β-MANNOSIDOSIS
β-Mannosidosis is an autosomal recessive genetic trait of Anglo-Nubian goats and Salers calves. Mannosidosis in goats has a worldwide distribution, with reported cases occurring in Australia, Canada, and the United States.1686-1690 The frequency of the condition is estimated at 1 in 2000 births of purebred Salers calves. The disease is seen in both the red and the black phenotype of Salers. The gene encoding for β-mannosidase has been characterized in both cattle and goats.1691,1692 A single base mutation in the complementary DNA (cDNA) coding for the enzyme results in premature termination of translation. PCR-based tests have been used to identify β-mannosidosis carriers of both species.1692,1693
The lesions of mannosidosis have been identified in aborted fetuses and fetuses in utero, and clinical signs are often present at birth.1694-1698
The clinical signs in goats include recumbency at birth, deafness, shortened sternum, narrowed palpebral fissures, decreased muscle mass, intentional head tremor, carpal contractures, pastern joint hyperextension, thickened skin, shortened head, excessive gingival tissue, short curled ears, and domed skull.1686,1699 Numerous ocular changes are seen, including pendular nystagmus, ventrolateral strabismus, thickened immovable eyelid, hazy vitreous humor, ptosis, and Horner’s syndrome.1700 The head movements are described as wide circular motions that culminate with the animal in lateral recumbency. Pupillary and corneal reflexes are intact, and the affected animals appear to have some vision. Compared with goats, affected calves respond to aural and visual stimuli. Other neurologic abnormalities include recumbency, depression, loss of suckle reflex, spontaneous chewing activity, head tremors, depression, and nystagmus.
The diagnosis of β-mannosidosis is based on observation of characteristic microscopic lesions in the central nervous system (CNS), demonstration of decreased tissue β-mannosidase activity, and demonstration of mannose-based oligosaccharides (Nanβ1-4G1cNAc and Manβ1-4G1cNAcβ1-4G1cNAc) in the CNS.1701 The concentration of serum thyroid hormones is decreased.1702 Some affected calves have concomitant colisepticemia or bovine viral diarrhea (BVD) infection.
In contrast to α-mannosidase, β-mannosidase has no isoenzymes.1703 The mean concentration of β-mannosidase in the plasma of normal goats ranges from 66 to 222 nmol/hr/mL of plasma.1695 There is no detectable β-mannosidase activity in the plasma of affected goats, and the activity in heterozygotes is intermediate between these ranges. The plasma concentrations of β-mannosidase in heterozygotes range from 43 to 64 nmol/hr/mL. These tests cannot be interpreted rigidly because there is significant variability among assays, storage conditions, and different age-groups of cattle.1690
In addition to the CNS abnormalities, cardiomegaly, thyromegaly, and pathologic fractures have been described in affected goats. Calves show a cerebral ventricular dilation and green discoloration of the renal cortices. Microscopic pathologic changes in the CNS include hypomyelination, axonal spheroids, and foamy-appearing neuronal cytoplasm.1703 The heat shock protein ubiquitin has been detected in the CNS of affected calves.1704 Cytoplasmic vacuolation also is present in the visceral organs.1705,1706 Although in utero bone marrow or stem cell transplantation may offer hope for alleviation of the disease in the postnatal animal, no practical treatment exists for animals with β-mannosidosis.1698
GENERALIZED GLYCOGENOSIS (GM1 GANGLIOSIDOSIS; β-GALACTOSIDASE DEFICIENCY)
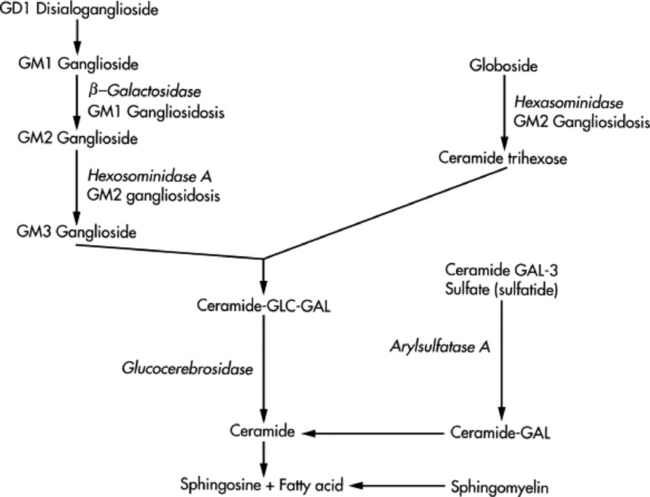
Generalized glycogenosis is a rare heritable defect of Holstein cattle and several breeds of sheep.1707-1712 Generalized glycogenosis results from deficient activity of β-galactosidase, resulting in accumulation of the GM1 ganglioside, asialo-GM1, and neutral long-chain oligosaccharides in the tissues1708 (Fig. 35-25).
A combined deficiency of β-galactosidase and α-neuraminidase has also been described.1713 This condition is thought to result from a defect of the structural gene of β-galactosidase. The loss of α-neuraminidase occurs because of an inability of the βα-galactosidase molecule to dimerize with α-neuraminidase, leading to deactivation of both molecules.
The clinical signs of all forms of β-galactosidase deficiency are similar. Affected animals tend to show lethargy and anorexia by 1 month of age. The head and neck are held low and rigidly extended. The animals are depressed, stiff, and ataxic, have a base-wide stance, appear to be blind, and eventually become recumbent. The animals tend to fall whenever the head is moved. The blindness is the result of dysmyelination in the optic nerve, which can be detected by observation of numerous small white spots on the retina. None of these signs are pathognomonic for the disease, so biochemical testing or lectin histochemistry to characterize the stored carbohydrates in affected cells is needed for specific diagnosis.1714,1715
The genetic disorder is thought to be caused by an autosomal recessive gene. A biochemical test for β-galactosidase using centrifugally purified bovine neutrophils has been described.1708 Animals with fewer than 3 IU of heat-stable activity are considered deficient. In herds in which GM1 gangliosidosis has already been substantiated, observation of slowness in feeding and lack of alertness has proved to be diagnostically significant.
Pathologic changes of GM1 gangliosidosis are present in the fetus as well as postnatally and include neuronal enlargement, vacuolation, accumulation of granular material in the nerve cells, spheroids, and loss of neurons without gliosis.1716,1717 The material stains strongly with periodic acid–Schiff (PAS)/alcian blue, Sudan black, and oil red O. The vacuolar contents are composed of complex lipopolysaccharides, including β-galactose, N-acetylneuraminic acid and N-acetylgalactosamine.1718
No treatment is currently available for this disease. Strategies such as in utero bone marrow transplantation or stem cell transplantation may hold promise for the future, but are unlikely to become a viable option in large animal species.1719
BOVINE GENERALIZED GLYCOGENOSIS (TYPE II GLYCOGENOSIS; POMPE’s DISEASE)
Generalized glycogenosis results from a dysfunction of α-glucosidase. The condition occurs in shorthorn and Brahman cattle1720-1722 and is controlled by a single recessive allele.1723 Several different mutations are described within the bovine α-glucosidase gene and are specific to the breeds.1724 Two separate clinical entities have been described: the cardiac (infantile) form and the late-onset form. Clinical signs of the infantile form are first seen at about 2 to 3 months of age and include growth failure, weakness, hyperesthesia, muscle tremors, ataxia, conscious proprioceptive deficits, and recumbency.1723 The cardiac (infantile) form is characterized by right-sided heart failure at 3 to 5 months of age. Brahman calves with the late-onset form die at age 8 to 9 months, whereas affected shorthorn calves may survive for more than a year. The tissues of affected cattle contain only 2% to 5% of the normal α-glucosidase activity.1723,1725 The concentration of glycogen in the liver and muscles is increased.
The pathologic lesions of the CNS in both forms are similar to those of cattle with α-mannosidosis, including cytoplasmic swelling and foamy cytoplasm in the CNS neurons. Lesions are found in the myocardium and skeletal muscle. These include vacuolation and swelling of Purkinje’s cells and myofibers.1726 Because of a gene dilution effect, the activity of α-glucosidase in peripheral blood lymphocytes can be used to detect asymptomatic heterozygotes.1727 However, the test result may be falsely positive if the animals are ingesting seeds of Castanospermum australe (Moreton Bay chestnut trees). The seeds of this tree have an α-glucosidase antagonist. Hematopoeitic chimerism in twin animals, on the other hand, can result in normal levels of enzyme activity in animals that are heterozygote carriers for the disease.1728 Enzymatic methods for detecting carriers have now been superseded by DNA testing of leukocytes or hair root samples.1729
GLOBOID CELL LEUKODYSTROPHY (KRABBE’S DISEASE)
Globoid cell leukodystrophy has been reported in polled Dorset sheep.1730 The genetic defect is the result of a lack of galactocerebrosidase, which produces a high concentration of galactocerebroside in myelin. The clinical signs are seen by 4 months of age. Affected animals show depression, hypersensitivity, conscious proprioceptive deficits, slight tremor of the head and neck, exaggerated patellar reflex, incoordination, and tetraplegia. The activity of the tissue galactocerebrosidase can be measured to substantiate the clinical diagnosis. Differential diagnoses include other heritable disorders, such as neuropathy and abiotrophy of the cerebellum and swainsonine (locoweed) toxicity (see later).1731
NEURONAL LIPODYSTROPHY
Neuronal lipodystrophy occurs in Angus and Beefmaster calves and in sheep.1732-1734 The biochemical lesion of the condition is unknown. The clinical signs are first seen at about 10 months of age and include depression, blindness, ataxia, circling, coma, and tonic-clonic convulsions. The pathologic lesions include neuronal vacuolation with eosinophilic and sudanophilic inclusions. The inclusions are cytoplasmic and perinuclear and are located in the axonal and dendritic zones. As with other neurovisceral storage diseases, the inclusions are bound by a single membrane. Involvement of the spleen and lymph nodes also can be demonstrated. The mode of inheritance is unknown. There is no effective treatment for the disease.
SHAKER CALF SYNDROME
Shaker calf syndrome is an inherited neurodegenerative disorder of newborn horned Hereford calves.1735 The condition is characterized by recumbency, fine tremors of the neck and hindlimbs, and hypermetria. The amplitude and frequency of the tremors are increased by stimulation. Other clinical signs include aphonia, loss of fine motor control of the tongue, hyperesthesia, exaggerated spinal reflexes, and hypertonia. Most affected calves die of starvation by 5 days of life; however, one case of remission followed by relapse after 2 weeks has been described. The cause of the disease is unknown. Limited breeding trials indicate a 12.5% inbreeding factor in affected calves, suggesting a hereditary etiology. The pathologic lesions include a neurofilamentous neuronal degeneration of multiple cell groups of the central nervous system (CNS) and of ganglion cells of the peripheral and autonomic nervous systems. The spinal cord is most severely affected. Neuronal degenerative changes include distention of axons and dendrites by a faintly fibrillar material, neuronophagia, and reactive gliosis. Wallerian degeneration of the spinal rootlets, spheroids, and empty fiber tracts in the spinal cord are noted. The pathologic appearance of the tissues differs from that of calves with hereditary neuraxial edema.
MAPLE SYRUP URINE DISEASE (SPONGIFORM ENCEPHALOPATHY)
Maple syrup urine disease (MSUD) is a hereditary spongiform encephalopathy characterized by severe CNS disturbance in newborn Hereford and polled shorthorn calves.1736 The disease has been reported in Australia and Canada and may have occurred in the United States.1737 The biochemical lesion is a deficiency of branched-chain 2-oxo acid dehydrogenase, which results in accumulation of the 2-oxo acids 4-methyl-2-oxopentanoate, 3-methyl-2-oxobutanoate, (S)-(S-KMV), and (R)-3-methyl-2-oxopentanoate, as well as their precursors leucine, valine, isoleucine, and alloisoleucine.1736,1738,1739 The urine becomes highly viscous, discolored, and malodorous because of the excretion of these substances through the kidneys. The buildup of the transamination product of isoleucine, α-keto-β-methylvaleric acid, probably gives the urine the odor of burnt maple syrup. Branched-chain α-keto acids have been shown to inhibit energy production in the brain and to cause morphologic changes and death of astrocytes in vitro.1740,1741 High levels of leucine are directly neurotoxic.1742 Pyruvate is a vital constituent of the Krebs cycle, which is important for the production of transmitter amino acids. The CNS hyperactivity probably is related to a decrease in GABA-mediated inhibitory transmission. The genetic defect in polled shorthorns and polled Herefords has been shown to be a thymidine-to-cytidine transition in the cDNA coding for a subunit of the branched-chain amino acid dehydrogenase, resulting in a substitution of leucine for proline.1743 PCR testing now can be used to detect both affected and carrier animals. A protocol for genotyping cattle for both MSUD and inherited neuraxial edema has been used to estimate the frequency of the alleles responsible for these diseases in Australian cattle (0.01 to 0.02).1744
Affected calves are born normal but are depressed by 2 to 3 days. They also are febrile (39.5° C to 42° C; 103.1° F to 107.6° F). They initially show ataxia and depression and become recumbent by the second to third day of life. At that time they show hyperesthesia, opisthotonos, muscular rigidity, myoclonic limb jerks, nystagmus, repetitive head tremors, stimulus-induced tetanic spasms, blepharospasm, generalized decrease of spinal reflexes, and convulsions. The urine has the characteristic color and odor reminiscent of burnt maple syrup. The calves usually die by 5 to 10 days of age. The presence of ketoacids can be detected by mixing urine with dinitrophenylhydrazine and observing a faint-yellow precipitate.1745
The clinical presentation of MSUD differs from that of hereditary neuraxial edema.1737 Calves with neuraxial edema have extensor rigidity but tend to be bright and alert, whereas MSUD calves have rigid extensor tonus and obtundation. These differential features appear to be significant because the original reports of hereditary neuraxial edema likely included calves with MSUD1746 (see following section).
Spongiform changes caused by intramyelinic vacuolation are present in the brains of affected calves in both white matter and gray matter.1747 MSUD can be definitively diagnosed by measuring the ratio of isoleucine, leucine, and valine to α-aminobutyric acid in fixed tissues and finding increased concentrations of these amino acids or their corresponding branched-chain 2-keto acids in urine or blood.1747 Heterozygotes have normal blood and urine levels of both amino acids and keto acids. PCR analysis of DNA extracted from hair root samples can identify both homozygote affected animals and the clinically normal heterozygote carriers of the disease.1748-1750 Pharmacologic dosages of thiamine are beneficial for treatment of MSUD in some human patients, probably by increasing mitochondrial thiamin diphosphate, which promotes the activity of the branched-chain α-keto acid dehydrogenase complex.1751 However, there is no known effective treatment for MSUD in calves.1736,1737
HEREDITARY NEURAXIAL EDEMA (CONGENITAL MYOCLONUS; DODDLER SYNDROME)
Neuraxial edema is an inherited neurologic disease of newborn calves. Polled and horned Herefords and Hereford-Friesian crossbred cattle are affected.1752-1759 An autosomal recessive genetic trait is thought to be responsible for the condition. The disease is well defined clinically and pathologically1756,1759; however, some investigators have reported a disease of polled Hereford calves that has similar neurologic signs but does not result in status spongiosus or CNS edema. These calves had a high frequency of bilateral slippage of the capital femoral epiphysis, subluxation of the femoral head, and acetabular articular cartilage fractures.1757 The authors named the condition “congenital myoclonus” to differentiate it from hereditary neuraxial edema. Calves with congenital myoclonus have a shorter-than-normal gestational length.
The earliest reports of hereditary neuraxial edema also described two clinical forms in which some calves were bright, alert, and responsive and others had a severely depressed sensorium. Subsequent studies suggested that the calves with systemic depression probably had MSUD (see previous section) and those with normal sensorium had hereditary neuraxial edema.
The clinical signs of hereditary neuraxial edema include hyperesthesia and myoclonic discharges of skeletal musculature that occur spontaneously or in response to tactile, visual, or auditory stimuli. The calves are stillborn or are affected at birth. Affected calves are of normal size but are unable to rise; they lie quietly without lifting the head.1752-1755 These calves develop marked extensor tonus and clonic spasms of the limbs and head when stimulated. During the spasm the animals become transiently apneic and remain dyspneic for several minutes.1756 The spasms are less severe after repeated stimulation. Between spasms the patients can stand with assistance, but the proprioceptive responses are greatly altered, and the animals fall when support is withdrawn.1755 The sensorium and suckling reflexes are unaltered when the calves are not in a spasmodic episode. Some authors have reported that vision and cranial nerve function are unimpaired, but others have reported nystagmus in some calves.1752,1756 Administration of anticonvulsant drugs does not ameliorate the clinical signs. Pathologic lesions usually are not seen in the CNS of affected calves.1753 The condition has an autosomal recessive mode of inheritance. The defect lies within the postsynaptic glycinergic receptors in the inhibitory interneurons of the spinal cord.1760 A protocol for genotyping cattle for both inherited neuraxial edema and MSUD has been used to estimate the frequency of the alleles responsible for these diseases in Australian cattle (0.01 to 0.02).1744
INHERITED MYOCLONUS OF PERUVIAN PASO FOALS
Inherited myoclonus is a disorder of Peruvian Paso foals that is characterized by myoclonic contractions of the musculature in response to auditory or tactile stimuli.1761 These contractions are sustained with repeated stimulation. Some animals are ambulatory but have a “rabbit hopping” gait. Some animals are recumbent. If assisted, the foals can rise and walk, and the animals are not depressed. Analeptic drugs and tranquilizers are ineffective for controlling this condition. Inherited myoclonus is associated with a specific deficiency of spinal glycine receptors, which are responsible for synaptic inhibition in the CNS. Glycine is a major inhibitory transmitter and works through the Ia afferent neurons in the ventral columns and the Renshaw cells. Loss of the receptors results in uninhibited synaptic transmission.
CONGENITAL ENCEPHALOMYELOPATHY IN QUARTER HORSES
Congenital encephalomyelopathy has been described in quarter horse foals.1762 The condition occurred in three foals born to two different mares and three unrelated stallions. The condition was seen at birth, and clinical signs include recumbency and coarse tremors of the hindlimbs. When assisted into a standing position, the hindquarters bounced off the ground. The forelimb function appeared normal; however, the patellar reflexes were exaggerated. Affected foals are bright, alert, and responsive and have intact pain perception. There are no macroscopic CNS lesions. Microscopic lesions include spongiform degeneration and axonal swelling of the white matter of the medulla, spinocerebellar tracts, and spinothalamic tracts. The lesions extend through the entire length of the ventral funiculi of the spinal cord.
LOCOWEED POISONING (ACQUIRED MANNOSIDOSIS, ASTRAGALUS And OXYTROPIS POISONING; LOCOISM; SWAINSONINE TOXICITY; IPOMOEA And SIDA CARPINIFOLIA TOXICITIES)
Definition and Etiology
Chronic ingestion of a variety of different plants worldwide can result in an acquired neurovisceral storage disease. These include plants of the Astragalus and Oxytropis genera (locoweeds) in the western United States, Canada, and Australia; Ipomoea (shrubby morning glory) in Mozambique; the darling pea in Australia (Swainsona species), and Sida carpinifolia in Brazil (Table 35-14).1763-1770 Horses are most susceptible to intoxication, but cattle, sheep, goats, and deer also can be affected.
Table 35-14 Species of Astragalus and Oxytropis Found in Western United States
| Scientific Name |
Common Name |
| Oxytropis sericea |
White locoweed, point locoweed |
| Oxytropis lambertii |
Purple locoweed |
| Oxytropis campestris |
Yellow locoweed |
| Astragalus argillophilus |
Half-moon locoweed |
| Astragalus bisculatus |
Two-grooved milk vetch |
| Astragalus earlei |
Earles locoweed |
| Astragalus lentiginosus |
Speckled, spotted locoweed |
| Astragalus mollissimus |
Wooly locoweed |
| Astragalus mothoxys |
Sheep locoweed |
| Astragalus wootnii |
Wooton locoweed |
From Knight AP: Compend Cont Educ (Pract Vet) 9:F418, 1987.
Conditions that promote locoweed poisoning are hot, dry weather and a scarcity of alternative forage. Horses may be more prone to graze on locoweed than cattle, particularly when other green forage is scarce, and may increase their consumption over a single season.1771 Chronically exposed livestock can become habituated and feed selectively on the plant over successive grazing seasons.1772
The following toxic components have been identified:

Locoine, swainsonine n-oxide, and indolizidine alkaloids, which interfere with the activity of α-mannosidase and of Golgi mannosidase II.
1764,1765,1773,1774 β-Galactosidase and β-glucosidase.
1775
Aminonitrile, a compound that caused abortion and teratogenesis.
Miserotoxin, a compound that causes a respiratory disease complex.
Selenium accumulation (selenium toxicity may contribute to the neurotoxicity and fetal deformities).
Oxytropis and Astragalus plants are legumes that have herbaceous stems and alternate pinnately compound leaves. 1776 The fruits are characteristic leguminous pods that contain kidney-shaped seeds with pods marked by characteristic longitudinal grooves.1776 The plant is eaten because it is the first vegetation available in the spring. The Oxytropis and Astragalus species found in the United States are listed in Table 35-14. Symptoms do not usually develop in cattle until 3 weeks after the animals first begin grazing the plant and may not occur until long after ingestion of the plant has stopped. The toxicity of the locoweed may vary from year to year and even within one season.1771 Despite its relative unpalatability, some sheep may become habituated to the plant and selectively eat forage containing up to 20% Astragalus plants.1772
Clinical Signs
Experimentally and naturally poisoned horses show clinical signs by 2 to 3 weeks after continuous ingestion of locoweed.1769,1771 The clinical signs include ataxia, conscious proprioceptive deficits, and depression, with alternating periods of frenzied or manic activity. There is sometimes a high-stepping, stringhalt-like gait.1777 At rest the horses show intentional head tremor, flaccidity of the nose and lips, repetitive movements with the lips and tongue, and dysphagia.1777 The clinical signs worsen when affected horses are handled or transported. Signs in goats are similar, including ataxia, hypermetria, hyperesthesia, and muscle tremors.1766 Forcing the head backward can result in falling, nystagmus, opisthotonos, seizures, and tetany.1778 Tranquilization usually is ineffective for controlling the apparent hyperexcitability. Horses that survive locoweed poisoning retain an altered behavior. Abortions, stillbirths, and neonatal deaths can occur in all species exposed to these toxic plants, regardless of whether the dams have clinical signs of neurologic disease.1764,1766-1768
Neurologic signs in adult cattle include conscious proprioceptive deficits, hypermetria, weakness, depression, dull staring eyes, and loss of herding instinct. Heavy losses from abortions or malformed calves have also been described. The indolizidine alkaloids are secreted in the milk and may cause unthriftiness and weak suckling behavior in calves. Calves that have been exposed to the toxin in utero are weak and fail to thrive. Some may have flexural contractions of the limbs and lateral rotation of the carpus.1779-1782 Ingestion of locoweed by certain cattle at high altitudes may result in the development of cor pulmonale.1779,1780 Many cattle with mild signs of locoweed poisoning recover completely by 60 days after removal from the offending pastures. Ruminants with advanced chronic intoxication apparently have permanent loss of neural tissue.
Poisoned sheep have a star-gazing attitude and appear to be blind, nervous, and stiff. The normal flocking behavior is absent.1765 Affected sheep may exhibit ptyalism. Testicular atrophy has been reported in rams.1783 Affected animals have intercurrent pyogenic infections such as pneumonia, keratoconjunctivitis, and foot rot. This is thought to be related to the immunosuppressive effect of the plants on the T lymphocytes.1784 Depression and reluctance to move induced by locoweed toxicity may predispose animals to other problems, such as water deprivation.1785
Clinical Pathology
Microscopic examination of stained blood smears may reveal the presence of vacuolation in the cytoplasm of the lymphocytes. These changes are found in the majority of lysosomal storage disorders, but might be considered diagnostic of locoweed poisoning in animals with characteristic clinical signs and historical evidence of exposure. Serum concentrations of alkaline phosphatase (ALP) and aspartate transaminase (AST) may be helpful markers for toxicity; elevated concentrations were detected in sheep exposed to swainsonine in Oxytropis sericea.1786 Definitive diagnosis can be established by assaying sera from affected animals for α-mannosidase and swainsonine.1787
Pathology
The microscopic abnormalities of the soft tissues of acutely poisoned animals are similar to those of the inherited lysosomal storage diseases of cattle, including cytoplasmic vacuolation of neurons, particularly Purkinje’s cells, and cells in various other tissues.1766,1777 Paraffin-embedded tissues can be examined using lectin histochemistry to characterize the stored material within the vacuoles.1766,1788 Vacuolation of renal tubular epithelial cells may occur as early as 4 days after the start of daily feeding of 0.34 kg of locoweed to horses and may be present in animals exposed to toxic plants but clinically normal.1766,1789 Pulmonary lesions associated with chronic ingestion of locoweed that may predispose to high-altitude disease (brisket disease) in cattle (see Chapter 31) include alveolar emphysema, bronchiolar constriction and hypertrophy, and interlobular edema and fibrosis. Pyloric or gastric ulcers have been reported in affected cattle.1782,1789 Placental edema, fetal ascites, and hydrops allantois have been described in exposed pregnant cattle.
Treatment
There is no known effective long-term therapy for locoweed poisoning. Animals remain affected for a prolonged period after removal from the plants and may be permanently afflicted. Some recommend either tranylcypromine (60 mg PO), a monoamine oxidase inhibitor, and protryptyline (60 mg PO) or reserpine (3.125 g/500 kg IM once or 1.25 mg PO per animal once daily) for treatment of chronically affected animals.1790 However, the efficacy of these treatments is unknown. Addition of a mineral supplement and a natural clay (clinoptilolite) to the diet of cattle ingesting locoweed did not prevent toxicity.1791
Prevention
Nonaddicted livestock normally do not eat locoweed if other forage is available. The intoxication may be prevented by supplemental feeding during the early spring and late summer. One report has described conditioning aversion to locoweed in horses using lithium chloride administered simultaneously with grazing of Oxytropis sericea.1792 Whether this is a practical management tool in large numbers of animals has yet to be determined.
GRASS STAGGERS
Grass staggers is caused by a number of related products of plant or fungal metabolism. These compounds appear to have universal activity at the γ-aminobutyric acid (GABA) receptor of the internuncial neurons; therefore, intoxication causes clinical signs characteristic of released inhibition. The structural backbone of these toxins permits the molecules to bind to GABA receptors, thereby inactivating them. Some associated plant or fungal toxins also induce other physiologic effects, including agalactia, fever, and low productivity, because of a prolactin-like effect.
RYEGRASS STAGGERS
Perennial Ryegrass Staggers
Ingestion of toxic stands of perennial ryegrass (Lolium perenne) results in ataxia and tremors in horses, cattle, and sheep. The condition is recognized in livestock of New Zealand, Australia, Northern Europe, United States, South America, and Great Britain.1793-1808 The case-attack rate may reach 100%, but the mortality rate is typically less than 50%.1802 Conditions that favor toxicity include late seasonal growth, ambient temperatures over 23° C (73.4° F), and closely grazed pastures. For these reasons, the disease is seen exclusively between June and September in the Northern Hemisphere and between December and June in the Southern Hemisphere. The condition may appear 5 to 10 days after grazing on highly toxic pastures. For a pasture to develop toxicity, the ryegrass must constitute a majority of the forage growth.
Perennial ryegrass produces tremorgenic toxins when infested with the endophytic fungus Acremonium loliae or Acremonium coenophialum. The fungal infection confers resistance to the Argentine stem weevil, so there is a selective pressure for toxigenic cultivars. Strains of Acremonium-resistant ryegrass have been propagated but are difficult to maintain because of the devastating effects of the stem weevil infestation. The chemicals produced by Acremonium-infected plants are classified as indole terpenes. These compounds are chemically related to the fungal tremorgens, penitrem A and fumotremogen. A number of separate toxic compounds have been isolated, including lolitrems A and B, paxilline, and peramine. Paxilline is a biosynthetic precursor of lolitrem B, which is related chemically to peramine. Peramine has the major antagonistic effects against the Argentine stem weevil. The lolitrems have the greatest tremorgenic effect on livestock.1809 Concentrations of more than 2000 ppb of lolitrem B in forage or 1.68 mg/kg of forage have been associated with toxicity for sheep and cattle, respectively.1810 The concentration of lolitrem varies seasonally in the same grass, and toxic pastures may become nontoxic over the course of the grazing season.1811
A relationship also exists between the frequency of poisonings and the proportion of plants infested by the Acremonium fungus. Infection rates below 25% are associated with sporadic cases, whereas plots containing 90% infection rates are associated with large outbreaks of staggers. Intoxication is most common on dry pastures where the perennial ryegrass is growing slowly under relatively low ambient temperatures. The Acremonium fungus can be identified by microscopic examination of boiled leaves. The fungus is in greatest prevalence in the summer and is found in the uppermost part of the leaf. To identify the fungus, the ryegrass leaves are immersed in a stain containing 0.06 g aniline blue in 50 mL of lactic acid in 250 mL of distilled water, 50 mL of glycerine, and 50 g of phenol. The mixture is boiled 5 minutes and mounted in lactophenol (20 g phenol, 16.7 mL lactic acid, 40 mL glycerine, and 20 mL water). For biologic assay of lolitrem, chloroform: methanol extracts of suspect plants are injected into mice. The recipients are then examined every few hours for tremors.
The endophyte-infested grasses also produce ergovaline or other ergopeptine alkaloids that exert prolactin-like activity. The resulting clinical signs are diarrhea, fever, tachypnea, and reduced weight gain.
High-peramine, low-lolitrem cultivars of Lolium* have been propagated. Such cultivars have partial protection against the stem weevil but do not cause staggers in pastured animals.1812
Annual Ryegrass Staggers
Annual ryegrass toxicity is caused by corynetoxin, which is manufactured in the seed heads of annual ryegrass (Lolium rigidium) and related grasses. The seed head is infested by the nematode Anguina agrostis (Anguina funesta). The parasitic infestation forms a gall that becomes secondarily infected by the bacterium Clavibacter toxicus (Corynebacterium rathayii).1813,1814 The Corynebacterium organisms produce corynetoxin; this neurotoxin has been purified using high-performance liquid chromatography (HPLC) and can be detected using an enzyme-linked immunosorbent assay (ELISA). The structure of corynetoxin is similar to that of the antibiotic tunicamycin.1815 The corynetoxin is a glycolipid that inhibits the synthesis of lipid-linked oligosaccharides and blocks protein glycosylation.1816,1817 Bacterial proliferation in the gall results in the formation of a yellow to orange exudate, which contains the toxin. The toxic material usually leaks out over the seed but occasionally remains encapsulated within the gall and cannot be detected by external examination. Galls that have a normal external appearance are toxic if the interior of the defect maintains a deep-orange color. Loss of color is associated with a decrease in the amount of toxicity. A method of evaluating toxic pastures based on enumeration of contaminated seed heads and ELISA to detect corynetoxin has been developed.
Outbreaks of staggers may occur in animals grazing the same pasture for months because the toxin is not inactivated by the rumen microflora, and daily doses may accumulate in sheep for as long as 9 weeks.1818 Thus, repeated exposure leads to an accumulation of the toxin and delayed onset of clinical disease. Also, the concentration of the toxin increases in the seed heads during the summer and is greatest as the plant dries and the seeds ripen.1819,1820 Finally, toxic ryegrass may occur only in patches in the pastures, and the grazing patterns of the animals is altered by changes in the climatic conditions, the growth of the ryegrass, or introduction of new sheep. This could explain why outbreaks occur shortly after onset of inclement weather or after introduction of new sheep to the pastures.
Pathologic changes associated with annual ryegrass staggers include hemorrhage in the cerebellum, liver, and spleen. Ultrastructural changes include swelling of the capillary endothelial cells, dilation of the endoplasmic reticulum in the endothelial cells, mitochondrial degeneration, swelling of the astrocytic end feet, protein leakage across the blood-brain barrier, pyknosis and death of granular cell nuclei of the cerebellum, and changes in the neuropil adjacent to the damaged capillaries. These changes indicate that the toxin may access the central nervous system (CNS) by damaging the blood-brain–cerebrospinal fluid (CSF) barrier. CNS neurons could be affected because of vascular damage or direct activity of the toxins.1821
Clinical Signs and Pathologic Lesions
The clinical signs of annual and perennial ryegrass staggers are similar. For both disorders the case-attack rate usually is high, but mortality varies and can range from 0% to over 90%. The clinical signs may occur within 48 hours to several weeks after cattle are introduced to toxic pastures. The animals appear normal at rest but tremble when they are excited. The gait is stiff, and limbs are hypermetric. There are fine and coarse tremors of all major muscle groups, especially those of the shoulder and flank areas. The tremors worsen as the animal becomes excited. Other clinical signs include intentional head tremor, truncal sway, and base-wide stance. With continued stimulation, affected animals kneel and then fall over. While down, animals have stiff extension of the legs with occasional flailing and may display opisthotonos or convulsions. Frothy exudate from the mouth also has been described. After approximately 10 to 20 minutes of struggle, the animal recovers, stands, and walks back to the herd or flock. New cases and deaths can continue for as long as 1 week after the animals have been removed from the toxic pasture.
Differential Diagnosis
Grass staggers is easily recognized by clinical signs. The specific plant involved must be identified by examination of the pasture forage. Tremorgenic diseases of adult cattle are common throughout most of the world. In addition to ryegrass pastures, tremorgenic plants include Swainsona luteola and Swainsona galegifola; Solanum dimidiatum and Solanum fastigiatum; Astragalus species; red buckeye; Phalaris species (canary and reed canary grass); Eupatorium rugosum (white snakeroot); Cynodon dactylon (Bermuda grass); Dallis grass infested with the fungus Claviceps paspali; Polypogon monospeliensis (annual beard grass); Pennisetum clandestinum (kikuyu grass); and the mycotoxins of Penicillium cyclopium.1820 Hypomagnesemia has been reported to cause cerebellar degeneration under some circumstances. The storage diseases (α- and β-mannosidosis, generalized glycogenosis, globoid cell leukodystrophy, neuronal lipodystrophy) may also be important differential diagnoses for ataxic animals with tremor and cerebellar signs.
Treatment
There is no specific treatment for grass staggers. If the animals are removed from toxic pastures as soon as signs are first seen, the mortality rate is low despite the high number of affected animals. Several months may elapse before the neurologic signs resolve completely. Treatment with high doses of magnesium chloride has been recommended, although others have shown it to be ineffective for controlling the muscular spasms.1822 Pastures may lose toxicity after rain and growth of new grass. In subtropical regions, cattle should not be introduced to toxic pastures until the late fall or winter, when less toxic growth becomes abundant.
Prevention
Ammoniation of dried feed has been recommended as a method of reducing the toxicity of hay. This treatment simultaneously increases the digestibility and protein equivalency of the forage.1823 For preventing annual ryegrass staggers, high-risk pastures can be identified by visually examining seed heads for infected galls. ELISA is sufficiently sensitive to identify one infected seed gall per 100 g of dried seed heads and also can accurately predict toxicity in pastures.1824 The most practical strategy for controlling annual ryegrass toxicity is to break the nematode’s life cycle by killing the ryegrass for two or three growing seasons. Otherwise, pastures remain perpetually toxic. Integrated control measures that have been recommended for prevention of annual ryegrass toxicity include applying herbicides in the spring, seeding the pastures with legumes, burning the infected pasture grasses during early autumn, and applying ryegrass-selective herbicides in the summer months, combined with heavy winter grazing.1825
Prevention of perennial ryegrass staggers using endophyte-free cultivars has been recommended. Such resistant biotypes of ryegrass lack resistance to the Argentine stem weevil and consequently are less productive than other biotypes. The most convenient solution has been to minimize exposure of the animals until fall rains stimulate less toxic pasture growth. Newer cultivars containing fewer tremorgens may be useful in the future.
BERMUDA GRASS STAGGERS
Bermuda grass (Cynodon dactylon) occasionally may become toxic for livestock.1228 Cattle are most susceptible, followed by sheep, goats, and horses. Although the nature of the toxic principle in Bermuda grass is unknown, several factors, including sooty mold (Pullularia species), endogenous basic alkaloids, and leaf hopper infestation, have been associated with toxic pastures.1826
Animals may develop clinical signs as early as 36 hours after consuming toxic forage. Experimentally poisoned goats have developed clinical signs 8 days after being fed 772 g/head/day of toxic hay.1826 The toxin survives drying. Hay that is cut from offending pastures may remain toxic for as long as 9 years.1827 The pharmacologic nature of the toxin is unknown; however, the sclerotia of Claviceps purpurea have been identified on the seed heads of toxic pastures.1828 Pastures that are toxic remain so for successive seasons unless the vegetation is burned off and the ground is tilled and reseeded.
The clinical signs of Bermuda grass intoxication occur suddenly, usually simultaneously, in several animals in the herd. In some cases, most of the animals on a single pasture may be affected, whereas animals on an adjacent pasture remain normal. The clinical disease is indistinguishable from ryegrass staggers (see preceding sections). The electroencephalograms of affected animals are normal, indicating that the biochemical lesion is below the cortical level.1829 The mortality rate is low, and deaths usually occur from self-inflicted trauma. Affected animals recover 2 days to 2 months after removal from the pasture. Tremors may be controlled using intravenous diazepam (0.1 to 1.0 mg/kg two or three times daily as needed).
KIKUYU GRASS POISONING
A nervous system disease characterized by depression, ataxia, drooling of saliva, and ruminal distention occurs in cattle and sheep of northern New Zealand that are grazing kikuyu grass (Pennisetum clandestinum).1830
DALLIS GRASS STAGGERS (PASPALUM STAGGERS; CLAVICEPS PASPALI TOXICITY; NERVOUS ERGOTISM)
Ingestion of Dallis grass infected with the ergot fungus Claviceps paspali produces a tremorgenic disease similar to that of Bermuda and ryegrass staggers. Other Paspalum-type grasses that may become toxic include Argentine bahia grass (Paspalum dilatatum) and water couch grass (Paspalum distichum). Horses are susceptible to the toxin, but the condition occurs most often in cattle. The disease has been recognized in the United States, Great Britain, Australia, and New Zealand.1831-1835
Claviceps paspali first attacks the pistil of the grass flower and replaces the ovary with fungal tissue. The fungus secretes a sticky fluid, the “honeydew,” which contains a large number of spores but little toxin. The fluid hardens into a mature sclerotia containing large amounts of toxin. Toxic stands of Dallis grass can be recognized by the presence of numerous small, reddish brown or black sclerotia measuring 3 to 5 mm in diameter on the seed head of the plant. The fungus produces a number of neurologically active agents. Some of the products resemble lysergic acid diethylamine (LSD) in structure and activity, whereas others may act as dopaminergic agonists. Animals apparently develop a craving for the infested seed heads and graze them selectively. The toxin remains active in cured hay. Toxin production is greatest when there is a wet period after formation of the seed heads. Mowing the toxic pastures and removing or burning the infested seed heads are effective for preventing further outbreaks of the disease. If the amount of rainfall diminishes after mowing, the new growth usually is nontoxic.
The clinical signs of Dallis grass staggers are similar to those described earlier for ryegrass, including coarse and fine muscular fasciculations, head tremors, spastic hypermetric gait, and falling. The clinical signs are exacerbated by fright or external stimulation. Clinical diagnosis is made by visible detection of the toxic agent in the feed or by using thin-layer chromatography (TLC). Animals recover spontaneously within 1 to 3 months after being removed from the pasture.
CANARY GRASS STAGGERS (PHALARIS STAGGERS)
Cattle or sheep that graze on certain Phalaris species of canary grass (P. arundinacea, P. tuberosa, P. acquatica, P. angusta, P. caroliniana, P. brachystachys) grown under specific environmental conditions may develop neurointoxication.1836-1841 Phalaris poisoning has been reported in Australia, New Zealand, South Africa, Norway,1842 South America,1843 and the United States,1844 where the plant can be found in Virginia, Colorado, Oregon, Florida, Texas, Georgia, Mississippi, Alabama, and California.1845 The case-attack rate may reach 80%,1839 and the mortality rate ranges from 4% to 40%.1837,1839 Acute deaths may occur as early as 4 to 12 hours after commencement of grazing on a toxic pasture.1837,1840 Animals usually recover by 8 days after removal from offending pastures, but signs can persist for as long as 1 month after removal, and relapses can occur for up to 5 months.1836,1837,1840
Continuous exposure to low concentrations of alkaloid (<0.001% of dry matter intake) over 40 days has resulted in severe toxicosis in sheep on a drylot.1846
The toxic principles of canary grass are tryptamine alkaloids (dimethylated indolealkyl amines), which are found in one or more Phalaris species (P. tuberosa, P. minor, and P. arundinacea).1839 The most potent of the toxins is the alkylamine 5-methoxydimethyltryptamine. Intravenous doses of this compound as low as 0.1 mg/kg can cause severe neurologic signs in sheep in 16 seconds.1841 The toxins competitively inhibit the initial step in the breakdown of serotonin by monoamine oxidase and act on midbrain and medullary nuclei via presynaptic serotonin type 1 cholinergic receptor sites. The overall activity of the toxin is to enhance response to excitatory inputs.1847
The dynamics of toxin production have been examined.1848 The concentration of alkaloid in the plant is increased by a reduction of light intensity (shade) but not by a decrease in length of the daylight.1848 If light intensities are high, the grass is unlikely to be toxic unless soil nitrate levels are also high.1848 Rapid growth of the grass also favors the formation and accumulation of toxin. Other factors that enhance the toxicity of a pasture include fog, humidity, or rain, followed by sunny, warm weather or sunshine on nitrogen-fertilized pastures.1848 Although there is no specific age-related susceptibility to the toxin, only weaned animals tend to be affected during an outbreak. Many outbreaks occur when hungry sheep ingest large amounts of toxic grass over a short period.1837 The disease has also occurred 3 to 5 days after a rainfall has ended a period of drought.1843
Electromyographic studies have indicated that the tremors and spasms probably originate from the spinal cord and the peripheral nervous system. Excitation leads to increased muscle tone and extensor rigidity.1849
There are at least two distinct clinical forms of the intoxication: acute death from cardiovascular collapse and a more chronic nervous form.1838 The cardiovascular form of the disease occurs by 12 to 72 hours after animals are placed on a toxic pasture.1838,1840 Affected animals die suddenly from heart failure while being herded off toxic pastures. Animals also may be found dead with the head fixed in opisthotonos and the legs in rigid extension. The ground surrounding the limbs is disturbed, indicating that the animal died in convulsions. Signs associated with cardiac collapse include acute dyspnea, cyanosis, pounding heart sounds, irregular heart rate with alternating periods of extreme tachycardia (170 to 240 beats/min), and then bradycardia.1841,1846
The nervous form of the disease is more prolonged and occurs after repeated exposures of 2 to 33 weeks’ duration. Signs may be delayed for as long as 4 months after removal from the toxic grass.1844 The clinical signs include hyperexcitability, exaggerated responses to auditory or tactile stimuli, fine muscular fasciculations (particularly of masseter muscles), licking of the lips, wrinkling of the facial muscles, repetitive chewing, inability to swallow, flaring of the nostrils, ptyalism, nystagmus, intentional head tremor, ear and tail twitching, base-wide stance, reduced menace response, and deficient pupillary reflexes.1841,1849 The gait of affected sheep is described as stiff legged, with both hindlimbs moving in unison (“rabbit hopping”). Affected sheep buckle at the knees and assume sternal recumbency with the hindquarters elevated. They then fall into lateral recumbency and flail wildly while attempting to stand.1838 Poisoned cattle also show incoordination and repeated stabbing movements with the tongue and are unable to grasp the forage. They salivate profusely and drop feed from the mouth. Eventually the animals die of starvation. There may be an increased protein concentration (40 to 100 mg/mL) and white blood cell count (4 to 50 mononuclear cells/μL) in CSF of affected animals.1836 Animals may survive the initial signs but have neurologic symptoms for as long as 10 months.1850
For confirmation of a diagnosis, the amount of tryptamine alkaloids in suspect grasses can be measured. Alkaloid concentrations greater than 30 to 50 mg/100 g dry weight of forage are considered toxic for sheep.1848,1849
The pathologic lesions in the CNS of affected animals include focal, demarcated, greenish or slate-gray discoloration in the pons, medulla, and corticomedullary junction of the kidney; intracytoplasmic accumulations of greenish brown pigment in the dorsal root ganglia and medullary nuclei; neuronal loss; focal gliosis; and swelling of the axonal sheaths in the ventromedial aspect of the spinal cord.1836,1839 The pigment is thought to originate from metabolism and deamination of the toxic alkylamines but is not thought to play a direct role in the development of the neurologic deficits.1845,1849 Other lesions in cattle that have died acutely include ulcerative abomasitis, jejunitis, and ileitis; subcapsular renal hemorrhage; and ecchymoses of the pericardium and epicardium.
Administration of cobalt to animals on toxic grass pastures is protective. The biochemical function of cobalt is thought to be related to increased ruminal inactivation of the toxins. Weekly administration of 28 mg of cobalt to each animal is recommended to prevent clinical signs in exposed sheep.1844,1845,1850-1852 This dosage is much higher than that delivered by standard supplementation. Additional recommendations include removal of affected animals from the offending pasture, sedation with a phenothiazine tranquilizer, and administration of sodium pentobarbital or diazepam to convulsive sheep. Phalaris plants may also contain potentially toxic concentrations of nitrate or cyanide. In any outbreak of suspected Phalaris toxicosis with acute signs of sudden death, cyanide and nitrate poisonings should also be considered.1853
To prevent Phalaris poisoning, animals should be removed from the toxic grasses. The concentration of dimethylindolealkyl amines may be reduced by curing the forage as hay. Ensiling the canary grass does not reduce the amount of toxins.1854
PENICILLIUM CYCLOPIUM (TREMORGEN) INTOXICATION
Ruminants that ingest toxic species of Penicillium develop clinical signs that are indistinguishable from those of Dallis, ryegrass, Phalaris, and Bermuda grass staggers.1855-1858 The tremors are caused by mycotoxins,1858 which can be classified into four major groups: the aflatrem-paxilline group, verruculogen-fumotremogen group, territrem group, and tryptoquivaline group. Of these, the most important fungal tremorgens are aflatrem, penitrem A, fumotremogen B, and verruculogen. Verruculogen and fumotremogen B can be isolated from cultures of Penicillium estinogenum. Penitrem A is elaborated from Penicillium nigricans, Penicillium anitellum, Penicillium cyclopium, Penicillium clavigerum, and Aspergillus canescens. Verriculogen has also been identified in pure cultures of Aspergillus fumigatus.1859,1860
Ingestion of moldy cornstalks constitutes the most common source of fungal tremors in livestock. The fungi proliferate in the corn but do not produce tremorgens until the stalks touch the ground. After production at or near the soil surface, the toxins translocate in plants through root absorption.1858
The pathophysiologic mechanisms by which mycotoxins affect the CNS are unknown, but there is increased release of the transmitter amino acids aspartate, glutamate, and γ-aminobutyric acid in the corpus striatum, indicating the presence of a reversible biochemical lesion.
Diagnosis is based on the clinical signs, demonstration of the mycotoxin in the feed, and identification of the fungal elements in the feces. There is no specific treatment for the intoxication. Affected animals recover completely when they are removed from infected pastures. The diagnosis of tremorgenic fungal intoxication is difficult. The mycelial elements survive degradative conditions in the gastrointestinal tract and can be isolated from the feces of intoxicated animals. Penitrem A and verriculogen can be demonstrated in the forage by TLC or mouse assay.
TREMORGENIC NEUROTOXICOSIS FROM ASPERGILLUS CLAVATUS
Ingestion by cattle and sheep of fodder contaminated by the fungus Aspergillus clavatus can result in ataxia, weakness, and tremors, similar to signs caused by ingestion of other neurotoxic molds.1861,1862 Clinical signs include ataxia, knuckling, muscle weakness and tremors that may be exacerbated by handling, hypersalivation and drooling, altered behavior, loss of appetite, reduced milk production, muscle spasms, recumbency, opisthotonos, and death. Cases have occurred worldwide, linked to feeding sprouted grains and the by-products of beer production, both of which provide an environment for growth of the mold. Factors that can encourage mold growth include high ambient temperature and high humidity.1861 Hematologic changes include evidence of dehydration, moderate neutrophilia, hypochromasia, and microcytic erythrocytes. Changes in clinical chemistry include elevated creatine kinase (possibly from recumbency), AST, γ-glutamyltransferase, and glutamate dehydrogenase activity. The toxicity of A. clavatus is attributed to a variety of tremorgenic neuromycotoxins, including patulin, tryptoquivaline, tryptoquivalone, nortryptoquivalone, cythochalasin E, cythochalasin K, escladiol, and clavatol.1862
DISEASES PRODUCING SPINAL CORD OR PERIPHERAL NERVE SIGNS
CERVICAL VERTEBRAL STENOTIC MYELOPATHY (WOBBLER SYNDROME; CERVICAL STENOTIC MYELOPATHY; CERVICAL VERTEBRAL INSTABILITY)
BONNIE R. RUSH
Clinical Signs
Cervical vertebral stenotic myelopathy (CVSM) is a common cause of symmetric spinal ataxia in horses. Neurologic gait deficits are caused by spinal cord compression by stenotic and malformed cervical vertebrae.1863 A neurologic examination is performed to assess the symmetry of deficits and the severity of weakness, ataxia, and spasticity.1864 Gait analysis is performed at the walk; neurologic deficits can be accentuated by circling, elevation of the head, and maneuvering over obstacles and inclines. Ataxia or proprioceptive loss is manifested by circumduction of the hindlimbs, posting (pivoting on the inside hindlimb during circling), and truncal sway. In most cases, pelvic limb ataxia is more pronounced than forelimb deficits. Moderately to severely affected horses have lacerations on the heel bulbs (wobbler heels) and medial aspect of the forelimbs from overreaching and interference. Stumbling and toe dragging indicate weakness. The hooves of horses with prolonged clinical signs of CVSM are chipped, worn, or squared at the toe. At rest, affected horses may have a base-wide stance and may demonstrate delayed responses to proprioceptive positioning. When prompted to back, horses may stand base wide, lean backward, drag their hindlimbs, and step on their hindfoot with a forelimb. The musculature of the neck may appear disproportionately thin compared to the rest of the body, and in some horses, prominent articular processes of the fifth and sixth cervical vertebrae may be evident.1865
Occasionally, weakness and stumbling are more pronounced in the forelimbs. This is usually observed in horses with stenosis of the caudal cervical vertebrae (C6-C7) caused by compression of the cervical intumescence. Alternatively, arthropathy of the caudal cervical vertebrae may produce cervical pain and forelimb lameness from peripheral nerve compression, without producing clinical signs of spinal cord compression.1866 Affected horses typically travel with a short cranial phase of the stride and a low foot arc of the forelimbs and may stand or travel with their head and neck extended. Rarely, diskospondylosis of the cervical vertebrae will produce a short-strided gait and cervical pain, with or without spinal ataxia. Horses with diskospondylosis or arthropathy of the caudal vertebrae may demonstrate increased rate and depth of respiration with cervical manipulation because of pain.
The condition has been reported in most light and draft breeds.1867 Thoroughbreds are particularly predisposed to CVSM, which affects approximately 2% of the population. Between 10% and 50% of thoroughbreds have characteristic developmental malformations of the cervical vertebrae without spinal cord compression.1868,1869 Male horses are more frequently affected than females. Most horses with CVSM are 6 months to 3 years of age at presentation. Nonetheless, age (≥4 years) does not preclude a diagnosis of CVSM; spinal cord compression caused by vertebral abnormalities is routinely diagnosed in adult horses, including geriatric horses.
The onset of neurologic gait deficits is typically insidious, with progression of ataxia for several weeks, followed by stabilization (plateau) of clinical signs.1867,1870 Owners may report a traumatic incident with the onset of clinical signs of CVSM. The event may be the result of mild neurologic deficits, with the injury exacerbating the clinical signs of spinal cord compression. Asymmetric ataxia and paresis may be occasionally observed in horses with dorsolateral compression of the spinal cord by proliferative, degenerative articular processes and periarticular soft tissue structures.
Pathophysiology
CVSM appears to be a manifestation of developmental orthopedic disease. Developmental disease of the appendicular skeleton, such as physitis, joint effusion, osteochondrosis, and flexural limb deformities, occurs more often in young horses with CVSM.1871 A direct cause-and-effect relationship between osteochondrosis and CVSM has not been identified; however, the association between the frequency of occurrence of osteochondrosis and CVSM indicates that the two conditions have a similar pathophysiology.
The etiology of osteochondrosis and CVSM appears multifactorial, consisting of genetic and environmental influences. It is unlikely that CVSM is heritable by simple mendelian dominant recessive patterns.1872 The mode of inheritance more likely involves multiple alleles and variable penetrance, which determine genetic predisposition to CVSM. A high plane of nutrition, micronutrient imbalance, rapid growth, trauma, and abnormal biomechanical forces probably contribute to the development of CVSM in genetically predisposed animals.
Dietary copper, zinc, and carbohydrates are thought to play a role in the pathogenesis of osteochondrosis and CVSM. Low dietary copper (12 ppm) and high dietary zinc (1000 to 2000 mg/kg of dry weight) concentrations cause osteochondrosis in foals, whereas copper supplementation (55 ppm) reduces the incidence of osteochondrosis of the axial and appendicular skeleton.1873,1874 Copper supplementation does not eliminate developmental orthopedic disease, suggesting the existence of other etiologic factors. Excessive carbohydrate in the diet is hypothesized to contribute to the pathogenesis of osteochondrosis through endocrine imbalance.1875,1876
Spinal cord compression can be dynamic or static in horses with CVSM.1870,1877 Dynamic compression occurs because of vertebral instability and causes intermittent spinal cord compression during ventroflexion of the neck; compression is relieved when the neck is in the neutral position. Pathologic changes most often observed in horses with dynamic compression are instability between adjacent vertebrae, malformation of the caudal vertebral epiphysis (caudal epiphyseal flare), and malformation or malarticulation of the articular processes. Osteochondrosis of the articular processes is not always present at the site of spinal cord compression in horses with dynamic compression.1870 The intervertebral sites most frequently affected by dynamic compression are C3-C4 and C4-C5.
Static compression is defined as continuous spinal cord impingement regardless of cervical position. It occurs predominantly in the caudal cervical region, C5-C6 and C6-C7. Static compression is exacerbated by thickening of the dorsal lamina, hypertrophy of the ligamentum flavum, and degenerative joint disease (DJD) of the articular processes. Both static and dynamic compression are associated with narrowing of the vertebral canal from C3-C6, regardless of the site of spinal cord compression, indicating that generalized vertebral canal stenosis is an important factor in the pathophysiology of CVSM.1878
Histopathologic examination of the spinal cord identifies myelin degeneration (ventral and lateral funiculi), malacia, focal neuronal loss, and fibrosis at the sites of compression. Wallerian degeneration occurs in ascending white matter tracts cranial to the affected site and in descending tracts distal to the site of spinal cord compression.1879
Diagnosis
Radiographic examination and cerebrospinal fluid (CSF) analysis are indicated in horses with symmetric tetraparesis and ataxia to differentiate CVSM from other spinal cord disorders. The most important differential diagnoses for horses with symmetric tetraparesis and ataxia include equine herpesvirus myeloencephalitis, equine protozoal myeloencephalitis, equine degenerative myeloencephalopathy, and spinal cord/vertebral trauma. Cytologic CSF findings usually are unremarkable in horses with CVSM. When CSF findings are abnormal, the alterations are consistent with acute spinal cord compression, such as mild xanthochromia and mild increases in protein concentration.
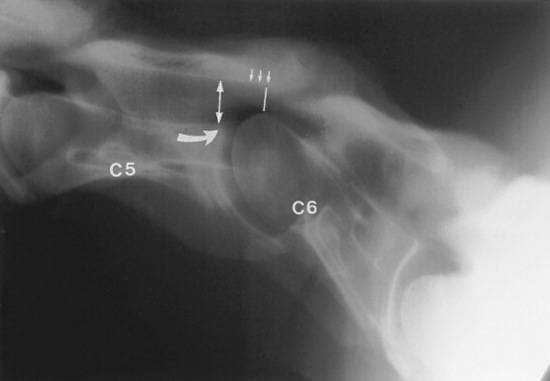
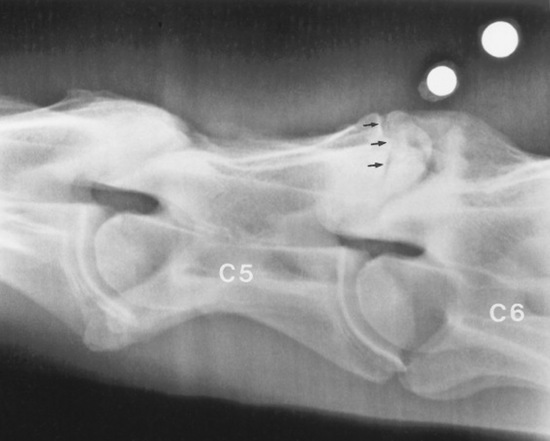
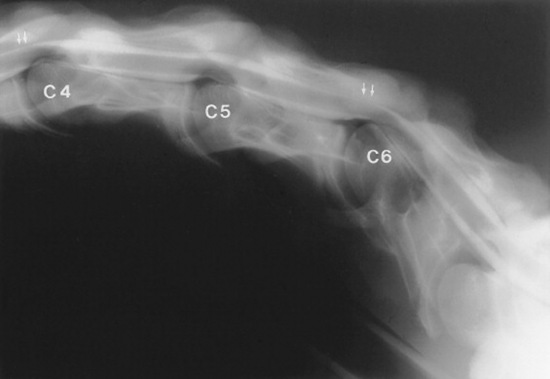
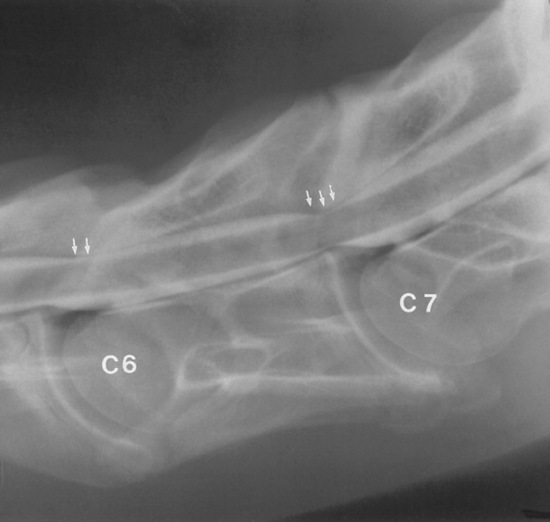
Survey radiographs of the cervical spine are obtained in standing, sedated horses. Cervical radiographs are evaluated by subjective assessment of vertebral malformation and objective determination of vertebral canal diameter.1878 The five categories of cervical malformation subjectively assessed in horses with CVSM are DJD of the articular processes, subluxation between adjacent vertebrae, flare of the caudal physis of the vertebral body, abnormal ossification patterns, and caudal extension of the dorsal laminae1878,1880 (Figs. 35-26 and 35-27). Although the presence of characteristic vertebral malformations supports the diagnosis of CVSM, subjective evaluation of survey radiographs does not reliably discriminate between horses affected and those unaffected by CVSM.1868,1878 DJD of the articular processes of the caudal cervical vertebrae is the most common and severe malformation observed in affected horses.1878 However, degenerative arthropathy occurs in 10% to 50% of nonataxic horses and is the most common and severe vertebral malformation in horses without CVSM.1869,1878 Subjective evaluation of degenerative arthropathy of the articular processes may lead to a false-positive diagnosis of CVSM.1868
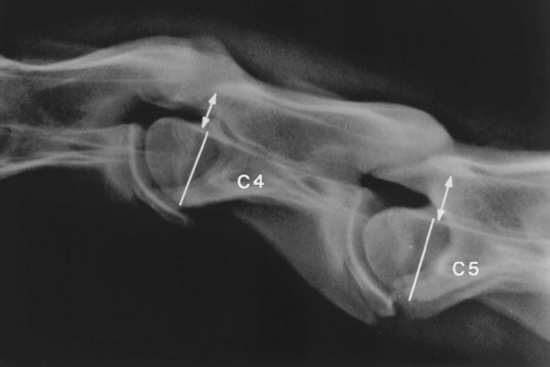
The vertebral canal diameter is objectively assessed by determining the sagittal ratio.1876 The sagittal ratio is obtained by dividing the minimum sagittal diameter of the vertebral canal by the width of the corresponding vertebral body. The minimum sagittal diameter is measured from the dorsal aspect of the vertebral body to the ventral border of the dorsal laminae, and the vertebral body width is measured perpendicular to the vertebral canal at the widest point of the cranial aspect of the vertebral body (Fig. 35-28). The sagittal ratio eliminates error caused by magnification because the vertebral canal and vertebral body are in the same anatomic plane. The sagittal ratio should exceed 52% from C4 through C6 and 56% at C7 in horses weighing more than 320 kg. The sensitivity and specificity of the sagittal ratio for identification of CVSM-affected horses are approximately 89% for vertebral sites C4 through C7.1881 Accurate measurement of the sagittal ratio requires a precise, lateral radiograph of the cervical vertebrae. Oblique views yield indistinct margins of the ventral aspect of the vertebral canal, resulting in erroneous values for minimum sagittal diameter and vertebral body width. Intervertebral ratios have been suggested to improve the ability to identify the site of spinal cord compression.1882 This measurement is obtained by determination of the minimum distance from the craniodorsal aspect of the vertebral body to the caudal aspect of the vertebral arch of the immediately rostral vertebra. This value is divided by the width of the vertebral body. Reference values for this technique have not been published.
The semiquantitative scoring system developed by Mayhew et al.1880 should be used in foals under 1 year of age to assess cervical radiographs for diagnosis of CVSM. The scoring system combines objective measurement of the vertebral canal diameter and subjective evaluation of vertebral malformation. Stenosis of the vertebral canal is assessed by determining the intervertebral and intravertebral minimum sagittal diameters. These values are corrected for radiographic magnification by dividing them by the length of the vertebral body (see Fig. 35-26). Foals that measure below the mean are assessed 5 points, and foals that measure 2 standard deviations (SD) below the mean or fall below the mean at multiple sites are assessed 6 to 10 points (Table 35-15). Cervical vertebral malformation is determined by subjective assessment of five categories: encroachment of the caudal epiphysis of the vertebral body dorsally into the vertebral canal; caudal extension of the dorsal lamina to the cranial physis of the next vertebra; angulation between adjacent vertebral bodies; abnormal ossification of the physis; and DJD of the articular processes. The maximum score allotted for each category of bony malformation is 5 points. A total score of 12 or higher (maximum, 25) confirms the radiographic diagnosis of CVSM. Stenosis of the vertebral canal and malalignment between adjacent vertebrae are the most discriminating parameters in this semiquantitative scoring system to differentiate CVSM-affected foals from normal foals.
Table 35-15 Mean Minimum Sagittal Diameters* and Corrected Minimum Sagittal Diameters of Cervical Vertebrae in Foals without Neurologic Disease
| Cervical Vertebral Site |
Minimum Sagittal Diameter (mm) ± SD |
Corrected Minimum Sagittal Diameter (%) ± SD |
| C2 |
23 ± 1 |
18 ± 1 |
| C2-C3 |
28 ± 4 |
33 ± 2 |
| C3 |
20 ± 1 |
24 ± 2 |
| C3-C4 |
25 ± 2 |
30 ± 2 |
| C4 |
20 ± 1 |
24 ± 2 |
| C4-C5 |
25 ± 2 |
31 ± 2 |
| C5 |
21 ± 1 |
25 ± 2 |
| C5-C6 |
26 ± 3 |
34 ± 3 |
| C6 |
21 ± 1 |
27 ± 2 |
| C6-C7 |
31 ± 5 |
46 ± 5 |
| C7 |
23 ± 1 |
35 ± 2 |
From Mayhew IG et al: Diagnosis and prediction of cervical vertebral malformation in thoroughbred foals based on semi-quantitative radiographic indicators, Equine Vet J 25:435, 1993.
SD, Standard deviation.
Survey radiographic examination of the cervical vertebrae determines the likelihood of spinal cord compression. Myelographic examination is required for definitive diagnosis of CVSM, identification of the location of affected vertebral sites, and classification of compressive lesions. The clinician should use radiographic interpretation to classify the patient into one of the following categories:
1
Low sagittal ratio (<48% at C4 to C6), moderate to severe bony malformation; myelographic examination to identify sites of spinal cord compression and to classify lesions as static or dynamic.
2
Marginal sagittal ratio (48% to 56%), mild to moderate bony malformation; myelographic examination to confirm or rule out CVSM.
3
High sagittal ratio (>56%), minimal bony malformation; other differential diagnoses pursued.
Myelographic examination is performed under general anesthesia with the patient in lateral recumbency.1883 The landmarks for cisternal puncture at the atlantooccipital site are the cranial border of the wings of the atlas, the caudal border of the occipital protuberance, and the dorsal midline. The poll region is aseptically prepared and the head flexed at a 90-degree angle to the cervical vertebral column. The spinal needle (3½-inch, 18-gauge needle with stylet) is introduced and directed toward the lower jaw. The needle is advanced until the dura mater is penetrated, which often produces a “popping” sensation. Clear CSF should drip rapidly or flow from the hub with successful placement of the needle. An equal volume (20 to 40 mL) of CSF is removed before injection of a contrast agent. From 20 to 40 mL of contrast medium produces sufficient positive-contrast opacity to identify spinal cord compression in adult horses.1883 The bevel of the spinal needle is directed caudally, and contrast medium is injected at a constant rate over 5 minutes. The head and neck are elevated under a wedged platform for 5 minutes at 30 to 45 degrees to facilitate caudal flow of the contrast medium. Iohexol (350 mg iodine/mL) and iopamidol (370 mg iodine/mL) are the most popular nonionic, water-soluble contrast media used for equine myelographic studies.1884-1886 These second-generation agents cause less neurotoxicity and meningeal irritation than metrizamide.1887
It is difficult for investigators to agree on myelographic criteria for definitive diagnosis of CVSM. In many cases the site of compression (or lack thereof) is obvious, and all recommended criteria would produce the same result. However, there is a population of horses for which myelographic interpretation is more difficult. Many reports recommend a 50% or greater decrease in the sagittal diameter of the dorsal contrast column, paired with obliteration of the ventral contrast column.1883 The decrease in the sagittal diameter of the contrast column is determined by comparing the value at the intervertebral space to a midvertebral site cranial or caudal to the suspected intervertebral space. The 50% reduction should be interpreted conservatively, given the propensity for false-positive diagnosis; a 70% reduction may be more reliable.1882 Some investigators prefer to use a diagnostic criterion of less than 2 mm of dorsal contrast column (or smaller) to reduce false-positive results on myelographic studies, but this criterion will increase the risk of false-negative diagnosis. Most recently, a 20% reduction in dural diameter (height of dural sac) at a given intervertebral junction, compared with the dural diameter at the level of the midvertebral body, has been suggested as the most reliable indication of spinal cord compression.1882
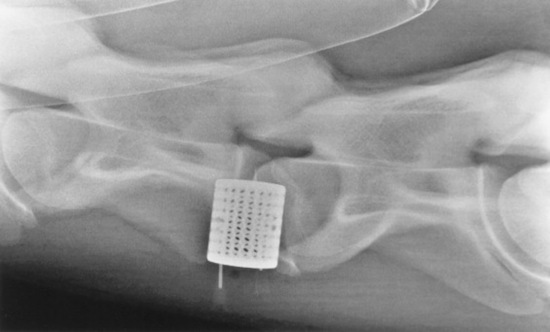
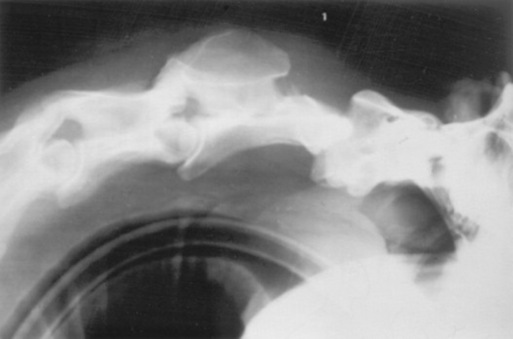
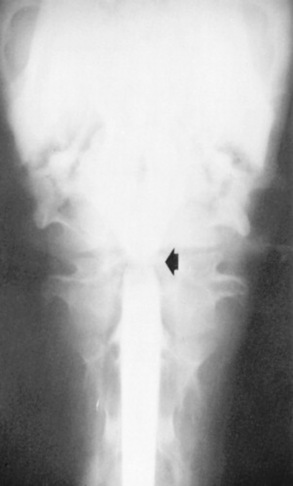
A complete myelographic examination should include neutral and stressed (flexed and extended) views of the cervical vertebrae.1863,1883 Horses with dynamic spinal cord compression show obliteration of the dorsal and ventral contrast columns during ventroflexion of the neck (Fig. 35-29), whereas spinal cord compression is not apparent with the neck in the neutral position. Static vertebral canal stenosis is characterized by constant spinal cord compression regardless of cervical position (Fig. 35-30). In some cases of static compression, ventroflexion of the neck stretches the ligamentum flavum and relieves spinal cord compression, whereas hyperextension exacerbates compression. In horses with obvious sites of spinal cord compression on neutral myelographic views, excessive flexion and extension of the neck should be avoided while obtaining dynamic views to prevent exacerbation of spinal cord injury.
Horses should be monitored for 24 hours after the myelographic procedure for depression, fever, seizure, and worsening of neurologic status.1888 Worsening of neurologic status after myelography may result from spinal cord trauma during hyperflexion, iatrogenic puncture of the spinal cord, or chemical meningitis. Administration of phenylbutazone (4.4 mg/kg PO every 24 hours) 1 day before through 1 day after myelographic examination attenuates fever and depression associated with chemical meningitis.
Treatment
Conservative management of CVSM-affected horses consists of administration of antiinflammatory therapy (glucocorticoids, dimethyl sulfoxide [DMSO], and nonsteroidal antiinflammatory drugs [NSAIDs]) and exercise restriction. Antiinflammatory therapy alone may reduce the edema associated with spinal cord compression; however, full recovery is unlikely without dietary or surgical intervention.
The most successful conservative treatment option for CVSM-affected foals (<1 year of age) is the “paced diet” program.1889 This program is designed to correct endocrine imbalance associated with high-carbohydrate diets. After a carbohydrate meal, high serum insulin and low serum thyroxine concentrations promote cartilage proliferation and retention without promoting maturation. This dietary program is restricted in energy and protein (65% to 75% of National Research Council [NRC] recommendations) but maintains a balanced vitamin and mineral intake (minimum 100% of NRC recommendations). Vitamins A and E are provided at three times the NRC recommendations, and selenium is supplemented to 0.3 ppm. Roughage is provided by pasture or low-quality grass hay (6% to 9% crude protein). Solitary stall confinement is recommended to minimize repetitive spinal cord compression from dynamic instability.
Horses with cervical pain and forelimb lameness caused by cervical vertebral arthropathy may benefit from intraarticular administration of corticosteroids and chondroprotective agents.1870,1890-1892 Arthrocentesis of the cervical vertebral articulations (facets) is performed with ultrasound guidance using a 6-inch, 18-gauge spinal needle in the standing, sedated, or recumbent horse.1893 The cranial facet of the caudal vertebrae will appear superficial to the caudal facet of the cranial vertebrae. The articular space is accessed at the cranioventral opening of the articular facet, which is angled approximately 60 degrees from the ultrasound beam. The needle should be introduced 5 cm cranial to the facet and inserted at a 30-degree angle to the skin surface. Joint penetration should be confirmed by aspiration of synovial fluid. If the neck is extended, the transverse process of the cranial vertebrae may obscure the path to the articulation. Intraarticular triamcinolone (6 mg/joint) or methylprednisolone (100 mg/joint) has produced a positive clinical response in approximately 50% of horses with arthrosis of the articular processes. An antimicrobial agent (e.g., amikacin, 250 mg) can be administered prophylactically with intraarticular corticosteroids or chondroprotective agents. The goal of intraarticular antiinflammatory therapy should be to improve cervical mobility, reduce cervical pain, and eliminate forelimb lameness. It is unlikely that intraarticular therapy will significantly improve clinical signs of spinal ataxia.
Surgical intervention is the most widely reported treatment for CVSM and is indicated to stop repetitive trauma to the spinal cord.1894-1897 The goals of surgical intervention are to stabilize the cervical vertebrae and decompress the spinal cord. Cervical vertebral interbody fusion (ventral stabilization) provides intervertebral stability for horses with dynamic spinal cord compression. The affected cervical vertebrae are fused in the extended position to provide immediate relief of compression and prevent repetitive spinal cord trauma (Fig. 35-31).
Dorsal laminectomy (subtotal Funkquist type B) is performed to decompress static lesions by removing portions of the dorsal lamina, ligamentum flavum, and joint capsule at the compressed site.1895 This procedure provides immediate decompression of the spinal cord; however, fatal postoperative complications may occur.1896 Interbody fusion in horses with static compression causes remodeling and atrophy of the articular processes, resulting in delayed decompression of the spinal cord over weeks to months.1898 Decompression is immediate with dorsal laminectomy. Because of the relative safety of interbody fusion, however, some surgeons believe it is the technique of choice for both dynamic and static compressive lesions.1896
Cervical vertebral interbody fusion improves the neurologic status in about half the horses with CVSM, with some horses returning to athletic function.1894,1896 An improvement in one or two neurologic grades of five is expected. The most important patient factor for determining the postoperative prognosis is the duration of clinical signs before surgical intervention; horses with clinical signs for less than 1 month before surgery are more likely to return to athletic function than those with clinical signs for longer than 3 months.1896 In addition, the number of compressive sites, severity of the compression, and severity of postoperative complications contribute to the long-term prognosis.1870 Subtotal laminectomy and cervical vertebral interbody fusion for static compression of the caudal cervical vertebrae are associated with fatal postoperative complications, including vertebral body fracture, spinal cord edema, and implant failure.1896
Postoperatively, horses should be maintained on strict stall rest for 3 weeks and fed from a hay net to minimize motion at the surgical site. Intraarticular injection of the intervertebral articulations with corticosteroids immediately after surgery may lead to more rapid decompression.1870 The duration of convalescence and rehabilitation after cervical vertebral interbody fusion is approximately 6 to 12 months. An individualized exercise program, determined by the projected use of the horse and the animal’s neurologic status, should be designed to promote muscular strength. Extended exercise at slow speed, including ponying and lunging on inclines, is recommended during rehabilitation. A neurologic examination should be performed to determine the horse’s ability to return to athletic function after surgery. It is unlikely that significant improvement in neurologic status will occur beyond the 1-year postoperative period.1896
EQUINE DEGENERATIVE MYELOENCEPHALOPATHY
Definition and Etiology
Equine degenerative myeloencephalopathy (EDM) is a symmetric, noncompressive spinal cord disease of young horses characterized by demyelination in the dorsal funiculi of the cervical spinal cord and in the brainstem.1899 A similar disorder in which lesions are found only in the gracile and cuneate nuclei of the medulla oblongata also has been described, termed neuraxonal dystrophy to differentiate it from the more common EDM.1900 Signs most often develop in horses less than 1 year of age (mean, 5 months), but onset of signs can occur as late as 12 years.1899,1901 The incidence of EDM in Arabian horses is disproportionately high.1899 The disease has been recognized in zebras, donkeys, Welsh ponies, and horses of the Przewalski, thoroughbred, standardbred, Appaloosa, quarter horse, Paso Fino, Paint, Haflinger, Norwegian Fjord, Trakehner, Hanoverian, and Morgan breeds.1902-1909 EDM has also been observed in Mongolian wild horses.1903
The disease may be related to a dietary deficiency of vitamin E, but EDM also has a familial pattern of distribution. These observations suggest a genetic etiology, but the mode of inheritance is not clear.1910
Clinical Signs
Affected horses show a symmetric proprioceptive (sensory) ataxia characterized by knuckling, stumbling, circumduction, abduction, interference, abnormal limb protraction, spasticity, hypermetria, and inability to turn sharply or lift the inside forefoot during sharp turns. When the animal is forced to turn sharply in a circle, the inside hindfoot pivots instead of lifting off the ground. Affected animals appear clumsy and cannot stop rapidly from a gallop. When stopped suddenly, they step on the rear of the forelimbs and end in a dog-sitting posture. Affected animals usually have severe conscious proprioceptive deficits that can be detected by postural placement tests. Some patients are unable to back or move easily down an incline. They may fall or stumble when light pressure is applied to the tuber coxae or withers. Signs are often more severe in the hindlimbs than in the forelimbs; in some cases, signs are only apparent in the hindlimbs.1911
Other clinical signs that may be observed with EDM are deficits of the local cervical and cervicoauricular reflexes, absence of response to the “slap test”1912 (see Chapter 8), and paralysis of the laryngeal adductor muscles. Absence of these findings, however, does not completely exclude a cervical spinal cord disease.1913 In rare cases, animals with EDM may present in acute recumbency without a prior history of neurologic disease (M.O. Smith, unpublished data).
The most important differential diagnoses for EDM are cervical stenotic myelopathy (“wobbler syndrome”), equine herpesvirus myeloencephalopathy, and equine protozoal myeloencephalitis.
Clinical Pathology and Radiographic Findings
EDM is best differentiated from cervical stenotic myelopathy by plain and contrast radiographic examination of the spinal column. The cervical spinal canal, CSF protein concentration, and white blood cell count of horses with degenerative myeloencephalopathy are normal. Serologic testing and CSF analysis is indicated to rule out herpesvirus and protozoal myelopathies. The acute clinical course of herpesvirus myelopathy and the presence of signs such as cranial nerve involvement and lower motor neuron lesions in protozoal disease help to differentiate these entities from EDM. The plasma vitamin E concentration may be below the reference range (1.70 to 10.50 μg/μL) in some unsupplemented affected horses.1903,1904 For analysis, optimal methods of blood collection include collection of unhemolyzed blood in a clot tube and storage in an upright position in a refrigerator. The refrigerated specimens should not be allowed to contact rubber stoppers and should not be stored for longer than 72 hours unless the red cells are removed and the plasma is gassed with nitrogen and frozen at −16° C (3.2° F). The concentration of vitamin E in specimens handled in this manner remains constant for at least 3 months.1914
Pathophysiology
Some suggest that EDM is associated with a low plasma concentration of vitamin E from 6 weeks to 10 months of age.1904,1915,1916 Oral absorption of vitamin E does not appear to be deficient in affected horses.1916 Vitamin-replete, clinically normal horses have a plasma concentration ranging from 1.7 to 9.5 μg/mL; vitamin E concentration is approximately 2 μg/mL higher in the spring and summer than in winter. Horses fed diets that lack fresh green forage have a low plasma concentration of vitamin E. Such diets include poor-quality, sun-baked hay and pelleted rations. In one study testing vitamin E supplementation, the case-attack rate declined from 40% to 10% in the first year after supplementation, and the severity of disease in subsequent foal crops was diminished. Moreover, supplementation of five affected horses with 6000 IU vitamin E daily improved their neurologic condition. Horses’ susceptibility to development of low vitamin E concentrations also appears to be age related. Foals sired by affected stallions tend to have lower plasma α-tocopherol concentrations than those sired by normal stallions. These lower concentrations are first noted by 6 weeks of age, but the differences disappear by 10 months. The causes of the differences in the two groups of animals are unknown. An alternative explanation for the role of vitamin E in EDM is a defect in vitamin E absorption, transportation, or metabolism.1980 The nature of such a defect has yet to be elucidated.
Vitamin E deficiency is apparently not the sole etiologic factor in EDM, however, because one report documented normal vitamin E levels in the sera of 40 affected horses examined at a veterinary hospital.1917 Subsequent epidemiologic studies indicated that risk factors for the disorder include heredity, application of insecticides on foals, exposure to dirt lots without pasture, and exposure to wood preservatives. Access to green pasture had a protective effect on foals.1916 This study may indicate that the changes in the central nervous system are related to the patient’s inability to respond adequately to oxidative stress such as phenols, anthelmintics, and insecticides. The key role of vitamin E as an antioxidant may explain the link between the disparate etiologies proposed for EDM.
The deficiency occurs most often in related horses, suggesting a genetic predisposition.1900,1916 The importance of heredity in the development of EDM is supported by studies in a number of breeds, including Appaloosa, Haflinger, and Morgan horses.1900,1907,1918 A higher incidence has been documented in both foals sired by certain stallions and foals born to mares that already produced an affected foal.1901,1904 However, the mode of inheritance is not clear.
Recent studies have indicated that a defect in axonal transport of proteins, particularly certain proteins vital to synaptic function, may be a key mechanism in the pathogenesis of EDM.1919
Pathology
The microscopic lesions are present throughout the spinal cord, involving both white matter and gray matter, and are most pronounced in the dorsal spinocerebellar tracts.1903 These lesions include diffuse axonal degeneration of ascending and descending spinal cord funiculi and prominent myelin loss. Gliosis and astrocytosis develop in response to myelin breakdown. Horses with acute, rapidly progressive disease have evidence of active myelin destruction and vaculoation, whereas those with a more gradual course of disease have prominent astrogliosis. Considerable accumulation of lipofucsin-like pigment has been reported in some affected horses.1909 The form of EDM termed neuraxonal dystrophy (e.g., in Morgan horses) is characterized by lesions almost entirely confined to the lateral caudate nucleus in the medulla. Because of the clinical signs in these horses, however, it is suspected that subtle lesions may be present within the spinal cord.
Treatment and Control
EDM is a chronically progressive disorder, although the clinical signs may stabilize after 2 to 3 years of age. The signs are irreversible, and most patients eventually are euthanized because they present a hazard to other livestock and humans.
Some improvement of clinical signs has been reported in horses treated with vitamin E. The current recommended vitamin E level for horse feed is 80 to 100 IU/kg/day.1920 In suspected cases of EDM, supplementation with 6000 IU of D,L-α-tocopherol acetate in feed is recommended, a level that appears to be safe. This dose should be mixed with 60 mL corn oil and fed in 1 L concentrate ration daily.1920 Once clinical signs are present, the prognosis for complete recovery is guarded to poor. Supplementation has been recommended for foals at risk of developing the disease (e.g., family history of EDM) and foals maintained on poor-quality pasture.1904,1916
EQUINE MOTOR NEURON DISEASE
THOMAS J. DIVERS
In 1990 an acquired neurodegenerative disease of adult horses was first described.1921 The equine disease appeared similar to one form of human motor neuron disease, amyotrophic lateral sclerosis (ALS, or Lou Gehrig’s disease). Equine motor neuron disease (EMND) is the only naturally occurring animal model for ALS. Experimental studies have revealed that the disease occurs secondary to chronic vitamin E deficiency and oxidative neuronal cell injury.1922 Reports of this disorder are now almost worldwide.
Clinical Signs
Affected animals are adults with a mean age of 9 years (range, 2 to 23 years).1923 Clinical signs vary depending on the stage or duration of the disorder; therefore the signs are best summarized by dividing the disease into subacute and chronic forms. A subclinical form also occurs, although these cases cannot be currently diagnosed antemortem.
SUBACUTE FORM
Horses develop acute onset of trembling, fasciculations, lying down more than normal, frequent shifting of weight in the hindlegs, and abnormal sweating. Head carriage may be abnormally low. Appetite and gait often are not noticeably affected, but some horses have a ravenous appetite. The owner may mention that the horse had been losing weight (loss of muscle mass) for a month before the other signs appeared.
CHRONIC FORM
The trembling and fasciculations subside, and the horse’s condition stabilizes, but with varying degrees of muscle atrophy. In some cases the atrophy is so severe that the horse looks emaciated. In other cases, muscle mass and fat deposition show noticeable improvement. The tailhead frequently is in an abnormally high, resting position.
SUBCLINICAL FORM
Experimental research has proved that horses maintained on prolonged diets low in vitamin E may have subclinical disease. This could have significant implications because unknown to the rider or owner, the affected horse would have diminished strength. Pathology of EMND without clinical signs specific for the disease has also been found in a few yearlings with equine degenerative myeloencephalopathy.
Clinical Pathology
Horses with acute or subacute EMND have mild to moderately elevated levels of creatine kinase (CK) and aspartate transaminase (AST). The serum vitamin E is low (<1 μg/mL) in almost all acute and subacute cases. Levels of α- and γ-tocopherol are low in the gray matter of affected horses. Superoxide dismutase (SOD) activity in red blood cells also is severely decreased, believed to result from increased consumption of SOD. Other clinicopathologic abnormalities may be present in particular individuals, including elevated serum γ-glutamyltransferase (GGT) activity; low vitamin A, β-carotene, and ascorbic acid levels; and high liver iron and spinal cord copper levels. Many horses have abnormal glucose absorption, and some have enhanced glucose utilization.1924 This supports the observation that many horses have ravenous appetites and that most have normal fat deposition at necropsy.
The protein CSF level is elevated in almost half of affected horses, and intrathecal immunoglobulin G (IgG) is increased in many cases. Increased IgG is thought be a response to neuronal death rather than a primary pathophysiologic event.
Pathophysiology
The sites of lesions in EMND are the ventral horn cells (lower motor neurons) of the spinal cord gray matter; the nuclei of cranial nerves V, VII, and XII; and the nucleus ambiguous; all undergo noninflammatory degeneration. Neurogenic atrophy occurs in muscles innervated by degenerate neurons, particularly those with predominantly type 1 myofibers, which have a higher oxidative requirement than type 2 fibers and thus are more susceptible to oxidative damage. The dysfunction and death of motor neurons is an oxidative disorder, presumably caused by vitamin E deficiency and the resulting inability to protect against oxidative (prooxidant) stress. Clinical signs occur when approximately 30% of the motor neurons are affected.1925 Some of these neurons may regain function with vitamin E treatment, which may explain why some horses do not have continual progression of clinical signs, as occurs in ALS in humans. Human motor neuron disease, although an oxidative disorder, is more complex that EMND, and the causes have yet to be determined.
Pathology
Gross lesions are limited to pallor of the medial head of the triceps brachii and vastus intermedius muscles. Severe atrophy is present in these muscles and many others, including the sacrocaudalis dorsalis medialis muscle of the tail, which shows severe denervation atrophy and fibrosis. Scattered areas of myofiber necrosis are present in many muscles. A pigment retinopathy1926 and deposition of lipopigment in the spinal cord vasculature are found in horses with EMND, apparently related to similar pigment depositions in other species with vitamin E deficiency. A vitamin E–supplemented diet may result in resolution of pigment deposition in the spinal cord vasculature but not the retina.
Epidemiology
EMND is a sporadic disease, with only single animals in a barn usually affected, although a few outbreaks of the disease have been reported. All breeds of horse can be affected, as can ponies. The apparent prevalence of EMND in certain breeds probably reflects the prevalence of the breed in specific populations of horses, and no familial or heritable predisposition has been identified. Affected horses usually are housed in facilities with little or no access to pasture for more than a year before signs appear.1923,1927 The diet of affected animals in North America is deficient in green foodstuffs and frequently consists of pelleted or sweet feed and poor-quality grass hay, without alfalfa or other sources of vitamin E supplementation.1928,1929 In the United Kingdom a few horses with EMND have been on pasture, have no laboratory or necropsy evidence to support malabsorption, but are deficient in vitamin E.1930 The explanation for this remains elusive. No other management practices, such as worming and vaccination regimens, insecticide use, amount of exercise, or type of bedding, have been related to development of the disease. The highest prevalence of EMND in North America occurs in the northeastern United States and Canada, although sporadic cases have been reported across the continent. This regional distribution is believed to relate to the frequency of predisposing management practices in high-incidence areas rather than other environmental factors.
Diagnosis
The diagnosis of EMND can be made from the following observations and testing:
1
Epidemiologic information.
a
Previous cases in the stable.
b
Historic information that suggests the horse might have been without green forage for an extended period (with a few exceptions in the United Kingdom).
2
Clinical signs (see earlier section).
3
Measurement of plasma vitamin E and muscle enzyme levels.
MUSCLE OR NERVE BIOPSY
Biopsy of the sacrocaudalis dorsalis muscle is the invasive test of choice, with almost 90% sensitivity and specificity.1931 The biopsy specimen should be placed in 10% formalin for laboratory submission. A positive result is the finding of characteristic abnormalities, including denervation atrophy of myofibers and scattered myofiber necrosis. Biopsy of the spinal accessory nerve is more technically demanding but is a more sensitive test in chronic cases.1931,1932 Nerve biopsy requires general anesthesia, whereas muscle biopsy can be performed using a local block in the sedated horse.
Differential diagnoses for EMND include equine protozoal myeloencephalitis, colic, laminitis, botulism, and tying-up.
Treatment
Oral vitamin E supplementation at 5000 to 7000 IU daily may result in improvement in some clinically affected horses, although full recovery is unlikely. This therapy cannot reverse the neuronal death, and all affected horses are permanently weakened. Use of a source of pure vitamin E is preferable to a multi–vitamin-mineral supplement. Natural vitamin E products are preferred over synthetic products, but both will raise serum levels.
Prevention
All horses without green forage (grass of good quality or green hay) for prolonged periods (>1 year) should be routinely tested for the plasma vitamin E concentration and supplemented with vitamin E. If this were a standard policy, most if not all cases of EMND could be prevented.
SPINAL FRACTURES AND LUXATIONS AND SPINAL CORD TRAUMA
Definition and Etiology
Vertebral fractures are relatively common causes of spinal cord injury in food animals. In horses with various neurologic disorders, however, spinal fractures represented only about 3% of cases in two studies.1933,1934 Because of differences in management, temperament, and regional spinal strength, the pathogenesis and predominant anatomic location of spinal injury sites differ among the livestock species.
HORSES
In a U.K. study examining 26 horses with spinal fractures, 16 had cervical fractures, 17 had thoracic fractures, five had lumbar fractures, and four had sacrococcygeal fractures.1935 The vertebrae most often injured were C1, T12, and L5.1935 Horses with lesions of the thoracic dorsal spinous processes did not show neurologic deficits, whereas a high proportion of horses that had lesions in the lumbar and cervical spine had ataxia or other associated neurologic deficits. Stress fractures of the lumbar vertebral laminae may be common in racehorses, frequently are undiagnosed during life,1936 and should be considered in the differential diagnoses for poor performance and hindlimb lameness in at-risk animals.
RUMINANTS
Fractures of the vertebral column in ruminants may result from abnormal bone mineralization. The common sites of spinal fractures in calves are C2 to C4, T10 to T13, and L3 to L6 vertebrae. Spinal fractures are especially common in 3- to 6-month-old ruminants as a result of nutritional deficiencies, including vitamin D, calcium, and copper. Differentiation of nutritional osteodystrophies from traumatic vertebral fractures is essential, because different preventive measures must be taken for the two conditions. Traumatic cervical vertebral fractures of cattle and small ruminants may be caused by injuries sustained in falls, roadway accidents, butting of other animals, predation, or squeeze chutes.1937,1938 Spontaneous fractures of vertebrae weakened by developmental defects (hemivertebrae) or spinal abscesses also are common problems in calves. Rarely, pathologic fracture may result from weakening of bone caused by bacterial osteomyelitis.1939 More often, bacterial infection is a sequela to spinal fracture.
Traumatic luxations or fractures of the atlantooccipital and atlantoaxial joints of pygmy goats may occur when the horns are held during restraint.
Fractures of the lumbosacral spine of cattle are frequently caused by slipping in cemented areas.1937 Many of the spinal fractures occur during mounting by herdmates exhibiting estral behavior. Thoracolumbar fractures may occur in calves during correction of a dystocia, particularly epiphyseal slippage at the middle to caudal thoracic vertebrae.1940,1941 The occurrence of spinal fractures during assisted delivery is mainly related to the excessive traction and rotational force used.1942 Luxation of the sacroiliac joint in the dam may occur with the use of excessive force during manual extraction of a calf. Traumatic lumbar vertebral fractures occur secondary to chronic ankylosing spondylosis in mature bulls and rams.1943
Trauma to the spinal cord without spinal fracture can result in concussive damage to the cord, including shearing forces and hemorrhage within the neuropil. Hemorrhage and inflammation around the cord can result in space-occupying lesions that cause clinical signs similar to those resulting from spinal fractures. Prognosis for recovery is generally more favorable with blunt trauma than spinal fractures, although outcome depends on the severity of the injury.
Clinical Signs
The clinical presentation of a spinal fracture varies, depending on the site of the traumatic lesion, severity of the spinal cord compression, and involvement of specific anatomic tracts.
CERVICAL SPINE
Acute vertebral fractures are painful, and the patient usually shows some distress in the early stages. Goats may bleat or cry when the spine is manipulated. Horses groan and, if recumbent, thrash wildly. Cattle and sheep may lie on their sides and groan. Animals with noncompressive cervical lesions maintain a stiff neck (“weather vane” attitude), refuse to lower the head, and eat from the ground while kneeling. To prehend food, they often keep the head away from the ground and extend the tongue. These animals have stiff necks and resist passive flexion of the head.1944 Similar signs may be seen with meningitis. Forelimb lameness with or without obvious neurologic deficits may be the presenting complaint when fractures are fairly stable and cause minimal disruption to the spinal cord.1945,1946 Cervical fracture should be a differential diagnosis for unexplained forelimb lamenesses.
Animals with severe lesions may be recumbent. Depending on the amount of pain and the secondary complications associated with the disease, the sensorium varies from bright, alert, and responsive to depressed and painful. Recumbent animals may not be able to lift the head from the floor if a high cervical lesion (C1 to C4) is present.1947 Crepitation may be palpable in some cases. Luxation of the atlantooccipital joint results in asymmetry of the wings of the atlas. These animals often have a wry neck as a result of the twisting forces on the displaced atlas.1948 Patients with mild lesions of the cervical spine may be able to stand and show varying degrees of ataxia and conscious proprioceptive deficits. Specific gait abnormalities include circumduction, interference, knuckling, incomplete limb protraction, crossing over, and excessive body sway. Animals with high partial cervical lesions may show hypalgesia of the entire body. Bilateral, severely compressive lesions of the cervical spine can result in rapid death from respiratory paralysis if neurologic injury causes paralysis of the phrenic nerve. If the spinal cord lesion is above the C6 segment (C6 to C7 vertebrae), the muscular tone and spinal reflexes (panniculus, triceps, and biceps) are normal to increased in all limbs. If the lesion is located at the C6 to C8 segments, forelimb reflexes are diminished or absent and hindlimb reflexes normal to increased. The panniculus response may be absent on one or both sides of the body if the lesion affects the C8 segment, the origin of the lateral thoracic nerves.
Conscious pain perception in the limbs often is diminished or absent, depending on the amount of damage in the sensory spinal cord tracts. In long-standing cases in which the ventral rootlets or motor neurons of C6 to C8 have been destroyed, the muscles of the neck and forelimbs may become atrophic. Regional or strip sweating may be observed in some horses. The anal tone and tail tone are normal. The bladder may be distended, but the tone of the urethral musculature is normal.
THORACIC SPINE
Animals with thoracic trauma have an attitude similar to those with cervical lesions, except that thoracic limbs are normal. Animals with severe spinal cord lesions intermittently lie on their sides and then arise to assume a dog-sitting posture. When lying in sternal recumbency, animals hold the hindlimbs extended rather than in the normal tucked-up position. The spinal reflexes of the forelimbs are normal. A crossed extensor reflex may be observed in the hindlimbs. Depending on the amount of damage to the sensory tracts, conscious perception of pain in the hindlimbs may be decreased or absent. The tail and anal tones are normal. The bladder is distended, but the tone of the urethral sphincter and penis is normal. Animals with acute lesions of the thoracic spine (<2 days’ duration) may display Schiff-Sherrington syndrome.1949 Forelimb postural reactions are normal but muscular tone is increased, resulting in a stiff forelimb gait. Postural reactions in the hindlimbs are decreased to absent, with normal to hyperreflexic myotactic reflexes (see Chapter 8). Schiff-Sherrington syndrome is rare in large animals. Most animals with thoracic injury have normal to exaggerated spinal reflexes and hypertonia of the rear limbs.
LUMBAR SPINE
Animals with lumbar injury have an attitude, gait, and posture similar to those with thoracic lesions. Fig. 35-32 shows the characteristic posture of a calf with a fracture at the L6 vertebra. Fig. 35-33 illustrates myelographic findings of cord compression at L6. The forelimbs are normal. Lesions at the L1 to L3 spinal cord segments result in normal or hypertonic and hyperreflexic hindlimbs. Conversely, hypotonia and hyporeflexia of the hindlimbs may be seen in animals with lesions at segments L4 to S2.1950 Marked spasm of the longissimus dorsi muscle has been described in horses with a lesion between the T11 and T12 segments.1951 Lesions of segments L4 to L6 result in cutaneous desensitization of the medial surface of the rear leg and diminished or absent patellar reflex because of dysfunction of the femoral and saphenous nerves. Lesions of segments L6 to S2 result in desensitization of the hindlimb and diminished withdrawal (flexor) reflexes. The panniculus response is absent when the skin posterior to the lesion is stimulated. Damage to the spinal rootlets may cause regional swelling. Tail tone and anal tone are normal. The bladder is distended, but the sphincter tone is normal.
SACROCOCCYGEAL SPINE
Lesions of spinal cord segments S1 to S2 result in decreased conscious proprioceptive responses of the rear limbs1952 and diminished flexor reflexes of those limbs. Anal tone is diminished to absent, and the bladder is distended and hypotonic. The patient is incontinent because of atonia of the urethral sphincter, and urine scalding may be seen over the perineum. The tail is flaccid and paralyzed. The anal sphincter is dilated, and the rectum is filled with dry fecal matter. There may be palpable abnormalities of the pelvis or sacrococcygeal joint. Crepitus of the pelvic bones may be noted when the animal is moved.
Clinical Pathology and Radiographic Findings
Radiography is the usual method of diagnosing a spinal fracture. In young horses, slipped physeal plates are often seen in the atlas.1951,1953 Shortening of the lumbar vertebrae is consistent with either an oblique overriding fracture of the vertebral body or a compression fracture.1950,1953 Abnormally shaped vertebrae may signify an anomalous vertebral arch. Myelographic examination may detect stenosis in segments distant from the fracture site.1954
Examination of cerebrospinal fluid (CSF) collected through a lumbosacral puncture may be useful for ancillary diagnosis of a spinal fracture. Fracture-induced changes in the CSF may be classified as either acute (0 to 1 day) or chronic (>1 day). The acute changes include diffuse blood contamination, a high red blood cell (RBC) count, a normal to high white blood cell (WBC) count, and a high protein concentration. The CSF changes in patients with more chronic injuries include a normal to slightly increased WBC count, normal to increased RBC count, increased protein concentration, and xanthochromia. These changes are not pathognomonic for spinal fracture, however, so should be interpreted with caution.
Pathology and Pathophysiology
Neurologic dysfunction after spinal trauma is similar to that of brain trauma and is a result of both immediate and secondary events. Immediate causes of injury include shearing forces that result in tearing of tissue, hemorrhage, and impingement of bone fragments on the spinal cord, particularly when the fracture is unstable. Trauma initiates a cascade of secondary biochemical events that further injure nervous system tissue, including adenosine triphosphate (ATP) depletion, which deprives cells of their ability to maintain their ionic environment through the action of sodium-potassium ATPase pumps. Rapid intracellular accumulation of calcium and sodium ions causes cytotoxic edema and neuronal depolarization. Release of the excitatory neurotransmitter glutamate and activation of the arachidonic acid and xanthine oxidase pathways further compound cellular damage. Other injurious substances generated after traumatic injury include nitric oxide, oxygen free radicals, lactic acid, arachidonic acid metabolites, and a variety of cytokines. Local vasogenic reflexes, mostly mediated by α-adrenergic receptors, result in decreased blood flow in the gray and white matter. Platelets aggregate in the hypoperfused capillaries and form microthrombi and infarcts. Regional ischemia caused by hypoperfusion results in lipid peroxidation of the axons, myelin degradation, and demyelination.1955
Treatment and Prognosis
It is important to recognize that the radiographic appearance of a spinal fracture is not a reliable prognostic indicator, because the vertebral components are likely to be in a different position from that at the time of injury. Not all vertebral body fractures result in neurologic disease. The prognosis is best judged on the basis of repeated neurologic examinations. Provided the animal is not suffering inhumanely and the pain can be adequately controlled, repeated neurologic examinations should be made over the first several hours. The longer the patient remains recumbent and neurologically impaired, the more unfavorable the prognosis.
The mainstay of immediate treatment of spinal injuries is maintaining whole-body homeostasis, particularly the maintenance of cardiovascular integrity and blood pressure.1956 Ischemia local to the spinal cord plays a key role in development of the injury, but global cardiovascular failure compounds this effect. Most recoveries from spinal cord contusion occur spontaneously and are not appreciably influenced by drug administration. Some recommend treating acutely affected animals with DMSO (0.25 to 1 g/kg IV in 5% dextrose as a 40% DMSO solution) and dexamethasone (0.1 to 0.2 mg/kg four times daily for 2 to 4 days), but supportive data from controlled studies are not available. Analgesics or tranquilizers should be administered with care to ambulatory patients that have thoracolumbar spinal lesions because an ataxic patient may slip and worsen the spinal cord contusion. If signs of pain are severe, NSAIDs or narcotic analgesics may be administered. Epidural catheterization can be an effective method for delivery of analgesics over several days in animals with severe pain.1957 In animals with pelvic lesions, the bladder should be evacuated either by manual palpation per rectum or by insertion of a catheter and use of a closed urinary drainage system. Attention to aseptic procedure during catheter maintenance may reduce the number of iatrogenic bacterial infections. The urine should be cultured and examined repeatedly. Bladder infections should be treated with an appropriate antibiotic. In animals with paralysis of the rectal musculature, the feces should be removed manually. Lubricants (1 to 2 quarts of warm detergent or methylcellulose) may be administered with an enema.
If the spinal fracture appears stable and the animal can stand with assistance, the patient may be placed in a water tank and supported for prolonged periods. Goats are most amenable to treatment for spinal fractures, whereas adult horses and cattle may present insurmountable nursing problems and should be euthanized if they are unable to rise after several days or have intractable pain. Slings are commercially available or can be fashioned from canvas or burlap and wool or cotton. The time spent in the sling varies, depending on the animal’s temperament and degree of neurologic dysfunction. Some horses become frantic while suspended in the device and cannot be supported without the risk of severe injury to the patient and the handlers. The sling should not be used for recumbent animals that cannot support themselves while harnessed. Such animals can sustain severe (even fatal) respiratory compromise or secondary myositis. Nevertheless, daily slinging of animals with mild neurologic signs may reduce secondary medical complications and facilitate recovery.
Recumbent cattle should be floated in a tub of warm water. Watertight tubs for use with large cattle are commercially available* (see Down Cows at end of chapter).
Cervical fractures and luxations of small ruminants may be stabilized by incorporation of the head, neck, and anterior thorax in a fiberglass cast. The cast should extend from the middle part of the thorax to the tip of the nose. The feed and water supply of all animals with cervical fractures should be placed so that the animal can reach it without bending the neck. Surgical methods for stabilization of cervical fractures have been described in horses. The effectiveness of such treatments for restoration of neurologic function after traumatic injury or fracture of the spinal column is variable, but good results may be achieved when presurgical signs are mild and the repair is stable.1958-1961
Fractures of the dorsal spinous processes, transverse processes, and some small, oblique fractures of vertebral bodies may produce minimal instability of the spine. Such injuries may respond well to conservative treatment, such as stall confinement. Bone sequestra can result in the formation of draining tracts, however, and may require surgical removal to facilitate healing.1962
The dietary intake of copper, molybdenum, and calcium should be measured in cattle. If the daily intake is inadequate, the minerals should be fed in supplements. Animals with metabolic bone disease should not be restrained because of the risk of inducing additional pathologic fractures.
ANKYLOSING SPONDYLITIS OF HOLSTEIN BULLS
Ankylosing spondylitis is an inflammatory disease of joint tissue associated with fusion of lumbar vertebrae in Holstein bulls. Approximately 4% of all Holstein bulls used for artificial insemination are culled each year because of spondylitis.1963 The clinical onset of the condition is insidious. The first signs of the disorder are a stilted gait, reluctance to move, and dragging of the toes of the rear limbs. Affected bulls are slow to mount teaser dummies for collection. The condition is progressive, and over several months the animals develop paraparesis and ataxia. With mounting, the ankylosed area of the spine may fracture, leading to acute recumbency. Pathologic changes associated with the condition include calcification of the ventral vertebral ligaments between the T11 and L3 vertebrae. The condition appears to be hereditary. All bulls with the condition possess the class I major histocompatibility complex (MHC) BoLA A8 phenotype. In comparison, the phenotypic frequency of BoLA A8 in the general population of normal Holstein bulls is only 44%.1964
SPINAL ABSCESSES
Definition and Etiology
Most abscesses of the spinal cord originate from a preexisting vertebral body osteomyelitis. The bone usually is infected hematogenously. Extension of bacteria from the lungs, the heart, or a septic injection site is common. Neonates frequently develop vertebral abscesses secondary to septicemia.1965,1966 Bone lesions may develop from sequestra broken from fractured vertebrae. Epizootics of spinal abscesses can result from injection of contaminated vaccines or mineral supplements near the spinal column. Similarly, spinal abscesses may be seen along with other diseases in groups of animals that are immunocompromised, such as cattle with bovine immunodeficiency virus infection.1967 Infectious agents isolated from spinal abscesses of ruminants include Corynebacterium pseudotuberculosis, Aracanobacterium pyogenes, Mannheimia haemolytica, Staphylococcus aureus, and Fusobacterium necrophorum.1965,1968-1970 Agents typically found in vertebral infections of foals include β-hemolytic streptococci, Salmonella species, Actinobacillus equuli, Escherichia coli, Rhodococcus equi, and Klebsiella pneumoniae.1970-1974 Agents less often associated with vertebral body osteomyelitis of horses and cattle include Mycobacterium bovis, Mycobacterium avium, Aspergillus species, Eikenella corrodens, and Brucella abortus.1975-1977 In rare cases, septic arthritis of the atlantooccipital joint may result from extension of a mycotic guttural pouch lesion.1978,1979
If the infectious agent remains localized in the vertebral body, the patient usually shows signs consistent with a transverse myelopathy. If the infection erodes through the dura mater, the animal develops signs of septic meningitis. If the bone infection is extensive, the vertebrae may fracture suddenly, resulting in signs characteristic of spinal trauma.
Clinical Signs
The neurologic deficits of animals with vertebral body abscesses without pachymeningitis are similar to those described previously for spinal fractures.1969 Animals with mildly compressive cervical abscesses show a characteristic “weather vane” attitude, appear stiff, and are reluctant to eat food from the ground.1969,1971,1972 Ruminants with this lesion hold the neck in extension and attempt to prehend the food with the tongue while the head is held more than 30 cm (12 inches) from the ground.1969 Additional signs of spinal abscess include heat, pain, swelling, or crepitus over the affected areas and associated signs of bacteremia. Abscesses in the thoracolumbar spine cause hindlimb weakness and ataxia of variable severity, from mild gait abnormalities to complete recumbency. Stud males may be unable to breed because of weakness and pain.1980 Animals with pachymeningitis show characteristic signs of meningeal inflammation such as hyperesthesia, intermittent spasmodic muscle contractions, and recurrent profuse sweating1971,1975 (see earlier section on meningitis). Differential diagnoses include trauma, aberrant parasite migration, tumor and pathologic fractures, hemorrhage into or around the spinal cord (e.g., postanesthetic myelopathy), myopathies, caprine arthritis-encephalitis, and fibrocartilaginous embolism.1981 Spinal abscesses can be mistaken for other painful conditions, such as orthopedic disorders or traumatic reticuloperitonitis.1982
Clinical Pathology and Radiographic Findings
Radiographs are the best method for obtaining a definitive diagnosis of spinal abscessation. A random pattern of hyperlucency and increased bone density characteristic of osteomyelitis is seen in the affected vertebrae, with or without adjacent soft tissue mass lesions indicative of abscessation1976,1983 (Fig. 35-34). Diskospondylitis usually results in detectable osteolysis in the intervertebral joints.1971 Occasional cases of extradural abscesses without radiographic evidence of osteomyelitis have been described in calves and lambs.1331 Nuclear scintigraphy can be used when the bone lesions are not well defined in plain film radiography.1977 In addition, myelography can be used to detect the specific site of the spinal cord compression.
A complete blood count (CBC) may indicate the presence of a chronic inflammatory focus. Specific changes in the CBC include hyperfibrinogenemia, neutrophilia, monocytosis, nonresponsive anemia, and left shift. The plasma globulin levels are increased in adults but may be increased or decreased in neonates, depending on the adequacy of colostral immunoglobulin transfer.
The changes in the cerebrospinal fluid (CSF) depend on the location of the abscess in the nervous system tissue and the meninges. Changes are present in CSF caudal to the lesion, but not cranial to the lesion; thus, CSF should be obtained by lumbar puncture and not cisternal puncture.1981 In most cases the abscess does not infiltrate through the pachymeninges, and the CSF is normal or shows xanthochromia and mild increases in the protein concentration (60 to 120 mg/dL), with mild to no increase in nucleated cell count.1984 The CSF of animals with pachymeningitis contains high numbers of WBCs (>100 neutrophils/μL) and a greatly increased protein concentration (>200 mg/dL). The CSF may clot after collection because of high concentrations of fibrinogen. Bacteria may be observed in a Gram-stained smear of CSF sediment. Horses with spinal brucellosis may have a rising serum agglutination titer or one above 1:160.1975 Because of a high number of nonspecific reactions in equine sera, titers below 1:40 are considered nondiagnostic.1985 Horses with spinal tuberculosis may be identified by an intradermal skin test using purified protein derivative or tuberculin.1976
Pathophysiology
Hematogenously derived abscesses arise because of embolization of septic thrombi into the metaphyseal arteries. These vessels have a sluggish blood flow because they become tortuous as they approach the physis. The metaphyseal vessels communicate with the ventral vertebral plexus, which in turn drains into the post cava, the portal vein, and the pulmonary veins. The ventral vertebral plexus does not have valves; blood flow reverses with an increase in abdominal or pleural pressure. Regurgitated blood from infected sites in the body cavities showers the vertebrae and spinal cord with bacteria.1968,1977
Necropsy Findings
The most common sites of involvement are the costovertebral and intervertebral articulations and the vertebral body epiphyses.1986 Lumbar vertebrae frequently are involved. The bone is uneven, deformed, and softened. The abscessed area is interspersed with calcified trabeculae and pockets of necrotic debris. Sequestration of necrotic bone may be seen in some cases. The meninges may be adherent to the abscessed site, and occasionally a fistulous tract may be seen from the center of the abscess pocket to the subarachnoid space. In other cases the abscess is compartmentalized away from the CSF, but the proliferating bone impinges on the spinal cord.
Treatment and Prevention
If spinal abscessation is recognized early, prolonged antimicrobial therapy generally is effective. Selection of the appropriate antimicrobial agents should be based on the results of cultures from the patient’s blood, urine, feces, and CSF. When bacteriologic culturing is inconclusive, a broad-spectrum antimicrobial should be chosen. Amikacin (7.5 to 10 mg/kg IM four times daily) or gentamicin (1 mg/kg IM three times daily) combined with potassium penicillin G (10,000 IU/kg IV three or four times daily) should be administered. After 1 or 2 weeks of this therapy, a trimethoprim-sulfonamide combination (2 to 3 mg/kg trimethoprim, 10 to 15 mg/kg sulfadiazine PO twice daily) in horses or procaine penicillin G (10,000 to 20,000 IU/kg SC or IM daily) in cattle can be administered for 2 to 3 months.
Small ruminants with cervical diskospondylitis may show a good response after 2 to 4 weeks of procaine penicillin G (10,000 IU/kg IM twice daily). Phenylbutazone, flunixin meglumine, or aspirin may be administered for pain relief. Immobilization of the head and neck in a fiberglass cast extending from the thorax to the nose may provide support to smaller patients with a cervical abscess. Surgical drainage of the abscess and curettage of the necrotic bone may be feasible in smaller animals with sufficient economic value to justify the procedure.1974,1987 Surgical intervention in adult cattle and horses is usually difficult because of the size of the epaxial musculature and the inaccessibility of the spine in large animals.
SPINAL TUMORS
With the exception of lymphosarcoma, spinal tumors in domestic animals are rare. Tumors reported to invade the spinal cord of horses include lymphosarcoma, plasma cell myeloma, meningioma, ependymoblastoma, fibrosarcoma, schwannoma, melanoma, carcinoma, angioma, angioblastoma, ganglioglioma, and neurofibroma.1988-1999 The most common tumor of the spine of ruminants is lymphosarcoma, but others, such as embryonal neuroectodermal tumors, have been reported.2000,2001
Clinical Signs
The clinical signs of tumorous invasion are indistinguishable from those described previously for spinal fractures. The onset of the neurologic dysfunction varies. Some neurofibromas, melanomas, and lymphosarcomas invade centripetally along the peripheral nerve rootlets. These patients develop slowly progressive dysfunction of the peripheral nerve or spinal cord, which eventually leads to tetraplegia or paraplegia (Figs. 35-35 and 35-36). In rare cases the onset of tetraplegia in cattle with a neurofibroma or lymphosarcoma may be peracute and unaccompanied by prodromal neurologic symptoms. Lymphosarcoma has a predilection for the lumbar segments of the spinal cord and the cauda equina in cattle over 5 years of age. A diagnosis of tumorous spinal invasion should be considered in cases of progressive neurologic disease characterized by flaccid tail and anus, dysuria, urine scalding, distended bladder, perineal analgesia or anesthesia, and paraparesis. Although most animals with spinal tumors are mature to older adults, immature animals can be affected rarely, mainly by embryonal tumors.1994,1999-2001
Clinical Pathology
Examination of CSF may be useful when the tumor has infiltrated the cauda equina and is located in the lumbosacral cistern. In these cases, tumor cells may be biopsied as the needle is inserted into the lumbosacral space. In other cases the CSF may be normal, or albuminocytologic dissociation can be expected (elevated CSF protein with normal to mildly elevated nucleated cell count) with or without variable degrees of hemorrhage and xanthochromia. After necropsy, various immunohistochemical techniques can be used to identify the tumor type when routine histologic examination is insufficient.2000,2001
Treatment
There is currently no treatment for most spinal tumors of large animals. One study reported survival of 57 days after three treatments with L-asparaginase at 10,000 IU/m2. The body surface area was estimated using the following formula:
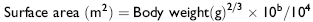
where 10b is a constant that is routinely used for the calculation of surface area in dogs.
The nearly 2-month period of survival allowed the investigators to successfully superovulate the cow. When treating food animals with L-asparaginase, the benefits of the antimetabolite drug must be weighed against the potential for teratogenicity of the fetus, toxicity for humans, and the certainty of relapse in the patient.2002
CEREBROSPINAL NEMATODIASIS
Migration of nematodes and insect larvae through the central nervous system (CNS) can cause acute CNS disease in all species of domestic livestock. The condition occurs in most countries. Parasitic agents that have been reported in the CNS of horses include Micronema deletrix, Hypoderma lineatum, Hypoderma bovis, Strongylus vulgaris, Draschia megastoma, Setaria species, and hydatid cysts.2003-2028 In cattle the condition is caused principally by Setaria species and Hypoderma bovis. Small ruminants that share pastures with the white-tailed deer may become infected with the meningeal worm Parelaphostrongylus tenuis.2013-2015
These parasites can attack any region of the CNS, but most clinical cases result from lesions of the brainstem and spinal cord. The clinical signs are similar for all parasitic CNS infestations and include tetraplegia/tetraparesis, paraplegia/paraparesis, asymmetric conscious proprioceptive deficit, hyperreflexia/areflexia, anesthesia or analgesia of dermatomes, and neurogenic atrophy. Cranial nerve deficits may be seen if the parasites migrate through the brainstem. The following sections discuss specific parasitic syndromes in livestock.
HORSES
Strongylus vulgaris Migration
Migration of S. vulgaris in the CNS causes two major clinical syndromes: acute embolization of parasitic emboli and slow perivascular migration of living parasites in the CNS. The two forms have a common pathogenesis. Aberrantly migrating fourth-stage or fifth-stage larvae in the intima of the aorta or left ventricle damage the endothelium, stimulate the clotting cascade, and cause formation of a thrombus that often contains the parasitic larva.2020 Embolization of the thrombus to the brain results in fulminating encephalitic signs.2009 As the embolus is degraded, the parasite migrates from the blood vessel into the CNS, resulting in the progressive brainstem disease. The cranial brainstem (diencephalon) is most often affected. The thrombosis and migration are accompanied by multifocal infarction, edema, hemorrhage, and necrosis2007 (Fig. 35-37). Microscopic findings include linear tracts of hemorrhage lined by neutrophils, macrophages, eosinophils, and reactive glial cells. Anesthesia of the hindquarters has been described in some affected horses.2010 Donkeys are also susceptible to the aberrant migration.2029 CSF changes associated with S. vulgaris migration in equids include xanthochromia, refractive index over 1.3353, protein concentration ranging from 32 to 550 mg/dL, and increased WBC count (42 to 10,000/μL). The differential counts range from 70% to 80% neutrophils, 12% to 19% mononuclear cells, and 1% to 2% eosinophils.
Hypoderma lineatum and Hypoderma bovis (Warble Flies)
Hypoderma lineatum and H. bovis are parasites that typically affect cattle but occasionally migrate aberrantly in the horse. Warble flies hatch from pupae in the early spring and mature during the summer. The flies deposit the eggs of H. lineatum on the lips, where they hatch and are swallowed. The ingested worms burrow through the intestine and along the adventitia of blood vessels until they reach the CNS.2008 The flies deposit the eggs of H. bovis on the legs, where they hatch and burrow into the skin. The subcutaneous parasites then migrate as first-instar larvae to the spinal column, where they penetrate the epidural space along the peripheral nerves.2004 Peroneal nerve paralysis from a local parasitic invasion has been reported in a pony.
Micronema deletrix
Micronema deletrix is thought to be a free-living rhabditid nematode that gains access to the CNS by penetration through the skin of the face and the lips, gums, and tongue.2005,2006 The parasites migrate into the brain through the vascular system and cause diffuse encephalitis. Nematodes are found in the tunica adventitia and tunica media of blood vessels.2005 Clinical signs of M. deletrix depend on the localization of the parasite. Spinal cord invasion is apparently less common than migration through the brainstem, cerebellum, thalamus, forebrain, and deeper layers of the cerebral cortex. Clinical signs include asymmetric ataxia, loss of conscious proprioception, depression, behavioral changes, propulsive walking, head pressing, head tilt, circling, nystagmus, recumbency, convulsions, and coma. Affected animals may have granulomatous masses in the nares, pharynx, and maxilla. These could be helpful in formulating a differential diagnosis when parasitic migration is suspected. CSF changes in affected horses include pleocytosis (25 to 80 nucleated cells/μL), a normal to high protein concentration (69 to 114 mg/dL), and xanthochromia.2003 The cellular types were mostly lymphocytes and macrophages ranging from 78% to 91% of the nucleated cells. Eosinophils were observed in the CSF of one horse.
Draschia megastoma
Draschia megastoma has been found in the brainstem of a horse from the southern United States.2011 The adult worm is embedded in a pyogranulomatous lesion of the equine stomach, and the eggs are shed into the stomach. They hatch in the small intestine to form first-stage larvae, which are passed in the feces and ingested by the larvae of Musca flies. The third-stage larvae migrate to the mouthparts of the fly and are deposited on the mucous membranes of the host as the fly feeds.
Setaria
Setaria parasites are common filarid parasites of cattle that migrate aberrantly when they infect horses, sheep, or goats. These parasites have a worldwide distribution, and clinical cases are especially common in India and the Orient, where the common name for the disease is “kumri” (weak back).2025 There are at least four Setaria species, of which S. equina, S. digitata, and S. labiatopapillosa are most common. The parasite is found in the connective tissues and peritoneal cavity of cattle, where it produces circulating microfilariae. Mosquitoes and possibly other bloodsucking insects become infected by the microfilariae and thus transmit the parasite.2024 The parasite has a predilection for the spinal cord in horses. The clinical signs of reported cases include hypotonic tail, bladder paralysis, ataxia, and conscious proprioceptive deficits.2012 Changes in the CSF associated with Setaria infestation include xanthochromia, pleocytosis (25 to 84 cells/dL), and a slightly increased protein concentration (∼114 mg/dL). The cells of one horse contained a small proportion of eosinophils and basophils, but this finding was inconsistent with other reports.2003
SHEEP AND GOATS
Setaria
The infection caused by Setaria species in sheep and goats is similar to that described previously for horses.
Parelaphostrongylus tenuis
Disease caused by P. tenuis occurs predominantly in sheep and goats of the northeastern United States and western Canada.2013,2016,2018,2019 The case-attack rate ranges from 10% to 59%.2014 The disease appears to be spreading because of the increased range of the primary host, the white-tailed deer. Migration of the parasite in the CNS of deer is relatively innocuous, but aberrant migration occurs in domestic small ruminants. The result of this migration is severe signs of spinal cord and brainstem disease.2014 The life cycle of the worm is complex. Adult worms are found in the cranial subarachnoid space, venous sinuses, and spinal subarachnoid space of the deer, where they reproduce.2015 Eggs are deposited into the venous blood and migrate into the lungs, where they embryonate. The larvae penetrate into the airways and are coughed into the pharynx, swallowed, and passed in the feces. They then penetrate into snails and slugs. Sheep and goats are infected when they eat the snails. After ingestion by the ruminant, the larvae penetrate the gastrointestinal wall and enter the CNS by migration along the nerve rootlets. Because of the complex life cycle of the parasite and the indirect life cycle in invertebrate hosts, the neurologic disease in sheep and goats is seen exclusively in late fall and winter. The pathologic lesions of the CNS of affected animals include asymmetric irregular tracts of disrupted necrotic tissue with macrophage infiltration. Coiled larvae occasionally may be seen in the tissues of some affected animals and may be excreted in the feces; however, these are difficult to distinguish from the larvae of Müllerius worms.
The clinical signs of P. tenuis infection are acute. In untreated animals the disease is progressive. The CSF of animals with Parelaphostrongylus infection contains increased concentrations of protein (56 to 157 mg/dL), RBCs (300 to 41,000/μL), and WBCs (17 to 700/μL). The differential cell counts contain a large number of eosinophils (7% to 97%).2030
CATTLE
Hypoderma Bovis
After hatching, the larvae of H. bovis burrow through the skin and migrate along the peripheral nerves to the spinal canal. When the larvae reach the spinal canal, they lie dormant for 2 to 3 months in the epidural fat. Most of the larvae lodge in the lumbosacral part of the spinal cord; very few are found in the cervicothoracic region. If the larvae are killed while lodged in the epidural fat, the host mounts a marked inflammatory response. The swelling and inflammation caused by the dead worms results in spinal cord disease. In clinical practice these signs most often occur by 2 days after administration of a systemic organophosphate grub treatment.2027,2028 Other drugs that kill Hypoderma larvae in the spinal cord could cause a similar problem. In most of North America the grub is located in the epidural space between the months of July and October.
The clinical signs of hypodermiasis include stiffness of the rear limbs, ataxia, paraparesis/paraplegia, hemiparesis/hemiplegia, or tetraparesis/tetraplegia. The conscious proprioceptive responses are greatly altered in the affected limbs. Reflex activity varies, depending on the level of the lesion in the spinal cord.
The CSF changes of hypodermiasis vary. Because of the epidural location of the grub, most affected animals have normal CSF values. If pressure changes induce vascular damage, CSF changes might include mild xanthochromia and slight increases in WBCs and the protein concentration.
Setaria digitata
Infection of the spinal cord of cattle by S. digitata has been described in India.2017
Diagnosis of Parasitic Infestation of Central Nervous System
Parasitic myeloencephalopathy must be considered in all cases of acute asymmetric disease of the spinal cord, cerebellum, or brainstem. Identification of eosinophils in the CSF may be helpful; however, this pattern is not seen in every neuroparasitic diseases. For example, Hypoderma infestations often are extradural, and CSF changes reflect only increased pressure. The CSF of horses with acute S. vulgaris migration could be normal or could show xanthochromia, increased concentrations of RBCs or WBCs, and increased protein concentration.2009
Treatment
Although severe reactions often are associated with death of the CNS parasites, administration of parasiticides in conjunction with heavy antiinflammatory therapy is recommended. Such treatment prevents further migration of the parasite yet mitigates the host inflammatory responses.
The recommended treatment for neural S. vulgaris infection is either thiabendazole (440 mg/kg PO daily for 2 days) or mebendazole (30 mg/kg daily for 2 days). Horses should also be given a combination of corticosteroids and NSAIDs for 10 days after administration of the parasiticides.
Some experts speculate that ivermectin may be a valuable broad-spectrum treatment for all CNS parasitic infections. The drug has a prolonged plasma half-life after parenteral or oral administration (2.7 days) and may exert an antiparasitic effect for as long as 14 to 21 days after subcutaneous administration.2031-2033 Although ivermectin diffuses across the blood-brain barrier, the plasma concentrations after oral administration are low, and the drug should be administered parenterally to achieve optimum efficacy. Unfortunately, significant side effects occur (0.92% overall adverse reaction rate) after parenteral administration to horses.2034,2035 Consequently, until ivermectin is proved to be effective by pharmacologic and clinical studies, alternative parasiticides should be considered for initial treatment of parasitic CNS disease in horses.
HYPODERMA BOVIS
Systemic organophosphate insecticides formulated for oral administration or pour-on application have been recommended for eliminating H. bovis from the CNS. These one-time formulations include crufomate (75 mg/kg as 13.5% Ruelene), trichlorfon (40 mg/kg PO), famphur* (13.2% 1 fluid ounce per 90 kg body weight, not to exceed total dosage of 4 oz for cattle), ronnel (100 mg/kg PO for cattle or horses), and ivermectin 0.5% solution (1 mL/10 kg). Although ivermectin kills the cattle grub in the subcutaneous tissues, its safety and efficacy in the treatment of clinical neurologic disease are uncertain.
It is important to remember that the treatment of affected animals with any of the systemic parasiticides may aggravate neurologic disturbances through release of toxic factors or the development of local immunologic responses to the dying worms.2019,2036 Concomitant treatment with corticosteroids (e.g., dexamethasone, 0.1 to 0.25 mg/kg IV every 6 hours) 1 day before and 5 days after treatment is recommended to reduce the inflammation. Dying cattle grubs also release a systemic toxin that lowers blood pressure and causes acute dyspnea and collapse. The systemic toxic effects of the dying grubs can be ameliorated by concomitant administration of phenylbutazone (4 mg/kg IV or PO twice daily in horses; 10 mg/kg IV or PO once every 36 hours in cattle), aspirin (100 mg/kg PO two or three times daily for cattle), or flunixin meglumine (0.5 to 2.2 mg/kg IV twice daily for horses or cattle). Naproxen (10 mg/kg IV twice daily for horses) may be a useful alternative to phenylbutazone therapy.
Hypoderma infestation can be controlled by prophylactic administration of the pour-on insecticides before worms have migrated into the nervous tissues. The appropriate time for application of the grubicide depends on the time of pupation and the emergence of adults. In most of North America the flies emerge by May, and the larvae reach the nervous tissues by November. Therefore, prophylactic treatment of cattle or horses with organophosphorus compounds should be completed by August or September in warmer climates and October in colder areas.
PARELAPHOSTRONGYLUS TENUIS
A number of drugs, including levamisole (7 mg/kg PO in a single dose), diethylcarbamazine (40 to 100 mg/kg twice in 72 hours), and thiabendazole (250 to 440 mg/kg PO on 2 consecutive days), may be effective for eliminating P. tenuis from the CNS.2013,2037 Ivermectin has been administered to some affected goats, but only one of three treated animals recovered.2030 The lack of response to ivermectin was related to its poor distribution in the CNS. Administration of ivermectin to animals before exposure protects them from larva migrans for 7 to 14 days.2038 All patients treated with anthelmintics should be given corticosteroids and NSAIDs concomitantly.
SETARIA SPECIES
Administration of a single dose of diethylcarbamazine (80 to 100 mg/kg PO) may be effective against migrating Setaria larvae and adults. The drug also has been shown to be effective in preventing infection in sheep and goats when given at 20-day intervals (40 mg/kg PO) during the vector season. The efficacy of diethylcarbamazine in the treatment and prevention of Setaria infection in horses is unclear. As with therapy for other parasitic nervous system infections, patients should be given corticosteroids and NSAIDs concomitantly with the parasiticide therapy.
FIBROCARTILAGINOUS EMBOLIZATION
Fibrocartilaginous embolization has been described in horses, lambs, and a calf.2039-2044 The clinical signs are those of an acute to peracute onset of myelopathy that usually is asymmetric. Paresis to paralysis of the limbs caudal to the lesion occurs, as does hyperreflexia (if the lesion is above the brachial or lumbosacral intumescences) or hyporeflexia to areflexia (if the lesion is in an intumescence). Differential diagnoses that should be considered include traumatic injuries and, in horses, equine herpesvirus infection. Other types of myelopathy usually have a more gradual onset and less acute course. Lambs may develop diffuse tremors that resemble the truncal ataxia of cerebellar disease. The index of suspicion is increased by ruling out other causes of myelopathy, but definitive diagnosis usually is only made at necropsy. There is no effective treatment for the condition, and affected large animals have not recovered. In dogs, however, in which a similar or identical condition is common, partial to complete recovery over several weeks to months is common and might be anticipated in some milder cases in large animals, particularly when the signs are the upper motor neuron type.2045,2046
The emboli are believed to originate from the nucleus pulposus of the intervertebral disks and can be identified histologically using an alcian blue stain.2042 The exact cause of the embolization and the associated mechanisms are unknown. Pressure changes or lesions associated with degenerative arthropathy might result in herniation of disk material into the marrow cavity of a vertebral body. From there the material is hypothesized to enter the basivertebral veins and pass retrograde along the valveless basivertebral plexus to the spinal veins, where it gains access to the vertebral arterial circuit. The manner in which the material enters the arteries is unknown, but some authors have postulated the presence of arteriovenous shunts in the vertebral vasculature.2039 Other mechanisms proposed to explain embolization include herniation of nucleus pulposus into persistent or anomalous embryonic vasculature, into neovasculature formed at the site of a chronically degenerated intervertebral disk, or through the vertebral endplate via Schmorl’s nodes into the marrow cavity of the vertebrae and then into the vascular system. Sudden increases in pressure within the disk, such as may occur during exercise or from trauma, may play a role in the formation of emboli. Once the material has embolized, the neurologic signs are related to swelling, infarction, necrosis, and hemorrhage of the neuropil. The emboli occur exclusively in the brainstem, spinal cord, and cerebellum. The CSF of affected animals has been reported to be normal,2040 but mild pleocytosis and elevations in the protein concentration could be expected in CSF obtained caudal to the lesion in some cases.
POSTANESTHETIC MYELOPATHY AND ENCEPHALOPATHY
A sudden-onset myelopathy has been described in several horses and in a calf after anesthesia.2047-2050 Most of the horses were young and in dorsal recumbency during anesthesia. The calf described with a similar syndrome was severely hypotensive during anesthesia. Paraparesis, ataxia, recumbency, or paraplegia occurs immediately after anesthesia or within a few days. Other reported signs include hypalgesia and scoliosis. Lesions occur mainly in the thoracolumbar spinal cord, although the cervical spine may be involved, and consist of malacia predominantly within gray matter (poliomyelomalacia), with severe hemorrhage in the spinal cord parenchyma (hemorrhagic myelomalacia) in some cases.
Rare cases of postanesthetic encephalopathy have been described in horses.2051,2052 Clinical signs included dementia, pacing and circling, cortical (“central”) blindness, ataxia, hypermetria, and recumbency. The onset of signs ranged from a few hours after recovery to several weeks later. The major pathologic finding in affected horses was cerebrocortical necrosis, particularly in regions of the cortex supplied by terminal arterial branches (“watershed zones”).
Although the pathogenesis of postanesthetic myelopathy is incompletely understood, it likely results from compromise of the vascular supply to the spinal cord caused by hypotension or pressure from the abdominal viscera on the great veins (caudal vena cava, azygous vein) resulting in venous stasis. Similarly, systemic hypoxia, hypovolemia, and hypercapnia may underlie postanesthetic encephalopathy.2052 Factors suspected to increase the risk of postanesthetic encephalopathy include positioning in dorsal recumbency during anesthesia, multiple anesthesias, endotoxemia, and shock. Differential diagnoses for myelopathy include orthopedic conditions such as limb fractures, spinal fracture, and myopathy secondary to recumbency. Differentials for encephalopathy include numerous metabolic, infectious, and toxic disorders. Prognosis for recovery in both conditions is poor. The risk of these complications probably can be reduced by supporting adequate blood pressure and ventilation in patients under anesthesia and avoiding placing patients in dorsal recumbency whenever possible.
OCCIPITOATLANTOAXIAL MALFORMATION
Occipitoatlantoaxial malformation appears to consist of a spectrum of cervical spinal abnormalities rather than a single specific anatomic defect.2053 It occurs in cattle, sheep, goats, and horses.2054-2056 At least five different types of defect have been reported in horses, as follows2057-2064:
1
A heritable condition in Arabians characterized by symmetric atlantooccipital fusion, atlantalization of the axis, and hypoplasia of the atlantal wings.
2058,2062,2065 A similar disorder has been reported in an Arabian-cross colt.
20662
A nonfamilial disease of quarter horses with spinal lesions similar to the lesions in Arabians.
2067,20683
Asymmetric malformations of the occipitoatlantoaxial area in Morgan horses and standardbred foals.
20534
Atlantal duplication in Arabian and Arabian crossbred horses.
2069,20705
Asymmetric occipitalization of the atlas and symmetric atlantalization of the axis.
Macroscopic pathologic lesions common to the five forms include loss or flattening of the occipital condyles, asymmetric flattening of the articular surfaces of the axis, and shortened dens. The pathogenesis of the vertebral defects is unknown, but the disease has been shown to be heritable in the Arabian horse.2071
The clinical signs vary considerably and range from normal neurologic function with or without torticollis to brainstem compression, sudden unexpected death, and stillbirth.2053 In typical cases, signs of tetraplegia or tetraparesis begin at or shortly after birth and progress at a variable rate. Foals may become suddenly tetraplegic, appear to stabilize for several days, but then die suddenly.2062 In rare cases, horses may not show nervous system signs until 3 years of age.2061
The signs are symmetric in most affected animals and include conscious proprioceptive deficits, tetraplegia, hyperreflexia, and hypertonia. Some affected animals may show a reluctance to move the neck and head and resist vigorously when the proximal cervical area is passively flexed. A clicking, creaking, or crepitation may be palpated over the cervical spine when the head is moved.2053,2057 Animals with asymmetric bone lesions often show torticollis, whereas patients with symmetric lesions hold their heads in extension and frequently display the “weather vane” attitude. Neurologic deficits may not be seen despite moderate torticollis.
The bone lesions are readily apparent on radiographic examination (Figs. 35-38 and 35-39), and computed tomography (CT) can further elucidate the anatomic abnormalities.2067 Affected animals may show subluxation of the atlantoaxial joint, ventral displacement of C2 in relation to C1, nonunited ossification center of the dens, shortened or elongated dens, shortened transverse process of the atlas, fusion of C1 with the occipital condyles, atlantal duplication, and deviation of the basilar bone.2072 In sheep, additional malformations of the cervical vertebrae have been seen concomitantly.
Some suggest that treatment could include surgical fusion of the atlantoaxial joints, with or without a laminectomy.2058,2061,2073-2075 Laminectomy alone has been used to alleviate spinal cord compression and clinical signs caused by occipitoatlantoaxial malformation.2076 However, long-term results of surgical intervention have been poor, with neurologic deficits persisting. Closed reduction of the luxated atlantoaxial joint has been similarly unsatisfactory.2067,2077 Arabian horses should not be treated because of the hereditary nature of the disease in that breed.
SYSTEMIC NEUROAXONAL DYSTROPHY
Systemic neuroaxonal dystrophy is seen in purebred Suffolk sheep.2078 The animals are born normal but show a hindlimb ataxia beginning at 1 to 5 months of age. The disease is progressive, and eventually the animals become recumbent and either die or are euthanized after 8 to 10 weeks. Pedigree analyses have indicated that an autosomal recessive trait may be responsible for the condition. The pathology of this disease is characterized by numerous focal axonal swellings in gray matter and adjacent white matter, particularly in areas of the spinal cord and brainstem involved in conscious proprioception (dorsal and intermediate horn gray matter in spinal cord, various nuclei in medulla and caudal midbrain). A condition that is clinically and pathologically similar to systemic neuroaxonal dystrophy has been described in 4- to 7-month-old Merino lambs.2079
WEAVER SYNDROME (BOVINE PROGRESSIVE DEGENERATIVE MYELOENCEPHALOPATHY)
Weaver syndrome is a progressive hereditary CNS disease of 5- to 10-month-old Brown Swiss and Angler cattle.2080,2081 The disease has been reported in the United States, Canada, Denmark, and Switzerland. The incidence of weaver syndrome in some countries may be as high as 563 per 100,000 registered Brown Swiss cattle.2082,2083 The disease affects both genders, but males are affected more often than females.2082
Affected animals develop clinical signs between 5 and 8 months of age. The animals are easily pushed around by herdmates and show marked proprioceptive deficits when forced to move. The clinical signs worsen until the animals become recumbent by 18 to 36 months of age.2080,2084,2085 The pelvic limbs are most severely affected. Specific signs include weakness, ataxia, conscious proprioceptive deficits (circumduction, crossing over, interference, knuckling), muscle tremors, and recumbency. In early stages, attempts to move rapidly may result in cessation of movement of gait in the hindlimbs while the animal continues to pull with the forelimbs, causing the hindlimbs to be pulled too far caudally. Some animals may show varying degrees of hypermetria (“goose stepping” gait) in the limbs. The spinal reflexes and cranial nerve function are normal. Anestrus in females and aspermatogenesis of affected bulls have been described.2085 The disease is progressive and leads to irreversible recumbency. The sensory nerve conduction velocity is reduced in affected calves, but motor nerve conduction velocity is normal. Electromyograms and electroencephalograms are normal in affected calves.2086 The CSF of affected cattle may show an increased concentration of protein (0 to 127 mg/dL) and creatine phosphokinase (2 to 89 mg/dL).2087
Except for muscular atrophy of the pelvic limbs (in long-standing cases), the small ovaries, and hypoplastic testicles, macroscopic lesions are not seen.2082,2088,2089 Although severe muscular changes are not observed, ultrastructural studies of muscle from affected cattle support the hypothesis that myopathic changes are a significant feature of bovine myeloencephalopathy.2090,2091 The primary microscopic abnormalities include degeneration of the rubrospinal spinocerebellar tracts, particularly in the ventral funiculi of the thoracic spinal cord. The lesions include axonal degeneration, vacuolation of the white matter, spheroids, phagocytosis of myelin debris, gliosis, and status spongiosus.2082 Axonal swellings have been observed in the brainstem nuclei and medulla oblongata. An ultrastructural study has shown a reduction of the height of the paramembranous densities of the synaptic junctions of affected cattle,2092 indicating impairment of transmitter releases, dysfunction of the synaptic endplates, or losses of specific cell populations from the motor cortex. Other ultrastructural changes include axonal swelling and vesiculation, swelling of the mitochondria of the Schwann cells, and membrane-bound vesicles.2085,2093 Cerebellar lesions include degeneration and loss of Purkinje’s cells and swelling of Purkinje cell axons.2088
The disease is thought to be transmitted by a simple autosomal recessive trait. Apparent association between the weaver condition and genetic predisposition for high milk yield would favor retention of weaver carriers in situations of intense genetic pressure for production.2094 A genetic test is available for this disease.2095 Carrier bulls identified by the American Brown Swiss registry are designated by the suffix “W” as an integral part of their registry name.2096
PROGRESSIVE SPINAL MYELINOPATHY OF BEEF CATTLE
Spinal myelinopathy of beef cattle has been reported in Australia.2097 The condition occurs mainly in animals of the Murray Grey breed and is inherited in an autosomal recessive manner.2098 Most of the affected animals are unable to stand at birth. Less severely affected calves usually exhibit severe conscious proprioceptive defects. The muscular tone of the hindlimbs is increased. The condition is progressive, and affected animals die or are euthanized by 12 months of age.
The pathologic changes are restricted to the spinal cord white matter, with swollen axons, dilated myelin sheath, wallerian degeneration, and ballooning of the axonal sheaths. There is mild chromatolysis. The animals have normal hepatic copper concentrations, indicating that the condition is not related to enzootic ataxia.2097
BOVINE SPINAL MUSCULAR ATROPHY
Bovine spinal muscular atrophy (SMA) is a heritable disorder of Brown Swiss cattle, certain breeds derived from the Brown Swiss (e.g., Braunvieh, Red Swiss), and Holstein-Friesians.2099-2103 Most affected calves appear normal immediately after birth but within a few weeks develop severe weakness and rapidly progressive muscle atrophy, particularly affecting proximal musculature. Calves deteriorate rapidly; most become recumbent and develop bronchopneumonia. SMA is characterized pathologically by degeneration of motor neurons in the ventral horn of the spinal cord gray matter (equivalent to human anterior horn cells). Pathology is most severe in the brachial and lumbosacral plexuses but occurs throughout the spinal cord. Abnormalities include neuronal loss, swelling and accumulation of phosphorylated neurofilaments, and neuronophagia.2104 These findings suggest that neuronal degeneration in SMA is a consequence of necrosis rather than apoptosis.2102,2104 Sensory neurons are unimpaired.2099,2105 The SMA-determining gene has recently been mapped to chromosome 24 in Brown Swiss cattle, similar to the equivalent gene in humans.2106,2107 Bovine SMA is inherited as an autosomal recessive trait.2099 Although this condition resembles weaver syndrome, the two diseases can be differentiated clinically. Signs of spinal muscular atrophy usually are first seen between 2 and 5 weeks of age. Calves with weaver syndrome first show signs at 5 months of age. The degree of muscular wastage is much greater in calves with spinal muscular atrophy than with weaver syndrome.2108 SMA also must be distinguished from the dysmyelination described in Braunvieh and Brown Swiss–Braunvieh calves (see next section).
SPINAL DYSMYELINATION OF BRAUNVIEH AND BRAUNVIEH-CROSS CALVES
A congenital spinal condition characterized by dysmyelination of the dorsal tracts of the spinal cord has been described in Braunvieh–Brown Swiss crossbred calves.2109 Test matings have shown that the disease is inherited in an autosomal recessive manner.2110 Calves are recumbent from birth. They have a coarse tremor of the head, neck, and body when stimulated and generalized muscular atrophy. Necropsy findings included deficient myelin production and demyelination of the dorsal sensory tracts of the spinal cord. The neurons of these calves are normal, which differentiates the condition from weaver syndrome and spinal muscular atrophy. A similar condition was described in purebred Braunvieh calves; recumbency from birth, opisthotonos, and muscular spasticity with increased reflexes were present in affected calves.2111 Bilaterally symmetric reduction of myelin was found in the spinal cord.
MYELOPATHY IN HOLSTEIN-GIR CALVES
A single report describes a myelopathy in Holstein-Gir calves in Brazil.2112 Calves were normal at birth but developed quadriparesis and ataxia at about 3 months of age, becoming recumbent within 1 to 2 weeks after onset of signs. All died from respiratory complications within a few weeks. Nervous system lesions were confined to the spinal cord in all except one calf examined; findings were consistent with a dying-back neuropathy. Lesions were broadly symmetric bilaterally and not limited to any specific tracts within the cord. In one calf, small glial nodules were found in the basal nuclei and medulla. Both male and female calves were affected, and all were offspring of a single Holstein bull, supporting a genetic but not sex-linked basis for this disorder.
NEUROPATHY, MYOPATHY, AND GLOMERULOPATHY OF GELBVIEH CATTLE
A disorder affecting peripheral nerves, spinal cord, kidneys, and muscles has been described in a small number of Gelbvieh calves.2113,2114 Animals present with progressive ataxia, stiffness, and weakness at a few weeks of age to slightly over 1 year old. Signs may start in the rear limbs but progress over weeks to months to affect all limbs. Animals may become recumbent but remain bright, with a good appetite. Muscle atrophy is frequently observed, as are flaccid paresis, hyporeflexia, and decreased anal tone.2114 Hypalgesia to analgesia may be present in affected limbs. Signs of cranial nerve dysfunction are rare. Proteinuria is the most consistent abnormality of clinical chemistry, whereas other changes, such as neutrophilia and elevated serum creatine kinase, are variable and may relate to secondary problems (e.g., muscle damage when recumbent). Histologic alterations include degenerative changes in peripheral nerves and spinal cord (particularly dorsal columns), renal lesions described as “interstitial nephritis” or “glomerulopathy,” and variable myopathic changes (e.g., necrosis, fibrosis, atrophy). Vascular lesions also have been described.2113 Controversy surrounds the relative severity and importance of neuropathy, myelopathy, and myopathy, but all agree that all these tissues are involved and that renal lesions are prominent. Studies of potential toxicities or nutritional deficiencies within affected herds tend not to support these etiologies, but rather suggest that this is a heritable disorder. The exact mechanism of inheritance and the molecular basis for this disease have not been elucidated. No treatment has been described, and the prognosis appears to be poor. Prevention likely would involve changes in breeding management.
PROGRESSIVE ATAXIA OF CHAROLAIS CALVES
Progressive ataxia occurs in purebred and crossbred Charolais calves of both genders between 6 and 36 months of age.2115-2120 The condition has a worldwide distribution. Affected calves develop posterior paresis and become recumbent by approximately 2 years of age. The disease is thought to be caused by a recessive genetic defect. Preliminary studies to locate the defect in the gene coding for myelin basic protein have been reported.2121 The clinical signs begin with posterior ataxia and end in lateral recumbency. Other neurologic signs of progressive ataxia include stiffness of the neck, aggressiveness, dragging of the rear toes, stumbling, and loss of conscious proprioception (abduction, knuckling, circumduction, abnormal leg placement at rest).2122 In the initial stages of the disease, the gait deficits worsen with exercise and improve after a period of rest. Muscular tremors or a jerking movement of the limbs and tail may be seen when the affected animal attempts to rise.2117,2123 Some animals may be found down acutely and primarily show signs of central vestibular disturbance.2122 Head bobbing may be observed, but affected animals have no other signs of brain involvement. Difficulty in assuming and maintaining a urination posture and prolonged pulsatile micturition are characteristic abnormalities in affected animals.2115,2119,2124 The major pathologic lesion is eosinophilic plaques in the white matter of the brain. Plaques also extend into the white matter of the cerebellar folia and peduncles. Ultrastructural changes include hypertrophy of oligodendrocytes and dysmyelination.2125 These lesions, which have been characterized as an oligodendrogial dysplasia, are almost unique to this particular disease of Charolais cattle, although similar pathology has recently been described in dogs.2126
SPASTIC PARESIS (ELSO HEEL)
Spastic paresis is characterized by marked asymmetric spasticity and hypertonia of the rear limbs. The etiology is uncertain; it may be genetic. Breeds in which the condition has been recognized include the Holstein, Brahman, Angus, shorthorn, Charolais, Simmental, Red Danish, crossbred shorthorn, Gelbvieh, Ayrshire, polled Hereford, Hungarian red spotted, Kankrej, Belgian blue, and other rare European breeds.2127-2139 The clinical signs of this condition are clinically similar to inherited periodic spasticity (see next section), except that spastic paresis is seen in young animals (onset at 3 weeks to 1 year of age) and occurs at all times when the animal stands.2127-2130 In comparison, inherited periodic spasticity occurs in adults in episodic fashion, with normal gait between episodes. Spastic paresis has been recognized in pygmy goats.2140
Clinical Signs
Spastic paresis is characterized by intermittently increased extensor tonus in the pelvic limb as the animal attempts to walk.2127,2131,2136,2141 The gastrocnemius and superficial digital extensor muscles are spastically contracted in all calves, whereas the biceps femoris, adductor, quadriceps, semitendinosus, and semimembranosus muscles are less often affected.2128,2131,2141,2142 The extensor tone is normal when the calf is recumbent and relaxed but becomes excessive when the animal stands and attempts to bear weight. The excessive extensor motor activity results in an inability to flex the hock during protraction of the pelvic limb. To prevent the toes from dragging, the limb is circumducted, resulting in a pendulum-like motion.2142 At rest the limb is held stiffly abducted and is repeatedly circumducted. The foot is held off the ground, and the gastrocnemius muscles appear to be underdeveloped. The tail is elevated from the ischiorectal fossa as the animal attempts to move. Eventually there is atrophy of the hindquarters. The spasticity is progressive, and affected animals experience difficulty rising and grazing. If untreated, the animals are stunted and usually are culled.
The excessive pull of the extensor tendons produces radiographically detectable changes in the bones of the hock, including osteoporosis, lipping of the dorsal aspect of the tibial epiphysis, plantar displacement of the proximal part of the tibial diaphysis and epiphysis, and excessive straightening of the tuber calcis.2127,2128,2143
The etiology and pathogenesis of spastic paresis are unknown. Some investigators have documented the presence of subtle demyelinating lesions of the red nucleus but were unclear about the contribution of these changes to the clinical syndrome.2132,2134 Changes observed in affected calves include nonsuppurative encephalitis and reduced concentrations of dopamine and 5-homovanillic acid in the cerebrospinal fluid.2144 There are no histologic or biochemical alterations of the myofibrils.2145 Neuropharmacologic studies have indicated that the disease may be related to overstimulation of the gamma motoneurons of the spinal cord.2146 A genetic basis for the disease has been postulated because affected offspring tend to have a common paternity2127,2130,2132; however, breeding experiments and progeny evaluation of cattle have not demonstrated heritability. The condition has been seen in female calves only, in one case, and occurred in a single year; calves with the same parentage born in previous and subsequent years were not affected, and calves in the same environment but with different parentage were unaffected.2139 This suggests that a conjunction of environmental and genetic factors may determine disease pathogenesis. Consequently, some authors do not recommend culling a bull merely because an offspring has developed the condition,2147 whereas others disagree with this recommendation.2148
Treatment
Surgical techniques for correction of the spasticity include sectioning of the spinal afferents at segments L4 through L6, neurectomy of the tibial nerve rootlets supplying the medial and lateral heads of the gastrocnemius muscle, and superficial digital flexor tenotomy proximal to the tuber calcis.2129,2141,2149-2151 The tenotomy procedure is performed by incising proximal to the tuber calcis and transecting the superficial head of the gastrocnemius tendon completely (this tendon twists around the superficial digital flexor tendon from medial to lateral, coursing distally) and partly nicking the superficial digital flexor tendon.2150,2152
To perform a tibial neurectomy, an incision is made in the groove separating the heads of the biceps femoris. The tibial nerve is identified as the more caudal of the branches off the ischiatic nerve. Some surgeons have recommended a concomitant sectioning of the caudal cutaneous sural nerve.
Success rates of 82% for the neurectomy technique and 40% for the tenotomy procedure have been reported.2149,2153 Although recurrences may be common, some authors found high rates of sustained improvement over several months with the neurectomy technique.2149 Results are poorer when animals under 2 months of age are treated.
Despite the reported success of these procedures, these treatments are not routinely performed in most countries because of the possible heritability of the condition and the need for specialized instruments and general anesthesia. Administration of lithium gluconate (4 mg/kg IM or PO daily for 10 to 30 days) is reportedly efficacious when used to treat calves in the early stages of spastic paresis.2135
INHERITED PERIODIC SPASTICITY (CRAMPY SYNDROME; STRETCHES; BARN CRAMPS; KRAMPFIGKEIT)
Inherited periodic spasticity is seen frequently in Holstein, Ayrshire, Jersey, Brown Swiss, and Guernsey cattle of either gender.2154 Beef cattle are rarely affected. The condition is thought to be transmitted by a single autosomal recessive factor.2155 Affected cattle are normal until they reach 3 to 7 years of age, when they develop marked muscular spasms of the hip and upper limb.2154 An earlier onset was reported in a Hereford bull.2156 The disease is mild for the first 2 to 3 years but progressively worsens over time. Eventually, affected animals are culled early because of weight loss or chronic foot problems. Specific pathologic changes are absent at necropsy.2157
Clinical Signs
During the attack the leg may be held spastically in flexion, but more often it is held in rigid extension. The attacks are episodic, which differentiates them from the spasms of spastic paresis. The two diseases are otherwise similar in appearance. Each spasm is accompanied by kyphosis, which initially lasts 15 to 30 seconds and often is terminated by a fine tremor in the hindquarters or the digit. The intensity and duration of the spasms progressively increase over time. Both hindlimbs are affected. During a spasm the animal usually extends or flexes only one leg at a time. Some animals with advanced disease arch the neck and back dorsally and lift the contralateral forelimb during an attack.2154 At first the signs appear to typify a response to a painful focus, and affected animals may be misdiagnosed as having laminitis, colic, or peritonitis. Gait and proprioceptive responses appear normal.
Treatment
There is no specific treatment for periodic spasticity, although mephenesin has been recommended, at a dosage of 30 to 40 mg/kg daily for 3 days. Although the pharmacologic effect of mephenesin lasts only 6 to 8 hours, severe symptoms were reportedly reduced for as long as several months after a single course of therapy.2158 Methocarbamol also has been recommended, but reports of efficacy are anecdotal only.
DODDLER SYNDROME (HEREDITARY LETHAL SPASMS)
Doddler syndrome is a rare congenital lethal trait of Jersey cattle.2159 The calves are down but appear bright and alert and usually are able to suckle. When stimulated, they develop severe intermittent spasms of the head and neck and convulsions. The animals can stand with assistance but are very ataxic and fall easily. They have a severe head tremor when forced to stand. The signs improve when the animal is allowed to rest but worsen again with restimulation.2160 Calcification of multiple neurons and small vessels in the brainstem and cerebellum is observed at necropsy. The condition probably is inherited as a lethal autosomal recessive trait.
CONGENITAL VERTEBRAL ANOMALIES (SPINA BIFIDA; BUTTERFLY VERTEBRAE; HEMIVERTEBRAE; ARNOLD-CHIARI SYNDROME)
A large number of spinal deformities have been reported in domestic livestock, including the following:
Spina bifida. Failure of closure of a vertebral neural arch; spina bifida usually occurs concomitantly with one of the forms of myelodysplasia.
2161,2162
Spina bifida cystica. Spina bifida associated with a cerebrospinal fluid cyst at the site of the bony defect.
Hemivertebra. A unilaterally incomplete vertebral segment; spina bifida and hemivertebrae have been described in calves, and spina bifida also has been reported in sheep and foals.
2161-2170
Arnold-Chiari syndrome. A complex disorder that occurs in lambs and calves
2171 and is characterized by a number of pathologic changes, including herniation of cerebellar tissue through the foramen magnum, caudal overgrowth and displacement of the brainstem, internal hydrocephalus, polymicrogyria of the cerebral cortex, malformation of the base of the skull, and enlargement of the foramen magnum. The strong correlation between the occurrence of spina bifida and Arnold-Chiari syndrome of calves indicates that the pathogenesis of the two conditions may be interrelated.
2161,2171
Animals with spina bifida may be asymptomatic or may show paraparesis and paraplegia or tetraparesis and tetraplegia. If the spina bifida is associated with syringomyelia, the calves may have a peculiar “rabbit hopping” gait. The clinical signs often are present at birth or develop in the first 2 postnatal months.2168,2172 Kyphoscoliosis and abnormalities of the rib cage may be seen at birth in some animals.2168 The skin over the abnormal vertebrae may be smooth and hairless. When examined microscopically, this tissue resembles meninges or ependyma.2162,2163
Spina bifida and hemivertebrae are easily diagnosed by examination of plain radiographs. The specific site of central nervous system stenosis can be detected by performing a myelogram. The etiology of spina bifida is unknown, but some suggest that it may have either genetic2163-2165 or toxic etiologies.2173,2174
Arthrogryposis occurs secondary to the spinal cord changes. Spina bifida and myelodysplasias are not the only causes of arthrogryposis, however, and clinicians should attempt to differentiate between these and other diseases that interfere in utero with motor neuron development in the spinal cord. Nongenetic arthrogrypotic conditions of cattle include perosomus elumbis, hydranencephaly, manganese deficiency, and ingestion of lupines or Nicotiana glauca (40 to 60 days of gestation).2175-2179
See also Chapters 51 and 52.
MYELODYSPLASIAS (SYRINGOMYELIA; SPINAL DYSRAPHISM; HYDROMYELIA)
The following types of myelodysplasia occur in livestock2180-2182:
1
Spinal dysraphism. A general term denoting arrested development of the spinal cord before complete differentiation of gray and white matter; the areas of agenesis form longitudinal cystic structures instead of differentiated nervous tissue.
2
Syringomyelia. Longitudinal canalicular cavitations of the spinal cord.
3
Hydromyelia. Abnormal dilation of the central canal.
4
Diastematomyelia. Duplication of the gray matter at one or more segments.
5
Rachischisis. Complete failure to close the neural tube; the central canal remains open and communicates with the integument.
6
Meningocele. Herniation of the dura mater through a spinal column defect.
7
Meningomyelocele. Herniation of the meninges through a spinal column defect and a concomitant myelodysplasia.
Myelodysplasias are most often seen in Charolais calves, in which the condition is associated with palatoschisis and arthrogryposis.2180,2183 The condition has also been recognized in 5-month-old thoroughbred foals and an Arabian foal.2184,2185
The neurologic deficits of myelodysplasia are difficult to differentiate from other spinal diseases unless the disorder is accompanied by spina bifida. Clinical recognition of myelodysplasia usually is based on the historical findings of paraplegia in a newborn calf without radiographic evidence of spinal fractures and with no myelographic evidence of spinal cord compression.
COYOTILLO POISONING (TULLIDORA TOXICITY, BUCKTHORN FRUIT POISONING)
Ingestion of the fruit of the coyotillo plant, Karwinskia humboldtiana, by domestic animals produces a stiff stilted gait, hypotonia, and hyperreflexia to areflexia.2186,2187 The condition occurs in the southwestern United States. The neurotoxin identified in and purified from the plant is called “tullidora” toxin, after another name for the plant itself.2188 Goats are susceptible to the effects of the intoxication. Daily doses of the fresh plant amounting to 0.04% to 0.05% of the body weight are sufficient to produce neurologic signs by 60 days. The intoxication results in a peripheral polyneuropathy characterized by degenerative changes in both axons and myelin.2188,2189 Higher dosages may result in neuroaxonal dystrophy characterized by axonal swelling and gliosis. Experimental studies in small animals have revealed that the plant toxin exerts effects both on the Schwann cell and peripheral motor neurons, but not on sensory neurons.2190 It induces a reversible inflammatory polyneuropathy with segmental demyelination and also decreases fast axonal transport.2191,2192
CYCAD PALM POISONING (ZAMIAPARALYSIS)
Ingestion of the palm cycad Cycas circinalis L., Bowenia serrulata, Macrozamia lucida, and Cycas media is associated with the development of posterior paresis in cattle and sheep.2193-2195 The condition is seen exclusively in the tropics. The toxic principles of the cycad palm are glycosides and methylazoxymethanol (aglycone). The clinical signs include curvature of the spine, elevation of the tailhead, paraparesis, and paraplegia. The anal sphincter and tail tone are normal. Cattle develop ataxia by 50 days after feeding of the plant (3.9 kg wet weight total intake). The cerebrospinal fluid of affected cattle is normal. The pathologic lesions include demyelination, spheroids, and cavitation of the spinal cord white matter. Changes relating to hepatic disease have also been reported.2196 These lesions included coagulative centrilobular hepatic necrosis, icterus, and petechial hemorrhages on the serous surfaces. Poisoning by Macrozamia reidlei can cause death from hepatic failure without signs of neurologic disease.2197
ACQUIRED TORTICOLLIS
Primary acquired torticollis with or without neurologic disease occurs in all species of domestic livestock. Causes include fracture or subluxation of the cervical vertebrae, basilar skull fractures, dystrophic muscle degeneration, unilateral cicatricial muscular contracture from injections, lupinosis, traumatic rupture of the cervical muscles, hydranencephaly, asymmetric neurodegeneration, and congenital vertebral deformity.2198-2202 Calves with torticollis have a deviated head and neck. One study hypothesized that physical constraint of late in utero development by narrow tips of uterine horns caused acquired torticollis and a variety of other deformities, such as head scoliosis and limb malformations, in more than 200 foals.2203 Draft horses may be predisposed to this problem, which frequently results in severe dystocia in horses. Provided the spinal cord is intact, no neurologic deficits result.
Treatment of traumatic torticollis should be directed at reducing the edema, relieving pain, and immobilizing the damaged structures. Muscular tears may be treated by incorporating the head, neck, and proximal thorax in a fiberglass cast. Ancillary supportive treatment may include dexamethasone (0.04 to 0.08 mg/kg daily for 2 to 3 days), methocarbamol (8 mg/kg IV daily for 5 days), and NSAIDs (e.g., phenylbutazone PO or IV or flunixin meglumine IV for 3 to 5 days after injury). A method of surgical correction of cervical muscle contractures using a muscle-splitting procedure has been described2204; however, this seems unnecessary because most animals recover with only medical treatment. When torticollis is the cause of equine dystocia, delivery of the foal by cesarean section often is followed by rapid and complete anatomic and functional recovery.2203
TETANUS (LOCKJAW)
Definition and Etiology
Tetanus is characterized by muscular rigidity and death from respiratory arrest or convulsions. The disease is caused by the exotoxins produced by the anaerobic, spore-forming, gram-positive bacterium Clostridium tetani. Tetanus has a worldwide distribution, and all species of domestic livestock are susceptible. The bacterium is typically isolated from the bowel contents of herbivores, but fecal contamination is considered to be only partly responsible for soil contamination.2205 The agent also can be found in dirt with no documented contact with domestic livestock. This indicates that C. tetani should be considered a primary soil contaminant. Tetanus usually is a disease of individual animals; however, herd outbreaks of tetanus after tail docking or castration have been described.2206,2207 During outbreaks of tetanus, C. tetani can be isolated from the feces of a large proportion of the cattle, indicating that in some cases the disease may be caused by proliferation of C. tetani in the patient’s gastrointestinal tract.2206,2207
Clinical Signs
The incubation period of tetanus varies and depends on the size of the wound, the redox potential in the contaminated tissue, the number of bacteria inoculated, and the host’s antitoxin titer. In most susceptible animals the signs occur from 2 weeks to 1 month after the bacterial inoculation. During the first 24 hours, horses may develop intractable colic, and ruminants may bloat.2208 The first signs in some animals may be a vague stiffness and lameness of the infected limb, which are related to a local effect of the absorbed toxin (localized tetanus). By 24 hours, generalized spasticity usually is evident. Affected animals display a stiff gait and an extended head posture.2209 The hypertonia is most evident in the antigravity muscles. Thus the limbs are held in a characteristic posture that resembles the legs of a sawhorse (sawhorse stance). The lips are retracted toward the poll, and the ears are pulled slightly down and caudal. The tail is elevated from the ischiorectal fossa. There is excessive muscle tone of all facial musculature. The jaws are clamped tightly shut (trismus), and the legs are held rigidly extended.
Muscular spasms can be elicited by auditory, ocular, or tactile stimulation. The limbs and head are very resistant to passive flexion. Retraction of the eye and a rapid flashing of the third eyelid across the cornea occurs after a menacing gesture or a slap over the neck.2208,2210 This sign is more consistently observed in horses than in ruminants. Aspiration pneumonia may develop as a result of impaired deglutition. Severely affected animals become recumbent and lie on their side with the head and legs in full extension and the ears held almost parallel to the thoracic spinal cord (Fig. 35-40). Progression of the disease is associated with increased tonic muscular activity, which results in pyrexia in all species and profuse sweating in horses.2207,2211 Frothy saliva accumulates at the commissures of the lips because the animals are unable to swallow, and respiratory incursions whip the mucinous saliva into a foam. The respiratory muscles (diaphragm and intercostals) are affected, and the animals develop hypoxia. Ventrolateral strabismus and dilated, fixed pupils may occur in advanced tetanus of cattle. Animals die while in a terminal convulsion. Death is attributable to hypoxemia and heart failure secondary to systemic hypertension and aspiration pneumonia. Survivors begin to show some improvement after 2 weeks, but the clinical signs may persist for as long as 1 month, and lameness may be permanent.
Clinical Pathology
There are no reliable clinicopathologic tests for confirmation of a diagnosis of tetanus. Attempts should be made to culture C. tetani from the suspected site of entry.
Pathophysiology
In horses, puncture wounds of the foot or the soft tissues are the most frequent sites of infection, whereas dairy cattle are infected most frequently through the uterus. Other sites for growth of C. tetani include lesions induced by elastrator bands, tail docking, dehorning, bull rings, or infected umbilical stalks.2212 Proliferation of C. tetani in the forestomachs of normal cattle may produce sufficient concentrations of toxin to result in clinical signs.2213,2214 Outbreaks of tetanus have been correlated with ingestion of millet. This diet has been postulated to promote the growth of C. tetani in the large bowel2215 and probably accounts for the lack of visible wounds in some animals affected with tetanus.2206,2210
When inoculated into the anaerobic site, C. tetani spores germinate into the vegetative form. Factors that enhance the sporulation and growth of C. tetani include necrotic tissue, pus, concomitant bacterial infection, and foreign bodies. Spores inoculated into the tissues are highly resistant to normal host defenses and may remain dormant for months or years before developing into the vegetative state. The production of the tetanus toxins occurs at the end of the logarithmic growth phase of the vegetative form and is governed by a plasmid-associated gene.2216,2217 The bacterium produces at least three toxic proteins: tetanospasmin, tetanolysin, and a nonspasmogenic toxin. Tetanolysin promotes the spread of the infection by increasing the amount of local tissue necrosis.2211
Tetanospasmin is a lipoprotein exotoxin that diffuses from the site of production into the vascular system, where it is distributed hematogenously to the presynaptic part of the motor endplates. Once bound to the nerves, the toxin is internalized and transported to the central nervous system along the axons of the alpha motoneurons through the membrane-bound smooth endoplasmic reticulum.2209,2218,2219 After reaching the ventral horn of the spinal cord, the toxin crosses the synaptic cleft to presynaptic inhibitory interneurons (Renshaw cells) in the intermediate gray column.2220 Tetanospasmin probably inhibits the release of glycine and γ-aminobutyric acid (GABA) from the Renshaw cells, resulting in disinhibition of the gamma motoneurons. The inhibition of these cells results in hypertonia and muscular spasms.
The nonspasmogenic toxin is thought to produce overstimulation of the sympathetic nervous system. Systemic hypertension seen in tetanus can be related to excessive catecholamine production by the adrenal medulla. Other physiologic changes identified in humans and laboratory animals include increased plasma cortisol concentrations and neuromuscular blockade. Whether these changes are caused by the effects of the toxin or these are secondary changes occurring in response to a painful and life-threatening problem is unclear. There are no characteristic postmortem lesions associated with tetanus.
Treatment
The six general medical principles for treating tetanus in large animals are as follows:
1
Provide muscular relaxation.
3
Eliminate the infection.
4
Neutralize the unbound toxin.
5
Maintain hydration and nutritional status.
6
Establish active antitoxic immunity.
Provide Muscular Relaxation
The patient should be sedated and placed in a quiet, darkened stall. Drugs are administered that may reduce the muscular spasms. These include promazine (0.5 to 1 mg/kg IV) or acetylpromazine (0.05 to 0.1 mg/kg IV) given at 4- to 6-hour intervals. Predictable muscular relaxation may be obtained inexpensively by concomitant IV administration of acetylpromazine (0.06 mg/kg) and 5% sodium pentobarbital (2 to 4 mL/50 kg). An IV catheter may be placed to minimize treatment-associated stimulation. Mephenesin (10 to 20 mg/kg IV three times daily) and guaifenesin have also been recommended. These drugs interfere with the internuncial neurons of the spinal cord that participate in reflex muscle activities. They do not have a high therapeutic efficacy for tetanus. Diazepam (0.01 to 0.4 mg/kg IV two to eight times daily) effectively reduces muscular spasms in large animals by enhancing GABA, but prolonged administration to a large ruminant or horse is expensive because of the short duration in the plasma and CNS. In addition to its enhancement of GABA, diazepam is efficacious because of its glycine-mimetic effects.2208 Packing the ears with cotton to minimize auditory stimulation also can help reduce muscle spasms.
Ensure Good Footing
Excellent footing is essential. Tetanic animals have difficulty rising because of increased spasms and muscular tone. The stall should be bedded deeply in shavings or straw to minimize decubital ulcers. Horses and small ruminants that cannot stand should be supported in a sling, provided they do not become frantic while suspended.
Eliminate the Infection
Because C. tetani grows in nonvascularized sites, the infection is best eliminated by surgical debridement of the affected area. Concomitant infiltration of penicillin G around the wound and parenteral administration of potassium penicillin G (22,000 IU/kg three or four times daily) or procaine penicillin G (22,000 IU/kg IM twice daily) also may be beneficial.
Neutralize the Unbound Toxin
Before the tetanus antitoxin has bound to the nerve cells, it is susceptible to neutralization with antitoxin. Although administration of antitoxin to animals several days after the onset of clinical signs seems to have little benefit in horses, increased survival rates have been documented in human patients who have received high dosages of tetanus antitoxin in the early phase of the disease. Infiltration of the area with 3000 to 9000 IU of tetanus antitoxin may effectively neutralize toxin that has not yet reached the peripheral vasculature. Although specific dosages have not been determined for domestic animals, suggested doses range from 1000 to 5000 IU/500-kg animal to 1000 to 5000 IU/kg.2221-2223 The limited therapeutic benefits of intravenous antitoxin must be compared to the cost of the biologic, the potential side effects of hepatic necrosis (see Chapter 33) or anaphylaxis, and the economic value of the animal.
Some claim that administration of 50 mL antitoxin (1000 IU/mL) intracysternally to horses resulted in stabilization of the clinical signs. However, this treatment did not reverse the condition.2224 The antitoxin is administered after slow removal of 30 mL of cerebrospinal fluid (CSF). Although a survival rate of 77% has been claimed,2224 the study failed to consider several cysternally injected horses that died of intercurrent diseases. When all cases were considered, there appeared to be no statistically significant difference between cysternally injected horses and conventionally treated controls. Complications of this procedure included iatrogenic CSF infections, anesthesia-related deaths, and sepsis from indwelling catheters. Because of severe reactions to intrathecal equine serum in ruminants, intrathecal administration of equine-origin tetanus antitoxin is contraindicated.
Maintain Hydration and Nutritional Status
The patient’s hydration and food intake should be monitored daily. The food should be placed off the ground in an elevated feed bunk or hay net to allow easier access. Intravenous fluids should be administered as needed to correct dehydration and electrolyte abnormalities. Alimentation with a nasogastric tube may be attempted in anorectic horses, but severe adverse reactions to this procedure in some tetanus patients may limit its usefulness. A rumenostomy in anorectic cattle relieves the chronic ruminal tympany and provides a convenient means of administering oral fluids and feed.
Establish Active Antitoxic Immunity
The concentration of tetanus toxin necessary to cause neurologic symptoms is less than that required to stimulate an active immunologic response. Affected animals should be immunized with tetanus toxoid at the time of treatment and given a second dose 1 to 2 months later. Concomitant injections of tetanus antitoxin and toxoid should be made at different sites of the body and should not be admixed before administration.
Prognosis
The mortality rate may reach 50% in cattle and 80% in horses. The rate of progression of clinical signs is indirectly related to the prognosis. Animals that survive for longer than 7 days have a fair to good chance of complete recovery.
Prevention
Because cattle appear to be more resistant to tetanus than horses and small ruminants, they are not routinely immunized against the disease unless outbreaks have occurred previously. Colostral antibodies may interfere with the active immunization of neonates. One report indicates that most foals from immunized dams (82.9%) lose passively acquired specific antitoxic antibodies by 4 months of age. However, other studies suggest that passive immunity may last as long as 6 to 12 months of age. Because some animals have low titers and others have high, persistent titers, a general recommendation might include vaccination of livestock at 2, 3, and 6 months of age, followed by a booster after 1 year. To ensure protective levels of colostral antibodies, mares, does, or ewes should receive an annual booster dose of the toxoid 1 to 2 months before the anticipated date of parturition. One study indicated that tetanus prophylaxis was not cost-effective for sheep, but vaccination recommendations would differ, depending on the economic value of the animal at risk.2225
Acute hepatic necrosis of horses (Theiler’s disease) has been associated with administration of certain lots of commercially prepared tetanus antitoxin (see Chapter 33) 1 to 3 months previously. Such findings indicate that administration of tetanus antitoxin should be limited to unvaccinated horses with tetanus-prone wounds. The recommended doses of tetanus antitoxin are 1500 IU subcutaneously (SC) or intramuscularly (IM) for adult horses or cattle2226 and 500 IU SC or IM for sheep and goats. Tetanus toxoid should be administered concomitantly. The antitoxin and the toxoid should not be mixed in the same syringe and should be administered at different sites of the body; a second toxoid dose should be given in 1 month.2226,2227 Previously immunized horses with tetanus-prone lesions should be given a booster dose of tetanus toxoid and should not be given antitoxin. Foals from unvaccinated dams should receive 1500 IU of tetanus antitoxin at birth.2226
Tetanus toxoid is effective, but not all horses develop protective immunity. Historical information citing a previous vaccination should not completely exclude the possibility of tetanus.2228