3 How drugs act
Molecular aspects
Overview
In this chapter, we move from the general principles of drug action outlined in Chapter 2 to the molecules that are involved in recognising chemical signals and translating them into cellular responses. Molecular pharmacology is advancing rapidly, and the new knowledge is changing our understanding of drug action and also opening up many new therapeutic possibilities, further discussed in other chapters.
First, we consider the types of target proteins on which drugs act. Next, we describe the main families of receptors and ion channels that have been revealed by cloning and structural studies. Finally, we discuss the various forms of receptor–effector linkage (signal transduction mechanisms) through which receptors are coupled to the regulation of cell function. The relationship between the molecular structure of a receptor and its functional linkage to a particular type of effector system is a principal theme. In the next two chapters, we see how these molecular events alter important aspects of cell function—a useful basis for understanding the effects of drugs on intact living organisms. We go into more detail than is necessary for understanding today’s pharmacology at a basic level, intending that students can, if they wish, skip or skim these chapters without losing the thread; however, we are confident that tomorrow’s pharmacology will rest solidly on the advances in cellular and molecular biology that are discussed here.
Targets for Drug Action
The protein targets for drug action on mammalian cells (Fig. 3.1) that are described in this chapter can be broadly divided into:
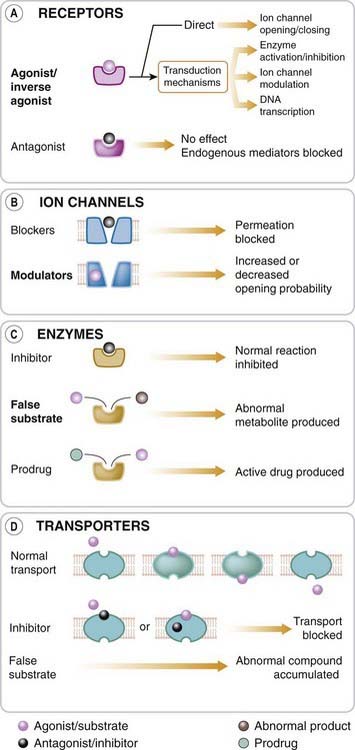
The great majority of important drugs act on one or other of these types of protein, but there are exceptions. For example, colchicine (Ch. 26) interacts with the structural protein tubulin, while several immunosuppressive drugs (e.g. ciclosporin, Ch. 26) bind to cytosolic proteins known as immunophilins. Therapeutic antibodies that act by sequestering cytokines (protein mediators involved in inflammation; see Ch. 26) are also used. Targets for chemotherapeutic drugs (Chs 49–55Chapter 49Chapter 50Chapter 51Chapter 52Chapter 53Chapter 54Chapter 55), where the aim is to suppress invading microorganisms or cancer cells, include DNA and cell wall constituents as well as other proteins.
Receptors
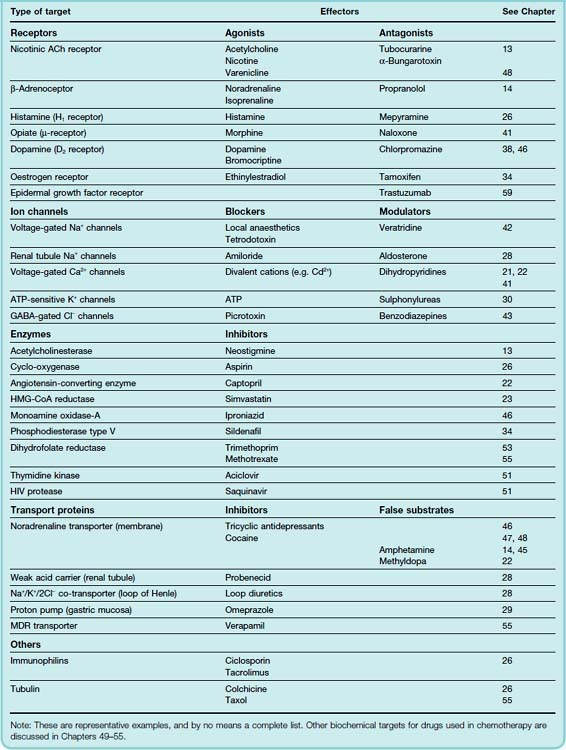
Receptors (Fig. 3.1A) are the sensing elements in the system of chemical communications that coordinates the function of all the different cells in the body, the chemical messengers being the various hormones, transmitters and other mediators discussed in Section 2. Many therapeutically useful drugs act, either as agonists or antagonists, on receptors for known endogenous mediators. Some examples are given in Table 3.1. In most cases, the endogenous mediator was discovered before—often many years before—the receptor was characterised pharmacologically and biochemically, but in recent years, many receptors have been identified initially on the basis of their pharmacological or molecular characteristics. In some cases, such as the cannabinoid receptors (see Ch. 18), the endogenous mediator was identified later; in many others, known as orphan receptors (see below) the mediator—if it exists—remains unknown.
Ion Channels
Ion channels1 are essentially gateways in cell membranes, which selectively allow the passage of particular ions, and which are induced to open or close by a variety of mechanisms. Two important types are ligand-gated channels and voltage-gated channels. The former open only when one or more agonist molecules are bound, and are properly classified as receptors, since agonist binding is needed to activate them. Voltage-gated channels are gated by changes in the transmembrane potential rather than by agonist binding.
In general, drugs can affect ion channel function either by binding to the channel protein itself (to the ligand-binding site of ligand-gated channels, or to other parts of the channel molecule), or they may affect channel function by an indirect interaction, involving a G-protein and other intermediaries (see below). In the simplest case, exemplified by the action of local anaesthetics on the voltage-gated sodium channel (see Ch. 42), the drug molecule plugs the channel physically (Fig. 3.1B), blocking ion permeation.
Examples of drugs that bind to accessory (allosteric) sites on the channel protein and thereby affect channel gating include:
A summary of the different ion channel families and their functions is given below (p. 43).
Enzymes
Many drugs are targeted on enzymes (Fig. 3.1C), examples being given in Table 3.1. Often, the drug molecule is a substrate analogue that acts as a competitive inhibitor of the enzyme (e.g. captopril, acting on angiotensin-converting enzyme; Ch. 22); in other cases, the binding is irreversible and non-competitive (e.g. aspirin, acting on cyclo-oxygenase; Ch. 26). The immunophilin to which ciclosporin binds (see above) has enzymic activity as an isomerase that catalyses the cis–trans isomerisation of proline residues in proteins, a reaction that is important in allowing expressed proteins to fold correctly. Inhibition of this enzymic activity is one of the mechanisms by which ciclosporin causes immunosuppression. Drugs may also act as false substrates, where the drug molecule undergoes chemical transformation to form an abnormal product that subverts the normal metabolic pathway. An example is the anticancer drug fluorouracil, which replaces uracil as an intermediate in purine biosynthesis but cannot be converted into thymidylate, thus blocking DNA synthesis and preventing cell division (Ch. 55).
It should also be mentioned that drugs may require enzymic degradation to convert them from an inactive form, the prodrug (see Ch. 9), to an active form. Examples are given in Table 9.3. Furthermore, as discussed in Chapter 57, drug toxicity often results from the enzymic conversion of the drug molecule to a reactive metabolite. Paracetamol (see Ch. 26) causes liver damage in this way. As far as the primary action of the drug is concerned, this is an unwanted side reaction, but it is of major practical importance.
Transport Proteins
The movement of ions and small organic molecules across cell membranes generally occurs either through channels (see above), or through the agency of a transport protein, because the permeating molecules are often too polar (i.e. insufficiently lipid soluble) to penetrate lipid membranes on their own (Fig. 3.1D). Many such carriers are known; examples of particular pharmacological importance include those responsible for the transport of ions and many organic molecules across the renal tubule, the intestinal epithelium and the blood–brain barrier, the transport of Na+ and Ca2+ out of cells, and the uptake of neurotransmitter precursors (such as choline) or of neurotransmitters themselves (such as noradrenaline, 5-hydroxytryptamine [5-HT], glutamate and peptides) by nerve terminals, and the transport of drug molecules and their metabolites across cell membranes and epithelial barriers. We shall encounter them frequently in later chapters.
In many cases, hydrolysis of ATP provides the energy for transport of substances against their electrochemical gradient. Such transport proteins include a distinct ATP binding site, and are termed ABC (ATP-binding cassette) transporters. Important examples include the sodium pump (Na+-K+-ATPase; see Ch. 4) and ‘multi-drug-resistance’ (MDR) transporters that eject cytotoxic drugs from cancer and microbial cells, conferring resistance to these therapeutic agents (see Ch. 55). In other cases, including the neurotransmitter transporters, the transport of organic molecules is coupled to the transport of ions (usually Na+), either in the same direction (symport) or in the opposite direction (antiport), and therefore relies on the electrochemical gradient for Na+ generated by the ATP-driven sodium pump. The carrier proteins embody a recognition site that makes them specific for a particular permeating species, and these recognition sites can also be targets for drugs whose effect is to block the transport system. Some examples are given in Table 3.1.
The importance of transport proteins as a source of individual variation in the pharmacokinetic characteristics of various drugs is becoming increasingly recognised (see Ch. 10).
Receptor Proteins
Isolation and Cloning of Receptors
In the 1970s, pharmacology entered a new phase when receptors, which had until then been theoretical entities, began to emerge as biochemical realities following the development of receptor-labelling techniques (see Ch. 2), which made it possible to extract and purify the receptor material. This approach was first used successfully on the nicotinic acetylcholine receptor (see Ch. 13), where advantage was taken of two natural curiosities. The first was that the electric organs of many fishes, such as rays (Torpedo sp.) and electric eels (Electrophorus sp.) consist of modified muscle tissue in which the acetylcholine-sensitive membrane is extremely abundant, and these organs contain much larger amounts of acetylcholine receptor than any other tissue. The second was that the venom of snakes of the cobra family contains polypeptides that bind with very high specificity to nicotinic acetylcholine receptors. These substances, known as α-toxins, can be labelled and used to assay the receptor content of tissues and tissue extracts. The best known is α-bungarotoxin, the main component of the venom of the Malayan banded krait (Bungarus multicinctus).2 Treatment of muscle or electric tissue with non-ionic detergents renders the membrane-bound receptor protein soluble, and it can then be purified by the technique of affinity chromatography. Similar approaches have now been used to purify a great many hormone and neurotransmitter receptors, as well as ion channels, carrier proteins and other kinds of target molecules.
 Once receptor proteins were isolated and purified, it was possible to analyse the amino acid sequence of a short stretch, allowing the corresponding base sequence of the mRNA to be deduced and full-length DNA to be isolated, by conventional cloning methods, starting from a cDNA library obtained from a tissue source rich in the receptor of interest. The first receptor clones were obtained in this way, but subsequently expression cloning and cloning strategies based on sequence homologies, which do not require prior isolation and purification of the receptor protein, were widely used, and now several hundred receptors of all four structural families (see below) have been cloned. Endogenous ligands for many of these ‘receptor-like’ molecules identified by gene cloning are so far unknown, and they are described as ‘orphan receptors’.3 Identifying ligands for these presumed receptors is often difficult. However, there are examples (e.g. the cannabinoid receptor; see Ch. 18) where important endogenous ligands have been linked to hitherto orphan receptors, and others, such as PPARs (peroxisome proliferator-activated receptors), which have emerged as the targets of important therapeutic drugs (see Ch. 30) though the endogenous ligand remains unknown. Several endogenous peptide ligands for orphan receptors have been identified (see Davenport, 2003), whose physiological and possible therapeutic significance is under investigation. There is optimism that novel therapeutic agents will emerge by targeting this pool of unclaimed receptors.
Once receptor proteins were isolated and purified, it was possible to analyse the amino acid sequence of a short stretch, allowing the corresponding base sequence of the mRNA to be deduced and full-length DNA to be isolated, by conventional cloning methods, starting from a cDNA library obtained from a tissue source rich in the receptor of interest. The first receptor clones were obtained in this way, but subsequently expression cloning and cloning strategies based on sequence homologies, which do not require prior isolation and purification of the receptor protein, were widely used, and now several hundred receptors of all four structural families (see below) have been cloned. Endogenous ligands for many of these ‘receptor-like’ molecules identified by gene cloning are so far unknown, and they are described as ‘orphan receptors’.3 Identifying ligands for these presumed receptors is often difficult. However, there are examples (e.g. the cannabinoid receptor; see Ch. 18) where important endogenous ligands have been linked to hitherto orphan receptors, and others, such as PPARs (peroxisome proliferator-activated receptors), which have emerged as the targets of important therapeutic drugs (see Ch. 30) though the endogenous ligand remains unknown. Several endogenous peptide ligands for orphan receptors have been identified (see Davenport, 2003), whose physiological and possible therapeutic significance is under investigation. There is optimism that novel therapeutic agents will emerge by targeting this pool of unclaimed receptors.
Much information has been gained by introducing the cloned DNA encoding individual receptors into cell lines, producing cells that express the foreign receptors in a functional form. Such engineered cells allow much more precise control of the expressed receptors than is possible with natural cells or intact tissues, and the technique is widely used to study the binding and pharmacological characteristics of cloned receptors. Expressed human receptors, which often differ in their sequence and pharmacological properties from their animal counterparts, can be studied in this way.
The cloning of receptors revealed many molecular variants (subtypes) of known receptors, which had not been evident from pharmacological studies. This produced some taxonomic confusion, but in the long term molecular characterisation of receptors is essential. Barnard, one of the high priests of receptor cloning, was undaunted by the proliferation of molecular subtypes among receptors that pharmacologists had thought that they understood. He quoted Thomas Aquinas: ‘Types and shadows have their ending, for the newer rite is here’. The newer rite, Barnard confidently asserted, was molecular biology. Analysis of the human and other mammalian genomes suggests that many hundreds of receptor-like genes are present, of which only a minority so far have a pharmacological identity. Now that the genes have been clearly identified, and the full molecular inventory established, the emphasis has shifted to characterising the receptors pharmacologically and determining their physiological functions.
Types of Receptor
Receptors elicit many different types of cellular effect. Some of them are very rapid, such as those involved in synaptic transmission, operating within milliseconds, whereas other receptor-mediated effects, such as those produced by thyroid hormone or various steroid hormones, occur over hours or days. There are also many examples of intermediate timescales—catecholamines, for example, usually act in a matter of seconds, whereas many peptides take rather longer to produce their effects. Not surprisingly, very different types of linkage between the receptor occupation and the ensuing response are involved. Based on molecular structure and the nature of this linkage (the transduction mechanism), we can distinguish four receptor types, or superfamilies (see Figs 3.2 and 3.3; Table 3.2).
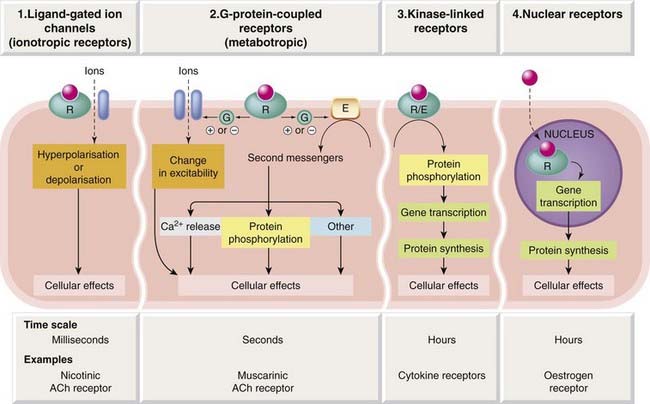
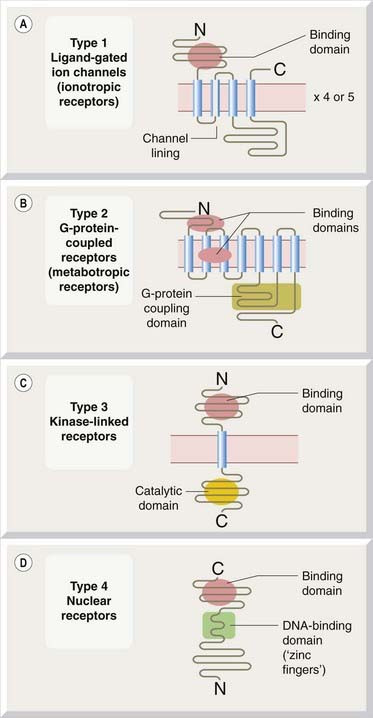
Fig. 3.3 General structure of four receptor families.
The rectangular segments represent hydrophobic α-helical regions of the protein comprising approximately 20 amino acids, which form the membrane-spanning domains of the receptors. [A] Type 1: ligand-gated ion channels. Many ligand-gated ion channels comprise four or five subunits of the type shown, the whole complex containing 16–20 membrane-spanning segments surrounding a central ion channel. Other structural types are shown in Fig. 3.18. [B] Type 2: G-protein-coupled receptors. [C] Type 3: kinase-linked receptors. Most growth factor receptors incorporate the ligand-binding and enzymatic (kinase) domains in the same molecule, as shown, whereas cytokine receptors lack an intracellular kinase domain but link to cytosolic kinase molecules. Other structural variants also exist. [D] Type 4: nuclear receptors that control gene transcription.
Molecular Structure of Receptors
The molecular organisation of typical members of each of these four receptor superfamilies is shown in Figure 3.3. Although individual receptors show considerable sequence variation in particular regions, and the lengths of the main intracellular and extracellular domains also vary from one to another within the same family, the overall structural patterns and associated signal transduction pathways are very consistent. The realisation that just four receptor superfamilies provide a solid framework for interpreting the complex welter of information about the effects of a large proportion of the drugs that have been studied has been one of the most refreshing developments in modern pharmacology.
Receptor Heterogeneity and Subtypes
Receptors within a given family generally occur in several molecular varieties, or subtypes, with similar architecture but significant differences in their sequences, and often in their pharmacological properties.6 Nicotinic acetylcholine receptors are typical in this respect; distinct subtypes occur in different brain regions (see Table 38.2), and these differ from the muscle receptor. Some of the known pharmacological differences (e.g. sensitivity to blocking agents) between muscle and brain acetylcholine receptors correlate with specific sequence differences; however, as far as we know, all nicotinic acetylcholine receptors respond to the same physiological mediator and produce the same kind of synaptic response, so why many variants should have evolved is still a puzzle.
Much of the sequence variation that accounts for receptor diversity arises at the genomic level, i.e. different genes give rise to distinct receptor subtypes. Additional variation arises from alternative mRNA splicing, which means that a single gene can give rise to more than one receptor isoform. After translation from genomic DNA, the mRNA normally contains non-coding regions (introns) that are excised by mRNA splicing before the message is translated into protein. Depending on the location of the splice sites, splicing can result in inclusion or deletion of one or more of the mRNA coding regions, giving rise to long or short forms of the protein. This is an important source of variation, particularly for GPCRs (see Kilpatrick et al., 1999), which produces receptors with different binding characteristics and different signal transduction mechanisms, although its pharmacological relevance remains to be clarified. Another process that can produce different receptors from the same gene is mRNA editing, which involves the mischievous substitution of one base in the mRNA for another, and hence a small variation in the amino acid sequence of the receptor.
Molecular heterogeneity of this kind is a feature of all kinds of receptors—indeed of functional proteins in general. New receptor subtypes and isoforms continue to be discovered, and regular updates of the catalogue are available (Alexander et al., 2009; IUPHAR Receptor Database and Channel Compendium). The problems of classification, nomenclature and taxonomy resulting from this flood of data have been mentioned earlier (p. 8). From the pharmacological viewpoint, where our concern is to understand individual drugs and what they do to living organisms, and to devise better ones, it is important that we keep molecular pharmacology in perspective. The ‘newer rite’ has proved revelatory in many ways, but the sheer complexity of the ways in which molecules behave means that we have a long way to go before reaching the reductionist Utopia that molecular biology promises. When we do, this book will get much shorter. In the meantime, we try to pick out the general principles without getting too bogged down in detail.
We will now describe the characteristics of each of the four receptor superfamilies.
Type 1: Ligand-Gated Ion Channels
Molecular Structure
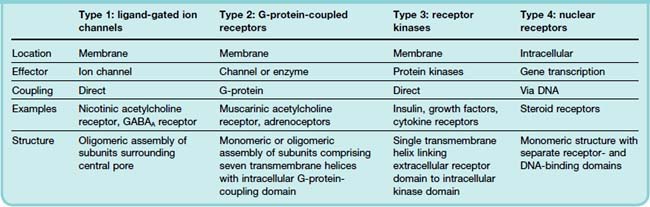
These molecules have structural features in common with other ion channels, described on p. 45 (Ashcroft, 2000). The nicotinic acetylcholine receptor (Fig. 3.4), the first to be cloned, has been studied in great detail (see Karlin, 1993). It consists of a pentameric assembly of different subunits, of which there are four types, termed α, β, γ and δ, each of molecular weight (Mr) 40–58 kDa. The subunits show marked sequence homology, and each contains four membrane-spanning α-helices, inserted into the membrane as shown in Figure 3.4B. The pentameric structure (α2, β, γ, δ) possesses two acetylcholine binding sites, each lying at the interface between one of the two α subunits and its neighbour. Both must bind acetylcholine molecules in order for the receptor to be activated. This receptor is sufficiently large to be seen in electron micrographs, and Figure 3.4B shows its structure, based mainly on a high-resolution electron diffraction study (Unwin, 1993, 1995; Miyazawa et al., 2003). Each subunit spans the membrane four times, so the channel comprises no fewer than 20 membrane-spanning helices surrounding a central pore.
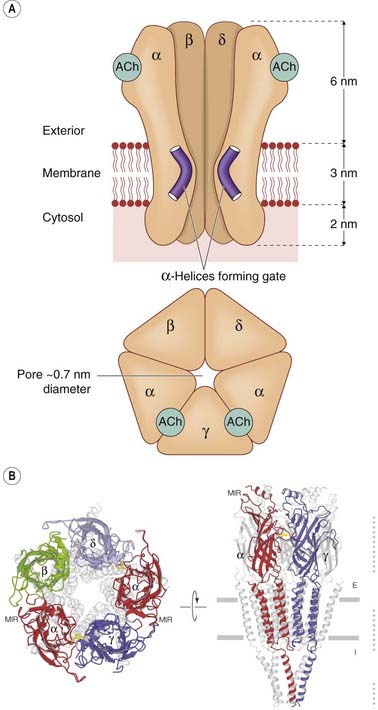
Fig. 3.4 Structure of the nicotinic acetylcholine receptor (a typical ligand-gated ion channel).
[A] Schematic diagram in side view (upper) and plan view (lower). The five receptor subunits (α2, β, γ, δ) form a cluster surrounding a central transmembrane pore, the lining of which is formed by the M2 helical segments of each subunit. These contain a preponderance of negatively charged amino acids, which makes the pore cation selective. There are two acetylcholine binding sites in the extracellular portion of the receptor, at the interface between the α and the adjoining subunits. When acetylcholine binds, the kinked α-helices either straighten out or swing out of the way, thus opening the channel pore.
(Based on Unwin N 1993 Nicotinic acetylcholine receptor at 9A resolution. J Mol Biol 229: 1101–1124, and Unwin N 1995 Acetylcholine receptor channel imaged in the open state. Nature 373: 37–43.) [B] High-resolution image showing revised arrangement of intracellular domains.
(Reproduced with permission from Unwin N 2005 Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol Mar 4;346(4):967–989.)
The two acetylcholine-binding sites lie on the extracellular parts of the two α subunits. One of the transmembrane helices (M2) from each of the five subunits forms the lining of the ion channel (Fig. 3.4). The five M2 helices that form the pore are sharply kinked inwards halfway through the membrane, forming a constriction. When acetylcholine molecules bind, a conformation change occurs in the extracellular part of the receptor (see review by Gay & Yakel, 2007), which twists the α subunits, causing the kinked M2 segments to swivel out of the way, thus opening the channel (Miyazawa et al., 2003). The channel lining contains a series of anionic residues, making the channel selectively permeable to cations.
The use of site-directed mutagenesis, which enables short regions, or single residues, of the amino acid sequence to be altered, has shown that a mutation of a critical residue in the M2 helix changes the channel from being cation selective (hence excitatory in the context of synaptic function) to being anion selective (typical of receptors for inhibitory transmitters such as GABA). Other mutations affect properties such as gating and desensitisation of ligand-gated channels.
Receptors for other fast transmitters, such as GABAA receptors (Ch. 37), 5-HT (Ch. 15) and glycine receptors (Ch. 37), are built on the same five-subunit pattern, and form the group of cys-loop receptors. Other ligand-gated ion channels, such as glutamate receptors (see Ch. 37) and the ‘capsaicin receptor’ (TRPV1; see Ch. 41), whose structures are shown in Figure 3.18, have a different (P-loop) architecture, in which the pore is built from loops rather than transmembrane helices (see p. 45), in common with many other (non-ligand-gated) ion channels.
The Gating Mechanism
Receptors of this type control the fastest synaptic events in the nervous system, in which a neurotransmitter acts on the postsynaptic membrane of a nerve or muscle cell and transiently increases its permeability to particular ions. Most excitatory neurotransmitters, such as acetylcholine at the neuromuscular junction (Ch. 12) or glutamate in the central nervous system (Ch. 37), cause an increase in Na+ and K+ permeability. This results in a net inward current carried mainly by Na+, which depolarises the cell and increases the probability that it will generate an action potential. The action of the transmitter reaches a peak in a fraction of a millisecond, and usually decays within a few milliseconds. The sheer speed of this response implies that the coupling between the receptor and the ionic channel is a direct one, and the molecular structure of the receptor–channel complex (see above) agrees with this. In contrast to other receptor families (see below), no intermediate biochemical steps are involved in the transduction process.
A breakthrough by Katz and Miledi in 1972 made it possible for the first time to study the properties of individual ligand-gated channels by the use of noise analysis. Studying the action of acetylcholine at the motor endplate, they observed that small random fluctuations of membrane potential were superimposed on the steady depolarisation produced by acetylcholine (Fig. 3.5). These fluctuations arise because, in the presence of an agonist, there is a dynamic equilibrium between open and closed ion channels. In the steady state, the rate of opening balances the rate of closing, but from moment to moment the number of open channels will show random fluctuations about the mean. By measuring the amplitude of these fluctuations, the conductance of a single ion channel can be calculated, and by measuring their frequency (usually in the form of a spectrum in which the noise power of the signal is plotted as a function of frequency), the average duration for which a single channel stays open (mean open time) can be calculated. In the case of acetylcholine acting at the endplate, the channel conductance is about 20 picosiemens (pS), which is equivalent to an influx of about 107 ions per second through a single channel under normal physiological conditions, and the mean open time is 1–2 ms. The magnitude of the single channel conductance confirms that permeation occurs through a physical pore through the membrane, because the ion flow is too large to be compatible with a carrier mechanism. The channel conductance produced by different acetylcholine-like agonists is the same, whereas the mean channel lifetime varies.
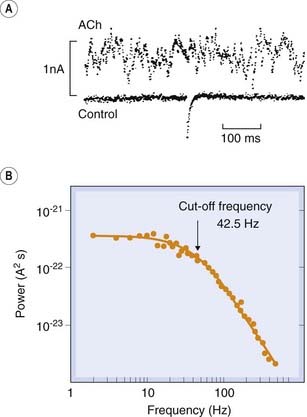
Fig. 3.5 Acetylcholine-induced noise at the frog motor endplate.
[A] Records of membrane current recorded at high gain under voltage clamp. The upper noise record was recorded during the application of acetylcholine (ACh) from a micropipette. The lower record was obtained in the absence of ACh, the blip in the middle being caused by the spontaneous release of a packet of ACh from the motor nerve. The steady (DC) component of the ACh signal has been removed by electronic filtering, leaving the high-frequency noise signal. [B] Power spectrum of ACh-induced noise recorded in a similar experiment to that shown above. The spectrum is calculated by Fourier analysis and fitted with a theoretical (Lorentzian) curve that corresponds to the expected behaviour of a single population of channels whose lifetime varies randomly. The cut-off frequency (at which the power is half of its limiting low-frequency value) enables the mean channel lifetime to be calculated.
(From [A] Anderson C R, Stevens C F 1973 J Physiol 235: 655; [B] Ogden D C et al. 1981 Nature 289: 596.)
The simple scheme shown in Fig. 2.1 is a useful model for ion channel gating. The conformation R*, representing the open state of the ion channel, is thought to be the same for all agonists, accounting for the finding that the channel conductance does not vary. Kinetically, the mean open time is determined mainly by the closing rate constant, α, and this varies from one drug to another. As explained in Chapter 2, an agonist of high efficacy that activates a large proportion of the receptors that it occupies will be characterised by β/α >> 1, whereas for a drug of low efficacy β/α has a lower value.
The patch clamp recording technique, devised by Neher and Sakmann, allows the very small current flowing through a single ionic channel to be measured directly (Fig. 3.6), and the results have fully confirmed the interpretation of channel properties based on noise analysis. This technique provides a view, unique in biology, of the physiological behaviour of individual protein molecules in real time, and has given many new insights into the gating reactions and permeability characteristics of both ligand-gated channels and voltage-gated channels (see p. 43). Single-channel recording has shown that many agonists cause individual channels to open to one or more of several distinct conductance levels. In the case of glutamate-activated channels, it appears that different agonists produce different receptor conformations associated with different channel conductances (Jin et al., 2003). Desensitisation of ligand-gated ion channels also involves one or more additional agonist-induced conformational states. These findings necessitate some elaboration of the simple scheme of Figure 2.1, in which only a single open state, R*, is represented, and are an example of the way in which the actual behaviour of receptors makes our theoretical models look a little threadbare.
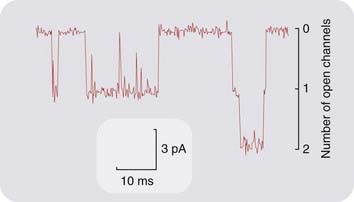
Fig. 3.6 Single acetylcholine-operated ion channels at the frog motor endplate recorded by the patch clamp technique.
The pipette, which was applied tightly to the surface of the membrane, contained 10 µmol/l ACh. The downward deflections show the currents flowing through single ion channels in the small patch of membrane under the pipette tip. Towards the end of the record, two channels can be seen to open simultaneously. The conductance and mean lifetime of these channels agrees well with indirect estimates from noise analysis (see Fig. 3.5).
(Courtesy of D Colquhoun and D C Ogden.)
Type 2: G-Protein-Coupled Receptors
The abundant GPCR family comprises many of the receptors that are familiar to pharmacologists, such as mAChRs, adrenoceptors, dopamine receptors, 5-HT receptors, opioid receptors, receptors for many peptides, purine receptors and many others, including the chemoreceptors involved in olfaction and pheromone detection, and also many ‘orphans’ (see Fredriksson & Schiöth, 2005). For most of these, pharmacological and molecular studies have revealed a variety of subtypes. All have the characteristic heptahelical structure.
Many neurotransmitters, apart from peptides, can interact with both GPCRs and ligand-gated channels, allowing the same molecule to produce a wide variety of effects. Individual peptide hormones, on the other hand, generally act either on GPCRs or on kinase-linked receptors (see below), but rarely on both, and a similar choosiness applies to the many ligands that act on nuclear receptors.7
The human genome includes genes encoding about 400 GPCRs (excluding odorant receptors), which constitute the commonest single class of targets for therapeutic drugs, and it is thought that many promising therapeutic drug targets of this type remain to be identified. For a short review, see Hill (2006).
Molecular Structure
The first GPCR to be fully characterised was the β-adrenoceptor (Ch. 14), which was cloned in 1986. Molecular biology caught up very rapidly with pharmacology, and all of the receptors that had been identified by their pharmacological properties have now been cloned. What seemed revolutionary in 1986 is now commonplace, and nowadays any aspiring receptor has to be cloned before it is taken seriously.
G-protein-coupled receptors consist of a single polypeptide chain of up to 1100 residues whose general anatomy is shown in Figure 3.3B. Their characteristic structure comprises seven transmembrane α-helices, similar to those of the ion channels discussed above, with an extracellular N-terminal domain of varying length, and an intracellular C-terminal domain.
GPCRs are divided into three distinct families (see Schwartz, 1996). There is considerable sequence homology between the members of one family, but none between different families. They share the same seven-helix (heptahelical) structure, but differ in other respects, principally in the length of the extracellular N terminus and the location of the agonist binding domain (Table 3.3). Family A is by far the largest, comprising most monoamine, neuropeptide and chemokine receptors. Family B includes receptors for some other peptides, such as calcitonin and glucagon (see Ch. 19). Family C is the smallest, its main members being the metabotropic glutamate and GABA receptors (Ch. 37) and the Ca2+-sensing receptors8 (see Ch. 35).
Table 3.3 G-protein-coupled receptor familiesa
| Family | Receptorsb | Structural features |
|---|---|---|
| A: rhodopsin family | The largest group. Receptors for most amine neurotransmitters, many neuropeptides, purines, prostanoids, cannabinoids, etc. | Short extracellular (N terminal) tail. Ligand binds to transmembrane helices (amines) or to extracellular loops (peptides) |
| B: secretin/glucagon receptor family | Receptors for peptide hormones, including secretin, glucagon, calcitonin | Intermediate extracellular tail incorporating ligand-binding domain |
| C: metabotropic glutamate receptor/calcium sensor family | Small group Metabotropic glutamate receptors, GABAB receptors, Ca2+-sensing receptors | Long extracellular tail incorporating ligand-binding domain |
a A fourth distinct family includes many receptors for pheromones but no pharmacological receptors.
b For full lists, see http://www.iuphar-db.org.
The understanding of the function of receptors of this type owes much to studies of a closely related protein, rhodopsin, which is responsible for transduction in retinal rods. This protein is abundant in the retina, and much easier to study than receptor proteins (which are anything but abundant); it is built on an identical plan to that shown in Figure 3.3 and also produces a response in the rod (hyperpolarisation, associated with inhibition of Na+ conductance) through a mechanism involving a G-protein (see below). The most obvious difference is that a photon, rather than an agonist molecule, produces the response. In effect, rhodopsin can be regarded as incorporating its own inbuilt agonist molecule, namely retinal, which isomerises from the trans (inactive) to the cis (active) form when it absorbs a photon.
Site-directed mutagenesis experiments show that the long third cytoplasmic loop is the region of the molecule that couples to the G-protein, because deletion or modification of this section results in receptors that still bind ligands but cannot associate with G-proteins or produce responses. Usually, a particular receptor subtype couples selectively with a particular G-protein, and swapping parts of the cytoplasmic loop between different receptors alters their G-protein selectivity.
For small molecules, such as noradrenaline (norepinephrine), the ligand-binding domain of class A receptors is buried in the cleft between the α-helical segments within the membrane (Fig. 3.3B), similar to the slot occupied by retinal in the rhodopsin molecule. Peptide ligands, such as substance P (Ch. 19) bind more superficially to the extracellular loops, as shown in Figure 3.3B. By single-site mutagenesis experiments, it is possible to map the ligand-binding domain of these receptors, and the hope is that it may soon be possible to design synthetic ligands based on knowledge of the receptor site structure—an important milestone for the pharmaceutical industry, which has relied up to now mainly on the structure of endogenous mediators (such as histamine) or plant alkaloids (such as morphine) for its chemical inspiration.9 Recently, the difficulties of crystallising type A GPCRs have been overcome, allowing the use of the powerful technique of X-ray crystallography to study the molecular structure of these receptors in detail (see Weis & Kobilka, 2008). Also, fluorescence methods have been developed to study the kinetics of ligand binding and subsequent conformational changes associated with activation (see Lohse et al., 2008). From such studies we should gain a clearer picture of the mechanism of activation of GPCRs and the factors determining agonist efficacy, as well as having a better basis for designing new GPCR ligands.
Protease-activated receptors
Although activation of GPCRs is normally the consequence of a diffusible agonist, it can be the result of protease activation. Four types of protease-activated receptors (PARs), have been identified (see review by Ramachandran & Hollenberg, 2008). Many proteases, such as thrombin (a protease involved in the blood-clotting cascade; see Ch. 24), activate PARs by snipping off the end of the extracellular N-terminal tail of the receptor (Fig. 3.7) to expose five or six N-terminal residues that bind to receptor domains in the extracellular loops, functioning as a ‘tethered agonist’. Receptors of this type occur in many tissues (see Ramachandran & Hollenberg, 2008), and they appear to play a role in inflammation and other responses to tissue damage where tissue proteases are released. One of the family of PARs, PAR-2, is activated by a protease released from mast cells, and is expressed on sensory neurons. It is thought to play a role in inflammatory pain (see Ch. 41). A PAR molecule can be activated only once, because the cleavage cannot be reversed, so continuous resynthesis of receptor protein is necessary. Inactivation occurs by a further proteolytic cleavage that frees the tethered ligand, or by desensitisation, involving phosphorylation (see below), after which the receptor is internalised and degraded, to be replaced by newly synthesised protein.
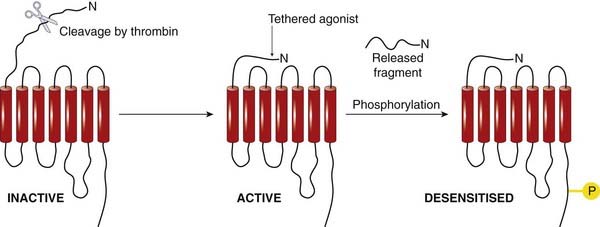
Fig. 3.7 Activation of a protease-activated receptor by cleavage of the N-terminal extracellular domain.
Inactivation occurs by phosphorylation. Recovery requires resynthesis of the receptor.
G-protein-coupled receptors 
G-Proteins and Their Role
G-proteins comprise a family of membrane-resident proteins whose function is to recognise activated GPCRs and pass on the message to the effector systems that generate a cellular response. They represent the level of middle management in the organisational hierarchy, intervening between the receptors—choosy mandarins alert to the faintest whiff of their preferred chemical—and the effector enzymes or ion channels—the blue-collar brigade that gets the job done without needing to know which hormone authorised the process. They are the go-between proteins, but were actually called G-proteins because of their interaction with the guanine nucleotides, GTP and GDP. For more detailed information on the structure and functions of G-proteins, see reviews by Milligan & Kostenis (2006) and Oldham & Hamm (2008). G-proteins consist of three subunits: α, β and γ (Fig. 3.8). Guanine nucleotides bind to the α subunit, which has enzymic activity, catalysing the conversion of GTP to GDP. The β and γ subunits remain together as a βγ complex. All three subunits are anchored to the membrane through a fatty acid chain, coupled to the G-protein through a reaction known as prenylation. G-proteins appear to be freely diffusible in the plane of the membrane, so a single pool of G-protein in a cell can interact with several different receptors and effectors in an essentially promiscuous fashion. In the ‘resting’ state (Fig. 3.8), the G-protein exists as an unattached αβγ trimer, with GDP occupying the site on the α subunit. When a GPCR is activated by an agonist molecule, a conformational change occurs, involving the cytoplasmic domain of the receptor (Fig. 3.3B), causing it to acquire high affinity for αβγ. Association of αβγ with the receptor occurs within about 50 ms, causing the bound GDP to dissociate and to be replaced with GTP (GDP–GTP exchange), which in turn causes dissociation of the G-protein trimer, releasing α-GTP and βγ subunits; these are the ‘active’ forms of the G-protein, which diffuse in the membrane and can associate with various enzymes and ion channels, causing activation of the target (Fig. 3.8). It was originally thought that only the α subunit had a signalling function, the βγ complex serving merely as a chaperone to keep the flighty α subunits out of range of the various effector proteins that they might otherwise excite. However, the βγ complexes actually make assignations of their own, and control effectors in much the same way as the α subunits (see Clapham & Neer, 1997). Association of α or βγ subunits with target enzymes or channels can cause either activation or inhibition, depending on which G-protein is involved (see Table 3.4).
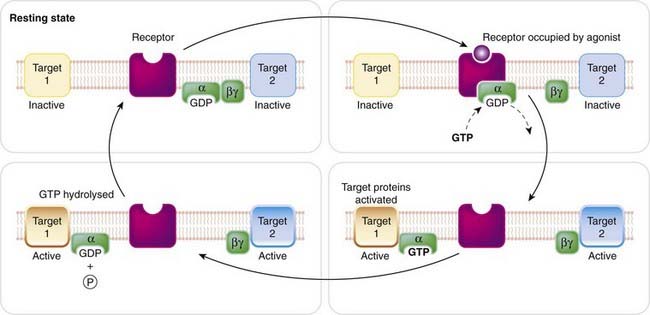
Fig. 3.8 The function of the G-protein.
The G-protein consists of three subunits (α, β, γ), which are anchored to the membrane through attached lipid residues. Coupling of the α subunit to an agonist-occupied receptor causes the bound GDP to exchange with intracellular GTP; the α–GTP complex then dissociates from the receptor and from the βγ complex, and interacts with a target protein (target 1, which may be an enzyme, such as adenylyl cyclase, or an ion channel). The βγ complex may also activate a target protein (target 2). The GTPase activity of the α subunit is increased when the target protein is bound, leading to hydrolysis of the bound GTP to GDP, whereupon the α subunit reunites with βγ.
Signalling is terminated when the hydrolysis of GTP to GDP occurs through the GTPase activity of the α subunit. The resulting α–GDP then dissociates from the effector, and reunites with βγ, completing the cycle. Attachment of the α subunit to an effector molecule actually increases its GTPase activity, the magnitude of this increase being different for different types of effector. Because GTP hydrolysis is the step that terminates the ability of the α subunit to produce its effect, regulation of its GTPase activity by the effector protein means that the activation of the effector tends to be self-limiting. The mechanism results in amplification because a single agonist–receptor complex can activate several G-protein molecules in turn, and each of these can remain associated with the effector enzyme for long enough to produce many molecules of product. The product (see below) is often a ‘second messenger’, and further amplification occurs before the final cellular response is produced.
How is specificity achieved so that each kind of receptor produces a distinct pattern of cellular responses? With a common pool of promiscuous G-proteins linking the various receptors and effector systems in a cell, it might seem that all specificity would be lost, but this is clearly not the case. For example, mAChRs and β-adrenoceptors, both of which occur in cardiac muscle cells, produce opposite functional effects (Chs 13 and 14). The main reason is molecular variation within the α subunits, of which more than 20 subtypes have been identified10 (see Wess, 1998; Table 3.4). Four main classes of G-protein (Gs, Gi, Go and Gq) are of pharmacological importance. As summarised in Table 3.4, they show selectivity with respect to both the receptors and the effectors with which they couple, having specific recognition domains in their structure complementary to specific G-protein-binding domains in the receptor and effector molecules. Gs and Gi produce, respectively, stimulation and inhibition of the enzyme adenylyl cyclase (Fig. 3.9).
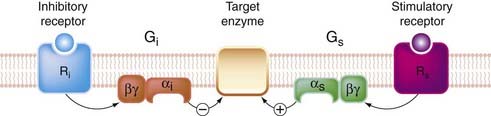
Fig. 3.9 Bidirectional control of a target enzyme, such as adenylate cyclase by Gs and Gi.
Heterogeneity of G-proteins allows different receptors to exert opposite effects on a target enzyme.
The α subunits of these G-proteins differ in structure. One functional difference that has been useful as an experimental tool to distinguish which type of G-protein is involved in different situations concerns the action of two bacterial toxins, cholera toxin and pertussis toxin (see Table 3.4). These toxins, which are enzymes, catalyse a conjugation reaction (ADP ribosylation) on the α subunit of G-proteins. Cholera toxin acts only on Gs, and it causes persistent activation. Many of the symptoms of cholera, such as the excessive secretion of fluid from the gastrointestinal epithelium, are due to the uncontrolled activation of adenylate cyclase that occurs. Pertussis toxin specifically blocks Gi and Go by preventing dissociation of the G-protein trimer.
Targets for G-Proteins
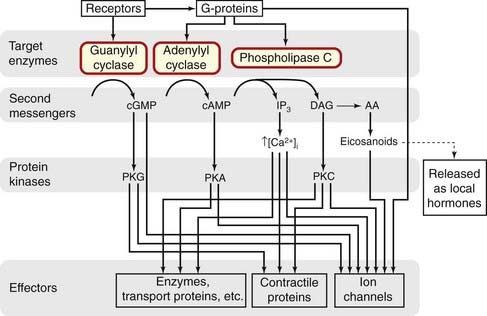
The main targets for G-proteins, through which GPCRs control different aspects of cell function (see Milligan, 1995; Nahorski, 2006; Table 3.4), are:
The adenylyl cyclase/cAMP system
The discovery by Sutherland and his colleagues of the role of cAMP (cyclic 3′,5′-adenosine monophosphate) as an intracellular mediator demolished at a stroke the barriers that existed between biochemistry and pharmacology, and introduced the concept of second messengers in signal transduction. cAMP is a nucleotide synthesised within the cell from ATP by the action of a membrane-bound enzyme, adenylyl cyclase. It is produced continuously and inactivated by hydrolysis to 5′-AMP by the action of a family of enzymes known as phosphodiesterases (PDEs). Many different drugs, hormones and neurotransmitters act on GPCRs and produce their effects by increasing or decreasing the catalytic activity of adenylyl cyclase, thus raising or lowering the concentration of cAMP within the cell. There are nine different molecular isoforms of the enzyme, some of which respond selectively to Gαs or Gαi (see Simonds, 1999).
Cyclic AMP regulates many aspects of cellular function including, for example, enzymes involved in energy metabolism, cell division and cell differentiation, ion transport, ion channels, and the contractile proteins in smooth muscle. These varied effects are, however, all brought about by a common mechanism, namely the activation of protein kinases by cAMP. Protein kinases regulate the function of many different cellular proteins by controlling protein phosphorylation (see p. 39) Figure 3.10 shows how increased cAMP production in response to β-adrenoceptor activation affects enzymes involved in glycogen and fat metabolism in liver, fat and muscle cells. The result is a coordinated response in which stored energy in the form of glycogen and fat is made available as glucose to fuel muscle contraction.
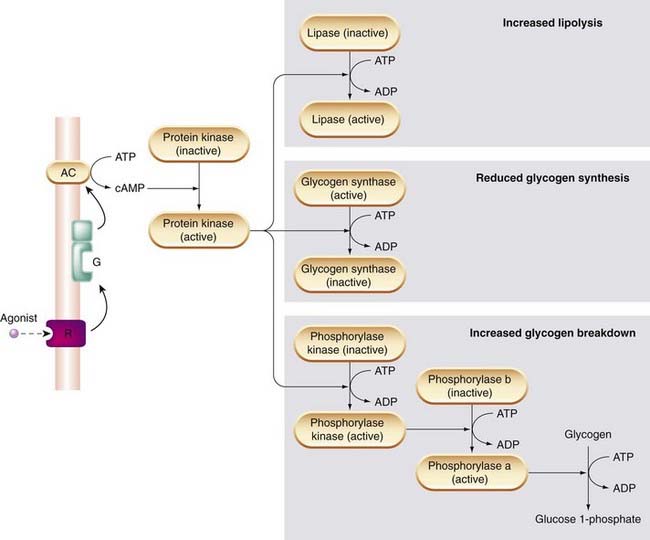
Other examples of regulation by cAMP-dependent protein kinases include the increased activity of voltage-gated calcium channels in heart muscle cells (see Ch. 21). Phosphorylation of these channels increases the amount of Ca2+ entering the cell during the action potential, and thus increases the force of contraction of the heart.
In smooth muscle, cAMP-dependent protein kinase phosphorylates (thereby inactivating) another enzyme, myosin-light-chain kinase, which is required for contraction. This accounts for the smooth muscle relaxation produced by many drugs that increase cAMP production in smooth muscle (see Ch. 4).
As mentioned above, receptors linked to Gi rather than Gs inhibit adenylyl cyclase, and thus reduce cAMP formation. Examples include certain types of mAChR (e.g. the M2 receptor of cardiac muscle; see Ch. 13), α2 adrenoceptors in smooth muscle (Ch. 14) and opioid receptors (see Ch. 41). Adenylyl cyclase can be activated directly by certain agents, including forskolin and fluoride ions, agents that are used experimentally to study the role of the cAMP system.
Cyclic AMP is hydrolysed within cells by phosphodiesterases (PDES), an important and ubiquitous family of enzymes (see Beavo, 1995, for review). Eleven PDE subtypes exist, of which some (e.g. PDE3 and PDE4) are cAMP selective, while others (e.g. PDE5) are cGMP selective. Most are weakly inhibited by drugs such as methylxanthines (e.g. theophylline and caffeine; see Chs 27 and 47). Rolipram (used to treat asthma; Ch. 27) is selective for PDE4, expressed in inflammatory cells; milrinone (used to treat heart failure; Ch. 21) is selective for PDE3, which is expressed in heart muscle; sildenafil (better known as Viagra; Ch. 34) is selective for PDE5, and consequently enhances the vasodilator effects of nitrous oxide (NO) and drugs that release NO, whose effects are mediated by cGMP (see Ch. 20). The similarity of some of the actions of these drugs to those of sympathomimetic amines (Ch. 14) probably reflects their common property of increasing the intracellular concentration of cAMP. Selective inhibitors of the various PDEs are being developed, mainly to treat cardiovascular and respiratory diseases.
The phospholipase C/inositol phosphate system
The phosphoinositide system, an important intracellular second messenger system, was first discovered in the 1950s by Hokin and Hokin, whose recondite interests centred on the mechanism of salt secretion by the nasal glands of seabirds. They found that secretion was accompanied by increased turnover of a minor class of membrane phospholipids known as phosphoinositides (collectively known as PIs; Fig. 3.11). Subsequently, Michell and Berridge found that many hormones that produce an increase in free intracellular Ca2+ concentration (which include, for example, muscarinic agonists and α-adrenoceptor agonists acting on smooth muscle and salivary glands, and vasopressin acting on liver cells) also increase PI turnover. Subsequently, it was found that one particular member of the PI family, namely phosphatidylinositol (4,5) bisphosphate (PIP2), which has additional phosphate groups attached to the inositol ring, plays a key role. PIP2 is the substrate for a membrane-bound enzyme, phospholipase Cβ (PLCβ), which splits it into diacylglycerol (DAG) and inositol (1,4,5) trisphosphate (IP3; Fig. 3.12), both of which function as second messengers as discussed below. The activation of PLCβ by various agonists is mediated through a G-protein (Gq, see Table 3.4). After cleavage of PIP2, the status quo is restored as shown in Figure 3.12, DAG being phosphorylated to form phosphatidic acid (PA), while the IP3 is dephosphorylated and then recoupled with PA to form PIP2 once again.11 Lithium, an agent used in psychiatry (see Ch. 46), blocks this recycling pathway (see Fig. 3.12).
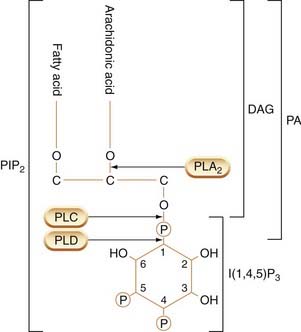
Fig. 3.11 Structure of phosphatidylinositol bisphosphate (PIP2), showing sites of cleavage by different phospholipases to produce active mediators.
Cleavage by phospholipase A2 (PLA2) yields arachidonic acid. Cleavage by phospholipase C (PLC) yields inositol trisphosphate (I(1,4,5)P3) and diacylglycerol (DAG). PA, phosphatidic acid; PLD, phospholipase D.
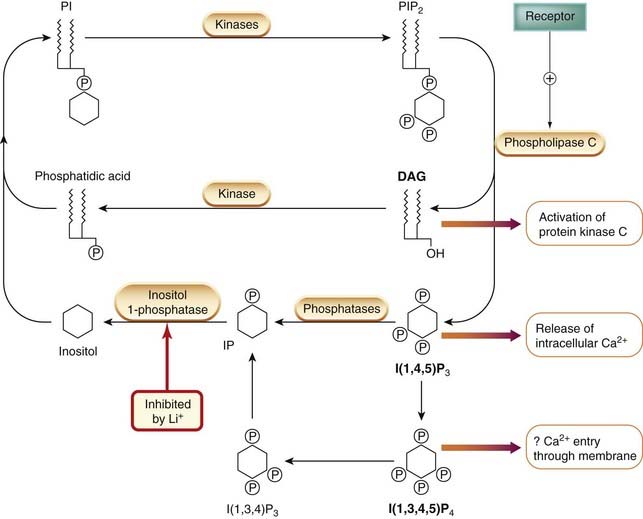
Fig. 3.12 The phosphatidylinositol (PI) cycle.
Receptor-mediated activation of phospholipase C results in the cleavage of phosphatidylinositol bisphosphate (PIP2), forming diacylglycerol (DAG) (which activates protein kinase C) and inositol trisphosphate (IP3) (which releases intracellular Ca2+). The role of inositol tetraphosphate (IP4), which is formed from IP3 and other inositol phosphates, is unclear, but it may facilitate Ca2+ entry through the plasma membrane. IP3 is inactivated by dephosphorylation to inositol. DAG is converted to phosphatidic acid, and these two products are used to regenerate PI and PIP2.
Inositol phosphates and intracellular calcium
Inositol (1,4,5) trisphosphate (IP3) is a water-soluble mediator that is released into the cytosol and acts on a specific receptor—the IP3 receptor—which is a ligand-gated calcium channel present on the membrane of the endoplasmic reticulum. The main role of IP3, described in more detail in Chapter 4, is to control the release of Ca2+ from intracellular stores. Because many drug and hormone effects involve intracellular Ca2+, this pathway is particularly important. IP3 is converted inside the cell to the (1,3,4,5) tetraphosphate, IP4, by a specific kinase. The exact role of IP4 remains unclear, but recent evidence suggests that it, and also higher inositol phosphates, plays a role in controlling gene expression.
Diacylglycerol and protein kinase C
Diacylglycerol is produced as well as IP3 whenever receptor-induced PI hydrolysis occurs. The main effect of DAG is to activate a membrane-bound protein kinase, protein kinase C (PKC), which catalyses the phosphorylation of a variety of intracellular proteins (see Nishizuka, 1988; Walaas & Greengard, 1991). DAG, unlike the inositol phosphates, is highly lipophilic and remains within the membrane. It binds to a specific site on the PKC molecule, which migrates from the cytosol to the cell membrane in the presence of DAG, thereby becoming activated. There are 10 different mammalian PKC subtypes, which have distinct cellular distributions and phosphorylate different proteins. Most are activated by DAG and raised intracellular Ca2+, both of which are produced by activation of GPCRs. PKCs are also activated by phorbol esters (highly irritant, tumour-promoting compounds produced by certain plants), which have been extremely useful in studying the functions of PKC. One of the subtypes is activated by the lipid mediator arachidonic acid (see Ch. 17) generated by the action of phospholipase A2 on membrane phospholipids, so PKC activation can also occur with agonists that activate this enzyme. The various PKC isoforms, like the tyrosine kinases discussed below (p. 37), act on many different functional proteins, such as ion channels, receptors, enzymes (including other kinases), transcription factors and cytoskeletal proteins. Kinases in general play a central role in signal transduction, and control many different aspects of cell function. The DAG–PKC link provides a channel whereby GPCRs can mobilise this army of control freaks.
Ion channels as targets for G-proteins
G-protein-coupled receptors can control ion channel function directly by mechanisms that do not involve second messengers such as cAMP or inositol phosphates. Direct G-protein–channel interaction was first shown for cardiac muscle, but appears to be a general mechanism for controlling K+ and Ca2+ channels (see Wickham & Clapham, 1995). In cardiac muscle, for example, mAChRs are known to enhance K+ permeability (thus hyperpolarising the cells and inhibiting electrical activity; see Ch. 21). Similar mechanisms operate in neurons, where many inhibitory drugs such as opioid analgesics reduce excitability by opening K+ channels or inhibiting Ca2+ channels (see Ch. 41). These actions are produced by direct interaction between the βγ subunit of G0 and the channel, without the involvement of second messengers.
Effectors controlled by G-proteins
Two key pathways are controlled by receptors via G-proteins. Both can be activated or inhibited by pharmacological ligands, depending on the nature of the receptor and G-protein.
The Rho/Rho kinase system
This recently discovered signal transduction pathway (see Bishop & Hall, 2000) is activated by certain GPCRs (and also by non-GPCR mechanisms), which couple to G-proteins of the G12/13 type. The free G-protein α subunit interacts with a guanosine nucleotide exchange factor, which facilitates GDP–GTP exchange at another GTPase, Rho. Rho–GDP, the resting form, is inactive, but when GDP–GTP exchange occurs, Rho is activated, and in turn activates Rho kinase. Rho kinase phosphorylates many substrate proteins and controls a wide variety of cellular functions, including smooth muscle contraction and proliferation, angiogenesis and synaptic remodelling. By enhancing hypoxia-induced pulmonary artery vasoconstriction, activation of Rho kinase is thought to be important in the pathogenesis of pulmonary hypertension (see Ch. 21). Specific Rho kinase inhibitors (e.g. fasudil) are in development for a wide range of clinical indications—an area to watch.
The MAP kinase system
This signal transduction pathway (see below and Fig. 3.15) is activated not only by various cytokines and growth factors acting on kinase-linked receptors (see p. 37), but also by GPCR ligands. It controls many processes involved in cell division, apoptosis and tissue regeneration.
The main postulated roles of GPCRs in controlling enzymes and ion channels are summarised in Figure 3.13.
Desensitisation
As described in Chapter 2, desensitisation is a feature of all GPCRs, and the mechanisms underlying it have been extensively studied. Two main processes are involved (see Koenig & Edwardson, 1997; Ferguson, 2001; Kelly et al., 2008):
The sequence of GPCRs includes certain residues (serine and threonine), mainly in the C-terminal cytoplasmic tail, which can be phosphorylated by kinases such as protein kinase A (PKA), PKC and specific membrane-bound GPCR kinases (GRKs).
Phosphorylation by PKA and PKC, which are activated by many GPCRs, generally leads to impaired coupling between the activated receptor and the G-protein, so the agonist effect is reduced. These kinases are not very selective, so receptors other than that for the desensitising agonist will also be affected. This effect, whereby one agonist can desensitise other receptors, is known as heterologous desensitisation, and is generally weak and short lasting (see Fig. 3.14).
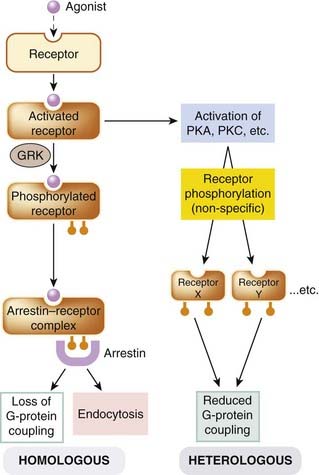
Fig. 3.14 Desensitisation of G-protein-coupled receptors (GPCRs).
Homologous (agonist-specific) desensitisation involves phosphorylation of the activated receptor by a specific kinase (GPCR kinase, GRK). The phosphorylated receptor (P-R) then binds to arrestin, causing it to lose its ability to associate with a G-protein, and to undergo endocytosis, which removes the receptor from the membrane. Heterologous (cross-) desensitisation occurs as a result of phosphorylation of one type of receptor as a result of activation of kinases by another. PKA and PKC, protein kinase A and C, respectively.
Phosphorylation by GRKs (Fig. 3.14) is receptor-specific to a greater or lesser degree, and affects mainly receptors in their activated (i.e. agonist-bound) state, resulting in homologous desensitisation. The residues that GRKS phosphorylate are different from those targeted by other kinases, and the phosphorylated receptor serves as a binding site for β-arrestins, intracellular proteins that block the interaction with G-proteins and also target the receptor for endocytosis, producing a more profound and long-lasting desensitisation. The first GRK to be identified was the β-adrenoceptor kinase, BARK, but several others have since been discovered, and this type of desensitisation seems to occur with most GPCRs. Besides initiating desensitisation, GRKs and arrestins are also involved as intermediates in various other GPCR-mediated signalling pathways that are distinct from those involving G-proteins (see Reiter & Lefkowitz, 2006). For example, binding of β-arrestin to GPCRs can recruit Src proteins, which in turn activate the MAP kinase cascade, which plays an important role in controlling cell division (see p. 39).
Further Developments in Gpcr Biology
By the early 1990s, we thought we had more or less got the measure of GPCR function, as described above. Since then, the plot has thickened, and recent developments (see review by Pierce et al., 2002) have necessitated a substantial overhaul of the basic model, whose implications for pharmacology in the future are not yet clear. Those wishing to stick to the basic story of GPCR function can safely skip this section.
GPCR dimerisation
The conventional view that GPCRs exist and function as monomeric proteins (in contrast to ion channels, which generally form multimeric complexes; see p. 44) was first overturned by work on the GABAB receptor. Two subtypes of this GPCR exist, encoded by different genes, and the functional receptor consists of a heterodimer of the two (see Ch. 37). It now seems likely that most, if not all, GPCRs exist as oligomers (Prinster et al., 2005). Within the opioid receptor family (see Ch. 41), stable and functional dimers of κ and δ receptors, whose pharmacological properties differ from those of either parent, have been created in cell lines. More diverse GPCR combinations have also been found, such as that between dopamine (D2) and somatostatin receptors, on which both ligands act with increased potency. Roaming even further afield in search of functional assignations, the dopamine receptor D5 can couple directly with a ligand-gated ion channel, the GABAA receptor, inhibiting the function of the latter without the intervention of any G-protein (Liu et al., 2000). These interactions have so far been studied mainly in engineered cell lines, but they also occur in native cells. Functional dimeric complexes between angiotensin (AT1) and bradykinin (B2) receptors occur in human platelets and show greater sensitivity to angiotensin than ‘pure’ AT1 receptors (AbdAlla et al., 2001). In pregnant women suffering from hypertension (pre-eclamptic toxaemia), the number of these dimers increases due to increased expression of B2 receptors, resulting—paradoxically—in increased sensitivity to the vasoconstrictor action of angiotensin. This is the first instance of the role of dimerisation in human disease.
It is too early to say what impact this newly discovered versatility of GPCRs in linking up with other receptors to form functional combinations will have on conventional pharmacology and therapeutics, but it could be considerable.
Constitutively active receptors
G-protein-coupled receptors may also be constitutively (i.e. spontaneously) active in the absence of any agonist (see Ch. 2 and review by Costa & Cotecchia, 2005). This was first shown for the β-adrenoceptor (see Ch. 14), where mutations in the third intracellular loop, or simply overexpression of the receptor, result in constitutive receptor activation. There are now many examples of native GPCRs that show constitutive activity when expressed in vitro (see Teitler et al., 2002). The histamine H3 receptor also shows constitutive activity in vivo, and this may prove to be a quite general phenomenon. It means that inverse agonists, which suppress this basal activity, may exert effects distinct from those of neutral antagonists, which block agonist effects without affecting basal activity.
Agonist specificity
It was thought that the linkage of a particular GPCR to a particular signal transduction pathway depends mainly on the structure of the receptor, particularly in the region of the third intracellular loop, which confers specificity for a particular G-protein, from which the rest of the signal transduction pathway follows. This would imply, in line with the two-state model discussed in Chapter 2, that all agonists acting on a particular receptor stabilise the same activated (R*) state and should activate the same signal transduction pathway, and produce the same type of cellular response. It is now clear that this is an oversimplification. In many cases, for example with agonists acting on opioid receptors, or with inverse agonists on β-adrenoceptors, the cellular effects are qualitatively different with different ligands, implying the existence of more than one—probably many—R* states (sometimes referred to as agonist trafficking or protean agonism, see Kenakin, 2002). This has profound implications—indeed heretical to many pharmacologists, who are accustomed to think of agonists in terms of their affinity and efficacy, and nothing else; it will add a new dimension to the way in which we think about drug efficacy and specificity (see Kelly et al., 2008).
RAMPs and RGS proteins
Receptor activity-modifying proteins (RAMPs) are a family of membrane proteins that associate with GPCRs and alter their functional characteristics. They were discovered in 1998 when it was found that the functionally active receptor for the neuropeptide calcitonin gene-related peptide (CGRP) (see Ch. 19) consisted of a complex of a GPCR—called calcitonin receptor-like receptor (CRLR)—that by itself lacked activity, with another membrane protein (RAMP1). More surprisingly, CRLR when coupled with another RAMP (RAMP2) showed a quite different pharmacology, being activated by an unrelated peptide, adrenomedullin. In other words, the agonist specificity is conferred by the associated RAMP as well as by the GPCR itself. More RAMPs have emerged, and so far (see Parameswaran & Spielman, 2006) nearly all the examples involve peptide receptors.
Regulators of G-protein signalling (RGS) proteins (see review by Xie & Palmer 2007) are a family of about 20 cellular proteins that possess a conserved sequence that binds specifically to Gα subunits. They increase greatly the GTPase activity of the active GTP–Gα complex, so hastening the hydrolysis of GTP and inactivating the complex. They thus exert an inhibitory effect on G-protein signalling, a mechanism that is thought to have a regulatory function in many situations. RAMPs and RGS proteins are two examples where protein–protein interactions influence the pharmacological behaviour of the receptors in a highly selective way.
G-protein-independent signalling
In using the term G-protein-coupled receptor to describe the class of receptors characterised by their heptahelical structure, we are following conventional textbook dogma but neglecting the fact that G-proteins are not the only link between GPCRs and the various effector systems that they regulate. The example of direct linkage between GPCRs and ion channels was mentioned above. There are also many examples where the various ‘adapter proteins’ that link receptors of the tyrosine kinase type to their effectors (see below) can also interact with GPCRs (see Brzostowski & Kimmel, 2001), allowing the same effector systems to be regulated by receptors of either type. In this context, the coupling of β-arrestins (see above), rather than G-proteins, to the activated GPCR, or phosphorylation of the C-terminal region of the GPCR by GRKs, produces a recognition site for molecules of the signal transduction pathway, analogous to the functioning of the kinase-linked receptors (see below; reviews by Bockaert & Pin, 1999; Delcourt et al., 2007).
In summary, the simple dogma that underpins much of our current understanding of GPCRs, namely,
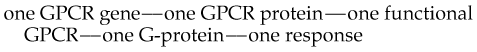
is showing distinct signs of wear. In particular:
G-protein-coupled receptors are evidently versatile and adventurous molecules around which much modern pharmacology revolves, and nobody imagines that we have reached the end of the story.
Type 3: Kinase-Linked and Related Receptors
These membrane receptors are quite different in structure and function from either the ligand-gated channels or the GPCRs. They mediate the actions of a wide variety of protein mediators, including growth factors and cytokines (see Chs 17, 19 17 19), and hormones such as insulin (see Ch. 30) and leptin (Ch. 31), whose effects are exerted mainly at the level of gene transcription. Most of these receptors are large proteins consisting of a single chain of up to 1000 residues, with a single membrane-spanning helical region, associated with a large extracellular ligand-binding domain, and an intracellular domain of variable size and function. The basic structure is shown in Figure 3.3C, but many variants exist (see below). Over 100 such receptors have been cloned, and many structural variations exist. For more detail, see reviews by Schenk & Snaar-Jakelska (1999) and Hubbard & Miller (2007). They play a major role in controlling cell division, growth, differentiation, inflammation, tissue repair, apoptosis and immune responses, discussed further in Chapters 5 and 17.
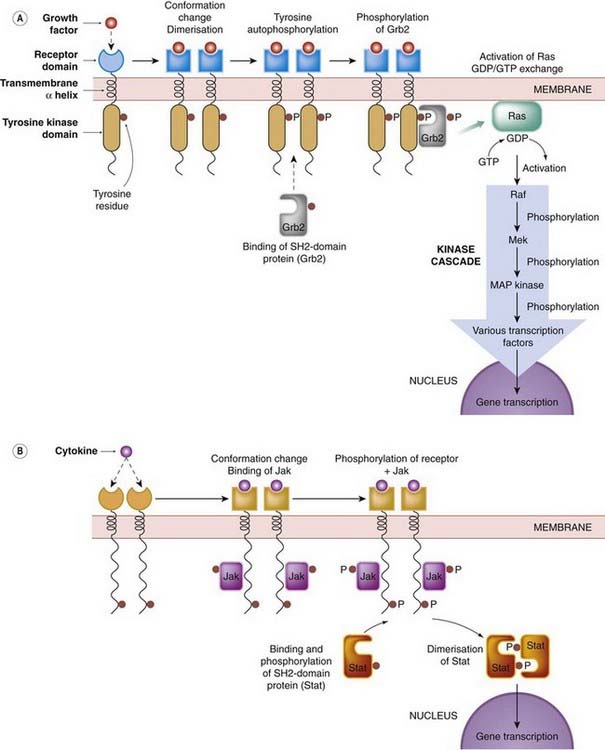
Fig. 3.15 Transduction mechanisms of kinase-linked receptors.
The first step following agonist binding is dimerisation, which leads to autophosphorylation of the intracellular domain of each receptor. SH2-domain proteins then bind to the phosphorylated receptor and are themselves phosphorylated. Two well-characterised pathways are shown: [A] the growth factor (Ras/Raf/mitogen-activated protein [MAP] kinase) pathway (see also Ch. 5); [B] the cytokine (Jak/Stat) pathway (see also Ch. 17). Several other pathways exist, and these phosphorylation cascades interact with components of G-protein systems.
Kinase-linked receptors
Protein Phosphorylation and Kinase Cascade Mechanisms
One of the major principles to emerge over the last 10–20 years (see Cohen, 2002) is that protein phosphorylation is a key mechanism for controlling the function of proteins (e.g. enzymes, ion channels, receptors, transport proteins) involved in regulating cellular processes. Phosphorylation and dephosphorylation are accomplished by kinases and phosphatases, respectively—enzymes of which several hundred subtypes are represented in the human genome—which are themselves subject to regulation dependent on their phosphorylation status. Much effort is currently being invested in mapping the complex interactions between signalling molecules that are involved in drug effects and pathophysiological processes such as oncogenesis, neurodegeneration, inflammation and much else. Here we can present only a few pharmacologically relevant aspects of what has become an enormous subject.
In many cases, ligand binding to the receptor leads to dimerisation. The association of the two intracellular kinase domains allows a mutual autophosphorylation of intracellular tyrosine residues to occur. The phosphorylated tyrosine residues then serve as high-affinity docking sites for other intracellular proteins that form the next stage in the signal transduction cascade. One important group of such ‘adapter’ proteins is known as the SH2 domain proteins (standing for Src homology, because it was first identified in the Src oncogene product). These possess a highly conserved sequence of about 100 amino acids, forming a recognition site for the phosphotyrosine residues of the receptor. Individual SH2 domain proteins, of which many are now known, bind selectively to particular receptors, so the pattern of events triggered by particular growth factors is highly specific. The mechanism is summarised in Figure 3.15.
What happens when the SH2 domain protein binds to the phosphorylated receptor varies greatly according to the receptor that is involved; many SH2 domain proteins are enzymes, such as protein kinases or phospholipases. Some growth factors activate a specific subtype of phospholipase C (PLCγ), thereby causing phospholipid breakdown, IP3 formation and Ca2+ release (see above). Other SH2-containing proteins couple phosphotyrosine-containing proteins with a variety of other functional proteins, including many that are involved in the control of cell division and differentiation. The end result is to activate or inhibit, by phosphorylation, a variety of transcription factors that migrate to the nucleus and suppress or induce the expression of particular genes. For more detail, see Pawson (2002). Nuclear factor kappa B (NFκB) is a transcription factor that plays a key role in inflammatory responses (see Ch. 17; Karin et al., 2004). It is normally present in the cytosol complexed with an inhibitor (IκB). Phosphorylation of IκB occurs when a specific kinase (IKK) is activated in response to various inflammatory cytokines and GPCR agonists. This results in dissociation of IκB from NFκB and migration of NFκB to the nucleus, where it switches on a wide variety of proinflammatory genes.
Two well-defined signal transduction pathways are summarised in Figure 3.15. The Ras/Raf pathway (Fig. 3.15A) mediates the effect of many growth factors and mitogens. Ras, which is a proto-oncogene product, functions like a G-protein, and conveys the signal (by GDP/GTP exchange) from the SH2-domain protein, Grb, which is phosphorylated by the RTK. Activation of Ras in turn activates Raf, which is the first of a sequence of three serine/threonine kinases, each of which phosphorylates, and activates, the next in line. The last of these, mitogen-activated protein (MAP) kinase, (which is also activated by GPCRs, see above), phosphorylates one or more transcription factors that initiate gene expression, resulting in a variety of cellular responses, including cell division. This three-tiered MAP kinase cascade forms part of many intracellular signalling pathways involved in a wide variety of disease processes, including malignancy, inflammation, neurodegeneration, atherosclerosis and much else. The kinases form a large family, with different subtypes serving specific roles. They are thought to represent an important target for future therapeutic drugs. Many cancers are associated with mutations in the genes coding for proteins involved in this cascade, leading to activation of the cascade in the absence of the growth factor signal (see Chs 5 and 55). For more details, see reviews by Marshall (1996), Schenk & Snaar-Jakelska (1999), Avruch (2007).
A second pathway, the Jak/Stat pathway (Fig. 3.15B) is involved in responses to many cytokines. Dimerisation of these receptors occurs when the cytokine binds, and this attracts a cytosolic tyrosine kinase unit (Jak) to associate with, and phosphorylate, the receptor dimer. Jaks belong to a family of proteins, different members having specificity for different cytokine receptors. Among the targets for phosphorylation by Jak are a family of transcription factors (Stats). These are SH2-domain proteins that bind to the phosphotyrosine groups on the receptor–Jak complex, and are themselves phosphorylated. Thus activated, Stat migrates to the nucleus and activates gene expression (see Ihle, 1995).
Other important mechanisms centre on phosphaditylinositol-3-kinase (PI3 kinases, see Vanhaesebroeck et al., 1997), a ubiquitous enzyme family that is activated both by GPCRs and RTKs and attaches a phosphate group to position 3 of PIP2 to form PIP3. Other kinases, particularly protein kinase B (PKB, also known as Akt), have recognition sites for PIP3 and are thus activated, controlling a wide variety of cellular functions, including apoptosis, differentiation, proliferation and trafficking. Akt also causes nitric oxide synthase activation in the vascular endothelium (see Ch. 20).
Recent work on signal transduction pathways has produced a bewildering profusion of molecular detail, often couched in a jargon that is apt to deter the faint-hearted. Perseverance will be rewarded, however, for there is no doubt that important new drugs, particularly in the areas of inflammation, immunology and cancer, will come from the targeting of these proteins (see Cohen, 2002). A recent breakthrough in the treatment of chronic myeloid leukaemia was achieved with the introduction of the first specific kinase inhibitor, imatinib, a drug that inhibits a specific tyrosine kinase involved in the pathogenesis of the disease (see Ch. 55).
The membrane-bound form of guanylyl cyclase, the enzyme responsible for generating the second messenger cGMP in response to the binding of natriuretic peptides (see Chs 19 and 21), resembles the tyrosine kinase family and is activated in a similar way by dimerisation when the agonist is bound (see Lucas et al., 2000).
Protein phosphorylation in signal transduction
Figure 3.16 illustrates the central role of protein kinases in signal transduction pathways in a highly simplified and schematic way. Many, if not all, of the proteins involved, including the receptors and the kinases themselves, are substrates for kinases, so there are many mechanisms for feedback and cross-talk between the various signalling pathways. Given that there are over 500 protein kinases, and similarly large numbers of receptors and other signalling molecules, the network of interactions can look bewilderingly complex. Dissecting out the details has become a major theme in cell biology. For pharmacologists, the idea of a simple connection between receptor and response, which guided thinking throughout the 20th century, is undoubtedly crumbling, although it will take some time before the complexities of signalling pathways are assimilated into a new way of thinking about drug action.
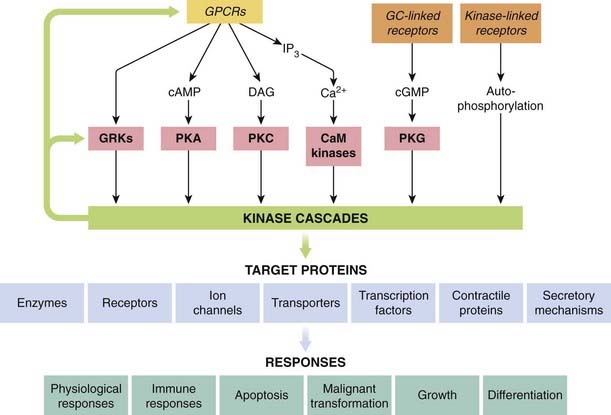
Fig. 3.16 Central role of kinase cascades in signal transduction.
Kinase cascades (e.g. those shown in Fig. 3.15) are activated by GPCRs, either directly or via different second messengers, by receptors that generate cGMP, or by kinase-linked receptors. The kinase cascades regulate various target proteins, which in turn produce a wide variety of short- and long-term effects. CaM kinase, Ca2+/calmodulin-dependent kinase; DAG, diacylglycerol; GC, guanylyl cyclase; GRK, GPCR kinase; IP3, inositol trisphosphate; PKA, cAMP-dependent protein kinase; PKC, protein kinase C; PKG, cGMP-dependent protein kinase.
Type 4: Nuclear Receptors
The fourth type of receptors we will consider belong to the nuclear receptor (NR) family. By the 1970s, it was clear that receptors for steroid hormones such as oestrogen and the glucocorticoids were present in the cytoplasm of cells and translocated into the nucleus after binding with their steroid partner. Other hormones, such as the thyroid hormone T3 (Ch. 33) and the fat-soluble vitamins D and A (retinoic acid), were found to act in a similar fashion. Comparisons of genome and protein sequence data led to the recognition that they were members of a much larger family of related proteins. As well as NRs such as the glucocorticoid and retinoic acid receptor, whose ligands were well characterised, this family includes a great many (40%) orphan receptors—receptors with no known well-defined ligands. The first of these to be described, in the 1990s, was RXR, a receptor cloned on the basis of its similarity with the vitamin A receptor and that was subsequently found to bind the vitamin A derivative 9-cis-retinoic acid. Over the intervening years, binding partners have been identified for many NRs (‘adopted orphans’; e.g. RXR) but the ligands of many others (’true orphans’) have yet to be identified, or perhaps do not exist as such.
Today, it is convenient to regard the entire NR family as ligand-activated transcription factors that transduce signals by modifying gene transcription. Unlike the receptors described in the preceding sections of this chapter, the nuclear receptors are not embedded in membranes (although see below), but are present in the soluble phase of the cell. Some, such as the steroid receptors, become mobile in the presence of their ligand and can translocate from the cytoplasm to the nucleus, while others such as the RXR probably dwell mainly within the nuclear compartment. Some NRs, while unliganded, act to constitutively repress some genes (e.g. RXR).
In man, there are at least 48 NR genes (although only about half code for liganded receptors) but more NR proteins may arise through alternative splicing events. While this represents a rather small proportion of all receptors (less than 10% of the total number of GPCRS), the NRs are very important drug targets, being responsible for the biological effects of approximately 10% of all prescription drugs. They can recognise an extraordinarily diverse group of substances (mostly small hydrophobic molecules), which may exhibit full or partial agonist, antagonist or inverse agonist activity. Some NRs are involved predominately in endocrine signalling but many act as lipid sensors and are thus crucial links between our dietary and metabolic status and the expression of genes that regulate the metabolism and disposition of lipids. They also regulate expression of many drug metabolic enzymes and transporters. Many illnesses are associated with malfunctioning of the NR system, including inflammation, cancer, diabetes, cardiovascular disease, obesity and reproductive disorders (see Murphy & Holder, 2000, Kersten et al., 2000).
Structure of Nuclear Receptors
All NRs are monomeric proteins that share a broadly similar structural design (see Fig. 3.17 and Bourguet et al., 2000, for further details). The N-terminal domain displays the most heterogeneity. It harbours the AF1 (activation function 1) site that binds to other cell-specific transcription factors in a ligand-independent way and modifies the binding or activity of the receptor itself. Alternative splicing of genes may yield several receptor isoforms each with slightly different N-terminal regions. The core domain of the receptor is highly conserved and consists of the structure responsible for DNA recognition and binding. At the molecular level, this comprises two zinc fingers—cysteine- (or cystine-/histidine-) rich loops in the amino acid chain that are held in a particular conformation by zinc ions. The main function of this portion of the molecule is to recognise and bind to the hormone response elements located in genes that are regulated by this family of receptors, but it also plays a part in regulating receptor dimerisation as well.
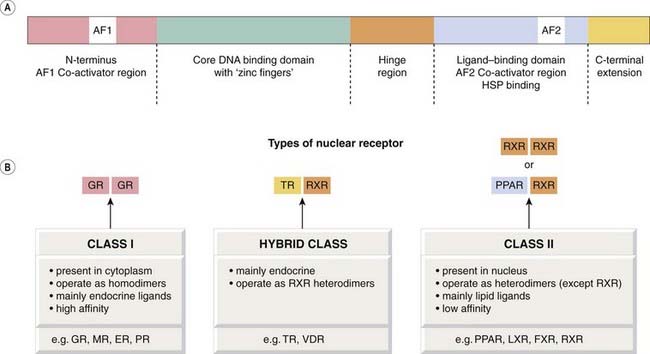
[A] Structure of a nuclear receptor, showing the different domains. [B] The two main classes of nuclear receptors. ER, oestrogen receptor; FXR, farnesoid receptor; GR, glucocorticoid receptor; HSP, heat shock protein; LXR, liver oxysterol receptor; MR, mineralocorticoid receptor; PPAR, peroxisome proliferator receptor; PR, prolactin receptor; RXR, retinoid receptor; TR, thyroid receptor; VDR, vitamin D receptor.
It is the highly flexible hinge region in the molecule that allows it to dimerise with other NRs and also to exhibit DNA binding in a variety of configurations. Finally, the C-terminal domain contains the ligand-binding module and is specific to each class of receptor. A highly conserved AF2 region is important in ligand-dependent activation. Also located near the C-terminal are motifs that contain nuclear localisation signals and others that may, in the case of some receptors, bind accessory heat shock and other proteins.
Classification of Nuclear Receptors
The NR superfamily consists of two main classes (I and II), together with a third that shares some of the characteristics of both (see Fig. 3.17 and Germain et al., 2006, for further details). Class I consists largely of receptors for the steroid hormones, including the glucocorticoid and mineralocorticoid receptors (GR and MR), as well as the oestrogen, progesterone and androgen receptors (ER, PR and AR, respectively). These receptors generally recognise hormones (e.g. glucocorticoids) that act in a negative feedback fashion to control biological events (see Ch. 32 for more details). In the absence of their ligand, these NRs are predominantly located in the cytoplasm, complexed with heat shock and other proteins and possibly reversibly attached to the cytoskeleton or other structures. Following diffusion (or possibly transportation) from the blood into the cell, their ligand partner binds to their NR with high affinity. These liganded receptors generally form homodimers and translocate to the nucleus, where they can transactivate or transrepress genes by binding to ‘positive’ or ‘negative’ hormone response elements (see below). Large numbers of genes can be regulated in this way by a single ligand. For example, it is estimated that the activated GR itself can regulate transcription of ~1% of the genome either directly or indirectly.
Class II NRs function in a slightly different way. Their ligands are generally lipids already present to some extent within the cell. This group includes the peroxisome proliferator-activated receptor (PPAR) that recognises fatty acids; the liver oxysterol receptor (LXR) that recognises and acts as a cholesterol sensor, the farnesoid (bile acid) receptor (FXR), a xenobiotic receptor (SXR; in rodents the PXR) that recognises a great many foreign substances, including therapeutic drugs, and the constitutive androstane receptor (CAR), which not only recognises the steroid androstane but also some drugs such as phenobarbital (see Ch. 43). These latter NRs are akin to airport security guards who alert the bomb disposal squad when suspicious luggage is found. They induce drug-metabolising enzymes such as CYP3A (which is responsible for metabolising about 60% of all prescription drugs; see Ch. 9 and Synold et al., 2001), and also bind some prostaglandins and non-steroidal drugs, as well as the antidiabetic thiazolidinediones (see Ch. 30) and fibrates (see Ch. 23). Unlike the receptors in class I, these NRs almost always operate as heterodimers together with the retinoid receptor (RXR). They tend to mediate positive feedback effects (e.g. occupation of the receptor amplifies rather than inhibits a particular biological event). When class II monomeric receptors bind to RXR, two types of heterodimer may be formed: a non-permissive heterodimer, which can be activated only by the RXR ligand itself, and the permissive heterodimer, which can be activated either by retinoic acid itself or by its partner’s ligand.
A third group of NRs is really a subgroup of class II in the sense that they form obligate heterodimers with RXR, but rather than sensing lipids, they play a part in endocrine signalling. The group includes the thyroid hormone receptor (TR), the vitamin D receptor (VDR) and the retinoic acid receptor (RAR).
Nuclear receptors
Control of Gene Transcription
Hormone response elements are the short (four or five base pairs) sequences of DNA to which the NRs bind to modify gene transcription. They are usually present symmetrically in pairs or half sites, although these may be arranged together in different ways (e.g. simple repeats or inverted repeats). Each NR exhibits a preference for a particular consensus sequence but because of the family homology, there is a close similarity between these sequences.
Once in the nucleus, the ligand-bound receptor recruits further proteins including co-activators or co-repressors to modify gene expression through its AF1 and AF2 domains. Some of these co-activators are enzymes involved in chromatin remodelling such as histone acetylase/deacetylase which, together with other enzymes, regulate the unravelling of the DNA to facilitate access by polymerase enzymes and hence gene transcription. Co-repressor complexes are recruited by some receptors and comprise histone deacetylase and other factors that cause the chromatin to become tightly packed, preventing further transcriptional activation. Some unliganded class II receptors such as TR and VDR are constitutively bound to these repressor complexes in the nucleus, thus ‘silencing’ the gene. The complex dissociates on ligand binding, permitting an activator complex to bind. The case of CAR is particularly interesting; like some types of G-proteins described earlier in this chapter, CAR also forms a constitutively active complex that is terminated when it binds its ligand.
The discussion here must be taken only as a broad guide to the action of NRs, as many other types of interaction have also been discovered. For example, some receptors may bring about non-genomic actions by directly interacting with factors in the cytosol, or they may be covalently modified by phosphorylation or by protein–protein interactions with other transcription factors such that their function is altered (see Falkenstein et al., 2000). In addition, there is good evidence for separate membrane and other types of receptor that can bind some steroid hormones such as oestrogen (see Walters & Nemere, 2004). This intricate network of receptors and their nuclear and cytosolic interactions serves as a subtle regulator of blood lipids as well as transducing the effects of hormones that have arrived from distant tissues. Much remains to be discovered about this interesting and complex family of receptor proteins.
Ion Channels as Drug Targets
We have discussed ligand-gated ion channels as one of the four main types of drug receptor. There are many other types of ion channel that represent important drug targets, even though they are not generally classified as ‘receptors’ because they are not the immediate targets of fast neurotransmitters.12
Here we discuss the structure and function of ion channels at the molecular level; their role as regulators of cell function is described in Chapter 4.
Ions are unable to penetrate the lipid bilayer of the cell membrane, and can get across only with the help of membrane-spanning proteins in the form of channels or transporters. The concept of ion channels was developed in the 1950s on the basis of electrophysiological studies on the mechanism of membrane excitation (see below). Electrophysiology, particularly the voltage clamp technique (see Ch. 4) remains an essential tool for studying the physiological and pharmacological properties of ion channels. Since the mid-1980s, when the first ion channels were cloned by Numa in Japan, much has been learned about the structure and function of these complex molecules. The use of tight-seal (‘patch clamp’) recording, which allows the behaviour of individual channels to be studied in real time, has been particularly valuable in distinguishing channels on the basis of their conductance and gating characteristics. Accounts by Hille (2001), Ashcroft (2000) and Catterall (2000) give more information.
Ion channels consist of protein molecules designed to form water-filled pores that span the membrane, and can switch between open and closed states. The rate and direction of ion movement through the pore is governed by the electrochemical gradient for the ion in question, which is a function of its concentration on either side of the membrane, and of the membrane potential. Ion channels are characterised by:
Ion Selectivity
Channels are generally either cation selective or anion selective. The main cation-selective channels are selective for Na+, Ca2+ or K+, or non-selective and permeable to all three. Anion channels are mainly permeable to Cl−, although other types also occur. The effect of modulation of ion channels on cell function is discussed in Chapter 4.
Gating
Voltage-Gated Channels
These channels open when the cell membrane is depolarised. They form a very important group because they underlie the mechanism of membrane excitability (see Ch. 4). The most important channels in this group are selective sodium, potassium or calcium channels.
Commonly, the channel opening (activation) induced by membrane depolarisation is short lasting, even if the depolarisation is maintained. This is because, with some channels, the initial activation of the channels is followed by a slower process of inactivation.
The role of voltage-gated channels in the generation of action potentials and in controlling other cell functions is described in Chapter 4.
Ligand-Gated Channels
These (see above) are activated by binding of a chemical ligand to a site on the channel molecule. Fast neurotransmitters, such as glutamate, acetylcholine, GABA and ATP (see Chs 13, 16 and 37) act in this way, binding to sites on the outside of the membrane. The vanilloid receptor TRPV1 mediates the pain-producing effect of capsaicin on sensory nerves (as well as responding to low pH and heat; see Ch. 41).
Some ligand-gated channels in the plasma membrane respond to intracellular rather than extracellular signals, the most important being the following:
Other examples of channels that respond to intracellular ligands include arachidonic acid-sensitive potassium channels and DAG-sensitive calcium channels, whose functions are not well understood.
Calcium Release Channels
These are present on the endoplasmic or sarcoplasmic reticulum rather than the plasma membrane. The main ones, IP3 and ryanodine receptors (see Ch. 4) are a special class of ligand-gated calcium channels that control the release of Ca2+ from intracellular stores.
Store-Operated Calcium Channels
When the intracellular Ca2+ stores are depleted, ‘store-operated’ channels (SOCs) in the plasma membrane open to allow Ca2+ entry. The mechanism by which this linkage occurs involves interaction of a Ca2+-sensor protein in the endoplasmic reticulum membrane with a dedicated Ca2+ channel in the plasma membrane (see Potier & Trebak, 2008). In response to GPCRs that elicit Ca2+ release, the opening of these channels allows [Ca2+]i to remain elevated even when the stores are running low, and also provides a route through which the stores can be replenished (see Ch. 4).
Molecular Architecture of Ion Channels
Ion channels are large and elaborate molecules. Their characteristic structural motifs have been revealed as knowledge of their sequence and structure has accumulated since the mid-1980s, when the first ligand-gated channel (the nicotinic acetylcholine receptor) and the first voltage-gated sodium channel were cloned. The main structural subtypes are shown in Figure 3.18. All consist of several (often four) domains, which are similar or identical to each other, organised either as an oligomeric array of separate subunits, or as one large protein. Each subunit or domain contains a bundle of two to six membrane-spanning helices. Most ligand-gated channels have the basic structure shown in Figure 3.18A, comprising a pentameric array of non-identical subunits, each consisting of four transmembrane helices, of which one—the M2 segment—from each subunit lines the pore. The large extracellular N-terminal region contains the ligand-binding region. Several exceptions to this basic design for ligand-gated channels have emerged recently. They include (see Fig. 3.18) the glutamate NMDA receptor (Ch. 37), and the vanilloid receptor (a channel that responds not only to chemicals of the vanilloid class, but also to heat and protons; see Ch. 41). In these, as in many other types of channel, the pore-forming part of the molecule consists of a hairpin loop—the pore (P) loop—between two of the helices.
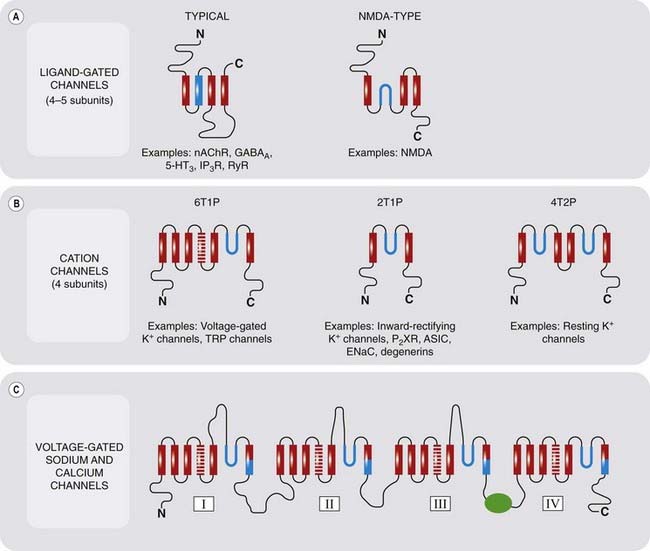
Fig. 3.18 Molecular architecture of ion channels.
Red and blue rectangles represent membrane-spanning α-helices. Blue hairpins are pore loop (P) domains, present in many channels, blue rectangles being the pore-forming regions of the membrane-spanning α-helices. Cross-shaded rectangles represent the voltage-sensing regions of voltage-gated channels. The green symbol represents the inactivating particle of voltage-gated sodium channels. Potassium channel nomenclature is based on the number of transmembrane helices (T) and pore-forming loops (P) in each subunit. Further information on ion channels is given in Chapter 4. 5-HT3, 5-hydroxytryptamine type 3 receptor; ASIC, acid-sensing ion channel; ENaC, epithelial sodium channel; GABAA, GABA type A receptor; IP3R, inositol trisphosphate receptor; nAChR, nicotinic acetylcholine receptor; P2XR, purine P2X receptor; RyR, ryanodine receptor.
Voltage-gated channels generally include one transmembrane helix that contains an abundance of basic (i.e. positively charged) amino acids. When the membrane is depolarised, so that the interior of the cell becomes less negative, this region—the voltage sensor—moves slightly towards the outer surface of the membrane, which has the effect of opening the channel (see Bezanilla, 2008). Many voltage-activated channels also show inactivation, which happens when an intracellular appendage of the channel protein moves to plug the channel from the inside. Voltage-gated sodium and calcium channels are remarkable in that the whole structure with four six-helix domains consists of a single huge protein molecule, the domains being linked together by intracellular loops of varying length. Potassium channels comprise the most numerous and heterogeneous class.13 Voltage-gated potassium channels resemble sodium channels, except that they are made up of four subunits rather than a single long chain. The class of potassium channels known as ‘inward rectifier channels’ because of their biophysical properties has the two-helix structure shown in Figure 3.18C, whereas others are classed as ‘two-pore domain’ channels, because each subunit contains two P loops.
The various architectural motifs shown in Figure 3.18 only scrape the surface of the molecular diversity of ion channels. In all cases, the individual subunits come in several molecular varieties, and these can unite in different combinations to form functional channels as hetero-oligomers (as distinct from homo-oligomers built from identical subunits). Furthermore, the channel-forming structures described are usually associated with other membrane proteins, which significantly affect their functional properties. For example, the ATP-gated potassium channel exists in association with the sulfonylurea receptor (SUR), and it is through this linkage that various drugs (including antidiabetic drugs of the sulfonylurea class; see Ch. 30) regulate the channel (see Ashcroft & Gribble, 2000). Good progress is being made in understanding the relation between molecular structure and ion channel function, but we still have only a fragmentary understanding of the physiological role of many of these channels. Many important drugs exert their effects by influencing channel function, either directly or indirectly.
Pharmacology of Ion Channels
Many drugs and physiological mediators described in this book exert their effects by altering the behaviour of ion channels. Here we outline the general mechanisms as exemplified by the pharmacology of voltage-gated sodium channels (Fig. 3.19). Ion channel pharmacology is likely to be a fertile source of future new drugs (see Clare et al., 2000).
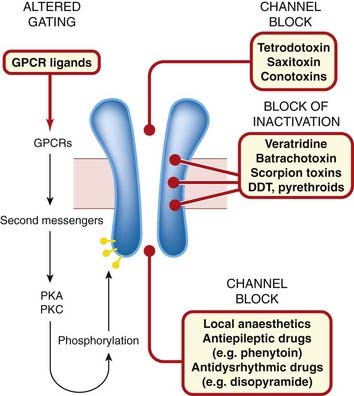
Fig. 3.19 Drug-binding domains of voltage-gated sodium channels (see Ch. 42).
The multiplicity of different binding sites and effects appears to be typical of many ion channels. DDT, dichlorodiphenyltrichloroethane (dicophane, a well-known insecticide); GPCR, G-protein-coupled receptor; PKA, protein kinase A; PKC, protein kinase C.
The gating and permeation of both voltage-gated and ligand-gated ion channels is modulated by many factors, including the following.
Figure 3.19 summarises the main sites and mechanisms by which drugs affect voltage-gated sodium channels, a typical example of this type of drug target.
Control of Receptor Expression
Receptor proteins are synthesised by the cells that express them, and the level of expression is itself controlled, via the pathways discussed above, by receptor-mediated events. We can no longer think of the receptors as the fixed elements in cellular control systems, responding to changes in the concentration of ligands, and initiating effects through the signal transduction pathway—they are themselves subject to regulation. Short-term regulation of receptor function generally occurs through desensitisation, as discussed above. Long-term regulation occurs through an increase or decrease of receptor expression. Examples of this type of control include the proliferation of various postsynaptic receptors after denervation (see Ch. 12), the upregulation of various G-protein-coupled and cytokine receptors in response to inflammation (see Ch. 17), and the induction of growth factor receptors by certain tumour viruses (see Ch. 5). Long-term drug treatment invariably induces adaptive responses, which, particularly with drugs that act on the central nervous system, are often the basis for therapeutic efficacy. They may take the form of a very slow onset of the therapeutic effect (e.g. with antidepressant drugs; see Ch. 46), or the development of drug dependence (Ch. 48). It is likely that changes in receptor expression, secondary to the immediate action of the drug, are involved in delayed effects of this sort—a kind of ‘secondary pharmacology’ whose importance is only now becoming clearer. The same principles apply to drug targets other than receptors (ion channels, enzymes, transporters, etc.) where adaptive changes in expression and function follow long-term drug administration, resulting, for example, in resistance to certain anticancer drugs (Ch. 55).
Receptors and Disease
Increasing understanding of receptor function in molecular terms has revealed a number of disease states directly linked to receptor malfunction. The principal mechanisms involved are:
An example of the former is myasthenia gravis (see Ch. 13), a disease of the neuromuscular junction due to autoantibodies that inactivate nicotinic acetylcholine receptors. Autoantibodies can also mimic the effects of agonists, as in many cases of thyroid hypersecretion, caused by activation of thyrotropin receptors. Activating antibodies have also been discovered in patients with severe hypertension (α-adrenoceptors), cardiomyopathy (β-adrenoceptors), and certain forms of epilepsy and neurodegenerative disorders (glutamate receptors).
Inherited mutations of genes encoding GPCRs account for various disease states (see Spiegel & Weinstein, 2004; Thompson et al., 2005). Mutated vasopressin and adrenocorticotrophic hormone receptors (see Chs 28 and 32) can result in resistance to these hormones. Receptor mutations can result in activation of effector mechanisms in the absence of agonists. One of these involves the receptor for thyrotropin, producing continuous oversecretion of thyroid hormone; another involves the receptor for luteinising hormone and results in precocious puberty. Adrenoceptor polymorphisms are common in humans, and recent studies suggest that certain mutations of the β2-adrenoceptor, although they do not directly cause disease, are associated with a reduced efficacy of β-adrenoceptor agonists in treating asthma (Ch. 27) and a poor prognosis in patients with cardiac failure (Ch. 22). Mutations in G-proteins can also cause disease (see Spiegel & Weinstein, 2004). For example, mutations of a particular Gα subunit cause one form of hypoparathyroidism, while mutations of a Gβ subunit result in hypertension. Many cancers are associated with mutations of the genes encoding growth factor receptors, kinases and other proteins involved in signal transduction (see Ch. 5).
Apart from these examples, the high expectations that the numerous polymorphisms described for GPCRs and other receptors would provide a clear understanding of the variability between individuals in their disease susceptibility and response to therapeutic drugs (see Chs 56 and 57) have been, so far, largely unfulfilled, but research activity in this area continues apace.
References and Further Reading
Alexander S.P.H., Mathie A., Peters J.A. Guide to Receptors and Channels (GRAC), fourth ed. Br. J. Pharmacol.. 2009;158(Suppl. 1):S1-S254. (Comprehensive catalogue of molecular and pharmacological properties of known receptors—also transporters and some enzymes involved in signal transduction)
Davenport A.P. Peptide and trace amine orphan receptors: prospects for new therapeutic targets. Curr. Opin. Pharmacol.. 2003;3:127-134. (Describes progress in identifying endogenous peptide ligands for ‘orphan’ GPCRs)
IUPHAR Receptor Database and Channel Compendium http://www.iuphar-db.org (Online catalogue and coding scheme for receptors and channels)
Nelson N. The family of Na+/Cl− neurotransmitter transporters. J. Neurochem.. 1998;71:1785-1803. (Review article describing the molecular characteristics of the different families of neurotransporters)
Walaas S.I., Greengard P. Protein phosphorylation and neuronal function. Pharmacol. Rev.. 1991;43:299-349. (Excellent general review)
Ashcroft F.M. Ion channels and disease. London: Academic Press; 2000. (A useful textbook covering all aspects of ion channel physiology and its relevance to disease, with a lot of pharmacological information for good measure)
Ashcroft F.M., Gribble F.M. New windows on the mechanism of action of KATP channel openers. Trends Pharmacol. Sci.. 2000;21:439-445.
Bezanilla F. How membrane proteins sense voltage. Nat. Rev. Mol. Cell Biol.. 2008;9:323-332. (Review of recent studies of how membrane proteins respond to changes in transmembrane potential)
Catterall W.A. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13-25. (General review of sodium channel structure, function and pharmacology)
Clare J.J., Tate S.N., Nobbs M., Romanos M.A. Voltage-gated sodium channels as therapeutic targets. Drug Discov. Today. 2000;5:506-520. (Useful review dealing at a basic level with the therapeutic potential of drugs affecting sodium channels)
Gay E.A., Yakel J.L. Gating of nicotinic ACh receptors: new insights into structural transitions triggered by agonist binding that induce channel opening. J. Physiol.. 2007;548:727-733.
Halliwell R.F. A short history of the rise of the molecular pharmacology of ionotropic drug receptors. Trends Pharmacol. Sci.. 2007;28:214-219. (Good account of the main discoveries in this active field)
Hille B. Ionic channels of excitable membranes. Sunderland: Sinauer Associates; 2001. (A clear and detailed account of the basic principles of ion channels, with emphasis on their biophysical properties)
Jin R., Banke T., Mayer M., et al. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat. Neurosci.. 2003;6:803-819. (Shows that partial agonists for glutamate receptors induce different conductance states of the channel)
Karlin A. Structure of nicotinic acetylcholine receptors. Curr. Opin. Neurobiol.. 1993;3:299-309. (Excellent general review)
Miyazawa A., Fujiyoshi Y., Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949-955. (Description of how the channel is opened by agonists, based on high-resolution crystallography)
Potier M., Trebak M. New developments in the signaling mechanisms of the store-operated calcium entry pathway. Pflugers Arch.. 2008;457:405-415. (Recent clarification of an old enigma)
Unwin N. Nicotinic acetylcholine receptor at 9A resolution. J. Mol. Biol.. 1993;229:1101-1124. (The first structural study of a channel-linked receptor)
Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37-43. (Refinement of 1993 paper, showing for the first time how channel opening occurs—a technical tour de force)
Wickham K.D., Clapham D.E. G-protein regulation of ion channels. Curr. Opin. Neurobiol.. 1995;5:278-285. (Discusses direct and indirect regulation of ion channels by G-protein-coupled receptors)
AbdAlla S., Lother H., El Massiery A., Quitterer U. Increased AT1 receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat. Med.. 2001;7:1003-1009. (The first instance of disturbed GPCR heterodimerisation in relation to human disease)
Bockaert J., Pin J.P. Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J.. 1999;18:1723-1729. (Short review covering some newer aspects of GPCR function)
Conigrave A.D., Quinn S.J., Brown E.M. Cooperative multi-modal sensing and therapeutic implications of the extracellular Ca2+-sensing receptor Trends Pharmacol. Sci. 2000;401-407 (Short account of the Ca2+-sensing receptor, an anomalous type of GPCR)
Costa T., Cotecchia S. Historical review: negative efficacy and the constitutive activity of G-protein-coupled receptors. Trends Pharmacol. Sci.. 2005;26:618-624. (A clear and thoughtful review of ideas relating to constitutive receptor activation and inverse agonists)
Ferguson S.S.G. Evolving concepts in G-protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol. Rev.. 2001;53:1-24. (Detailed account of the role of phosphorylation of receptors in fast and slow desensitisation mechanisms)
Fredriksson R., Schiöth H.B. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol. Pharmacol.. 2005;67:1414-1425. (Estimation of the number of GPCR genes in different species—nearly 500 more in mouse than in human!)
Hill S.J. G-protein-coupled receptors: past, present and future. Br. J. Pharmacol.. 2006;147(Suppl.):27-37. (Good introductory review)
Kelly E., Bailey C.P., Henderson G. Agonist-selective mechanisms of GPCR desensitization. Br. J. Pharmacol.. 2008;153(Supp. 1):S379-S388. (Short review of main mechanisms of GPCR desensitisation)
Kenakin T. Efficacy at G-protein-coupled receptors. Nat. Rev. Drug Discov.. 2002;1:103-110. (Mainly theoretical discussion of the implications of agonist trafficking)
Kilpatrick G., Dautzenberg F.M., Martin G.R., Eglen R.M. 7TM receptors: the splicing on the cake. Trends Pharmacol. Sci.. 1999;20:294-301. (Review of the importance of mRNA splicing as a source of variation among GPCRs—a salutary reminder that cloning genes is not the last word in defining receptor diversity)
Koenig J.A., Edwardson J.M. Endocytosis and recycling of G-protein-coupled receptors. Trends Pharmacol. Sci.. 1997;18:276-287. (Excellent review of the complex life cycle of a receptor molecule)
Liu F., Wan Q., Pristupa Z., et al. Direct protein–protein coupling enables cross-talk between dopamine D5 and γ-aminobutyric acid A receptors. Nature. 2000;403:274-280. (The first demonstration of direct coupling of a GPCR with an ion channel. Look, no G-protein!)
Lohse M.J., Hein P., Hoffmann C., et al. Kinetics of G-protein-coupled receptor signals in intact cells. Br. J. Pharmacol.. 2008;153(Suppl. 1):S125-S132. (Describes the use of fluorescence methods to measure GPCR signalling events in real time—an important step forward)
Milligan G. Signal sorting by G-protein-linked receptors. Adv. Pharmacol.. 1995;32:1-29. (More on the selectivity problem)
Milligan G., Kostenis E. Heterotrimeric G-proteins: a short history. Br. J. Pharmacol.. 2006;147(Suppl.):46-55.
Offermanns S. G-proteins as transducers in transmembrane signalling. Prog. Biophys. Mol. Biol.. 2003;83:101-130. (Detailed review of G-protein subtypes and their function in signal transduction)
Oldham W.M., Hamm H.E. Heterotrimeric G-protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol.. 2008;9:60-71. (Useful review of the current state of knowledge about the structure and function of G-proteins)
Parameswaran N., Spielman W.S. RAMPs: the past, present and future. Trends Biochem. Sci.. 2006;31:631-638. (Review on knowledge about RAMPs—still a confusing picture)
Pierce K.L., Premont R.T., Lefkowitz R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol.. 2002;3:639-650. (Useful general review of GPCRs, including desensitisation mechanisms, signal transduction pathways and dimerisation)
Prinster S.C., Hague C., Hall R.A. Heterodimerization of G-protein-coupled receptors: specificity and functional significance. Pharmacol. Rev.. 2005;57:289-298. (Good short review of what the unexpected finding of GPCR dimerisation may signify)
Ramachandran R.M.D., Hollenberg M.D. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br. J. Pharmacol.. 2008;153(Suppl. 1):S263-S282. (Useful short review of PAR mechanisms and relevance to disease states)
Reiter E., Lefkowitz R.J. GRKs and β-arrestins: roles in receptor silencing, trafficking and signaling. Trends Pharmacol. Sci.. 2006;17:159-165. (Review of the role of these proteins in GPCR function, independent of G-protein involvement)
Schwartz T.W. Molecular structure of G-protein-coupled receptors. In: Foreman J.C., Johanesen T., editors. Textbook of Receptor Pharmacology. Boca Raton: CRC Press, 1996. (Useful account without unnecessary detail)
Simonds W.F. G-protein regulation of adenylate cyclase. Trends Pharmacol. Sci.. 1999;20:66-72. (Review of mechanisms by which G-proteins affect adenylate cyclase at the level of molecular structure)
Spiegel A.M., Weinstein L.S. Inherited diseases involving G proteins and G protein-coupled receptors. Annu. Rev. Med.. 2004;55:27-39. (Short review article)
Teitler M., Herrick-Davis K., Purohit A. Constitutive activity of G-protein coupled receptors: emphasis on serotonin receptors. Curr. Top Med. Chem.. 2002;2:529-538. (Review of evidence that spontaneous activity is common among both native and mutated GPCRs)
Thompson M.D., Burnham W.M., Cole D.E.C. The G-protein coupled receptors: pharmacogenetics and disease. Crit. Rev. Clin. Lab. Sci.. 2005;42:311-389. (Extensive review with many examples of GPCR polymorphisms associated with disease)
Weis W.I., Kobilka B.K. Structural insights into G-protein-coupled receptor activation. Curr. Opin. Struct. Biol.. 2008;18:734-740.
Wess J. Molecular basis of receptor/G-protein-coupling selectivity. Pharmacol. Ther.. 1998;80:231-264. (Detailed review of molecular biology of GPCRs and G-proteins, emphasising what is known about the thorny question of how selectivity is achieved)
Xie G.-X., Palmer P.P. How regulators of G protein signalling achieve selective regulation. J. Mol. Biol.. 2007;366:349-365. (General review about RGS proteins and how they work)
Cohen P. Protein kinases—the major drug targets of the twenty-first century. Nat. Rev. Drug Discov.. 2002;1:309-315. (General review on pharmacological aspects of protein kinases)
Cook D.N., Pisetsky D.S., Schwartz D.A. Toll-like receptors in the pathogenesis of human disease. Nat. Immunol.. 2004;5:975-979. (Review emphasising the role of this class of receptor tyrosine kinases in many human disease states)
Delcourt N., Bockaert J., Marin P. GPCR-jacking: from a new route in RTK signalling to a new concept in GPCR activation. Trends Pharmacol. Sci.. 2007;28:602-607. (Gives examples of ‘cross-talk’ between GPCR and RTK signalling pathways)
Hubbard S.R., Miller W.T. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr. Opin. Cell Biol.. 2007;19:117-123. (Reviews recent structural results showing mechanism of RTK dimerisation and signalling)
Ihle J.N. Cytokine receptor signalling. Nature. 1995;377:591-594.
Pawson T. Regulation and targets of receptor tyrosine kinases. Eur. J. Cancer. 2002;38:S3-S10. (Short review of RTK signalling)
Schenk P.W., Snaar-Jakelska B.E. Signal perception and transduction: the role of protein kinases. Biochim. Biophys Acta.. 1999;1449:1-24. (General review of receptor–protein kinase interactions)
Bourguet W., Germain P., Gronemeyer H. Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol. Sci.. 2000;21:381-388. (Accessible review concentrating on distinction between agonist and antagonist effects at the molecular level)
Chawla A., Repa J.J., Evans R.M., Mangelsdorf D.J. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866-1870. (Accessible review that deals mainly with the role of nuclear receptors in lipid metabolism)
Falkenstein E., Tillmann H.C., Christ M., et al. Multiple actions of steroid hormones—a focus on rapid non-genomic effects. Pharm. Rev.. 2000;52:513-553. (Comprehensive review article describing the non-classical effects of steroids)
Germain P., Staels B., Dacquet C., Spedding M., Laudet V. Overview of nomenclature of nuclear receptors. Pharmacol. Rev.. 2006;58:685-704. (Comprehensive and authoritative review that deals with receptor biology as well as nomenclature. Recommended)
Giguere V. Orphan nuclear receptors: from gene to function. Endocr. Rev.. 1999;20:689-725. (For the serious reader)
Kersten S., Desvergne B., Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421-424. (General review of an important class of nuclear receptors)
Murphy G.J., Holder J.C. PPAR-γ agonists: therapeutic role in diabetes, inflammation and cancer. Trends Pharmacol. Sci.. 2000;21:469-474. (Account of the emerging importance of nuclear receptors of the PPAR family as therapeutic targets)
Synold T.W., Dussault I., Forman B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nature Med.. 2001;7:584-590. (Rather advanced research paper which highlights the role of the SXR receptor in regulating the metabolism of drugs)
Walters M.R., Nemere I. Receptors for steroid hormones: membrane-associated and nuclear forms. Cell Mol. Life Sci.. 2004;61:2309-2321. (Good discussion about alternative types of steroid hormone receptors)
Avruch J. MAP kinase pathways: the first twenty years. Biochim. Biophys. Acta.. 2007;1773:1150-1160. (Short general review. One of a series of articles on MAP kinases in this issue)
Beavo J.A. Cyclic nucleotide phosphodiesterase. Functional implications of multiple isoforms. Physiol. Rev.. 1995;75:725-748. (Useful account of the numerous PDE subtypes, selective inhibitors of which have many potential therapeutic applications)
Bishop A.L., Hall R.A. Rho-GTPases and their effector proteins. Biochem. J.. 2000;348:241-255. (General review article on the Rho/Rho kinase system and the various pathways and functions that it controls)
Brzostowski J.A., Kimmel A.R. Signaling at zero G: G-protein-independent functions for 7TM receptors. Trends Biochem. Sci.. 2001;26:291-297. (Review of evidence for GPCR signalling that does not involve G-proteins, thus conflicting with the orthodox dogma)
Clapham D., Neer E. G-protein βγ subunits. Annu. Rev. Pharmacol. Toxicol.. 1997;37:167-203. (On the diversity and role in signalling of G-protein βγ subunits—the poor relations of the α subunits)
Hollinger S., Hepler J.R. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Physiol. Rev.. 2002;54:527-559. (Describes the nature of this family of proteins that bind to α subunits and modulate G-protein signalling in many situations)
Karin M., Yamamoto Y., Wang M. The IKK-NFκB system: a treasure trove for drug development. Nat. Rev. Drug Discov.. 2004;3:17-26. (Describes the transcription factor NFκB, which plays a key role in inflammation, and its control by kinase cascades)
Lucas K.A., Pitari J.M., Kazerounian S., et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol. Rev.. 2000;52:375-414. (Detailed review of guanylyl cyclase and its role in signalling. Covers both membrane receptors linked to guanylyl cyclase, and soluble guanylyl cyclases that respond to nitric oxide, etc.)
Marshall C.J. Ras effectors. Curr. Opin. Cell Biol.. 1996;8:197-204. (Account of one of the most important signal transduction pathways)
Nahorski S.R. Pharmacology of intracellular signalling pathways. Br. J. Pharmacol.. 2006;147(Suppl.):38-45. (Useful short review)
Nishizuka Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature. 1988;334:661-665. (As above)
Vanhaesebroeck B., Leevers S.J., Panayotou G., Waterfield M.D. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem. Sci.. 1997;22:267-272. (Review by the group that discovered PI-3 kinases, summarising the multiple roles of this signal transduction mechanism—much expanded since 1997)
1‘Ion channels and the electrical properties they confer on cells are involved in every human characteristic that distinguishes us from the stones in a field.’ (Armstrong C M 2003 Voltage-gated K channels; http://www.stke.org.)
2Nature has had the good sense to keep these heavily armed fishes and snakes well apart. Ironically enough, B. multicinctus is now officially an endangered species, threatened by scientists’ demand for its venom. Evolution for survival can go one step too far.
3An oddly Dickensian term that seems inappropriately condescending, because we can assume that these receptors play defined roles in physiological signalling—their ‘orphanhood’ reflects our ignorance, not their status.
4Here, focusing on receptors, we include ligand-gated ion channels as an example of a receptor family. Other types of ion channels are described later (p. 43); many are also drug targets, although not receptors in the strict sense.
5There are 865 human GPCRs comprising 1.6% of the genome (Fredricksson & Schiöth, 2005). Nearly 500 of these are believed to be odorant receptors involved in smell and taste sensations, the remainder being receptors for known or unknown endogenous mediators—enough to keep pharmacologists busy for some time yet.
6Receptors for 5-HT (see Ch. 15) are currently the champions with respect to diversity, with 14 cloned subtypes.
7Examples of promiscuity are increasing, however. Steroid hormones, normally faithful to nuclear receptors, make the occasional pass at ion channels and other targets (see Falkenstein et al., 2000), and some eicosanoids act on nuclear receptors as well as GPCRs. Nature is quite open minded, although such examples are liable to make pharmacologists frown and students despair.
8The Ca2+-sensing receptor (see Conigrave et al., 2000) is an unusual GPCR that is activated, not by conventional mediators, but by extracellular Ca2+ in the range of 1–10 mM—an extremely low affinity in comparison with other GPCR agonists. It is expressed by cells of the parathyroid gland, and serves to regulate the extracellular Ca2+ concentration by controlling parathyroid hormone secretion (Ch. 35). This homeostatic mechanism is quite distinct from the mechanisms for regulating intracellular Ca2+ discussed in Chapter 4.
9Many lead compounds in recent years have come from screening huge chemical libraries (see Ch. 56). No inspiration is required, just robust assays, large computers and efficient robotics.
10In humans there are 21 known subtypes of Gα, there are 6 of Gβ and 12 of Gγ, providing, in theory, about 1500 variants of the trimer. We know little about the role of different α, β and γ subtypes, but it would be rash to assume that the variations are functionally irrelevant. By now, you will be unsurprised (even if somewhat bemused) by such a display of molecular heterogeneity, for it is the way of evolution.
11Alternative abbreviations for these mediators are PtdIns (PI), PtdIns (4,5)-P2 (PIP2), Ins (1,4,5)-P3 (IP3), and Ins (1,2,4,5)-P4 (IP4).
12In truth, the distinction between ligand-gated channels and other ion channels is an arbitrary one. In grouping ligand-gated channels with other types of receptor in this book, we are respecting the historical tradition established by Langley and others, who first defined receptors in the context of the action of acetylcholine at the neuromuscular junction. The advance of molecular biology may force us to reconsider this semantic issue in the future, but for now we make no apology for upholding the pharmacological tradition.
13The human genome encodes more than 70 distinct potassium channel subtypes—either a nightmare or a golden opportunity for the pharmacologist, depending on one’s perspective.