14 Noradrenergic transmission
Overview
The peripheral noradrenergic neuron and the structures that it innervates are important targets for drug action, both as objects for investigation in their own right and as points of attack for many clinically useful drugs. In this chapter, we describe the physiology and function of noradrenergic neurons and the properties of adrenoceptors (the receptors on which noradrenaline and adrenaline act), and discuss the various classes of drugs that affect them. For convenience, much of the pharmacological information is summarised in tables later in the chapter.
Catecholamines
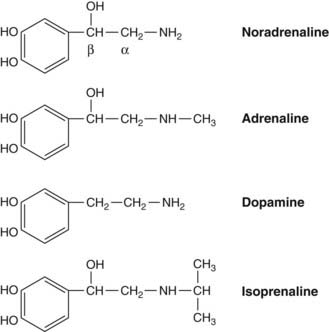
Catecholamines are compounds containing a catechol moiety (a benzene ring with two adjacent hydroxyl groups) and an amine side chain (Fig. 14.1). Pharmacologically, the most important ones are:
Classification of Adrenoceptors
In 1896, Oliver and Schafer discovered that injection of extracts of adrenal gland caused a rise in arterial pressure. Following the subsequent isolation of adrenaline as the active principle, it was shown by Dale in 1913 that adrenaline causes two distinct kinds of effect, namely vasoconstriction in certain vascular beds (which normally predominates and, together with its actions on the heart—see below—causes the rise in arterial pressure) and vasodilatation in others. Dale showed that the vasoconstrictor component disappeared if the animal was first injected with an ergot derivative2 (see p. 198), and noticed that adrenaline then caused a fall, instead of a rise, in arterial pressure. This result paralleled Dale’s demonstration of the separate muscarinic and nicotinic components of the action of acetylcholine (see Ch. 13). He avoided interpreting it in terms of different types of receptor, but later pharmacological work, beginning with that of Ahlquist, showed clearly the existence of several subclasses of adrenoceptor.
Ahlquist found in 1948 that the rank order of the potencies of various catecholamines, including adrenaline, noradrenaline and isoprenaline, fell into two distinct patterns, depending on what response was being measured. He postulated the existence of two kinds of receptor, α and β, defined in terms of agonist potencies as follows:
It was then recognised that certain ergot alkaloids, which Dale had studied, act as selective α-receptor antagonists, and that Dale’s adrenaline reversal experiment reflected the unmasking of the β effects of adrenaline by α-receptor blockade. Selective β-receptor antagonists were not developed until 1955, when their effects fully confirmed Ahlquist’s original classification and also suggested the existence of further subdivisions of both α and β receptors. Subsequently (see Bylund et al., 1994) it has emerged that there are two α-receptor subtypes (α1 and α2), each comprising three further subclasses (α1A, α1B, α1D and α2A, α2B, α2C) and three β-receptor subtypes (β1, β2 and β3)—altogether nine distinct subtypes—all of which are typical G-protein-coupled receptors (Table 14.1). Evidence from specific agonists and antagonists, as well as studies on receptor knockout mice (Philipp & Hein, 2004), has shown that α1 receptors are particularly important in the cardiovascular system and lower urinary tract, while α2 receptors are predominantly neuronal, acting to inhibit transmitter release both in the brain and at autonomic nerve terminals in the periphery. The distinct functions of the different subclasses of α1- and α2 adrenoceptors remain for the most part unclear; they are frequently co-expressed in the same tissues, and may form heterodimers, making pharmacological analysis difficult.
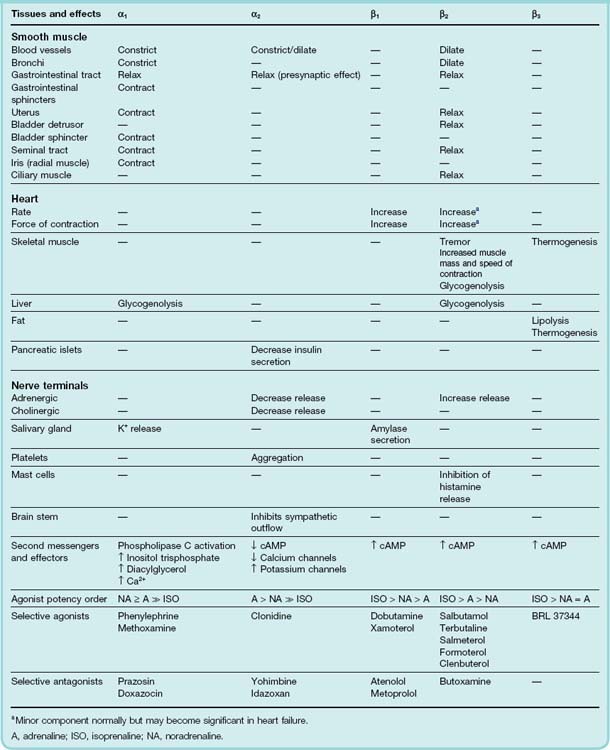
Each of the three main receptor subtypes is associated with a specific second messenger system (Table 14.1). Thus α1 receptors are coupled to phospholipase C and produce their effects mainly by the release of intracellular Ca2+; α2 receptors are negatively coupled to adenylyl cyclase, and reduce cAMP formation as well as inhibiting Ca2+ channels and activating K+ channels ; and all three types of β receptor act by stimulation of adenylyl cyclase. The major effects that are produced by these receptors, and the main drugs that act on them, are shown in Table 14.1.
The distinction between β1 and β2 receptors is an important one, for β1 receptors are found mainly in the heart, where they are responsible for the positive inotropic and chronotropic effects of catecholamines (see Ch. 21). β2 receptors, on the other hand, are responsible for causing smooth muscle relaxation in many organs. The latter is often a useful therapeutic effect, while the former is more often harmful; consequently, considerable efforts have been made to find selective β2 agonists, which would relax smooth muscle without affecting the heart, and selective β1 antagonists, which would exert a useful blocking effect on the heart without at the same time blocking β2 receptors in bronchial smooth muscle (see Table 14.1). It is important to realise that the available drugs are not completely selective, and that compounds used as selective β1 antagonists invariably have some action on β2 receptors as well, which can cause unwanted effects such as bronchoconstriction.
In relation to vascular control, it is broadly true that α1 and β2 receptors act mainly on the smooth muscle cells themselves, while α2 receptors act on presynaptic terminals, but different vascular beds deviate from this general rule. Both α- and β-receptor subtypes are expressed in smooth muscle cells, nerve terminals and endothelial cells, and their role in physiological regulation and pharmacological responses of the cardiovascular system is only partly understood (see Guimaraes & Moura, 2001).
Classification of adrenoceptors 
Physiology of Noradrenergic Transmission
The Noradrenergic Neuron
Noradrenergic neurons in the periphery are postganglionic sympathetic neurons whose cell bodies lie in sympathetic ganglia. They generally have long axons that end in a series of varicosities strung along the branching terminal network. These varicosities contain numerous synaptic vesicles, which are the sites of synthesis and release of noradrenaline and of co-released mediators such as ATP and neuropeptide Y (see Ch. 12), which are stored in vesicles and release by exocytosis (Ch. 4). In most peripheral tissues, the tissue content of noradrenaline closely parallels the density of the sympathetic innervation. With the exception of the adrenal medulla, sympathetic nerve terminals account for all the noradrenaline content of peripheral tissues. Organs such as the heart, spleen, vas deferens and some blood vessels are particularly rich in noradrenaline (5–50 nmol/g of tissue) and have been widely used for studies of noradrenergic transmission. For detailed information on noradrenergic neurons, see Robertson (2004) and Cooper et al. (1996).
Noradrenaline Synthesis
The biosynthetic pathway for noradrenaline synthesis is shown in Figure 14.2. The metabolic precursor for noradrenaline is L-tyrosine, an aromatic amino acid that is present in the body fluids and is taken up by adrenergic neurons. Tyrosine hydroxylase, a cytosolic enzyme that catalyses the conversion of tyrosine to dihydroxyphenylalanine (dopa), is found only in catecholamine-containing cells. It is a rather selective enzyme; unlike other enzymes involved in catecholamine metabolism, it does not accept indole derivatives as substrates, and so is not involved in 5-hydroxytryptamine (5-HT) metabolism. This first hydroxylation step is the main control point for noradrenaline synthesis. Tyrosine hydroxylase is inhibited by the end product of the biosynthetic pathway, noradrenaline, and this provides the mechanism for the moment-to-moment regulation of the rate of synthesis; much slower regulation, taking hours or days, occurs by changes in the rate of production of the enzyme.
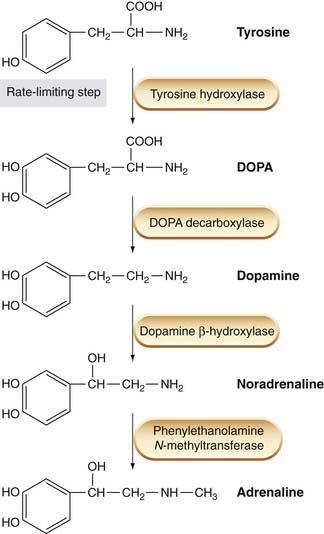
The tyrosine analogue α-methyltyrosine strongly inhibits tyrosine hydroxylase and may be used experimentally to block noradrenaline synthesis.
The next step, conversion of dopa to dopamine, is catalysed by dopa decarboxylase, a cytosolic enzyme that is by no means confined to catecholamine-synthesising cells. It is a relatively non-specific enzyme, and catalyses the decarboxylation of various other L-aromatic amino acids, such as L-histidine and L-tryptophan, which are precursors in the synthesis of histamine (Ch. 17) and 5-HT (Ch. 15), respectively. Dopa decarboxylase activity is not rate limiting for noradrenaline synthesis. Although various factors, including certain drugs, affect the enzyme, it is not an effective means of regulating noradrenaline synthesis.
Dopamine-β-hydroxylase (DBH) is also a relatively non-specific enzyme, but is restricted to catecholamine-synthesising cells. It is located in synaptic vesicles, mainly in membrane-bound form. A small amount of the enzyme is released from adrenergic nerve terminals in company with noradrenaline, representing the small proportion in a soluble form within the vesicle. Unlike noradrenaline, the released DBH is not subject to rapid degradation or uptake, so its concentration in plasma and body fluids can be used as an index of overall sympathetic nerve activity.
Many drugs inhibit DBH, including copper-chelating agents and disulfiram (a drug used mainly for its effect on ethanol metabolism; see Chs 9 and 48). Such drugs can cause a partial depletion of noradrenaline stores and interference with sympathetic transmission. A rare genetic disorder, DBH deficiency, causes failure of noradrenaline synthesis resulting in severe orthostatic hypotension (see Ch. 22).
Phenylethanolamine N-methyl transferase (PNMT) catalyses the N-methylation of noradrenaline to adrenaline. The main location of this enzyme is in the adrenal medulla, which contains a population of adrenaline-releasing (A) cells separate from the smaller proportion of noradrenaline-releasing (N) cells. The A cells, which appear only after birth, lie adjacent to the adrenal cortex, and the production of PNMT is induced by an action of the steroid hormones secreted by the adrenal cortex (see Ch. 32). PNMT is also found in certain parts of the brain, where adrenaline may function as a transmitter, but little is known about its role.
Noradrenaline turnover can be measured under steady-state conditions by measuring the rate at which labelled noradrenaline accumulates when a labelled precursor, such as tyrosine or dopa, is administered. The turnover time is defined as the time taken for an amount of noradrenaline equal to the total tissue content to be degraded and resynthesised. In peripheral tissues, the turnover time is generally about 5–15 h, but it becomes much shorter if sympathetic nerve activity is increased. Under normal circumstances, the rate of synthesis closely matches the rate of release, so that the noradrenaline content of tissues is constant regardless of how fast it is being released.
Noradrenaline Storage
Most of the noradrenaline in nerve terminals or chromaffin cells is contained in vesicles; only a little is free in the cytoplasm under normal circumstances. The concentration in the vesicles is very high (0.3–1.0 mol/l) and is maintained by the vesicular monoamine transporter (VMAT), which is similar to the amine transporter responsible for noradrenaline uptake into the nerve terminal (see Ch. 12), but uses the transvesicular proton gradient as its driving force. Certain drugs, such as reserpine (see below; Table 14.2) block this transport and cause nerve terminals to become depleted of their vesicular noradrenaline stores. The vesicles contain two major constituents besides noradrenaline, namely ATP (about four molecules per molecule of noradrenaline) and a protein called chromogranin A. These substances are released along with noradrenaline, and it is generally assumed that a reversible complex, depending partly on the opposite charges on the molecules of noradrenaline and ATP, is formed within the vesicle. This would serve both to reduce the osmolarity of the vesicle contents and also to reduce the tendency of noradrenaline to leak out of the vesicles within the nerve terminal.
ATP itself has a transmitter function at noradrenergic synapses (see Lundberg, 1996; Fig. 12.5; Ch. 16), being responsible for the fast excitatory synaptic potential and the rapid phase of contraction produced by sympathetic nerve activity in many smooth muscle tissues.
Noradrenaline Release
The processes linking the arrival of a nerve impulse at a nerve terminal to Ca2+ entry and the release of transmitter are described in Chapter 4.
An unusual feature of the release mechanism at the varicosities of noradrenergic nerves is that the probability of release, even of a single vesicle, when a nerve impulse arrives at a varicosity is very low (less than 1 in 50; see Cunnane, 1984). A single neuron possesses many thousand varicosities, so one impulse leads to the discharge of a few hundred vesicles, scattered over a wide area. This contrasts sharply with the neuromuscular junction (Ch. 13), where the release probability at a single terminal is high, and release of acetylcholine is sharply localised.
Regulation of noradrenaline release
Noradrenaline release is affected by a variety of substances that act on presynaptic receptors (see Ch. 12). Many different types of nerve terminal (cholinergic, noradrenergic, dopaminergic, 5-HT-ergic, etc.) are subject to this type of control, and many different mediators (e.g. acetylcholine acting through muscarinic receptors, catecholamines acting through α and β receptors, angiotensin II, prostaglandins, purine nucleotides, neuropeptides, etc.) can act on presynaptic terminals. Presynaptic modulation represents an important physiological control mechanism throughout the nervous system.
Furthermore, noradrenaline, by acting on presynaptic β2 receptors, can regulate its own release, and also that of co-released ATP (see Ch. 12). This is believed to occur physiologically, so that released noradrenaline exerts a local inhibitory effect on the terminals from which it came—the so-called autoinhibitory feedback mechanism (Fig. 14.3; see Starke et al., 1989). Agonists or antagonists affecting these presynaptic receptors can have large effects on sympathetic transmission. The physiological significance of presynaptic autoinhibition in the sympathetic nervous system is still somewhat contentious, and there is evidence that, in most tissues, it is less influential than biochemical measurements of transmitter overflow would imply. Thus, although blocking autoreceptors causes large changes in noradrenaline overflow—the amount of noradrenaline released into the bathing solution or the bloodstream when sympathetic nerves are stimulated—the associated changes in the tissue response are often rather small. This suggests that what is measured in overflow experiments may not be the physiologically important component of transmitter release.
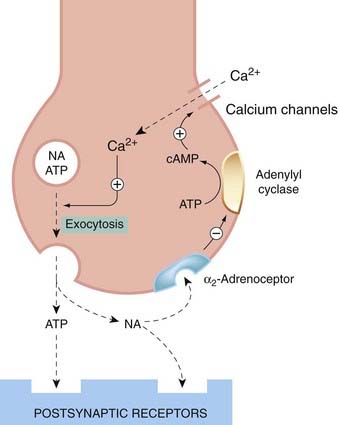
Fig. 14.3 Feedback control of noradrenaline release.
The presynaptic α2 receptor inhibits adenylyl cyclase, thereby reducing intracellular cAMP. cAMP acts to promote Ca2+ influx in response to membrane depolarisation, and hence to promote the release of noradrenaline and ATP.
The inhibitory feedback mechanism operates through α2 receptors, which inhibit adenylyl cyclase and prevent the opening of calcium channels. Sympathetic nerve terminals also possess β2 receptors, coupled to activation of adenylyl cyclase, which cause an increased noradrenaline release. Whether they have any physiological function is not yet clear.
Uptake and Degradation of Catecholamines
The action of released noradrenaline is terminated mainly by reuptake of the transmitter into noradrenergic nerve terminals. Some is also sequestered by other cells in the vicinity. Circulating adrenaline and noradrenaline are degraded enzymically, but much more slowly than acetylcholine (see Ch. 13), where synaptically located acetylcholinesterase inactivates the transmitter in milliseconds. The two main catecholamine-metabolising enzymes are located intracellularly, so uptake into cells necessarily precedes metabolic degradation.
Uptake of Catecholamines
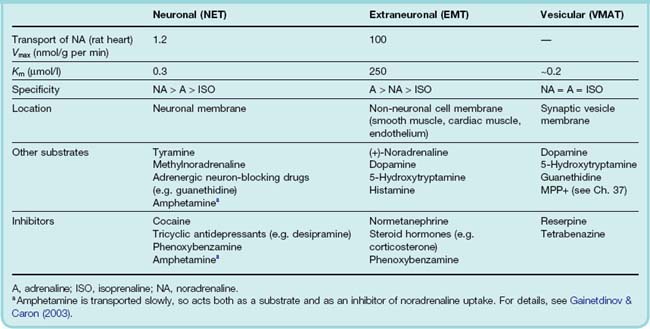
About 75% of the noradrenaline released by sympathetic neurons is recaptured and repackaged into vesicles. This serves to cut short the action of the released noradrenaline, as well as recycling it. The remaining 25% is captured by non-neuronal cells in the vicinity, limiting its local spread. These two uptake mechanisms depend on distinct transporter molecules. Neuronal uptake is performed by the plasma membrane noradrenaline transporter (generally known as NET, the norepinephrine transporter), which belongs to the family of neurotransmitter transporter proteins (NET, DAT, SERT, etc.) specific for different amine transmitters, described in Chapter 12; these act as co-transporters of Na+, Cl− and the amine in question, using the electrochemical gradient for Na+ as a driving force. Packaging into vesicles occurs through the vesicular monoamine transporter (VMAT), driven by the proton gradient between the cytosol and the vesicle contents. Extraneuronal uptake is performed by the extraneuronal monoamine transporter (EMT), which belongs to a large and widely distributed family of organic cation transporters. NET is relatively selective for noradrenaline, with high affinity and a low maximum rate of uptake, and it is important in maintaining releasable stores of noradrenaline. EMT has lower affinity and higher transport capacity than NET, and transports adrenaline and isoprenaline as well as noradrenaline. The effects of several important drugs that act on noradrenergic neurons depend on their ability either to inhibit NET or to enter the nerve terminal with its help. Table 14.2 summarises the properties of neuronal and extraneuronal uptake.
Metabolic Degradation of Catecholamines
Endogenous and exogenous catecholamines are metabolised mainly by two enzymes: monoamine oxidase (MAO) and catechol-O-methyl transferase (COMT). MAO (of which there are two distinct isoforms, MAO-A and MAO-B; see Chs 38 and 46) occurs within cells, bound to the surface membrane of mitochondria. It is abundant in noradrenergic nerve terminals but is also present in many other places, such as liver and intestinal epithelium. MAO converts catecholamines to their corresponding aldehydes,3 which, in the periphery, are rapidly metabolised by aldehyde dehydrogenase to the corresponding carboxylic acid (3,4-dihydroxyphenylglycol being formed from noradrenaline; Fig. 14.4). MAO can also oxidise other monoamines, including dopamine and 5-HT. It is inhibited by various drugs (see Table 14.3), which are used mainly for their effects on the central nervous system, where these three amines all have transmitter functions (see Ch. 38). These drugs have important side effects that are related to disturbances of peripheral noradrenergic transmission. Within sympathetic neurons, MAO controls the content of dopamine and noradrenaline, and the releasable store of noradrenaline increases if the enzyme is inhibited. MAO and its inhibitors are discussed in more detail in Chapter 46.
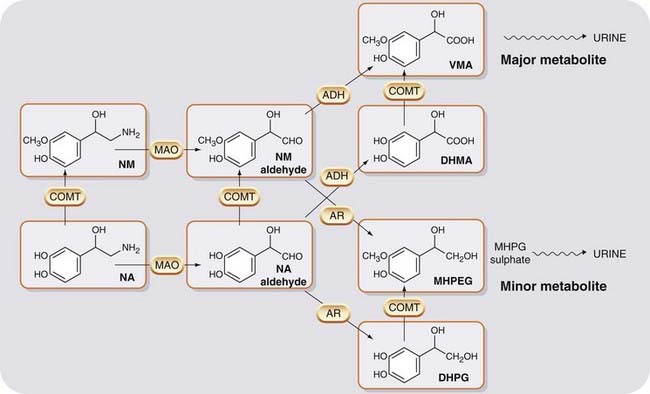
Fig. 14.4 The main pathways of noradrenaline metabolism.
The oxidative branch (catalysed by ADH) predominates, giving VMA as the main urinary metabolite. The reductive branch (catalysed by AR) produces the less abundant metabolite, MHPG, which is conjugated to MHPG sulfate before being excreted. ADH, aldehyde dehydrogenase; AR, aldehyde reductase; COMT, catechol-O-methyl transferase; DHMA, 3,4-dihydroxymandelic acid; DHPG, 3,4-dihydroxyphenylglycol; MAO, monoamine oxidase; MHPG, 3-methoxy-4-hydroxyphenylglycol; NA, noradrenaline; NM, normetanephrine; VMA, vanillylmandelic acid.
The second major pathway for catecholamine metabolism involves methylation of one of the catechol hydroxyl groups by COMT to give a methoxy derivative. COMT is absent from noradrenergic neurons but present in the adrenal medulla and many other cells and tissues. The final product formed by the sequential action of MAO and COMT is 3-methoxy-4-hydroxyphenylglycol (MHPG; see Fig. 14.4). This is partly conjugated to sulfate or glucuronide derivatives, which are excreted in the urine, but most of it is converted to vanillylmandelic acid (VMA; Fig. 14.4) and excreted in the urine in this form.4 In patients with tumours of chromaffin tissue that secrete these amines (a rare cause of high blood pressure), the urinary excretion of VMA is markedly increased, this being used as a diagnostic test for this condition.
In the periphery, neither MAO nor COMT is primarily responsible for the termination of transmitter action, most of the released noradrenaline being quickly recaptured by NET. Circulating catecholamines are sequestered and inactivated by a combination of NET, EMT and COMT, the relative importance of these processes varying according to the agent concerned. Thus circulating noradrenaline is removed mainly by NET, whereas adrenaline is more dependent on EMT. Isoprenaline, on the other hand, is not a substrate for NET, and is removed by a combination of EMT and COMT.
In the central nervous system (see Ch. 38), MAO is more important as a means of terminating transmitter action than it is in the periphery, and MAO knockout mice show a greater enhancement of noradrenergic transmission in the brain than do NET knockouts, in which neuronal stores of noradrenaline are much depleted (see Gainetdinov & Caron, 2003). The main excretory product of noradrenaline released in the brain is MHPG.
Noradrenergic transmission
Drugs Acting on Noradrenergic Transmission
Many clinically important drugs, particularly those used to treat cardiovascular, respiratory and psychiatric disorders (see Chs 21, 22, 27 and 46), act by affecting noradrenergic neuron function, acting on adrenoceptors, transporters or catecholamine-metabolising enzymes. The properties of the most important drugs in this category are summarised in Table 14.3.
Drugs Acting on Adrenoceptors
The overall activity of these drugs is governed by their affinity, efficacy and selectivity with respect to different types of adrenoceptor, and intensive research has been devoted to developing drugs with the right properties for specific clinical indications. As a result, the pharmacopoeia is awash with adrenoceptor ligands. Many clinical needs are met, it turns out, by drugs that relax smooth muscle in different organs of the body5 and those that block the cardiac stimulant effects of the sympathetic nervous system; on the other hand, cardiac stimulation is generally undesirable.
Broadly speaking, β-adrenoceptor agonists are useful as smooth muscle relaxants (especially in the airways), while β-adrenoceptor antagonists (often called β-blockers) are used mainly for their cardiodepressant effects. α-Adrenoceptor antagonists are used mainly for their vasodilator effects in cardiovascular indications and also for the treatment of prostatic hyperplasia. α-Adrenoceptor agonists have few clinical uses.
Adrenoceptor Agonists
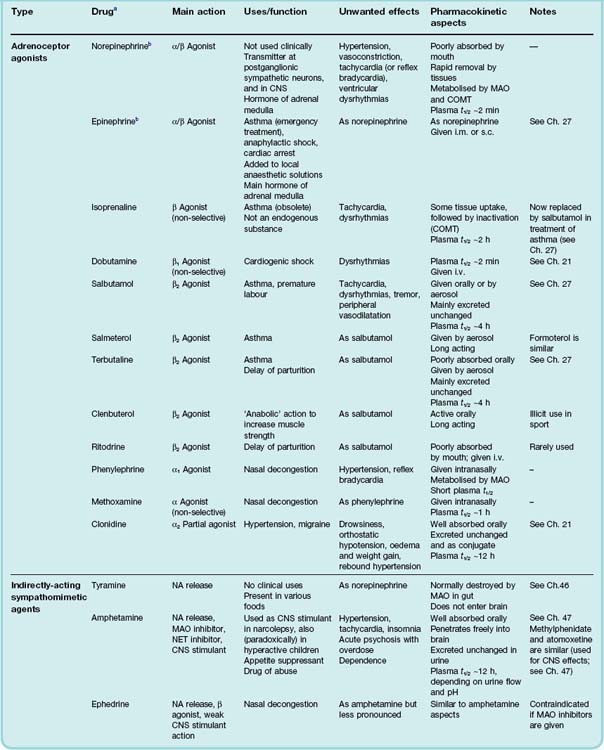
Examples of adrenoceptor agonists (also known as directly-acting sympathomimetic drugs) are given in Table 14.1, and the characteristics of individual drugs are summarised in Table 14.3.
Actions
The major physiological effects mediated by different types of adrenoceptor are summarised in Table 14.1.
Smooth muscle
All types of smooth muscle, except that of the gastrointestinal tract, contract in response to stimulation of α1-adrenoceptors, through activation of the signal transduction mechanism described in Chapter 4.
When α agonists are given systemically to experimental animals or humans, the most important action is on vascular smooth muscle, particularly in the skin and splanchnic vascular beds, which are strongly constricted. Large arteries and veins, as well as arterioles, are also constricted, resulting in decreased vascular compliance, increased central venous pressure and increased peripheral resistance, all of which contribute to an increase in systolic and diastolic arterial pressure and increased cardiac work. Some vascular beds (e.g. cerebral, coronary and pulmonary) are relatively little affected.
In the whole animal, baroreceptor reflexes are activated by the rise in arterial pressure produced by α agonists, causing reflex bradycardia and inhibition of respiration.
Smooth muscle in the vas deferens, spleen capsule and eyelid retractor muscles (or nictitating membrane, in some species) is also stimulated by α agonists, and these organs are often used for pharmacological studies.
The α receptors involved in smooth muscle contraction are mainly α1 in type, although vascular smooth muscle possesses both α1 and α2 receptors. It appears that α1 receptors lie close to the sites of release (and are mainly responsible for neurally mediated vasoconstriction), while α2 receptors lie elsewhere on the muscle fibre surface and are activated by circulating catecholamines.
Stimulation of β receptors causes relaxation of most kinds of smooth muscle by increasing cAMP formation (see Ch. 4). Additionally, β-receptor activation enhances Ca2+ extrusion and intracellular Ca2+ binding, both effects acting to reduce intracellular Ca2+ concentration.
Relaxation is usually produced by β2 receptors, although the receptor that is responsible for this effect in gastrointestinal smooth muscle is not clearly β1 or β2. In the vascular system, β2-mediated vasodilatation is (particularly in humans) mainly endothelium dependent and mediated by nitric oxide release (see Ch. 20). It occurs in many vascular beds and is especially marked in skeletal muscle.
The powerful inhibitory effect of the sympathetic system on gastrointestinal smooth muscle is produced by both α and β receptors, this tissue being unusual in that α receptors cause relaxation in most regions. Part of the effect is due to stimulation of presynaptic α2 receptors (see below), which inhibit the release of excitatory transmitters (e.g. acetylcholine) from intramural nerves, but there are also α receptors on the muscle cells, stimulation of which hyperpolarises the cell (by increasing the membrane permeability to K+) and inhibits action potential discharge. The sphincters of the gastrointestinal tract are contracted by α-receptor activation.
Bronchial smooth muscle is relaxed by activation of β2-adrenoceptors, and selective β2 agonists are important in the treatment of asthma (see Ch. 27). Uterine smooth muscle responds similarly, and these drugs are also used to delay premature labour (Ch. 34).
α1-Adrenoceptors also mediate a long-lasting trophic response, stimulating smooth muscle proliferation in various tissues, for example in blood vessels and in the prostate gland, which is of pathological importance. Benign prostatic hyperplasia (see Ch. 34) is commonly treated with α-adrenoceptor antagonists (see the clinical box on p. 187). ‘Cross-talk’ between the α1-adrenoceptor and the growth factor signalling pathways (see Ch. 3) probably accounts for this effect.
Nerve terminals
Presynaptic adrenoceptors are present on both cholinergic and noradrenergic nerve terminals (see Chs 4 and 12). The main effect (α2 mediated) is inhibitory, but a weaker facilitatory action of β receptors on noradrenergic nerve terminals has also been described.
Heart
Catecholamines, acting on β1 receptors, exert a powerful stimulant effect on the heart (see Ch. 21). Both the heart rate (chronotropic effect) and the force of contraction (inotropic effect) are increased, resulting in a markedly increased cardiac output and cardiac oxygen consumption. The cardiac efficiency (see Ch. 21) is reduced. Catecholamines can also cause disturbance of the cardiac rhythm, culminating in ventricular fibrillation. (Paradoxically, but importantly, adrenaline is also used to treat ventricular fibrillation arrest as well as other forms of cardiac arrest; Ch. 21, Table 21.1, p. 255.). In normal hearts, the dose required to cause marked dysrhythmia is greater than that which produces the chronotropic and inotropic effects, but in ischaemic conditions dysrhythmias are produced much more readily. Figure 14.5 shows the overall pattern of cardiovascular responses to catecholamine infusions in humans, reflecting their actions on both the heart and vascular system.
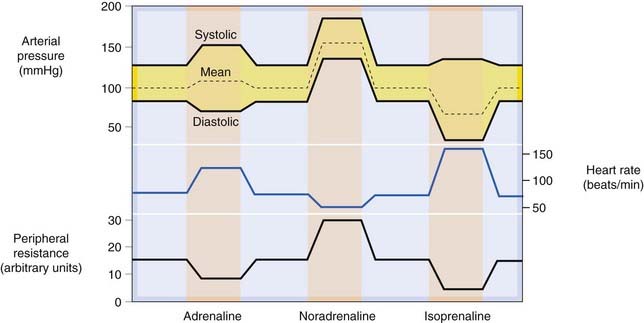
Fig. 14.5 Schematic representation of the cardiovascular effects of intravenous infusions of adrenaline, noradrenaline and isoprenaline in humans.
Noradrenaline (predominantly α agonist) causes vasoconstriction and increased systolic and diastolic pressure, with a reflex bradycardia. Isoprenaline (β agonist) is a vasodilator, but strongly increases cardiac force and rate. Mean arterial pressure falls. Adrenaline combines both actions.
Cardiac hypertrophy occurs in response to activation of both β1 and α1 receptors, probably by a mechanism similar to the hypertrophy of vascular and prostatic smooth muscle. This may be important in the pathophysiology of hypertension and cardiac failure, conditions associated with sympathetic overactivity (see Ch. 21).
Metabolism
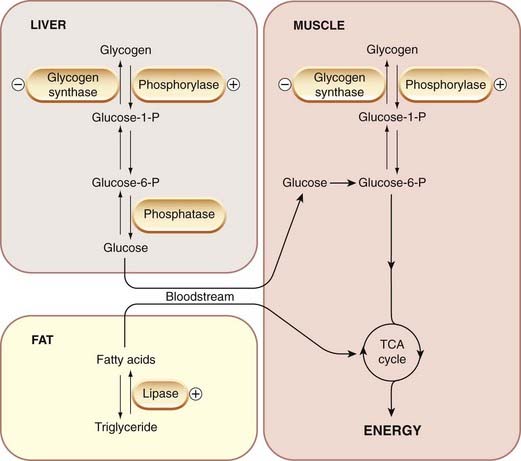
Catecholamines encourage the conversion of energy stores (glycogen and fat) to freely available fuels (glucose and free fatty acids), and cause an increase in the plasma concentration of the latter substances. The detailed biochemical mechanisms (see review by Nonogaki, 2000) vary from species to species, but in most cases the effects on carbohydrate metabolism of liver and muscle (Fig. 14.6) are mediated through β1 receptors (although hepatic glucose release can also be produced by α agonists), and the stimulation of lipolysis is produced by β3 receptors (see Table 14.1). Activation of α2 receptors inhibits insulin secretion, an effect that further contributes to the hyperglycaemia. The production of leptin by adipose tissue (see Ch. 31) is also inhibited. Adrenaline-induced hyperglycaemia in humans is blocked completely by a combination of α and β antagonists but not by either on its own. Selective β3-receptor agonists (e.g. BRL 37344) have been developed as possible treatments for obesity, but their action is too transient for them to be clinically useful.
Other effects
Skeletal muscle is affected by adrenaline, acting on β2 receptors, although the effect is far less dramatic than that on the heart. The twitch tension of fast-contracting fibres (white muscle) is increased by adrenaline, particularly if the muscle is fatigued, whereas the twitch of slow (red) muscle is reduced. These effects depend on an action on the contractile proteins, rather than on the membrane, and the mechanism is poorly understood. In humans, adrenaline and other β2 agonists cause a marked tremor, the shakiness that accompanies fear, excitement or the excessive use of β2 agonists (e.g. salbutamol) in the treatment of asthma being examples of this. It probably results from an increase in muscle spindle discharge, coupled with an effect on the contraction kinetics of the fibres, these effects combining to produce an instability in the reflex control of muscle length. β-Receptor antagonists are sometimes used to control pathological tremor. The tendency to cardiac dysrhythmias associated with β2 agonists is thought to be partly due to hypokalaemia, caused by an increase in K+ uptake by skeletal muscle. β2 Agonists also cause long-term changes in the expression of sarcoplasmic reticulum proteins that control contraction kinetics, and thereby increase the rate and force of contraction of skeletal muscle (see Zhang et al., 1996). Clenbuterol, an ‘anabolic’ drug used illicitly by athletes to improve performance (see Ch. 58), is a β2 agonist that acts in this way.
Histamine release by human and guinea pig lung tissue in response to anaphylactic challenge (see Ch. 17) is inhibited by catecholamines, acting on β2 receptors.
Lymphocytes and other cells of the immune system also express adrenoceptors (mainly β-adrenoceptors). Lymphocyte proliferation, lymphocyte-mediated cell killing, and production of many cytokines are inhibited by β-adrenoceptor agonists. The physiological and clinical importance of these effects has not yet been established. For a review of the effects of the sympathetic nervous system on immune function, see Elenkov et al., 2000.
Adrenoceptor agonists
Clinical use
The main clinical uses of adrenoceptor agonists are summarised in the clinical box above and Table 14.3, the most important being the use of β-adrenoceptor agonists for the treatment of asthma (Ch. 27).
Clinical uses of adrenoceptor agonists 
Adrenoceptor Antagonists
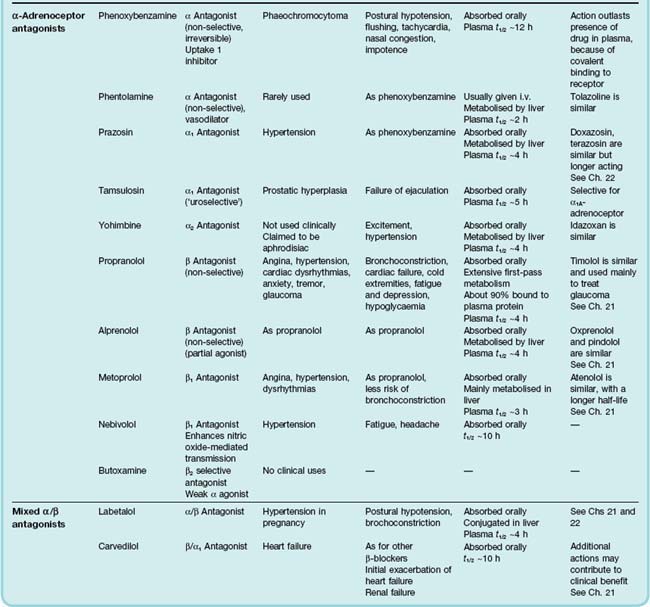
The main drugs are listed in Table 14.1, and further information is given in Table 14.3. Most are selective for α or β receptors, and many are also subtype selective.
α-Adrenoceptor antagonists
The main groups of α-adrenoceptor antagonists are:
In addition, ergot derivatives (e.g. ergotamine, dihydroergotamine) block α receptors as well as having many other actions, notably on 5-HT receptors. They are described in Chapter 15. Their action on α-adrenoceptors is of pharmacological interest (see p. 174) but not used therapeutically.
Non-selective α-adrenoceptor antagonists
Phenoxybenzamine is not specific for α receptors, and also antagonises the actions of acetylcholine, histamine and 5-HT. It is long lasting because it binds covalently to the receptor. Phentolamine is more selective, but it binds reversibly and its action is short lasting. In humans, these drugs cause a fall in arterial pressure (because of block of α-receptor-mediated vasoconstriction) and postural hypotension. The cardiac output and heart rate are increased. This is a reflex response to the fall in arterial pressure, mediated through β receptors. The concomitant block of α2 receptors tends to increase noradrenaline release, which has the effect of enhancing the reflex tachycardia that occurs with any blood pressure-lowering agent. Phenoxybenzamine retains a niche (but vital) use in preparing patients with phaeochromocytoma (see below) for surgery; its irreversible antagonism and the resultant depression in the maximum of the agonist dose–response curve (see Ch. 2, Fig. 2.4, p. 10) are desirable in a situation where surgical manipulation of the tumour may release a large bolus of pressor amines into the circulation.
Labetalol and carvedilol are mixed α1- and β-receptor-blocking drugs, although clinically they act predominantly on β receptors. Much has been made of the fact that they combine both activities in one molecule. To a pharmacologist, accustomed to putting specificity of action high on the list of pharmacological saintly virtues, this may seem like a step backwards rather than forwards. Carvedilol is used mainly to treat hypertension and heart failure (see Chs 21 and 22); labetalol is used to treat hypertension in pregnancy.
Selective α1 antagonists
Prazosin was the first α1-selective antagonist. Similar drugs with longer half-lives (e.g. doxazosin, terazosin), which have the advantage of allowing once-daily dosing, are now available. They are highly selective for α1-adrenoceptors and cause vasodilatation and fall in arterial pressure, but less tachycardia than occurs with non-selective α-receptor antagonists, presumably because they do not increase noradrenaline release from sympathetic nerve terminals. Some postural hypotension may occur.
The α1-receptor antagonists cause relaxation of the smooth muscle of the bladder neck and prostate capsule, and inhibit hypertrophy of these tissues, and are therefore useful in treating urinary retention associated with benign prostatic hypertrophy. Tamsulosin, an α1A-receptor antagonist, shows some selectivity for the bladder, and causes less hypotension than drugs such as prazosin, which act on α1B receptors to control vascular tone.
It is believed that α1A receptors play a part in the pathological hypertrophy not only of prostatic and vascular smooth muscle, but also in the cardiac hypertrophy that occurs in hypertension and heart failure, and the use of selective α1A-receptor antagonists to treat these chronic conditions is under investigation.
Selective α2 antagonists
Yohimbine is a naturally occurring alkaloid; various synthetic analogues have been made, such as idazoxan. These drugs are used experimentally to analyse α-receptor subtypes, and yohimbine, probably by virtue of its vasodilator effect, historically enjoyed notoriety as an aphrodisiac, but they are not used therapeutically.
α-Adrenoceptor antagonists
Clinical uses and unwanted effects of α-adrenoceptor antagonists
The main uses of α-adrenoceptor antagonists are related to their cardiovascular actions, and are summarised in the clinical box above. They have been tried for many purposes, but have only limited therapeutic applications. In hypertension, non-selective α-blocking drugs are unsatisfactory, because of their tendency to produce tachycardia and cardiac dysrhythmias, and increased gastrointestinal activity. Selective α1-receptor antagonists (especially the longer-acting compounds doxazosin and terazosin) are, however, useful. They do not affect cardiac function appreciably, and postural hypotension is less troublesome than with prazosin or non-selective α-receptor antagonists. They have a place in treating severe hypertension, where they are added to treatment with first- and second-line drugs, but are not used as first-line agents (see Ch. 22). Unlike other antihypertensive drugs, they cause a modest decrease in low-density lipoprotein, and an increase in high-density lipoprotein cholesterol (see Ch. 23), although the clinical importance of these ostensibly beneficial effects is uncertain. They are also used to control urinary retention in patients with benign prostatic hypertrophy.
Phaeochromocytoma is a catecholamine-secreting tumour of chromaffin tissue, which causes episodes of severe hypertension. A combination of α- and β-receptor antagonists is the most effective way of controlling the blood pressure. The tumour may be surgically removable, and it is essential to block α and β receptors before surgery is begun, to avoid the effects of a sudden release of catecholamines when the tumour is disturbed. A combination of phenoxybenzamine and atenolol is effective for this purpose.
Clinical uses of α-adrenoceptor antagonists
β-Adrenoceptor antagonists
The β-adrenoceptor antagonists are an important group of drugs. They were first discovered in 1958, 10 years after Ahlquist had postulated the existence of β-adrenoceptors. The first compound, dichloroisoprenaline, had fairly low potency and was a partial agonist. Further development led to propranolol, which is much more potent and a pure antagonist that blocks β1 and β2 receptors equally. The potential clinical advantages of drugs with some partial agonist activity, and/or with selectivity for β1 receptors, led to the development of practolol (selective for β1 receptors but withdrawn because of its toxicity), oxprenolol and alprenolol (non-selective with considerable partial agonist activity), and atenolol (β1 selective with no agonist activity). Two newer drugs are carvedilol (a non-selective β-adrenoceptor antagonist with additional α1-blocking activity) and nebivolol (a β1-selective antagonist that also causes vasodilatation by inducing endothelial nitric oxide production; see Ch. 20). Both of these drugs have proven more effective than conventional β-adrenoceptor antagonists in treating heart failure (see Ch. 21). The characteristics of the most important compounds are set out in Table 14.3. Most β-receptor antagonists are inactive on β3 receptors so do not affect lipolysis.
Actions
The pharmacological actions of β-receptor antagonists can be deduced from Table 14.1. The effects produced in humans depend on the degree of sympathetic activity and are slight in subjects at rest. The most important effects are on the cardiovascular system and on bronchial smooth muscle (see Chs 21, 22 and 27).
In a subject at rest, propranolol causes little change in heart rate, cardiac output or arterial pressure, but reduces the effect of exercise or excitement on these variables (Fig. 14.7). Drugs with partial agonist activity, such as oxprenolol, increase the heart rate at rest but reduce it during exercise. Maximum exercise tolerance is considerably reduced in normal subjects, partly because of the limitation of the cardiac response, and partly because the β-mediated vasodilatation in skeletal muscle is reduced. Coronary flow is reduced, but relatively less than the myocardial oxygen consumption, so oxygenation of the myocardium is improved, an effect of importance in the treatment of angina pectoris (see Ch. 21). In normal subjects, the reduction of the force of contraction of the heart is of no importance, but it may have serious consequences for patients with heart disease (see below).
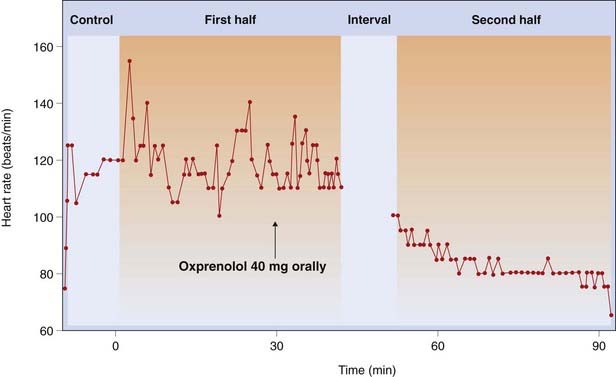
Fig. 14.7 Heart rate recorded continuously in a spectator watching a live football match, showing the effect of the β-adrenoceptor antagonist oxprenolol.
(From Taylor S H, Meeran M K 1973. In: Burley et al. (eds) New perspectives in beta-blockade. CIBA Laboratories, Horsham.)
An important, and somewhat unexpected, effect of β-receptor antagonists is their antihypertensive action (see Ch. 22). Patients with hypertension (although not normotensive subjects) show a gradual fall in arterial pressure that takes several days to develop fully. The mechanism is complex and involves the following:
Carvedilol and nebivolol (see above) are particularly effective in lowering blood pressure, because of their additional vasodilator properties.
Blockade of the facilitatory effect of presynaptic β receptors on noradrenaline release (see Table 14.1) may also contribute to the antihypertensive effect. The antihypertensive effect of β-receptor antagonists is clinically very useful. Because reflex vasoconstriction is preserved, postural and exercise-induced hypotension (see Ch. 21) are less troublesome than with many other antihypertensive drugs.
Many β-receptor antagonists have an antidysrhythmic effect on the heart, which is of clinical importance (see Ch. 21).
Airways resistance in normal subjects is only slightly increased by β-receptor antagonists, and this is of no consequence. In asthmatic subjects, however, non-selective β-receptor antagonists (such as propranolol) can cause severe bronchoconstriction, which does not, of course, respond to the usual doses of drugs such as salbutamol or adrenaline. This danger is less with β1-selective antagonists, but none is so selective that this danger can be ignored.
Despite the involvement of β receptors in the hyperglycaemic actions of adrenaline, β-receptor antagonists cause only minor metabolic changes in normal subjects. They do not affect the onset of hypoglycaemia following an injection of insulin, but somewhat delay the recovery of blood glucose concentration. In diabetic patients, the use of β-receptor antagonists increases the likelihood of exercise-induced hypoglycaemia, because the normal adrenaline-induced release of glucose from the liver is diminished.
Clinical use
The main uses of β-receptor antagonists are connected with their effects on the cardiovascular system, and are discussed in Chapters 21 and 22. They are as summarised in the clinical box above.
The use of β-receptor antagonists in cardiac failure deserves special mention, as clinical opinion has undergone a U-turn in recent years. Patients with heart disease may rely on a degree of sympathetic drive to the heart to maintain an adequate cardiac output, and removal of this by blocking β receptors can exacerbate cardiac failure, so using these drugs in patients with cardiac failure was considered ill-advised. In theory, drugs with partial agonist activity (e.g. oxprenolol, alprenolol) offer an advantage because they can, by their own action, maintain a degree of β1-receptor activation, while at the same time blunting the cardiac response to increased sympathetic nerve activity or to circulating adrenaline. Clinical trials, however, have not shown a clear advantage of these drugs measurable as a reduced incidence of cardiac failure.
Paradoxically, β-receptor antagonists are increasingly being used in low doses to treat cardiac failure, although at the outset there is a danger of exacerbating the problem. Several mechanisms may contribute, including inhibition of central sympathetic outflow, direct vasodilator effects (see review by Pfeffer & Stevenson, 1996) and prevention of cardiac hypertrophy by interference with signalling pathways other than the major cAMP pathway—a phenomenon still poorly understood. Carvedilol is often used for this purpose.
Unwanted effects
The main side effects of β-receptor antagonists result from their receptor-blocking action.
Bronchoconstriction
This is of little importance in the absence of airways disease, but in asthmatic patients the effect can be dramatic and life-threatening. It is also of clinical importance in patients with other forms of obstructive lung disease (e.g. chronic bronchitis, emphysema).
Cardiac depression
Cardiac depression can occur, leading to signs of heart failure, particularly in elderly people. Patients suffering from heart failure who are treated with β-receptor antagonists (see above) often deteriorate in the first few weeks before the beneficial effect develops.
Bradycardia
This side effect can lead to life-threatening heart block and can occur in patients with coronary disease, particularly if they are being treated with antiarrhythmic drugs that impair cardiac conduction (see Ch. 21).
Hypoglycaemia
Glucose release in response to adrenaline is a safety device that may be important to diabetic patients and to other individuals prone to hypoglycaemic attacks. The sympathetic response to hypoglycaemia produces symptoms (especially tachycardia) that warn patients of the urgent need for carbohydrate (usually in the form of a sugary drink). β-Receptor antagonists reduce these symptoms, so incipient hypoglycaemia is more likely to go unnoticed by the patient. The use of β-receptor antagonists is generally to be avoided in patients with poorly controlled diabetes. There is a theoretical advantage in using β1-selective agents, because glucose release from the liver is controlled by β2 receptors.
Fatigue
This is probably due to reduced cardiac output and reduced muscle perfusion in exercise. It is a frequent complaint of patients taking β-receptor-blocking drugs.
Cold extremities
This is due to a loss of β-receptor-mediated vasodilatation in cutaneous vessels, and is a common side effect. Theoretically, β1-selective drugs are less likely to produce this effect, but it is not clear that this is so in practice.
Other side effects associated with β-receptor antagonists are not obviously the result of β-receptor blockade. One is the occurrence of bad dreams, which occur mainly with highly lipid-soluble drugs such as propranolol, which enter the brain easily.
 There are several additional factors that make β-adrenoceptor pharmacology more complicated than it appears at first sight, and may have implications for the clinical use of β-adrenoceptor antagonists:
There are several additional factors that make β-adrenoceptor pharmacology more complicated than it appears at first sight, and may have implications for the clinical use of β-adrenoceptor antagonists:
Drugs That Affect Noradrenergic Neurons
Emphasis in this chapter is placed on peripheral sympathetic transmission. The same principles, however, are applicable to the central nervous system (see Ch. 36), where many of the drugs mentioned here also act.
Drugs That Affect Noradrenaline Synthesis
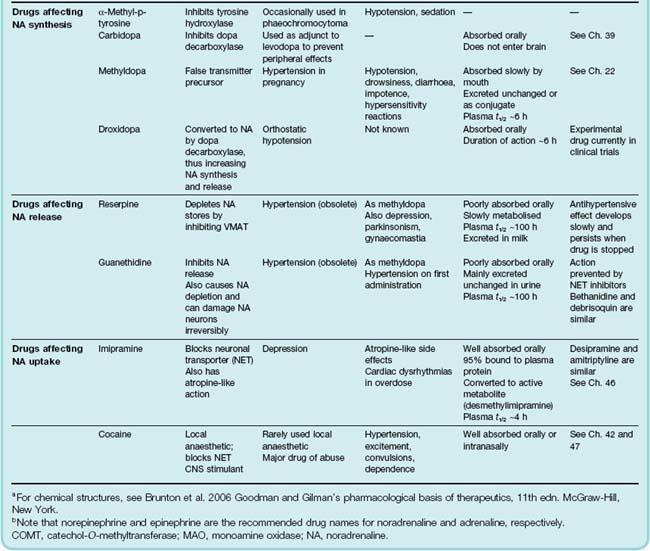
Only a few clinically important drugs affect noradrenaline synthesis directly. Examples are α-methyltyrosine, which inhibits tyrosine hydroxylase (used rarely to treat phaeochromocytoma), and carbidopa, a hydrazine derivative of dopa, which inhibits dopa decarboxylase and is used in the treatment of parkinsonism (see Ch. 39).
Methyldopa, a drug still used in the treatment of hypertension during pregnancy (see Ch. 22), is taken up by noradrenergic neurons, where it is converted to the false transmitter α-methylnoradrenaline. This substance is not deaminated within the neuron by MAO, so it accumulates and displaces noradrenaline from the synaptic vesicles. α-Methylnoradrenaline is released in the same way as noradrenaline, but is less active than noradrenaline on α1 receptors and thus is less effective in causing vasoconstriction. On the other hand, it is more active on presynaptic (α2) receptors, so the autoinhibitory feedback mechanism operates more strongly than normal, thus reducing transmitter release below the normal levels. Both of these effects (as well as a central effect, probably caused by the same cellular mechanism) contribute to the hypotensive action. It produces side effects typical of centrally acting antiadrenergic drugs (e.g. sedation), as well as carrying a risk of immune haemolytic reactions and liver toxicity, so it is now little used, except for hypertension in late pregnancy.
6-Hydroxydopamine (identical with dopamine except that it possesses an extra ring hydroxyl group) is a neurotoxin of the Trojan horse kind. It is taken up selectively by noradrenergic nerve terminals, where it is converted to a reactive quinone, which destroys the nerve terminal, producing a ‘chemical sympathectomy’. The cell bodies survive, and eventually the sympathetic innervation recovers. The drug is useful for experimental purposes but has no clinical uses. If injected directly into the brain, it selectively destroys those nerve terminals (i.e. dopaminergic, noradrenergic and adrenergic) that take it up, but it does not reach the brain if given systemically. MPTP (1-methyl-4-phenyl-1,2,3,5-tetrahydropyridine; see Ch. 39) is a similar selective neurotoxin.
Droxidopa (dihydroxyphenylserine, DOPS) is currently under investigation for treating hypotensive states associated with reduced noradrenaline synthesis. It can be regarded as β-hydroxy-dopa, which is converted to noradrenaline directly by dopa decarboxylase, bypassing the DBH-catalysed hydroxylation step, which is normally rate limiting. It raises blood pressure by increasing noradrenaline release.
Drugs That Affect Noradrenaline Storage
Reserpine is an alkaloid from the shrub Rauwolfia, which has been used in India for centuries for the treatment of mental disorders. Reserpine, at very low concentration, blocks the transport of noradrenaline and other amines into synaptic vesicles, by blocking the vesicular monoamine transporter. Noradrenaline accumulates instead in the cytoplasm, where it is degraded by MAO. The noradrenaline content of tissues drops to a low level, and sympathetic transmission is blocked. Reserpine also causes depletion of 5-HT and dopamine from neurons in the brain, in which these amines are transmitters (see Ch. 38). Reserpine is now used only experimentally, but was at one time used as an antihypertensive drug. Its central effects, especially depression, which probably result from impairment of noradrenergic and 5-HT-mediated transmission in the brain (see Ch. 46), are a serious disadvantage.
Drugs That Affect Noradrenaline Release
Drugs can affect noradrenaline release in four main ways:
Noradrenergic Neuron-Blocking Drugs
Noradrenergic neuron-blocking drugs (e.g. guanethidine) were first discovered in the mid-1950s when alternatives to ganglion-blocking drugs, for use in the treatment of hypertension, were being sought. The main effect of guanethidine is to inhibit the release of noradrenaline from sympathetic nerve terminals. It has little effect on the adrenal medulla, and none on nerve terminals that release transmitters other than noradrenaline. Drugs very similar to it include bretylium, bethanidine and debrisoquin (which is of interest mainly as a tool for studying drug metabolism; see Ch. 11).
Actions
Drugs of this class reduce or abolish the response of tissues to sympathetic nerve stimulation, but do not affect (or may potentiate) the effects of circulating noradrenaline.
The action of guanethidine on noradrenergic transmission is complex (see Broadley, 1996). It is selectively accumulated by noradrenergic nerve terminals, being a substrate for NET (see above). Its initial blocking activity is due to block of impulse conduction in the nerve terminals that selectively accumulate the drug. Its action is prevented by drugs, such as tricyclic antidepressants (see Ch. 46), which block NET.
Guanethidine is also concentrated in synaptic vesicles by means of the vesicular transporter VMAT, possibly interfering with their ability to undergo exocytosis, and also displacing noradrenaline. In this way, it causes a gradual and long-lasting depletion of noradrenaline in sympathetic nerve endings, similar to the effect of reserpine.
Given in large doses, guanethidine causes structural damage to noradrenergic neurons, which is probably due to the fact that the terminals accumulate the drug in high concentration. It can therefore be used experimentally as a selective neurotoxin.
Guanethidine, bethanidine and debrisoquin are no longer used clinically, now that better antihypertensive drugs are available. Although extremely effective in lowering blood pressure, they produce severe side effects associated with the loss of sympathetic reflexes. The most troublesome are postural hypotension, diarrhoea, nasal congestion and failure of ejaculation.
Indirectly Acting Sympathomimetic Amines
Mechanism of action and structure–activity relationships
The most important drugs in the indirectly acting sympathomimetic amine category are tyramine, amphetamine and ephedrine, which are structurally related to noradrenaline. Drugs that act similarly and are used for their central effects (see Ch. 47) include methylphenidate and atomoxetine.
These drugs have only weak actions on adrenoceptors, but sufficiently resemble noradrenaline to be transported into nerve terminals by NET. Once inside the nerve terminals, they are taken up into the vesicles by VMAT, in exchange for noradrenaline, which escapes into the cytosol. Some of the cytosolic noradrenaline is degraded by MAO, while the rest escapes via NET, in exchange for the foreign monoamine, to act on postsynaptic receptors (Fig. 14.8). Exocytosis is not involved in the release process, so their actions do not require the presence of Ca2+. They are not completely specific in their actions, and act partly by a direct effect on adrenoceptors, partly by inhibiting NET (thereby enhancing the effect of the released noradrenaline) and partly by inhibiting MAO.
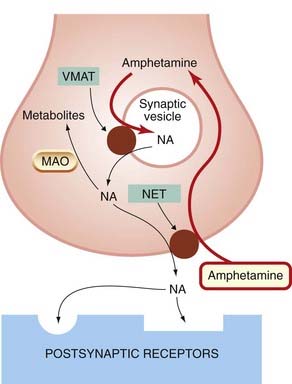
Fig. 14.8 The mode of action of amphetamine, an indirectly acting sympathomimetic amine.
Amphetamine enters the nerve terminal via the noradrenaline transporter (NET) and enters synaptic vesicles via the vesicular monoamine transporter (VMAT), in exchange for NA, which accumulates in the cytosol. Some of the NA is degraded by monoamine oxidase (MAO) within the nerve terminal and some escapes, in exchange for amphetamine via the noradrenaline transporter, to act on postsynaptic receptors. Amphetamine also reduces NA reuptake via the transporter, so enhancing the action of the released NA.
As would be expected, the effects of these drugs are strongly influenced by other drugs that modify noradrenergic transmission. Thus reserpine and 6-hydroxydopamine abolish their effects by depleting the terminals of noradrenaline. MAO inhibitors, on the other hand, strongly potentiate their effects by preventing inactivation, within the terminals, of the transmitter displaced from the vesicles. MAO inhibition particularly enhances the action of tyramine, because this substance is itself a substrate for MAO. Normally, dietary tyramine is destroyed by MAO in the gut wall and liver before reaching the systemic circulation. When MAO is inhibited this is prevented, and ingestion of tyramine-rich foods such as fermented cheese (e.g. ripe Brie) can then provoke a sudden and dangerous rise in blood pressure. Inhibitors of NET, such as imipramine (see below), interfere with the effects of indirectly acting sympathomimetic amines by preventing their uptake into the nerve terminals.
These drugs, especially amphetamine, have important effects on the central nervous system (see Ch. 47) that depend on their ability to release not only noradrenaline, but also 5-HT and dopamine from nerve terminals in the brain. An important characteristic of the effects of indirectly acting sympathomimetic amines is that marked tolerance develops. Repeated doses of amphetamine or tyramine, for example, produce progressively smaller pressor responses. This is probably caused by a depletion of the releasable store of noradrenaline. A similar tolerance to the central effects also develops with repeated administration, which partly accounts for the liability of amphetamine and related drugs to cause dependence.
Actions
The peripheral actions of the indirectly acting sympathomimetic amines include bronchodilatation, raised arterial pressure, peripheral vasoconstriction, increased heart rate and force of myocardial contraction, and inhibition of gut motility. They have important central actions, which account for their significant abuse potential and for their limited therapeutic applications (see Chs 47 and 58). Apart from ephedrine, which is still sometimes used as a nasal decongestant because it has much less central action, these drugs are no longer used for their peripheral sympathomimetic effects.
Inhibitors of Noradrenaline Uptake
Reuptake of released noradrenaline by NET is the most important mechanism by which its action is brought to an end. Many drugs inhibit NET, and thereby enhance the effects of both sympathetic nerve activity and circulating noradrenaline. NET is not responsible for clearing circulating adrenaline, so these drugs do not affect responses to this amine.
The main class of drugs whose primary action is inhibition of NET are the tricyclic antidepressants (see Ch. 46), for example imipramine. These drugs have their major effect on the central nervous system but also cause tachycardia and cardiac dysrhythmias, reflecting their peripheral effect on sympathetic transmission. Cocaine, known mainly for its abuse liability (Ch. 48) and local anaesthetic activity (Ch. 42), enhances sympathetic transmission, causing tachycardia and increased arterial pressure. Its central effects of euphoria and excitement (Ch. 47) are probably a manifestation of the same mechanism acting in the brain. It strongly potentiates the actions of noradrenaline in experimental animals or in isolated tissues provided the sympathetic nerve terminals are intact.
Many drugs that act mainly on other steps in sympathetic transmission also inhibit NET to some extent, presumably because the carrier molecule has structural features in common with other noradrenaline recognition sites, such as receptors and degradative enzymes.
The extraneuronal monoamine transporter EMT, which is important in clearing circulating adrenaline from the bloodstream, is not affected by most of the drugs that block NET. It is inhibited by phenoxybenzamine, however, and also by various corticosteroids (see Ch. 26). This action of corticosteroids may have some relevance to their therapeutic effect in conditions such as asthma, but is probably of minor importance.
The main sites of action of drugs that affect adrenergic transmission are summarised in Figure 14.9.
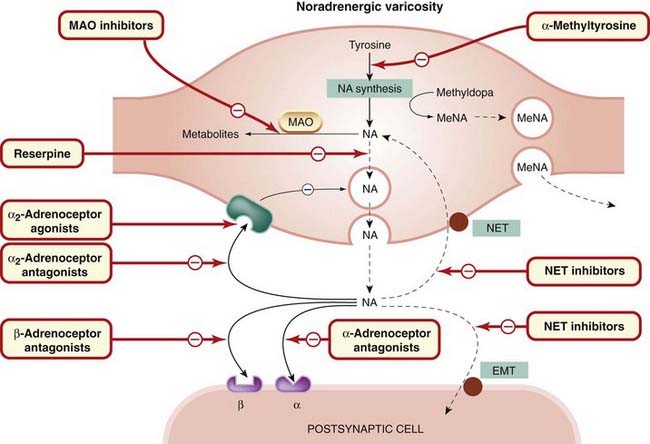
Fig. 14.9 Generalised diagram of a noradrenergic nerve terminal, showing sites of drug action.
EMT, extraneuronal monoamine transporter; MAO, monoamine oxidase; MeNA, methylnoradrenaline; NA, noradrenaline; NET, neuronal noradrenaline transporter.
Drugs acting on noradrenergic nerve terminals
References and Further Reading
Broadley K.J. Autonomic pharmacology. London: Taylor & Francis; 1996. (Detailed textbook)
Cooper J.R., Bloom F.E., Roth R.H. The biochemical basis of neuropharmacology. New York: Oxford University Press; 1996. (Excellent standard textbook)
Robertson D.W., editor. Primer on the autonomic nervous system. New York: Academic Press, 2004. (An excellent comprehensive textbook on all aspects, including pharmacology, of the autonomic nervous system. By no means elementary despite its title)
Trendelenburg U., Weiner N. Catecholamines. Handbook of experimental pharmacology vol. 90. 1988 Springer-Verlag Berlin. (Massive compilation of knowledge to date) parts 1 and 2
Baker J.G., Hall I.P., Hill S.J. Agonist and inverse agonist actions of β-blockers at the human β2-adrenoceptor provide evidence for agonist-directed signalling. Mol. Pharmacol.. 2003;64:1357-1369. (Recent studies showing that β-blockers differ in their ability to activate and block cAMP and mitogen-activated protein kinase pathways, possibly explaining why some are better than others in treating heart disease)
Brodde O. β1- and β2-Adrenoceptor polymorphisms and cardiovascular diseases. Fund. Clin. Pharmacol.. 2008;22:107-125. (Comprehensive review of possible genetic influences on human response to drugs acting on β-adrenoceptors)
Guimaraes S., Moura D. Vascular adrenoceptors: an update. Pharmacol. Rev.. 2001;53:319-356. (Review describing the complex roles of different adrenoceptors in blood vessels)
Insel P.A. Adrenergic receptors—evolving concepts and clinical implications. New Engl. J. Med.. 1996;334:580-585. (Excellent review focusing on applications)
Bylund D.B., Eikenberg D.C., Hieble J.P., et al. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol. Rev.. 1994;46:121-136. (Rationalisation of the taxonomy of adrenoceptors)
Cunnane T.C. The mechanism of neurotransmitter release from sympathetic nerves. Trends Neurosci.. 1984;7:248-253. (Points out important differences between adrenergic and cholinergic neurons)
Elenkov I.J., Wilder R.L., Chrousos G.P., et al. The sympathetic nerve—an integrative interface between two supersystems: the brain and the immune system. Pharmacol. Rev.. 2000;52:595-638. (Detailed catalogue of effects of catecholamines and the sympathetic nervous system on the immune system)
Gainetdinov R.R., Caron M.G. Monoamine transporters: from genes to behaviour. Annu. Rev. Pharmacol. Toxicol.. 2003;43:261-284. (Review article focusing on the characteristics of transgenic mice lacking specific monoamine transporters)
Liu Y., Edwards R.H. The role of vesicular transport proteins in synaptic transmission and neural degeneration. Annu. Rev. Neurosci.. 1997;20:125-156. (Review of recent ideas about the functional role of transporters)
Lundberg J.M. Pharmacology of co-transmission in the autonomic nervous system: integrative aspects on amines, neuropeptides, adenosine triphosphate, amino acids and nitric oxide. Pharmacol. Rev.. 1996;48:114-192. (Comprehensive and informative review)
Philipp M., Hein L. Adrenergic receptor knockout mice: distinct functions of 9 receptor subtypes. Pharm. Ther.. 2004;101:65-74.
Starke K., Göthert M., Kilbinger H. Modulation of transmitter release by presynaptic autoreceptors. Physiol. Rev.. 1989;69:864-989. (Comprehensive review)
Eisenhofer G., Kopin I.J., Goldstein D.S. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol. Rev.. 2004;56:331-349. (Review that dismisses a number of fallacies concerning the routes by which catecholamines from different sources are metabolised and excreted)
Nonogaki K. New insights into sympathetic regulation of glucose and fat metabolism. Diabetologia. 2000;43:533-549. (Review of the complex adrenoceptor-mediated effects on the metabolism of liver, muscle and adipose tissue; up to date, but not a particularly easy read)
Pfeffer M.A., Stevenson L.W. β-Adrenergic blockers and survival in heart failure. New Engl. J. Med.. 1996;334:1396-1397. (Shows that β-adrenergic blockers in low doses can be beneficial in heart failure)
Zhang K.-M., Hu P., Wang S.-W., et al. Salbutamol changes the molecular and mechanical properties of canine skeletal muscle. J. Physiol.. 1996;496:211-220. (Surprising finding that salbutamol affects muscle function by non-receptor mechanisms)
1The conventional British names (e.g. adrenaline, noradrenaline) are used, although the recommended international non-proprietary names (rINNs) are now epinephrine and norepinephrine.
2Dale was a new recruit in the laboratories of the Wellcome pharmaceutical company, given the job of checking the potency of batches of adrenaline coming from the factory. He tested one batch at the end of a day’s experimentation on a cat that he had earlier injected with an ergot preparation. Because it produced a fall in blood pressure rather than the expected rise, he advised that the whole expensive consignment should be rejected. Unknown to him, he was given the same sample to test a few days later, and reported it to be normal. How he explained this to Wellcome’s management is not recorded.
3Aldehyde metabolites are potentially neurotoxic, and are thought to play a role in certain degenerative CNS disorders (see Ch. 39).
4The amounts of MHPG and VMA excreted are often taken to reflect noradrenaline release from sympathetic neurons and central nervous system neurons, respectively, but this is now believed to be unreliable (see Eisenhofer et al., 2004).
5And conversely, contracting smooth muscle is usually bad news. This bald statement must not be pressed too far, but the exceptions (such as nasal decongestants and drugs acting on the eye) are surprisingly few.