Chapter 4 The drug discovery process
General principles and some case histories
Introduction
The creation of a new drug can be broadly divided into three main phases (Figure 4.1):
• Drug discovery – from therapeutic concept to molecule
• Drug development – from molecule to registered product
• Commercialization – from product to therapeutic application to sales.
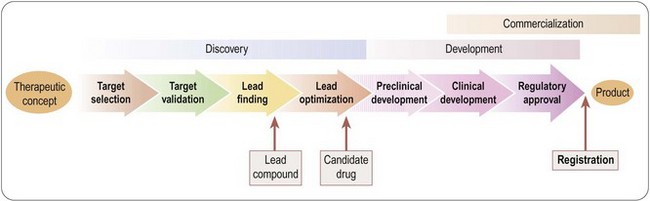
Fig. 4.1 Three main phases of the creation of a new drug: discovery, development and commercialization.
Traditionally, these functions are performed by Research, Development and Marketing, respectively, reflecting the different professional training and expertise required to do the job. Figure 4.1 greatly oversimplifies what is actually a very complex process. For example, development activities, in the form of additional clinical trials, or testing of new formulations, continue well beyond the point of registration, with the aim of extending the range of applications of the compound or complying with regulatory requirements. The discovery team, having delivered the first candidate drug, will carry on looking for others, to serve as back-ups in case the lead compound should fail in development, or as follow-up compounds intended to have advantages over the lead compound. The three components of the overall process are not independent and consecutive stages, but have to be closely coordinated at all stages of the project. At the outset of any new project, the criteria against which the plan will be judged include not only its scientific strength and originality but, importantly, development and marketing issues. For example, if the therapeutic target is an ill-defined clinical disorder, such as chronic fatigue syndrome, will it be possible to measure clinical efficacy objectively? Does the project face stiff competition from other companies working in the same area, or from drugs already in clinical use? Is it likely that an esoteric drug delivery system will be required, and if so, can this be developed? If the drug is successfully developed, is the expected market sufficient to justify the cost of development? The answers to questions of this kind are likely to change, for better or for worse, during the course of the project, so it is essential to keep such issues constantly under review, and to adapt the project plan if necessary.
To integrate successfully the different interests – and cultures – of research, development and marketing is one of the major challenges for a pharmaceutical company, and the need for such integration is a relatively modern development in the industry. As recently as 25–30 years ago in most companies, the process was much more compartmentalized: scientists produced molecules with interesting pharmacological properties, development functions were responsible for checking their safety and turning them into registrable drugs, and the marketing department generated sales and turned them into revenues. At the time this worked well, and many companies prospered. The drop-out rate was not excessive, because regulatory requirements were less stringent, and the failures that did occur were not unduly expensive in terms of time and resources lost. Since then, biomedical science has advanced dramatically, drug discovery and development have become more technology-driven and, hence, expensive, regulatory requirements are much more stringent, and the competition is more intense. With bigger teams, and more complex multidisciplinary tasks, effective project management has become much more important than it used to be to keep costs and delays to a minimum. An additional complication is that of the increased amount of work being done in partnership with other companies or with academic groups, adding the need for alliance as well as project management (see also Chapter 22).
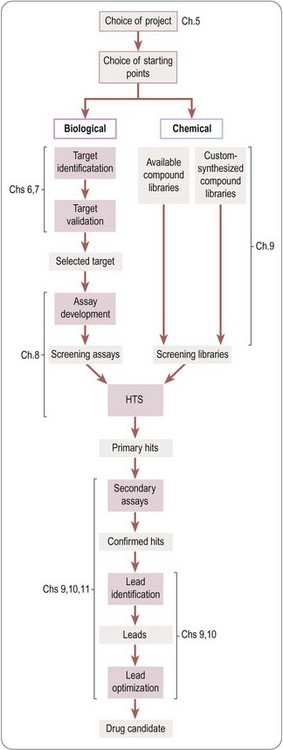
A more detailed overview of the drug discovery phase of a typical project aimed at producing a new synthetic drug is shown in Figure 4.2. It starts with the choice of a disease area and defining the therapeutic need that is to be met. It proceeds to the identification of the biochemical, cellular or pathophysiological mechanism that will be targeted, and, if possible, the identification and validation of a molecular ‘drug target’. Next comes the identification of a lead structure, followed by the design, testing and fine-tuning of the drug molecule to the point where it is deemed suitable for development, discussed in more detail in subsequent chapters.
The strong emphasis on defined molecular (normally protein) drug targets as a starting point is too recent to have culminated so far in many actual drugs on the market. Many drugs now being registered have their origins in research going back 20 years or more, in the ‘premolecular’ drug discovery era, when the selected targets were mainly pathophysiological or biochemical mechanisms, such as blood pressure regulation, inflammation or cholesterol metabolism, of which the molecular components were not yet defined. This earlier period, roughly from 1960 to 1980, was actually highly productive in terms of drug discovery, representing a return on R&D investment considerably greater than what can be achieved today, and the discovery approaches used then remain very much alive despite the increasing emphasis on molecular targets. Nevertheless, we now think increasingly in terms of defined molecular targets as the necessary starting point for drug discovery, and turn automatically to molecular technologies to provide the necessary tools. Until about 1980 this was rarely feasible; even when the ‘target’ was defined, for example as an enzyme or a receptor, it was seldom available in sufficient quantities in a purified functional form to be used as the basis for screening assays. Instead, the functional activity of the target was measured by indirect means in isolated tissue preparations, or even in whole animals, methods which we nowadays regard as too slow, laborious and error prone to place at the front end of a drug discovery project.
The foregoing remarks apply to the discovery of conventional ‘small-molecule’ therapeutics, and the strategy for developing biopharmaceuticals – an increasing proportion of new drugs appearing on the market – is generally different. Biopharmaceutical agents (see Chapter 3) are very diverse, including endogenous mediators, monoclonal antibodies and vaccines, and in the future, no doubt, products for siRNA and gene therapy applications. Where endogenous molecules are involved, the concept of targets and lead compounds has much less relevance, as nature has done the discovery part of the work, so once the therapeutic relevance of the substance has been established, the problems mainly revolve around the production, purification and formulation of the material in a form suitable for the market. With other kinds of biopharmaceuticals, such as therapeutic antibodies, the molecular target will generally be chosen in advance, and the main task is to obtain an antibody with the required properties.
A glance at the pharmacopoeia will show that many therapeutic agents, particularly anti-infective and anti-tumour drugs, originate from natural products, rather than synthetic molecules (Table 4.1). Until about 1950, when synthetic chemistry really came into its own as a source of new drugs, most of the pharmacopoeia consisted of natural products, and they continue to be important, as the example of paclitaxel described below shows. It is reasonable to suppose that such ready-made, highly evolved biomolecules stand a better chance of interacting with selected drug targets than do random synthetic molecules, and the pool from which they come is huge and largely untapped. Exploiting such a ready-made compound library is seen as an attractive strategy which has led to some important therapeutic breakthroughs, such as the anti-malarial drug artemesinin, immunosuppressants such as ciclosporin fujimycin (FK506) and rapamycin, as well as paclitaxel and other recently introduced anticancer drugs such as epothilones. In 2008–2010, 8 out of 64 novel compounds registered were natural products or derived from natural products. In practice, the theoretical advantages of natural products are balanced by several practical disadvantages. Access to source material in remote places can be troublesome for geographical reasons, as well as being politically sensitive, and the continuing availability of the active compound, if it cannot be synthesized on a commercial basis, may be uncertain. Microorganisms have an advantage over higher species in this regard, but initial positive test data on microbial samples frequently cannot be replicated, presumably because of inconsistencies in the culture conditions. Purification and structure determination of natural products is now fairly routine, but is often difficult and time-consuming. A recent example is the introduction of Sativex, a standardized preparation of cannabinoids, given as intra oral drops for the treatment of spasticity associated with multiple sclerosis.
Table 4.1 Examples of therapeutic drugs derived from natural products
| Warfarin | Anticoagulant. Synthetic compound derived from dicoumarol, found in spoiled sweet clover |
| Heparin | Anticoagulant, occurring naturally in mammalian tissues |
| Hirudin | Anticoagulant from leech, now produced by genetic engineering |
| Opiates | Analgesic compounds from poppies |
| Methylxanthines (caffeine, theophylline) | Phosphodiesterase inhibitors and adenosine receptor antagonists. Produced by tea, coffee and coca plants |
| Statins | HMG CoA reductase inhibitors used to reduce plasma cholesterol. Lovastatin is a fungal metabolite. Later compounds (mevastatin, pravastatin) synthesized from lovastatin |
| Cromoglycate | Asthma prohylaxis. Synthetic compound based on khellin, a plant product used as a herbal medicine |
| Vinca alkaloids (vincristine, vinblastine) | Anticancer drugs produced by plants of the periwinkle family |
| Paclitaxel | Anticancer drug from yew tree |
| Etoposide | Anticancer drug synthesized from podophyllotoxin, produced by mandrake plant; used in folk medicine |
| Artemether | Antimalarial drug, semisynthetic derivative of artemesin, produced by Chinese herb |
| Ivermectin | Antihelminthic drug, semisynthetic derivative of avermectin, a fungal metabolite |
| Antibiotics | Too numerous to list. The majority of current antibiotics are derived from fungal metabolites |
In earlier times the pharmacopoeia consisted very largely of plant-derived compounds (e.g. opiates, atropine, ephedrine, ergot alkaloids, strychnine, tubocurarine, digoxin, quinine, veratridine, reserpine, etc.), many of which remain in therapeutic use or provide valuable research tools.
Some case histories
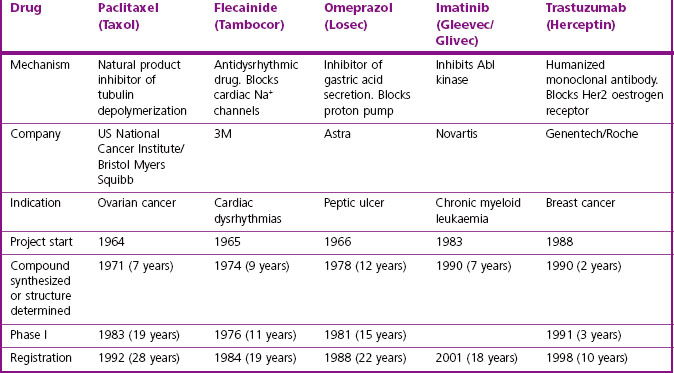
It is clear that there are many starting points and routes to success in drug discovery projects (Lednicer, 1993; Drews, 2000). The brief case histories of five successful drugs, paclitaxel (Taxol), flecainide (Tambocor), omeprazol (Losec), imatinib (Gleevec/Glivec) and trastuzumab (Herceptin), are summarized in Figure 4.3 and Table 4.2, and described in more detail below. Each represents a highly innovative ‘breakthrough’ project rather than an incremental development based on an existing therapy, and they illustrate the variety of different approaches taken by successful projects over the past 35 years. However, for several reasons we should avoid interpreting these as guidelines for success in the future. For one thing, the approach changes as the underlying technologies advance; furthermore, pharmaceutical companies generally publicize only their successes, and even then the accounts are often somewhat sanitized, and fail to describe the errors that were made, the deadlines missed and the blind alleys that were encountered – the full ‘shaggy drug stories’ generally remain discreetly hidden. It must be remembered that, of drug discovery projects begun, only about 1 in 50 is successful in terms of bringing a compound to market. Only at the point when official approval for trials in man is granted does the project become visible to the outside world, so data on success rates, timelines, etc., are much more accessible for the minority of projects that progress to Phase I or beyond than for the majority that never get that far. Analysing the success factors for early-stage drug discovery projects is therefore difficult.
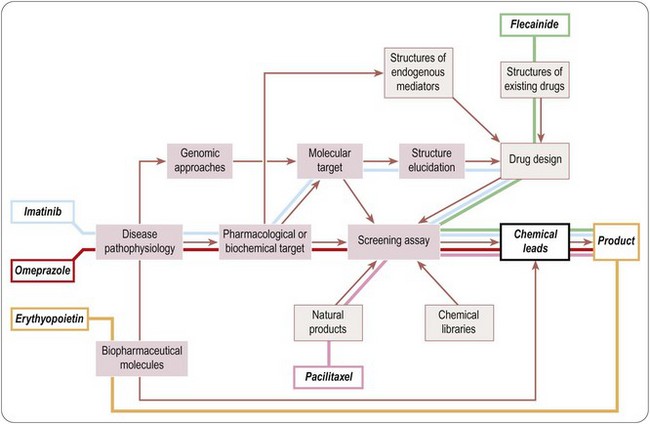
Paclitaxel (Taxol)
Paclitaxel is an interesting example of a project based on the development of a natural product (Cragg, 1998). It began in the early 1960s, when the US National Cancer Institute, responding to the Nixon-inspired ‘war on cancer’, set up one of the first directed screening programmes – still running – to seek new anticancer drugs from plant sources. The sample of bark from the Pacific Yew was collected in 1962 and found to have modest activity against various tumour cell lines. The active substance was isolated in 1969 and joined a collection of moderately active, but not particularly interesting, lead compounds. When this collection was dusted off in 1975 and tested on a new assay, a melanoma cell line, paclitaxel stood out as highly active. Its activity was confirmed in animal models, and it was soon chosen as a development candidate. Interest was further stimulated when its novel mechanism of action, the promotion of microtubule polymerization, was very elegantly demonstrated. Development was difficult, for two main reasons. Paclitaxel is insoluble in water, and the early formulations for injection used in Phase I trials contained a high proportion of the solubilizing agent Cremophor EL, causing frequent severe allergic reactions when given as a bolus intravenous injection. After considerable delay, the problem was overcome by the use of slow infusions and development was resumed. The second problem was the supply of material for clinical trials, and the uncertainty that it could ever be produced on a commercial basis. The Pacific Yew grows slowly and has a restricted habitat, and conservationists were opposed to commercial harvesting. As a result, there was only enough material for limited Phase II studies, on patients with ovarian cancer. The improvement in these patients was dramatic, but continuation of the project, now in collaboration with Bristol Myers Squibb, was seriously hindered by the limited supplies of yew tree bark. The conservation concerns were overcome when a census showed that the tree population was not in fact threatened, and industrial supplies of bark were collected to support the trials programme right through to 1992, when the drug was officially approved.
Commercialization of the material extracted from bark was seen as a major problem, but was solved when it was found that the needles of many yew species contain baccatin, from which paclitaxel can be produced. This semisynthetic paclitaxel, made from an abundant and renewable source, was officially approved in 1999 and is a highly successful and clinically valuable form of cancer therapy. The obstacles to progress in this case were (a) the failure of the primary screen to reveal the compound as anything out of the ordinary; (b) the appearance of serious side effects resulting from the properties of the excipient; and (c) the supply problem.
Flecainide (Tambocor)
The story of flecainide (Banitt and Schmid, 1993) represents a completely different route to success, variations of which gave rise to many innovative drugs (e.g. antihypertensive drugs, antidepressants and antipsychotics) during the 1960s. In the early 1960s, the drugs used to treat cardiac dysrhythmias were mainly quinidine, procainamide, digoxin (for supraventricular tachycardias) and lidocaine (given i.v. for ventricular dysrhythmias). The first three had many troublesome side effects, whereas lidocaine’s use was largely confined to intensive care settings. In 1964, the 3M company decided to seek better antidysrhythmic drugs. Their chemists had developed a new synthetic pathway for introducing –CF3 groups, and they started a chemistry programme based on fluorinated derivatives of known local anaesthetic and antidysrythmic drugs. Assays for antidysrhythmic activity at the time involved elaborate studies on anaesthetized dogs, which were quite unsuitable for screening, and so the group developed a simple primary screening assay based on the ability of compounds to prevent ventricular fibrillation induced by chloroform inhalation in mice, which was used to screen hundreds of compounds. Secondary assays on selected compounds were carried out on anaesthetized dogs in the then conventional fashion. Questions of mechanism were not addressed, it being (correctly) assumed that efficacy in these animal models would serve as a good predictor of clinical efficacy irrespective of the cellular mechanisms involved. A potential development compound was synthesized in 1969, but abandoned on account of CNS side effects. After a further 5 years of painstaking chemistry, during which many different structural classes were tested, flecainide was synthesized (1974) and found to have a much improved therapeutic window compared to its predecessors. The first clinical studies were performed in 1976, and development proceeded quite smoothly until the compound was registered in 1984. It was the first deliberate effort to develop an improved antidysrhythmic drug and proved highly successful in the clinic, now accepted as the standard Class 1c antidysrhythmic agent according to the current classification.
With the benefit of hindsight, we can see that the main delaying factor in the flecainide project was simply slow chemistry, guided largely by empiricism. One result of this was that, after encountering side-effect problems with the lead compound, it took 5 years to find the solution (during which, one suspects, the biologists on the team were growing a little bored!). This model of drug discovery research, where chemistry was both the driving force and the rate-limiting factor for the whole project, is typical of many projects conducted in the 1970s (including many that were, like flecainide, ultimately very successful).
Omeprazole (Losec)
Omeprazole, developed by Astra, was the first proton pump inhibitor, and transformed the treatment of peptic ulcers when it was launched in 1988, quickly becoming the company’s best-selling drug. The project, however, graphically described by Östholm (1995) had a chequered and death-defying history. In 1966 Astra started a project aimed at developing inhibitors of gastric acid secretion, having previously developed profitable antacid preparations. They started a chemistry programme based on carbamates, and collaborated with an academic group to develop a suitable in vivo screening assay in rats. Compounds with weak activity were quickly identified; initial hepatotoxicity problems were overcome, and a potential development compound was tested in humans in 1968. It had no effect on acid secretion, and the project narrowly escaped termination. In the meantime, good progress was being made by Smith, Kline and French in developing histamine H2 antagonists for the same indication, thereby adding to the anxiety within Astra. At the same time Searle reported a new class of inhibitory compounds, benzimidazoles, which were active but toxic. Astra began a new chemistry programme based on this series, and in 1973 produced a highly active compound which was proposed for further development. To their dismay, they found that a Hungarian company had a patent on this compound (for a completely different application). However, upon entering licensing negotiations they found that the Hungarian patent had actually lapsed because the company had defaulted on payment of the fees to the patent office! Further studies with this compound revealed problems with thyroid toxicity, however, and more demands to terminate this hapless project were narrowly fought off. The thyroid toxicity was thought to be associated with the thiouracil structure, and further chemistry aimed at eliminating this resulted, in 1976, in the synthesis of picoprazole, the forerunner of omeprazole. After yet another toxicological alarm – this time vasculitis in dogs – which turned out to be an artefact, picoprazole was tested in human patients suffering from Zollinger–Ellison syndrome and was found to be highly effective in reducing acid secretion. At around the same time, an academic group showed that acid secretion involved a specific transport mechanism, the proton pump, which was strongly inhibited by the Astra compounds, so their novel mechanism of action was established. Omeprazole, an analogue of picoprazole, was synthesized in 1979, and was chosen for development instead of picoprazole. The chemistry team had by then made over 800 compounds during the 13-year lifetime of the project. The chemical development of omeprazole was complicated by the compound’s poor stability and sensitivity to light, requiring special precautions in formulation. Phase II/III clinical trials began in 1981, but were halted for 2 years as a result of yet another toxicology scare – carcinogenicity – which again proved to be a false alarm. Omeprazole was finally registered in 1988.
That omeprazole, one of the most significant new drugs to appear in the early 1990s, should have survived this frightful odyssey is something of a miracle. One setback after another was faced and overcome, a tribute to the sheer determination and persuasive skills of the discovery team. Nowadays, when research managers pride themselves on their decisiveness and courage in terminating projects at the first hint of trouble, omeprazole would surely stand little chance.
Imatinib (Gleevec/Glivec)
Imatinib (Druker and Lydon, 2000; Capdeville et al., 2002), registered in 2001, is the most recent example in these brief histories, and exemplifies the shift towards defined molecular targets that has so altered the approach to drug discovery over the last 20 years. In the mid-1980s, it was discovered that a rare form of cancer, chronic myeloid leukaemia (CML), was almost invariably associated with the expression of a specific oncogene product, Bcr-Abl kinase. The enhanced tyrosine kinase activity of this mutated protein was shown to underlie the malignant transformation of the white blood cells. The proven association between the gene mutation1, the enhanced kinase activity and the distinct clinical phenotype, provided a particularly clear example of cancer pathogenesis. On this basis, the oncology team of Ciba-Geigy (later Novartis) began a project seeking specific inhibitors of Abl kinase. It is known that there are many different kinases involved in cellular regulatory mechanisms, all using ATP as a phosphate donor and possessing highly conserved ATP-binding domains. Interest in kinase inhibitors as drugs was, and remains, high (Cohen, 2002), but at the time the known inhibitors were all relatively non-specific and distinctly toxic, and the widely held view was that, as the known compounds all acted at the highly conserved ATP-binding site, specific subtype selective kinase inhibitors would be difficult to produce. The commercial potential also appeared weak, as CML is a rare disease. Undaunted, the team started by developing routine biochemical assays for this and other kinases, based on purified enzymes produced in quantity by a genetic engineering technique based on the baculovirus expression system. Screening of synthetic compound libraries revealed that compounds of the 2-phenylaminopyrimidine class showed selectivity in blocking Abl and PDGF-receptor kinases, and systematic chemical derivatization led to the synthesis of imatinib in 1992, roughly 8 years after starting the project. Although crystallographic analysis of the kinase structure played no part in guiding the chemistry that produced imatinib, a later structural study (Schindler et al., 2000) provided an explanation for its selectivity for Abl kinase by showing that its binding site extends beyond the ATP site to other, less conserved domains. Imatinib proved to have no major shortcomings in relation to pharmacokinetics or toxicology, and was highly effective in suppressing the growth of cells engineered to express Bcr-Abl, and of human tumour cells transplanted into mice. Importantly, it also inhibited the growth in culture of peripheral blood or bone marrow cells from CML patients (Druker et al., 1996). The latter result was particularly valuable for the project, as it is rarely possible to carry out such ex vivo tests on material from patients – normally, it is necessary to wait until the compound enters Phase II trials before any evidence relating to clinical efficacy emerges. On that basis the project was given high priority and an accelerated clinical trials programme was devised. The first trials (Druker et al., 2001), beginning in 1998, were performed not on normal subjects, but on 83 CML patients who had failed to respond to treatment with interferon. Different doses were tested in groups of six to eight patients, and the pharmacokinetic parameters, adverse effects and clinical response were measured in parallel. These highly streamlined studies showed an unequivocal clinical effect, with 100% of patients receiving the higher doses showing a good haematological response. As a result, and because the regulatory procedures were dispatched particularly rapidly, the drug was registered in record time, in May 2001, just 3 years after being tested for the first time in humans. Imatinib is the first ‘designer’ kinase inhibitor to be registered (other drugs, such as rapamycin, probably act by kinase inhibition, but this was not known at the time). Imatinib has proved efficacious also in certain gastrointestinal tumours, and is the first of a number of kinase inhibitors now used for treating cancer.
In retrospect, the imatinib project owes its success to several factors, most obviously to the selection of a precisely defined molecular target which was known to be disease relevant, and was amenable to modern assay technologies. Setting up the various kinase assays took 4–5 years, but thereafter screening produced the lead series of compounds rather quickly, and imatinib itself was made within about 4 years of starting the screening programme. Avoiding the pitfalls of pharmacokinetics and toxicology, which so often hinder development, was very fortunate. What was quite exceptional was the speed of clinical development and registration. This was possible partly because CML is resistant to conventional anticancer drugs, and so imatinib did not need to be compared with other treatments. Also, the designation of imatinib as an ‘orphan drug’ (see Chapter 20), based on the rarity of CML, allowed the trials programme to be simplified and accelerated. Its action is readily monitored by haematological tests, permitting a rapid clinical readout. The therapeutic effect of the drug on circulating white cells is directly related to its plasma level, which is often not the case for drugs acting on solid tumours. It is an example where the choice of indication, initially made on the basis of a solid biological hypothesis, proved highly advantageous in allowing the clinical development to progress rapidly.
Trastuzumab (Herceptin)
Trastuzumab is a humanized monoclonal antibody which selectively blocks the oestrogen receptor Her2. This project, which took 8 years from compound discovery to registration, shows the speed with which biopharmaceuticals, under the right conditions, can be developed. The Her2 receptor was first cloned in 1985, and 2 years later it was found to be strongly over-expressed in the most aggressive breast cancers. Genentech used its in-house technology to develop a humanized mouse monoclonal antibody that blocked the function of the receptor and suppressed the proliferation of receptor-bearing cells. Compared with conventional lead finding and lead optimization of synthetic molecules, this took very little time – only 2 years from the start of the project. Antibodies generally exhibit much simpler and more predictable pharmacological effects than synthetic compounds, and run into fewer problems with chemical development, formulation and toxicology, so that trastuzumab was able to enter Phase I within 2 years. Clear-cut efficacy was evident in Phase II, and the rest of the clinical development was rapid and straightforward. Trastuzumab represents a significant step forward in the treatment of breast cancer, as well as a commercial success. Given the right circumstances, biopharmaceuticals can be developed more quickly and more cheaply than conventional drugs, a fact reflected in the growing proportion (approximately 35% in 2001 but approaching 50% in 2011) of biopharmaceuticals among new chemical entities being registered.
Comments and conclusions
One common feature that emerges from a survey of the many anecdotal reports of drug discovery projects is that they often have outcomes quite different from what was originally intended. The first tricyclic antidepressant drug, imipramine, emerged from a project aimed at developing antihistamines based on the structure of promethazine. Clonidine was synthesized in the early 1960s as part of a project intended to develop α-adrenoceptor agonists as vasoconstrictors for use as decongestant nose drops. The physician involved tested the nose drops on his wife, who had a cold, and was surprised by the fact that her blood pressure plummeted. She also slept for 24 hours. It turned out that the dose was about 30 times what was later found effective in humans. The experiment revealed the unexpected hypotensive action of clonidine upon which its subsequent commercial development was based. More recently, it is well known that sildenafil (Viagra) was originally intended as a vasodilator for treating angina, and only during clinical testing did its erection-inducing effect become evident and matters have gone full circle with the recent realization that it can be used to treat pulmonary hypertension as well.
It might be supposed that the increasing emphasis now being placed on defined molecular targets as starting points for drug discovery projects would reduce the likelihood of such therapeutic surprises. However, the molecular targets used for screening nowadays lie further, in the functional sense, from the therapeutic response that is being sought than do the physiological responses relied on previously (see Chapter 2, Figure 2.3). Thus compounds aimed with precision at well-defined targets commonly fail to produce the desired therapeutic effect, evidently because we do not sufficiently understand the pathophysiological pathway linking the two. Recent examples of such failures include ondansetron, a 5HT3-receptor antagonist conceived as an antimigraine drug but ineffective in this indication (developed instead as an antiemetic), and substance P receptor antagonists which were expected to have analgesic properties in humans, but which proved ineffective, although again these agents have found a useful niche as antiemetics. Lack of efficacy in clinical trials – a measure of our inability to predict therapeutic efficacy on the basis of pharmacological properties – remains one of the commonest causes of project failure, accounting for 30% of failures (Kennedy, 1997), second only to pharmacokinetic shortcomings (39%).
The stages of drug discovery
In this section we are concerned with the initial stages of the overall project outlined in Figure 4.1, up to the point at which a molecule makes its solemn rite of passage from research to development. Figure 4.4 summarizes the main stages that make up a ‘typical’ drug discovery project, from the identification of a target to the production of a candidate drug2.
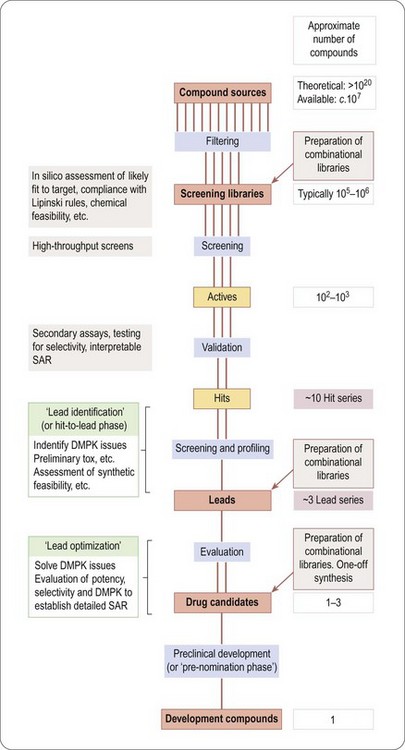
A huge number of ‘theoretical’ compounds (far too many to be physically accessible) is first ‘filtered’ in silico to reduce it to a practicable number of compounds that is available, or can be synthesized, in screening libraries. High-throughput screening is then used to identify ‘hits’, which show significant activity in the chosen screen. This may throw up hundreds or thousands of compounds, depending on the nature of the screen and the size and quality of the library. Normally, a significant proportion of these prove to be artefacts of one sort or another, for example results that cannot be repeated when the compound is resynthesized, positives in the assay resulting from non-specific effects, or simply ‘noise’ in the assay system. ‘Validation’ of hits is, therefore, necessary to eliminate artefacts, and this may involve repeating the screening of the hits, confirming the result in a different assay designed to measure activity on the chosen target, as well as resynthesis and retesting of the hit compounds. Validation will also entail assessment of the structure–activity relationships within the screening library, and whether the hit belongs to a family of compounds – a ‘hit series’ – which represents a reasonable starting point for further chemistry.
In the next stage, lead identification, the validated hits are subjected to further scrutiny, particularly in respect of their pharmacokinetic properties and toxicity as well as addressing in more detail the feasibility of building a synthetic chemistry programme. In this process, the handful of ‘hit series’ is further reduced to one or a few ‘lead series’. Up to this point, the aim has been to reduce the number of compounds in contention from millions to a few – ‘negative chemistry’, if you will.
Synthetic chemistry then begins, in the ‘lead optimization’ stage. This usually involves parallel synthesis to generate derivatives of the lead series, which are screened and profiled with respect to pharmacology, pharmacokinetics and toxicology, to home in on a small number of ‘drug candidates’ (often a single compound, or if the project is unlucky, none at all) judged suitable for further development, at which point they are taken into preclinical development.
The flow diagram in Figure 4.4 provides a useful basis for the more detailed accounts of the various activities that contribute to the drug discovery process, described in the chapters that follow. It should be realized, though, that many variations on this basic scheme take place in real life. A project may, for example, work on one lead series and, at the same time, go back to library screening to find new starting points. New biological discoveries, such as the identification of a new receptor subtype, may cause the project to redefine its objectives midstream.
Increasingly, large companies have tended to centralize the early stages of drug discovery, up to the identification of lead series, and to carry out many screens in parallel with very large compound libraries, focusing on putative drug targets often identified on the basis of genomics, rather than on pharmacology and pathophysiology relating to specific disease indications. In this emerging scenario, the work of the drug discovery team effectively begins from the chemical lead. In the last few years there has been a return to a more intuitive approach to drug discovery in some companies as it has been realized that mass screening is not always the best way to generate good-quality chemical leads.
Trends in drug discovery
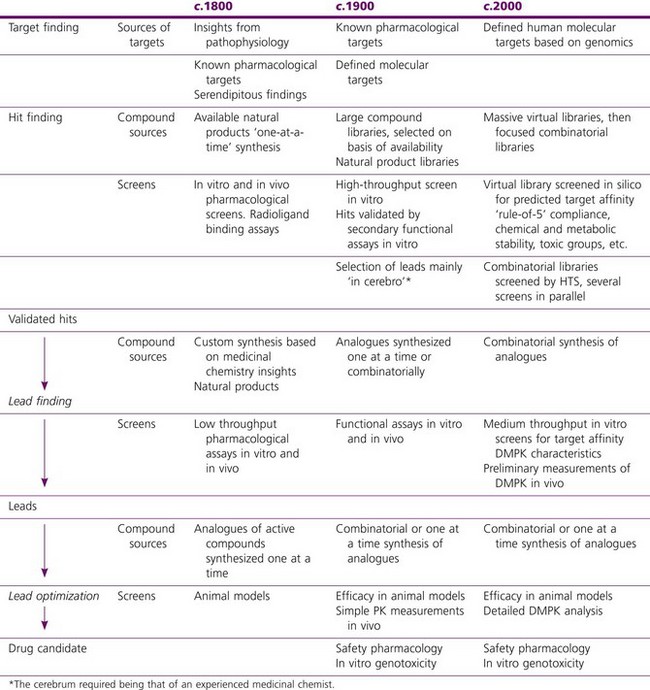
Increasingly, drug discovery has become focused on cloned molecular targets that can be incorporated into high-throughput screens. Target selection, discussed in Chapter 6, has, therefore, assumed a significance that it rarely had in the past: of the projects described above, three began without any molecular target in mind, which would seldom be the case nowadays.
The examples summarized in Table 4.2 show that it took 7–12 years from the start of the project to identificaton of the compound that was finally developed. That interval has now been substantially reduced, often to 3 years or less once a molecular target has been selected, mainly as a result of (a) high-throughput screening of large compound libraries (including natural product libraries) to identify initial lead compounds; (b) improvements at the lead optimization stage, including the use of automated synthesis to generate large families of related compounds, and increased use of molecular modelling techniques, whereby the results of compound screening are analysed to reveal the molecular configurations that are associated with biological activity.
There is strong motivation to improve not only the speed of lead optimization, but also the ‘quality’ of the compound selected for development. Quality, in this context, means a low probability that the compound will fail later in development. The main reasons that compounds fail, apart from lack of efficacy or unexpected side effects, are that they show toxicity, or that they have undesirable pharmacokinetic properties (e.g. poor absorption, too long or too short plasma half-life, unpredictable metabolism, accumulation in tissues, etc.). In the past, these aspects of a drug’s properties were often not investigated until the discovery/development hurdle had been crossed, as investigating them was traditionally regarded as a responsibility of ‘development’ rather than ‘research’. Frequently a compound would fail after several months in preclinical development – too late for the problem to be addressed by the drug discovery team, which had by then moved on. This highlighted the need to incorporate pharmacokinetic and toxicological studies, as well as pharmacological ones, at an earlier stage of the project, during the lead optimization phase. As described in Chapter 10, studies of this kind are now routinely included in most drug discovery projects. Inevitably this has a cost in terms of time and money, but this will be more than justified by a reduction in the likelihood of compounds failing during clinical development.
The main trends that have occurred over the last two decades are summarized in Table 4.3, the key ones being, as discussed above:
• A massive expansion of the compound collections used as starting points, from large ‘white-powder’ libraries of 100 000 or more compounds created during the 1990s, to massive virtual libraries of tens of millions
• Use of automated synthesis methods to accelerate lead optimization
• To deal with large, compound libraries, the introduction of high-throughput screens for actual compounds, and in silico screens for virtual compounds
• Increasing reliance on in silico predictions to generate leads
• Focus on cloned human targets as starting points
• Progressive ‘front-loading’ of assessment of DMPK (drug metabolism and pharmacokinetic) characteristics, including in silico assessment of virtual libraries.
Table 4.3 Trends in drug discovery
Project planning
When a project moves from the phase of exploratory research to being an approved drug discovery project to which specific resources are assigned under the direction of a project leader, its objectives, expected timelines and resource requirements need to be agreed by the members of the project team and approved by research management.
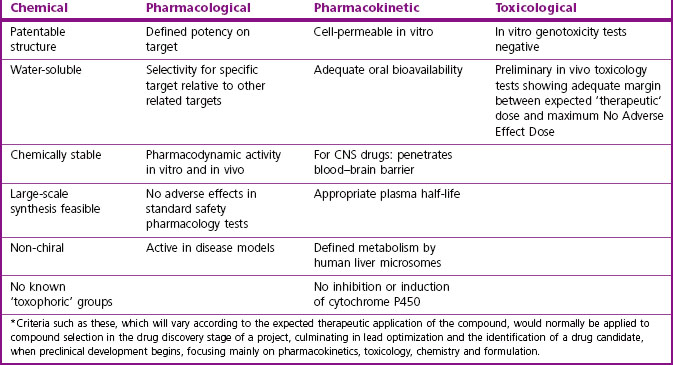
The early drug discovery phase of the project will typically begin when a target has been selected and the necessary screening methods established, and its aim will be to identify one or a few ‘drug candidates’ suitable for progressing to the next stage of preclinical development. The properties required for a drug candidate will vary from project to project, but will invariably include chemical, pharmacological, pharmacokinetic and toxicological aspects of the compound. Table 4.4 summarizes the criteria that might apply to a typical drug acting on a target such as an enzyme or receptor, and intended for oral use. Where appropriate (e.g. potency, oral bioavailability), quantitative limits will normally be set. Such a list of necessary or desirable features, based on results from many independent assessments and experimental tests, provides an essential focus for the project. Some of these, such as potency on target, or oral bioavailability, will be absolute requirements, whereas others, such as water solubility or lack of in vitro genotoxicity, may be highly desirable but not essential. There are, in essence, two balancing components of a typical project:
• Designing and synthesizing novel compounds
• Filtering, to eliminate compounds that fail to satisfy the criteria.
Whereas in an earlier era of drug discovery these two activities took place independently – chemists made compounds and handed over white powders for testing, while pharmacologists tried to find ones that worked – nowadays the process is invariably an iterative and interactive one, whereby the design and synthesis of new compounds continuously takes into account the biological findings and shortcomings that have been revealed to date. Formal project planning, of the kind that would be adopted for the building of a new road, for example, is therefore inappropriate for drug discovery. Experienced managers consequently rely less on detailed advance planning, and more on good communication between the various members of the project team, and frequent meetings to review progress and agree on the best way forward.
An important aspect of project planning is deciding what tests to do when, so as to achieve the objectives as quickly and efficiently as possible. The factors that have to be taken into account for each test are:
• Amount of compound required in relation to amount available
• Time required. Some in vivo pharmacodynamic tests (e.g. bone density changes, tumour growth) are inherently slow. Irrespective of compound throughput, they cannot provide rapid feedback
• ’Salience’ of result (i.e. is the criterion an absolute requirement, or desirable but non-essential?)
• Probability that the compound will fail. In a typical high-throughput screen more than 99% of compounds will be eliminated, so it is essential that this is done early. In vitro genotoxicity, in contrast, will be found only occasionally, so it would be wasteful to test for this early in the sequence.
The current emphasis on fast drug discovery, to increase the time window between launch and patent expiry, and on decreasing the rate of failure of compounds during clinical development, is having an important effect on the planning of drug discovery projects. As discussed above, there is increasing emphasis on applying fast-result, high-throughput methods of testing for pharmacokinetic and toxicological properties at an early stage (‘front loading’; see Chapter 10), even though the salience (i.e. the ability to predict properties needed in the clinic) of such assays may be limited. The growth of high-throughput test methods has had a major impact on the work of chemists in drug discovery (see Chapter 9), where the emphasis has been on preparing ‘libraries’ of related compounds to feed the hungry assay machines. These changes have undoubtedly improved the performance of the industry in finding new lead compounds of higher quality for new targets. The main bottlenecks now in drug discovery are in lead optimization (see Chapter 9) and animal testing (see Chapter 11), areas so far largely untouched by the high-throughput revolution.
Research in the pharmaceutical industry
Pharmaceutical companies perform research for commercial reasons and seek to ensure that it produces a return on investment. The company owns the data, and is free to publish it or keep it secret as it sees fit. Although most pharmaceutical companies include altruistic as well as commercial aims in their mission statements, the latter necessarily take priority. The company will, therefore, wish to ensure that the research it supports is in some way relevant to its commercial objectives. Clearly, studies aimed directly at drug discovery present no problems. At the other end of the spectrum lies pure curiosity-driven (‘blue-skies’) research. Although such work may – and often does – lead to progress in drug discovery in the long term, the commercial pay-off is highly uncertain and inevitably long delayed. Generally speaking, only a minimal amount of such long-term research is performed in a commercial setting. Between the two lies a large territory of ‘applied’ research, more clearly focused on drug discovery, though still medium-term and somewhat uncertain in its applicability. Many technological projects come into this category, such as novel high-throughput screening methods, imaging technologies, etc., as well as research into pathophysiological mechanisms aimed at the identification of new drug targets. The many applications of genomics and molecular biology in drug discovery make up an increasing proportion of work in this applied research category. Small biotechnology companies which started during the 1980s and 1990s moved quickly into this territory, and several large pharmaceutical companies, notably SmithKlineBeecham (now part of GSK), also made major investments. The extent to which pharmaceutical companies invest in medium-term applied research projects varies greatly. A growing tendency seems to be for larger companies to set up what amounts to an in-house biotechnology facility, often incorporating one or more acquired biotech companies. The technological revolution in drug discovery, referred to frequently in this book, has yet to pay dividends in terms of improved performance, so it is uncertain whether this level of commitment to medium-term applied research can be sustained. More recently the view has emerged that much of the information obtained from industry driven genomic studies of disease should be freely available to all and, for example, Merck in the USA has spun out its database to a not-for-profit organization called Sage (see http://www.sagebase.org) for this purpose.
The cultural difference between academic and commercial research is real, but less profound than is often thought. Quality, in the sense of good experimental design, methodology and data interpretation, is equally important to both, as is creativity, the ability to generate new ideas and see them through. Key differences are that, in the industry environment, freedom to choose projects and to publish is undoubtedly more limited, and effective interdisciplinary teamwork is obligatory, rather than a matter of personal choice: there is little room in industry for the lone genius. These differences are, however, becoming narrower as the bodies funding academic research take a more ‘corporate’ approach to its management. Individual researchers in academia are substantially constrained to work on projects that attract funding support, and are increasingly being required to collaborate in interdisciplinary projects. They are certainly more free to publish, but are also under much more pressure to do so, as, in contrast to the situation in industry, publications are their only measure of research achievement. Pharmaceutical companies also have incentives to publish their work: it gives their scientists visibility in the scientific community and helps them to establish fruitful collaborations, as well as strengthening the company’s attractiveness as an employer of good scientists. The main restrictions to publication are the company’s need to avoid compromising its future patent position, or giving away information that might help its competitors. Companies vary in their attitude to publication, some being much more liberal than others.
Scientists in industry have to learn to work, and communicate effectively, with the company’s management. Managers are likely to ask ‘Why do we need this information?’ which, to scientists in academia, may seem an irritatingly silly question. To them, the purpose of any research is to add to the edifice of human knowledge, and working towards this aim fully justifies the time, effort and money that support the work. The long-term benefits to humanity are measured in terms of cultural richness and technological progress. In the short term the value of what has been achieved is measured by peer recognition and grant renewal; the long-term judgment is left to history.
Within a pharmaceutical company, research findings are aimed at a more limited audience and their purpose is more focused. The immediate aim is to provide the data needed for prediction of the potential of a new compound to succeed in the clinic. The internal audiences are the research team itself and research management; the external audiences are the external scientific community and, importantly, the regulatory authorities.
We should, however, resist the tendency to think of drug discovery as a commercial undertaking comparable developing a new sports car, where the outcome is assured provided that operatives with the necessary skill and experience fulfil what is asked of them. They are not likely by chance to invent a new kind of aeroplane or vacuum cleaner.
Like all research, drug discovery is more likely than not to come up with unexpected findings, and these can have a major impact on the plan and its objectives. So, frequent review – and, if necessary, amendment – of the plan is essential, and research managers need to understand (as most do, having their own research experiences to draw on) that unexpected results, and the problems that may follow for the project, reflect the complexity of the problem, rather than the failings of the team. Finding by experiment that a hypothesis is wrong is to be seen as a creditable scientific achievement, not at all the same as designing a car with brakes that do not work.
Banitt EH, Schmid JR. Flecainide. In: Lednicer D, ed. Chronicles of drug discovery, vol. 3. Washington DC: American Chemical Society; 1993.
Capdeville R, Buchdunger E, Zimmermann J, et al. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nature Reviews Drug Discovery. 2002;1:493–502.
Cohen P. Protein kinases – the major drug targets of the twenty-first century? Nature Reviews Drug Discovery. 2002;1:309–316.
Cragg GM. Paclitaxel (Taxol): a success story with valuable lessons for natural product drug discovery and development. Medical Research Review. 1998;18:315–331.
Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960–1964.
Druker BJ, Lydon NB. Lessons learned from the development of an Abl tyrosine kinase inhibitor for chronic myelognous leukemia. Journal of Clinical Investigation. 2000;105:3–7.
Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature Medicine. 1996;2:561–566.
Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the Bcr-Abl tyrosine kinase in chronic myeloid leukemia. New England Journal of Medicine. 2001;344:1031–1037.
Kennedy T. Managing the drug discovery/development interface. Drug Discovery Today. 1997;2:436–444.
Lednicer D. Chronicles of drug discovery. vol. 3. Washington DC: American Chemical Society; 1993.
Östholm I. Drug discovery: a pharmacist’s view. Stockholm: Swedish Pharmaceutical Press; 1995.
Schindler T, Bornmann W, Pellicana P, et al. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science. 2000;289:1938–1942.