 CHAPTER 37 Acid-Base Disorders
CHAPTER 37 Acid-Base Disorders
Close regulation of pH is necessary for cellular enzymes and other metabolic processes, which function optimally at a normal pH (7.35 to 7.45). Chronic, mild derangements in acid-base status may interfere with normal growth and development, whereas acute, severe changes in pH can be fatal. Control of acid-base balance depends on the kidneys, the lungs, and intracellular and extracellular buffers.
The lungs and the kidneys maintain a normal acid-base balance. Carbon dioxide (CO2) generated during normal metabolism is a weak acid. The lungs prevent an increase in the partial pressure of CO2 (PCO2) in the blood by excreting the CO2 that the body produces. CO2 production varies depending on the body’s metabolic needs. The rapid pulmonary response to changes in CO2 concentration occurs via central sensing of the PCO2 and a subsequent increase or decrease in ventilation to maintain a normal PCO2 (35 to 45 mm Hg).
The kidneys excrete endogenous acids. An adult normally produces about 1 to 2 mEq/kg/day of hydrogen ions. Children normally produce 2 to 3 mEq/kg/day of hydrogen ions. The hydrogen ions from endogenous acid production are neutralized by bicarbonate, potentially causing the bicarbonate concentration to fall. The kidneys regenerate this bicarbonate by secreting hydrogen ions, maintaining the serum bicarbonate concentration in the normal range (20 to 28 mEq/L).
CLINICAL ASSESSMENT OF ACID-BASE DISORDERS
Acidemia is a pH below normal (<7.35), and alkalemia is a pH above normal (>7.45). An acidosis is a pathologic process that causes an increase in the hydrogen ion concentration, and an alkalosis is a pathologic process that causes a decrease in the hydrogen ion concentration. A simple acid-base disorder is a single primary disturbance. During a simple metabolic disorder, there is respiratory compensation; the PCO2 decreases during a metabolic acidosis and increases during a metabolic alkalosis. With metabolic acidosis, the decrease in the pH increases the ventilatory drive, causing a decrease in the PCO2. The fall in the CO2 concentration leads to an increase in the pH. This appropriate respiratory compensation for a metabolic process happens quickly and is complete within 12 to 24 hours.
During a primary respiratory process, there is a slower metabolic compensation, mediated by the kidneys. The kidneys respond to a respiratory acidosis by increasing hydrogen ion excretion, increasing bicarbonate generation, and raising the serum bicarbonate concentration. The kidneys increase bicarbonate excretion to compensate for a respiratory alkalosis; the serum bicarbonate concentration decreases. In contrast to a rapid respiratory compensation, it takes 3 to 4 days for the kidneys to complete appropriate metabolic compensation. However, there is a small and rapid compensatory change in the bicarbonate concentration during a primary respiratory process. The expected appropriate metabolic compensation for a respiratory disorder depends on whether the process is acute or chronic.
A mixed acid-base disorder is present when there is more than one primary acid-base disturbance. An infant with bronchopulmonary dysplasia may have a respiratory acidosis from chronic lung disease and a metabolic alkalosis from a diuretic used to treat the chronic lung disease. Formulas are available for calculating the appropriate metabolic or respiratory compensation for the six primary simple acid-base disorders (Table 37-1). Appropriate compensation is expected in a simple disorder; it is not optional. If a patient does not have appropriate compensation, a mixed acid-base disorder is present.
TABLE 37-1 Appropriate Compensation During Simple Acid-Base Disorders
| Disorder | Expected Compensation* |
|---|---|
| Metabolic acidosis | PCO2 = 1.5 × [HCO3−] 8 ± 2 |
| Metabolic alkalosis | PCO2 increases by 7 mm Hg for each 10-mEq/L increase in the serum [HCO3−] |
| Respiratory acidosis | |
| Acute | [HCO3−] increases by 1 for each 10-mm Hg increase in the PCO2 |
| Chronic | [HCO3−] increases by 3.5 for each 10-mm Hg increase in the PCO2 |
| Respiratory alkalosis | |
| Acute | [HCO3−] falls by 2 for each 10-mm Hg decrease in the Pco2 |
| Chronic | [HCO3−] falls by 4 for each 10-mm Hg decrease in the Pco2 |
METABOLIC ACIDOSIS
Metabolic acidosis occurs frequently in hospitalized children; diarrhea is the most common cause. For a patient with an unknown medical problem, the presence of a metabolic acidosis is often helpful diagnostically because it suggests a relatively narrow differential diagnosis (Table 37-2).
TABLE 37-2 Causes of Metabolic Acidosis
NORMAL ANION GAP
Diarrhea
Renal tubular acidosis
Urinary tract diversions
Posthypocapnia
Ammonium chloride intake
INCREASED ANION GAP
Lactic acidosis (shock)
Ketoacidosis (diabetic, starvation, or alcoholic)
Kidney failure
Poisoning (e.g., ethylene glycol, methanol, or salicylates)
Inborn errors of metabolism
Etiology
Diarrhea causes a loss of bicarbonate from the body. The amount of bicarbonate lost in the stool depends on the volume of diarrhea and the bicarbonate concentration of the stool, which tends to increase with more severe diarrhea. Diarrhea often causes volume depletion because of losses of sodium and water, potentially exacerbating the acidosis by causing hypoperfusion (shock) and a lactic acidosis.
There are three forms of renal tubular acidosis (RTA):
In distal RTA, children may have accompanying hypokalemia, hypercalciuria, nephrolithiasis, and nephrocalcinosis; rickets is a less common finding. Failure to thrive, resulting from chronic metabolic acidosis, is the most common presenting complaint. Autosomal dominant and autosomal recessive forms of distal RTA exist. The autosomal dominant form is relatively mild. Autosomal recessive distal RTA is more severe and often associated with deafness secondary to a defect in the gene for a H+-ATPase that is present in the kidney and the inner ear. Distal RTA also may be secondary to medications or congenital or acquired renal disease. Patients with distal RTA cannot acidify their urine and have a urine pH greater than 5.5, despite a metabolic acidosis.
Proximal RTA is rarely present in isolation. In most patients, proximal RTA is part of Fanconi syndrome, a generalized dysfunction of the proximal tubule. Along with renal wasting of bicarbonate, Fanconi syndrome causes glycosuria, aminoaciduria, and excessive urinary losses of phosphate and uric acid. The chronic hypophosphatemia is more clinically significant because it ultimately leads to rickets in children. Rickets or failure to thrive may be the presenting complaint. Fanconi syndrome is rarely an isolated genetic disorder, with pediatric cases usually secondary to an underlying genetic disorder, most commonly cystinosis. Toxic medications, such as ifosfamide or valproate, may cause Fanconi syndrome. The ability to acidify the urine is intact in proximal RTA, and untreated patients have a urine pH less than 5.5. However, bicarbonate therapy increases bicarbonate losses in the urine, and the urine pH increases.
In hyperkalemic RTA, renal excretion of acid and potassium is impaired due to either an absence of aldosterone or an inability of the kidney to respond to aldosterone. In severe aldosterone deficiency, as occurs with congenital adrenal hyperplasia secondary to 21α-hydroxylase deficiency, the hyperkalemia and metabolic acidosis are accompanied by hyponatremia and volume depletion from renal salt wasting. Incomplete aldosterone deficiency causes less severe electrolyte disturbances; children may have isolated hyperkalemic RTA, hyperkalemia without acidosis, or isolated hyponatremia.
Lactic acidosis most commonly occurs when inadequate oxygen delivery to the tissues leads to anaerobic metabolism and excess production of lactic acid. Lactic acidosis may be secondary to shock, severe anemia, or hypoxemia. Inborn errors of carbohydrate metabolism produce a severe lactic acidosis (see Chapter 52). In diabetes mellitus, inadequate insulin leads to hyperglycemia and diabetic ketoacidosis (see Chapter 171). Renal failure (see Chapter 165) causes a metabolic acidosis because the kidneys are unable to excrete the acid produced by normal metabolism.
A variety of toxic ingestions cause a metabolic acidosis. Acute salicylate intoxication occurs after a large overdose. Chronic salicylate intoxication is possible because of the gradual buildup of the drug. In addition to a metabolic acidosis, some patients may have a respiratory alkalosis. Other symptoms of salicylate intoxication include fever, seizures, lethargy, and coma. Hyperventilation may be particularly marked. Tinnitus, vertigo, and hearing impairment are more likely with chronic salicylate intoxication. Ethylene glycol, a component of antifreeze, is converted in the liver to glyoxylic and oxalic acids, causing a severe metabolic acidosis. Excessive oxalate excretion causes calcium oxalate crystals to appear in the urine, and calcium oxalate precipitation in the kidney tubules can cause renal failure. The toxicity of methanol ingestion also depends on liver metabolism; formic acid is the toxic end product that causes the metabolic acidosis and other sequelae, which include damage to the optic nerve and central nervous system.
There are many inborn errors of metabolism that may cause a metabolic acidosis (see Section 10). The metabolic acidosis may be due to excessive production of ketoacids, lactic acid, or other organic anions. Some patients have accompanying hyperammonemia. In most patients, acidosis occurs only episodically during acute decompensations, which may be precipitated by ingestion of specific dietary substrates (proteins), the stress of a mild illness (fasting, catabolism), or poor compliance with dietary or medical therapy.
Clinical Manifestations
The underlying disorder usually produces most of the signs and symptoms in children with a mild or moderate metabolic acidosis. The clinical manifestations of the acidosis are related to the degree of acidemia; patients with appropriate respiratory compensation and less severe acidemia have fewer manifestations than patients with a concomitant respiratory acidosis. At a serum pH less than 7.20, there is impaired cardiac contractility and an increased risk of arrhythmias, especially if underlying heart disease or other predisposing electrolyte disorders are present. With acidemia, there is a decrease in the cardiovascular response to catecholamines, potentially exacerbating hypotension in children with volume depletion or shock. Acidemia causes vasoconstriction of the pulmonary vasculature, which is especially problematic in newborns with persistent fetal circulation (see Chapter 61). The normal respiratory response to metabolic acidosis—compensatory hyperventilation—may be subtle with mild metabolic acidosis, but it causes discernible increased respiratory effort with worsening acidemia. Chronic metabolic acidosis causes failure to thrive.
Diagnosis
The plasma anion gap is useful for evaluating patients with a metabolic acidosis. It divides patients into two diagnostic groups: normal anion gap and increased anion gap. The following formula determines the anion gap:
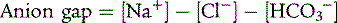
A normal anion gap is 3 to 11. A decrease in the albumin concentration of 1 g/dL decreases the anion gap by roughly 4 mEq/L. Similarly, albeit less commonly, an increase in unmeasured cations, such as calcium, potassium, or magnesium, decreases the anion gap. Conversely, a decrease in unmeasured cations is a rare cause of an increased anion gap. Because of these variables, the broad range of a normal anion gap, and other factors, the presence of a normal or increased anion gap is not always reliable in differentiating the causes of a metabolic acidosis, especially when the metabolic acidosis is mild. Some patients have more than one explanation for their metabolic acidosis, such as a child with diarrhea and lactic acidosis secondary to hypoperfusion. The anion gap should not be interpreted in dogmatic isolation; consideration of other laboratory abnormalities and the clinical history improves its diagnostic utility.
Treatment
The most effective therapeutic approach for patients with a metabolic acidosis is correction of the underlying disorder, if possible. The administration of insulin in diabetic ketoacidosis or restoration of adequate perfusion in lactic acidosis eventually results in normalization of acid-base balance. The use of bicarbonate therapy is indicated when the underlying disorder is irreparable; examples include RTA and chronic renal failure. In other disorders, the cause of the metabolic acidosis eventually resolves, but base therapy may be necessary during the acute illness. In salicylate poisoning, alkali administration increases renal clearance of salicylate and decreases the amount of salicylate in brain cells. Short-term base therapy is often necessary in other poisonings and inborn errors of metabolism.
METABOLIC ALKALOSIS
Etiology
The causes of a metabolic alkalosis are divided into two categories based on the urinary chloride (Table 37-3). The alkalosis in patients with a low urinary chloride is maintained by volume depletion. They are called chloride responsive because volume repletion with fluid containing sodium chloride and potassium chloride is necessary to correct the metabolic alkalosis. Emesis, which causes loss of hydrochloride and volume depletion, is the most common cause of a metabolic alkalosis. Diuretic use increases chloride excretion in the urine. Consequently, while a patient is receiving diuretics, the urinary chloride is typically high (>20 mEq/L). After the diuretic effect has worn off, the urinary chloride is low (<15 mEq/L), because of appropriate renal chloride retention in response to volume depletion. Categorization of diuretics based on urinary chloride depends on the timing of the measurement. The metabolic alkalosis from diuretics is clearly chloride responsive; it corrects only after adequate volume repletion. This is the rationale for including it among the chloride-responsive causes of a metabolic alkalosis.
TABLE 37-3 Causes of Metabolic Alkalosis
CHLORIDE RESPONSIVE (URINARY CHLORIDE <15 MEQ/L)
Gastric losses (emesis or nasogastric suction)
Pyloric stenosis
Diuretics (loop or thiazide)
Chloride-losing diarrhea
Chloride-deficient formula
Cystic fibrosis (sweat losses of chloride)
Posthypercapnia (chloride loss during respiratory acidosis)
CHLORIDE RESISTANT (URINARY CHLORIDE <20 MEQ/L)
High blood pressure
Adrenal adenoma or hyperplasia
Glucocorticoid-remediable aldosteronism
Renovascular disease
Renin-secreting tumor
17α-Hydroxylase deficiency
11β-Hydroxylase deficiency
Cushing syndrome
11β-Hydroxysteroid dehydrogenase deficiency
Licorice ingestion
Liddle syndrome
Normal blood pressure
Gitelman syndrome
Bartter syndrome
Base administration
The chloride-resistant causes of metabolic alkalosis can be subdivided based on blood pressure. Patients with the rare disorders that cause a metabolic alkalosis and hypertension either have increased aldosterone or act like they have increased aldosterone (owing to a genetic defect causing overproduction of another mineralocorticoid or a constitutively overactive sodium channel, as occurs in Liddle syndrome). Patients with Bartter syndrome or Gitelman syndrome (Chapter 36) have metabolic alkalosis, hypokalemia, and normal blood pressure secondary to renal tubular defects that cause continuous urinary losses of chloride.
Clinical Manifestations
The symptoms in patients with a metabolic alkalosis often are related to the underlying disease and associated electrolyte disturbances. Hypokalemia is often present, and occasionally severe, in all the diseases that cause a metabolic alkalosis (see Chapter 36). Children with chloride-responsive causes of metabolic alkalosis often have symptoms related to volume depletion (see Chapter 33). In contrast, children with chloride-unresponsive causes may have symptoms related to hypertension. Alkalemia may cause arrhythmias, hypoxia secondary to hypoventilation, or decreased cardiac output.
Diagnosis
Measurement of the urinary chloride concentration is the most helpful test in differentiating among the causes of a metabolic alkalosis. The history usually suggests a diagnosis, although no obvious explanation may be present in the patient with bulimia, surreptitious diuretic use, or an undiagnosed genetic disorder, such as Bartter syndrome or Gitelman syndrome.
Treatment
The approach to therapy of metabolic alkalosis depends on the severity of the alkalosis and the underlying etiology. In children with a mild metabolic alkalosis ([HCO3−] <32 mEq/L), intervention is often unnecessary. Patients with chloride-responsive metabolic alkalosis respond to correction of hypokalemia and volume repletion with sodium and potassium chloride, but aggressive volume repletion may be contraindicated if mild volume depletion is medically necessary in the child receiving diuretic therapy. In children with chloride-resistant causes of a metabolic alkalosis that are associated with hypertension, volume repletion is contraindicated because it exacerbates the hypertension and does not repair the metabolic alkalosis. Treatment focuses on eliminating or blocking the action of the excess mineralocorticoid. In children with Bartter syndrome or Gitelman syndrome, therapy includes oral potassium supplementation and potassium-sparing diuretics.
RESPIRATORY ACID-BASE DISTURBANCES
During a respiratory acidosis, there is a decrease in the effectiveness of CO2 removal by the lungs. The causes of a respiratory acidosis are either pulmonary or nonpulmonary (Table 37-4). A respiratory alkalosis is an inappropriate reduction in the blood CO2 concentration. A variety of stimuli can increase the ventilatory drive and cause a respiratory alkalosis (Table 37-5). Treatment of respiratory acid-base disorders focuses on correction of the underlying disorder. Mechanical ventilation may be necessary in a child with a refractory respiratory acidosis.
TABLE 37-4 Causes of Respiratory Acidosis
Central nervous system depression (encephalitis or narcotic overdose)
Disorders of the spinal cord, peripheral nerves, or neuromuscular junction (botulism or Guillain-Barré syndrome)
Respiratory muscle weakness (muscular dystrophy)
Pulmonary disease (pneumonia or asthma)
Upper airway disease (laryngospasm)
Fry A.C., Karet F.E. Inherited renal acidosis. Physiology. 2007;22:202-211.
Greenbaum L.A. Pathophysiology of body fluids and fluid therapy. In: Kliegman R.M., Behrman R.E., Jenson H.B., et al, editors. Nelson Textbook of Pediatrics. 18th ed. Philadelphia: Elsevier Science; 2007:267-319.
Kleta R., Bockenhauer D. Bartter syndromes and other salt-losing tubulopathies. Nephron Physiol. 2006;104:73-80.
Schaefer T.J., Wolford R.W. Disorders of potassium. Emerg Med Clin North Am. 2005;23:723-747.
Spandorfer P.R., Alessandrini E.A., Joffe M.D., et al. Oral versus intravenous rehydration of moderately dehydrated children: a randomized, controlled trial. Pediatrics. 2005;115:295-301.
Wessel J.J., Kocoshis S.A. Nutritional management of infants with short bowel syndrome. Semin Perinatol. 2007;31:104-111.