 CHAPTER 51 Metabolic Assessment
CHAPTER 51 Metabolic Assessment
Although each inborn error of metabolism is rare, in the aggregate, they significantly contribute to the causes of mental retardation, seizures, sudden infant death, and neurologic impairment. Inborn errors of metabolism are a cause of acute neonatal illness in which prompt diagnosis and treatment make a difference in outcome and even survival. They affect all age groups. Prompt recognition is the responsibility of primary care physicians, who then need the expertise of metabolic teams that specialize in the diagnosis and management of these disorders.
Inborn errors of metabolism result from a genetic deficiency in a metabolic pathway; signs and symptoms result from the accumulation of metabolites related to the pathway. These metabolites may be toxic or may destroy cells because of storage in organelles. Deficiency of downstream metabolites also plays a role in pathogenesis. The process can involve one or multiple systems (Fig. 51-1). All mechanisms of inheritance occur (see Section 9).
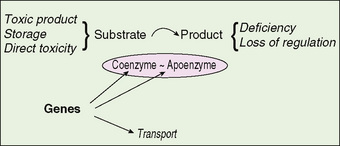
FIGURE 51-1 Depiction of the basic paradigm in inherited disorders of metabolism. Deficiency of an enzyme complex results in accumulation of metabolites proximal to the blocked metabolism and deficiency of the product of the reaction. Sites of genetic control are indicated.
The approach to diagnosis and management requires knowledge of the pathophysiology, the typical presentations in each age group, and the clinical consequences in body organs and systems. Evaluation includes clinical and laboratory assessment, as well as specific laboratory testing. Management strategies depend on understanding the pathophysiology and use of laboratory analyses to provide the best clinical control.
SIGNS AND SYMPTOMS THAT SHOULD SUGGEST AN INBORN ERROR OF METABOLISM
The signs and symptoms of an inborn error are protean. Any organ system can be involved; the presentation varies among different age groups. Inborn errors of metabolism usually do not present immediately after birth but often occur after a few days to weeks, during which the infant appears well. Infants who survive the neonatal period without developing recognized symptoms often experience intermittent illness separated by periods of being well. Family history may be helpful, if positive. A history of early infant deaths is particularly suggestive. A negative family history does not exclude an inborn error of metabolism because this may be the first affected child in the family. The presentations may include toxicity, specific organ involvement, energy deficiency, dysmorphic findings, and appearance of organ storage.
TYPES OF CLINICAL PRESENTATION OF INBORN ERRORS
Toxic Presentation
Although some aspects of the presentation are specific to the abnormal pathway, many features are nonspecific and occur in a wide range of disorders. The toxic presentation often presents as an encephalopathy. Fever, infection, fasting, or other catabolic stresses may precipitate the symptom complex. A metabolic acidosis, vomiting, lethargy, and other neurologic findings may be present. During the acute presentation, diagnostic testing is most effective when metabolites are present in highest concentration in blood and urine during this time. Abnormal metabolism of amino acids, organic acids, ammonia, or carbohydrates may be at fault. Hyperammonemia is an important diagnostic possibility if an infant or child presents with features of toxic encephalopathy. Symptoms and signs depend on the underlying cause of the hyperammonemia, the age at which it develops, and its degree. The severity of hyperammonemia may provide a clue to the etiology (Tables 51-1 and 51-2).
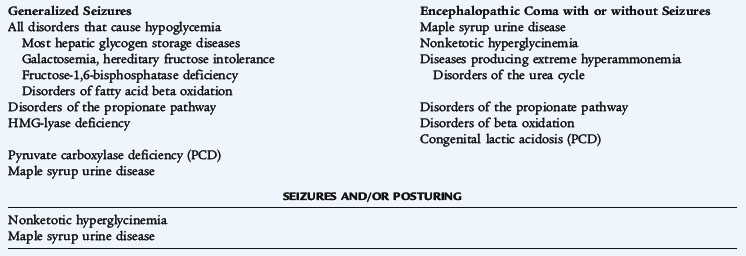
TABLE 51-2 Etiologies of Hyperammonemia in Infants
| Etiology of Hyperammonemia | Comments |
|---|---|
| Disorders of the urea cycle | Lethal hyperammonemia is common |
| Disorders of the propionate pathway | Severe hyperammonemia may precede acidosis |
| Disorders of fatty acid catabolism and of ketogenesis | Reye-like syndrome possible |
| Transient neonatal hyperammonemia | Idiopathic, self-limited |
| Portal-systemic shunting | Thrombosis of portal vein, cirrhosis, hepatitis |
| Idiopathic Reye syndrome | Rare |
| Drug intoxication: salicylate, valproic acid, acetaminophen | Obtain drug levels |
| Hyperinsulinism/hyperammonemia syndrome | Clinical hypoglycemia, subclinical hyperammonemia |
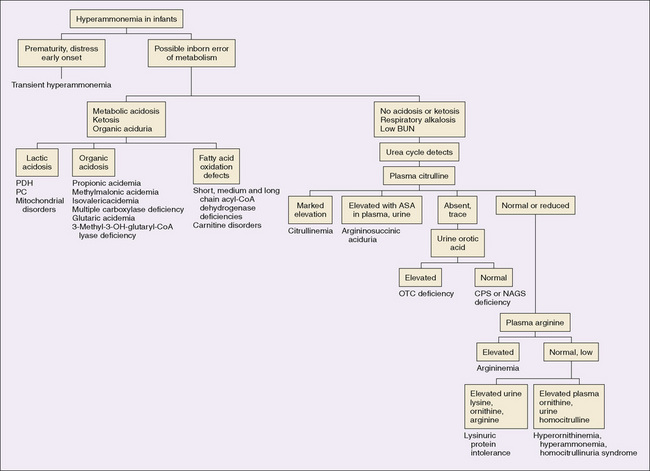
Severe Neonatal Hyperammonemia
Infants with genetic defects in urea synthesis, transient neonatal hyperammonemia, and impaired synthesis of urea and glutamine secondary to genetic disorders of organic acid metabolism can have levels of blood ammonia (>1000 μmol/L) more than 10 times normal in the neonatal period. Poor feeding, hypotonia, apnea, hypothermia, and vomiting rapidly give way to coma and occasionally to intractable seizures. Respiratory alkalosis is common. Death occurs within days if the condition remains untreated.
Moderate Neonatal Hyperammonemia
Moderate neonatal hyperammonemia (range, 200 to 400 μmol/L) is associated with depression of the central nervous system, poor feeding, and vomiting. Seizures are not characteristic. Respiratory alkalosis may occur. This type of hyperammonemia may be caused by partial blocks in urea synthesis and commonly is caused by disorders of organic acid metabolism that secondarily interfere with the elimination of nitrogen.
Clinical Hyperammonemia in Later Infancy and Childhood
Infants who are affected by defects in the urea cycle may continue to do well while receiving the low-protein intake of breast milk, only to develop clinical hyperammonemia when dietary protein is increased or when catabolic stress occurs. Vomiting and lethargy frequently progress to coma. As protein intake is restricted by anorexia and vomiting or if intravenous glucose is given, the sensorium clears, and the infant recovers, but symptoms may recur again when metabolically stressed or ingesting an increased protein intake. Seizures are not typical. During a crisis, the plasma ammonia level is usually 200 to 500 μmol/L, but as it decreases with decreased intake the condition may go unrecognized for years, especially in the absence of central nervous system symptoms. If a crisis occurs during an epidemic of influenza, the child mistakenly may be thought to have Reye syndrome. Older children may have neuropsychiatric or behavioral abnormalities (Fig. 51-2).
Specific Organ Presentation
Any organ or system can be injured by toxic accumulation of any of the metabolites involved in inborn errors. Symptoms relate to organ-specific or system-specific toxicity and injury. Examples include nervous system (seizures, coma, ataxia), liver (hepatocellular damage), eye (cataracts, dislocated lenses), renal (tubular dysfunction, cysts), and heart (cardiomyopathy, pericardial effusion) (Table 51-3; see Table 51-1).

Energy Deficiency
Disorders whose pathophysiology results in energy deficiency may manifest myopathy; central nervous system dysfunction, including mental retardation and seizures; cardiomyopathy; vomiting; hypoglycemia; or renal tubular acidosis. Examples include disorders of fatty acid oxidation, disorders of mitochondrial function/oxidative phosphorylation, and disorders of carbohydrate metabolism.
Ketosis and Ketotic Hypoglycemia
Acidosis is often found in children without metabolic diseases, with fasting associated with anorexia, vomiting, and diarrhea in the course of a viral illness. The clinical manifestations of the causative illness result in mild acidosis and ketonuria; administration of carbohydrate restores balance. In this normal response to fasting, the blood glucose level is relatively low. Severe ketosis may also be the result of disorders of ketone utilization such as ketothiolase deficiency (see Fig. 54-1) or glycogen synthase deficiency, a disorder of glycogen synthesis. In these conditions, hypoglycemia is significant, and the ketosis clears slowly, requiring significant amounts of intravenous glucose. These disorders frequently present in the context of fasting, infection with fever, or decreased intake secondary to vomiting and diarrhea. Ketotic hypoglycemia is a common condition in which tolerance for fasting is impaired to the extent that symptomatic hypoglycemia with seizures or coma occurs when the child encounters a ketotic stress. The stress may be significant (viral infection with vomiting) or minor (a prolongation by several hours of the normal overnight fast). Ketotic hypoglycemia first appears in the second year of life and occurs in otherwise healthy children. It is treated by frequent snacks and the provision of glucose during periods of stress. The pathophysiology is poorly understood (see Chapter 172). Although ketonuria is a normal response to prolonged (not overnight) fasting in older infants and children, it indicates metabolic disease in neonates. A high anion gap metabolic acidosis with or without ketosis suggests a metabolic disorder (Table 51-4). Although ketone production may not be robust in fatty acid oxidation disorders, the presence of ketonuria does not exclude this group of disorders.
TABLE 51-4 Etiologies of Metabolic Acidosis Caused by Inborn Errors of Metabolism in Infants
| Disorder | Comment |
|---|---|
| Methylmalonic acidemia (MMA) | Hyperammonemia, ketosis, neutropenia, thrombocytopenia |
| Propionic acidemia | Similar to MMA |
| Isovaleric acidemia | Similar to MMA; odor of sweaty feet |
| Pyruvate dehydrogenase deficiency | Lactic acidosis, hyperammonemia |
| Pyruvate carboxylase deficiency | Lactic acidosis, hypoglycemia, and ketosis |
| Respiratory chain (mitochondrial) disorders | Lactic acidosis, ketosis |
| Medium-chain acyl-CoA dehydrogenase deficiency (MCAD) | Moderate acidosis, hypoglycemia, decreased ketosis, possible hyperammonemia |
| Other fatty acid oxidation defects | Similar to MCAD |
| Galactosemia | Renal tubular acidosis, Escherichia coli sepsis, hypoglycemia |
| 3-Hydroxy-3-methyl-glutaryl-CoA lyase deficiency | Severe lactic acidosis, hyperammonemia, hypoglycemia |
| 3-Methylcrotonyl-CoA carboxylase deficiency | Severe lactic acidosis, hyperammonemia, hypoglycemia, ketosis |
| Multiple acyl-CoA dehydrogenase deficiency (glutaric aciduria 2) | Metabolic acidosis, hypoglycemia, lethal renal malformations |
Disorders Associated with Dysmorphic Findings
Congenital malformations or dysmorphic features are not intuitively thought of as symptoms and signs of inborn errors. Conditions that cause congenital malformations include carbohydrate-deficient glycoprotein syndrome, disorders of cholesterol biosynthesis (Smith-Lemli-Opitz syndrome), disorders of copper transport (Menkes syndrome, occipital horn syndrome), maternal phenylketonuria syndrome, glutaric aciduria II (also called multiple acyl-coenzyme A [CoA] dehydrogenase deficiency), and several storage diseases.
Storage Disorders
Storage disorders are caused by accumulation of incompletely metabolized large molecules. This storage often occurs in subcellular organelles, such as lysosomes. The glycogen storage diseases and mucopolysaccharide disorders are examples of storage disorders.
Different Age Groups and Differing Clinical Phenotypes
A neonate no longer has the protective functions of the placenta to detoxify metabolites that accumulate because of an inborn error. In addition, maternal metabolism no longer provides nutrition to the neonate who cannot metabolize substrates such as glycogen and fatty acids. Introduction of new foods in older infancy and frequency of metabolic stress with fasting and fever in that age group make the infant vulnerable. Increased protein intake associated with growth in older children and adolescents may stress deficient pathways that previously were compensated. Hormonal factors in adolescence influence intermediary metabolism in unpredictable ways. In neonates, galactosemia may present with the introduction of milk, fatty acid disorders during fasting or breastfeeding, and organic acid and urea cycle disorders following the loss of maternal detoxification. In infants, hereditary fructose intolerance occurs after introduction of fructose or sucrose in the diet, and fatty acid disorders present with fasting or illness. In children, increased protein intake may unmask disorders of ammonia detoxification. In adolescents, hormonal changes may trigger changes that allow diagnosis of cobalamin C methylmalonic aciduria (cblC MMA).
CLINICAL ASSESSMENT AND CLINICAL LABORATORY TESTING
The assessment begins with a careful history and clinical evaluation. Clinical laboratory testing can define the metabolic derangement (Table 51-5). The results generate a differential diagnosis and a list of more specific laboratory testing to confirm the diagnosis.
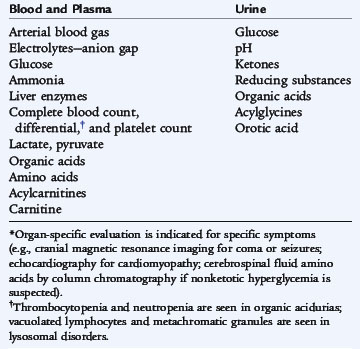
The combination of symptoms and abnormal clinical laboratory findings demands urgent metabolic evaluation. A metabolic emergency often presents with vomiting, acidosis, hypoglycemia, ketosis (or lack of appropriate ketosis), intercurrent infection, anorexia/failure to feed, lethargy proceeding to coma, and hyperventilation or hypoventilation. Clinical evaluation should focus on the cardiac, renal, neurologic, and developmental assessment and look for change in mental status, seizures, abnormal tone, visual symptoms, poor developmental progress, global developmental delay, loss of milestones, cardiomyopathy, cardiac failure, cystic renal malformation, and renal tubular dysfunction.
Clinical laboratory testing should begin with tests that are available in most hospital clinical laboratories. Care in the collection and handling of laboratory specimens is crucial to obtaining accurate results. Plasma measurements of lactate and ammonia are particularly subject to spurious results if not handled correctly. Significant ketosis in the neonate is unusual and suggests an organic acid disorder. Ketosis out of proportion to fasting status in an older child occurs in disorders of ketone usage. Lack of severe ketosis in an older child under conditions of metabolic stress is a feature of fatty acid oxidation disorders.
GENETIC ASPECTS OF INBORN ERRORS
Mechanisms of Inheritance
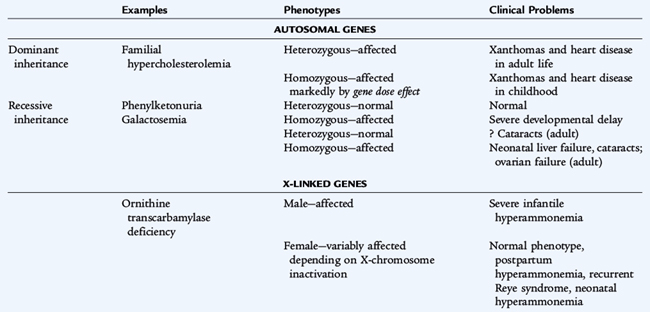
Although all of the classic mechanisms of inheritance are represented, most inborn errors of metabolism are autosomal recessive (Table 51-6). Isolation or founder effect may make a specific recessive condition common in some populations (e.g., maple syrup urine disease in the Old Order Mennonite population in Pennsylvania). X-linked conditions exhibit the usual increased prevalence in males. Carriers of recessive genes or X-linked genes (females) are usually asymptomatic except in a few conditions. In ornithine transcarbamylase deficiency, females can be symptomatic if they have a significant distribution of abnormal cells in the liver. Mitochondrial inheritance, caused by a mutation in 1 of 13 mitochondrial genes coding for proteins involved in oxidative phosphorylation, shows a maternal pattern of inheritance. The clinical disorders produced by abnormalities of these maternal genes depend on the specific defects and the tissue distribution of affected and normal mitochondria over time. Dominantly inherited diseases may be sporadic or evident in the family. When parents are closely related (consanguineous), there is a significant chance that a child has inherited two copies of the same abnormal allele, acquired from a common ancestor and resulting in a recessive condition.

Identification of Molecular Pathology
If the molecular basis of an inborn error of metabolism is known (i.e., the gene or genes has/have been mapped and mutations defined), specific molecular testing may be clinically available. A defined set of mutations comprises most of the mutations for many disorders. In other conditions, there may be many hundreds of different mutations. In recessive disorders, it is common for a patient to be doubly heterozygous for two different mutations at the gene locus involved. There is a good correlation between specific mutations and clinical outcome for some disorders; the result of this testing can be an additional guide to treatment. The ability to perform genetic testing in other at-risk family members can provide important genetic information for them, enabling decision making throughout the rest of the family.
IDENTIFICATION OF INBORN ERRORS BY NEONATAL SCREENING
Disorders Identified by Neonatal Screening
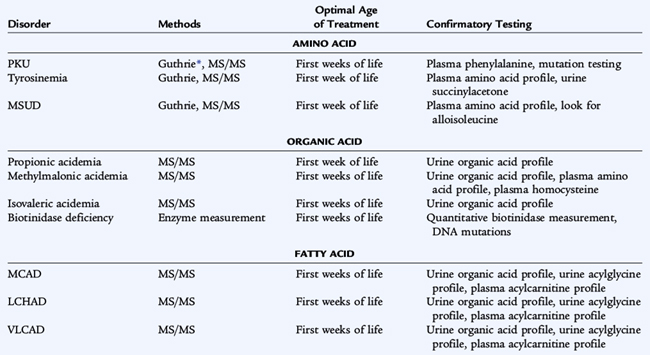
Nearly all states screen for at least eight or nine disorders. In most states, tandem mass spectrometry is used, and 30 metabolic disorders may be identifiable (Table 51-7). The disorders of amino acid metabolism, phenylketonuria, homocystinuria, maple syrup urine disease, and tyrosinemia all need to be treated early in infancy for treatment to be effective. The disorders of organic acid metabolism, propionic acidemia, methylmalonic acidemia, and isovaleric academia may result in vomiting and ketoacidosis early in life; some forms of methylmalonic acidemia present later, but central nervous system damage already may have occurred. Galactosemia is the disorder of carbohydrate metabolism that can be detected by screening. The initial presentation of galactosemia not identified by screening includes jaundice, bleeding, cataract, sepsis, and liver failure. Biotinidase deficiency is screened for by enzyme measurement. Presymptomatic treatment affects outcome in all of these conditions; in some it is lifesaving.

Strategy of Neonatal Screening
The purpose of neonatal screening is the early detection and rapid treatment of disorders before onset of symptoms, early enough to prevent the morbidity and mortality of the disorders. The screening tests for inborn errors of metabolism historically have used microbiologic (Guthrie) assays for single analytes or semiquantitative fluorometric methods. The adaptation of tandem mass spectrometry of blood specimens to neonatal screening has been adopted in most states, allowing rapid neonatal diagnosis of numerous metabolic diseases with a single blood specimen.
In most states, infants are tested just before discharge or by 7 days of age if the infant remains hospitalized (see Chapter 58). A positive test demands prompt evaluation. Specific follow-up testing and treatment of an affected child depends on the disorder. Many infants who have a positive neonatal screening test do not have a metabolic disorder. Parents may need to be reassured that this false-positive test in no way reflects the infant’s health and is the result of establishing cutoff values for the screening test to ensure that no affected infant is missed.
Confirmatory Testing Principles
Neonatal screening is designed not to miss affected infants but is not diagnostic. Cutoff values for each analyte are established carefully to identify all infants with an elevated concentration of the analyte or decreased activity of an enzyme without having an unacceptable number of false-positive results. A positive screening test must be followed by specific clinical evaluation and laboratory testing to confirm the disorder. Protocols for the evaluation of an infant with an abnormal screening result clarify which infants need to be treated and which have had a false-positive test.
A positive screening test result causes anxiety for new parents. Definitive testing must be carried out promptly and accurately. If the diagnosis is confirmed, treatment must be begun immediately. If an inborn error of metabolism is excluded, parents need a thorough explanation and reassurance that the infant is well. Widespread newborn screening for these new disorders has shown that there are milder forms of these conditions than previously recognized by clinical presentation. Clarifying this after a positive newborn screening test requires access to molecular testing to define specific mutations. Some of these milder forms do not need aggressive treatment.
Specialized Laboratory and Clinical Testing
Specialized testing for inherited disorders of metabolism is effective in confirming a diagnosis suspected on the basis of an abnormal newborn screening result or on the basis of clinical suspicion. The analytes that are helpful and examples of diagnoses made using these measurements depend on the deficient pathway in the disorder under consideration (Table 51-8).
TABLE 51-8 Specialized Metabolic Testing
| Test | Analytes Measured | Test Helpful in Identifying Disorders |
|---|---|---|
| Plasma amino acid profile | Amino acids, including alloisoleucine | PKU, tyrosinemias, MSUD, homocystinuria |
| Plasma total homocysteine | Protein-bound and free homocysteine | Homocystinuria, some forms of methylmalonic acidemia |
| Urine amino acid profile | Amino acids | Disorders of amino acid renal transport |
| Plasma acylcarnitine profile | Acylcarnitine derivatives of organic and fatty acid catabolism | Organic acid disorders, fatty acid oxidation disorders |
| Urine acylglycine profile | Acylglycine derivatives of organic and fatty acid catabolism | Organic acid disorders, fatty acid oxidation disorders |
| Plasma carnitines | Free, total, and acylated carnitine | Primary and secondary carnitine deficiency; abnormal in many organic acid and fatty acid disorders |
| Urine organic acid profile | Organic acids | Organic acid, mitochondrial and fatty acid disorders |
| Urine or blood succinylacetone | Succinylacetone | Tyrosinemia I |
| Urine oligosaccharide chromatography | Glycosaminoglycans, mucopolysaccharides | Lysosomal storage disorders |
MSUD, maple syrup urine disease; PKU, phenylketonuria.
Amino acid analysis is performed in plasma, urine, and cerebrospinal fluid. The plasma amino acid profile is most useful in identifying disorders of amino acid catabolism. Amino acids in the deficient pathway of the organic acid disorders may be abnormal, but often they are normal or may not be diagnostic.
The urine amino acid profile is helpful in diagnosing primary disorders of renal tubular function, such as Lowe syndrome and cystinuria, as well as secondary disorders of renal tubular function, such as cystinosis and Fanconi syndrome of any cause. The urine amino acid profile is not the test of choice for diagnosing disorders of amino acid or organic acid metabolism.
Markers of disordered fatty acid oxidation are measured in urine and plasma. Excessive intermediates of fatty acid oxidation and organic acid catabolism are conjugated with glycine and carnitine. The urine acylglycine profile and the plasma acylcarnitine profile reflect this accumulation. In organic acid disorders and fatty acid oxidation disorders, measurement of plasma carnitine reveals the secondary deficiency of carnitine and abnormal distribution of free and acylated carnitine. The plasma free fatty acid profile is helpful in diagnosis of disorders of fatty acid oxidation. Excess 3-OH-butyrate suggests a disorder of ketone metabolism; absence of ketones or decreased amounts of 3-OH-butyrate suggest a fatty acid oxidation disorder. Profiling of fatty acid intermediates in cultured skin fibroblasts may be informative.
The urine organic acid profile is a useful test. Disorders of organic acid metabolism, such as propionic acidemia and methylmalonic acidemia, have typical urine organic acid profiles. Although the analytes in blood and urine usually suggest the specific diagnosis, more targeted testing may be needed, in which enzymatic activity in the pathway is measured, or molecular abnormalities in the gene are sought.
Disorders of creatine biosynthesis are reflected by an increase in guanidinoacetic acid in blood and urine. Disorders of purine and pyrimidine metabolism are suggested by the presence of an abnormal urinary profile of purines, such as xanthine, hypoxanthine, inosine, guanosine, adenosine, adenine, and succinyladenosine. Similarly, disorders of pyrimidine metabolism are identified by an abnormal profile of pyrimidines, including uracil, uridine, thymine, thymidine, orotic acid, orotidine, dihydrouracil, dihydrothymine, pseudouridine, N-carbamoyl-β-alanine, and N-carbamoyl-β-amino-isobutyrate, in the urine.
Storage disorders show abnormalities in urine mucopolysaccharides (glycosaminoglycans, glycoproteins), sialic acid, heparan sulfate, dermatan sulfate, and chondroitin sulfate. Specific enzymology depends on the disorder; tissue can be either white blood cells or cultured skin fibroblasts, depending on assay. In several disorders, cerebrospinal fluid is the most helpful specimen, including glycine encephalopathy (amino acid profile), disorders of neurotransmitter synthesis (biogenic amine profile), glucose transporter (GLUT1) deficiency (plasma–to–cerebrospinal fluid glucose ratio), and serine synthesis defect (amino acid profile).
In many disorders, an abnormal metabolic profile is consistently present during illness and when the child is well. In some cases, during an episode of illness is the time when metabolic profiles are most likely to be diagnostic.
OVERVIEW OF TREATMENT
There are several basic principles to treatment of inborn errors of metabolism. Syndromes with toxicity often present with encephalopathy, and the removal of toxic compounds is the first goal of therapy. Strategies include hemodialysis, hemovenovenous filtration, and administration of trapping agents (see Chapter 53). A second strategy is to enhance deficient enzyme activity through administration of enzyme cofactors (e.g., pyridoxine in homocystinuria). If deficiency of a pathway product plays an important role, providing missing products is helpful (e.g., tyrosine in the treatment of phenylketonuria). A major principle is to decrease flux through the deficient pathway by restricting precursors in the diet. Examples include the restriction of phenylalanine in phenylketonuria, of protein in disorders of ammonia detoxification, and of amino acid precursors in the organic acid disorders.