Neurophysiology
Desirable Properties of Local Anesthetics
Local anesthesia has been defined as loss of sensation in a circumscribed area of the body caused by depression of excitation in nerve endings or inhibition of the conduction process in peripheral nerves.1 An important feature of local anesthesia is that it produces this loss of sensation without inducing loss of consciousness. In this one major area, local anesthesia differs dramatically from general anesthesia.
Many methods are used to induce local anesthesia:
However, only those methods or substances that induce a transient and completely reversible state of anesthesia have application in clinical practice. Following are those properties deemed most desirable for a local anesthetic:
1. It should not be irritating to the tissue to which it is applied.
2. It should not cause any permanent alteration of nerve structure.
3. Its systemic toxicity should be low.
4. It must be effective regardless of whether it is injected into the tissue or is applied locally to mucous membranes.
5. The time of onset of anesthesia should be as short as possible.
6. The duration of action must be long enough to permit completion of the procedure yet not so long as to require an extended recovery.
Most local anesthetics discussed in this section meet the first two criteria: They are (relatively) nonirritating to tissues and are completely reversible. Of paramount importance is systemic toxicity, because all injectable and most topical local anesthetics are eventually absorbed from their site of administration into the cardiovascular system. The potential toxicity of a drug is an important factor in its consideration for use as a local anesthetic. Toxicity varies greatly among the local anesthetics currently in use. Toxicity is discussed more thoroughly in Chapter 2. Although it is a desirable characteristic, not all local anesthetics in clinical use today meet the criterion of being effective, regardless of whether the drug is injected or applied topically. Several of the more potent injectable local anesthetics (e.g., procaine, mepivacaine) prove to be relatively ineffective when applied topically to mucous membranes. To be effective as topical anesthetics, these drugs must be applied in concentrations that prove to be locally irritating to tissues while increasing the risk of systemic toxicity. Dyclonine, a potent topical anesthetic, is not administered by injection because of its tissue-irritating properties. Lidocaine and tetracaine, on the other hand, are effective anesthetics when administered by injection or topical application in clinically acceptable concentrations. The last factors—rapid onset of action and adequate duration of clinical action—are met satisfactorily by most of the clinically effective local anesthetics in use today. Clinical duration of action does vary considerably among drugs and also among different preparations of the same drug, as well as by the type of injection administered (e.g., nerve block vs. supraperiosteal). The duration of anesthesia necessary to complete a procedure is a major consideration in the selection of a local anesthetic.
In addition to these qualities, Bennett2 lists other desirable properties of an ideal local anesthetic:
7. It should have potency sufficient to give complete anesthesia without the use of harmful concentrated solutions.
8. It should be relatively free from producing allergic reactions.
9. It should be stable in solution and should readily undergo biotransformation in the body.
10. It should be sterile or capable of being sterilized by heat without deterioration.
No local anesthetic in use today satisfies all of these criteria; however, all anesthetics do meet a majority of them. Research is continuing in an effort to produce newer drugs that possess a maximum of desirable factors and a minimum of negative ones.
Fundamentals of Impulse Generation and Transmission
The discovery in the late 1800s of a group of chemicals with the ability to prevent pain without inducing loss of consciousness was one of the major steps in the advancement of the medical and dental professions. For the first time, medical and dental procedures, could be carried out easily and in the absence of pain, a fact that is virtually taken for granted by contemporary medical and dental professionals and their patients.
The concept behind the actions of local anesthetics is simple: They prevent both the generation and the conduction of a nerve impulse. In effect, local anesthetics set up a chemical roadblock between the source of the impulse (e.g., the scalpel incision in soft tissues) and the brain. Therefore the aborted impulse, prevented from reaching the brain, cannot be interpreted by the patient as pain.
This is similar to the effect of lighting the fuse on a stick of dynamite. The fuse is the “nerve,” whereas the dynamite is the “brain.” If the fuse is lit and the flame reaches the dynamite, an explosion occurs (Fig. 1-1). When a nerve is stimulated, an impulse is propagated that will be interpreted as pain when it reaches the brain. If the fuse is lit, but “water” (e.g., local anesthetic) is placed somewhere between the end of the fuse and the dynamite stick, the fuse will burn up to the point of water application and then die out. The dynamite does not explode. When a local anesthetic is placed at some point between the pain stimulus (e.g., the drill) and the brain, the nerve impulse is still propagated and travels up to the point of local anesthetic application and then “dies,” never reaching the brain, and pain does not occur (Fig. 1-2).
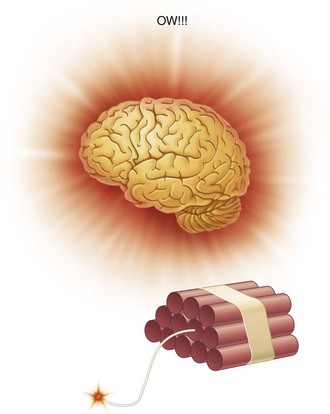
Figure 1-1 The fuse is lit and the flame reaches the dynamite; an explosion occurs, and the patient experiences pain.
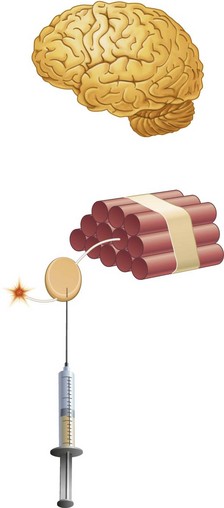
Figure 1-2 Local anesthetic is placed at some point between the pain stimulus and the brain (dynamite). The nerve impulse travels up to the point of local anesthetic application and then “dies,” never reaching the brain, and pain does not occur.
How, in fact, do local anesthetics, the most used drugs in dentistry, function to abolish or prevent pain? Following is a discussion of current theories seeking to explain the mode of action of local anesthetic drugs. To understand their action better, however, the reader must have an acquaintance with the fundamentals of nerve conduction. A review of the relevant characteristics and properties of nerve anatomy and physiology follows.
The Neuron
The neuron, or nerve cell, is the structural unit of the nervous system. It is able to transmit messages between the central nervous system (CNS) and all parts of the body. There are two basic types of neuron: sensory (afferent) and motor (efferent). The basic structure of these two neuronal types differs significantly (Fig. 1-3).
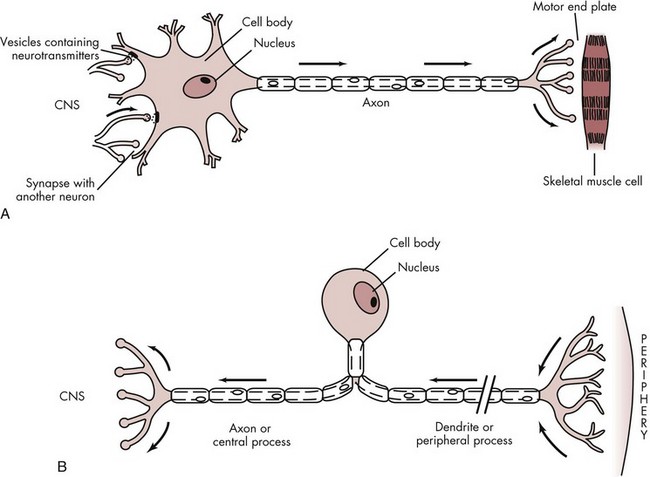
Figure 1-3 A, Multipolar motor neuron. B, Unipolar sensory neuron. (From Liebgott B: Anatomical basis of dentistry, ed 2, St Louis, 2001, Mosby.)
Sensory neurons that are capable of transmitting the sensation of pain consist of three major portions.3 The peripheral process (also known as the dendritic zone), which is composed of an arborization of free nerve endings, is the most distal segment of the sensory neuron. These free nerve endings respond to stimulation produced in the tissues in which they lie, provoking an impulse that is transmitted centrally along the axon. The axon is a thin cable-like structure that may be quite long (the giant squid axon has been measured at 100 to 200 cm). At its mesial (or central) end is an arborization similar to that seen in the peripheral process. However, in this case the arborization forms synapses with various nuclei in the CNS to distribute incoming (sensory) impulses to their appropriate sites within the CNS for interpretation. The cell body is the third part of the neuron. In the sensory neuron described here, the cell body is located at a distance from the axon, the main pathway of impulse transmission in this nerve. The cell body of the sensory nerve therefore is not involved in the process of impulse transmission, its primary function being to provide vital metabolic support for the entire neuron (Fig. 1-3, B).
Nerve cells that conduct impulses from the CNS toward the periphery are termed motor neurons and are structurally different from the sensory neurons just described in that their cell body is interposed between the axon and dendrites. In motor neurons, the cell body not only is an integral component of the impulse transmission system but also provides metabolic support for the cell. Near its termination, the axon branches with each branch, ending as a bulbous axon terminal (or bouton). Axon terminals synapse with muscle cells (Fig. 1-3, A).
The Axon
The single nerve fiber, the axon, is a long cylinder of neural cytoplasm (axoplasm) encased in a thin sheath, the nerve membrane, or axolemma. Neurons have a cell body and a nucleus, as do all other cells; however, neurons differ from other cells in that they have an axonal process from which the cell body may be at a considerable distance. The axoplasm, a gelatinous substance, is separated from extracellular fluids by a continuous nerve membrane. In some nerves, this membrane is itself covered by an insulating lipid-rich layer of myelin.
Current thinking holds that both sensory nerve excitability and conduction are attributable to changes developing within the nerve membrane. The cell body and the axoplasm are not essential for nerve conduction. They are important, however. The metabolic support of the membrane is probably derived from the axoplasm.
The nerve (cell) membrane itself is approximately 70 to 80 Å thick. (An angstrom unit is 1/10,000 of a micrometer.) Figure 1-4 represents a currently acceptable configuration. All biological membranes are organized to block the diffusion of water-soluble molecules; to be selectively permeable to certain molecules via specialized pores or channels; and to transduce information through protein receptors responsive to chemical or physical stimulation by neurotransmitters or hormones (chemical) or light, vibration, or pressure (physical).4 The membrane is described as a flexible nonstretchable structure consisting of two layers of lipid molecules (bilipid layer of phospholipids) and associated proteins, lipids, and carbohydrates. The lipids are oriented with their hydrophilic (polar) ends facing the outer surface and their hydrophobic (nonpolar) ends projecting to the middle of the membrane (Fig. 1-4, A). Proteins are visualized as the primary organizational elements of membranes (Fig. 1-4, B).5 Proteins are classified as transport proteins (channels, carriers, or pumps) and receptor sites. Channel proteins are thought to be continuous pores through the membrane, allowing some ions (Na+, K,+ Ca++) to flow passively, whereas other channels are gated, permitting ion flow only when the gate is open.4 The nerve membrane lies at the interface between extracellular fluid and axoplasm. It separates highly diverse ionic concentrations within the axon from those outside. The resting nerve membrane has an electrical resistance about 50 times greater than that of the intracellular and extracellular fluids, thus preventing the passage of sodium, potassium, and chloride ions down their concentration gradients. However, when a nerve impulse passes, electrical conductivity of the nerve membrane increases approximately 100-fold. This increase in conductivity permits the passage of sodium and potassium ions along their concentration gradients through the nerve membrane. It is the movement of these ions that provides an immediate source of energy for impulse conduction along the nerve.
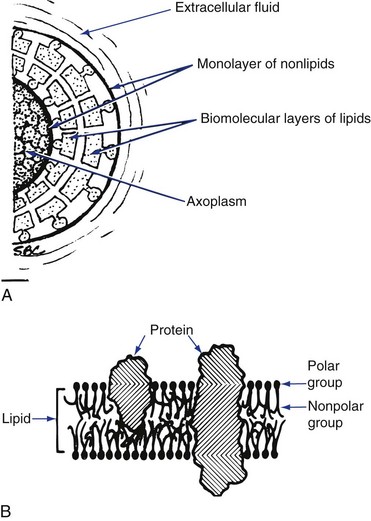
Figure 1-4 A, Configuration of a biological membrane. B, Heterogeneous lipoprotein membrane as suggested by Singer and Nicholson. (Redrawn from Covino BG, Vassalo HG: Local anesthetics: mechanisms of action and clinical use, New York, 1976, Grune & Stratton.)
Some nerve fibers are covered by an insulating lipid layer of myelin. In vertebrates, myelinated nerve fibers include all but the smallest of axons (Table 1-1).6 Myelinated nerve fibers (Fig. 1-5) are enclosed in spirally wrapped layers of lipoprotein myelin sheaths, which are actually a specialized form of Schwann cell. Although primarily lipid (75%), the myelin sheath also contains some protein (20%) and carbohydrate (5%).7 Each myelinated nerve fiber is enclosed in its own myelin sheath. The outermost layer of myelin consists of the Schwann cell cytoplasm and its nucleus. Constrictions are located at regular intervals (approximately every 0.5 to 3 mm) along the myelinated nerve fiber. These are nodes of Ranvier, and they form a gap between two adjoining Schwann cells and their myelin spirals.8 At these nodes, the nerve membrane is exposed directly to the extracellular medium.
TABLE 1-1
Classification of Peripheral Nerves According to Fiber Size and Physiologic Properties
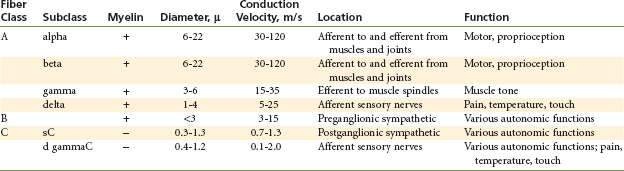
From Berde CB, Strichartz GR: Local anesthetics. In Miller RD, editor: Anesthesia, ed 5, Philadelphia, 2000, Churchill Livingstone, pp 491–521.
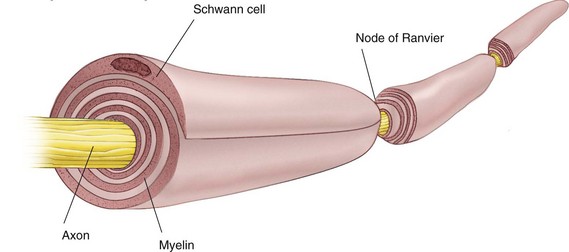
Figure 1-5 Structure of a myelinated nerve fiber. (Redrawn from de Jong RH: Local anesthetics, St Louis, 1994, Mosby.)
Unmyelinated nerve fibers (Fig. 1-6) are also surrounded by a Schwann cell sheath. Groups of unmyelinated nerve fibers share the same sheath. The insulating properties of the myelin sheath enable a myelinated nerve to conduct impulses at a much faster rate than an unmyelinated nerve of equal size.
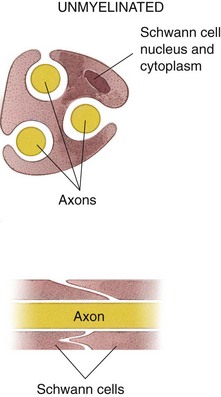
Physiology of the Peripheral Nerves
The function of a nerve is to carry messages from one part of the body to another. These messages, in the form of electrical action potentials, are called impulses. Action potentials are transient depolarizations of the membrane that result from a brief increase in the permeability of the membrane to sodium, and usually also from a delayed increase in its permeability to potassium.9 Impulses are initiated by chemical, thermal, mechanical, or electrical stimuli.
Once an impulse is initiated by a stimulus in any particular nerve fiber, the amplitude and shape of that impulse remain constant, regardless of changes in the quality of the stimulus or in its strength. The impulse remains constant without losing strength as it passes along the nerve because the energy used for its propagation is derived from energy that is released by the nerve fiber along its length and not solely from the initial stimulus. de Jong has described impulse conduction as being like the active progress of a spark along a fuse of gunpowder.10 Once lit, the fuse burns steadily along its length, with one burning segment providing the energy necessary to ignite its neighbor. Such is the situation with impulse propagation along a nerve.
Electrophysiology of Nerve Conduction
Following is a description of electrical events that occur within a nerve during the conduction of an impulse. Subsequent sections describe the precise mechanisms for each of these steps.
A nerve possesses a resting potential (Fig. 1-7, Step 1). This is a negative electrical potential of −70 mV that exists across the nerve membrane, produced by differing concentrations of ions on either side of the membrane (Table 1-2). The interior of the nerve is negative relative to the exterior.
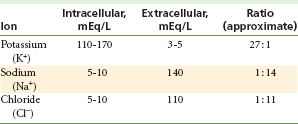
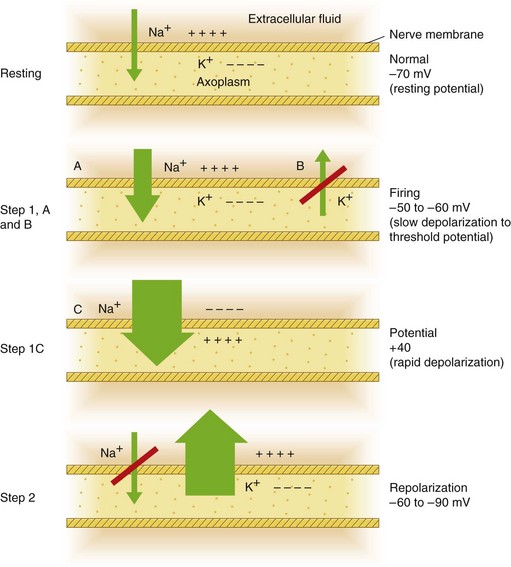
Figure 1-7 Top, Resting potential. Step 1, A and B, Slow depolarization to threshold. Step 1, C, Rapid depolarization. Step 2, Repolarization.
Step 1
A stimulus excites the nerve, leading to the following sequence of events:
A An initial phase of slow depolarization. The electrical potential within the nerve becomes slightly less negative (see Fig. 1-7, Step 1A).
B When the falling electrical potential reaches a critical level, an extremely rapid phase of depolarization results. This is termed threshold potential, or firing threshold (see Fig. 1-7, Step 1B).
C This phase of rapid depolarization results in a reversal of the electrical potential across the nerve membrane (see Fig. 1-7, Step 1C). The interior of the nerve is now electrically positive in relation to the exterior. An electrical potential of +40 mV exists on the interior of the nerve cell.11
Step 2
After these steps of depolarization, repolarization occurs (Fig. 1-7, Step 2). The electrical potential gradually becomes more negative inside the nerve cell relative to outside until the original resting potential of −70 mV is again achieved.
The entire process (Steps 1 and 2) requires 1 millisecond (msec); depolarization (Step 1) takes 0.3 msec; repolarization (Step 2) takes 0.7 msec.
Electrochemistry of Nerve Conduction
The preceding sequence of events depends on two important factors: the concentrations of electrolytes in the axoplasm (interior of the nerve cell) and extracellular fluids, and the permeability of the nerve membrane to sodium and potassium ions.
Table 1-2 shows the differing concentrations of ions found within neurons and in the extracellular fluids. Significant differences exist for ions between their intracellular and extracellular concentrations. These ionic gradients differ because the nerve membrane exhibits selective permeability.
Resting State
In its resting state, the nerve membrane is
Potassium remains within the axoplasm, despite its ability to diffuse freely through the nerve membrane and its concentration gradient (passive diffusion usually occurs from a region of greater concentration to one of lesser concentration), because the negative charge of the nerve membrane restrains the positively charged ions by electrostatic attraction.
Chloride remains outside the nerve membrane instead of moving along its concentration gradient into the nerve cell, because the opposing, nearly equal, electrostatic influence (electrostatic gradient from inside to outside) forces outward migration. The net result is no diffusion of chloride through the membrane.
Sodium migrates inwardly because both the concentration (greater outside) and the electrostatic gradient (positive ion attracted by negative intracellular potential) favor such migration. Only the fact that the resting nerve membrane is relatively impermeable to sodium prevents a massive influx of this ion.
Membrane Excitation
Depolarization: Excitation of a nerve segment leads to an increase in permeability of the cell membrane to sodium ions. This is accomplished by a transient widening of transmembrane ion channels sufficient to permit the unhindered passage of hydrated sodium ions. The rapid influx of sodium ions to the interior of the nerve cell causes depolarization of the nerve membrane from its resting level to its firing threshold of approximately −50 to −60 mV (see Fig. 1-7, Steps 1A and 1B).12 The firing threshold is actually the magnitude of the decrease in negative transmembrane potential that is necessary to initiate an action potential (impulse).
A decrease in negative transmembrane potential of 15 mV (e.g., from −70 to −55 mV) is necessary to reach the firing threshold; a voltage difference of less than 15 mV will not initiate an impulse. In a normal nerve, the firing threshold remains constant. Exposure of the nerve to a local anesthetic raises its firing threshold. Elevating the firing threshold means that more sodium must pass through the membrane to decrease the negative transmembrane potential to a level where depolarization occurs.
When the firing threshold is reached, membrane permeability to sodium increases dramatically and sodium ions rapidly enter the axoplasm. At the end of depolarization (the peak of the action potential), the electrical potential of the nerve is actually reversed; an electrical potential of +40 mV exists (see Fig. 1-7, Step 1C). The entire depolarization process requires approximately 0.3 msec.
Repolarization: The action potential is terminated when the membrane repolarizes. This is caused by the extinction (inactivation) of increased permeability to sodium. In many cells, permeability to potassium also increases, resulting in the efflux of K+, and leading to more rapid membrane repolarization and return to its resting potential (see Fig. 1-7, Step 2).
Movement of sodium ions into the cell during depolarization and subsequent movement of potassium ions out of the cell during repolarization are passive (not requiring the expenditure of energy), because each ion moves along its concentration gradient (higher → lower). After the return of the membrane potential to its original level (−70 mV), a slight excess of sodium exists within the nerve cell, along with a slight excess of potassium extracellularly. A period of metabolic activity then begins in which active transfer of sodium ions out of the cell occurs via the sodium pump. An expenditure of energy is necessary to move sodium ions out of the nerve cell against their concentration gradient; this energy comes from the oxidative metabolism of adenosine triphosphate (ATP). The same pumping mechanism is thought to be responsible for the active transport of potassium ions into the cell against their concentration gradient. The process of repolarization requires 0.7 msec.
Immediately after a stimulus has initiated an action potential, a nerve is unable, for a time, to respond to another stimulus regardless of its strength. This is termed the absolute refractory period, and it lasts for about the duration of the main part of the action potential. The absolute refractory period is followed by a relative refractory period, during which a new impulse can be initiated but only by a stronger than normal stimulus. The relative refractory period continues to decrease until the normal level of excitability returns, at which point the nerve is said to be repolarized.
During depolarization, a major proportion of ionic sodium channels are found in their open (O) state (thus permitting the rapid influx of Na+). This is followed by a slower decline into a state of inactivation (I) of the channels to a nonconducting state. Inactivation temporarily converts the channels to a state from which they cannot open in response to depolarization (absolute refractory period). This inactivated state is slowly converted back, so that most channels are found in their closed (C) resting form when the membrane is repolarized (−70 mV). Upon depolarization, the channels change configuration, first to an open ion-conducting (O) state and then to an inactive nonconducting (I) state. Although both C and I states correspond to nonconducting channels, they differ in that depolarization can recruit channels to the conducting O state from C but not from I. Figure 1-8 describes the sodium channel transition stages.13
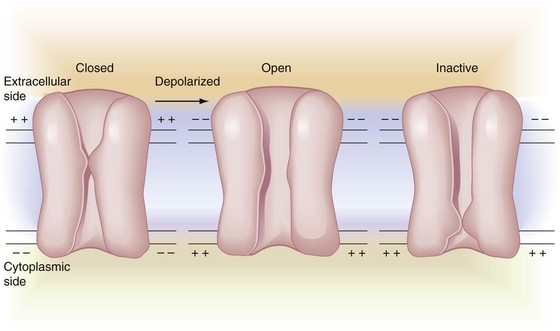
Figure 1-8 Sodium channel transition stages. Depolarization reverses resting membrane potential from interior negative (left) to interior positive (center). The channel proteins undergo corresponding conformational changes from resting state (closed) to ion-conducting stage (open). State changes continue from open (center) to inactive (right), where channel configuration assumes a different, but still impermeable, state. With repolarization, the inactivated refractory channel reverts to the initial resting configuration (left), ready for the next sequence. (Redrawn from Siegelbaum SA, Koester F: Ion channels. In Kandel ER, editor: Principles of neural science, ed 3, Norwalk, Conn, 1991, Appleton-Lange.)
Membrane Channels
Discrete aqueous pores through the excitable nerve membrane, called sodium (or ion) channels, are molecular structures that mediate its sodium permeability. A channel appears to be a lipoglycoprotein firmly situated in the membrane (see Fig. 1-4). It consists of an aqueous pore spanning the membrane that is narrow enough at least at one point to discriminate between sodium ions and others; Na+ passes through 12 times more easily than K+. The channel also includes a portion that changes configuration in response to changes in membrane potential, thereby gating the passage of ions through the pore (C, O, and I states are described). The presence of these channels helps explain membrane permeability or impermeability to certain ions. Sodium channels have an internal diameter of approximately 0.3 × 0.5 nm.14
A sodium ion is thinner than a potassium or chloride ion and therefore should diffuse freely down its concentration gradient through membrane channels into the nerve cell. However, this does not occur, because all these ions attract water molecules and thus become hydrated. Hydrated sodium ions have a radius of 3.4 Å, which is approximately 50% greater than the 2.2 Å radius of potassium and chloride ions. Sodium ions therefore are too large to pass through narrow channels when a nerve is at rest (Fig. 1-9). Potassium and chloride ions can pass through these channels. During depolarization, sodium ions readily pass through the nerve membrane because configurational changes that develop within the membrane produce transient widening of these transmembrane channels to a size adequate to allow the unhindered passage of sodium ions down their concentration gradient into the axoplasm (transformation from the C to the O configuration). This concept can be visualized as the opening of a gate during depolarization that is partially occluding the channel in the resting membrane (C) (Fig. 1-10).
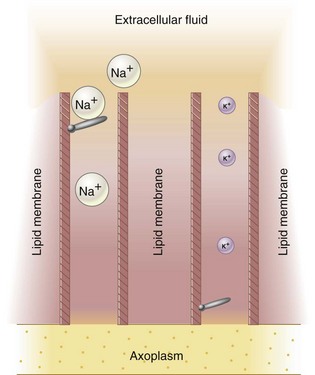
Figure 1-9 Membrane channels are partially occluded; the nerve is at rest. Hydrated sodium ions (Na+) are too large to pass through channels, although potassium ions (K+) can pass through unimpeded.
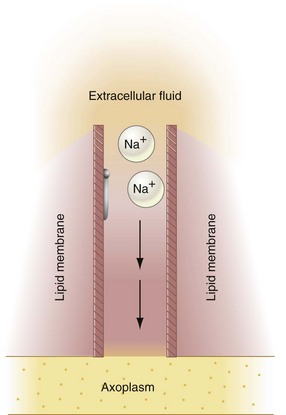
Figure 1-10 Membrane channels are open; depolarization occurs. Hydrated sodium ions (Na+) now pass unimpeded through the sodium channel.
Evidence indicates that channel specificity exists in that sodium channels differ from potassium channels.15 The gates on the sodium channel are located near the external surface of the nerve membrane, whereas those on the potassium channel are located near the internal surface of the nerve membrane.
Impulse Propagation
After initiation of an action potential by a stimulus, the impulse must move along the surface of the axon. Energy for impulse propagation is derived from the nerve membrane in the following manner.
The stimulus disrupts the resting equilibrium of the nerve membrane; the transmembrane potential is reversed momentarily, with the interior of the cell changing from negative to positive, and the exterior changing from positive to negative. This new electrical equilibrium in this segment of nerve produces local currents that begin to flow between the depolarized segment and the adjacent resting area. These local currents flow from positive to negative, extending for several millimeters along the nerve membrane.
As a result of this current flow, the interior of the adjacent area becomes less negative and its exterior less positive. Transmembrane potential decreases, approaching firing threshold for depolarization. When transmembrane potential is decreased by 15 mV from resting potential, a firing threshold is reached and rapid depolarization occurs. The newly depolarized segment sets up local currents in adjacent resting membrane, and the entire process starts anew.
Conditions in the segment that has just depolarized return to normal after the absolute and relative refractory periods. Because of this, the wave of depolarization can spread in only one direction. Backward (retrograde) movement is prevented by the inexcitable, refractory segment.
Impulse Spread
The propagated impulse travels along the nerve membrane toward the CNS. The spread of this impulse differs depending on whether or not a nerve is myelinated.
Unmyelinated Nerves
An unmyelinated nerve fiber is basically a long cylinder with a high–electrical resistance cell membrane surrounding a low-resistance conducting core of axoplasm, all of which is bathed in low-resistance extracellular fluid.
The high-resistance cell membrane and low-resistance intracellular and extracellular media produce a rapid decrease in the density of current within a short distance of the depolarized segment. In areas immediately adjacent to this depolarized segment, local current flow may be adequate to initiate depolarization in the resting membrane. Farther away it will prove to be inadequate to achieve a firing threshold.
The spread of an impulse in an unmyelinated nerve fiber therefore is characterized as a relatively slow forward-creeping process (Fig. 1-11). The conduction rate in unmyelinated C fibers is 1.2 m/sec compared with 14.8 to 120 m/sec in myelinated A-alpha and A-delta fibers.16
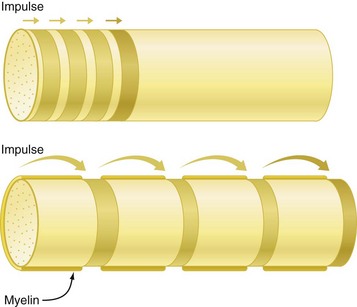
Figure 1-11 Saltatory propagation. Comparison of impulse propagation in nonmyelinated (upper) and myelinated (lower) axons. In nonmyelinated axons, the impulse moves forward by sequential depolarization of short adjoining membrane segments. Depolarization in myelinated axons, on the other hand, is discontinuous; the impulse leaps forward from node to node. Note how much farther ahead the impulse is in the myelinated axon after four depolarization sequences. (Redrawn from de Jong RH: Local anesthetics, St Louis, 1994, Mosby.)
Myelinated Nerves
Impulse spread within myelinated nerves differs from that in unmyelinated nerves because of the layer of insulating material separating the intracellular and extracellular charges. The farther apart are the charges, the smaller is the current necessary to charge the membrane. Local currents thus can travel much farther in a myelinated nerve than in an unmyelinated nerve before becoming incapable of depolarizing the nerve membrane ahead of it.
Impulse conduction in myelinated nerves occurs by means of current leaps from node to node, a process termed saltatory conduction (see Fig. 1-11) (saltare is the Latin verb “to leap”). This form of impulse conduction proves to be much faster and more energy efficient than that employed in unmyelinated nerves. The thickness of the myelin sheath increases with increasing diameter of the axon. In addition, the distance between adjacent nodes of Ranvier increases with greater axonal diameter. Because of these two factors, saltatory conduction is more rapid in a thicker axon.
Saltatory conduction usually progresses from one node to the next in a stepwise manner. However, it can be demonstrated that the current flow at the next node still exceeds that necessary to reach the firing threshold of the nodal membrane. If conduction of an impulse is blocked at one node, the local current skips over that node and proves adequate to raise the membrane potential at the next node to its firing potential, producing depolarization. A minimum of perhaps 8 to 10 mm of nerve must be covered by anesthetic solution to ensure thorough blockade.17
Mode and Site of Action of Local Anesthetics
How and where local anesthetics alter the processes of impulse generation and transmission needs to be discussed. It is possible for local anesthetics to interfere with the excitation process in a nerve membrane in one or more of the following ways:
1. Altering the basic resting potential of the nerve membrane
2. Altering the threshold potential (firing level)
It has been established that the primary effects of local anesthetics occur during the depolarization phase of the action potential.18 These effects include a decrease in the rate of depolarization, particularly in the phase of slow depolarization. Because of this, cellular depolarization is not sufficient to reduce the membrane potential of a nerve fiber to its firing level, and a propagated action potential does not develop. There is no accompanying change in the rate of repolarization.
Where Do Local Anesthetics Work?
The nerve membrane is the site at which local anesthetics exert their pharmacologic actions. Many theories have been promulgated over the years to explain the mechanism of action of local anesthetics, including the acetylcholine, calcium displacement, and surface charge theories. The acetylcholine theory stated that acetylcholine was involved in nerve conduction, in addition to its role as a neurotransmitter at nerve synapses.19 No evidence indicates that acetylcholine is involved in neural transmission along the body of the neuron. The calcium displacement theory, once popular, maintained that local anesthetic nerve block was produced by the displacement of calcium from some membrane site that controlled permeability to sodium.20 Evidence that varying the concentration of calcium ions bathing a nerve does not affect local anesthetic potency has diminished the credibility of this theory. The surface charge (repulsion) theory proposed that local anesthetics act by binding to the nerve membrane and changing the electrical potential at the membrane surface.21 Cationic (RNH+) (p. 16) drug molecules were aligned at the membrane–water interface, and because some of the local anesthetic molecules carried a net positive charge, they made the electrical potential at the membrane surface more positive, thus decreasing the excitability of the nerve by increasing the threshold potential. Current evidence indicates that the resting potential of the nerve membrane is unaltered by local anesthetics (they do not become hyperpolarized), and that conventional local anesthetics act within membrane channels rather than at the membrane surface. Also, the surface charge theory cannot explain the activity of uncharged anesthetic molecules in blocking nerve impulses (e.g., benzocaine).
Two other theories, membrane expansion and specific receptor, are given credence today. Of the two, the specific receptor theory is more widely held.
The membrane expansion theory states that local anesthetic molecules diffuse to hydrophobic regions of excitable membranes, producing a general disturbance of the bulk membrane structure, expanding some critical region(s) in the membrane, and preventing an increase in permeability to sodium ions.22,23 Local anesthetics that are highly lipid soluble can easily penetrate the lipid portion of the cell membrane, producing a change in configuration of the lipoprotein matrix of the nerve membrane. This results in a decreased diameter of sodium channels, which leads to inhibition of both sodium conductance and neural excitation (Fig. 1-12). The membrane expansion theory serves as a possible explanation for the local anesthetic activity of a drug such as benzocaine, which does not exist in cationic form yet still exhibits potent topical anesthetic activity. It has been demonstrated that nerve membranes, in fact, do expand and become more fluid when exposed to local anesthetics. However, no direct evidence suggests that nerve conduction is entirely blocked by membrane expansion per se.
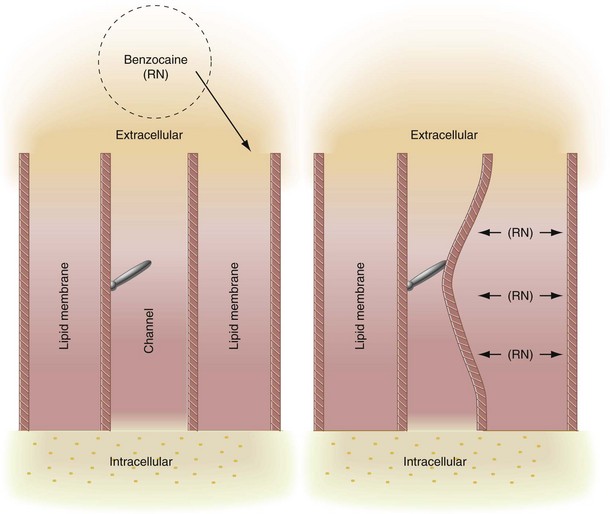
The specific receptor theory, the most favored today, proposes that local anesthetics act by binding to specific receptors on the sodium channel (Fig. 1-13).24 The action of the drug is direct, not mediated by some change in the general properties of the cell membrane. Both biochemical and electrophysiologic studies have indicated that a specific receptor site for local anesthetics exists in the sodium channel either on its external surface or on the internal axoplasmic surface.25,26 Once the local anesthetic has gained access to the receptors, permeability to sodium ions is decreased or eliminated, and nerve conduction is interrupted.
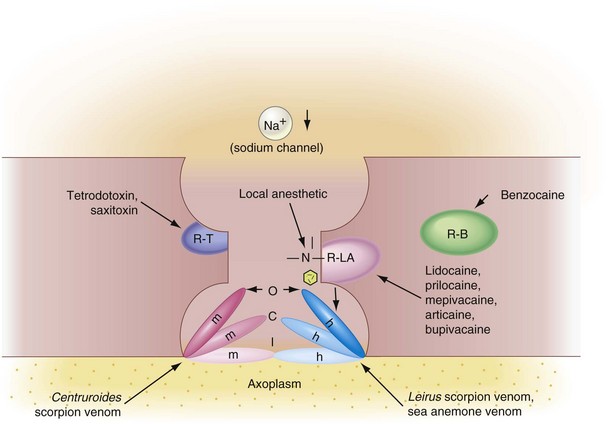
Figure 1-13 A, Tertiary amine local anesthetics inhibit the influx of sodium during nerve conduction by binding to a receptor within the sodium channel (R-LA). This blocks the normal activation mechanism (O gate configuration, depolarization) and also promotes movement of the activation and inactivation gates (m and h) to a position resembling that in the inactivated state (I). B, Biotoxins (R-T) block the influx of sodium at an outer surface receptor; various venoms do it by altering the activity of the activation and inactivation gates; and benzocaine (R-B) does it by expanding the membrane. C, Channel in the closed configuration. (Redrawn from Pallasch TJ: Dent Drug Serv Newsletter 4:25, 1983.)
Local anesthetics are classified by their ability to react with specific receptor sites in the sodium channel. It appears that drugs can alter nerve conduction in at least four sites within the sodium channel (see Fig. 1-13):
1. Within the sodium channel (tertiary amine local anesthetics)
2. At the outer surface of the sodium channel (tetrodotoxin, saxitoxin)
3 and 4. At the activation or the inactivation gate (scorpion venom)
Table 1-3 is a biological classification of local anesthetics based on their site of action and the active form of the compound. Drugs in Class C exist only in the uncharged form (RN), whereas Class D drugs exist in both charged and uncharged forms. Approximately 90% of the blocking effects of Class D drugs are caused by the cationic form of the drug; only 10% of blocking action is produced by the base (Fig. 1-14).
TABLE 1-3
Classification of Local Anesthetic Substances According to Biological Site and Mode of Action
| Classification | Definition | Chemical Substance |
| Class A | Agents acting at receptor site on external surface of nerve membrane | Biotoxins (e.g., tetrodotoxin, saxitoxin) |
| Class B | Agents acting at receptor site on internal surface of nerve membrane | Quaternary ammonium analogs of lidocaine Scorpion venom |
| Class C | Agents acting by a receptor-independent physico-chemical mechanism | Benzocaine |
| Class D | Agents acting by combination of receptor and receptor-independent mechanisms | Most clinically useful local anesthetic agents (e.g., articaine, lidocaine, mepivacaine, prilocaine) |
Modified from Covino BG, Vassallo HG: Local anesthetics: mechanisms of action and clinical use, New York, 1976, Grune & Stratton. Used by permission.
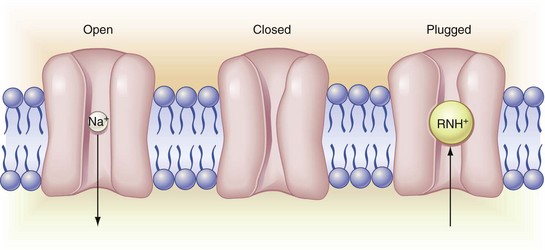
Figure 1-14 Channel entry. On the left is an open channel, inward permeant to sodium ion. The center channel is in the resting closed configuration; although impermeant to sodium ion here, the channel remains voltage responsive. The channel on the right, although in open configuration, is impermeant because it has local anesthetic cation bound to the gating receptor site. Note that local anesthetic enters the channel from the axoplasmic (lower) side; the channel filter precludes direct entry via the external mouth. Local anesthetic renders the membrane impermeant to sodium ion, hence inexcitable by local action currents. (Redrawn from de Jong RH: Local anesthetics, St Louis, 1994, Mosby.)
Myelinated Nerve Fibers
One additional factor should be considered with regard to the site of action of local anesthetics in myelinated nerves. The myelin sheath insulates the axon both electrically and pharmacologically. The only site at which molecules of local anesthetic have access to the nerve membrane is at the nodes of Ranvier, where sodium channels are found in abundance. Ionic changes that develop during impulse conduction arise only at the nodes.
Because an impulse may skip over or bypass one or two blocked nodes and continue on its way, it is necessary for at least two or three nodes immediately adjacent to the anesthetic solution to be blocked to ensure effective anesthesia—a length of approximately 8 to 10 mm.
Sodium channel densities differ in myelinated and unmyelinated nerves. In small unmyelinated nerves, the density of sodium channels is about 35/µm, whereas at the nodes of Ranvier in myelinated fibers, it may be as high as 20,000/µm. On an average nerve length basis, relatively few sodium channels are present in unmyelinated nerve membranes. For example, in the garfish olfactory nerve, the ratio of sodium channels to phospholipid molecules is 1 : 60,000, corresponding to a mean distance between channels of 0.2 µm, whereas at densely packed nodes of Ranvier, the channels are separated by only 70 Å.27,28
How Local Anesthetics Work
The primary action of local anesthetics in producing a conduction block is to decrease the permeability of ion channels to sodium ions (Na+). Local anesthetics selectively inhibit the peak permeability of sodium, whose value is normally about five to six times greater than the minimum necessary for impulse conduction (e.g., there is a safety factor for conduction of 5× to 6×).29 Local anesthetics reduce this safety factor, decreasing both the rate of rise of the action potential and its conduction velocity. When the safety factor falls below unity,10 conduction fails and nerve block occurs.
Local anesthetics produce a very slight, virtually insignificant decrease in potassium (K+) conductance through the nerve membrane.
Calcium ions (Ca++), which exist in bound form within the cell membrane, are thought to exert a regulatory role on the movement of sodium ions across the nerve membrane. Release of bound calcium ions from the ion channel receptor site may be the primary factor responsible for increased sodium permeability of the nerve membrane. This represents the first step in nerve membrane depolarization. Local anesthetic molecules may act through competitive antagonism with calcium for some site on the nerve membrane.
The following sequence is a proposed mechanism of action of local anesthetics1:
1. Displacement of calcium ions from the sodium channel receptor site, which permits …
2. Binding of the local anesthetic molecule to this receptor site, which produces …
3. Blockade of the sodium channel, and a …
4. Decrease in sodium conductance, which leads to …
5. Depression of the rate of electrical depolarization, and …
6. Failure to achieve the threshold potential level, along with …
7. Lack of development of propagated action potentials, which is called …
The mechanism whereby sodium ions gain entry to the axoplasm of the nerve, thereby initiating an action potential, is altered by local anesthetics. The nerve membrane remains in a polarized state because the ionic movements responsible for the action potential fail to develop. Because the membrane’s electrical potential remains unchanged, local currents do not develop, and the self-perpetuating mechanism of impulse propagation is stalled. An impulse that arrives at a blocked nerve segment is stopped because it is unable to release the energy necessary for its continued propagation. Nerve block produced by local anesthetics is called a nondepolarizing nerve block.
Active Forms of Local Anesthetics
Most injectable local anesthetics are tertiary amines. Only a few (e.g., prilocaine, hexylcaine) are secondary amines. The typical local anesthetic structure is shown in Figures 1-15 and 1-16. The lipophilic part is the largest portion of the molecule. Aromatic in structure, it is derived from benzoic acid, aniline, or thiophene (articaine). All local anesthetics are amphipathic, that is, they possess both lipophilic and hydrophilic characteristics, generally at opposite ends of the molecule. The hydrophilic part is an amino derivative of ethyl alcohol or acetic acid. Local anesthetics without a hydrophilic part are not suited for injection but are good topical anesthetics (e.g., benzocaine). The anesthetic structure is completed by an intermediate hydrocarbon chain containing an ester or an amide linkage. Other chemicals, especially histamine blockers and anticholinergics, share this basic structure with local anesthetics and commonly exhibit weak local anesthetic properties.
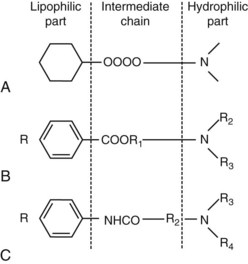
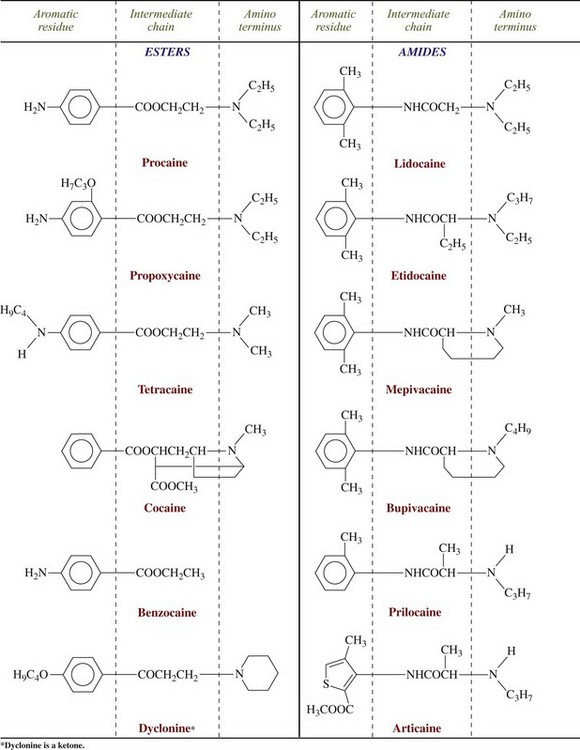
Figure 1-16 Chemical configuration of local anesthetics. (From Yagiela JA, Neidle EA, Dowd FJ: Pharmacology and therapeutics for dentistry, ed 6, St Louis, 2010, Mosby.)
Local anesthetics may be classified as amino esters or amino amides according to their chemical linkages. The nature of the linkage is important in defining several properties of the local anesthetic, including the basic mode of biotransformation. Ester-linked local anesthetics (e.g., procaine) are readily hydrolyzed in aqueous solution. Amide-linked local anesthetics (e.g., lidocaine) are relatively resistant to hydrolysis. A greater percentage of an amide-linked drug than of an ester-linked drug is excreted unchanged in the urine. Procainamide, which is procaine with an amide linkage replacing the ester linkage, is as potent a local anesthetic as procaine, yet because of its amide linkage, it is hydrolyzed much more slowly. Procaine is hydrolyzed in plasma in only a few minutes, but just approximately 10% of procainamide is hydrolyzed in 1 day.
As prepared in the laboratory, local anesthetics are basic compounds that are poorly soluble in water and unstable on exposure to air.30 Their pKa values range from 7.5 to 10. In this form, they have little or no clinical value. However, because they are weakly basic, they combine readily with acids to form local anesthetic salts, in which form they are quite soluble in water and comparatively stable. Thus local anesthetics used for injection are dispensed as acid salts, most commonly the hydrochloride salt (e.g., lidocaine HCl, articaine HCl), dissolved in sterile water or saline.
It is well known that the pH of a local anesthetic solution (as well as the pH of the tissue into which it is injected) greatly influences its nerve-blocking action. Acidification of tissue decreases local anesthetic effectiveness. Inadequate anesthesia results when local anesthetics are injected into inflamed or infected areas. The inflammatory process produces acidic products: The pH of normal tissue is 7.4; the pH of an inflamed area is 5 to 6. Local anesthetics containing epinephrine or other vasopressors are acidified by the manufacturer to inhibit oxidation of the vasopressor (p. 18). The pH of solutions without epinephrine is about 6.5; epinephrine-containing solutions have a pH of about 3.5. Clinically, this lower pH is more likely to produce a burning sensation on injection, as well as a slightly slower onset of anesthesia.
Elevating the pH (alkalinization) of a local anesthetic solution speeds its onset of action, increases its clinical effectiveness, and makes its injection more comfortable. However, the local anesthetic base, because it is unstable, precipitates out of alkalinized solutions, making these preparations ill-suited for clinical use. Buffered (e.g., carbonated) local anesthetics have received much attention in recent years both in medicine and, more recently, in dentistry.31 Sodium bicarbonate or carbon dioxide (CO2) added to the anesthetic solution immediately before injection provides greater comfort and more rapid onset of anesthesia (see Chapter 19).32,33 The use of buffered local anesthetics in dentistry is reviewed in depth in Chapter 20.
Despite potentially wide pH variation in extracellular fluids, the pH at the interior of a nerve remains stable. Normal functioning of a nerve therefore is affected very little by changes in the extracellular environment. However, the ability of a local anesthetic to block nerve impulses is profoundly altered by changes in extracellular pH.
Dissociation of Local Anesthetics
As discussed, local anesthetics are available as acid salts (usually hydrochloride) for clinical use. The local anesthetic salt, both water soluble and stable, is dissolved in sterile water or saline. In this solution, it exists simultaneously as uncharged molecules (RN), also called the base, and as positively charged molecules (RNH+), called the cation.
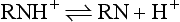
The relative proportion of each ionic form in the solution varies with the pH of the solution or surrounding tissues. In the presence of a high concentration of hydrogen ions (low pH), the equilibrium shifts to the left, and most of the anesthetic solution exists in cationic form:
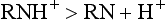
As hydrogen ion concentration decreases (higher pH), the equilibrium shifts toward the free base form:
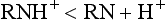
The relative proportion of ionic forms also depends on the pKa, or dissociation constant, of the specific local anesthetic. The pKa is a measure of the affinity of a molecule for hydrogen ions (H+). When the pH of the solution has the same value as the pKa of the local anesthetic, exactly 50% of the drug exists in the RNH+ form and 50% in the RN form. The percentage of drug existing in either form can be determined from the Henderson-Hasselbalch equation.
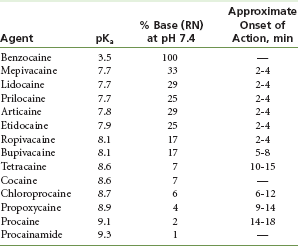
Table 1-4 lists the pKa values for commonly used local anesthetics.
Actions on Nerve Membranes
The two factors involved in the action of a local anesthetic are (1) diffusion of the drug through the nerve sheath, and (2) binding at the receptor site in the ion channel. The uncharged, lipid-soluble, free base form (RN) of the anesthetic is responsible for diffusion through the nerve sheath. This process is explained in the following example:
1. One thousand molecules of a local anesthetic with a pKa of 7.9 are injected into the tissues outside a nerve. The tissue pH is normal (7.4) (Fig. 1-17).
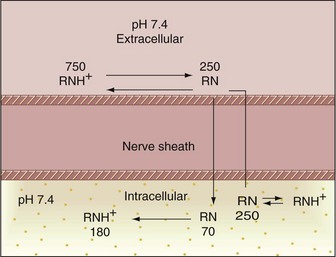
Figure 1-17 Mechanism of action of the local anesthetic molecule. Anesthetic pKa of 7.9; tissue pH of 7.4.
2. From Table 1-4 and the Henderson-Hasselbalch equation, it can be determined that at normal tissue pH, 75% of local anesthetic molecules are present in the cationic form (RNH+) and 25% in the free base form (RN).
3. In theory then, all 250 lipophilic RN molecules will diffuse through the nerve sheath to reach the interior (axoplasm) of the neuron.
4. When this happens, the extracellular equilibrium between RNH+ 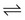 RN has been disrupted by passage of the free base forms into the neuron. The remaining 750 extracellular RNH+ molecules will now reequilibrate according to the tissue pH and the pKa of the drugs:
RN has been disrupted by passage of the free base forms into the neuron. The remaining 750 extracellular RNH+ molecules will now reequilibrate according to the tissue pH and the pKa of the drugs:
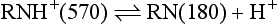
5. The 180 newly created lipophilic RN molecules diffuse into the cell, starting the entire process (Step 4) again. Theoretically, this will continue until all local anesthetic molecules diffuse into the axoplasm.
The reality, however, is somewhat different. Not all the local anesthetic molecules will eventually reach the interior of the nerve, because of the process of diffusion (drugs will diffuse in all possible directions, not just toward the nerve), and because some will be absorbed into blood vessels and extracellular soft tissues at the injection site.
6. The inside of the nerve should be viewed next. After penetration of the nerve sheath and entry into the axoplasm by the lipophilic RN form of the anesthetic, reequilibration takes place inside the nerve, because a local anesthetic cannot exist in only the RN form at an intracellular pH of 7.4. Seventy-five percent of those RN molecules present within the axoplasm revert into the RNH+ form; the remaining 25% of molecules remain in the uncharged RN form.
7. From the axoplasmic side, the RNH+ ions enter into the sodium channels, bind to the channel receptor site, and ultimately are responsible for the conduction blockade that results (see Figs. 1-13 and 1-14).
Of the two factors—diffusibility and binding—responsible for local anesthetic effectiveness, the former is extremely important in actual practice. The ability of a local anesthetic to diffuse through the tissues surrounding a nerve is of critical significance, because in clinical situations the local anesthetic cannot be applied directly to the nerve membrane, as it can in a laboratory setting. Local anesthetic solutions better able to diffuse through soft tissue provide an advantage in clinical practice.
A local anesthetic with a high pKa value has very few molecules available in the RN form at a tissue pH of 7.4. The onset of anesthetic action of this drug is slow because too few base molecules are available to diffuse through the nerve membrane (e.g., procaine, with a pKa of 9.1). The rate of onset of anesthetic action is related to the pKa of the local anesthetic (see Table 1-4).
A local anesthetic with a lower pKa (<7.5) has a greater number of lipophilic free base molecules available to diffuse through the nerve sheath; however, the anesthetic action of this drug is inadequate because at an intracellular pH of 7.4, only a very small number of base molecules dissociate back to the cationic form necessary for binding at the receptor site.
In actual clinical situations with the local anesthetics currently available, the pH of the extracellular fluid determines the ease with which a local anesthetic moves from the site of its administration into the axoplasm of the nerve cell. The intracellular pH remains stable and independent of the extracellular pH, because hydrogen ions (H+), such as the local anesthetic cations (RNH+), do not readily diffuse through tissues. The pH of extracellular fluid therefore may differ from that of the nerve membrane. The ratio of anesthetic cations to uncharged base molecules (RNH+/RN) also may vary greatly at these sites. Differences in extracellular and intracellular pH are highly significant in pain control when inflammation or infection is present.34 The effect of a decrease in tissue pH on the actions of a local anesthetic is described in Figure 1-18. This can be compared with the example in Figure 1-17, involving normal tissue pH:
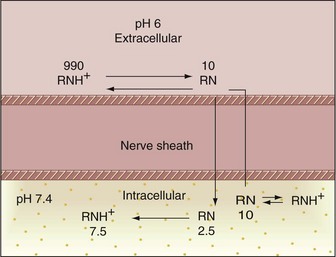
1. Approximately 1000 molecules of a local anesthetic with a pKa of 7.9 are deposited outside a nerve. The tissue is inflamed and infected and has a pH of 6.
2. At this tissue pH, approximately 99% of local anesthetic molecules are present in the charged cationic (RNH+) form, with approximately 1% in the lipophilic free base (RN) form.
3. Approximately 10 RN molecules diffuse across the nerve sheath to reach the interior of the cell (contrasting with 250 RN molecules in the healthy example). The pH of the interior of the nerve cell remains normal (e.g., 7.4).
4. Extracellularly, the equilibrium between RNH+  RN, which has been disrupted, is reestablished. The relatively few newly created RN molecules diffuse into the cell, starting the entire process again. However, a sum total of fewer RN molecules succeed in eventually crossing the nerve sheath than would succeed at a normal pH because of greatly increased absorption of anesthetic molecules into the blood vessels in the region (increased vascularity is noted in the area of inflammation and infection).
RN, which has been disrupted, is reestablished. The relatively few newly created RN molecules diffuse into the cell, starting the entire process again. However, a sum total of fewer RN molecules succeed in eventually crossing the nerve sheath than would succeed at a normal pH because of greatly increased absorption of anesthetic molecules into the blood vessels in the region (increased vascularity is noted in the area of inflammation and infection).
5. After penetration of the nerve sheath by the base form, reequilibrium occurs inside the nerve. Approximately 75% of the molecules present intracellularly revert to the cationic form (RNH+), 25% remaining in the uncharged free base form (RN).
6. The cationic molecules bind to receptor sites within the sodium channel, resulting in conduction blockade.
Adequate blockade of the nerve is more difficult to achieve in inflamed or infected tissues because of the relatively small number of molecules able to cross the nerve sheath (RN) and the increased absorption of remaining anesthetic molecules into dilated blood vessels in this region. Although it presents a potential problem in all aspects of dental practice, this situation is seen most often in endodontics. Possible remedies are described in Chapter 16.
Clinical Implications of pH and Local Anesthetic Activity
Most commercially prepared solutions of local anesthetics without a vasoconstrictor have a pH between 5.5 and 7. When injected into tissue, the vast buffering capacity of tissue fluids returns the pH at the injection site to a normal 7.4. Local anesthetic solutions containing a vasopressor (e.g., epinephrine) are acidified by the manufacturer through the addition of sodium (meta)bisulfite to retard oxidation of the vasoconstrictor, thereby prolonging the period of effectiveness of the drug. (See Chapter 3 for a discussion of the appropriate use of vasoconstrictors in local anesthetics.)
Epinephrine may be added to a local anesthetic solution immediately before its administration without the addition of antioxidants; however, if the solution is not used in a short time, it will oxidize, slowly turning yellow then brown (much like a sliced apple oxidizing).
Rapid oxidation of the vasopressor may be delayed, thereby increasing the shelf life of the product, through the addition of antioxidants. Sodium bisulfite in a concentration between 0.05% and 0.1% is commonly used. A 2% solution of lidocaine HCl, with a pH of 6.8, is acidified to 4.2 by the addition of sodium bisulfite.
Even in this situation, the enormous buffering capacity of the tissues tends to maintain a normal tissue pH; however, it does require a longer time to do so after injection of a pH 4.2 solution than with a pH 6.8 solution. During this time, the local anesthetic is not able to function at its full effectiveness, resulting in a slower onset of clinical action for local anesthetics with vasoconstrictors compared with their plain counterparts.
Local anesthetics are clinically effective on both axons and free nerve endings. Free nerve endings lying below intact skin may be reached only by injection of anesthetic beneath the skin. Intact skin forms an impenetrable barrier to the diffusion of local anesthetics. EMLA (eutectic mixture of local anesthetics lidocaine and prilocaine) enables local anesthetics to penetrate intact skin, albeit slowly.35
Mucous membranes and injured skin (e.g., burns, abrasions) lack the protection afforded by intact skin, permitting topically applied local anesthetics to diffuse through to reach free nerve endings. Topical anesthetics can be employed effectively wherever skin is no longer intact because of injury, as well as on mucous membranes (e.g., cornea, gingiva, pharynx, trachea, larynx, esophagus, rectum, vagina, bladder).36
The buffering capacity of mucous membrane is poor; thus topical application of a local anesthetic with a pH between 5.5 and 6.5 lowers the regional pH to below normal, and less local anesthetic base is formed. Diffusion of the drug across the mucous membrane to free nerve endings is limited, and nerve block is ineffective. Increasing the pH of the drug provides more RN form, thereby increasing the potency of the topical anesthetic; however, the drug in this form is more rapidly oxidized. The effective shelf life of the local anesthetic is decreased as the pH of the drug is increased.30
To enhance the clinical efficacy of topical anesthetics, a more concentrated form of the drug is commonly used (5% or 10% lidocaine) than for injection (2% lidocaine). Although only a small percentage of the drug is available in the base form, raising the concentration provides additional RN molecules for diffusion and dissociation to the active cation form at free nerve endings.
Some topical anesthetics (e.g., benzocaine) are not ionized in solution; thus their anesthetic effectiveness is unaffected by pH. Because of the poor water solubility of benzocaine, its absorption from the site of application is minimal, and systemic reactions (e.g., overdose) are rarely encountered.
Kinetics of Local Anesthetic Onset And Duration of Action
Barriers to Diffusion of the Solution
A peripheral nerve is composed of hundreds to thousands of tightly packed axons. These axons are protected, supported, and nourished by several layers of fibrous and elastic tissues. Nutrient blood vessels and lymphatics course throughout the layers.
Individual nerve fibers (axons) are covered with, and are separated from each other by, the endoneurium. The perineurium then binds these nerve fibers together into bundles called fasciculi. The radial nerve, located in the wrist, contains between 5 and 10 fasciculi. Each fasciculus contains between 500 and 1000 individual nerve fibers. Five thousand nerve fibers occupy approximately 1 mm2 of space.
The thickness of the perineurium varies with the diameter of the fasciculus it surrounds. The thicker the perineurium, the slower the rate of local anesthetic diffusion across it.37 The innermost layer of perineurium is the perilemma. It is covered with a smooth mesothelial membrane. The perilemma represents the main barrier to diffusion into a nerve.
Fasciculi are contained within a loose network of areolar connective tissue called the epineurium. The epineurium constitutes between 30% and 75% of the total cross-section of a nerve. Local anesthetics are readily able to diffuse through the epineurium because of its loose consistency. Nutrient blood vessels and lymphatics traverse the epineurium. These vessels absorb local anesthetic molecules, removing them from the site of injection.
The outer layer of the epineurium surrounding the nerve is denser and is thickened, forming what is termed the epineural sheath or nerve sheath. The epineural sheath does not constitute a barrier to diffusion of local anesthetic into a nerve.
Table 1-5 summarizes the layers of a typical peripheral nerve.
TABLE 1-5
Organization of a Peripheral Nerve
| Structure | Description |
| Nerve fiber | Single nerve cell |
| Endoneurium | Covers each nerve fiber |
| Fasciculi | Bundles of 500 to 1000 nerve fibers |
| Perineurium* | Covers fasciculi |
| Perilemma* | Innermost layer of perineurium |
| Epineurium | Alveolar connective tissue supporting fasciculi and carrying nutrient vessels |
| Epineural sheath | Outer layer of epineurium |
*The perineurium and perilemma constitute the greatest anatomic barriers to diffusion in a peripheral nerve.
Induction of Local Anesthesia
Following administration of a local anesthetic into the soft tissues near a nerve, molecules of the local anesthetic traverse the distance from one site to another according to their concentration gradient. During the induction phase of anesthesia, the local anesthetic moves from its extraneural site of deposition toward the nerve (as well as in all other possible directions). This process is termed diffusion. It is the unhindered migration of molecules or ions through a fluid medium under the influence of the concentration gradient. Penetration of an anatomic barrier to diffusion occurs when a drug passes through a tissue that tends to restrict free molecular movement. The perineurium is the greatest barrier to penetration of local anesthetics.
Diffusion
The rate of diffusion is governed by several factors, the most significant of which is the concentration gradient. The greater the initial concentration of the local anesthetic, the faster the diffusion of its molecules and the more rapid its onset of action.
Fasciculi that are located near the surface of the nerve are termed mantle bundles (Fig. 1-19, A). Mantle bundles are the first ones reached by the local anesthetic and are exposed to a higher concentration of it. Mantle bundles usually are blocked completely shortly after injection of a local anesthetic (Fig. 1-19, B).
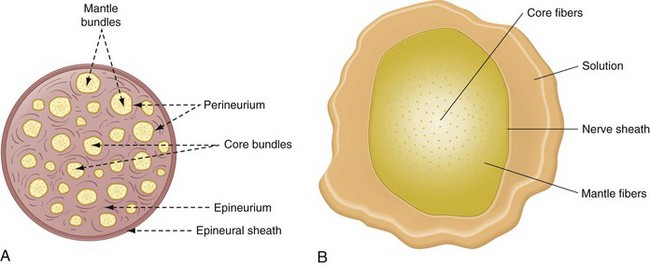
Figure 1-19 A, Composition of nerve fibers and bundles within a peripheral nerve. B, In a large peripheral nerve (containing hundreds or thousands of axons), local anesthetic solution must diffuse inward toward the nerve core from the extraneural site of injection. Local anesthetic molecules are removed by tissue uptake while tissue fluid mixes with the carrier solvent. This results in gradual dilution of the local anesthetic solution as it penetrates the nerve toward the core. A concentration gradient occurs during induction so that the outer mantle fibers are solidly blocked, whereas the inner core fibers are not yet blocked. Not only are core fibers exposed to a lower local anesthetic concentration, but the drug arrives later. Delay depends on the tissue mass to be penetrated and the diffusivity of the local anesthetic. (Redrawn from B, de Jong RH: Local anesthetics, St Louis, 1994, Mosby.)
Fasciculi found closer to the center of the nerve are called core bundles. Core bundles are contacted by a local anesthetic only after much delay and by a lower anesthetic concentration because of the greater distance that the solution must traverse and the greater number of barriers it must cross.
As the local anesthetic diffuses into the nerve, it becomes increasingly diluted by tissue fluids with some being absorbed by capillaries and lymphatics. Ester anesthetics undergo almost immediate enzymatic hydrolysis. Thus core fibers are exposed to a decreased concentration of local anesthetic, a fact that may explain the clinical situation of inadequate pulpal anesthesia developing in the presence of subjective symptoms of adequate soft tissue anesthesia. Complete conduction blockade of all nerve fibers in a peripheral nerve requires that an adequate volume, as well as an adequate concentration, of the local anesthetic be deposited. In no clinical situation are 100% of the fibers within a peripheral nerve blocked, even in cases of clinically excellent pain control.38 Fibers near the surface of the nerve (mantle fibers) tend to innervate more proximal regions (e.g., the molar area with an inferior alveolar nerve block), whereas fibers in the core bundles innervate the more distal points of nerve distribution (e.g., the incisors and canine with an inferior alveolar block).
Blocking Process
After deposition of local anesthetic as close to the nerve as possible, the solution diffuses in all directions according to prevailing concentration gradients. A portion of the injected local anesthetic diffuses toward the nerve and into the nerve. However, a significant portion of the injected drug also diffuses away from the nerve. The following reactions then occur:
1. Some of the drug is absorbed by nonneural tissues (e.g., muscle, fat).
2. Some is diluted by interstitial fluid.
3. Some is removed by capillaries and lymphatics from the injection site.
The sum total effect of these factors is to decrease the local anesthetic concentration outside the nerve; however, the concentration of local anesthetic within the nerve continues to rise as diffusion progresses. These processes continue until an equilibrium results between intraneural and extraneural concentrations of anesthetic solution.
Induction Time
Induction time is defined as the period from deposition of the anesthetic solution to complete conduction blockade. Several factors control the induction time of a given drug. Those under the operator’s control are the concentration of the drug and the pH of the local anesthetic solution. Factors not under the clinician’s control include the diffusion constant of the anesthetic drug and the anatomic diffusion barriers of the nerve.
Physical Properties and Clinical Actions
Other physicochemical factors of a local anesthetic may influence its clinical characteristics.
The effect of the dissociation constant (pKa) on the rate of onset of anesthesia has been described. Although both molecular forms of the anesthetic are important in neural blockade, drugs with a lower pKa possess a more rapid onset of action than those with a higher pKa.39
Lipid solubility of a local anesthetic appears to be related to its intrinsic potency. The estimated lipid solubilities of various local anesthetics are presented in Table 1-6. Greater lipid solubility permits the anesthetic to penetrate the nerve membrane (which itself is 90% lipid) more easily. This is reflected biologically in increased potency of the anesthetic. Local anesthetics with greater lipid solubility produce more effective conduction blockade at lower concentrations (lower percentage solutions or smaller volumes deposited) than is produced by less lipid-soluble local anesthetics.
TABLE 1-6
Chemical Structure, Physicochemical Properties, and Pharmacologic Properties of Local Anesthetic Agents
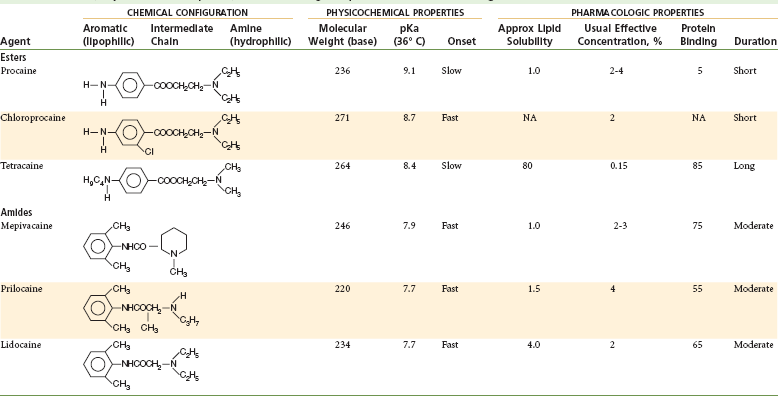
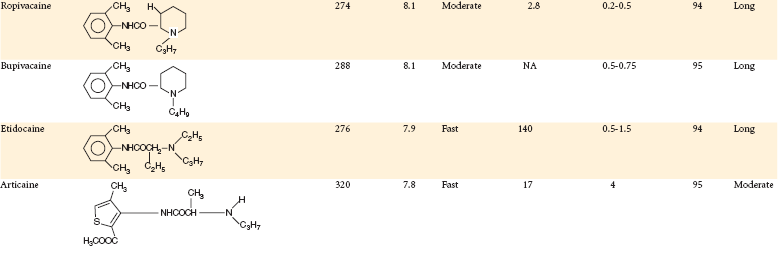
Modified from Rogers MC, et al, editors: Principles and practice of anesthesiology, St Louis, 1993, Mosby.
The degree of protein binding of the local anesthetic molecule is responsible for the duration of anesthetic activity. After penetration of the nerve sheath, a reequilibrium occurs between the base and cationic forms of the local anesthetic according to the Henderson-Hasselbach equation. Now, in the sodium channel itself, RNH+ ions bind at the receptor site. Proteins constitute approximately 10% of the nerve membrane, and local anesthetics (e.g., etidocaine, ropivacaine, bupivacaine) possessing a greater degree of protein binding (see Table 1-6) than others (e.g., procaine) appear to attach more securely to the protein receptor sites and to possess a longer duration of clinical activity.40
Vasoactivity affects both the anesthetic potency and the duration of anesthesia provided by a drug. Injection of local anesthetics, such as procaine, with greater vasodilating properties increases perfusion of the local site with blood. The injected local anesthetic is absorbed into the cardiovascular compartment more rapidly and is carried away from the injection site and from the nerve, thus providing for a shortened duration of anesthesia, as well as decreased potency of the drug. Table 1-7 summarizes the influence of various factors on local anesthetic action.
TABLE 1-7
Factors Affecting Local Anesthetic Action
| Factor | Action Affected | Description |
| pKa | Onset | Lower pKa = More rapid onset of action, more RN molecules present to diffuse through nerve sheath; thus onset time is decreased |
| Lipid solubility | Anesthetic potency | Increased lipid solubility = Increased potency (e.g., procaine = 1; etidocaine = 140) |
| Etidocaine produces conduction blockade at very low concentrations, whereas procaine poorly suppresses nerve conduction, even at higher concentrations | ||
| Protein binding | Duration | Increased protein binding allows anesthetic cations (RNH+) to be more firmly attached to proteins located at receptor sites; thus duration of action is increased |
| Nonnervous tissue diffusibility | Onset | Increased diffusibility = Decreased time of onset |
| Vasodilator activity | Anesthetic potency and duration | Greater vasodilator activity = Increased blood flow to region = Rapid removal of anesthetic molecules from injection site; thus anesthetic potency and duration are decreased |
From Cohen S, Burns RC: Pathways of the pulp, ed 6, St Louis, 1994, Mosby.
Recovery from Local Anesthetic Block
Emergence from a local anesthetic nerve block follows the same diffusion patterns as induction; however, it does so in the reverse order.
The extraneural concentration of local anesthetic is continually depleted by diffusion, dispersion, and uptake of the drug, whereas the intraneural concentration of local anesthetic remains relatively stable. The concentration gradient is reversed, the intraneural concentration exceeds the extraneural concentration, and anesthetic molecules begin to diffuse out of the nerve.
Fasciculi in the mantle begin to lose the local anesthetic much sooner than do the core bundles. Local anesthetic within the core then diffuses into the mantle, so that the first nerve fibers to entirely lose anesthesia are those centermost in the nerve. Mantle fibers remain anesthetized the longest, and core fibers the shortest, time. Recovery from anesthesia is a slower process than induction because the local anesthetic is bound to the drug receptor site in the sodium channel and therefore is released more slowly than it is absorbed.
Readministration of Local Anesthetic
Occasionally a dental procedure outlasts the duration of clinically effective pain control, and a repeat injection of local anesthetic is necessary. Usually this repeat injection immediately results in a return of profound anesthesia. On some occasions, however, the clinician may encounter greater difficulty in reestablishing adequate pain control with subsequent injections.
Recurrence of Immediate Profound Anesthesia
At the time of reinjection, the concentration of local anesthetic in the core fibers is less than that in the mantle fibers. Partially recovered core fibers still contain some local anesthetic, although not enough to provide complete anesthesia. After deposition of a new high concentration of anesthetic near the nerve, the mantle fibers are once again exposed to a concentration gradient directed inward toward the nerve; this eventually leads to an increased concentration in the core fibers. This combination of residual local anesthetic (in the nerve) and the newly deposited supply results in rapid onset of profound anesthesia and administration of a smaller volume of local anesthetic drug.
Difficulty Reachieving Profound Anesthesia
In this second situation, as in the first, the dental procedure has outlasted the clinical effectiveness of the local anesthetic drug, and the patient is experiencing pain. The doctor readministers a volume of local anesthetic, but unlike in the first scenario, effective control of pain does not occur.
Tachyphylaxis: In this second clinical situation, a process known as tachyphylaxis occurs. Tachyphylaxis is defined as increasing tolerance to a drug that is administered repeatedly. It is much more likely to develop if nerve function is allowed to return before reinjection (e.g., if the patient complains of pain). The duration, intensity, and spread of anesthesia with reinjection are greatly reduced.41
Although difficult to explain, tachyphylaxis is probably brought about by some or all of the following factors: edema, localized hemorrhage, clot formation, transudation, hypernatremia, and decreased pH of tissues. The first four factors isolate the nerve from contact with the local anesthetic solution. The fifth, hypernatremia, raises the sodium ion gradient, thus counteracting the decrease in sodium ion conduction brought about by the local anesthetic. The last factor, a decrease in pH of the tissues, is brought about by the first injection of the acidic local anesthetic. The ambient pH in the area of injection may be somewhat lower, so that fewer local anesthetic molecules are transformed into the free base (RN) on reinjection.
Duration of Anesthesia
As the local anesthetic is removed from the nerve, the function of the nerve returns rapidly at first, but then it gradually slows. Compared with onset of the nerve block, which is rapid, recovery from the nerve block is much slower because the local anesthetic is bound to the nerve membrane. Longer-acting local anesthetics (e.g., bupivacaine, etidocaine, ropivacaine, tetracaine) are more firmly bound in the nerve membrane (increased protein binding) than are shorter-acting drugs (e.g., procaine, lidocaine) and therefore are released more slowly from receptor sites in the sodium channels. The rate at which an anesthetic is removed from a nerve has an effect on the duration of neural blockade; in addition to increased protein binding, other factors that influence the rate of removal of a drug from the injection site are the vascularity of the injection site and the presence or absence of a vasoactive substance. Anesthetic duration is increased in areas of decreased vascularity (e.g., Gow-Gates mandibular nerve block vs. inferior alveolar nerve block), and the addition of a vasopressor decreases tissue perfusion to a local area, thus increasing the duration of the block.
References
1. Covino, BG, Vassallo, HG. Local anesthetics: mechanisms of action and clinical use. New York: Grune & Stratton; 1976.
2. Bennett, CR. Monheim’s local anesthesia and pain control in dental practice, ed 5. St Louis: Mosby; 1974.
3. Fitzgerald, MJT. Neuroanatomy: basic and clinical. London: Baillière Tyndall; 1992.
4. Noback, CR, Strominger, NL, Demarest, RJ. The human nervous system: introduction and review, ed 4. Philadelphia: Lea & Febiger; 1991.
5. Singer, SJ, Nicholson, GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731.
6. Guyton, AC. Basic neuroscience: anatomy and physiology, ed 2. Philadelphia: WB Saunders; 1991.
7. Guidotti, G. The composition of biological membranes. Arch Intern Med. 1972;129:194–201.
8. Denson, DD, Maziot, JX. Physiology, pharmacology, and toxicity of local anesthetics: adult and pediatric considerations. In: Raj PP, ed. Clinical practice of regional anesthesia. New York: Churchill Livingstone, 1991.
9. Heavner, JE. Molecular action of local anesthetics. In: Raj PP, ed. Clinical practice of regional anesthesia. New York: Churchill Livingstone, 1991.
10. de Jong, RH. Local anesthetics, ed 2. Springfield, Ill: Charles C Thomas; 1977.
11. Hodgkin, AL, Huxley, AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol (London). 1954;117:500–544.
12. Noback, CR, Demarest, RJ. The human nervous system: basic principles of neurobiology, ed 3. New York: McGraw-Hill; 1981. [pp 44–45].
13. Keynes, RD. Ion channels in the nerve-cell membrane. Sci Am. 1979;240:326–335.
14. Cattarall, WA. Structure and function of voltage-sensitive ion channels. Science. 1988;242:50–61.
15. Hille, B. Ionic selectivity, saturation, and block in sodium channels: a four-barrier model. J Gen Physiol. 1975;66:535–560.
16. Ritchie, JM. Physiological basis for conduction in myelinated nerve fibers. In: Morell P, ed. Myelin. ed 2. New York: Plenum Press; 1984:117–145.
17. Franz, DN, Perry, RS. Mechanisms for differential block among single myelinated and non-myelinated axons by procaine. J Physiol. 1974;235:193–210.
18. de Jong, RH, Wagman, IH. Physiological mechanisms of peripheral nerve block by local anesthetics. Anesthesiology. 1963;24:684–727.
19. Dettbarn, WD. The acetylcholine system in peripheral nerve. Ann N Y Acad Sci. 1967;144:483–503.
20. Goldman, DE, Blaustein, MP. Ions, drugs and the axon membrane. Ann N Y Acad Sci. 1966;137:967–981.
21. Wei, LY. Role of surface dipoles on axon membrane. Science. 1969;163:280–282.
22. Lee, AG. Model for action of local anesthetics. Nature. 1976;262:545–548.
23. Seeman, P. The membrane actions of anesthetics and tranquilizers. Pharmacol Rev. 1972;24:583–655.
24. Strichartz, GR, Ritchie, JM. The action of local anesthetics on ion channels of excitable tissues. In: Strichartz GR, ed. Local anesthetics. New York: Springer-Verlag, 1987.
25. Butterworth, JF, IV., Strichartz, GR. Molecular mechanisms of local anesthesia: a review. Anesthesiology. 1990;72:711–734.
26. Ritchie, JM. Mechanisms of action of local anesthetic agents and biotoxins. Br J Anaesth. 1975;47:191–198.
27. Rasminsky, M. Conduction in normal and pathological nerve fibers. In: Swash M, Kennard C, eds. Scientific basis of clinical neurology. Edinburgh: Churchill Livingstone, 1985.
28. Ritchie, JM. The distribution of sodium and potassium channels in mammalian myelinated nerve. In: Ritchie JM, Keyes RD, Bolis L, eds. Ion channels in neural membranes. New York: Alan R Liss, 1986.
29. Hille, B, Courtney, K, Dum, R. Rate and site of action of local anesthetics in myelinated nerve fibers. In: Fink BR, ed. Molecular mechanisms of anesthesia. New York: Raven Press; 1975:13–20.
30. Setnikar, I. Ionization of bases with limited solubility: investigation of substances with local anesthetic activity. J Pharm Sci. 1990;55:1190–1195.
31. Malamed, SF. Buffering local anesthetics in dentistry. ADSA Pulse. 2011;44(3):8–9.
32. Stewart, JH, Chinn, SE, Cole, GW, Klein, JA. Neutralized lidocaine with epinephrine for local anesthesia-II. J Dermatol Surg Oncol. 1990;16:942–945.
33. Bokesch, PM, Raymond, SA, Strichartz, GR. Dependence of lidocaine potency on pH and pCO2. Anesth Analg. 1987;66:9–17.
34. Bieter, RN. Applied pharmacology of local anesthetics. Am J Surg. 1936;34:500–510.
35. Buckley, MM, Benfield, P. Eutectic lidocaine/prilocaine cream: a review of the topical anesthetic/analgesic efficacy of a eutectic mixture of local anesthetics (EMLA). Drugs. 1993;46:126–151.
36. Campbell, AH, Stasse, JA, Lord, GH, Willson, JE. In vivo evaluation of local anesthetics applied topically. J Pharm Sci. 1968;57:2045–2048.
37. Noback, CR, Demarest, RJ. The human nervous system: basic principles of neurobiology, ed 3. New York: McGraw-Hill; 1981.
38. de Jong, RH. Local anesthetics, ed 2. Springfield, Ill: Charles C Thomas; 1977. [pp 66–68].
39. Ritchie, JM, Ritchie, B, Greengard, P. The active structure of local anesthetics. J Pharmacol Exp Ther. 1965;150:152.
40. Tucker, GT. Plasma binding and disposition of local anesthetics. Int Anesthesiol Clin. 1975;13:33.
41. Cohen, EN, Levine, DA, Colliss, JE, Gunther, RE. The role of pH in the development of tachyphylaxis to local anesthetic agents. Anesthesiology. 1968;29:994–1001.