patients with type II HIT. It is administered intravenously, the dose being adjusted depending on the APTT, and can cause bleeding or hypersensitivity reactions (rash or fever). Bivalirudin, another hirudin analogue, is used by cardiologists in selected patients undergoing percutaneous coronary interventions. Treatment is initiated with an intravenous bolus followed by an infusion during and up to 4 h after the procedure. It can cause bleeding and hypersensitivity reactions.
Orally active inhibitors
This field has had more than one false dawn, but hope springs eternal as thrombin inhibitors could replace warfarin, a venerable but inconvenient drug that is a common cause of serious adverse effects (see below). Dabigatran is a synthetic serine protease inhibitor; dabigatran etexilate, a prodrug with a hydrophobic tail, is orally active as a direct thrombin inhibitor and is licensed for prevention of venous thromboembolism following hip or knee replacement. It works rapidly and is administered shortly after surgery and then once daily for up to a month. Rivaroxaban, also a synthetic inhibitor, is selective for factor Xa rather than for thrombin, but similar to dabigatran in other respects These drugs are administered in standard doses without laboratory monitoring of their anticoagulant effects. The commonest adverse effects of both drugs are predictable (bleeding, anaemia); rivaroxaban also commonly causes nausea. Other indications for both drugs are being investigated, and if they prove safe and effective for a range of indications, this could transform the clinical management of the large group of patients currently maintained on warfarin (see the clinical box on the clinical use of anticoagulants).
Various other approaches are being explored. These include several naturally occurring anticoagulants (tissue factor pathway inhibitor, thrombomodulin and protein C) synthesised by recombinant technology. A particularly ingenious approach is the development of thrombin agonists that are selective for the anticoagulant properties of thrombin. One such modified thrombin, differing by a single amino acid substitution, has substrate specificity for protein C. It produces anticoagulation in monkeys without prolonging bleeding times, suggesting that it may be less likely than standard anticoagulants to cause bleeding (Bah et al., 2009).
Vitamin K Antagonists: Warfarin
Oral anticoagulants were discovered as an indirect result of a change in agricultural policy in North America in the 1920s. Sweet clover was substituted for corn in cattle feed, and an epidemic of deaths of cattle from haemorrhage ensued. This turned out to be caused by bishydroxycoumarin in spoiled sweet clover, and it led to the discovery of warfarin (named for the Wisconsin Alumni Research Foundation). One of the first uses to which this was put was as a rat poison, but for more than 50 years it has been the standard anticoagulant for the treatment and prevention of thromboembolic disease.
Warfarin (Fig. 24.3) is the most important oral anticoagulant; alternatives with a similar mechanism of action, for example phenindione, are now used only in rare patients who experience idiosyncratic adverse reactions to warfarin. Warfarin and other vitamin K antagonists require frequent blood tests to individualise dose, and are consequently inconvenient as well as having a low margin of safety.
Mechanism of action
Vitamin K antagonists act only in vivo and have no effect on clotting if added to blood in vitro. They interfere with the post-translational γ-carboxylation of glutamic acid residues in clotting factors II, VII, IX and X. They do this by inhibiting vitamin K epoxide reductase component 1 (VKORC1), thus inhibiting the reduction of vitamin K epoxide to its active hydroquinone form (Fig. 24.5). Inhibition is competitive (reflecting the structural similarity between warfarin and vitamin K; Fig. 24.3). The VKORC1 gene is polymorphic (see Ch. 11), and different haplotypes have different affinities for warfarin. Genotyping to determine the haplotype, combined with genotyping CYP2C9 (see below), while not yet routine, can reduce the variability in response to warfarin by around one-third. The effect of warfarin takes several days to develop because of the time taken for degradation of preformed carboxylated clotting factors. Onset of action thus depends on the elimination half-lives of the relevant factors. Factor VII, with a half-life of 6 h, is affected first, then IX, X and II, with half-lives of 24, 40 and 60 h, respectively.
Administration and pharmacokinetic aspects
Warfarin is absorbed rapidly and completely from the gut after oral administration. It has a small distribution volume, being strongly bound to plasma albumin (see Ch. 8). The peak concentration in the blood occurs within an hour of ingestion, but because of the mechanism of action this does not coincide with the peak pharmacological effect, which occurs about 48 h later. The effect on prothrombin time (PT, see below) of a single dose starts after approximately 12–16 h and lasts 4–5 days. Warfarin is metabolised by CYP2C9, which is polymorphic (see Ch. 11). Partly in consequence of this, its half-life is very variable, being of the order of 40 h in many individuals.
Warfarin crosses the placenta and is not given in the first months of pregnancy because it is teratogenic (see below), nor in the later stages because it can cause intracranial haemorrhage in the baby during delivery. It appears in milk during lactation. This could theoretically be important because newborn infants are naturally deficient in vitamin K. However, infants are routinely prescribed vitamin K to prevent haemorrhagic disease, so warfarin treatment of the mother does not generally pose a risk to the breastfed infant.
The therapeutic use of warfarin requires a careful balance between giving too little, leaving unwanted coagulation unchecked, and giving too much, thereby causing haemorrhage. Therapy is complicated not only because the effect of each dose is maximal some 2 days after its administration, but also because numerous medical and environmental conditions modify sensitivity to warfarin, including interactions with other drugs (see Ch. 56). The effect of warfarin is monitored by measuring PT, which is expressed as an international normalised ratio (INR).
The PT is the time taken for clotting of citrated plasma after the addition of Ca2+ and standardised reference thromboplastin; it is expressed as the ratio (PT ratio) of the PT of the patient to the PT of a pool of plasma from healthy subjects on no medication. Because of the variability of thromboplastins, different results are obtained in different laboratories. To standardise PT measurements internationally, each thromboplastin is assigned an international sensitivity index (ISI), and the patient’s PT is expressed as an INR, where INR = (PT ratio)ISI. This kind of normalisation procedure shocks purists but provides similar results when a patient moves from, say, Birmingham to Baltimore, permitting warfarin dose adjustment independent of laboratory. Pragmatic haematologists argue that the proof of the pudding is in the eating!
The dose of warfarin is usually adjusted to give an INR of 2–4, the precise target depending on the clinical situation. The duration of treatment also varies, but for several indications (e.g. to prevent thromboembolism in chronic atrial fibrillation), treatment is long term, with the logistical challenge of providing a worldwide network of anticoagulant clinics and demands on the patient in terms of repeat visits and blood tests.
Factors that Potentiate Oral Anticoagulants
Various diseases and drugs potentiate warfarin, increasing the risk of haemorrhage.
Disease
Liver disease interferes with the synthesis of clotting factors; conditions in which there is a high metabolic rate, such as fever and thyrotoxicosis, increase the effect of anticoagulants by increasing degradation of clotting factors.
Drugs (see also Chs 9 and 56)
Many drugs potentiate warfarin.
Agents that inhibit hepatic drug metabolism
Examples include co-trimoxazole, ciprofloxacin, metronidazole, amiodarone and many antifungal azoles. Stereoselective effects (warfarin is a racemate, and its isomers are metabolised differently from one another) are described in Chapter 56.
Drugs that inhibit platelet function
Aspirin increases the risk of bleeding if given during warfarin therapy, although this combination can be used safely with careful monitoring. Other non-steroidal anti-inflammatory drugs (NSAIDs) also increase the risk of bleeding, partly by their effect on platelet thromboxane synthesis (Ch. 26) and, in the case of some NSAIDs, also by inhibiting warfarin metabolism as above. Some antibiotics, including moxalactam and carbenicillin, inhibit platelet function.
Drugs that displace warfarin from binding sites on plasma albumin
Some of the NSAIDs and chloral hydrate cause a transient increase in the concentration of free warfarin in plasma by competing with it for binding to plasma albumin. This mechanism seldom causes clinically important effects, unless accompanied by inhibition of warfarin metabolism, as with phenylbutazone (Ch. 56).
Drugs that decrease the availability of vitamin K
Broad-spectrum antibiotics and some sulfonamides (see Ch. 49) depress the intestinal flora that normally synthesise vitamin K2 (see above); this has little effect unless there is concurrent dietary deficiency.
Factors that Lessen the Effect of Oral Anticoagulants
Physiological state/disease
There is a decreased response to warfarin in conditions (e.g. pregnancy) where there is increased coagulation factor synthesis. Similarly, the effect of oral anticoagulants is lessened in hypothyroidism, which is associated with reduced degradation of coagulation factors.
Drugs (see also Chs 9 and 56)
Several drugs reduce the effectiveness of warfarin; this leads to increased doses being used to achieve the target INR. Furthermore, the dose of warfarin must be reduced when the interacting drug is discontinued, to avoid haemorrhage.
Drugs that induce hepatic P450 enzymes
Enzyme induction (e.g. by rifampicin, carbamazepine) increases the rate of degradation of warfarin. Induction may wane only slowly after the inducing drug is discontinued, making it difficult to adjust the warfarin dose appropriately.
Drugs that reduce absorption
Drugs that bind warfarin in the gut, for example colestyramine, reduce its absorption.
Drugs affecting blood coagulation
Injectable anticoagulants (e.g. heparin, low-molecular-weight heparins)
Oral anticoagulants (e.g. warfarin)
Unwanted Effects of Oral Anticoagulants
Haemorrhage (especially into the bowel or the brain) is the main hazard. Depending on the urgency of the situation, treatment may consist of withholding warfarin (for minor problems), administration of vitamin K, or fresh plasma or coagulation factor concentrates (for life-threatening bleeding).
Oral anticoagulants are teratogenic, causing disordered bone development which is believed to be related to binding to the vitamin K-dependent protein osteocalcin.
Hepatotoxicity occurs but is uncommon.
Necrosis of soft tissues (e.g. breast or buttock) owing to thrombosis in venules occurs shortly after starting treatment and is attributed to inhibition of biosynthesis of protein C, which has a shorter elimination half-life than do the vitamin K-dependent coagulation factors; this results in a procoagulant state soon after starting treatment. This is a rare but serious adverse effect. Treatment with a heparin is usually started at the same time as warfarin, avoiding this problem except in individuals experiencing HIT as an adverse effect of heparin (see above).
The clinical use of anticoagulants is summarised in the box.
Platelet Adhesion and Activation
Platelets maintain the integrity of the circulation: a low platelet count results in thrombocytopenic purpura.5
When platelets are activated, they undergo a sequence of reactions that are essential for haemostasis, important for the healing of damaged blood vessels, and play a part in inflammation (see Ch. 17). These reactions, several of which are redundant (in the sense that if one pathway of activation is blocked another is available) and several autocatalytic, include:
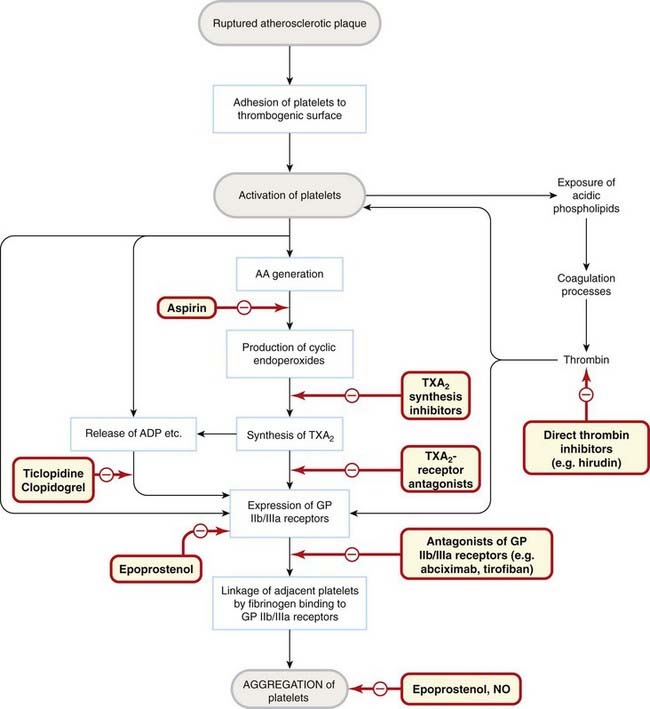
Fig. 24.7 Platelet activation.
Events involved in platelet adhesion and aggregation are shown, with the sites of action of drugs and endogenous mediators. AA, arachidonic acid; ADP, adenosine bisphosphate; GP, glycoprotein; NO, nitric oxide; TXA2, thromboxane A2.
These processes are essential for haemostasis but may be inappropriately triggered if the artery wall is diseased, most commonly with atherosclerosis, resulting in thrombosis (Fig. 24.7).
Platelet function
Antiplatelet Drugs
Platelets play such a critical role in thromboembolic disease that it is no surprise that antiplatelet drugs are of great therapeutic value. Clinical trials of aspirin radically altered clinical practice, and more recently drugs that block ADP receptors and GPIIb/IIIa have also been found to be therapeutically useful. Sites of action of antiplatelet drugs are shown in Figure 24.7.
Aspirin
Low-dose aspirin (see Ch. 26) profoundly (> 95%) inhibits platelet TXA2 synthesis, by irreversible acetylation of a serine residue in the active site of cyclo-oxygenase I (COX-I). Oral administration is relatively selective for platelets because of presystemic elimination (Ch. 9). Unlike nucleated cells, platelets cannot synthesise proteins, so after administration of aspirin, TXA2 synthesis does not recover until the affected cohort of platelets is replaced in 7–10 days. Clinical trials have demonstrated the efficacy of aspirin in several clinical settings (e.g. Fig. 24.8). Adverse effects of aspirin, mainly on the gastrointestinal tract, are, however, clearly dose related, so a low dose (often 75 mg once daily) is usually recommended for thromboprophylaxis. Thromboprophylaxis is reserved for people at high cardiovascular risk (e.g. survivors of myocardial infarction), in whom the cardiovascular benefit of aspirin usually outweighs the risk of gastrointestinal bleeding.
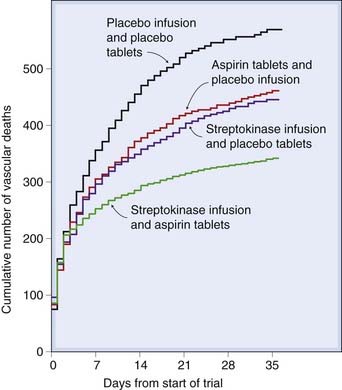
Fig. 24.8 Efficacy of aspirin and streptokinase for myocardial infarction.
The curves show cumulative vascular mortality in patients treated with placebo, aspirin alone, streptokinase alone or a combined aspirin–streptokinase regimen.
(ISIS-2 trial 1988 Lancet ii: 350–360.)
Treatment failure can occur despite taking aspirin, and there is current interest in the possibility that some patients exhibit a syndrome of ‘aspirin resistance’, although the mechanism and possible importance of this remains controversial (see Goodman et al., 2008). Other non-steroidal drugs which inhibit platelet TXA2 synthesis > 95% (e.g. sulfinpyrazone, for which there is also supportive clinical trial evidence) may have antithrombotic effects, but where inhibition of platelet TXA2 synthesis does not reach this threshold there is evidence that such drugs are proaggregatory, related to inhibition of COX-2, possibly due to inhibition of antiaggregatory PGI2.
Dipyridamole
Dipyridamole inhibits platelet aggregation by several mechanisms, including inhibition of phosphodiesterase, block of adenosine uptake into red cells (see Ch. 16) and inhibition of TXA2 synthesis (see Ch. 26). Clinical effectiveness has been uncertain, but one study showed that a modified-release form of dipyridamole reduced the risk of stroke and death in patients with transient ischaemic attacks by around 15%—similar to aspirin (25 mg twice daily).7 The beneficial effects of aspirin and dipyridamole were additive. The main side effects of dipyridamole are dizziness, headache and gastrointestinal disturbances; unlike aspirin, it does not increase the risk of bleeding.
Adenosine (P2y) Receptor Antagonists (Thienopyridines)
Ticlopidine was the first to be introduced, but causes neutropenia and thrombocytopenia and is now little used. The main agent is currently clopidogrel; prasugrel has been introduced recently.
These drugs inhibit ADP-induced platelet aggregation by irreversible inhibition of P2Y12 receptors (Ch. 16) to which they link via a disulfide bond.
Pharmacokinetics and unwanted effects
Clopidogrel is well absorbed when administered by mouth, and in urgent situations is given orally as a loading dose of 300 mg followed by maintenance dosing of 75 mg once daily. It is a prodrug and is converted into its active sulfhydryl metabolite by CYP enzymes in the liver including CYP2C19. Patients with variant alleles of CYP2C19 (poor metabolisers) are at increased risk of therapeutic failure. There is a potential for interaction with other drugs, such as omeprazole (Ch. 29), that are metabolised by CYP2C19 and current labelling recommends against use with proton pump inhibitors for this reason.
Clopidogrel can cause dyspepsia, rash or diarrhoea. The serious blood dyscrasias caused by ticlopidine are very rare with clopidogrel.
Clinical use
Clopidogrel was slightly more effective than aspirin in reducing a composite outcome of ischaemic stroke, myocardial infarction or vascular death in one large trial; it can be used instead of aspirin in patients with symptomatic atheromatous disease, but, because of cost, is usually reserved for patients who are intolerant of aspirin. Clinical trials of adding clopidogrel to aspirin in patients with acute coronary syndromes (see Fig. 24.9) and (in a megatrial of over 45 000 patients) in patients with acute myocardial infarction (COMMIT Collaborative Group, 2005) demonstrated that combined treatment reduces mortality. Treatment with clopidogrel for this indication is given for 4 weeks. Prasugrel is more effective than clopidogrel in acute coronary syndromes, but more often causes serious bleeding (Wiviott et al., 2007). Pretreatment with clopidogrel and aspirin followed by longer-term therapy is also effective in patients with ischaemic heart disease undergoing percutaneous coronary interventions.
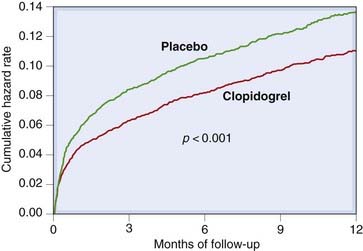
Glycoprotein Iib/Iiia Receptor Antagonists
Antagonists of the GPIIb/IIIa receptor have the theoretical attraction that they inhibit all pathways of platelet activation (because these all converge on activation of GPIIb/IIIa receptors). A hybrid murine–human monoclonal antibody Fab fragment directed against the GPIIb/IIIa receptor, which rejoices in the catchy little name of abciximab,8 is licensed for use in high-risk patients undergoing coronary angioplasty, as an adjunct to heparin and aspirin. It reduces the risk of restenosis at the expense of an increased risk of bleeding. Immunogenicity limits its use to a single administration.
Tirofiban is a synthetic non-peptide and eptifibatide is a cyclic peptide based on the Arg–Gly–Asp (‘RGD’) sequence that is common to ligands for GPIIb/IIIa receptors. Neither is absorbed if administered by mouth. Given intravenously as an adjunct to aspirin and a heparin preparation, they reduce early events in acute coronary syndrome, but long-term oral therapy with GPIIb/IIIa receptor antagonists is not effective and may be harmful. Unsurprisingly, they increase the risk of bleeding.
Other Antiplatelet Drugs
Epoprostenol (PGI2), an agonist at prostanoid IP receptors (see Ch. 17), causes vasodilatation as well as inhibiting platelet aggregation. It is added to blood entering the dialysis circuit in order to prevent thrombosis during haemodialysis, especially in patients in whom heparin is contraindicated. It is also used in severe pulmonary hypertension (Ch. 22) and circulatory shock. It is unstable under physiological conditions and has a half-life of around 3 min, so it is administered by an intravenous infusion pump. Adverse effects related to its vasodilator action include flushing, headache and hypotension.
The clinical use of antiplatelet drugs is summarised in the clinical box below.
Antiplatelet drugs
Clinical uses of antiplatelet drugs 
The main drug is aspirin. Other drugs with distinct actions (e.g. dipyridamole, clopidogrel) can have additive effects, or be used in patients who are intolerant of aspirin. Uses of antiplatelet drugs relate mainly to arterial thrombosis and include:
Other antiplatelet drugs such as epoprostenol (PGI2; see Ch. 17) have specialised clinical applications (e.g. in haemodialysis or haemofiltration, Ch. 28, or in pulmonary hypertension, Ch. 22).
Fibrinolysis (Thrombolysis)
When the coagulation system is activated, the fibrinolytic system is also set in motion via several endogenous plasminogen activators, including tissue plasminogen activator (tPA), urokinase-type plasminogen activator, kallikrein and neutrophil elastase. tPA is inhibited by a structurally related lipoprotein, lipoprotein(a), increased concentrations of which constitute an independent risk factor for myocardial infarction (Ch. 23). Plasminogen is deposited on the fibrin strands within a thrombus. Plasminogen activators are serine proteases and are unstable in circulating blood. They diffuse into thrombus and cleave plasminogen, a zymogen present in plasma, to release plasmin (see Fig. 24.10).
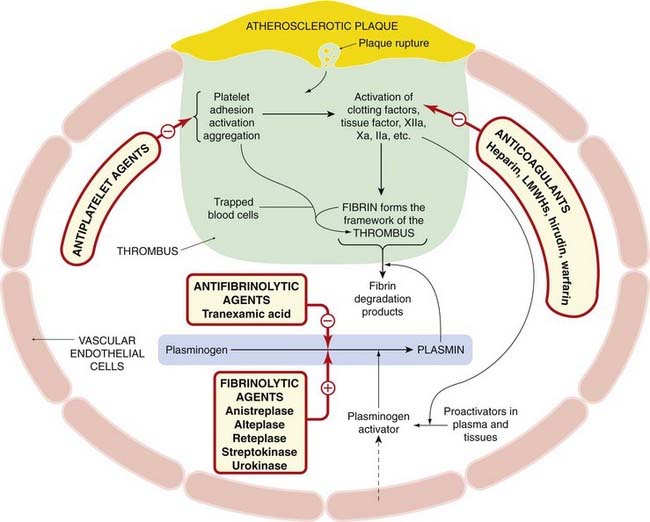
Fig. 24.10 Fibrinolytic system.
The schematic shows interactions with coagulation and platelet pathways and sites of action of drugs that modify these systems. LMHs, low-molecular-weight heparins. For more details of platelet activation and the coagulation cascade, refer to Figures 24.1, 24.2 and 24.7.
Plasmin is trypsin-like, acting on Arg–Lys bonds, and thus digests not only fibrin but fibrinogen; factors II, V and VIII; and many other proteins. It is formed locally and acts on the fibrin meshwork, generating fibrin degradation products and lysing the clot. Its action is localised to the clot, because plasminogen activators are effective mainly on plasminogen adsorbed to fibrin; any plasmin that escapes into the circulation is inactivated by plasmin inhibitors, including PAI-1 (see above and Ch. 22), which protect us from digesting ourselves from within.
Drugs affect this system by increasing or inhibiting fibrinolysis (fibrinolytic and antifibrinolytic drugs, respectively).
Fibrinolytic Drugs
Figure 24.10 summarises the interaction of the fibrinolytic system with the coagulation cascade and platelet activation, and the action of drugs that modify this. Several fibrinolytic (thrombolytic) drugs are used clinically, principally to reopen the occluded arteries in patients with acute myocardial infarction9 or stroke, less commonly in patients with life-threatening venous thrombosis or pulmonary embolism.
Streptokinase is a protein extracted from cultures of streptococci. It activates plasminogen. Infused intravenously, it reduces mortality in acute myocardial infarction, and this beneficial effect is additive with aspirin (Fig. 24.8). Its action is blocked by antibodies, which appear 4 days or more after the initial dose: its use should not be repeated after this time has elapsed.
Alteplase and duteplase are, respectively, single- and double-chain recombinant tPA. They are more active on fibrin-bound plasminogen than on plasma plasminogen, and are therefore said to be ‘clot selective’. Recombinant tPA is not antigenic, and can be used in patients likely to have antibodies to streptokinase. Because of their short half-lives, they must be given as intravenous infusions. Reteplase is similar but has a longer elimination half-life, allowing for bolus administration and making for simplicity of administration. It is available for clinical use in myocardial infarction.
Unwanted Effects and Contraindications
The main hazard of all fibrinolytic agents is bleeding, including gastrointestinal haemorrhage and haemorrhagic stroke. If serious, this can be treated with tranexamic acid (see below), fresh plasma or coagulation factors. Streptokinase can cause allergic reactions and low-grade fever. Streptokinase causes a burst of plasmin formation, generating kinins (see Ch. 17), and can cause hypotension by this mechanism.
Contraindications to the use of these agents are active internal bleeding, haemorrhagic cerebrovascular disease, bleeding diatheses, pregnancy, uncontrolled hypertension, invasive procedures in which haemostasis is important, and recent trauma—including vigorous cardiopulmonary resuscitation.
Which Fibrinolytic Agent is Best?
Much has been written as to which drug is best, but an authoritative review (Collins et al., 1997) concluded that:
… the choice of fibrinolytic drug makes little difference to the overall probability of stroke-free survival, because the regimens that dissolve coronary thrombi more rapidly produce greater risks of cerebral haemorrhage … It is … important that any uncertainties about which fibrinolytic regimen or dose of aspirin to use do not engender uncertainty about whether to use fibrinolytic and antiplatelet therapies routinely.
Clinical Use
Several large placebo-controlled studies in patients with myocardial infarction have shown convincingly that fibrinolytic drugs reduce mortality if given within 12 h of the onset of symptoms, and that the sooner they are administered the better is the result. Similar considerations apply to their use in thrombotic stroke. Scanning to exclude haemorrhagic stroke is advisable, though not always practicable in an emergency situation. Other uses of fibrinolytic agents are listed in the clinical box.
Fibrinolysis and drugs modifying fibrinolysis
Antifibrinolytic and Haemostatic Drugs
Tranexamic acid inhibits plasminogen activation and thus prevents fibrinolysis. It can be given orally or by intravenous injection. It is used to treat various conditions in which there is bleeding or risk of bleeding, such as haemorrhage following prostatectomy or dental extraction, in menorrhagia (excessive menstrual blood loss) and for life-threatening bleeding following thrombolytic drug administration. It is also used in patients with the rare disorder of hereditary angio-oedema.
References and Further Reading
Blood coagulation and anticoagulants
Bah A., Carrell C.J., Chen Z.W., et al. Stabilization of the E* form turns thrombin into an anticoagulant. J. Biol. Chem.. 2009;284:20034-20040. (The anticoagulant profile caused by a mutation of the thrombin gene is due to stabilisation of the inactive E* form of thrombin that is selectively shifted to the active E form upon thrombomodulin and protein C binding)
Bates S.M., Weitz J.I. Emerging anticoagulant drugs. Arterioscler. Thromb. Vasc. Biol.. 2003;23:1491-1500.
Gurm H.S., Bhatt D.L. Thrombin, an ideal target for pharmacological inhibition: a review of direct thrombin inhibitors. Am. Heart J.. 2005;149:S43-S53.
Hirsh J., O’Donnell M., Weitz J.I. New anticoagulants. Blood. 2005;105:453-463. (‘Limitations of existing anticoagulants, vitamin K antagonist and heparins, have led to the development of newer anticoagulant therapies … New anticoagulants under evaluation include: inhibitors of the factor VIIa/tissue factor pathway; factor Xa inhibitors, both indirect and direct; activated protein C and soluble thrombomodulin; and direct thrombin inhibitors. Several of the direct inhibitors of factor Xa and thrombin are orally active. The greatest clinical need is for an oral anticoagulant to replace warfarin for long-term prevention and treatment of patients with venous and arterial thrombosis.’)
Ibbotson T., Perry C.M. Danaparoid—a review of its use in thromboembolic and coagulation disorders. Drugs. 2002;62:2283-2314. (Danaparoid is an effective anticoagulant that has undergone clinical evaluation in a wide range of disease indications; discusses use in HIT)
Shearer M.J., Newman P. Metabolism and cell biology of vitamin K. Thromb. Haemost. 2008;100:530-547. (Review)
Endothelium, platelets and antiplatelet agents
Chew D.P., Bhatt D., Sapp S., et al. Increased mortality with oral platelet glycoprotein IIb/IIIa antagonists: a meta-analysis of phase III multicenter trials. Circulation. 2001;103:201-206.
COMMIT Collaborative Group. Addition of clopidogrel to aspirin in 45 852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1607-1621. (Clopidogrel reduced the risk of death, myocardial infarction or stroke combined, and of mortality alone; see accompanying comment by Sabatine, M.S., pp. 1587–1589 in the same issue)
CURE Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N. Engl. J. Med.. 2001;345:494-502. (A total of 12 562 patients randomised; primary outcome occurred in 9.4% of patients in the clopidogrel + aspirin group and in 11.3% of those in the placebo + aspirin group, a relative risk of 0.72–0.90, P < 0.001)
Goodman T., Ferro A., Sharma P. Pharmacogenetics of aspirin resistance: a comprehensive systematic review. Br. J. Clin. Pharmacol.. 2008;66:222-232. (Supports a genetic association between the PlA1/A2 molecular variant and aspirin resistance in healthy subjects, with the effect diminishing in the presence of cardiovascular disease)
Patrono C., Coller B., FitzGerald G.A., et al. Platelet-active drugs: the relationships among dose, effectiveness, and side effects. Chest. 2004;126:234S-264S.
Wiviott S.D., Braunwald E., McCabe C.H., et alFor the TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med.. 2007;357:2001-2015. (Prasugrel reduced ischaemic events, including stent thrombosis, but with an increased risk of major bleeding, including fatal bleeding. Overall mortality did not differ significantly between treatment groups)
Aster R.H. Heparin-induced thrombocytopenia and thrombosis. N. Engl. J. Med.. 1995;332:1374-1376. (Succinct and lucid editorial; see also accompanying paper, pp. 1330–1335)
Collins R., Peto R., Baigent C., et al. Aspirin, heparin and thrombolytic therapy in suspected acute myocardial infarction. N. Engl. J. Med.. 1997;336:847-860. (Unbiased and authoritative overview; includes a section on ‘general problems of unduly selective emphasis’—fighting stuff!)
Diener H., Cunha L., Forbes C., et al. European Stroke Prevention Study 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J. Neurol. Sci.. 1996;143:1-14. (Slow-release dipyridamole 200 mg twice daily was as effective as aspirin 25 mg twice daily, and the effects of aspirin and dipyridamole were additive)
Goldhaber S.Z. Pulmonary embolism. Lancet. 2004;363:1295-1305.
Kyrle P.A., Eichinger S. Deep vein thrombosis. Lancet. 2005;365:1163-1174.
Levine M. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep vein thrombosis. N. Engl. J. Med.. 1995;334:677-681. (Concludes that LMWH can be used safely and effectively at home; this has potentially very important implications for patient care)
Markus H.S. Current treatments in neurology: stroke. J. Neurol.. 2005;252:260-267.
Warkentin T.E. Management of heparin-induced thrombocytopenia: a critical comparison of lepirudin and argatroban. Thromb. Res.. 2003;110:73-82. (A direct thrombin inhibitor should be given alone during acute HIT, with oral anticoagulants deferred until substantial resolution of the thrombocytopenia has occurred)
1Coagulation of 100 ml of blood requires 0.2 mg of factor VIII, 2 mg of factor X, 15 mg of prothrombin and 250 mg of fibrinogen.
2Mr Hageman (the patient deficient in factor XII after whom it was named) died not from excessive bleeding but from a pulmonaory embolism: factor XII deficiency does not give rise to a bleeding disorder.
3Von Willebrand factor is a glycoprotein that is missing in a hereditary haemorrhagic disorder called von Willebrand’s disease. It is synthesised by vascular endothelial cells (the presence of immunoreactive von Willebrand factor is an identifying feature of these cells in culture) and is also present in platelets.
4This kind of good fortune also favoured Vane and his colleagues in their discovery of PGI2 (Ch. 17), where they were looking for one kind of biological activity and found another. More specific chemical assays (Ch. 7), for all their strengths, cannot throw up this kind of unexpected discovery.
5Purpura means a purple rash caused by multiple spontaneous bleeding points in the skin. When this is caused by reduced circulating platelets, bleeding can occur into other organs, including the gut and brain.
6Various platelet membrane glycoproteins act as receptors or binding sites for adhesive proteins such as von Willebrand factor or fibrinogen.
7This dose regimen of aspirin is unconventional, being somewhat lower than the 75 mg once daily commonly used in thromboprophylaxis.
8The convention for naming monoclonals is: -momab = mouse monoclonal antibody; -umab = human; -zumab = humanised; -ximab = chimeric—a kind of medieval mouse–man nightmare.
9Fibrinolytic drugs are now less widely used in acute myocardial infarction since many units throughout the world provide an emergency angioplasty service (the blocked artery is identified angiographically, opened with a balloon catheter and, if necessary, kept open by means of a stent, Ch. 21). The important thing is to open up the thrombosed artery as swiftly as possible. If facilities are available to do this surgically, this is at least as good as using a lytic drug.