51 Human Herpesviruses
(a) A vesicular lesion erupts at the corner of a 27-year-old man’s mouth 3 days after returning from a skiing trip.
(b) A 26-year-old pediatric medical resident develops serious pneumonia, and then vesicular lesions erupt in crops on his head, trunk, and elsewhere.
(c) Several high-school cheerleaders developed a sore throat, fever, swollen glands, and were very tired. They shared a water bottle at the football game.
(d) A 57-year-old heart transplant recipient had an outbreak of herpes simplex virus lesions, cytomegalovirus pneumonitis, and subsequently, an Epstein-Barr virus–related lymphoma. The lymphoma resolved after immunosuppressive therapy was decreased.
1. Which viruses cause these diseases?
2. What features are similar/different for these viruses?
3. How were each of these infections obtained?
4. What are the risk factors for serious herpes disease?
5. Which of the infections can be prevented by vaccine or treated with antiviral drugs?
Answers
1. (a) HSV-1 and HSV-2. Both viruses cause similar presentations and it depends on which virus was placed at the site.
(b) Varicella-zoster virus (VZV)
(d) HSV, cytomegalovirus (CMV), and EBV
2. (a) The viruses are very similar and can cause the same diseases, except HSV-2 is usually transmitted and occurs below the belt and HSV-1 above the belt. HSV-1 can cause encephalitis, and HSV-2 meningitis. The two viruses can be discriminated antigenically, by antiviral drug susceptibility, protein patterns, restriction fragment polymorphism, and DNA sequence.
(b) VZV resembles HSV in that it is neurotropic and expresses a thymidine kinase. Unlike HSV, it is transmitted by aerosol, acquired in the lungs, and then spread by viremia to the target tissues (e.g., skin). Like HSV, VZV establishes latency in neurons, but unlike HSV, recurrence (zoster) results in replication and release along an entire dermatome, whereas HSV is released only at the terminus of the nerve.
(c) EBV lacks thymidine kinase and has a very specific receptor specificity, which defines its tropism to B cells and some epithelial cells. Once in the B cell, it uses the natural cell biology of the B cell to promote its latent and recurrent cycles.
(d) All establish lytic, latent, and recurrent infections. HSV is neurotropic; CMV and EBV are lymphotropic, but unlike EBV, CMV can infect many different cell types.
3. (a) The infection was initially acquired by contact with another person with an active lesion (kissing) or their saliva. This presentation is likely to be a recurrence of an HSV infection after exposure to ultraviolet B radiation, a common trigger of recurrence.
(b) The patient breathed in an aerosol containing the virus. The virus can also be obtained by contact with active lesions, but this route is not efficient.
(c) EBV is acquired by sharing saliva (e.g., kissing)—in this case, from the water bottle.
(d) All of the disease presentations are recurrences from latent virus present in neurons for HSV, in macrophages and other cells for CMV, and in B cells for EBV as a result of the immunosuppression.
4. (a) Immunocompromised individuals are at risk to disseminated disease. For neonates, HSV infection can be lethal because of their limited cell-mediated immunity. Also, HSV spreads extensively in individuals with eczema because of their compromised skin.
(b) Adults suffer more severe disease than children and are prone to pneumonia during the initial infection stage of the lung. Immunocompromised individuals and neonates are at risk because of the lack of protective cell-mediated immunity.
(c) Immunocompromised individuals are at risk for a leukemia/lymphoma-like disease of B cells because of the ability of EBV to immortalize these cells.
(d) HSV and EBV cause disease in normal individuals, whereas CMV disease is usually asymptomatic or limited except in the immunocompromised individual. The immunocompromised individual is at risk for serious disease for all herpesviruses.
5. (a) There is no vaccine for HSV, but there are antiviral drugs, including acyclovir, valcyclovir, penciclovir, and famciclovir.
(b) There is a live vaccine for varicella, given on the same schedule as the measles-mumps-rubella vaccine. The zoster vaccine is a stronger version of the varicella vaccine and is administered to adults greater than 60 years of age. There are antiviral drugs, including acyclovir, valcyclovir, penciclovir, and famciclovir.
(c) There is no vaccine and no true antiviral drug for EBV.
(d) There are no vaccines for these viruses. HSV and CMV can be treated with antiviral drugs, CMV with ganciclovir, valganciclovir, foscarnet, and cidofovir.
The herpesviruses are an important group of large, deoxyribonucleic acid (DNA)–enveloped viruses with the following features in common: virion morphology, basic mode of replication, and capacity to establish latent and recurrent infections. Cell-mediated immunity is important for causing symptoms and controlling infection with these viruses. Herpesviruses encode proteins and enzymes that facilitate the replication and interaction of the virus with the host. Epstein-Barr virus (EBV) and human herpesvirus 8 (HHV-8) are associated with human cancers (Box 51-1).
Box 51-1
Unique Features of Herpesviruses
Herpesviruses have large, enveloped icosadeltahedral capsids containing double-stranded DNA genomes.
Herpesviruses encode many proteins that manipulate the host cell and immune response.
Herpesviruses encode enzymes (DNA polymerase) that promote viral DNA replication and are good targets for antiviral drugs.
DNA replication and capsid assembly occurs in the nucleus.
Virus is released by exocytosis, cell lysis, and through cell-to-cell bridges.
Herpesviruses can cause lytic, persistent, latent, and (for Epstein-Barr virus) immortalizing infections.
The human herpesviruses are grouped into three subfamilies on the basis of differences in viral characteristics (genome structure, tissue tropism, cytopathologic effect, and site of latent infection), as well as the pathogenesis of the disease and disease manifestation (Table 51-1). The human herpesviruses are herpes simplex viruses types 1 and 2 (HSV-1 and HSV-2), varicella-zoster virus (VZV), Epstein-Barr virus, cytomegalovirus (CMV), human herpesviruses 6 and 7 (HHV-6 and HHV-7), and the more recently discovered HHV-8, associated with Kaposi sarcoma.
Herpesvirus infections are common, and the viruses, except HHV-8, are ubiquitous. Although these viruses usually cause benign disease, especially in children, they can also cause significant morbidity and mortality, especially in immunosuppressed people. Fortunately, some herpesviruses encode targets for antiviral agents, and there is a live-virus vaccine for VZV.
Structure of Herpesviruses
The herpesviruses are large, enveloped viruses that contain double-stranded DNA. The virion is approximately 150 nm in diameter and has the characteristic morphology shown in Figure 51-1. The DNA core is surrounded by an icosadeltahedral capsid containing 162 capsomeres. This capsid is enclosed by a glycoprotein-containing envelope. Herpesviruses encode several glycoproteins for viral attachment, fusion, and for escaping immune control. Attached to the capsid and in the space between the envelope and the capsid (the tegument) are viral proteins and enzymes that help initiate replication. As enveloped viruses, the herpesviruses are sensitive to acid, solvents, detergents, and drying.
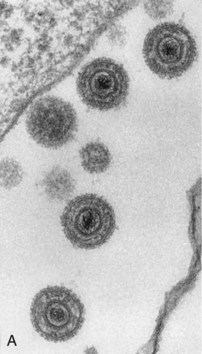
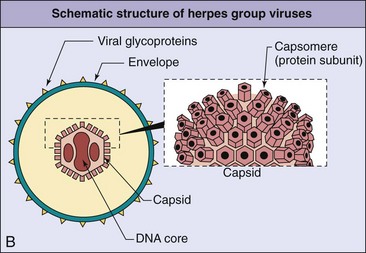
Figure 51-1 Electron micrograph (A) and general structure (B) of the herpesviruses. The DNA genome of the herpesvirus in the core is surrounded by an icosadeltahedral capsid and an envelope. Glycoproteins are inserted into the envelope.
(A, From Armstrong D, Cohen J: Infectious diseases, St Louis, 1999, Mosby.)
Herpesviral genomes are linear, double-stranded DNA, but they differ in size and gene orientation (Figure 51-2). Direct or inverted repeat sequences bracket unique regions of the genome (unique long [UL], unique short [US]), allowing circularization and recombination within the genome. Recombination among inverted repeats of HSV, CMV, and VZV allows large portions of the genome to flip the orientation of their UL and US gene segments with respect to each other to form isomeric genomes.
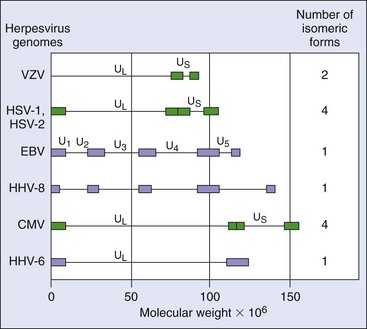
Figure 51-2 Herpesvirus genomes. The genomes of the herpesvirus are doubled-stranded DNA. The length and complexity of the genome differ for each virus. Inverted repeats in herpes simplex virus (HSV), varicella-zoster virus (VZV), and cytomegalovirus (CMV) allow the genome to recombine with itself to form isomers. Large genetic repeat sequences are boxed. The genomes of HSV and CMV have two sections, the unique long (UL) and the unique short (US), each of which is bracketed by two sets of inverted repeats of DNA. The inverted repeats facilitate the replication of the genome but also allow the UL and US regions to invert independently of each other to yield four different genomic configurations, or isomers. VZV has only one set of inverted repeats and can form two isomers. Epstein-Barr virus (EBV) exists in only one configuration, with several unique regions surrounded by direct repeats. Purple bars indicate direct repeat DNA sequences; green bars indicate inverted repeated DNA sequences. HHV-6, Human herpesvirus 6; HHV-8, human herpesvirus 8.
Herpesvirus Replication
Herpesvirus replication is initiated by the interaction of viral glycoproteins with cell surface receptors (see Chapter 44, Figure 44-12). The tropism of some herpesviruses (e.g., EBV) is highly restricted because of the species and tissue-specific expression of their receptors. The virus can fuse its envelope with the plasma membrane, releasing the nucleocapsid into the cytoplasm. Enzymes and transcription factors are carried into the cell in the tegument of the virion. The nucleocapsid docks with the nuclear membrane and delivers the genome into the nucleus, where the genome is transcribed and replicated.
Transcription of the viral genome and viral protein synthesis proceeds in a coordinated and regulated manner in the following three phases:
1. Immediate early proteins (α), consisting of proteins important for the regulation of gene transcription and takeover of the cell
2. Early proteins (β), consisting of more transcription factors and enzymes, including the DNA polymerase
3. Late proteins (γ), consisting mainly of structural proteins, which are generated after viral genome replication has begun
The viral genome is transcribed by the cellular DNA-dependent ribonucleic acid (RNA) polymerase and is regulated by viral-encoded and cellular nuclear factors. The interplay of these factors determines whether a lytic, persistent, or latent infection occurs. Cells that promote latent infection transcribe a special set of viral genes without genome replication. Progression to early and late gene expression results in cell death and lytic infection.
The viral-encoded DNA polymerase, which is a target of antiviral drugs, replicates the viral genome. Viral-encoded scavenging enzymes provide deoxyribonucleotide substrates for the polymerase. These and other viral enzymes facilitate replication of the virus in nongrowing cells that lack sufficient deoxyribonucleotides and enzymes for viral DNA synthesis (e.g., neurons). Other proteins manipulate cellular machinery to optimize replication, inhibit immune responses, and inhibit apoptosis or establish latency.
Empty procapsids assemble in the nucleus, are filled with DNA, acquire an envelope at the nuclear or Golgi membrane, and exit the cell by exocytosis or by lysis of the cell. Transcription, protein synthesis, glycoprotein processing, and exocytotic release from the cell are performed by cellular machinery. The replication of HSV is discussed in more detail as the prototype of the herpesviruses.
Herpes Simplex Virus
HSV was the first human herpesvirus to be recognized. The name herpes is derived from a Greek word meaning “to creep.” “Cold sores” were described in antiquity, and their viral etiology was established in 1919.
The two types of herpes simplex virus, HSV-1 and HSV-2, share many characteristics, including DNA homology, antigenic determinants, tissue tropism, and disease signs. However, they can still be distinguished by subtle but significant differences in these properties.
Herpes Simplex Virus Proteins
The HSV genome is large enough to encode approximately 80 proteins. Only half the proteins are required for viral replication; the others facilitate HSV’s interaction with different host cells and the immune response. The HSV genome encodes enzymes, including a DNA-dependent DNA polymerase and scavenging enzymes, such as deoxyribonuclease, thymidine kinase, ribonucleotide reductase, and protease. Ribonucleotide reductase converts ribonucleotides to deoxyribonucleotides, and thymidine kinase phosphorylates the deoxyribonucleosides to provide substrates for replication of the viral genome. The substrate specificities of these enzymes and the DNA polymerase differ significantly from those of their cellular analogues and thus represent potentially good targets for antiviral chemotherapy.
HSV encodes at least 10 glycoproteins that serve as viral attachment proteins (gB, gC, gD, gE/gI), fusion proteins (gB, gH/gL), structural proteins, immune escape proteins (gC, gE, gI), and other functions. For example, the C3 component of the complement system binds to gC and is depleted from serum. The Fc portion of immunoglobulin G (IgG) binds to a gE/gI complex, thereby camouflaging the virus and virus-infected cells. These actions reduce the antiviral effectiveness of antibody.
Replication
HSV can infect most types of human cells and even cells of other species. The virus generally causes lytic infections of fibroblasts and epithelial cells and latent infections of neurons (see Chapter 44, Figure 44-12, for a diagram).
HSV-1 binds quickly and efficiently to cells through an initial interaction with heparan sulfate, a proteoglycan found on the outside of many cell types, and then through a tighter interaction with receptor proteins at the cell surface. Penetration into the cell requires interaction with nectin-1 (herpesvirus entry mediator C), an intercellular adhesion molecule that is a member of the immunoglobulin protein family and similar to the poliovirus receptor. Nectin-1 is found on most cells and neurons. Another receptor is HveA, a member of the tumor necrosis factor receptor family that is expressed on activated T cells, neurons, and other cells. HSV can penetrate the host cell by fusion of its envelope with the cell surface membrane. On fusion, the virion releases its capsid into the cytoplasm, along with a protein that promotes the initiation of viral gene transcription, a viral-encoded protein kinase, and cytotoxic proteins. The capsid docks with a nuclear pore and delivers the genome into the nucleus.
The immediate early gene products include DNA-binding proteins, which stimulate DNA synthesis and promote the transcription of the early viral genes. During a latent infection of neurons, the only region of the genome to be transcribed generates the latency-associated transcripts (LATs). These RNAs are not translated into protein but encode micro-RNAs that inhibit expression of important immediate early and other genes.
The early proteins include the DNA-dependent DNA polymerase and a thymidine kinase. As catalytic proteins, relatively few copies of these enzymes are required to promote replication. Other early proteins inhibit the production and initiate the degradation of cellular messenger RNA (mRNA) and DNA. Expression of the early and late genes generally leads to cell death.
The genome is replicated as soon as the polymerase is synthesized. Circular, end-to-end concatameric forms of the genome are made initially. Later in the infection, the DNA is replicated by a rolling-circle mechanism to produce a linear string of genomes that, in concept, resembles a roll of toilet paper. The concatamers are cleaved into individual genomes as the DNA is sucked into a procapsid.
Genome replication triggers transcription of the late genes from which structural and other proteins are encoded. Many copies of the structural proteins are required. The capsid proteins are then transported to the nucleus, where they are assembled into empty procapsids and filled with DNA. DNA-containing capsids associate with viral protein-disrupted nuclear membranes and bud into and then out of the endoplasmic reticulum into the cytoplasm. The viral glycoproteins are synthesized and processed like cellular glycoproteins. Tegument proteins associate with the viral capsid in the cytoplasm, and then the capsid buds into a portion of the trans-Golgi network to acquire their glycoprotein-containing envelope. The virus is released by exocytosis or cell lysis. Virus can also spread between cells through intracellular bridges, which allows the virus to escape antibody detection. Virus-induced syncytia formation also spreads the infection.
HSV infection of neurons may result in virus replication or establishment of latency, depending on which viral genes the neuron is capable of transcribing. Transcription of the LAT and no other viral gene will result in latency. As for other alphaherpesviruses, HSV encodes a thymidine kinase (scavenging enzyme) to facilitate replication in nondividing cells, such as neurons. HSV also encodes ICP34.5, a unique protein that has multiple functions to facilitate virus growth in neurons. ICP34.5 removes a cellular block to protein synthesis activated in response to virus infection or as part of the response to interferon-α.
Pathogenesis and Immunity
The mechanisms involved in the pathogenesis of HSV-1 and HSV-2 are very similar (Box 51-2). Both viruses initially infect, replicate in mucoepithelial cells, cause disease at the site of infection, and then establish latent infection of the innervating neurons. HSV-1 is usually associated with infections above the waist, and HSV-2 with infections below the waist (Figure 51-3), consistent with the means of spread for these viruses. HSV-1 and HSV-2 also differ in growth characteristics and antigenicity, and HSV-2 has a greater potential to cause viremia with associated systemic flulike symptoms.
Box 51-2
Disease Mechanisms for Herpes Simplex Viruses
Disease is initiated by direct contact and depends on infected tissue (e.g., oral, genital, brain).
Virus causes direct cytopathologic effects.
Virus avoids antibody by cell-to-cell spread and syncytia.
Virus establishes latency in neurons (hides from immune response).
Virus is reactivated from latency by stress or immune suppression.
Cell-mediated immunity is required for resolution, with a limited role for antibody.
Cell-mediated immunopathologic effects contribute to symptoms.
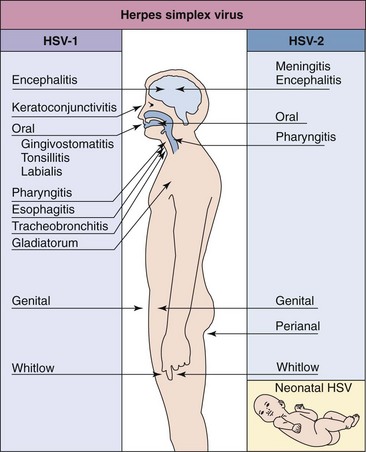
Figure 51-3 Disease syndromes of herpes simplex virus (HSV). HSV-1 and HSV-2 can infect the same tissues and cause similar diseases but have a predilection for the sites and diseases indicated.
HSV can cause lytic infections of most cells and latent infection of neurons. Cytolysis generally results from the virus-induced inhibition of cellular macromolecular synthesis, the degradation of host cell DNA, membrane permeation, cytoskeletal disruption, and senescence of the cell. Visible changes in the nuclear structure and margination of the chromatin occur, and Cowdry type A acidophilic intranuclear inclusion bodies are produced. Many strains of HSV also initiate syncytia formation. In tissue culture, HSV rapidly kills cells, causing them to appear rounded.
HSV initiates infection through mucosal membranes or breaks in the skin. The virus replicates in the cells at the base of the lesion and infects the innervating neuron, traveling by retrograde transport to the ganglion (the trigeminal ganglia for oral HSV and the sacral ganglia for genital HSV) (see Figure 51-5 later). CD8 T cells and interferon-γ are important to maintaining HSV in latency. Upon reactivation, the virus then returns to the initial site of infection, and the infection may be inapparent or may produce vesicular lesions. The vesicle fluid contains infectious virions. Tissue damage is caused by a combination of viral pathology and immunopathology. The lesion generally heals without producing a scar.
Innate protections, including interferon and natural killer cells, may be sufficient to limit the initial progression of the infection. T-helper 1 (TH1)–associated and CD8 cytotoxic killer T-cell responses are required to kill infected cells and resolve the current disease. The immunopathologic effects of the cell-mediated and inflammatory responses are also a major cause of the disease signs. Antibody directed against the glycoproteins of the virus neutralizes extracellular virus, limiting its spread, but is not sufficient to resolve the infection. In the absence of functional cell-mediated immunity, HSV infection is likely to recur, be more severe, and may disseminate to the vital organs and the brain.
HSV has several ways to escape host protective responses. The virus blocks the interferon-induced inhibition of viral protein synthesis and encodes a protein to plug the transporter-associated-with-processing (TAP) channel, preventing delivery of peptides into the endoplasmic reticulum (ER), which blocks their association with class I major histocompatibility complex (MHC I) molecules and prevents CD8 T-cell recognition of infected cells. The virus can escape antibody neutralization and clearance by direct cell-to-cell spread and by going into hiding during latent infection of the neuron. In addition, the virion and virus-infected cells express antibody (Fc) and complement receptors that weaken these humoral defenses.
Latent infection occurs in neurons and results in no detectable damage. A recurrence can be activated by various stimuli (e.g., stress, trauma, fever, sunlight [ultraviolet B]) (Box 51-3). These events trigger virus replication in an individual nerve cell within the bundle and allow the virus to travel back down the nerve to cause lesions to develop at the same dermatome and location each time. The stress triggers reactivation by promoting replication of the virus in the nerve, by transiently depressing cell-mediated immunity, or by inducing both processes. The virus can be reactivated despite the presence of antibody. However, recurrent infections are generally less severe, more localized, and of shorter duration than the primary episodes because of the nature of the spread and the existence of memory immune responses.
Epidemiology
Because HSV can establish latency with the potential for asymptomatic recurrence, the infected person is a lifelong source of contagion (Box 51-4). As an enveloped virus, HSV is transmitted in secretions and by close contact. The virus is very labile and is readily inactivated by drying, detergents, and the conditions of the gastrointestinal tract. Although HSV can infect animal cells, HSV infection is exclusively a human disease.
Box 51-4
Epidemiology of Herpes Simplex Virus (HSV)
Transmission
Virus is transmitted in saliva, in vaginal secretions, and by contact with lesion fluid (mixing and matching of mucous membranes).
Virus is transmitted orally and sexually and by placement into eyes and breaks in skin.
HSV-1 is generally transmitted orally; HSV-2 is generally transmitted sexually, but not exclusively.
Who Is at Risk?
Children and sexually active people are at risk for primary disease of HSV-1 and HSV-2, respectively.
Physicians, nurses, dentists, and others in contact with oral and genital secretions are at risk for infections of fingers (herpetic whitlow).
Immunocompromised people and neonates are at risk for disseminated, life-threatening disease.
HSV is transmitted in vesicle fluid, saliva, and vaginal secretions (the “mixing and matching of mucous membranes”). The site of infection, and hence the disease, is determined primarily by which mucous membranes are mixed. Both types of HSV can cause oral and genital lesions.
HSV-1 is usually spread by oral contact (kissing) or through the sharing of drinking glasses, toothbrushes, or other saliva-contaminated items. HSV-1 can infect the fingers or body through a cut or abraded skin. Autoinoculation may also cause infection of the eyes or fingers.
HSV-1 infection is common. More than 90% of people living in underdeveloped areas have the antibody to HSV-1 by 2 years of age.
HSV-2 is spread mainly by sexual contact or autoinoculation or from an infected mother to her infant at birth. Depending on a person’s sexual practices and hygiene, HSV-2 may infect the genitalia, anorectal tissues, or oropharynx. The incidence of HSV-1 genital infection is approaching that of HSV-2. HSV may cause symptomatic or asymptomatic primary genital infection or recurrences. Neonatal infection usually results from the excretion of HSV-2 from the cervix during vaginal delivery (Clinical Case 51-1) but can occur from an ascending in utero infection during a primary infection of the mother. Neonatal infection results in disseminated and neurologic disease with severe consequences.
Clinical Case 51-1
Neonatal Herpes Simplex Virus (HSV)
Parvey and Ch’ien (Pediatrics 65:1150–1153, 1980) reported a case of neonatal HSV contracted during birth. During a breech presentation, a fetal monitor was placed on the buttocks of the baby, and because of the greatly prolonged labor, the baby was delivered by cesarean section. The 5-pound boy had minor difficulties that were successfully treated, but on the sixth day, vesicles with an erythematous base appeared at the site where the fetal monitor had been placed. HSV was grown from the vesicle fluid and from spinal fluid, cornea, saliva, and blood. The baby became moribund, with frequent apneic episodes and seizures. Intravenous treatment with adenosine arabinoside (ara-A; vidarabine) was initiated. The baby also developed bradycardia and occasional vomiting. The vesicles spread to cover the lower extremities and were also on the back, palm, nares, and right eyelid. Within 72 hours of ara-A treatment, the baby’s condition started to improve. Treatment was continued for 11 days but discontinued because of a low platelet count. The baby was discharged on the 45th day after his birthday, and normal development was reported at 1 and 2 years of age. At 6 weeks after the birth, a herpes lesion was found on the mother’s vulva. This was a fortunate case of neonatal HSV infection in which the baby was successfully treated with ara-A and was able to overcome the damage caused by the infection. The virus, most likely HSV-2, was probably acquired through an abrasion caused by the fetal monitor while the neonate was in the birth canal. Ara-A has since been replaced with the better, less toxic, and easier to administer antiviral drugs: acyclovir, valacyclovir, and famciclovir.
Initial infection with HSV-2 occurs later in life than infection with HSV-1 and correlates with increased sexual activity. The current statistics indicate that 25% of adults in the United States are infected with HSV-2, which amounts to approximately 45 million people, with up to 1 million newly infected people per year.
Clinical Syndromes
HSV-1 and HSV-2 are common human pathogens that can cause painful but benign manifestations and recurrent disease. In the classic manifestation, the lesion is a clear vesicle on an erythematous base (“dewdrop on a rose petal”) and then progresses to pustular lesions, ulcers, and crusted lesions (Figure 51-4). Both viruses can cause significant morbidity and mortality on infection of the eye or brain and on disseminated infection of an immunosuppressed person or a neonate.
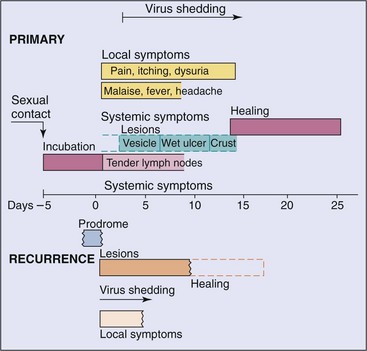
Figure 51-4 Clinical course of genital herpes infection. The time course and symptoms of primary and recurrent genital infection with herpes simplex virus type 2 are compared. Top, Primary infection; bottom, recurrent disease.
(Data from Corey L, et al: Genital herpes simplex virus infection: clinical manifestations, course and complications, Ann Intern Med 98:958–973, 1983.)
Oral herpes can be caused by HSV-1 or HSV-2. The lesions of herpes labialis or gingivostomatitis begin as clear vesicles that rapidly ulcerate. The vesicles may be widely distributed around or throughout the mouth, involving the palate, pharynx, gingivae, buccal mucosa, and tongue (Figure 51-5). Many other conditions (e.g., Coxsackie virus lesions, canker sores, acne) may resemble HSV lesions.
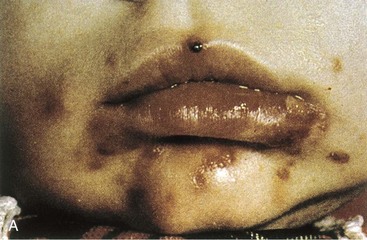
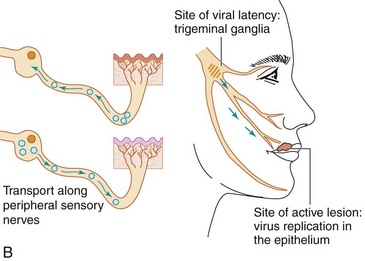
Figure 51-5 A, Primary herpes gingivostomatitis. B, Herpes simplex virus establishes latent infection and can recur from the trigeminal ganglia.
(A, From Hart CA, Broadhead RL: A color atlas of pediatric infectious diseases, London, 1992, Wolfe. B, Modified from Straus SE: Herpes simplex virus and its relatives. In Schaechter M, Eisenstein BI, Medoff G, editors: Mechanisms of microbial disease, ed 2, Baltimore, 1993, Williams & Wilkins.)
Infected people may experience recurrent mucocutaneous HSV infection (cold sores, fever blisters) (Figure 51-6), even though they never had a clinically apparent primary infection. The lesions usually occur at the corners of the mouth or next to the lips. Recurrent facial herpes infections are generally activated from the trigeminal ganglia. As noted earlier, the symptoms of a recurrent episode are less severe, more localized, and of shorter duration than those of a primary episode. Herpes pharyngitis is becoming a prevalent diagnosis in young adults with sore throats.
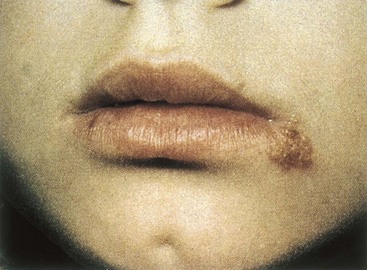
Figure 51-6 Cold sore of recurrent herpes labialis. It is less severe than that of primary disease.
(From Hart CA, Broadhead RL: A color atlas of pediatric infectious diseases, London, 1992, Wolfe.)
Herpetic keratitis is almost always limited to one eye. It can cause recurrent disease, leading to permanent scarring, corneal damage, and blindness.
Herpetic whitlow is an infection of the finger, and herpes gladiatorum is an infection of the body. The virus establishes infection through cuts or abrasions in the skin. Herpetic whitlow often occurs in nurses or physicians who attend patients with HSV infections, in thumb-sucking children (Figure 51-7), and in people who have genital HSV infections. Herpes gladiatorum is often acquired during wrestling or rugby.
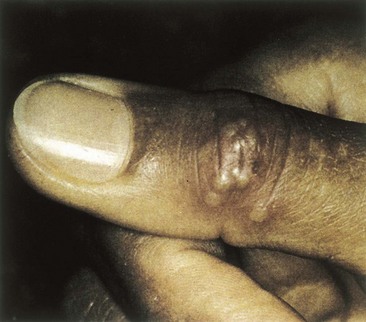
Eczema herpeticum is acquired by children with active eczema. The underlying disease promotes the spread of the infection along the skin and potentially to the adrenal glands, liver, and other organs.
Genital herpes can be caused by HSV-1 or HSV-2. In male patients, the lesions typically develop on the glans or shaft of the penis and occasionally in the urethra. In female patients, the lesions may be seen on the vulva, vagina, cervix, perianal area, or inner thigh and are frequently accompanied by itching and a mucoid vaginal discharge. Anal sex can lead to HSV proctitis, a condition in which the lesions are found in the lower rectum and anus. The lesions are usually painful. In patients of both sexes, a primary infection may be accompanied by fever, malaise, and myalgia, which are symptoms related to a transient viremia. The symptoms and time course of primary and recurrent genital herpes are compared in Figure 51-4.
Recurrent genital HSV disease is shorter in duration and less severe than the primary episode. In approximately 50% of patients, recurrences are preceded by a characteristic prodrome of burning or tingling in the area in which the lesions eventually erupt. Episodes of recurrence may be as frequent as every 2 to 3 weeks or may be infrequent. Unfortunately, any infected person may shed virus asymptomatically. Such individuals may be important vectors for spread of this virus.
Herpes encephalitis is usually caused by HSV-1. The lesions are generally limited to one of the temporal lobes. The viral pathology and immunopathology cause the destruction of the temporal lobe and give rise to erythrocytes in the cerebrospinal fluid, seizures, focal neurologic abnormalities, and other characteristics of viral encephalitis. HSV is the most common viral cause of sporadic encephalitis and results in significant morbidity and mortality, even in patients who receive appropriate treatment. The disease occurs at all ages and at any time of the year. HSV meningitis may be a complication of genital HSV-2 infection; symptoms resolve by themselves.
HSV infection in the neonate is a devastating and often fatal disease caused most often by HSV-2. It may be acquired in utero but more commonly is contracted either during passage of the infant through the vaginal canal (possibly at the baby’s scalp-monitor site) because the mother is shedding herpesvirus at the time of delivery, or it is acquired postnatally from family members or hospital personnel. The baby initially appears septic, and vesicular lesions may or may not be present. Because the cell-mediated immune response is not yet developed in the neonate, HSV disseminates to the liver, lung, and other organs, as well as to the central nervous system (CNS). Progression of the infection to the CNS results in death, mental retardation, or neurologic disability, even with treatment.
Laboratory Diagnosis
Direct Analysis of a Clinical Sample
Characteristic cytopathologic effects (CPEs) can be identified in a Tzanck smear (a scraping of the base of a lesion), Papanicolaou (Pap) smear, or biopsy specimen (Table 51-2). CPEs include syncytia, “ballooning” cytoplasm, and Cowdry type A intranuclear inclusions (see Chapter 47, Figure 47-2). A definitive diagnosis can be made by demonstrating viral antigen (using immunofluorescence or the immunoperoxidase method) or DNA (using in situ hybridization or polymerase chain reaction [PCR]) in the tissue sample or vesicle fluid.
Table 51-2 Laboratory Diagnosis of Herpes Simplex Virus (HSV) Infections
| Approach | Test/Comment |
|---|---|
| Direct microscopic examination of cells from base of lesion | Tzanck smear shows multinucleated giant cells and Cowdry type A inclusion bodies |
| Cell culture | HSV replicates and causes identifiable cytopathologic effect in most cell cultures |
| Assay of tissue biopsy, smear, cerebrospinal fluid, or vesicular fluid for HSV antigen or genome | Enzyme immunoassay, immunofluorescent stain, in situ DNA probe analysis, and PCR |
| HSV type distinction (HSV-1 vs. HSV-2) | Type-specific antibody, DNA maps of restriction enzyme fragments, sodium dodecyl sulfate–gel protein patterns, DNA probe analysis, and PCR |
| Serology | Serology is not useful except for epidemiology |
DNA, Deoxyribonucleic acid; PCR, polymerase chain reaction.
Virus Isolation
Virus isolation is the most definitive assay for the diagnosis of HSV infection. Virus can be obtained from vesicles but not crusted lesions. Specimens are collected by aspiration of the lesion fluid or by application of a cotton swab to the vesicles and direct inoculation of the sample into cell cultures.
HSV produces CPEs within 1 to 3 days in HeLa cells, human embryonic fibroblasts, and other cells. Infected cells become enlarged and appear ballooned (see Chapter 47, Figure 47-4). Some isolates induce fusion of neighboring cells, giving rise to multinucleated giant cells (syncytia). A new, sensitive approach to isolation and identification uses a cell line that expresses β-galactosidase upon HSV infection of cells (enzyme-linked viral-inducible system [ELVIS]). Addition of the appropriate substrate produces color and allows detection of the enzyme in the infected cells.
Genome Detection
HSV type-specific DNA probes, specific DNA primers for PCR and quantitative PCR, are used to detect and differentiate HSV-1 and HSV-2. PCR analysis of cerebrospinal fluid has replaced immunofluorescence analysis of a brain biopsy in the diagnosis for herpes encephalitis. The distinction between HSV-1 or HSV-2 and different strains of either virus can also be made by restriction endonuclease cleavage patterns of the viral DNA.
Treatment, Prevention, and Control
HSV encodes several target enzymes for antiviral drugs (Box 51-5) (see Chapter 48). Most antiherpes drugs are nucleoside analogues that inhibit the viral DNA polymerase, an enzyme essential for viral replication and the best antiviral drug target. Treatment prevents or shortens the course of primary or recurrent disease. None of the drug treatments can eliminate latent infection.
Box 51-5
FDA-Approved Antiviral Treatments for Herpesvirus Infections
* Also inhibits herpes simplex and varicella-zoster viruses.
The prototype anti-HSV drug is acyclovir (ACV). Valacyclovir (the valyl ester of ACV), penciclovir, and famciclovir (a derivative of penciclovir) are related to ACV in their mechanisms of action but have different pharmacologic properties. Vidarabine (adenosine arabinoside [Ara A]), idoxuridine (iododeoxyuridine), and trifluridine, also U.S. Food and Drug Administration (FDA)-approved for treatment of HSV, are less effective. Although cidofovir and adefovir are active against HSV, cidofovir is only approved for treatment of CMV.
ACV is the most prescribed anti-HSV drug. Phosphorylation of ACV and penciclovir by the viral thymidine kinase and cellular enzymes activates the drug as a substrate for the viral DNA polymerase. These drugs are then incorporated into and prevent the elongation of the viral DNA (see Chapter 48, Figure 48-2). ACV, valacyclovir, penciclovir, and famciclovir are (1) relatively nontoxic, (2) effective in treating serious presentations of HSV disease and first episodes of genital herpes, and (3) also used for prophylactic treatment.
The most prevalent form of resistance to these drugs results from mutations that inactivate the thymidine kinase, thereby preventing conversion of the drug to its active form. Mutation of the viral DNA polymerase also produces resistance. Fortunately, resistant strains appear to be less virulent.
Ara-A is less soluble, less potent, and more toxic than ACV. Trifluridine, penciclovir, and ACV have replaced iododeoxyuridine as topical agents for the treatment of herpetic keratitis. Tromantadine, an amantadine derivative, is approved for topical use in countries other than the United States. It works by inhibiting penetration and syncytia formation. Various nonprescription treatments may be effective for specific individuals.
Avoidance of direct contact with lesions reduces the risk of infection. Unfortunately, the symptoms may be inapparent, and thus the virus can be transmitted unknowingly. Physicians, nurses, dentists, and technicians must be especially careful when handling potentially infected tissue or fluids. The wearing of gloves can prevent the acquisition of infections of the fingers (herpetic whitlow). People with recurrent herpetic whitlow disease are very contagious and can spread the infection to patients.
HSV is readily inactivated by soap, disinfectants, bleach, and 70% ethanol. Washing with soap readily disinfects the virus.
Patients who have a history of genital HSV infection must be instructed to refrain from sexual intercourse while they have prodromal symptoms or lesions and to resume sexual intercourse only after lesions are completely reepithelialized because virus may be transmitted from lesions that have crusted over. Condoms may be useful and are undoubtedly better than nothing but may not be fully protective.
A pregnant woman who has active genital HSV infection or who is asymptomatically shedding the virus in the vagina at term may transmit HSV to the neonate if the infant is delivered vaginally. Such transmission can be prevented by cesarean section.
No vaccine is currently available for HSV. However, killed, subunit, vaccinia hybrid, and DNA vaccines are being developed to prevent acquisition of the virus or to treat infected people. The glycoprotein D is being used in several of these experimental vaccines.
Varicella-Zoster Virus
VZV causes chickenpox (varicella) and, upon recurrence, causes herpes zoster, or shingles. As an alphaherpesvirus, VZV shares many characteristics with HSV, including (1) the ability to establish latent infection of neurons and recurrent disease, (2) the importance of cell-mediated immunity in controlling and preventing serious disease, and (3) the characteristic blister-like lesions. Like HSV, VZV encodes a thymidine kinase and is susceptible to antiviral drugs. Unlike HSV, VZV spreads predominantly by the respiratory route and, after local replication of the virus in the respiratory tract, by viremia to form skin lesions over the entire body.
Structure and Replication
VZV has the smallest genome of the human herpesviruses. VZV replicates in a similar manner but slower and in fewer types of cells than HSV. Human diploid fibroblasts in vitro and activated T cells, epithelial cells, and epidermal cells in vivo support productive VZV replication. Like HSV, VZV establishes a latent infection of neurons, but unlike HSV, several viral RNAs and specific viral proteins can be detected in the cells.
Pathogenesis and Immunity
VZV is generally acquired by inhalation, and primary infection begins in the tonsils and mucosa of the respiratory tract. The virus then progresses via the bloodstream and lymphatic system to the cells of the reticuloendothelial system (Box 51-6; Figures 51-8 and 51-9). A secondary viremia occurs after 11 to 13 days and spreads the virus throughout the body and to the skin. The virus infects T cells, and these cells can home to the skin and transfer virus to skin epithelial cells. The virus overcomes inhibition by interferon-α, and vesicles are produced in the skin. The virus remains cell associated and is transmitted on cell-to-cell interaction, except for terminally differentiated epithelial cells in the lungs and keratinocytes of skin lesions, which can release infectious virus. Virus replication in the lung is a major source of contagion. The virus causes a dermal vesiculopustular rash that develops over time in successive crops. Fever and systemic symptoms occur with the rash.
Box 51-6
Disease Mechanisms of Varicella-Zoster Virus (VZV)
Initial replication is in the respiratory tract.
VZV infects epithelial cells, fibroblasts, T cells, and neurons.
VZV can form syncytia and spread directly from cell to cell.
Virus is spread by viremia to skin and causes lesions in successive crops.
VZV can evade antibody clearance, and cell-mediated immune response is essential to control infection. Disseminated, life-threatening disease can occur in immunocompromised people.
Virus establishes latent infection of neurons, usually dorsal root and cranial nerve ganglia.
Herpes zoster is a recurrent disease; it results from virus replication along the entire dermatome.
Herpes zoster may result from depression of cell-mediated immunity and other mechanisms of viral activation.
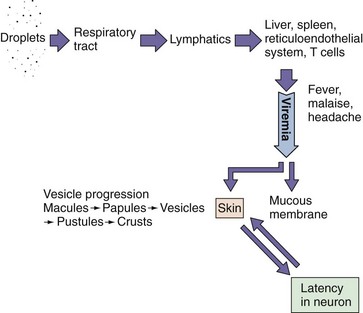
Figure 51-8 Mechanism of spread of varicella-zoster virus (VZV) within the body. VZV initially infects the respiratory tract and is spread to the reticuloendothelial system and T cells and then by cell-associated viremia to the skin.
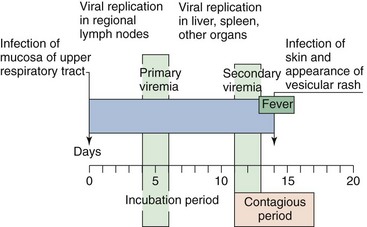
Figure 51-9 Time course of varicella (chickenpox). The course in young children, as presented in this figure, is generally shorter and less severe than that in adults.
The virus becomes latent in the dorsal root or cranial nerve ganglia after the primary infection. The virus can be reactivated in older adults when immunity wanes or in patients with impaired cellular immunity. On reactivation, the virus replicates and is released along the entire neural pathway to infect the skin, causing a vesicular rash along the entire dermatome, known as herpes zoster, or shingles. This damages the neuron and may result in postherpetic neuralgia.
Interferon-α, interferon-stimulated protections, and natural killer and T cells limit the spread of the virus in tissue, but antibody is important for limiting the viremic spread of VZV. Passive immunization with varicella-zoster immune globulin (VZIG) within 4 days of exposure is protective. Cell-mediated immunity is essential for resolving the disease. The virus causes more disseminated and more serious disease in the absence of cell-mediated immunity (e.g., in children with leukemia) and may recur on immunosuppression. Although important for protection, cell-mediated immune responses contribute to the symptomatology. An overzealous response in adults is responsible for causing more extensive cell damage and a more severe manifestation (especially in the lung) in primary infection than that seen in children. T-cell and antibody levels decrease later in life, allowing VZV recurrence and herpes zoster disease.
Epidemiology
VZV is extremely communicable, with rates of infection exceeding 90% among susceptible household contacts (Box 51-7). The disease is spread principally by the respiratory route but may also be spread through contact with skin vesicles. Patients are contagious before and during symptoms. More than 90% of adults in developed countries have the VZV antibody. Herpes zoster results from the reactivation of a patient’s latent virus. The disease develops in approximately 10% to 20% of the population infected with VZV, and the incidence rises with age. Herpes zoster lesions contain viable virus and therefore may be a source of varicella infection in a nonimmune person (child).
Box 51-7
Epidemiology of Varicella-Zoster Virus
Who Is at Risk?
Children (ages 5 to 9) experience mild classic disease.
Teenagers and adults are at risk for more severe disease with potential pneumonia.
Immunocompromised people and newborns are at risk for life-threatening pneumonia, encephalitis, and progressive disseminated varicella.
Elderly and immunocompromised people are at risk for recurrent disease (herpes zoster [shingles]).
Modes of Control
Antiviral drugs are available.
Immunity may wane in the elderly population.
Varicella-zoster immunoglobulin is available for immunocompromised people and staff exposed to virus, as well as newborns of mothers showing symptoms within 5 days of birth.
Live vaccine (Oka strain) is available for children (varicella) and adults (zoster).
Clinical Syndromes
Varicella (chickenpox) is one of the five classic childhood exanthems (along with rubella, roseola, fifth disease, and measles). The disease results from a primary infection with VZV; it is usually a mild disease of childhood and is normally symptomatic, although asymptomatic infection can occur (see Figure 51-9). Varicella characteristics include fever and a maculopapular rash that appear after an incubation period of approximately 14 days (Figure 51-10). Within hours, each maculopapular lesion forms a thin-walled vesicle on an erythematous base (“dewdrop on a rose petal”) that measures approximately 2 to 4 mm in diameter. This vesicle is the hallmark of varicella. Within 12 hours, the vesicle becomes pustular and begins to crust, after which scabbed lesions appear. Successive crops of lesions appear for 3 to 5 days, and at any given time, all stages of skin lesions can be observed.
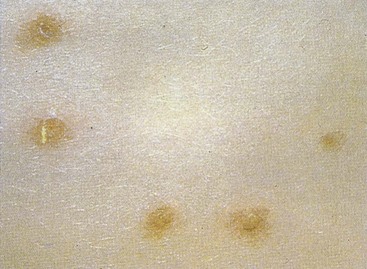
Figure 51-10 Characteristic rash of varicella in all stages of its evolution.
(From Hart CA, Broadhead RL: A color atlas of pediatric infectious diseases, London, 1992, Wolfe.)
The rash spreads across the entire body but is more prevalent on the trunk and head than on the extremities. Its presence on the scalp distinguishes it from many other rashes. The lesions itch and cause scratching, which may lead to bacterial superinfection and scarring. Lesions on the mucous membrane typically occur in the mouth, conjunctivae, and vagina.
Primary infection is usually more severe in adults than in children. Interstitial pneumonia may occur in 20% to 30% of adult patients and may be fatal. The pneumonia results from inflammatory reactions at this primary site of infection.
As noted earlier, herpes zoster (zoster means “belt” or “girdle”) is a recurrence of a latent varicella infection acquired earlier in the patient’s life. Severe pain in the area innervated by the nerve usually precedes the appearance of the chickenpox-like lesions. The rash is limited to a dermatome and resembles varicella (Figure 51-11). A chronic pain syndrome called postherpetic neuralgia, which can persist for months to years, occurs in as many as 30% of patients in whom herpes zoster develops.
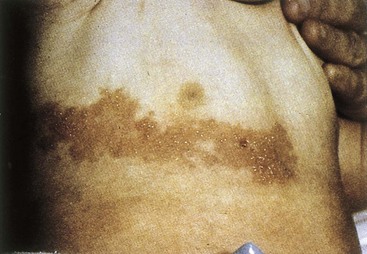
VZV infection in immunocompromised patients or neonates can result in serious, progressive, and potentially fatal disease. Defects of cell-mediated immunity in such patients increase the risk for dissemination of the virus to the lungs, brain, and liver, which may be fatal. The disease may occur in response to a primary exposure to varicella or because of recurrent disease.
Laboratory Diagnosis
The CPEs in VZV-infected cells are similar to those seen in HSV-infected cells and include Cowdry type A intranuclear inclusions and syncytia. These cells may be seen in skin lesions, respiratory specimens, or organ biopsy specimens. Syncytia may also be seen in Tzanck smears of scrapings from a vesicle’s base. A direct fluorescent antibody to membrane antigen (FAMA) test can also be used to examine skin lesion scrapings or biopsy specimens. Antigen and genome detection are sensitive means of diagnosing VZV infection. PCR techniques are especially useful for systemic and neuronal disease.
Isolation of VZV is not routinely done because the virus is labile during transport to the laboratory and replicates poorly in vitro.
Serologic tests that detect antibodies to VZV are used to screen populations for immunity to VZV. However, antibody levels are normally low, so sensitive tests such as immunofluorescence and enzyme-linked immunosorbent assay (ELISA) must be performed to detect the antibody. A significant increase in antibody level can be detected in people experiencing herpes zoster.
Treatment, Prevention, and Control
Treatment may be appropriate for adults and immunocompromised patients with VZV infections and for people with shingles, but no treatment is usually necessary for children with varicella. ACV, famciclovir, and valacyclovir have been approved for the treatment of VZV infections. The VZV DNA polymerase is much less sensitive to ACV treatment than the HSV enzyme, requiring large doses of ACV or the improved pharmacodynamics of famciclovir and valacyclovir (see Box 51-5). There is no good treatment, but analgesics and other painkillers, topical anesthetics, or capsaicin cream may provide some relief from the postherpetic neuralgia that follows zoster.
As with other respiratory viruses, it is difficult to limit the transmission of VZV. Because VZV infection in children is generally mild and induces lifelong immunity, exposure of children to VZV early in life is often encouraged. However, high-risk people (e.g., immunosuppressed children) should be protected from exposure to VZV.
Immunosuppressed patients susceptible to severe disease may be protected from serious disease through the administration of varicella-zoster immunoglobulin (VZIG). VZIG is prepared through the pooling of plasma from seropositive people. VZIG prophylaxis can prevent viremic spread leading to disease but is ineffective as a therapy for patients already suffering from active varicella or herpes zoster disease.
A live attenuated vaccine for VZV (Oka strain) has been licensed for use in the United States and elsewhere and is administered after 2 years of age on the same schedule as the measles, mumps, and rubella vaccine. The vaccine induces the production of protective antibody and cell-mediated immunity. A stronger version of this vaccine is available for adults older than 60 years; it boosts antiviral responses to limit the onset of zoster.
Epstein-Barr Virus
EBV is the ultimate B-lymphocyte parasite, and the diseases it causes reflect this association. EBV was discovered by electron-microscopic observation of characteristic herpes virions in biopsy specimens of a B-cell neoplasm, African Burkitt lymphoma (AfBL). Its association with infectious mononucleosis was discovered accidentally when serum collected from a laboratory technician convalescing from infectious mononucleosis was found to contain the antibody that recognized AfBL cells. This finding was later confirmed in a large serologic study performed on college students.
EBV causes heterophile antibody-positive infectious mononucleosis and stimulates the growth and immortalizes B cells in tissue culture. EBV has been causally associated with AfBL (endemic Burkitt lymphoma), Hodgkin disease, and nasopharyngeal carcinoma. EBV has also been associated with B-cell lymphomas in patients with acquired or congenital immunodeficiencies.
Structure and Replication
EBV is a member of the subfamily Gammaherpesvirinae, with a very limited host range and a tissue tropism defined by the limited cellular expression of its receptor. The primary receptor for EBV is also the receptor for the C3d component of the complement system (also called CR2 or CD21). It is expressed on B cells of humans and New World monkeys and on some epithelial cells of the oropharynx and nasopharynx.
EBV infection has the following three potential outcomes:
1. EBV can replicate in B cells or epithelial cells permissive for EBV replication.
2. EBV can cause latent infection of memory B cells in the presence of competent T cells.
EBV encodes more than 70 proteins, different groups of which are expressed for the different types of infections.
EBV in saliva infects epithelial cells and then naïve resting B cells in the tonsils. The growth of the B cells is stimulated first by virus binding to the C3d receptor, a B-cell growth-stimulating receptor, and then by expression of the transformation and latency proteins. These include Epstein-Barr nuclear antigens (EBNAs) 1, 2, 3A, 3B, and 3C; latent proteins (LPs); latent membrane proteins (LMPs) 1 and 2; and two small Epstein-Barr–encoded RNA (EBER) molecules, EBER-1 and EBER-2. The EBNAs and LPs are DNA-binding proteins that are essential for establishing and maintaining the infection (EBNA-1), immortalization (EBNA-2), and other purposes. The LMPs are membrane proteins with oncogene-like activity. The genome becomes circularized; the cells proceed to follicles that become germinal centers in the lymph node, where the infected cells differentiate into memory cells. EBV protein synthesis ceases, and the virus establishes latency in these memory B cells. EBNA-1 will be expressed only at cell division to hold onto and retain the genome in the cells.
Antigen stimulation of the B cells and infection of certain epithelial cells allow the transcription and translation of the ZEBRA (peptide encoded by the Z-gene region) transcriptional activator protein, which activates the immediate early genes of the virus and the lytic cycle. After synthesis of the DNA polymerase and replication of DNA, the structural and other late proteins are synthesized. They include gp350/220 (related glycoproteins of 350,000 and 220,000 Da), which is the viral attachment protein, and other glycoproteins. These glycoproteins bind to CD21 and MHC II molecules, receptors on B cells and epithelial cells, and also promote fusion of the envelope with cell membranes.
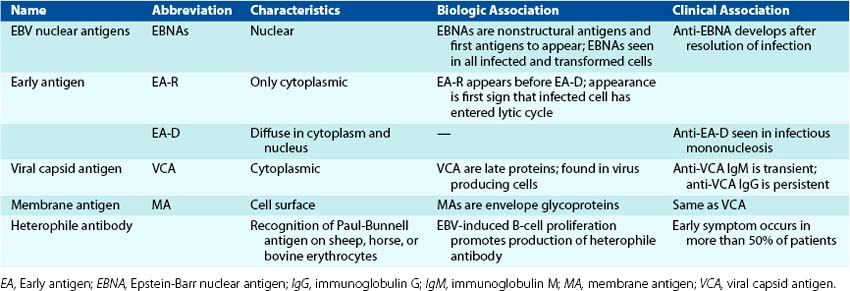
The viral proteins produced during a productive infection are serologically defined and grouped as early antigen (EA), viral capsid antigen (VCA), and the glycoproteins of the membrane antigen (MA) (Table 51-3). An early protein mimics a cellular inhibitor of apoptosis, and a late protein mimics the activity of human interleukin-10, which enhances B-cell growth and inhibits TH1 immune responses.
Pathogenesis and Immunity
EBV has adapted to the human B cell and manipulates and uses the different phases of B-cell development to establish a lifelong infection in the individual, and it still promotes its transmission. The diseases of EBV result from either an overactive immune response (infectious mononucleosis) or the lack of effective immune control (lymphoproliferative disease and hairy cell leukoplakia).
The productive infection of B cells and epithelial cells of the oropharynx, such as in the tonsils (Box 51-8 and Figure 51-12), promotes virus shedding into saliva to transmit the virus to other hosts and establishes a viremia to spread the virus to other B cells in lymphatic tissue and blood.
Box 51-8
Disease Mechanisms of Epstein-Barr Virus (EBV)
Virus in saliva initiates infection of oral epithelia and tonsillar B cells.
There is productive infection of epithelial and B cells.
Virus promotes growth of B cells (immortalizes).
T cells kill and limit B-cell outgrowth. T cells are required for controlling infection. Antibody role is limited.
EBV establishes latency in memory B cells and is reactivated when the B cell is activated.
T-cell response (lymphocytosis) contributes to symptoms of infectious mononucleosis.
There is causative association with lymphoma in immunosuppressed people and African children living in malarial regions (African Burkitt lymphoma) and with nasopharyngeal carcinoma in China.
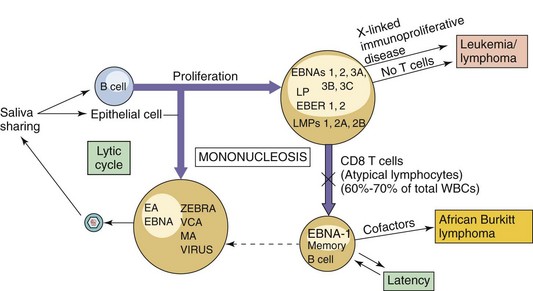
Figure 51-12 Progression of Epstein-Barr virus (EBV) infection. Infection may result in lytic, latent, or immortalizing infection, which can be distinguished on the basis of production of virus and expression of different viral proteins and antigens. T cells limit the outgrowth of the EBV-infected cells and maintain the latent infection. CD, Cluster of differentiation; EA, early antigen; EBER, Epstein-Barr–encoded RNA; EBNA, Epstein-Barr nuclear antigen; LMPs, latent membrane proteins; LP, latent protein; MA, membrane antigen; VCA, viral capsid antigen; WBCs, white blood cells; ZEBRA, peptide encoded by the Z gene region.
EBV proteins replace host factors that normally activate B-cell growth and development. In the absence of T cells (e.g., in tissue culture), EBV can immortalize B cells and promote the development of B-lymphoblastoid cell lines. In vivo, B-cell activation and proliferation occurs and is indicated by the spurious production of an IgM antibody to the Paul-Bunnell antigen, termed the heterophile antibody (see later discussion of serology).
The outgrowth of the B cell is normally controlled by T cells responding to proliferation indicators on the B cell and to EBV antigenic peptides. B cells are excellent antigen-presenting cells and present EBV antigens on both MHC I and MHC II molecules. The activated T cells appear as atypical lymphocytes (also called Downey cells) (Figure 51-13). They increase in number in the peripheral blood during the second week of infection, accounting for 10% to 80% of the total white blood cell count at this time (hence the “mononucleosis”).
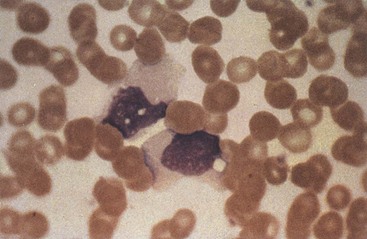
Figure 51-13 Atypical T-cell (Downey cell) characteristic of infectious mononucleosis. The cells have a more basophilic and vacuolated cytoplasm than normal lymphocytes, and the nucleus may be oval, kidney shaped, or lobulated. The cell margin may seem to be indented by neighboring red blood cells.
Infectious mononucleosis is essentially a “civil war” between the EBV-infected B cells and the protective T cells. The classic lymphocytosis (increase in mononuclear cells), swelling of lymphoid organs (lymph nodes, spleen, and liver), and malaise associated with infectious mononucleosis results mainly from the activation and proliferation of T cells. A great amount of energy is required to power the T-cell response, leading to great fatigue. The sore throat of infectious mononucleosis is a response to EBV-infected epithelium and B cells in the tonsils and throat. Children have a less active immune response to EBV infection and therefore have very mild disease.
During productive infection, antibody is first developed against the components of the virion, VCA, and MA, and later against the EA. After resolution of the infection (lysis of the productively infected cells), antibody against the nuclear antigens (EBNAs) is produced. T cells are essential for limiting the proliferation of EBV-infected B cells and controlling the disease (Figure 51-14). EBV counteracts some of the protective action of TH1 CD4 T-cell responses during productive infection by producing an interleukin-10 analogue (BCRF-1) that inhibits the protective TH1 CD4 T-cell responses and also stimulates B-cell growth.
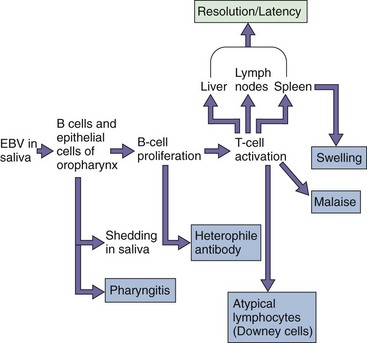
Figure 51-14 Pathogenesis of Epstein-Barr virus (EBV). EBV is acquired by close contact between persons through saliva and infects the B cells. The resolution of the EBV infection and many of the symptoms of infectious mononucleosis result from the activation of T cells in response to the infection.
The virus persists in at least one memory B cell per milliliter of blood for the infected person’s lifetime. EBV may be reactivated when the memory B cell is activated (especially in the tonsils or oropharynx) and may be shed in saliva.
Epidemiology
EBV is transmitted in saliva (Box 51-9). More than 90% of EBV-infected people intermittently shed the virus for life, even when totally asymptomatic. Children can acquire the virus at an early age by sharing contaminated drinking glasses. Children generally have subclinical disease. Saliva sharing between adolescents and young adults often occurs during kissing; thus EBV mononucleosis has earned the nickname “the kissing disease.” Disease in these people may go unnoticed or may manifest in varying degrees of severity. At least 70% of the population of the United States is infected by age 30.
Box 51-9
Epidemiology of Epstein-Barr Virus
The geographic distribution of some EBV-associated neoplasms indicates a possible association with cofactors. Malaria appears to be a cofactor in the progression of chronic or latent EBV infection to AfBL. The restriction of nasopharyngeal carcinoma to people living in certain regions of China indicates a possible genetic predisposition to the cancer or the presence of cofactors in the food or environment. More subtle mechanisms may facilitate the role of EBV in 30% to 50% of cases of Hodgkin disease.
Transplant recipients, patients with the acquired immunodeficiency syndrome (AIDS), and genetically immunodeficient people are at high risk for lymphoproliferative disorders initiated by EBV. These disorders may appear as polyclonal and monoclonal B-cell lymphomas. Such people are also at high risk for a productive EBV infection in the form of hairy oral leukoplakia.
Clinical Syndromes (Clinical Case 51-2)
Heterophile Antibody–Positive Infectious Mononucleosis
The triad of classic symptoms for infectious mononucleosis is lymphadenopathy (swollen glands), splenomegaly (large spleen), and exudative pharyngitis accompanied by high fever, malaise, and often hepatosplenomegaly (large liver and spleen). A rash may occur, especially after ampicillin treatment (for the sore throat). The major complaint of people with infectious mononucleosis is fatigue (Figure 51-15). The disease is rarely fatal in healthy people but can cause serious complications resulting from neurologic disorders, laryngeal obstruction, or rupture of the spleen. Neurologic complications include meningoencephalitis and the Guillain-Barré syndrome. Mononucleosis-like syndromes can also be caused by CMV, HHV-6, Toxoplasma gondii, and human immunodeficiency virus (HIV). Similar to infections caused by other herpesviruses, EBV infection in a child is much milder than infection in an adolescent or adult. In fact, infection in children is usually subclinical.
Clinical Case 51-2
Epstein-Barr Virus (EBV) in the Immunocompromised Individual
Purtilo and associates (Ann Intern Med 101:180–186, 1984) reported on a boy with Duncan disease who presented with reduced levels of IgA, a history of thrush, and recurrent episodes of otitis media. This member of the Duncan family had an X-linked recessive, progressive, combined, variable immunodeficiency disease caused by a mutation in the SH2D1A protein, which prevents proper communication between B and T cells. After exposure to EBV at age 11 years, the boy did not develop antibodies to EBV, but generic serum IgM levels increased, and EBNA-positive immortalized B-cell lines readily grew from his peripheral blood. Establishment of the B-cell lines is indicative of aberrant T-cell control of the virus-induced B-cell proliferation. At age 18 years, he was treated with packed red cells for red cell aplasia, and then 9 weeks later, he developed infectious mononucleosis with fever, generalized lymphadenomegaly, tender liver and swollen spleen, lymphocytosis with a predominance of atypical lymphocytes, and a positive monospot test. Within another 6 months, he was agammaglobulinemic with no detectable B cells and suffered from Haemophilus influenzae and Mycobacterium tuberculosis pneumonias. After an additional 5 months, B cells were again detected. The onset of infectious mononucleosis at age 18 years may have resulted from new infection or a reactivation of the earlier infection. This case illustrates the unusual nature of EBV and other virus infections when the immune response is compromised.
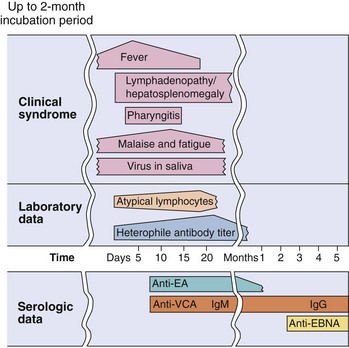
Figure 51-15 Clinical course of infectious mononucleosis and laboratory findings of those with the infection. Epstein-Barr virus infection may be asymptomatic or may produce the symptoms of mononucleosis. The incubation period can last as long as 2 months. EA, Early antigen; EBNA, Epstein-Barr nuclear antigen; VCA, viral capsid antigen.
Chronic Disease
EBV can cause cyclic recurrent disease in some people. These patients experience chronic tiredness and may also have low-grade fever, headaches, and a sore throat. This disorder is different from chronic fatigue syndrome, which has an unknown etiology.
Epstein-Barr Virus–Induced Lymphoproliferative Diseases
On infection with EBV, people lacking T-cell immunity are likely to suffer life-threatening polyclonal leukemia-like B-cell proliferative disease and lymphoma instead of infectious mononucleosis. Men with congenital deficiencies of T-cell function are likely to suffer life-threatening X-linked lymphoproliferative disease. One such X-linked genetic defect in a T-cell gene (SLAM [signaling lymphocyte activation molecule]–associated protein) prevents the T cell from controlling B-cell growth during a normal immune response to antigen or because of EBV. Transplant recipients undergoing immunosuppressive treatment are at high risk for posttransplant lymphoproliferative disease, instead of infectious mononucleosis, after exposure to the virus or on reactivation of latent virus. Similar diseases are seen in patients with AIDS.
EBV was first associated with African Burkitt lymphoma (endemic lymphoma) (AfBL) and then Burkitt lymphoma elsewhere in the world, Hodgkin lymphoma, and several other lymphoproliferative diseases. AfBL is a poorly differentiated monoclonal B-cell lymphoma of the jaw and face that is endemic in children living in the malarial regions of Africa. EBV infection facilitates the survival of cells that undergo a chromosomal translocation that juxtaposes the c-MYC oncogene to a very active promoter, such as an immunoglobulin gene promoter [t(8;14), t(8;22), t(8;2)], to allow tumor growth. The Burkitt tumors contain EBV DNA sequences but express only the EBNA-1 viral antigen. Virions can occasionally be seen on electron micrographs of infected material. The tumor cells are also relatively invisible to immune control. Malaria may enhance the development of AfBL by promoting the proliferation of EBV-bearing memory B cells.
EBV is also associated with nasopharyngeal carcinoma, which is endemic in adults in Asia. The tumor cells contain EBV DNA, but unlike Burkitt lymphoma, in which the tumor cells are derived from lymphocytes, the tumor cells of nasopharyngeal carcinoma are of epithelial origin.
Laboratory Diagnosis
EBV-induced infectious mononucleosis is diagnosed on the basis of the symptoms (Box 51-10), the finding of atypical lymphocytes, the presence of lymphocytosis (mononuclear cells constituting 60% to 70% of the white blood cell count, with 30% atypical lymphocytes), heterophile antibody, and antibody to viral antigens. Virus isolation is not practical. PCR and DNA probe analysis for the viral genome and immunofluorescent identification of viral antigens are used to detect evidence of infection.
Atypical lymphocytes are probably the earliest detectable indication of an EBV infection. These cells appear with the onset of symptoms and disappear with resolution of the disease.
Heterophile antibody results from the nonspecific, mitogen-like activation of B cells by EBV and the production of a wide repertoire of antibodies. These antibodies include an IgM heterophile antibody that recognizes the Paul-Bunnell antigen on sheep, horse, and bovine erythrocytes but not that on guinea pig kidney cells. The heterophile antibody response can usually be detected by the end of the first week of illness and lasts for as long as several months. It is an excellent indication of EBV infection in adults but is not as reliable in children or infants. The horse cell (Monospot) test and ELISA are rapid and widely used for the detection of the heterophile antibody.
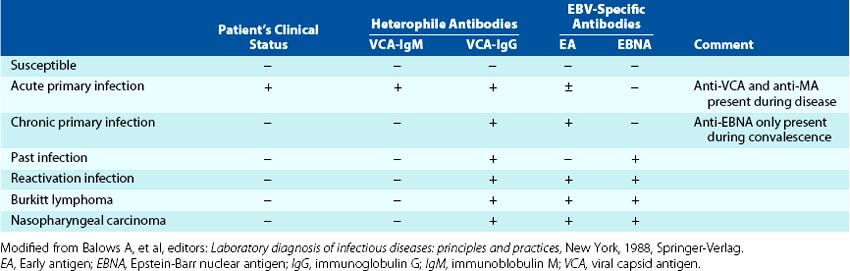
Serologic tests for antibody to viral antigens are a more dependable method than heterophile antibody to confirm the diagnosis of EBV mononucleosis (Table 51-4; see Figure 51-15). EBV infection is indicated by the finding of any of the following: (1) IgM antibody to the VCA, (2) the presence of VCA antibody and the absence of EBNA antibody, or (3) elevation of antibodies to VCA and early antigen. The finding of both VCA and EBNA antibodies in serum indicates that the person had a previous infection. Generation of antibody to EBNA requires lysis of the infected cell and usually indicates T-cell control of active disease.
Treatment, Prevention, and Control
No effective treatment or vaccine is available for EBV disease (see Box 51-5). The ubiquitous nature of the virus and the potential for asymptomatic shedding make control of infection difficult. However, infection elicits lifelong immunity. Therefore, the best means of preventing infectious mononucleosis is exposure to the virus early in life because the disease is more benign in children.
Cytomegalovirus
CMV is a common human pathogen, infecting 0.5% to 2.5% of all newborns and approximately 40% of women visiting clinics for sexually transmitted diseases. It is the most common viral cause of congenital defects. Although usually causing mild or asymptomatic disease in children and adults, CMV is particularly important as an opportunistic pathogen in immunocompromised patients.
Structure and Replication
CMV is a member of the subfamily Betaherpesvirinae. It has the largest genome of the human herpesviruses. In contrast to the traditional definition of a virus, which states that a virion particle contains DNA or RNA, CMV carries specific mRNAs into the cell in the virion particle to facilitate infection. Human CMV replicates only in human cells. Fibroblasts, epithelial cells, granulocytes, macrophages, and other cells are permissive for CMV replication. Virus replication is much slower than for HSV, and CPE may not be seen for 7 to 14 days. This may facilitate the establishment of latent infection in myeloid stem cells, monocytes, lymphocytes, the stromal cells of the bone marrow, or other cells.
Pathogenesis and Immunity
The pathogenesis of CMV is similar to that of other herpesviruses in many respects (Box 51-11). CMV is an excellent parasite and readily establishes persistent and latent infections rather than an extensive lytic infection. CMV is highly cell associated and is spread throughout the body within infected cells, especially lymphocytes and leukocytes. The virus is reactivated by immunosuppression (e.g., corticosteroids, infection with HIV) and possibly by allogeneic stimulation (i.e., the host response to transfused or transplanted cells).
Box 51-11
Disease Mechanisms of Cytomegalovirus (CMV)
CMV is acquired from blood, tissue, and most body secretions.
CMV causes productive infection of epithelial and other cells.
CMV establishes latency in T cells, macrophages, and other cells.
Cell-mediated immunity is required for resolution and maintenance of latency and contributes to symptoms.
The role of antibody is limited.
Suppression of cell-mediated immunity allows recurrence and severe disease.
Cell-mediated immunity is essential for resolving and controlling the outgrowth of CMV infection. However, CMV is an expert at immune evasion and has several means for evading innate and immune responses. CMV infection alters the function of lymphocytes and leukocytes. The virus prevents antigen presentation to both CD8 cytotoxic T cells and CD4 T cells by preventing the expression of MHC I molecules on the cell surface and by interfering with cytokine-induced expression of MHC II molecules on antigen-presenting cells (including the infected cells). A viral protein also blocks natural-killer-cell attack of CMV-infected cells. Similar to EBV, CMV also encodes an interleukin-10 analogue that would inhibit TH1 protective immune responses.
Epidemiology and Clinical Syndromes
In most cases, CMV replicates and is shed without causing symptoms (Table 51-5). Activation and replication of CMV in the kidney and secretory glands promote its secretion in urine and bodily secretions. CMV can be isolated from urine, blood, throat washings, saliva, tears, breast milk, semen, stool, amniotic fluid, vaginal and cervical secretions, and tissues obtained for transplantation (Table 51-6 and Box 51-12). Virus can be transmitted to other individuals by means of blood transfusions and organ transplants. The congenital, oral, and sexual routes; blood transfusion; and tissue transplantation are the major means by which CMV is transmitted. CMV disease is an opportunistic disorder, rarely causing symptoms in the immunocompetent host but causing serious disease in an immunosuppressed or immunodeficient person, such as a patient with AIDS or a neonate (Figure 51-16).
Table 51-5 Sources of Cytomegalovirus Infection
| Age Group | Source |
|---|---|
| Neonate | Transplacental transmission, intrauterine infections, cervical secretions |
| Baby or child | Body secretions: breast milk, saliva, tears, urine |
| Adult | Sexual transmission (semen), blood transfusion, organ graft |
Table 51-6 Cytomegalovirus Syndromes
| Tissue | Children/Adults | Immunosuppressed Patients |
|---|---|---|
| Predominant presentation | Asymptomatic | Disseminated disease, severe disease |
| Eyes | — | Chorioretinitis |
| Lungs | — | Pneumonia, pneumonitis |
| Gastrointestinal tract | — | Esophagitis, colitis |
| Nervous system | Polyneuritis, myelitis | Meningitis and encephalitis, myelitis |
| Lymphoid system | Mononucleosis syndrome, posttransfusion syndrome | Leukopenia, lymphocytosis |
| Major organs | Carditis,* hepatitis* | Hepatitis |
| Neonates | Deafness, intracerebral calcification, microcephaly, mental retardation | — |
* Complication of mononucleosis or posttransfusion syndrome.
Box 51-12
Epidemiology of Cytomegalovirus Infection
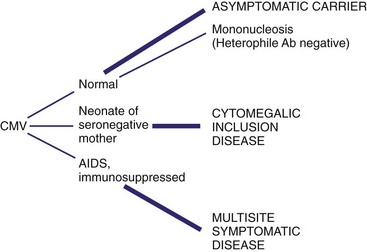
Figure 51-16 Outcomes of cytomegalovirus (CMV) infections. The outcome of CMV infection depends very heavily on the immune status of the patient. Ab, Antibody; AIDS, acquired immunodeficiency syndrome.
Congenital Infection
CMV is the most prevalent viral cause of congenital disease. Approximately 15% of stillborn babies are infected with CMV. A significant percentage (0.5% to 2.5%) of all newborns in the United States is infected with CMV before birth, and a large percentage of babies is infected within the first months of life. Disease signs include small size, thrombocytopenia, microcephaly, intracerebral calcification, jaundice, hepatosplenomegaly, and rash (cytomegalic inclusion disease). Unilateral or bilateral hearing loss and mental retardation are common consequences of congenital CMV infection. The risk for serious birth defects is extremely high for infants born to mothers who had primary CMV infections during their pregnancies.
Fetuses are infected by virus in the mother’s blood (primary infection) or by virus ascending from the cervix (after a recurrence). The symptoms of congenital infection are less severe or can be prevented by the immune response of a seropositive mother. Congenital CMV infection is best documented by isolation of the virus from the infant’s urine during the first week of life.
Perinatal Infection
In the United States, at least 20% of pregnant women harbor CMV in the cervix at term and are likely to experience reactivation of the virus during pregnancy. Approximately half the neonates born through an infected cervix acquire CMV infection and become excreters of the virus at 3 to 4 weeks of age. Neonates may also acquire CMV from maternal milk or colostrum. Perinatal infection causes no clinically evident disease in healthy full-term infants.
Another means by which neonates can acquire CMV is through blood transfusions. Of the seronegative babies who are exposed to blood from seropositive donors, 13.5% acquire CMV infection in the immediate postnatal period. Significant clinical infection may occur in premature infants who acquire CMV from transfused blood, usually resulting in pneumonia and hepatitis.
Infection in Children and Adults
Approximately 40% of adolescents are infected with CMV, but this number increases to 70% to 85% of adults in the United States by the age of 40. CMV is more prevalent among people in low socioeconomic brackets living in crowded conditions and in people living in developing countries. CMV is a sexually transmitted disease, and 90% to 100% of patients attending sexually transmitted disease clinics are infected. The titer of the CMV in semen is the highest of that in any body secretion.
Although most CMV infections acquired in young adulthood are asymptomatic, patients may show a heterophile-negative mononucleosis syndrome. The symptoms of CMV disease are similar to those of EBV infection but with less severe pharyngitis and lymphadenopathy (see Figure 51-16). Although the presence of CMV-infected cells promotes a T-cell outgrowth (atypical lymphocytosis) similar to that seen in EBV infection, heterophile antibody is not present. The absence of this antibody reflects the differences in the target cell and the action of the viruses on the target cell. CMV disease should be suspected in a patient who has heterophile-negative mononucleosis or in whom there are signs of hepatitis, but results of tests for hepatitis A, B, and C are negative.
Transmission via Transfusion and Transplantation
Transmission of CMV by blood most often results in an asymptomatic infection; if symptoms are present, they typically resemble those of mononucleosis. Fever, splenomegaly, and atypical lymphocytosis usually begin 3 to 5 weeks after transfusion. Pneumonia and mild hepatitis may also occur. CMV may also be transmitted by organ transplantation (e.g., kidneys, bone marrow), and CMV infection is often reactivated in transplant recipients during periods of intense immunosuppression.
Infection in the Immunocompromised Host
CMV is a prominent opportunistic infectious agent. In immunocompromised people, it causes symptomatic primary or recurrent disease (see Table 51-6).
CMV disease of the lung (pneumonia and pneumonitis) is a common outcome in immunosuppressed patients and can be fatal if not treated. CMV often causes retinitis, colitis, or esophagitis in patients who are severely immunodeficient (e.g., patients with AIDS). Interstitial pneumonia and encephalitis may also be caused by CMV but may be difficult to distinguish from infections caused by other opportunistic agents. CMV esophagitis may mimic candidal esophagitis. A smaller percentage of immunocompromised patients may experience CMV infection of the gastrointestinal tract. Patients with CMV colitis usually have diarrhea, weight loss, anorexia, and fever. Effective anti-HIV therapy has reduced the incidence of these diseases.
CMV is also responsible for the failure of many kidney transplants. This may be the result of virus replication in the graft after reactivation in the transplanted kidney or infection from the host.
Laboratory Diagnosis
Histology
The histologic hallmark of CMV infection is the cytomegalic cell, which is an enlarged cell (25 to 35 mm in diameter) that contains a dense, central, “owl’s eye,” basophilic intranuclear inclusion body (Table 51-7; Figure 51-17). Such infected cells may be found in any tissue of the body and in urine and are thought to be epithelial in origin. The inclusions are readily seen with Papanicolaou or hematoxylin-eosin staining.
Table 51-7 Laboratory Tests for Diagnosing Cytomegalovirus Infection
| Test | Finding |
|---|---|
| Cytology and histology* | “Owl’s-eye” inclusion body Antigen detection |
| In situ DNA probe hybridization | |
| PCR | |
| Cell culture | Cytologic effect in human diploid fibroblasts (slow) |
| Immunofluorescence detection of early antigens (faster) | |
| PCR (faster) | |
| Serology | Primary infection |
PCR, Polymerase chain reaction.
* Samples taken for analysis include urine, saliva, blood, bronchoalveolar lavage specimens, and tissue biopsy specimens.
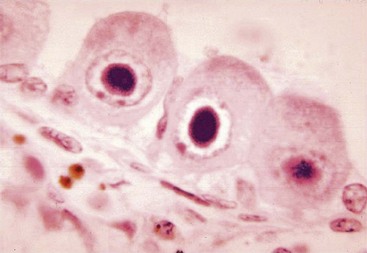
Antigen and Genome Detection
A rapid, sensitive diagnosis can be obtained by detection of viral antigen, using immunofluorescence or an ELISA, or the viral genome, using PCR and related techniques in cells of a biopsy, blood, bronchoalveolar lavage, or urine sample (see Chapter 5, Figure 5-3).
Culture
CMV is grown in diploid fibroblast cell cultures and normally must be maintained for at least 4 to 6 weeks because the characteristic CPE develops very slowly in specimens with very low titers of the virus. Isolation of CMV is especially reliable in immunocompromised patients, who often have high titers of virus in their secretions. For example, in the semen of patients with AIDS, titers of viable virus may be greater than 106.
More rapid results are achieved by centrifuging a patient’s sample onto cells grown on a coverslip within a shell vial. Specimens are examined after 1 to 2 days of incubation by indirect immunofluorescence for the presence of one or more of the immediate early viral antigens.
Serology
Seroconversion is usually an excellent marker for primary CMV infection. Titers of CMV-specific IgM antibody may be very high in patients with AIDS. However, CMV-specific IgM antibody may also develop during the reactivation of CMV and is therefore not a dependable indicator of primary infection.
Treatment, Prevention, and Control
Ganciclovir (dihydroxypropoxymethyl guanine), valganciclovir (valyl ester of ganciclovir), cidofovir, and foscarnet (phosphonoformic acid) have been approved by the FDA for the treatment of specific diseases resulting from CMV infections of immunosuppressed patients (see Box 51-5). Ganciclovir is structurally similar to ACV; it is phosphorylated and activated by a CMV-encoded protein kinase, inhibits the viral DNA polymerase, and causes DNA termination (see Chapter 48). Ganciclovir is more toxic than ACV. Ganciclovir can be used to treat severe CMV infections in immunocompromised patients. Valganciclovir is a prodrug of ganciclovir that can be taken orally, is converted to ganciclovir in the liver, and has better bioavailability than ganciclovir. Cidofovir is a phosphorylated cytidine nucleoside analogue that does not require a viral enzyme for activation. Foscarnet is a simple molecule that inhibits the viral DNA polymerase by mimicking the pyrophosphate portion of nucleotide triphosphates.
CMV spreads mainly by the sexual, tissue transplantation, and transfusion routes, and spread by these means is preventable. Semen is a major vector for the sexual spread of CMV to both heterosexual and homosexual contacts. The use of condoms or abstinence would limit viral spread. Transmission of the virus can also be reduced through the screening of potential blood and organ donors for CMV seronegativity. Screening is especially important for donors of blood transfusions to be given to infants. Although congenital and perinatal transmission of CMV cannot effectively be prevented, a seropositive mother is least likely to produce a baby with symptomatic CMV disease. No vaccine for CMV is available.
Human Herpesviruses 6 and 7
The two variants of HHV-6, HHV-6A and HHV-6B, and HHV-7, are members of the genus Roseolovirus of the subfamily Betaherpesvirinae. HHV-6 was first isolated from the blood of patients with AIDS and grown in T-cell cultures. It was identified as a herpesvirus because of its characteristic morphology within infected cells. Similar to CMV, HHV-6 is lymphotropic and ubiquitous. At least 45% of the population is seropositive for HHV-6 by age 2 years, and almost 100% by adulthood. In 1988, HHV-6 was serologically associated with a common disease of children, exanthem subitum, commonly known as roseola. HHV-7 was isolated in a similar manner from the T cells of a patient with AIDS who was also infected with HHV-6, and later it was also shown to cause exanthem subitum.
Pathogenesis and Immunity
HHV-6 infection occurs very early in life. The virus replicates in the salivary gland, is shed, and transmitted in saliva.
HHV-6 primarily infects lymphocytes, especially CD4 T cells. HHV-6 establishes a latent infection in T cells and monocytes but may replicate on activation of the cells. Cells in which the virus is replicating appear large and refractile and have occasional intranuclear and intracytoplasmic inclusion bodies.
Similar to the replication of CMV, the replication of HHV-6 is controlled by cell-mediated immunity. Similar to CMV, the virus is likely to become activated in patients with AIDS or other lymphoproliferative and immunosuppressive disorders and cause opportunistic disease.
Clinical Syndromes (Box 51-13)
Exanthem subitum, or roseola, is caused by either HHV-6B or HHV-7 and is one of the five classic childhood exanthems previously mentioned (Figure 51-18). It is characterized by the rapid onset of high fever of a few days’ duration, which is followed by a rash on the trunk and face, and then it spreads and lasts only 24 to 48 hours. The presence of infected T cells or the activation of delayed-type hypersensitivity T cells in the skin may be the cause of the rash. The disease is effectively controlled and resolved by cell-mediated immunity, but the virus establishes a lifelong latent infection of T cells. Although usually benign, HHV-6 is the most common cause of febrile seizures in childhood (age 6 to 24 months).
Box 51-13
Clinical Summaries
Herpes Simplex Virus (HSV)
Primary oral herpes: A 5-year-old boy has an ulcerative rash with vesicles around the mouth. Vesicles and ulcers are also present within the mouth. Results of a Tzanck smear show multinucleated giant cells (syncytia) and Cowdry type A inclusion bodies. The lesions resolve after 18 days.
Recurrent oral HSV: A 22-year-old medical student studying for examinations feels a twinge at the crimson border of his lip and 24 hours later has a single vesicular lesion at the site.
Recurrent genital HSV: A sexually active 32-year-old woman has a recurrence of ulcerative vaginal lesions with pain, itching, dysuria, and systemic symptoms 48 hours after being exposed to ultraviolet B light while skiing. The lesions resolve within 8 days. Results of a Papanicolaou smear shows multinucleated giant cells (syncytia) and Cowdry type A inclusion bodies.
Encephalitis HSV: A patient has focal neurologic symptoms and seizures. Magnetic resonance imaging results show destruction of a temporal lobe. Erythrocytes are present in the cerebrospinal fluid, and polymerase chain reaction is positive for viral DNA.
Varicella-Zoster Virus
Varicella (chickenpox): A 5-year-old boy develops a fever and a maculopapular rash on his abdomen 14 days after meeting with his cousin, who also developed the rash. Successive crops of lesions appeared for 3 to 5 days, and the rash spread peripherally.
Zoster (shingles): A 65-year-old woman has a belt of vesicles along the thoracic dermatome and experiences severe pain localized to the region.

Figure 51-18 Time course of symptoms of exanthem subitum (roseola) caused by human herpesvirus 6 (HHV-6). Compare these symptoms and this time course with those of fifth disease, which is caused by parvovirus B19 (see Chapter 53).
HHV-6 may also cause a mononucleosis syndrome and lymphadenopathy in adults and may be a cofactor in the pathogenesis of AIDS. Similar to CMV, HHV-6 may reactivate in transplant patients and contribute to the failure of the graft. HHV-6 has also been associated with multiple sclerosis and chronic fatigue syndrome.
Other Human Herpesviruses
Human Herpesvirus 8 (Kaposi Sarcoma–Associated Herpesvirus)
HHV-8 DNA sequences were discovered in biopsy specimens of Kaposi sarcoma, primary effusion lymphoma (a rare type of B-cell lymphoma), and multicentric Castleman disease through the use of PCR analysis. Kaposi sarcoma is one of the characteristic opportunistic diseases associated with AIDS. Genome sequence analysis showed that the virus was unique and a member of the subfamily Gammaherpesvirinae. Similar to EBV, the B cell is the primary target cell for HHV-8, but the virus also infects a limited number of endothelial cells, monocytes, and epithelial and sensory nerve cells. Within the Kaposi sarcoma tumors, endothelial spindle cells contain the virus.
HHV-8 encodes several proteins that resemble human proteins and promote the growth and prevent apoptosis of the infected and surrounding cells. These proteins include an interleukin-6 homologue (growth and antiapoptosis), a Bcl-2 analogue (antiapoptosis), chemokines, and a chemokine receptor. These proteins can promote the growth and development of polyclonal Kaposi sarcoma cells in AIDS patients and others. HHV-8 DNA is present and is associated with peripheral blood lymphocytes, most likely B cells, in approximately 10% of immunocompetent people. HHV-8 is more prevalent in certain geographic areas (Italy, Greece, Africa) and in patients with AIDS. Kaposis sarcoma is the most common cancer in sub-Saharan Africa. The virus is most likely a sexually transmitted disease but may be spread by other means.
Herpesvirus simiae (B virus) (subfamily Alphaherpesvirinae, the simian counterpart of HSV), is indigenous to Asian monkeys. The virus is transmitted to humans by monkey bites or saliva, or even by tissues and cells widely used in virology laboratories. Once infected, a human may have pain, localized redness, and vesicles at the site where the virus entered. An encephalopathy develops and is often fatal; most people who survive have serious brain damage. PCR or serologic tests can be used to establish the diagnosis of B-virus infections. Virus isolation requires special facilities.
A 2-year-old child with fever for 2 days has not been eating and has been crying often. On examination, the physician notes that the mucous membranes of the mouth are covered with numerous shallow, pale ulcerations. A few red papules and blisters are also observed around the border of the lips. The symptoms worsen over the next 5 days and then slowly resolve, with complete healing after 2 weeks.
1. The physician suspects that this is an HSV infection. How would the diagnosis be confirmed?
2. How could you determine whether this infection was caused by HSV-1 or HSV-2?
3. What immune responses were most helpful in resolving this infection, and when were they activated?
4. HSV escapes complete immune resolution by causing latent and recurrent infections. What was the site of latency in this child, and what might promote future recurrences?
5. What were the most probable means by which the child was infected with HSV?
6. Which antiviral drugs are available for the treatment of HSV infections? What are their targets? Were they indicated for this child? Why or why not?
A 17-year-old high school student has had low-grade fever and malaise for several days, followed by sore throat, swollen cervical lymph nodes, and increasing fatigue. The patient also notes some discomfort in the left upper quadrant of the abdomen. The sore throat, lymphadenopathy, and fever gradually resolve over the next 2 weeks, but the patient’s full energy level does not return for another 6 weeks.
7. What laboratory tests would confirm the diagnosis of EBV-induced infectious mononucleosis and distinguish it from CMV infection?
8. To what characteristic diagnostic feature of the disease does mononucleosis refer?
9. What causes the swollen glands and fatigue?
10. Who is at greatest risk for a serious outcome of an EBV infection? What is the outcome? Why?
1. The diagnosis can be confirmed by taking a Tzanck smear and analyzing the cells taken from the base of a lesion for syncytia and Cowdry type A inclusion bodies. The sample can also be analyzed by immunofluorescence. A sample of vesicle fluid can be put into cell culture and observed for characteristic cytopathologic effects or analyzed by PCR for the HSV genome.
2. Immunofluorescence, using type-specific antibodies or PCR analysis of the samples indicated in question 1, can distinguish HSV-1 from HSV-2.
3. Innate responses, such as interferon-α and natural killer cells, are activated early to limit the spread of virus, followed later by T-cell responses and antibody. T cells are essential for resolution of infection, but antibody assists in the cleanup of the infection, although it is not sufficient for protection or control of the infection.
4. Latency is established in the trigeminal ganglia. Future recurrences will be triggered by stresses, such as ultraviolet B light and emotional or physical stress.
5. The child was infected by contact with an infected person or by sharing an item with someone bearing an active lesion.
6. Most of the effective anti-HSV drugs are nucleotide analogues, which are activated by the viral-encoded thymidine kinase and then will inhibit the viral DNA-dependent DNA polymerase. These drugs include valacyclovir, acyclovir, penciclovir, and famciclovir. They are not indicated for this child because the infection is not life threatening, and the disease has progressed beyond the time within which the drugs would be effective.
7. The most simple test would be a heterophile antibody test, which is specific for EBV and not CMV. Serology for EBV antigens could confirm the diagnosis. These tests will also distinguish between a current and previous course of EBV disease.
8. The mononucleosis results from the expansion in numbers of T cells upon stimulation by the EBV-infected B cells. Mononucleosis-like syndromes accompany other infections of lymphocytes, including CMV and HIV.
9. Swollen glands and fatigue are caused by the large-scale activation of the immune response, as indicated by the expansion of the numbers of T cells.
10. Immunocompromised individuals are at risk for EBV-induced leukemia and lymphoma-like diseases because EBV-stimulated B cells will grow out of control in the absence of functional T cells. Boys with Duncan disease (X-linked immunodeficiency) die of leukemia-like immunoproliferation caused by the inability of their T cells to control the outgrowth of B cells (this function is normally used to limit the outgrowth of B cells in response to antigen).