Chapter 11 Pharmacology
Its role in drug discovery
Introduction
Pharmacology as an academic discipline, loosely defined as the study of the effects of chemical substances on living systems, is so broad in its sweep that it encompasses all aspects of drug discovery, ranging from the molecular details of the interaction between the drug molecule and its target to the economic and social consequences of placing a new therapeutic agent on the market. In this chapter we consider the more limited scope of ‘classical’ pharmacology, in relation to drug discovery. Typically, when a molecular target has been selected, and lead compounds have been identified which act on it selectively, and which are judged to have ‘drug-like’ chemical attributes (including suitable pharmacokinetic properties), the next stage is a detailed pharmacological evaluation. This means investigation of the effects, usually of a small number of compounds, on a range of test systems, up to and including whole animals, to determine which, if any, is the most suitable for further development (i.e. for nomination as a drug candidate). Pharmacological evaluation typically involves the following:
• Selectivity screening, consisting of in vitro tests on a broad range of possible drug targets to determine whether the compound is sufficiently selective for the chosen target to merit further investigation
• Pharmacological profiling, aimed at evaluating in isolated tissues or normal animals the range of effects of the test compound that might be relevant in the clinical situation. Some authorities distinguish between primary pharmacodynamic studies, concerning effects related to the selected therapeutic target (i.e. therapeutically relevant effects), and secondary pharmacodynamic studies, on effects not related to the target (i.e. side effects). At the laboratory level the two are often not clearly distinguishable, and the borderline between secondary pharmacodynamic and safety pharmacology studies (see below) is also uncertain. Nevertheless, for the purposes of formal documentation, the distinction may be useful
• Testing in animal models of disease to determine whether the compound is likely to produce therapeutic benefit
• Safety pharmacology, consisting of a series of standardized animal tests aimed at revealing undesirable side effects, which may be unrelated to the primary action of the drug. This topic is discussed in Chapter 15.
The pharmacological evaluation of lead compounds does not in general follow a clearly defined path, and often it has no clearcut endpoint but will vary greatly in its extent, depending on the nature of the compound, the questions that need to be addressed and the inclinations of the project team. Directing this phase of the drug discovery project efficiently, and keeping it focused on the overall objective of putting a compound into development, is one of the trickier management tasks. It often happens that unexpected, scientifically interesting data are obtained which beg for further investigation even though they may be peripheral to the main aims of the project. From the scientists’ perspective, the prospect of opening up a new avenue of research is highly alluring, whether the work contributes directly to the drug discovery aims or not. In this context, project managers need to bear in mind the question: Who needs the data and why? – a question which may seem irritatingly silly to a scientist in academia but totally obvious to the commercial mind. The same principles apply, of course, to all parts of a drug discovery and development project, but it tends to be at the stage of pharmacological evaluation that conflicts first arise between scientific aspiration and commercial need.
An important principle in pharmacological evaluation is the use of a hierarchy of test methods, covering the range from the most reductionist tests on isolated molecular targets to much more elaborate tests of integrated physiological function. Establishing and validating such a series of tests appropriate to the particular target and indication being addressed is one of the most important functions of pharmacologists in the drug discovery team. In general, assays become more complicated, slow and expensive, and more demanding of specialist skills as one moves up this hierarchy.
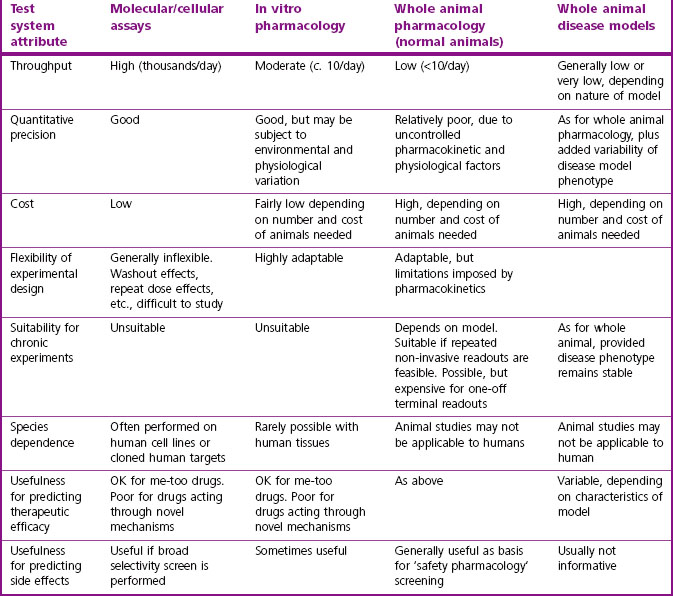
The strengths and weaknesses of these test systems are summarized in Table 11.1.
Pharmacological characterization of a candidate compound often has to take into account active metabolites, based on information from drug metabolism and pharmacokinetics (DMPK) studies (see Chapter 10). If a major active metabolite is identified, it will be necessary to synthesize and test it in the same way as the parent compound in order to determine which effects (both wanted and unwanted) relate to each. Particular problems may arise if the metabolic fate of the compound shows marked species differences, making it difficult to predict from animal studies what will happen in humans.
Although most of the work involved in pharmacological characterization of a candidate drug takes place before clinical studies begin, it does not normally end there. Both ongoing toxicological studies and early trials in man may reveal unpredicted effects that need to be investigated pharmacologically, and so the discovery team needs to remain actively involved and be able to perform experiments well into the phase of clinical development. They cannot simply wave the compound goodbye once the discovery phase is completed.
Screening for selectivity
The selectivity of a compound for the chosen molecular target needs to be assessed at an early stage. Compounds selected for their potency, for example on a given amine receptor, protease, kinase, transporter or ion channel, are very likely to bind also to related – or even unrelated – molecular targets, and thereby cause unwanted side effects. Selectivity is, therefore, as important as potency in choosing potential development candidates, and a ‘selectivity screen’ is usually included early in the project. The range of targets included in such a screen depends very much on the type of compound and the intended clinical indication. Ligands for monoamine receptors and transporters form a large and important group of drugs, and several contract research organizations (e.g. CEREP, MDL) offer a battery of assays – mainly binding assays, but also a range of functional assays – designed to detect affinity for a wide range of receptors, transporters and channels. In the field of monoamine receptors, for example, it is usually important to avoid compounds that block or activate peripheral muscarinic receptors, adrenergic receptors or histamine (particularly H1) receptors, because of the side effects that are associated with these actions, and a standard selectivity test battery allows such problems to be discovered early. Recently, several psychotropic and anti-infective drugs have been withdrawn because of sudden cardiac deaths, probably associated with their ability to block a particular type of potassium channel (known as the hERG channel; see Chapter 16) in myocardial cells. This activity can be detected by electrophysiological measurements on isolated myocardial cells, and such a test is now usually performed at an early stage of development of drugs of the classes implicated in this type of adverse reaction.
Interpretation of binding assays
Binding assays, generally with membrane preparations made from intact tissues or receptor-expressing cell lines, are widely used in drug discovery projects because of their simplicity and ease of automation. Detailed technical manuals describing the methods used for performing and analysing drug binding experiments are available (Keen, 1999; Vogel, 2002). Generally, the aim of the assay is to determine the dissociation constant, KD, of the test compound, as a measure of its affinity for the receptor. In most cases, the assay (often called a displacement assay) measures the ability of the test compound to inhibit the binding of a high-affinity radioligand which combines selectively with the receptor in question, correction being made for ‘non-specific’ binding of the radioligand.
In the simplest theoretical case, where the radioligand and the test compound bind reversibly and competitively to a homogeneous population of binding sites, the effect of the test ligand on the amount of the radioligand specifically bound is described by the simple mass-action equation:
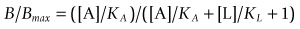 (1)
(1)where B = the amount of radioligand bound, after correcting for non-specific binding, Bmax = the maximal amount of radioligand bound, i.e. when sites are saturated, [A] = radioligand concentration, KA = dissociation constant for the radioligand, [L] = test ligand concentration, and KL = dissociation constant for the test ligand.
By testing several concentrations of L at a single concentration of A, the concentration, [L]50, needed for 50% inhibition of binding can be estimated. By rearranging equation 1, KL is given by:
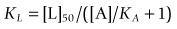 (2)
(2)This is often known as the Cheng–Prusoff equation, and is widely used to calculate KL when [L]50, [A] and KA are known. It is important to realize that the Cheng–Prusoff equation applies only (a) at equilibrium, (b) when the interaction between A and L is strictly competitive, and (c) when neither ligand binds cooperatively. However, an [L]50 value can be measured for any test compound that inhibits the binding of the radioligand by whatever mechanism, irrespective of whether equilibrium has been reached. Applying the Cheng–Prusoff equation if these conditions are not met can yield estimates of KL that are quite meaningless, and so it should strictly be used only if the conditions have been shown experimentally to be satisfied – a fairly laborious process. Nevertheless, Cheng–Prusoff estimates of ligand affinity constants are often quoted without such checks having been performed. In most cases it would be more satisfactory to use the experimentally determined [L]50 value as an operational measure of potency. A further important caveat that applies to binding studies is that they are often performed under conditions of low ionic strength, in which the sodium and calcium concentrations are much lower than the physiological range. This is done for technical reasons, as low [Na+] commonly increases both the affinity and the Bmax of the radioligand, and omitting [Ca2+] avoids clumping of the membrane fragments. Partly for this reason, ligand affinities estimated from binding studies are often considerably higher than estimates obtained from functional assays (Hall, 1992), although the effect is not consistent, presumably because ionic bonding, which will be favoured by the low ionic strength medium, contributes unequally to the binding of different ligands. Consequently, the correlation between data from binding assays and functional assays is often rather poor (see below). Figure 11.1 shows data obtained independently on 5HT3 and 5HT4 receptors; in both cases the estimated KD values for binding are on average about 10 times lower than estimates from functional assays, and the correlation is very poor.
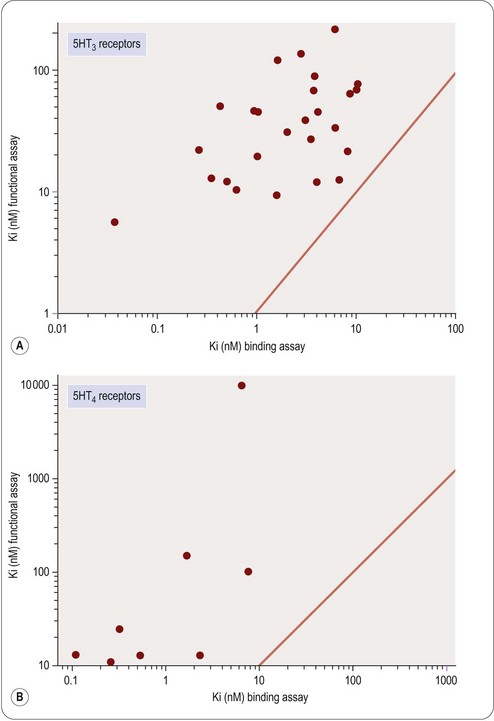
Fig. 11.1 Correlation of binding and functional data for 5HT receptor ligands. (A) 5HT3 receptors, (B) 5HT4 receptors.
Data from Heidempergher et al., 1997. Data from Yang et al., 1997.
Pharmacological profiling
Pharmacological profiling aims to determine the pharmacodynamic effects of the new compound – or more often of a small family of compounds – on in vitro model systems, e.g. cell lines or isolated tissues, normal animals, and animal models of disease. The last of these is particularly important, as it is intended to give the first real pointer to therapeutic efficacy as distinct from pharmacodynamic activity. It is valuable to assess the activity of the compounds in a series of assays representing increasingly complex levels of organization. The choice of test systems depends, of course, on the nature of the target. For example, characterization of a novel antagonist of a typical G-protein-coupled receptor might involve the following:
• Ligand-binding assay on membrane fragments from a cell line expressing the cloned receptor
• Inhibition of agonist activity in a cell line, based on a functional readout (e.g. raised intracellular calcium)
• Antagonism of a selective agonist in an isolated tissue (e.g. smooth muscle, cardiac muscle). Such assays will normally be performed with non-human tissue, and so interspecies differences in the receptor need to be taken into account. Sometimes specific questions have to be asked about effects on human tissues for particular compounds and then collecting viable tissues to use becomes a major challenge
• Antagonism of the response (e.g. bronchoconstriction, vasoconstriction, increased heart rate) to a selective receptor agonist in vivo. Prior knowledge about species specificity of the agonist and antagonist is important at this stage.
Pharmacological profiling is designed as a hypothesis-driven programme of work, based on the knowledge previously gained about the activity of the compound on its specific target or targets. In this respect it differs from safety pharmacology (see below), which is an open-minded exercise designed to detect unforeseen effects. The aim of pharmacological profiling is to answer the following questions:
• Do the molecular and cellular effects measured in screening assays actually give rise to the predicted pharmacological effects in intact tissues and whole animals?
• Does the compound produce effects in intact tissues or whole animals not associated with actions on its principal molecular target?
• Is there correspondence between the potency of the compound at the molecular level, the tissue level and the whole animal level?
• Do the in vivo potency and duration of action match up with the pharmacokinetic properties of the compound?
• What happens if the drug is given continuously or repeatedly to an animal over the course of days or weeks? Does it lose its effectiveness, or reveal effects not seen with acute administration? Is there any kind of ‘rebound’ after effect when it is stopped?
In vitro profiling
Measurements on isolated tissues
Studies on isolated tissues have been a mainstay of pharmacological methodology ever since the introduction of the isolated organ bath by Magnus early in the 20th century. The technique is extremely versatile and applicable to studies on smooth muscle (e.g. gastrointestinal tract, airways, blood vessels, urinary tract, uterus, biliary tract, etc.) as well as cardiac and striated muscle, secretory epithelia, endocrine glands, brain slices, liver slices, and many other functional systems. In most cases the tissue is removed from a freshly killed or anaesthetized animal and suspended in a chamber containing warmed oxygenated physiological salt solution. With smooth muscle preparations the readout is usually mechanical (i.e. tension, recorded with a simple strain gauge). For other types of preparation, various electrophysiological or biochemical readouts are often used. Vogel (2002) and Enna et al. (2003) give details of a comprehensive range of standard pharmacological assay methods, including technical instructions.
Studies of this kind have the advantage that they are performed on intact normal tissues, as distinct from isolated enzymes or other proteins. The recognition molecules, signal transduction machinery and the mechanical or biochemical readout are assumed to be a reasonable approximation to the normal functioning of the tissue. There is abundant evidence to show that tissue responses to GPCR activation, for example, depend on many factors, including the level of expression of the receptor, the type and abundance of the G proteins present in the cell, the presence of associated proteins such as receptor activity-modifying proteins (RAMPs; see Morfis et al., 2003), the state of phosphorylation of various constituent proteins in the signal transduction cascade, and so on. For compounds acting on intracellular targets, functional activity depends on permeation through the membrane, as well as affinity for the target. For these reasons – and probably also for others that are not understood – the results of assays on isolated tissues often differ significantly from results found with primary screening assays. The discrepancy may simply be a quantitative one, such that the potency of the ligand does not agree in the two systems, or it may be more basic. For example, the pharmacological efficacy of a receptor ligand, i.e. the property that determines whether it is a full agonist, a partial agonist, or an antagonist, often depends on the type of assay used (Kenakin, 1999), and this may have an important bearing on the selection of possible development compounds. Examples that illustrate the poor correlation that may exist between measurements of target affinity in cell-free assay systems, and functional activity in intact cell systems, are shown in Figures 11.1 and 11.2. Figure 11.1 shows the relationship between binding and functional assay data for 5HT3 and 5HT4 receptor antagonists. In both cases, binding assays overestimate the potency in functional assays by a factor of about 10 (see above), but more importantly, the correlation is poor, despite the fact that the receptors are extracellular, and so membrane penetration is not a factor. Figure 11.2 shows data on tyrosine kinase inhibitors, in which activity against the isolated enzyme is plotted against inhibition of tyrosine phosphorylation in intact cells, and inhibition of cell proliferation for a large series of compounds. Differences in membrane penetration can account for part of the discrepancy between enzyme and cell-based data, but the correlation between intracellular kinase inhibition and blocking of cell proliferation is also weak, which must reflect other factors.

Fig. 11.2 Correlation of cellular activity of EGFR receptor kinase inhibitors with enzyme inhibition.
Data from Traxler et al., 1997.
It is worth noting that these examples come from very successful drug discovery projects. The quantitative discrepancies that we have emphasized, though worrying to pharmacologists, should not therefore be a serious distraction in the context of a drug discovery project.
A very wide range of physiological responses can be addressed by studies on isolated tissues, including measurements of membrane excitability, synaptic function, muscle contraction, cell motility, secretion and release of mediators, transmembrane ion fluxes, vascular resistance and permeability, and epithelial transport and permeability. This versatility and the relative technical simplicity of many such methods are useful attributes for drug discovery. Additional advantages are that concentration–effect relationships can be accurately measured, and the design of the experiments is highly flexible, allowing rates of onset and recovery of drug effects to be determined, as well as measurements of synergy and antagonism by other compounds, desensitization effects, etc.
The main shortcomings of isolated tissue pharmacology are (a) that tissues normally have to be obtained from small laboratory animals, rather than humans or other primates; and (b) that preparations rarely survive for more than a day, so that only short-term experiments are feasible.
In vivo profiling
As already mentioned, experiments on animals have several drawbacks. They are generally time-consuming, technically demanding and expensive. They are subject to considerable ethical and legal constraints, and in some countries face vigorous public opposition. For all these reasons, the number of experiments is kept to a bare minimum, and experimental variability is consequently often a problem. Animal experiments must, therefore, be used very selectively and must be carefully planned and designed so as to produce the information needed as efficiently as possible. In the past, before target-directed approaches were the norm, routine in vivo testing was often used as a screen at a very early stage in the drug discovery process, and many important drugs (e.g. thiazide diuretics, benzodiazepines, ciclosporin) were discovered on the basis of their effects in vivo. Nowadays, the use of in vivo methods is much more limited, and will probably decline further in response to the pressures on time and costs, as alternative in vitro and in silico methods are developed, and as public attitudes to animal experimentation harden. An additional difficulty is the decreasing number of pharmacologists trained to perform in vivo studies1.
Imaging technologies (Rudin and Weissleder, 2003; see also Chapter 18) are increasingly being used for pharmacological studies on whole animals. Useful techniques include magnetic resonance imaging (MRI), ultrasound imaging, X-ray densitometry tomography, positron emission tomography (PET) and others. They are proving highly versatile for both structural measurements (e.g. cardiac hypertrophy, tumour growth) and functional measurements (e.g. blood flow, tissue oxygenation). Used in conjunction with radioactive probes, PET can be used for studies on receptors and other targets in vivo. Many of these techniques can also be applied to humans, providing an important bridge between animal and human pharmacology. Apart from the special facilities and equipment needed, currently the main drawback of imaging techniques is the time taken to capture the data, during which the animal must stay still, usually necessitating anaesthesia. With MRI and PET, which are currently the most versatile imaging techniques, data capture normally takes a few minutes, so they cannot be used for quick ‘snapshots’ of rapidly changing events.
A particularly important role for in vivo experiments is to evaluate the effects of long-term drug administration on the intact organism. ‘Adaptive’ and ‘rebound’ effects (e.g. tolerance, dependence, rebound hypertension, delayed endocrine effects, etc.) are often produced when drugs are given continuously for days or weeks. Generally, such effects, which involve complex physiological interactions, are evident in the intact functioning organism but are not predictable from in vitro experiments.
The programme of in vivo profiling studies for characterization of a candidate drug depends very much on the drug target and therapeutic indication. A comprehensive catalogue of established in vivo assay methods appropriate to different types of pharmacological effect is given by Vogel (2002). Charting the appropriate course through the plethora of possible studies that might be performed to characterize a particular drug can be difficult.
A typical example of pharmacological profiling is summarized in Box 11.1. The studies were carried out as part of the recent development of a cardiovascular drug, beraprost (Melini and Goa, 2002). Beraprost is a stable analogue of prostaglandin I2 (PGI2) which acts on PGI2 receptors of platelets and blood vessels, thereby inhibiting platelet aggregation (and hence thrombosis) and dilating blood vessels. It is directed at two therapeutic targets, namely occlusive peripheral vascular disease and pulmonary hypertension (a serious complication of various types of cardiovascular disease, drug treatment or infectious diseases), resulting in hypertrophy and often contractile failure of the right ventricle. The animal studies were, therefore, directed at measuring changes (reduction in blood flow, histological changes in vessel wall) associated with peripheral vascular disease, and with pulmonary hypertension. As these are progressive chronic conditions, it was important to establish that long-term systemic administration of beraprost was effective in retarding the development of the experimental lesions, as well as monitoring the acute pharmacodynamic effects of the drug.
Box 11.1
Pharmacological profiling of beraprost
In vitro studies
Binding to PGI2 receptors of platelets from various species, including human
PGI2 agonist activity (cAMP formation) in platelets
Dilatation of arteries and arterioles in vitro, taken from various species
Increased red cell deformability (hence reduced blood viscosity and increased blood flow) in blood taken from hypercholesterolaemic rabbits
In vivo studies
Increased peripheral blood flow in various vascular regions (dogs)
Cutaneous vasodilatation (rat)
Reduced pulmonary hypertension in rat model of drug-induced pulmonary hypertension (measured by reduction of right ventricular hypertrophy)
Reduced tissue destruction (gangrene) of rat tail induced by ergotamine/epinephrine infusion
Reduction of vascular occlusion resulting from intra-arterial sodium laureate infusion in rats
Reduction of vascular occlusion and thrombosis following electrical stimulation of femoral artery in anaesthetized dogs and rabbits
Reduction of vascular damage occurring several weeks after cardiac allografts in immunosuppressed rats
Species differences
It is important to take species differences into account at all stages of pharmacological profiling. For projects based on a defined molecular target – nowadays the majority – the initial screening assay will normally involve the human isoform. The same target in different species will generally differ in its pharmacological specificity; commonly, there will be fairly small quantitative differences, which can be allowed for in interpreting pharmacological data in experimental animals, but occasionally the differences are large, so that a given class of compounds is active in one species but not in another. An example is shown in Figure 11.3, which compares the activities of a series of bradykinin receptor antagonists on cloned human and rat receptors. The complete lack of correlation means that, for these compounds, tests of functional activity in the rat cannot be used to predict activity in man.
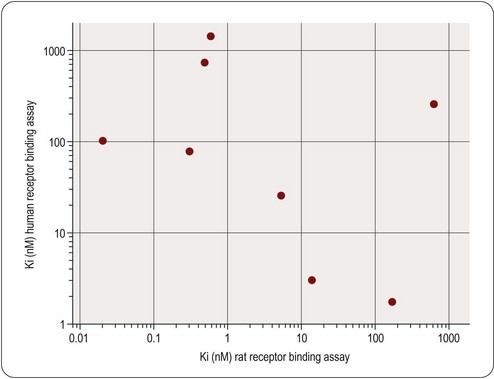
Species differences are, in fact, a major complicating factor at all stages of drug discovery and preclinical development. The physiology of disease processes such as inflammation, septic shock, obesity, atherosclerosis, etc., differs markedly in different species. Most importantly (see Chapter 10), drug metabolism often differs, affecting the duration of action, as well as the pattern of metabolites, which can in turn affect the observed pharmacology and toxicity.
Species differences are, of course, one of the main arguments used by animal rights activists in opposing the use of animals for the purpose of drug discovery. Their claim – misleading when examined critically (see Understanding Animal Research website) – is that animal data actually represent disinformation in this context. While being aware of the pitfalls, we should not lose sight of the fact that non-human data, including in vivo experiments, have actually been an essential part of every major drug discovery project to date. The growing use of transgenic animal models has led to an increase, rather than a decrease, in animal experimentation, as even breeding such animals is counted as an experiment for statistical purposes.
Animal models of disease
The animal models discussed earlier were used to investigate the pharmacodynamic effects of the drug and to answer the question: How do the effects observed at the molecular and cellular levels of organization translate into physiological effects in the whole animal?
The next, crucial, question is: Can these physiological effects result in therapeutic benefit? Animal experiments can never answer this conclusively – only clinical trials can do that – but the use of animal models of human disease provides a valuable link in the chain of evidence, and there is strong pressure on drug discovery teams to produce data of this sort as a basis for the important decision to test a new compound in man. Despite the immense range and diversity of animal models that have been described, this is often the most problematic aspect of a drug discovery project, particularly where a novel target or mechanism is involved, so that there is no mechanistic precedent among established drugs. The magnitude of the difficulties varies considerably among different therapeutic areas. Many inflammatory conditions, for example, are straightforward to model in animals, as are some cancers. Animal models of hypertension generally predict very well the ability of compounds to lower blood pressure in man. Endocrine disorders involving over- or undersecretion of particular hormones can also be simply modelled in animals. Psychiatric disorders are much more difficult, as the symptoms that characterize them are not observable in animals. In most therapeutic areas there are certain disorders, such as migraine, temporal lobe epilepsy, asthma or irritable bowel syndrome, for which animal models, if they exist at all, are far from satisfactory in predicting clinical efficacy.
Here we consider, with a few selected examples, the main experimental approaches to generating animal models, and the criteria against which their ‘validity’ as models of human disease need to be assessed.
Types of animal model
Animal models of disease can be divided broadly into acute and chronic physiological and pharmacological models, and genetic models.
Acute physiological and pharmacological models are intended to mimic certain aspects of the clinical disorder. There are many examples, including:
• Seizures induced by electrical stimulation of the brain as a model for epilepsy (see below)
• Histamine-induced bronchoconstriction as a model for asthma
• The hotplate test for analgesic drugs as a model for pain
• Injection of lipopolysaccharide (LPS) and cytokines as a model for septic shock
• The elevated maze test as a model for testing anxiolytic drugs.
Chronic physiological or pharmacological models involve the use of drugs or physical interventions to induce an ongoing abnormality similar to the clinical condition. Examples include:
• The use of alloxan to inhibit insulin secretion as a model for Type I diabetes
• Procedures for inducing brain or coronary ischaemia as models for stroke and ischaemic heart disease
• ‘Kindling’ and other procedures for inducing ongoing seizures as models for epilepsy
• Self-administration of opiates, nicotine or other drugs as a model for drug-dependence
• Cholesterol-fed rabbits as a model for hypercholesterolaemia and atherosclerosis
• Immunization with myelin basic protein as a model for multiple sclerosis
• Administration of the neurotoxin MPTP, causing degeneration of basal ganglia neurons as a model of Parkinson’s disease
• Transplantation of malignant cells into immunodeficient animals to produce progressive tumours as a model for certain types of cancer.
Details of these and many other examples of physiological and pharmacological models can be found in Vogel (2002). As discussed above, species differences need to be taken into account in the selection of animal models, and in the interpretation of results. In septic shock, for example, rodents show a much larger elevation of nitric oxide (NO) metabolites than do humans, and respond well to NO synthesis inhibitors, which humans do not. Rodents and rabbits transgenically engineered to favour cholesterol deposition nevertheless develop atherosclerosis only when fed high-cholesterol diets, whereas humans often do so even on low-cholesterol diets. Genetically obese mice are deficient in the hormone leptin and lose weight when treated with it, whereas obese humans frequently have high circulating leptin concentrations and do not respond to treatment with it. It is often not clear whether such discrepancies reflect inherent species differences, or simply failure of the model to replicate satisfactorily the predominant human disease state (see Validity criteria below).
Genetic models
There are many examples of spontaneously occurring animal strains that show abnormalities phenotypically resembling human disease. In addition, much effort is going into producing transgenic strains with deletion or over-expression of specific genes, which also exhibit disease-like phenotypes.
Long before genetic mapping became possible, it was realized that certain inbred strains of laboratory animal were prone to particular disorders, examples being spontaneously hypertensive rats, seizure-prone dogs, rats insensitive to antidiuretic hormone (a model for diabetes insipidus), obese mice and mouse strains exhibiting a range of specific neurological deficits. Many such strains have been characterized (see Jackson Laboratory website, www.jaxmice.jax.org) and are commercially available, and are widely used as models for testing drugs.
The development of transgenic technology has allowed inbred strains to be produced that over- or under-express particular genes. In the simplest types, the gene abnormality is present throughout the animal’s life, from early development onwards, and throughout the body. More recent technical developments allow much more control over the timing and location of the transgene effect. For reviews of transgenic technology and its uses in drug discovery, see Polites (1996), Rudolph and Moehler (1999), Törnell and Snaith (2002) and Pinkert (2002).
The genetic analysis of disease-prone animal strains, or of human families affected by certain diseases, has in many cases revealed the particular mutation or mutations responsible (see Chapters 6 and 7), thus pointing the way to new transgenic models. Several diseases associated with single-gene mutations, such as cystic fibrosis and Duchenne muscular dystrophy, have been replicated in transgenic mouse strains. Analysis of the obese mouse strain led to the identification of the leptin gene, which is mutated in the ob/ob mouse strain, causing the production of an inactive form of the hormone and overeating by the mouse. Transgenic animals closely resembling ob/ob mice have been produced by targeted inactivation of the gene for leptin or its receptor. Another example is the discovery that a rare familial type of Alzheimer’s disease is associated with mutations of the amyloid precursor protein (APP). Transgenic mice expressing this mutation show amyloid plaque formation characteristic of the human disease. This and other transgenic models of Alzheimer’s disease (Yamada and Nabeshima, 2000) represent an important tool for drug discovery, as there had hitherto been no animal model reflecting the pathogenesis of this disorder.
The number of transgenic animal models, mainly mouse, that have been produced is already large and is growing rapidly. Creating and validating a new disease model is, however, a slow business. Although the methodology for generating transgenic mice is now reliable and relatively straightforward, it is both time-consuming and labour-intensive. The first generation of transgenic animals are normally hybrids, as different strains are used for the donor and the recipient, and it is necessary to breed several generations by repeated back-crossings to create animals with a uniform genetic background. This takes 1–2 years, and is essential for consistent results. Analysis of the phenotypic changes resulting from the transgene can also be difficult and time-consuming, as the effects may be numerous and subtle, as well as being slow to develop as the animal matures. Despite these difficulties, there is no doubt that transgenic disease models are playing an increasing part in drug testing, and many biotechnology companies have moved into the business of developing and providing them for this purpose. The fields in which transgenic models have so far had the most impact are cancer, atherosclerosis and neurodegenerative diseases, but their importance as drug discovery tools extends to all areas.
Producing transgenic rat strains proved impossible until recently, as embryonic stem (ES) cells cannot be obtained from rats. Success in producing gene knockout strains by an alternative method has now been achieved (Zan et al., 2003), and the use of transgenic rats is increasing, this being the favoured species for pharmacological and physiological studies in many laboratories.
The choice of model
Apart from resource limitations, regulatory constraints on animal experimentation, and other operational factors, what governs the choice of disease model?
As discussed in Chapter 2, naturally occurring diseases produce a variety of structural biochemical abnormalities, and these are often displayed separately in animal models. For example, human allergic asthma involves: (a) an immune response; (b) increased airways resistance; (c) bronchial hyperreactivity; (d) lung inflammation; and (e) structural remodelling of the airways. Animal models, mainly based on guinea pigs, whose airways behave similarly to those of humans, can replicate each of these features, but no single model reproduces the whole spectrum. The choice of animal model for drug discovery purposes, therefore, depends on the therapeutic effect that is being sought. In the case of asthma, existing bronchodilator drugs effectively target the increased airways resistance, and steroids reduce the inflammation, and so it is the other components for which new drugs are particularly being sought.
A similar need for a range of animal models covering a range of therapeutic targets applies in many disease areas.
Validity criteria
Obviously an animal model produced in a laboratory can never replicate exactly a spontaneous human disease state, so on what basis can we assess its ‘validity’ in the context of drug discovery?
Three types of validity criteria were originally proposed by Willner (1984) in connection with animal models of depression. These are:
Face validity refers to the accuracy with which the model reproduces the phenomena (symptoms, clinical signs and pathological changes) characterizing the human disease.
Construct validity refers to the theoretical rationale on which the model is based, i.e. the extent to which the aetiology of the human disease is reflected in the model. A transgenic animal model in which a human disease-producing mutation is replicated will have, in general, good construct validity, even if the manifestations of the human disorder are not well reproduced (i.e. it has poor face validity).
Predictive validity refers to the extent to which the effect of manipulations (e.g. drug treatment) in the model is predictive of effects in the human disorder. It is the most pragmatic of the three and the most directly relevant to the issue of predicting therapeutic efficacy, but also the most limited in its applicability, for two main reasons. First, data on therapeutic efficacy are often sparse or non-existent, because no truly effective drugs are known (e.g. for Alzheimer’s disease, septic shock). Second, the model may focus on a specific pharmacological mechanism, thus successfully predicting the efficacy of drugs that work by that mechanism but failing with drugs that might prove effective through other mechanisms. The knowledge that the first generation of antipsychotic drugs act as dopamine receptor antagonists enabled new drugs to be identified by animal tests reflecting dopamine antagonism, but these tests cannot be relied upon to recognize possible ‘breakthrough’ compounds that might be effective by other mechanisms. Thus, predictive validity, relying as it does on existing therapeutic knowledge, may not be a good basis for judging animal models where the drug discovery team’s aim is to produce a mechanistically novel drug. The basis on which predictive validity is judged carries an inevitable bias, as the drugs that proceed to clinical trials will normally have proved effective in the model, whereas drugs that are ineffective in the model are unlikely to have been developed. As a result, there are many examples of tests giving ‘false positive’ expectations, but very few false negatives, giving rise to a commonly held view that conclusions from pharmacological tests tend to be overoptimistic.
Some examples
We conclude this discussion of the very broad field of animal models of disease by considering three disease areas, namely epilepsy, psychiatric disorders and stroke. Epilepsy-like seizures can be produced in laboratory animals in many different ways. Many models have been described and used successfully to discover new anti-epileptic drugs (AEDs). Although the models may lack construct validity and are weak on face validity, their predictive validity has proved to be very good. With models of psychiatric disorders, face validity and construct validity are very uncertain, as human symptoms are not generally observable in animals and because we are largely ignorant of the cause and pathophysiology of these disorders; nevertheless, the predictive validity of available models of depression, anxiety and schizophrenia has proved to be good, and such models have proved their worth in drug discovery. In contrast, the many available models of stroke are generally convincing in terms of construct and face validity, but have proved very unreliable as predictors of clinical efficacy. Researchers in this field are ruefully aware that despite many impressive effects in laboratory animals, clinical successes have been negligible.
Epilepsy models
The development of antiepileptic drugs, from the pioneering work of Merritt and Putnam, who in 1937 developed phenytoin, to the present day, has been highly dependent on animal models involving experimentally induced seizures, with relatively little reliance on knowledge of the underlying physiological, cellular or molecular basis of the human disorder. Although existing drugs have significant limitations, they have brought major benefits to sufferers from this common and disabling condition – testimony to the usefulness of animal models in drug discovery.
Human epilepsy is a chronic condition with many underlying causes, including head injury, infections, tumours and genetic factors. Epileptic seizures in humans take many forms, depending mainly on where the neural discharge begins and how it spreads.
Some of the widely used animal models used in drug discovery are summarized in Table 11.2. The earliest models, namely the maximal electroshock (MES) test and the pentylenetetrazol-induced seizure (PTZ) test, which are based on acutely induced seizures in normal animals, are still commonly used. They model the seizure, but without distinguishing its localization and spread, and do not address either the chronicity of human epilepsy or its aetiology (i.e. they score low on face validity and construct validity). But, importantly, their predictive validity for conventional antiepileptic drugs in man is very good, and the drugs developed on this basis, taken regularly to reduce the frequency of seizures or eliminate them altogether, are of proven therapeutic value. Following on from these acute seizure models, attempts have been made to replicate the processes by which human epilepsy develops and continues as a chronic condition with spontaneous seizures, i.e. to model epileptogenesis (Löscher, 2002; White, 2002) by the use of models that show greater construct and face validity. This has been accomplished in a variety of ways (see Table 11.2) in the hope that such models would be helpful in developing drugs capable of preventing epilepsy. Such models have thrown considerable light on the pathogenesis of epilepsy, but have not so far contributed significantly to the development of improved antiepileptic drugs. Because there are currently no drugs known to prevent epilepsy from progressing, the predictive validity of epileptogenesis models remains uncertain.
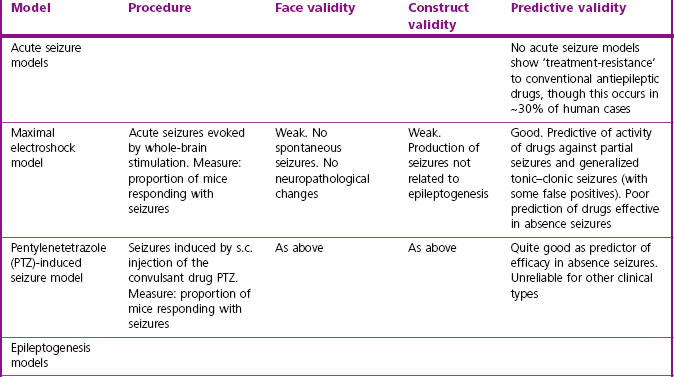
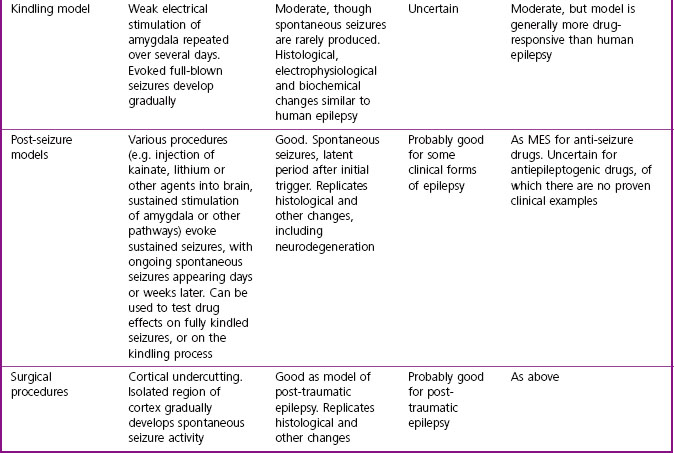
Psychiatric disorders
Animal models of psychiatric disorders are in general problematic, because in many cases the disorders are defined by symptoms and behavioural changes unique to humans, rather than by measurable physiological, biochemical or structural abnormalities. This is true in conditions such as schizophrenia, Tourette’s syndrome and autism, making face validity difficult to achieve. Depressive symptoms, in contrast, can be reproduced to some extent in animal models (Willner and Mitchell, 2002), and face validity is therefore stronger. The aetiology of most psychiatric conditions is largely unknown2, making construct validity questionable.
Models are therefore chosen largely on the basis of predictive validity, and suffer from the shortcomings mentioned above. Nonetheless, models for some disorders, particularly depression, have proved very valuable in the discovery of new drugs. Other disorders, such as autism and Tourette’s syndrome, have proved impossible to model so far, whereas models for others, such as schizophrenia (Lipska and Weinberger, 2000; Moser et al., 2000), have been described but are of doubtful validity. The best prediction of antipsychotic drug efficacy comes from pharmacodynamic models reflecting blockade of dopamine and other monoamine receptors, rather than from putative disease models, with the result that drug discovery has so far failed to break out of this mechanistic straitjacket.
Stroke
Many experimental procedures have been devised to produce acute cerebral ischaemia in laboratory animals, resulting in long-lasting neurological deficits that resemble the sequelae of strokes in humans (Small and Buchan, 2000). Interest in this area has been intense, reflecting the fact that strokes are among the commonest causes of death and disability in developed countries, and that there are currently no drugs that significantly improve the recovery process. Studies with animal models have greatly advanced our understanding of the pathophysiological events. Stroke is no longer seen as simple anoxic death of neurons, but rather as a complex series of events involving neuronal depolarization, activation of ion channels, release of excitatory transmitters, disturbed calcium homeostasis leading to calcium overload, release of inflammatory mediators and nitric oxide, generation of reactive oxygen species, disturbance of the blood–brain barrier and cerebral oedema (Dirnagl et al., 1999). Glial cells, as well as neurons, play an important role in the process. Irreversible loss of neurons takes place gradually as this cascade builds up, leading to the hope that intervention after the primary event – usually thrombosis – could be beneficial. Moreover, the biochemical and cellular events involve well-understood signalling mechanisms, offering many potential drug targets, such as calcium channels, glutamate receptors, scavenging of reactive oxygen species and many others. Ten years ago, on the basis of various animal models with apparently good construct and face validity and a range of accessible drug targets, the stage seemed to be set for major therapeutic advances. Drugs of many types, including glutamate antagonists, calcium and sodium channel blocking drugs, anti-inflammatory drugs, free radical scavengers and others, produced convincing degrees of neuroprotection in animal models, even when given up to several hours after the ischaemic event. Many clinical trials were undertaken (De Keyser et al., 1999), with uniformly negative results. The only drug currently known to have a beneficial – albeit small – effect is the biopharmaceutical ‘clot-buster’ tissue plasminogen activator (TPA), widely used to treat heart attacks. Stroke models thus represent approaches that have revealed much about pathophysiology and have stimulated intense efforts in drug discovery, but whose predictive validity has proved to be extremely poor, as the drug sensitivity of the animal models seems to be much greater than that of the human condition. Surprisingly, it appears that whole-brain ischaemia models show better predictive validity (i.e. poor drug responsiveness) than focal ischaemia models, even though the latter are more similar to human strokes.
Good laboratory practice (GLP) compliance in pharmacological studies
GLP comprises adherence to a set of formal, internationally agreed guidelines established by regulatory authorities, aimed at ensuring the reliability of results obtained in the laboratory. The rules (see GLP Pocketbook, 1999; EEC directives 87/18/EEC, 88/320/EEC, available online: pharmacos.eudra.org/F2/eudralex/vol-7/A/7AG4a.pdf) cover all stages of an experimental study, from planning and experimental design to documentation, reporting and archiving. They require, among other things, the assignment of specific GLP-compliant laboratories, certification of staff training to agreed standards, certified instrument calibration, written standard operating procedures covering all parts of the work, specified standards of experimental records, reports, notebooks and archives, and much else. Standards are thoroughly and regularly monitored by an official inspectorate, which can halt studies or require changes in laboratory practice if the standards are thought not to be adequately enforced. Adherence to GLP standards carries a substantial administrative overhead and increases both the time and cost of laboratory studies, as well as limiting their flexibility.
The regulations are designed primarily to minimize the risk of errors in studies that relate to safety. They are, therefore, not generally applied to pharmacological profiling as described in this chapter. They are obligatory for toxicological studies that are required in submissions for regulatory approval. Though not formally required for safety pharmacology studies, most companies and contract research organizations choose to do such work under GLP conditions.
Current protocols in pharmacology. New York: John Wiley and Sons. [Published as looseleaf binder and CD-ROM, and regularly updated.)
De Keyser J, Sulter G, Luiten PG. Clinical trials with neuroprotective drugs in ischaemic stroke: are we doing the right thing? Trends in Neurosciences. 1999;22:535–540.
Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends in Neurosciences. 1999;22:391–397.
Dziadulewicz EK, Ritchie TJ, Hallett A, et al. Nonpeptide bradykinin B2 receptor antagonists: conversion of rodent-selective bradyzide analogues into potent orally active human bradykinin B2 receptor antagonists. Journal of Medicinal Chemistry. 2002;45:2160–2172.
Enna S, Williams M, Ferkany JW, et al. Current protocols in pharmacology. Hoboken, NJ: Wiley, 2003.
GLP Pocketbook. London: MCA Publications; 1999.
Hall JM. Bradykinin receptors: pharmacological properties and biological roles. Pharmacology and Therapeutics. 1992;56:131–190.
Heidempergher F, Pillan A, Pinciroli V, et al. Phenylimidazolidin-2-one derivatives as selective 5-HT3 receptor antagonists and refinement of the pharmacophore model for 5-HT3 receptor binding. Journal of Medicinal Chemistry. 1997;40:3369–3380.
Keen M, ed. Receptor binding techniques. Totowa, NJ: Humana Press, 1999.
Kenakin T. The measurement of efficacy in the drug discovery agonists selection process. Journal of Pharmacologic and Toxicologic Methods. 1999;42:177–187.
Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: schizophrenia as a reality test. Neuropsychopharmacology. 2000;23:223–239.
Löscher W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Research. 2002;50:105–123.
Melini EB, Goa KL. Beraprost: a review of its pharmacology and therapeutic efficacy in the treatment of peripheral arterial disease and pulmonary hypertension. Drugs. 2002;62:107–133.
Morfis M, Christopoulos A, Sexton PM. RAMPs: 5 years on, where to now? Trends in Pharmacological Sciences. 2003;24:596–601.
Moser PC, Hitchcock JH, Lister S, et al. The pharmacology of latent inhibition as an animal model of schizophrenia. Brain Research Reviews. 2000;33:275–307.
Pinkert CA. Transgenic animal technology, 2nd ed. San Diego, CA: Academic Press; 2002.
Polites HG. Transgenic model applications to drug discovery. International Journal of Experimental Pathology. 1996;77:257–262.
Research Defense Society website. www.rds-online.org.uk/ethics/arclaims. – a reasoned rebuttal of the view put forward by opponents of animal experimentation that the use of such experiments in drug discovery is at best unnecessary, if not positively misleading
Rudin M, Weissleder R. Molecular imaging in drug discovery and development. Nature Reviews Drug Discovery. 2003;2:122–131.
Rudolph U, Moehler H. Genetically modified animals in pharmacological research: future trends. European Journal of Pharmacology. 1999;375:327–337.
Small DL, Buchan AM. Stroke: animal models. British Medical Bulletin. 2000;56:307–317.
Törnell J, Snaith M. Transgenic systems in drug discovery: from target identification to humanized mice. Drug Discovery Today. 2002;7:461–470.
Traxler P, Bold G, Frei J, et al. Use of a pharmacophore model for the design of EGF-R tyrosine kinase inhibitors: 4-(phenylamino)pyrazolo[3,4-d]pyrimidines. Journal of Medicinal Chemistry. 1997;40:3601–3616.
Vogel WH. Drug discovery and evaluation: pharmacological assays. Heidelberg: Springer-Verlag; 2002.
White HS. Animal models of epileptogenesis. Neurology. 2002;59:S7–14.
Willner P. The validity of animal models of depression. Psychopharmacology. 1984;83:1–16.
Willner P, Mitchell PJ. The validity of animal models of predisposition to depression. Behavioural Pharmacology. 2002;13:169–188.
Yamada K, Nabeshima T. Animal models of Alzheimer’s disease and evaluation of anti-dementia drugs. Pharmacology and Therapeutics. 2000;88:93–113.
Yang D, Soulier J-L, Sicsic S, et al. New esters of 4-amino-5-chloro-2-methoxybenzoic acid as potent agonists and antagonists for 5-HT4 receptors. Journal of Medicinal Chemistry. 1997;40:608–621.
Zan Y, Haag JD, Chen KS, et al. Production of knockout rats using ENU mutagenesis and a yeast-based screening assay. Nature Biotechnology. 2003;21:645–651.