Chapter 9 The role of medicinal chemistry in the drug discovery process
Introduction
Clinically the unmet need for novel and safe drugs is becoming more significant due to increasing longevity in the developed world and the consequential increased prevalence of age-related diseases such as cancer and dementia. Whilst few would dispute that the role of the medicinal chemist is pivotal to the discovery of new medicines to address this growing need, and will continue to be so for the foreseeable future, the environment in which the medicinal chemist operates has changed dramatically over the last 5–10 years. Despite many years of rising investment in R&D, the productivity of major Pharma, in terms of New Chemical Entities (NCEs) delivered to the patient, has not improved over the past 10 years. In 2009 only nineteen NCEs were approved by the FDA, two less than in 2008 (Hughes, 2010) (Figure 9.1; see also Chapter 22). The approval of six biological licence applications in 2009, compared with three in 2008, is perhaps the first indication of the greater emphasis which Pharma is placing on the discovery of biological therapeutic agents. However, the challenges and current limitations of such treatments are such that small molecule discovery is likely to remain the mainstay of therapeutic innovation for at least the next 20 years (but see also Chapter 22).
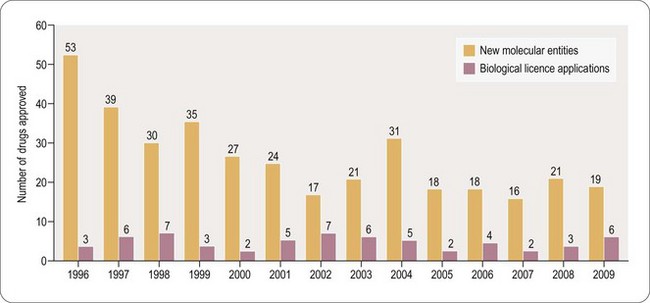
Fig. 9.1 Recent trends in small molecule approvals and biological licience applications –
reproduced with the kind permission of Nature publishing.
Whilst a detailed discussion of the reasons behind Pharma’s poor level of productivity is beyond the scope of this chapter (see Chapter 22), it is clear that the high level of attrition in the clinic, due to lack of clinical efficacy and insufficient safety margins, is a major component and it is appropriate to consider what role the medicinal chemist might play in addressing this challenge in the future. The high level of attrition in the clinic due to lack of sufficient efficacy reflects an inadequate understanding of disease pathophysiology in man and the poor predictability of many of the preclinical animal models of human disease. Thus there is a pressing need to improve the quality of validation of novel targets, placing less emphasis on preclinical in vivo models and single gene ‘knock-outs’, and a greater emphasis on cellular pathways, genetically associated targets and innovative clinical trial design. This approach will require the availability of selective chemical tools with which to validate targets in human tissue and thus the role of the medicinal chemist in the future may incorporate a greater component of chemical biology. The increasing trend towards academic target and drug discovery is also likely to place greater emphasis on chemical biology skills.
Despite stringent toxicity evaluation during the discovery phase, an unacceptable level of failure due to safety issues remains. The present situation is arguably worse than 20 years ago as there has been a trend towards molecules failing later in the safety evaluation process, incurring greater cost and time. If significant impact is to be made on this source of attrition it will be important for the medicinal chemist to develop a rational understanding of possible structural and physicochemical determinants of toxicity in much the same way as the determinants of oral bioavailability have been recognized. However, this objective is likely to be much more challenging due to the diverse and complex nature of the multiple pathologies. Preliminary studies to evaluate this approach have been recently reported and will be discussed later in the chapter.
It is clear that addressing the challenges described above and raising productivity will require long-term investment and persistence. However, due to the immediate financial pressures, the industry has responded to the productivity challenge by minimizing costs, shifting focus to cheaper sources of chemistry, predominantly in the Far East, and in-licensing assets. Thus today’s medicinal chemist is required to optimally integrate a network of both internal and external resources to prosecute the identification of development candidates.
Whilst medicinal chemistry is facing unprecedented pressures due to the volatility of the sector, it is clear that small molecule therapeutics will be required for the foreseeable future and that, whatever infrastructure evolves within the industry, the medicinal chemist will have a key role to play. This chapter will discuss some of the recent scientific advances in the field which will enable the medicinal chemist to rise to the challenges ahead.
Target selection and validation

A key challenge in increasing the industry’s productivity will be the selection of targets which have compelling validation in terms of both efficacy and safety in the human context and, which, consequently, have a greater probability of achieving a successful clinical proof of concept. The role of the medicinal chemist has previously focused primarily on ensuring the quality of preclinical candidate molecules in terms of potency, selectivity and physicochemical properties such as those highlighted initially by Lipinski et al. (Lipinski CA, 1997) and, more recently, by others (Leeson PD and Springthorpe B, 2007). As a result, the level of attrition due to poor ADME properties fell dramatically between 1991 and 2000 (Kola I and Landis J, 2004), but the level of clinical success with new mechanisms remains poor. However, chemists are now increasingly becoming engaged, along with their biology and clinical counterparts, in the process of selecting and prioritizing future protein targets. In particular the development of an in-depth understanding of the structural biology of the target and the biophysics of its interaction with low-molecular-weight ligands is a critical component at the outset of a project and very much in the chemist’s domain. The necessity for a rigorous analysis of potential targets cannot be over-emphasized since this occurs at the start of the 12–15-year period of intensive effort and investment required to achieve the launch of a new medicine into large scale clinical usage. The post genomic era has provided the industry with a plethora of potential drug targets; however, the selection of tractable targets with a high probability of delivering safe and effective treatments represents a huge challenge requiring a multidisciplinary approach.
Whilst a detailed discussion of the complex facets of target validation is beyond the scope of this chapter (see Chapter 6), it is clear that targets with strong genetic associations, such as the voltage-gated sodium channel NaV1.7 (Dib-Hajj SD et al., 2007), with potential utility in the treatment of pain, and the chemokine CCR5 receptor (Westby M and van der Ryst E, 2005), which has led to the discovery of treatments for HIV infection, exemplify an aspirational level of target validation.
Whilst genetic disease-association data provide strong evidence of target involvement, target modulation, ideally within a human cellular pathway, provides compelling validation, and the identification of early chemical probes in addition to subsequent lead molecules will be an important challenge for the medicinal chemist in the future.
Frye has recently described (Frye SV, 2010) essential features of a quality chemical probe which are summarised below:
Molecular profiling. Sufficient in vitro potency and selectivity data to confidently associate its in vitro profile to its cellular or in vivo profile.
Mechanism of action. Activity in a cell-based or cell-free assay influences a physiologic function of the target in a dose-dependent manner.
Identity of the active species. Has sufficient chemical and physical property data to interpret results as due to its intact structure or a well-characterized derivative.
Proven utility as a probe. Cellular activity data available to confidently address at least one hypothesis about the role of the molecular target in a cell’s response to its environment.
Availability. Is readily available to the academic community with no restrictions on use.
The first four criteria attempt to describe ideal properties of a probe whilst the last point addresses a more philosophical viewpoint on the sharing of precompetitive information. Further evolution of this ‘wish list’ will inevitably take place in the future.
It is well known that target classes are associated with different levels of tractability with respect to the probability of being able to identify quality lead molecules. For example the G-protein coupled receptor (GPCR) super-family is generally considered to be one of the most tractable target classes with approximately 40% of prescription drugs in current practice producing efficacy via modulation of a GPCR. In contrast, voltage-gated ion channels are generally regarded as one of the more challenging variety of protein targets to modulate, particularly with respect to subunit specificity. Progress in this area was for many years limited by lack of suitable screening platforms to facilitate drug discovery; however, recent technological developments offer the potential to more fully explore this target class (Kaczorowski GJ et al., 2008).
Various criteria for the selection and prioritization of targets are utilized within major Pharma in the process of building sustainable portfolios of viable potential targets for the discovery scientists to address. A recent illustration (Wehling M, 2009) has highlighted one such approach which objectively applies weighted scores to available target information.
With the emergence of exciting new insights into disease pathologies, the importance of rigorous target selection will become even more important in the future. The availability of the human genome sequence coupled with impressive advances in biology is uncovering challenging targets for the medicinal chemist to address. As an example, epigenetic phenomena are increasingly being recognized as a potentially important area in the development of chronic diseases (Gluckman PD et al., 2008) and the discovery and characterization of specific histone-modifying enzyme subfamilies (Cole PA, 2008) offers the medicinal chemist the opportunity to design specific enzyme modulators with which to regulate gene expression. The availability of selective chemical probes for specific domains within these enzyme classes will be a critical factor in the identification and selection of the most relevant targets. The promise of this area is illustrated by the successful discovery of the histone deacetylase (HDAC) inhibitor Vorinostat (McGuire C and Lee J, 2010; Figure 9.2).
Historically natural products have been considered as a source of both new drugs and potential targets. More recently this approach has suffered demise due to its disappointing productivity. However, there have been a number of recent success stories which are illustrated by the following examples. Ziconotide (Prialt®); (Schmidtko A et al., 2010) is a potent and selective N-type calcium channel blocker approved by the FDA in December 2004 for the treatment of severe chronic pain. Ziconotide has subsequently demonstrated efficacy in patients with refractory pain, i.e. pain which has proven difficult to control using conventional analgesics. Ziconotide is a synthetic peptide derived from a toxin extracted from the marine snail Conus magus. The success of ziconotide has highlighted the potential of natural toxins in drug discovery and has stimulated renewed interest from a number of groups in the area (Halai R and Craik DJ, 2009). Peptides offer high levels of both potency and selectivity which make them valuable tools for target validation and the toxins of a number of venomous species have proved to be a rich source of such molecules. The identification of the endogenous peptide hormone ghrelin (Helstroem PM, 2009), and subsequent understanding of its function, has highlighted the potential clinical value of ghrelin receptor agonists in a number of conditions such as gastroparesis (Ejskjaer N et al., 2009) and postoperative ileus (Popescu I, Fleschner PR et al., 2010) in which the ghrelin receptor agonist TZP-101 (Figure 9.3) has recently demonstrated clinical efficacy. Several of the major disease challenges facing society, such as Alzheimer’s disease, cancer and heart disease, etc., have been shown to be associated, at least in part, with aberrant protein folding and/or protein–protein interactions and the identification of both small molecule and peptidic therapeutics to address these pathologies will require significant innovation.
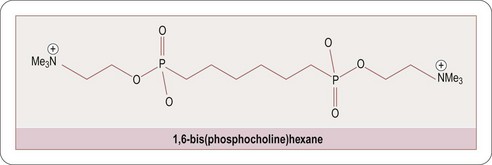
Encouragingly great strides have recently been made in our understanding of the factors which cause proteins to misfold, primarily through the use of biophysical and computational techniques that enable systematic and quantitative analysis of the effects of a range of different perturbations in proteins (Luheshi LM et al., 2008). Recent advances in the design of small molecule inhibitors which inhibit protein–protein interactions has been amply demonstrated by the discovery of the serum amyloid P cross-linking agent CPHPC (Pepys MB et al., 2002; Figure 9.4) and the C-reactive protein inhibitor 1,6-bis(phosphocholine)-hexane (Pepys MB et al., 2006; Figure 9.5).
It is clear from the above selected examples that there is likely to be a plethora of novel biological targets to challenge the innovation of medicinal chemists for the foreseeable future.
Lead identification/generation

The identification of high-quality lead molecules is a critical phase in the discovery of drug candidates since decisions made at this point in the process are likely to have a significant impact on the outcome of the project as it sets the framework for future lead optimization and development (Bleicher KH et al., 2003).
Reducing attrition has already been alluded to as a major challenge in the discovery of new therapeutic agents and the lead generation phase is the earliest point in the drug discovery process where the incorporation of appropriate physicochemical properties can be addressed. It is important to establish a clear target lead profile at this early stage of the process which takes into account the critical properties required of the ultimate preclinical candidate and extrapolates back into an appropriate minimum acceptable profile for a lead series. Such a profile should not simply take into account target potency and selectivity but should also include consideration of a wide range of physicochemical characteristics which would facilitate favourable ADMET properties later in the process.
The initial aim at the lead identification/generation stage is to identify ‘hits’ which are molecules that interact with the chosen biological target in an initial screen, often carried out at a high concentration, to elicit a measurable response in a reproducible assay. Once validated by establishing the molecular integrity of the hit molecule(s), and confirming the robustness of the biological response, a ‘validated hit’ may be further investigated to establish whether it possesses the potential to be regarded as a lead series.
Several strategies have been adopted to identify these early hits as listed below:
The widespread use of high-throughput screening has generally been reported to have enabled the identification of leads for approximately 50% of the projects addressed by this approach (Fox S et al., 2006), although a more recent review suggests that, over the past 3 years, the success rate may have increased to approximately 60% (Macarron R et al., 2011). However, it is noteworthy that, despite the massive growth in the numbers of compounds screened over the past 20 years, no corresponding increase in the number of successful registered NCEs has resulted. It has been argued that, in view of increasing development times, it is still too early to evaluate the true success of high-throughput screening (Macarron R et al., 2011). Nevertheless, there has been greater focus on improving the ‘drug and lead-like’ properties and the structure/property diversity of screening collections. ‘Drug-likeness’ can be broadly defined as the overall profile of biophysicochemical properties of a molecule which facilitate its access to and effective mode of interaction at the site of action at a biologically relevant and safe concentration for sufficient duration to elicit the desired therapeutic effect. Leeson and Springthorpe discuss the importance of considering the properties conferring ‘drug-likeness’, particularly lipophilicity, in a recent survey of selected development candidates in leading drug companies (Leeson PD and Springthorpe B, 2007). An analysis of the structural relationship of launched drugs to their corresponding lead molecules revealed that in general the structure of the marketed molecule was very closely related to the lead series (Proudfoot JR, 2002). Thus one could infer that the apparent disappointing success rate of high-throughput screening has been related to the lack of ‘drug likeness’ in the compound collections and significant effort is now being devoted to address this.
Major companies have, therefore, filtered their screening collections to exclude compounds which do not possess physicochemical properties that are generally accepted to confer ‘drug-like’ attributes (Bleicher KH et al., 2003). However, it should be noted that, although the discovery of drug-like preclinical development candidates requires the initial identification of quality leads, the properties which confer ‘drug-likeness’ and those associated with ‘lead-likeness’ are not the same (Rishton GM, 2003). It is, therefore, important to understand the components conferring ‘lead-likeness’ when assessing screening collections. A publication from the Astra-Zeneca group addressed the design of lead-like combinatorial libraries and suggested that leads suitable for further optimization are most likely to be relatively polar, low molecular weight (MW = 200–350) and of relatively low lipophilicity (clogP ca.1.0–3.0) (Oprea TI et al., 2001). The authors point out that when beginning from such a tractable low-molecular-weight lead molecule, subsequent optimization to increase potency and selectivity is likely to increase molecular weight and lipophilicity to values in the order of the accepted ‘drug-likeness’ parameters.
Whilst significant progress is being made in improving the quality of compound collections with respect to the enrichment of ‘lead-like’ components and the elimination of potentially toxic or undesirable functionality, improving chemical diversity is an ongoing preoccupation since the concept of diversity is open to a variety of interpretations. Whilst in its original conception combinatorial chemistry aimed to generate large numbers of molecules providing a high level of diversity, the early libraries tended to be bolstered with molecules having significant molecular complexity and only modest diversity due to the limitations of the synthetic methodologies which could be utilized in a high-throughput mode. Furthermore, compound collections with less than optimal physicochemical properties were previously employed in high-throughput screening campaigns. Thus more attention is now being paid to the generation of focused libraries containing subsets of compounds having so-called ‘privileged scaffolds’ for specific target classes. This has in turn shifted the emphasis from combinatorial synthesis to rapid automated synthesis of small quantities of target compounds.
High-throughput screening (Chapter 8) continues to be a valuable strategy where there is little information around the structure of the biological target or its ligands; however, where such knowledge exists, it has generally been more successful to utilize this information to identify potential molecular starting points. For example, structural information on ligands which are known to interact with a target can be used to construct a pharmacophore, which may be used in an initial virtual screening programme of proprietary or commercial compound collections to enrich the sample set of compounds ultimately selected for actual screening. A critical review assessing the past effectiveness of virtual screening and proposing considerations for future improvements has recently appeared (Schneider G, 2010). Computational tools are widely used to generate in silico pharmacophores, using either molecular or electronic overlays. The introduction of electronic field overlay technology has allowed chemists to interrogate electronic properties in a relatively accessible manner. Similarly the latter technology may be usefully applied to ‘scaffold hopping’ where a structurally distinct compound class has been derived from an active series via application of a field pharmacophore (Cheeseright TJ et al., 2008). An extreme example of utilizing structural information is where an existing known drug molecule is used as the starting point to discover an improved agent (Naik P et al., 2010).
Although the majority of high-throughput screens are aimed at known biological targets with supporting target validation in the disease of interest, there has been significant activity in establishing screens based on the cell phenotype, i.e. where biochemical pathways are targeted as opposed to specific proteins within the pathway. The major challenge associated with this approach is the subsequent identification of the specific molecular target or targets with which the agents interact to produce the desired cellular response (Hart CP, 2005). Whilst in some cases further optimization of leads has been carried using the phenotypic assay itself, because of the numerous variables involved, e.g. multiple targets and cellular distribution, etc., precise SAR can be difficult to obtain from the phenotypic assay. Thus a detailed analysis and elucidation of the mechanism of action is often required to enable rapid progress to be achieved using a protein specific assay.
Over the past few years fragment-based drug discovery has become an established technique for lead generation, often in conjunction with other strategic approaches (Whittaker M et al., 2010). Fragment-based screening consists of screening a chemical library of low-molecular-weight compounds, usually at high concentration, using sensitive assay methods. In addition to biochemical assays such campaigns often incorporate biophysical methodology such as NMR, surface plasmon resonance (SPR), isothermal titration calorimetry (ITC) and X-ray crystallography (Carr R and Jhoti H, 2002; Rees DC et al., 2004; Orita M et al., 2009). The considerable progress achieved with in silico design of fragment libraries using a variety of techniques has recently been reviewed (Konteatis ZD, 2010). Essentially the fragment-based approach aims to identify ligands for the target protein which have relatively low affinity but high ligand/ligand-lipophilicity efficiency. Ligand efficiency (LE) is a measure of the binding energy of a molecule normalized by its size. This is often calculated by dividing the pKi or IC50 by the molecular weight or number of heavy atoms. Similarly ligand-lipophilicity efficiency (LLE) can be calculated by determining the difference between the pKi or pIC50 value and the clogP, which provides a measure of the binding energy of a molecule normalized by its lipophilicity (Smith GF, 2009). Thus compounds with high LE and LLE interact highly efficiently with the biological target. Subsequent optimization is then focused on increasing potency whilst maintaining high LE/LLE, thus avoiding the pitfall of increasing potency by increasing size and lipophilicity which often leads to promiscuous ‘off-target’ interactions and subsequent toxicity. Although the concepts of LE and LLE have been particularly applied in fragment-based design they are now widely applied across all lead generation strategies. There are now numerous examples of a fragment-base approach being employed in drug discovery projects and several have been reviewed recently (Congreve M, Chessari G et al., 2008).
In stark contrast to the fragment-based approach to lead generation, natural products have been, and arguably continue to be, a fruitful source of lead molecules and drugs over the past 25–30 years (Newman DJ and Cragg JM, 2007). However, despite the demonstrable historical successes of natural product research, major Pharma has largely reduced its investment in natural product screening in favour of the small molecule library approaches described above. The anticipation that combinatorial chemistry/automated synthesis would provide an excess of small molecule leads for most biological targets was partly responsible for the demise of this strategy. Furthermore the technical challenges associated with the deconvolution of multicomponent natural product mixtures and subsequent chemical simplification of large and complex structures has also deterred continued investment in this field. Nevertheless a number of smaller companies have recognized this as an opportunity and are pursuing this strategy for lead identification and it has recently been argued that the technical challenges associated with this approach have been lessened (Harvey AI, 2008; Harvey AI, 2010).
Despite the relative demise of natural product screening over the past few years, natural products have inspired the development of new strategies in the identification of privileged structures from which small molecule libraries have been constructed. Herbert Waldmann has received notable acclaim for his biology-oriented synthesis (BIOS) strategy which encompasses the development of small molecular entities derived from natural product and drug families, the so-called ‘structural classification of natural products’ (SCONP) (Keiser M et al., 2008) and an associated ‘scaffold hunter’ approach (Wetzel S et al., 2009).
X-ray crystallography has been effectively applied over many years to optimize small molecule ligands primarily for soluble enzyme targets. With the introduction of fragment-based screening high-throughput X-ray crystallography has over the past decade also assumed an important role in the identification of lead molecules (Carr R and Jhoti H, 2002). Similarly, NMR is proving to be an effective, accessible, complementary technique in identifying ligand-protein interactions particularly for membrane bound proteins such as GPCRs, for which detailed X-ray structures are relatively rare (Bartoschek S et al., 2010).
Recent developments in the acquisition of bioengineered protein and subsequent X-ray crystal structures of stabilized GPCRs (Tate GC and Schertler GFX, 2009) may provide exciting future opportunities for medicinal chemists to actively use ligand-bound co-crystal structures of GPCRs in an analogous fashion to the enzyme co-crystallization structures which have greatly facilitated the discovery of selective enzyme inhibitors.
Industry pressure to improve productivity and efficiency in the identification of new chemical entities has added stimulus to the exploration of new technologies in the identification of lead molecules. In particular miniaturization and automation of chemical synthesis and biological screening assays are currently under intense focus (Lombardi D and Dittrich PS, 2010).
The identification of screening platforms which obviate the requirement for artificial labels or reporter systems is also receiving considerable attention (Shiau AK et al., 2008). Biophysical methods such as surface-plasmon resonance and impedance-based technologies are developing rapidly and will become more widely used, particularly since these approaches are less likely to identify false positives which result from labelling artefacts and aggregation phenomena (Giannetti AM et al., 2008). The successful development of these new technologies has the potential to transform the lead identification process and address, in part, the challenge of improving productivity.
Lead optimization
The lead optimization stage of drug discovery is at the pivotal interface between lead identification and the early development phase of a molecule. The ultimate aim of the lead optimization phase is the identification of a compound which possesses the required properties of the targeted preclinical development candidate. Thus the medicinal chemist must be cognizant of the wide range of parameters which will need to be optimized in the lead thus ensuring a high probability of success during subsequent development. It is important that the desired candidate profile is clearly defined as early as possible, ideally before the initiation of a programme, thus giving the medicinal chemist and the programme team a clear focus. From the candidate profile a focused screening strategy can be developed and should only contain screens which facilitate decision-making. Careful consideration should be given to the components and order of assays within the screening cascade which should include higher throughput assays, for which high attrition can be expected, early in the triage and lower throughput and more discerning assays, including those involving animals, later in the cascade. A major consideration in constructing an effective screening cascade is the choice of selectivity assays which need to be included. The selectivity panel normally consists of proteins both within the target family and selected ‘off-target’ proteins which, if modulated, would result in undesirable toxicity or side effects, discussed in more detail below.
In addition to fulfilling potency and selectivity criteria a potential development candidate should possess appropriate ADME properties to deliver acceptable exposure of the compound at the target site by the preferred route of administration allowing data obtained from subsequent in vivo profiling in preclinical disease models to be interpreted with confidence and facilitate predictions for future clinical efficacy. Much of this section will focus on molecules intended for oral administration, but a brief discussion on the different requirements of drugs intended for administration via other routes will also be included.
A number of excellent reviews have recently appeared which describe the lead optimization process in detail (Baringhaus KH and Matter H 2005; Lindsley GW et al., 2009). This section will focus on recent discoveries and developments that have enhanced or have the potential to further improve the traditional process.
The process commences with the careful selection of lead series which, as previously stated, is of great importance since this selection has consequences for the future lengthy and expensive discovery and development programme. It is essential that great attention is paid to selecting robust leads with the appropriate physicochemical properties. Recent analyses (Kola I and Landis J, 2004; Keserü GM and Makara GM, 2009) have clearly shown that leads chosen from the ‘wrong’ area of chemical space have a much higher probability of failure than those from the ‘correct’ area. Often failure occurs late in development and can be extremely costly. Far too often medicinal chemists have been seduced by high levels of potency in ‘non drug-like’ lead molecules only to find that the physical properties of the molecule are incompatible with the desired candidate profile.
Lipinski has highlighted (Lipinski CA et al., 1997) the importance of the physical properties of a molecule and, whilst this paper is often quoted, the guidelines contained within are too often disregarded.
It is important to measure key physical properties such as logD, solubility and pKa (where appropriate), etc., of novel compounds throughout the drug discovery process to ensure that molecules continue to reside within ‘drug-like’ chemical space and to regularly analyse data to investigate potential correlations between physicochemical properties, ADME parameters and biological activities (see also Chapter 10).
In addition to having the desired physicochemical properties a lead molecule should also have the following characteristics: (a) be a member of a distinct structural series with confirmed structure–activity relationships, (b) show evidence of the desired selectivity profile, (c) have activity in cellular systems if necessary, (d) have acceptable metabolic stability in vitro, (e) be free of any structural toxicity alerts, and (f) not have any major cytotoxicity issues. It is highly advantageous for a lead to demonstrate some evidence of oral exposure even at this early stage.
It is important also to consider how many lead series should be investigated in the early stages of the optimization process and the requisite level of structural diversity in order to mitigate against failure of a particular series. Ideally three structurally distinct lead series would be selected for lead optimization at the commencement of the process.
A lead molecule will have a predefined level of potency at the chosen target together with the initial target selectivity for both related targets and common ‘off-target’ liabilities which are considered important to avoid. The primary assays in most screening cascades focus on activity at the target protein and selectivity to allow the medicinal chemist to quickly optimize these important parameters early in the process. At this stage rudimentary structure–activity relationships will have already been established during the lead identification phase providing an insight as to which moieties of the lead compound may be modified to achieve the desired result. Furthermore the availability of other information such as protein X-ray crystal structures or docking predictions from target protein homology models can provide strategic guidance for the optimization process. In addition to considering selectivity within the target protein family it is important to consider selectivity more broadly (Bass AS et al., 2009) since a growing number of ‘liability targets’ are being identified as being potentially responsible for safety issues which are often only detected later in the development process.
Screening compounds against recombinant proteins has become widely accepted since this allows the configuration of robust high-throughput assays, which perform consistently over time. However, caution should be exercised when interpreting data generated from such assays since the biological activity data from these assays do not necessarily correlate well with activity from native tissue systems (Eglen RM et al., 2008). It is therefore important, where feasible, to periodically interrogate the relationship between activities derived from each assay system. Ideally the native tissue used should be closely related to the target tissue and from the relevant disease situation wherever possible.
A further consideration when designing a screening cascade is the potential for species differences to exist in which the biological activity at the target protein varies between different species. Species differences can lead to problems in the extrapolation of in vitro to in vivo data and the interpretation of data from in vivo efficacy and safety studies (Swanson R and Beasley JR, 2010). Whilst an indication of potential species differences can be obtained by performing a bioinformatics analysis early in the project, differences in activity can occur even when there is a high degree of homology between proteins. It is therefore important to consider the incorporation of animal orthologue assays in the screening cascade which would typically be the species of choice for the primary animal model.
Much of the focus in lead optimization is on optimization of ADME parameters which are clearly important to (a) select the most appropriate compounds for evaluation in in vivo disease models, (b) ensure that the drug is optimally delivered to its intended site of action, (c) allow a prediction of the human pharmacokinetics and estimation of clinical dose, (d) minimize the potential for drug–drug interactions, (e) avoid selecting compounds with the potential to form reactive metabolites and thus reduce the potential for idiosyncratic toxicity, and (f) provide confidence that safety studies can be performed in several species.
Several in vitro ADME screens feature early in virtually all screening strategies. The introduction of cytochrome P450 (CYP450) inhibition assays has allowed potent inhibitors of these key metabolizing enzymes to be identified early in the optimization process. The elucidation of crystal structures of a number of CYP450 enzymes has alerted medicinal chemists to specific chemical modifications with which to mitigate such liabilities from candidate molecules, thus reducing the risk of drug–drug interactions in the clinic (Ekroos M and Sjögren T, 2006; Foti RS et al., 2010). For most CYP450 isoforms a strong correlation exists between the lipophilicity of a compound and its CYP450 inhibitory potential (Lewis DFV et al., 2007).
In addition to determining the potential of a molecule to inhibit CYP450 enzymes, it is also important to assess the potential of a compound to induce CYP450 expression. CYP450 induction can potentially lead to increased clearance of the compound on chronic dosing and toxicity due to the possible formation of reactive oxygen species. For some years the pregnane X receptor (PXR), a nuclear hormone receptor assay has been used to determine the potential of a compound to induce CYP3A4. More recently, additional assays have become available for mechanisms by which other CYP isoforms are induced (Chu V et al, 2009) and could enter more routine use in the future. Currently such CYP450 induction assays are not commonly included early in the screening cascade unless the particular liability has been identified, but are employed in the later stages of lead optimization, particularly if the level of systemic exposure falls following chronic dosing.
Much attention is currently being given to the role of transporter proteins (Chu V et al, 2009), which have been implicated in the unfavourable disposition of drugs, either by active efflux from target tissue, potentially limiting efficacy, or through accumulation leading to toxicity and potential drug–drug interactions (Lai Y et al, 2010). Currently only P-glycoprotein (Pgp) assays are used in routine screening, but assays for many other transporters are being developed and are anticipated to be in routine use as screening assays in the near future.
The assessment of potential metabolic instability is also an important early evaluation in a screening cascade. Thus, typically compounds are incubated with human and/or rodent liver microsomes and their metabolic stability is expressed as the percentage of compound remaining after a given time or, more commonly, as an intrinsic clearance parameter (Nagai N, 2010). For a more thorough evaluation of in vitro metabolic turnover human and rodent hepatocytes may be used which possess the capacity to carry out both phase 1 and 2 metabolic processing. In addition an assessment of plasma protein binding is often included since the degree to which a molecule binds to plasma proteins can influence both the efficacy and the tissue distribution of the compound (Chang G et al, 2010).
As molecules progress to the later stages of lead optimization an understanding of the route and specific sites of metabolism of the compounds can be important information to assist the medicinal chemist to further improve the metabolic stability of the compound. For example, the introduction of blocking groups at sites of metabolism or electron withdrawing substituents at appropriate sites on the molecule, which will reduce electrophilicity, can significantly reduce the rate of metabolic turnover. An early assessment of the potential of a compound to form reactive and, therefore, potentially toxic metabolites is also valuable and can be achieved by using a combination of time-dependent CYP450 inhibition (Howard M et al, 2010) and trapping experiments, typically with either glutathione or cyanide (Riley RJ et al., 2007). For molecules with the potential for reactive metabolite formation additional in vitro and in vivo studies to detect covalent binding to tissue protein, using radiolabelled compound, may be required (Evans DC et al., 2004).
A preclinical pharmacokinetic study (see Chapter 10) is often the first occasion at which a compound is tested in an in vivo experiment. These studies are normally performed in rats since this is the species of choice for the majority of disease models and for rodent safety assessment studies. Compounds targeted as oral therapies are administered by both intravenous (i.v.) and oral (p.o.) routes, each experiment providing valuable information for the medicinal chemist. An i.v. pharmacokinetic study allows the clearance (the rate by which the drug is eliminated from the system) of the drug to be measured together with the volume of distribution (a measure of how well the drug distributes into tissue). Both the clearance and the volume of distribution determine the half-life of the drug. A p.o. study is used to determine the circulating levels that can be achieved after a given p.o. dose. Studies frequently use both i.v. and p.o. routes of administration to gain a full profile of a compound and allow the absolute bioavailability to be determined (White RE, 2009; Chapter 10).
The volume of distribution is a parameter which quantifies the distribution of the drug between the plasma and body tissue, and ideal molecules should have low clearance from the blood and a volume of distribution indicative of distribution into total body water (i.e. > 1). It is thus important to understand the properties which affect the volume of distribution of a compound. Common factors which can affect this parameter are plasma protein binding, the physical properties (particularly the presence or absence of either an acidic or basic centre) and the involvement of transporters (Grover A and Benet LZ, 2009). Common strategies to increase the volume of distribution include the introduction of a basic centre to increase the pKa, if appropriate, and to increase the LogD. However, unless the lead molecule is hydrophilic in nature, increasing lipophilicity can result in increased metabolism and CYP450 inhibition as discussed above and thus the identification of the optimal properties is often a trade-off of physicochemical properties. In addition to blocking sites of metabolism to reduce clearance, reducing the lipophilicity of a molecule is often effective at reducing the rate of metabolism. Drugs which are specifically aimed at targets within the central nervous system (CNS) require more stringent control of their physicochemical properties to facilitate passive permeation of the blood–brain barrier which is comprised of endothelial tight junctions. In comparison with Lipinski guidelines, molecules designed to cross the blood–brain barrier would typically have a lower molecular weight (<450), lower clogP (1–3), reduced hydrogen bond acceptors (<6) and donors (<2) and lower total polar surface area (TPSA) (<75A2). The blood–brain barrier also contains efflux transporters such as P-glycoprotein which serve to prevent access of substrates into the CNS. The free fraction of the compound, i.e. that proportion which is not bound to plasma protein, has also been reported to have a profound effect on both access to the CNS and subsequent distribution to the site of action (Jeffrey P and Summerfield S, 2010).
The desired pharmacokinetic profile of a drug will depend on the site of action and the nature of the target. Most commonly used drugs are required to penetrate tissue to reach their target protein and to be present at therapeutically efficacious concentrations for long enough to support once, or at most twice, daily dosing.
The majority of discovery programmes have targeted orally well-absorbed molecules with high metabolic stability to maintain high circulating blood levels for sufficient time to deliver the required duration of action. Thus the duration of action for these agents is related to their pharmacokinetic properties, particularly the rate of clearance from the plasma. An alternative approach has been to identify molecules which have an extended residence time, due to a slow ‘off rate’, at the target protein which is significantly longer than the time over which an efficacious plasma concentration of the compound is maintained. In such cases there is a clear mismatch between the pharmacokinetic and pharmacodynamic half-lives for the compounds, thus obviating the need to maintain high drug levels in the plasma over an extended time period to produce an efficacious response. A potential key advantage of these so-called ‘tight binders’ is that they may possess an improved safety profile (Packeu A et al., 2010), as the total body burden of drug is reduced.
Molecules which fulfil the targeted potency, selectivity and ADME properties will usually progress to in vivo studies in animal models to assess their potential for in vivo efficacy. The relevance of animal models for both efficacy and, in some cases, safety is a matter of much debate within the drug discovery community. With an increasing number of compounds failing to demonstrate clinical efficacy, despite having shown excellent results in preclinical models of disease, it is clear that the animal models are not totally predictive and a better understanding of the translation of data from animal model to human disease for a particular mechanism is required. Within areas where validated predictive models exist for specific mechanisms in vivo data can be very powerful both in aiding the selection of advanced molecules as potential development candidates and in predicting the efficacious clinical dose through the use of pharmacokinetic/pharmacodynamic (PK/PD) studies which correlate the systemic exposure of the compound with the degree of efficacy.
In addition to the optimization of ADME properties during the lead optimization phase, compounds are also evaluated for potential toxicity with subsequent chemical modification being applied to mitigate potential risks. A detailed discussion of toxicological evaluation in drug discovery is beyond the scope of this section (see Chapter 15); however, it is pertinent to mention the function of such assays in the process. Cytotoxicity assays are commonly used alongside cellular functional screens to identify molecules which produce biological activity by virtue of cellular toxicity. An area of intense current interest is that of the potential for compounds to produce adverse cardiovascular effects in the clinic, since cardiovascular toxicity is responsible for a significant number of drug withdrawals. An increasing number of targets, currently confined to ion channels, have been identified in recent years and are now commonly included in ‘off-target’ selectivity panels during lead optimization, the most well characterized of these being the hERG channel (Hancox JC et al., 2008; see also Chapter 15). The panel of cardiac liability channels being incorporated into screening cascades is increasing with many groups routinely including other channels such as Nav1.5, Kv1.5, Cav1.2, KCNQ1 (hmink) amongst others (Cao X et al., 2010). Later in the optimization phase, when potential candidates have been identified, assays to detect genotoxicity are routinely employed. Typically compounds are tested in both a bacterial mutagenicity assay such as the Ames test initially and also in a mammalian screen such as the mouse micronucleus assay (Reifferscheid G and Buchinger S, 2010). Prior to being accepted into preclinical development it is usual to conduct a preliminary in vivo safety study, typically dosing for 7 or 14 days, to provide an initial assessment of the maximum tolerated dose in preparation for subsequent more precise regulatory safety studies.
Much of this section has focused on the lead optimization process for oral drugs and it is important to recognize other routes of administration where the molecule profiles will be significantly different, particularly with respect to ADME and physical properties. Other common routes of administration are inhaled or intranasal, intravenous and topical.
For inhaled drugs, whose site of action is in the lung, a poorly soluble molecule which is highly metabolized in the circulation and of low membrane permeability is often the preferred profile. This profile is aimed at ensuring maximum residence time in the lung for optimal efficacy and minimal systemic exposure thus reducing the potential for any side effects (Ritchie TJ et al., 2009). A similar profile is desirable for topical drugs for application to the skin (Sloan KB et al., 2006).
For intravenously administered drugs a very different profile is required where a highly soluble molecule is preferred (Shi Y et al., 2009) to allow a low dose volume to be employed. In the latter case the focus of optimization is generally on the identification of highly potent agents (to facilitate low dose), with high metabolic stability and an acceptable duration of action. For drugs projected to be administered via routes other than the oral route, considerations such as Lipinski guidelines, which address oral bioavailability, are inappropriate.
Addressing attrition
Attrition is currently the major issue facing scientists engaged in the drug discovery process. During the 1990s the pharmaceutical industry took great steps to identify the reasons responsible for the high attrition rate in development and introduced measures to overcome the issues (Kola I and Landis J, 2004). The incorporation of both in vitro and in vivo ADME assays in discovery screening cascades significantly reduced the subsequent level of attrition due to poor pharmacokinetic properties during clinical evaluation between 1990 and 2000. However, in 2000 an increased number of molecules were failing due to toxicity (Kola I and Landis J, 2004). Ten years later the level of attrition due to toxicity is still high in the discovery phase and much effort is currently being devoted to identifying the key reasons responsible for this.
Of more concern is the level of late stage attrition, i.e. that occurring subsequent to the discovery and development phase and a worrying number of registered drugs continue to be withdrawn from the market at an unacceptable level. In the last 10 years 17 drugs have been withdrawn (http://en.wikipedia.org/wiki/List_of_withdrawn_drugs) due to adverse drug reactions. Furthermore, adverse drug reactions have been estimated to account for approximately 5% of deaths amongst hospital patients and 3% of the general population (Wester K et al., 2008). The most common toxicities are associated with the liver, the cardiovascular system, skin and blood. Significant efforts are being made to understand the reasons responsible for these toxicities and thus to develop assays capable of identifying molecules possessing the potential to cause these adverse events.
There are four main factors which can give rise to organ toxicities: the overall physical properties of a drug molecule, the primary pharmacology, the secondary or ‘off-target’ pharmacology and the presence of structural elements known to cause toxicity. The medicinal chemist needs to consider all of these factors when designing potential drug molecules.
Medicinal chemists have long been aware of the relationship between physical properties and ‘drug likeness’ (Lipinski CA et al., 1997); however, recent reviews (Leeson PD and Springthorpe B, 2007) suggest that a significant percentage of compounds being prepared by major pharmaceutical companies are too highly lipophilic and it has been proposed that high lipophilicity is likely to be a major cause of toxicity. A study by Pfizer (Price DA et al., 2009) which investigated potential correlations between physical properties and observed in vivo toxicity for 245 of their preclinical development compounds showed a marked correlation between cLogP and toxicity and an inverse correlation between total polar surface area (TPSA). They concluded that ideally a molecule should have a cLogP < 3 and TPSA > 75 to reduce the probability of producing toxicity. The importance of reducing lipophilicity cannot be over-emphasized as it has been shown to influence all parameters that the medicinal chemist needs to control in discovering an orally bioavailable drug including solubility, membrane permeability, plasma protein binding, metabolism (Waring MJ, 2010) and the potential to cause drug–drug interactions (Lewis D and Ito Y, 2010). High lipophilicity has been postulated to increase the probability for promiscuous binding of the compound to ‘off-target’ proteins thus increasing the likelihood of toxicity (Leeson PD and Springthorpe B, 2007).
Adverse drug reactions have been characterized into three groups (Pirmohamed M et al., 1998). Type A (augmented) adverse reactions are the so-called target-mediated toxicities and are related to the pharmacological effect of the drug and such adverse events are predictable and dose-related. Type B (idiosyncratic) adverse reactions are unpredictable, often non-dose related, less common, but frequently more severe than Type A. Type C (chemical) adverse reactions are related to structural elements in the drug molecule and may be predictable from the molecular structure.
Type A, augmented or target-mediated, toxicity is an important factor to take into account, particularly when choosing a target or during a target validation exercise. If interacting with the target is likely to cause an adverse event then the implications of the adverse event in the context of the disease requires careful consideration based on the potential benefit of the entity versus its risk. For example many anticancer agents are cytotoxic and would not be considered suitable treatments for less serious conditions.
Type B, idiosyncratic toxicity is a growing area of considerable interest (Ulrich RG, 2007) and the major cause of drug withdrawals. This type of toxicity is often severe and can result in hospitalization and, in extreme cases, death. Recent data suggest that some of the mechanisms responsible are becoming better understood, but there is still a need for much further work. In particular the formation of reactive metabolites which may bind covalently to cellular protein and precipitate an immunological response is currently a leading hypothesis to explain at least some of the idiosyncratic toxicity (Kaplowitz N, 2005). The potential for a compound to form reactive metabolites can be assessed by a combination of time-dependent CYP450 inhibition studies and electrophile trapping experiments with an agent such as glutathione (Kalgutkar AS and Didiuk MT, 2009) followed, if necessary, by detailed covalent protein binding studies (Evans D et al., 2004). The major organs affected by idiosyncratic toxicity are the heart, liver, kidneys and skin (Caldwell, 2006). It is important that any potential for idiosyncratic toxicity should be considered in the context of the predicted efficacious plasma concentration of the drug and the targeted indication. Empirically it is assumed that if the dose is 10 mg or less then the probability of idiosyncratic toxicity is reduced significantly (Uetrecht J, 2001).
Cardiovascular toxicity still remains one of the major causes of drug withdrawal and the recent removal of clobutinol from clinical usage as a result of cardiac QT interval prolongation (Takahara A et al., 2009) highlights the need to study potential drug molecules carefully to assess the risk of this phenomenon. The hERG channel, a potassium channel involved in the repolarization phase of the heart, has been known for some time to be associated with cardiac QT interval prolongation and there is a good correlation between compounds which have high hERG binding activity and clinical arrhythmogenic activity, particularly the torsade des pointes syndrome (Polak S et al., 2009). A variety of assays are available to assess the potential of compounds to inhibit the hERG channel, but it is important to choose the most appropriate in vitro assay and not to rely on data from one assay in isolation (Pollard CE et al., 2010). It is advisable, for advanced compounds, to obtain data in both in vitro and in vivo assays to allow an informed decision on the potential progression of the molecule (see Chapter 14). The hERG channel is one of several ion channels (Witchel HJ, 2010) involved in the repolarization phase of the heart, interference with any of which could lead to an adverse cardiovascular effect. Many groups are now screening compounds against a panel of cardiac ion channels throughout the discovery process (see Lead optimization section).
A number of medicinal chemistry strategies have been adopted to reduce the potential for hERG inhibition, which include reducing lipophilicity, modulating the pKa to reduce π-cationic interactions and reducing the potential for π-π interactions. The relative success of these strategies has been recently reviewed (Jamieson C et al., 2006). There is currently no available X-ray crystal structure of the hERG channel; however, the value of homology models using known potassium channel structures to guide structural modifications has been effectively demonstrated in the discovery of the CCR5 receptor antagonist, maraviroc (Price DA et al., 2006).
Drug-induced liver injury is also a major cause of compound attrition, often through the formation of reactive metabolites and this will be discussed later in this section. A greater understanding is emerging on the possible role of drug–transporter interactions in the development of hepatotoxicity (Lai Y et al., 2010) which may result in cellular accumulation, impaired efflux, alteration of nutrient transport and altered drug disposition leading to drug–drug interactions. For example the toxicity of troglitazone has been partially attributed to its interaction with the bile salt export pump (BSEP) (Funk C et al., 2001). The number of known transporters is currently growing and assays are being developed to assess the potential of drugs to cause toxicity through interaction with these proteins (Greer MI et al., 2010).
Another area of current growing interest, which has been implicated in liver toxicity, and, indeed, toxicity in other organs, is the potential of a drug to impair mitochondrial function (Dykens JA and Will Y, 2010). This is a complex and developing area which is beyond the scope of this chapter, however 80% of drugs which have received FDA ‘Black Box Warnings’ for hepatotoxicity and cardiovascular toxicity have been shown to inhibit mitochondrial function (Dykens JA and Will Y, 2007). Assay systems are currently available and a suggested protocol for their use in drug discovery has been published (Dykens JA and Will Y, 2010).
Recent studies have suggested biomarkers that are potentially predictive of liver injury that could be used both preclinically and clinically. Two such biomarkers are the high mobility group box1 and keratin 18 proteins (Antoine DJ et al., 2009).
Whilst renal toxicity has not been responsible for a large number of drug withdrawals, it is still a major cause for concern. Interaction with transporter proteins has recently been implicated in the renal toxicity of a number of substances. Rosuvastatin (Crestor®) was shown to cause proteinuria at a dose of 80 mg in Phase 3 clinical trials. This phenomenon has subsequently been shown to be due to a combination of the target pharmacology of the drug (inhibition of protein endocytosis) and the fact that it is a substrate for the organic anion transporter 3 (OAT3), which causes it to accumulate in the kidney (Windass AS et al., 2007). Until recently there has been a lack of predictive biomarkers for renal toxicity; however, a recent study by an international consortium, has identified seven biomarkers which can be used both preclinically and in human studies (Hewitt P and Herget T, 2009).
Type C or structural-based toxicity is clearly an area that the medicinal chemist should particularly consider before embarking on a chemical modification. Structural alerts, of which the chemist should be aware, can be divided into four groups, chemically reactive functionality, structural elements which present a risk following metabolic activation, DNA binders and CYP450 inhibitors. There are several excellent reviews in the recent literature covering this area (Blagg J, 2006; Kalgutkar AS and Didiuk MT, 2009) in detail and describe many structural features which should be given careful consideration before incorporation within a drug molecule. The medicinal chemist is advised to read these carefully and to be aware of new toxicophores as they appear in the literature.
Despite the increasing volume of information, the theoretical prediction of toxicity is still imprecise. It is, therefore, important to incorporate ‘predictive’ assays early in screening strategies. Despite recent progress, current in vitro safety testing is conducted in animal cells or, at best, in recombinant human cell lines. It would be preferable to conduct these studies in native human cells and recent advances in stem cell research facilitate this opportunity in the future (Balls M, 2010). Whilst toxicity is a major cause of compound failure, the failure of potential drug molecules to demonstrate efficacy upon clinical evaluation is a more difficult issue to address since the factors which contribute to these failures are complex. Inadequate target validation, poor predictability of animal models, poor clinical trial design and patient selection are potential causes. Whilst the medicinal chemist cannot address all of these issues, it is important that potent, selective and safe molecules with appropriate physical properties are identified for such studies to ensure that it is the mechanism of action of the drug that is being tested and not the quality of the molecule. Greater involvement of the medicinal chemist at the initial target identification and validation stage may provide better tool or probe compounds (see Target selection and validation section) with which to validate novel targets.
Summary
The environment in which drug discovery is taking place is currently undergoing considerable change and will continue to do so for some time to come. The downsizing of major Pharma and the increased focus on academic drug discovery are attempts to deal with the escalating costs, high attrition rate and patent expiries which are threatening the viability of the current discovery model (see Chapter 22).
This chapter is intended to convey the message that the medicinal chemist has an extremely important role to play in all aspects of the discovery process to address the challenges of increasing efficiency and productivity and reducing attrition. Whilst traditionally the medicinal chemist has not become engaged with the drug discovery process until the lead identification phase, there is a compelling argument for their involvement at the target selection and validation stage. The identification of high-quality chemical probes as early as possible will enable robust target validation and the selection of those biological targets which are more likely to succeed.
Furthermore, an increasing array of technological advances are becoming available which will enable the chemist to develop a detailed understanding of the biophysics associated with compound-target engagement, which can be effectively incorporated into the design of suitable preclinical candidates.
Whilst significant progress has been made over the past 10–15 years in reducing attrition due to poor ADME properties, toxicity remains to be a major source of attrition and the chemist has an important role to play in reducing the late-stage attrition due to compound-related toxicity. Whilst the current predictive packages are imperfect, there is a developing understanding of the impact of chemical structure and physicochemical properties on toxicity. It is anticipated that an increasing number of screening assays will become available for incorporation into project screening cascades which will help to identify potential risks as early as possible in the discovery process.
Thus, whilst the industry is entering uncharted territory, there is an unprecedented opportunity for the medicinal chemist to make a major contribution to the future success of the discovery of new and important medicines to meet the growing level of unmet clinical need.
Antoine DJ, Williams DP, Kipar A, et al. High-mobility group box-1 protein and keratin-18, circulating serum proteins informative of acetaminophen-induced necrosis and apoptosis in vivo. Toxicological Sciences. 2009;112:521–531.
Balls M. Adult human stem cells and toxicity – realising the potential. Alternatives to Laboratory Animals. 2010;38:91.
Baringhaus KH, Matter H. Efficient strategies for lead optimization by simultaneously addressing affinity, selectivity and pharmacokinetic parameters. Methods and Principles in Medicinal Chemistry. 2005;23:333–379. Chemoinformatics in Drug Discovery
Bartoschek S, Klabunde T, Defossa E, et al. Drug design for G-protein-coupled receptors by a ligand-based NMR method. Angewandte Chemie, International Edition. 2010;49:1426–1429. S1426/1–S1426/5
Bass AS, Cartwright ME, Mahon C, et al. Exploratory drug safety – a drug discovery strategy to reduce attrition in development. Journal of Pharmacological and Toxicological Methods. 2009;60:69–78.
Blagg J. Structure-activity relationships for in vitro and in vivo toxicity. Annual Reports in Medical Chemistry. 2006;41:358.
Bleicher KH, Boehm HJ, Mueller K, et al. A guide to drug discovery: hit and lead generation: beyond high-throughput screening. Nature Reviews Drug Discovery. 2003;2:369–378.
Caldwell GW, Yan Z. Screening for reactive intermediates and toxicity assessment in drug discovery. Current Opinion in Drug Discovery & Development. 2006;9:47–60.
Cao X, Lee YT, Holmqvist M, et al. Cardiac ion channel safety profiling on the IonWorks Quattro automated patch clamp system. Assay and Drug Development Technologies. 2010;8:766–780.
Carr R, Jhoti H. Structure-based screening of low-affinity compounds. Drug Discovery Today. 2002;7:522–527.
Chang G, Steyn SJ, Umland JP, et al. Strategic use of plasma and microsome binding to exploit in vitro clearance in early drug discovery. ACS Medicinal Chemistry Letters. 2010;1:50–53.
Cheeseright TJ, Mackey MD, Melville JL, et al. FieldScreen: virtual screening using molecular fields. Application to the DUD data set. Journal of Chemical Information and Modeling. 2008;48:2108–2117.
Chu V, Einolf HJ, Evers R, et al. In vitro and in vivo induction of Cytochrome P450: A survey of the current practices and recommendations: a pharmaceutical research and manufacturers of America perspective. Drug MetAbolism and Disposition. 2009;37:1339.
Cole PA. Chemical probes for histone-modifying enzymes. Nature Chemical Biology. 2008;4:590–597.
Congreve M, Chessari G, Tisi D, et al. Recent developments in fragment-based drug discovery. Journal of Medicinal Chemistry. 2008;51:3661–3680.
Dib-Hajj SD, Cummins TR, Black JA, et al. From genes to pain: Nav1.7 and human pain disorders. Trends in Neurosciences. 2007;30:555–563.
Dykens JA, Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discovery Today. 2007;12:777–785.
Dykens JA, Will Y. Drug-induced mitochondrial dysfunction: an emerging model of idiosyncratic drug toxicity. International Drug Discovery. 2010;5:32–36.
Eglen RM, Gilchrist A, Reisine T. The use of immortalized cell lines in GPCR screening: the good, bad and ugly. Combinatorial Chemistry & High Throughput Screening. 2008;11:560–565.
Ejskjaer N, Vestergaard ET, Hellström PM, et al. Ghrelin receptor agonist (TZP-101) accelerates gastric emptying in adults with diabetes and symptomatic gastroparesis. Alimentary Pharmacology and Therapeutics. 2009;29:1179–1187.
Ekroos M, Sjögren T. Structural basis for ligand promiscuity in cytochrome P 450 3A4. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13682–13687.
Evans DC, Watt AP, Nicoll-Griffith DA, et al. Drug-protein adducts: an industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chemical Research in Toxicology. 2004;17:3–16.
Foti RS, Wienkers LC, Wahlstrom JL. Application of cytochrome P450 drug interaction screening in drug discovery. Combinatorial Chemistry & High Throughput Screening. 2010;13:145–158.
Fox S, Farr-Jones S, Sopchak L, et al. High-throughput screening: update on practices and success. Journal of biomolecular screening. 2006;11:864–869.
Frye SV. The art of the chemical probe. Nature Chemical Biology. 2010;6:159–161.
Funk C, Ponelle C, Scheuermann G, et al. Cholestatic potential of troglitazone as a possible factor contributing to troglitazone-induced hepatotoxicity: in vivo and in vitro interaction at the canalicular bile salt export pump (BSEP) in the rat. Molecular Pharmacology. 2001;59:627–635.
Giannetti AM, Koch BD, Browner MF. Surface plasmon resonance based assay for the detection and characterization of promiscuous inhibitors. Journal of Medicinal Chemistry. 2008;51:574–580.
Gluckman PD, Hanson MA, Cooper C, et al. In utero and early-life conditions and adult health and disease – in reply. New England Journal of Medicine. 2008;359:61–63.
Greer ML, Barber J, Eakins J, et al. Cell based approaches for evaluation of drug induced liver injury. Toxicology. 2010;268:125.
Grover A, Benet LZ. Effects of drug transporters on volume of distribution. AAPS Journal. 2009;11:250–261.
Halai R, Craik DJ. Conotoxins: natural product drug leads. Natural Product Reports. 2009;26:526–536.
Hancox JC, McPate MJ, El Harchi A, et al. The hERG potassium channel and hERG screening for drug-induced torsades de pointes. Pharmacology & Therapeutics. 2008;119:118–132.
Hart CP. Finding the target after screening the phenotype. Drug Discovery Today. 2005;10:513–519.
Harvey AL. Natural products in drug discovery. Drug Discovery Today. 2008;13:894–901.
Harvey AL, Clark RL, Mackay SP, et al. Current strategies for drug discovery through natural products. Expert Opinion on Drug Discovery. 2010;5:559–568.
Helstroem PM. Faces of ghrelin – research for the 21st century. Neurogastroenterology and Motility. 2009;21:2.
Hewitt P, Herget T. Value of new biomarkers for safety testing in drug development. Expert Review of Molecular Diagnostics. 2009;9:531–536.
Howard ML, Hill JJ, Galluppi GR, et al. Plasma protein binding in drug discovery and development. Combinatorial Chemistry & High Throughput Screening. 2010;13:170–187.
Hughes B. 2009 FDA drug approvals. Nature Reviews Drug Discovery. 2010;9:89–92.
Jamieson C, Moir EM, Rankovic Z, et al. Medicinal chemistry of hERG optimizations: highlights and hang-ups. Journal of Medicinal Chemistry. 2006;49:5029–5046.
Jeffrey P, Summerfield S. Assessment of the blood–brain barrier in CNS drug discovery. Neurobiology of Disease. 2010;37:33–37.
Kaczorowski GJ, McManus OB, Priest BT, et al. Ion channels as drug targets: the next GPCRs. Journal of General Physiology. 2008;131:399–405.
Kalgutkar AS, Didiuk MT. Structural alerts, reactive metabolites, and protein covalent binding: how reliable are these attributes as predictors of drug toxicity? Chemistry & Biodiversity. 2009;6:2115–2137.
Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nature Reviews Drug Discovery. 2005;4:489–499.
Keiser M, Wetzel S, Kumar K, et al. Biology-inspired synthesis of compound libraries. Cellular and Molecular Life Sciences. 2008;65:1186–1201.
Keserü GM, Makara GM. The influence of lead discovery strategies on the properties of drug candidates. Nature Reviews Drug Discovery. 2009;8:203–212.
Kola I, Landis J. Opinion: can the pharmaceutical industry reduce attrition rates? Nature Reviews Drug Discovery. 2004;3:711–716.
Konteatis ZD. In silico fragment-based drug design. Expert Opinion on Drug Discovery. 2010;5:1047–1065.
Lai Y, Sampson KE, Stevens JC. Evaluation of drug transporter interactions in drug discovery and development. Combinatorial Chemistry & High Throughput Screening. 2010;13:112–134.
Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nature Reviews Drug Discovery. 2007;6:881–890.
Lewis D, Ito Y. Human CYPs involved in drug metabolism, structures, substrates and binding affinities. Expert Opinion on Drug Metabolism and Toxicology. 2010;6:661–674.
Lewis DFV, Lake BG, Dickins M. Quantitative structure-activity relationships (QSARs) in inhibitors of various cytochromes P450: the importance of compound lipophilicity. Journal of Enzyme Inhibition and Medicinal Chemistry. 2007;22:1–6.
Lindsley CW, Weaver D, Bridges TM, et al. Lead optimization in drug discovery. Wiley Encyclopedia of Chemical Biology. 2009;2:511–519.
Lipinski CA, Lombado F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 1997;23:3–25.
Lombardi D, Dittrich PS. Advances in microfluidics for drug discovery. Expert Opinion on Drug Discovery. 2010;5:1081–1094.
Luheshi LM, Crowther DC, Dobson CM. Protein misfolding and disease: from the test tube to the organism. Current Opinion in Chemical Biology. 2008;12:25–31.
Macarron R, Banks MN, Bojanic D, et al. Impact of high-throughput screening in biomedical research. Nature Reviews Drug Discovery. 2011;10:188–195.
McGuire C, Lee J. Brief review of vorinostat. Clinical Medicine Insights: Therapeutics. 2010;2:83–88.
Nagai N. Drug interaction studies on new drug applications – current situation and regulatory view in Japan. Drug Metabolism and Pharmacokinetics. 2010;25:3–15.
Naik P, Murumkar P, Giridhar R, et al. Angiotensin II receptor type 1 (AT1) selective nonpeptidic antagonists – a perspective. Bioorganic & Medicinal Chemistry. 2010;18:8418–8456.
Newman DJ, Cragg JM. Natural products as sources of new drugs over the last 25 years. Journal of Natural Products. 2007;70:461–477.
Oprea TI, Davis AM, Teague SJ, et al. Is there a difference between leads and drugs? A historical perspective. Journal of Chemical Information and Computer Sciences. 2001;41:1308–1315.
Orita M, Warizaya M, Amano Y, et al. Advances in fragment-based drug discovery platforms. Expert Opinion on Drug Discovery. 2009;4:1125–1144.
Packeu A, Wennerberg M, Balendran A. Estimation of the dissociation rate of unlabelled ligand-receptor complexes by a ‘two-step’ competition binding approach. British Journal of Pharmacology. 2010;161:1311–1328.
Pepys MB, Herbert J, Hutchinson WL, et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature (London, United Kingdom). 2002;417:254–259.
Pepys MB, Hirschfield GM, Tennent GA, et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature (London, United Kingdom). 2006;440:1217–1221.
Pirmohamed M, Breckenridge AM, Kitteringham NR, et al. Adverse drug reactions. British Medical Journal (Clinical Research Edition). 1998;316:1295–1298.
Polak S, Wisniowska B. Brandys Collation, assessment and analysis of literature in vitro data on hERG receptor blocking potency for subsequent modeling of drugs cardiotoxic properties. Journal of Applied Toxicology. 2009;29(3):183–206.
Pollard CE, Abi Gerges N, Bridgland-Taylor MH, et al. An introduction to QT interval prolongation and non-clinical approaches to assessing and reducing risk. British Journal of Pharmacology. 2010;159:12–21.
Popescu I, Fleshner PR, Pezzullo JC, et al. The ghrelin agonist TZP-101 for management of postoperative ileus after partial colectomy: a randomized, dose-ranging, placebo-controlled clinical trial. Diseases of the Colon and Rectum. 2010;53:126–134.
Price DA, Armour D, de Groot M, et al. Overcoming HERG affinity in the discovery of the CCR5 antagonist maraviroc. Bioorganic & Medicinal Chemistry Letters. 2006;16:4633–4637.
Price DA, Blagg J, Jones L, et al. Physicochemical drug properties associated with toxicological outcomes – a review. Expert Opinion on Drug Metabolism & Toxicology. 2009;5:921–931.
Proudfoot JR. Drugs, leads, and drug-likeness: an analysis of some recently launched drugs. Bioorganic & Medicinal Chemistry Letters. 2002;12:1647–1650.
Rees DC, Congreve M, Murray CW, et al. Fragment-based lead discovery. Nature Reviews Drug Discovery. 2004;3:660–672.
Reifferscheid G, Buchinger S. Cell-based genotoxicity testing. Genetically modified and genetically engineered bacteria in environmental genotoxicology. Advances in Biochemical Engineering/Biotechnology. 2010;118:85–112. Whole Cell Sensing Systems II
Riley RJ, Grime K, Weaver R. Time-dependent CYP inhibition. Expert Opinion on Drug Metabolism & Toxicology. 2007;3:51–66.
Rishton GM. Reactive compounds and in vitro false positives in HTS. Drug Discovery Today. 2003;8:86–96.
Ritchie TJ, Luscombe CN, Macdonald SJ. Analysis of the calculated physicochemical properties of respiratory drugs: can we design for inhaled drugs yet? Journal of Chemical Information and Modeling. 2009;49:1025–1032.
Schmidtko A, Lötsch J, Freynhagen R, et al. Ziconotide for treatment of severe chronic pain. Lancet. 2010;375:1569–1577.
Schneider G. Virtual screening: an endless staircase? Nature Reviews Drug Discovery. 2010;9:273–276.
Shi Y, Merdan T, Li LC. Recent advances in intravenous delivery of poorly water-soluble compounds. Expert Opinion on Drug Delivery. 2009;6:1261–1282.
Shiau AK, Massari ME, Ozbal CC. Back to basics: label-free technologies for small molecule screening. Combinatorial Chemistry & High Throughput Screening. 2008;11:231–237.
Sloan KB, Wasdo SC, Rautio J. Design for optimized topical delivery: prodrugs and a paradigm change. Pharmaceutical Research. 2006;23:2729–2747.
Smith GF. Medicinal chemistry by the numbers: the physicochemistry, thermodynamics and kinetics of modern drug design. Progress in Medicinal Chemistry. 2009;48:1–29.
Source http://en.wikipedia.org/wiki/List_of_withdrawn_drugs
Swanson R, Beasley JR. Pathway-specific, species, and sub-type counterscreening for better GPCR hits in high throughput screening. Current Pharmaceutical Biotechnology. 2010;11:757–763.
Takahara A, Sasaki R, Nakamura M, et al. Clobutinol delays ventricular repolarisation in the guinea pig heart – comparison UIT cardiac effects of hERG K+ channel inhibitor E-4031. Journal of Cardiovascular Pharmacology. 2009;54:552–559.
Tate CG, Schertler GFX. Engineering G protein-coupled receptors to facilitate their structure determination. Current Opinion in Structural Biology. 2009;19:386–395.
Uetrecht J. Prediction of a new drug’s potential to cause idiosyncratic reactions. Current Opinion in Drug Discovery & Development. 2001;4:55–59.
Ulrich RG. Idiosyncratic toxicity: a convergence of risk factors. Annual Review of Medicine. 2007;58:17–34.
Waring MJ. Lipophilicity in drug discovery. Expert Opinion on Drug Discovery. 2010;5:235–248.
Wehling M. Assessing the translatability of drug projects: what needs to be scored to predict success? Nature Reviews Drug Discovery. 2009;8:541–546.
Westby M, van der Ryst E. CCR5 antagonists: host-targeted antivirals for the treatment of HIV infection. Antiviral Chemistry & Chemotherapy. 2005;16(6):339–354.
Wester K, Jönsson AK, Spigset O, et al. Incidence of fatal adverse drug reactions: a population based study. British journal of Clinical Pharmacology. 2008;65:573–579.
Wetzel S, Klein K, Renner S, et al. Interactive exploration of chemical space with scaffold hunter. Nature Chemical Biology. 2009;5:581–583.
White RE. Pharmacokinetics of drug candidates. Wiley Encyclopedia of Chemical Biology. 2009;3:652–661.
Whittaker M, Law RJ, Ichihara O, et al. Fragments: past, present and future. Drug Discovery Today: Technologies. 2010;7:E163–E171.
Windass AS, Lowes S, Wang Y, et al. The contribution of organic anion transporters OAT1 and OAT3 to the renal uptake of rosuvastatin. Journal of Pharmacology and Experimental Therapeutics. 2007;322:1221–1227.
Witchel HJ. Emerging trends in ion channel-based assays for predicting the cardiac safety of drugs. IDrugs. 2010;13:90–96.