 CHAPTER 47 Patterns of Inheritance
CHAPTER 47 Patterns of Inheritance
TYPES OF GENETIC DISORDERS
Among infants born in the United States, 2% to 4% have congenital malformations, abnormalities of form or function identifiable at birth. At 1 year of age, the number approaches 7%, because some anomalies, such as ventriculoseptal defects, may not be identifiable until after the neonatal period. The incidence of congenital malformations is much greater in inpatient pediatric populations; 30% to 50% of hospitalized children have congenital anomalies or genetic disorders.
The clinical geneticist attempts to identify the etiology, mode of inheritance, and risk that a disorder might occur in the affected child's siblings. In evaluating children with congenital malformations, the clinical geneticist attempts to classify the patient's condition into one of five different categories:
INTRODUCTION TO GENETICS AND GENOMICS
DNA is composed of four nucleotide building blocks: adenine, guanine, cytosine, and thymine. Each nucleotide is linked to other nucleotides, forming a chain. The DNA molecule consists of two chains of nucleotides held together by hydrogen bonds. The purine nucleotides, adenine and guanine, cross-link by hydrogen bonds to the pyrimidines, thymine and cytosine. Because of this cross-linking, the nucleotide sequence of one strand sets the other strand's sequence. Separating the two strands permits complementary nucleotides to bind to each DNA strand; this copies the DNA and replicates the sequence.
DNA exists as multiple fragments that, together with a protein skeleton (chromatin), form chromosomes. Human cells have 23 pairs of chromosomes, with one copy of each chromosome inherited from each parent. Twenty-two pairs of chromosomes are autosomes; the remaining pair is called the sex chromosomes. Females have two X chromosomes; males have one X and one Y.
Spread along the chromosomes, like beads on a string, DNA sequences form genes, the basic units of heredity. A typical gene contains a promoter sequence, an untranslated region, and an open reading frame, all arrayed from the 5′ to the 3′ end of the DNA. In the open reading frame, every three nucleotides represent a single codon, coding for a particular amino acid. In this way, the sequence of bases dictates the sequence of amino acids in the corresponding protein. Some codons, rather than coding for a specific amino acid, act as a “start” signal, whereas others serve as “stop” signals. Between the start and stop codons, genes consist of two major portions: exons, regions containing the code that ultimately corresponds to a sequence of amino acids, and introns (intervening sequence), which do not become part of the amino acid sequence.
Genes are transcribed into messenger RNA (mRNA), then translated into proteins. During transcription, RNA is processed to remove introns. The mRNA serves as a template to construct the protein.
Human genetic material contains 3.1 billion bases. Less than 2% of the DNA codes for proteins, comprising the genome's approximately 20,000 genes. Through a mechanism called alternative splicing, these 20,000 genes may create more than 100,000 proteins. The remainder of the DNA, the portion not involved in protein formation, has been termed junk DNA; more than 50% appears as repeat sequences, in which two or three bases are repeated. The purpose of these repeat sequences and of the junk DNA is not yet known. However, it is believed to play a role in regulation of the remainder of the genome.
Disease may be caused by changes or mutations in the DNA sequence, with the point mutation, a change in a single DNA base, being the most common type. A point mutation that changes a codon and the resulting amino acid that goes into the protein is referred to as a missense mutation. A nonsense mutation is a point mutation that changes the codon to a “stop” codon so that transcription stops prematurely. A frameshift mutation often stems from the loss or addition of one or more DNA bases; this causes a shift in how the DNA is transcribed and generally leads to premature stop codons.
Single gene mutations have an autosomal dominant (AD), autosomal recessive (AR), or X-linked pattern of inheritance. If a mutation in only one of the two copies of a gene is sufficient to cause disease, that disorder is considered AD (Table 47-1); if mutation in both copies of the gene must be present for disease to result, the condition is termed AR (Table 47-2). If a condition results from a mutation in a gene on the X chromosome, it usually causes disease in males, but generally not in females. This pattern of inheritance is termed X-linked (Table 47-3). Occasionally, as a result of skewing of inactivation of the X chromosome, an X-linked disorder may manifest symptoms of a disease in a heterozygous female. Genetic disorders also may result from abnormalities in the amount of genetic material present (termed chromosomal disorders), from interplay between genetic factors and environmental factors (called multifactorial inheritance), from unusual modes of inheritance, and as the result of exposure to certain drugs and chemicals known to cause birth defects (called teratogens).
TABLE 47-1 Autosomal Dominant Diseases
| Disease | Frequency | Comments |
|---|---|---|
| Achondroplasia Thanatophoric dysplasia |
~1:12,000 | Mutations are in the gene for fibroblast growth factor receptor-3 on chromosome 4p16.3 |
| Crouzon syndrome with acanthosis nigricans Nonsyndromic craniosynostosis |
40% of cases are new mutations (different mutations in the same gene cause achondroplasia, thanatophoric dysplasia, Crouzon syndrome with acanthosis, and nonsyndromic craniosynostosis) | |
| Neurofibromatosis 1 | 1:3500 | About 50% of cases result from new mutations in the gene for neurofibromin, a tumor suppressor gene located at 17q11.2. Expression is quite variable |
| Neurofibromatosis 2 (NF2, bilateral acoustic neuromas, Merlin) | Genotype at birth, 1:33,000 Phenotype prevalence, 1:200,000 |
The NF2 gene is a tumor suppressor gene located at 22q12.2. The protein is called “Merlin” |
| Huntington disease (HD) | Variable in populations, 1:5000–1:20,000 | The disease is caused by a (CAG) repeat expansion in the “Huntington” protein gene on chromosome 4p16.3 |
| Myotonic dystrophy (DM, Steinert disease) | 1:500 in Quebec 1:25,000 Europeans |
The disease is caused by a (CTG) repeat expansion in the DM kinase gene at chromosome 19q13.2. The condition shows genetic anticipation with successive generations |
| Marfan syndrome (FBN-1) | 1:10,000 | The syndrome is caused by mutations in the fibrillin 1 (FBN 1) gene on chromosome 15q21.1; there is variable expression |
| Hereditary angioneurotic edema (HANE) (C-1 esterase inhibitor that regulates the C-1 component of complement) | 1:10,000 | The gene is located on chromosome 11q11-q13.2. The phenotype of episodic and variable subcutaneous and submucosal swelling and pain is caused by diminished or altered esterase inhibitor protein, which can result from any one of many mutations in the gene |
TABLE 47-2 Autosomal Recessive Diseases
| Disease | Frequency | Comments |
|---|---|---|
| Adrenal hyperplasia, congenital (CAH, 21-hydroxylase deficiency, CA21H, CYP21, cytochrome P450, subfamily XXI) | 1:5000 | Phenotype variation corresponds roughly to allelic variation. A deficiency causes virilization in females. The gene is located at 6p21.3 within the HLA complex and within 0.005 centimorgans (cM) of HLA B |
| Phenylketonuria (PKU, phenylalanine hydroxylase deficiency, PAH) | 1:12,000–1:17,000 | There are hundreds of disease-causing mutations in the PAH gene located on chromosome 12q22-q24.1. The first population-based newborn screening was a test for PKU because the disease is treatable by diet. Women with elevated phenylalanine have infants with damage to the CNS because high phenylalanine is neurotoxic and teratogenic |
| Cystic fibrosis (CF) | 1:2500 whites | The gene CF transmembrane conductance regulator (CFTR) is on chromosome 7q31.2 |
| Friedreich ataxia (FA, frataxin) | 1:25,000 | Frataxin is a mitochondrial protein involved with iron metabolism and respiration. The gene is on chromosome 9q13-q21, and the common mutation is a GAA expanded triplet repeat located in the first intron of the gene. FA does not show anticipation |
| Gaucher disease, all types (glucocerebrosidase deficiency, acid β-glucosidase deficiency) (a lysosomal storage disease) | 1:2500 Ashkenazi Jews | The gene is located on chromosome 1q21. There are many mutations; some mutations lead to neuropathic disease, but most are milder in expression. The phenotypes correspond to the genotypes, but the latter are difficult to analyze |
| Sickle cell disease (hemoglobin beta locus, beta 6 glu → val mutation) | 1:625 African Americans | This is the first condition with a defined molecular defect (1959). A single base change results in an amino acid substitution of valine for glutamic acid at position 6 of the beta chain of hemoglobin with resulting hemolytic anemia. The gene is on chromosome 11p15.5. Penicillin prophylaxis reduces death from pneumococcal infections in affected persons, especially in infants |
TABLE 47-3 X-Linked Recessive Diseases
| Disease | Frequency | Comments |
|---|---|---|
| Fragile X syndrome (FRAXA; numerous other names) | 1:4000 males | The gene is located at Xq27.3 The condition is attributable to a CGG triplet expansion that is associated with localized methylation (inactivation) of distal genes. Females may have some expression. Instability of the site may lead to tissue mosaicism; lymphocyte genotype and phenotype may not correlate |
| Duchenne muscular dystrophy (DMD, pseudohypertrophic progressive MD, dystrophin, Becker variants) | 1:4000 males | The gene is located at Xp21 The gene is relatively large, with 79 exons, and mutations and deletions may occur anywhere. The gene product is called dystrophin. Dystrophin is absent in DMD but abnormal in Becker MD |
| Hemophilia A (factor VIII deficiency, classic hemophilia) | 1:5000–1:10,000; males | The gene is located at Xq28 Factor VIII is essential for normal blood clotting. Phenotype depends on genotype and the presence of any residual factor VIII activity |
| Rett syndrome (RTT, RTS autism, dementia, ataxia, and loss of purposeful hand use); gene MECP2 (methyl-CpG-binding protein 2) | 1:10,000–1:15,000; girls | The gene is located at locus Xq28 These diseases are a subset of autism. There is a loss of regulation (repression) for other genes, including those in trans positions. The disease is lethal in males. Cases represent new mutations or parental gonadal mosaicism |
| Colorblindness (partial deutan series, green color blindness [75%]; partial protan series, red color blindness [25%]) | 1:12; males | The gene is located at Xq28 (proximal) for deutan color blindness and at Xq28 (distal) for protan color blindness |
| Adrenoleukodystrophy (ALD, XL-ALD, Addison disease, and cerebral sclerosis) | Uncommon | The gene is located at Xq28 The disease involves a defect in peroxisome function relating to very long-chain fatty acid CoA synthetase with accumulation of C-26 fatty acids. Phenotype is variable, from rapid childhood progression to later onset and slow progression |
| Glucose-6-phosphate dehydrogenase deficiency (G6PD) | 1:10 African Americans 1:5 Kurdish Jews A heteromorphism in these and other populations |
The gene is located at Xq28 There are numerous variants in which oxidants cause hemolysis. Variants can confer partial resistance to severe malaria |
Pedigree Drawing
To identify specific patterns of inheritance, geneticists construct and analyze pedigrees, which are pictorial representations of a family history. Males are represented by squares and females by circles. Matings are connected with a solid line between each partner's symbols. Unmarried couples are often connected by a dashed line. Children from a couple are represented below their parents and are the next generation.
Grandparents, uncles, and aunts are added in similar fashion. Children of aunts and uncles also may be included. Ages or birthdays may be written next to or underneath each symbol. The proband (the patient who is the initial contact) is indicated with an arrow. Affected individuals are indicated by shading, or some other technique, which should be explained in a key. Carriers for a disorder (e.g., sickle cell disease) usually are indicated by a dot in the center of their symbol (Fig. 47-1).
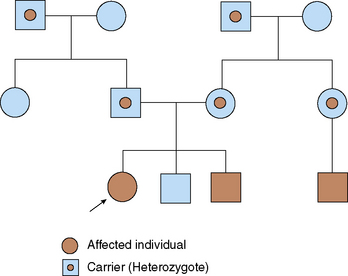
To be useful, pedigrees should include representatives of at least three generations of family members.
Autosomal Dominant Disorders
If a single copy of a gene bearing a mutation is sufficient to cause disease, that condition is inherited in an AD fashion (see Table 47-1). In AD disorders, an affected parent may have one or more children who are affected with the same disorder. The parent, who has a mutation in one gene, has a 50% chance of passing the mutated gene to each child (Fig. 47-2). Possessing one working gene and one nonworking gene is termed heterozygous. If both copies are the same, they are referred to as homozygous (Table 47-4).
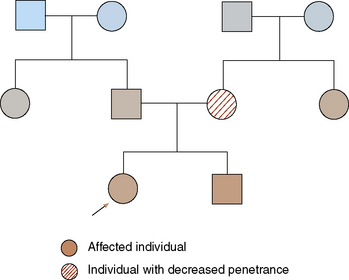
FIGURE 47-2 Pedigree showing decreased penetrance for an autosomal dominant disorder. Proband (arrow) is affected. Maternal grandfather also is affected. The individual's mother is presumed to be the carrier of the gene, even though she may show only slight symptoms of disease.
TABLE 47-4 Rules of Autosomal Dominant Inheritance
Trait appears in every generation
Each child of an affected parent has a one in two chance of being affected
Males and females are equally affected
Male-to-male transmission occurs
Traits generally involve mutations in genes that code for regulatory or structural proteins (collagen)
Some people who are obligate carriers of a mutation known to cause an AD disorder may not show clinical signs of the disorder, whereas other such individuals manifest symptoms. This phenomenon is referred to as penetrance. If all individuals who carry a mutation for an AD disorder show signs of that disorder, the gene is said to have complete penetrance. Many AD disorders show decreased penetrance.
Often, AD disorders show variability in symptoms expressed in different individuals carrying the same mutated gene. Some individuals have only mild clinical symptoms, whereas others have more severe disease. This phenomenon is referred to as variable expressivity. To illustrate the differences between penetrance and expressivity, consider the condition neurofibromatosis type 1 (NF1). Caused by a mutation in the NF1 gene on chromosome 17, NF1 is an AD disorder that causes café au lait macules, neurofibromas, and other findings. Although 100% penetrant, NF1 has marked variability in expression: some affected individuals have only café au lait spots, whereas others in the same family may experience life-threatening complications.
Sometimes, seemingly unrelated clinical findings, such as café au lait spots and neurofibromas in NF1, may be caused by a mutation in a single gene or gene pair. Known as pleiotropy, this phenomenon occurs in numerous single gene disorders.
AD disorders sometimes appear in a child of unaffected parents because of a spontaneous mutation. Known in some cases to be associated with advanced paternal age (>35 years of age), spontaneous mutations may account for most individuals with some disorders. Approximately 80% of patients with achondroplasia are believed to have experienced a mutation in the fibroblast growth factor receptor type 3 (FGFR3) gene.
Examples of Some Autosomal Dominant Disorders
Achondroplasia
Caused by a defect in cartilage-derived bone, achondroplasia (ACH) is the most common skeletal dysplasia in humans. The bony abnormalities lead to short stature, macrocephaly, a flat midface with a prominent forehead, and rhizomelic shortening of the limbs. The disorder occurs in approximately 1 in 12,000 births.
ACH is caused by mutations in the FGFR3 gene. Early in development, FGFR3 is expressed during endochondral bone formation. More than 95% of cases of ACH are caused by one of two mutations in the same base pair (site 1138). This site, extremely active for mutations, is known as a mutational hot spot.
As they grow, children with ACH often develop associated medical and psychological problems. Hydrocephalus and central apnea may occur due to narrowing of the foramen magnum and compression of the brainstem. Bowing of the legs may occur later in childhood because of unequal growth of the tibia and fibula. Dental malocclusion, obstructive apnea, and hearing loss due to middle ear dysfunction are common in later childhood. During later childhood and adolescence, the psychological effects of the short stature may manifest. In adulthood, further complications include compression of nerve roots and sciatica. People with ACH have normal life spans and normal intelligence.
The diagnosis of ACH is made on the basis of clinical findings; characteristic x-ray abnormalities confirm the diagnosis. Molecular testing is available but is usually reserved for cases that are difficult to diagnose or those in which prenatal diagnosis is requested. Prenatal diagnosis is possible by molecular testing, using fetal cells obtained through amniocentesis or chorionic villus sampling.
Neurofibromatosis Type 1
One of the most common AD disorders, NF1 is estimated to be present in 1 in 3500 individuals. NF1 is caused by a mutation in the gene NF1, which codes for the protein neurofibromin (see Chapter 186).
Although the penetrance of NF1 is 100%, the expression is extremely variable. Many affected individuals have features so mild that they are never diagnosed; such individuals may manifest only café au lait spots, axillary or inguinal freckling (smaller and darker than the café au lait spots and always found in clusters), Lisch nodules (pigmented hamartomas of the iris, best seen on slit-lamp examination), and neurofibromas (Schwann cell tumors). Between 10% and 20% of individuals have more severe symptoms, including brain tumors (optic gliomas and astrocytomas), renal vascular hypertension, skeletal involvement (scoliosis, pseudarthrosis of the tibia), and craniofacial disfigurement. Although diagnosis is made using clinical criteria, molecular diagnostic testing is also available. It is usually reserved for special cases.
Marfan Syndrome
A condition that occurs in approximately 1 in 10,000 individuals, Marfan syndrome (MS) shows pleiotropy. Caused by a mutation in the FBN1 gene, clinical symptoms mostly involve three systems: cardiac, ophthalmologic, and skeletal. Skeletal findings include a tall, thin body habitus (dolichostenomelia), spider-like fingers and toes (arachnodactyly), abnormalities of the sternum (pectus excavatum or carinatum), scoliosis, pes planus, and joint laxity. Eye findings include high myopia, which can lead to vitreoretinal degeneration; an abnormal suspensory ligament of the lens, which can lead to ectopia lentis (dislocation of the lens); and cataracts. Cardiac findings include progressive dilation of the aortic root. Aortic insufficiency followed by aortic dissection is a common complication. Other clinical features of MS include dural ectasia, abnormal pulmonary septation, and striae. Diagnostic criteria for MS are summarized in Table 47-5.
TABLE 47-5 Diagnostic Criteria for Marfan Syndrome*
| System | Major Criteria | Minor Criteria |
|---|---|---|
| Skeletal | Presence of at least 4 of the following manifestations: | Pectus excavatum of moderate severity Joint hypermobility Highly arched palate with crowding of teeth Facial appearance (dolichocephaly, malar hypoplasia, enophthalmos, retrognathia, downslanting palpebral fissures) |
| Ocular | Ectopia lentis (dislocated lens) | Abnormally flat cornea (as measured by keratometry) Increased axial length of globe (as measured by ultrasound) |
| Cardiac | Dilation of the ascending aorta with or without aortic regurgitation and involving at least the sinuses of Valsalva, or Dissection of the ascending aorta |
Mitral valve prolapse with or without mitral valve regurgitation Dilation of the main pulmonary artery, in the absence of valvular or peripheral pulmonic stenosis or any other obvious cause (<40 years of age) Calcification of the mitral annulus (<40 years of age) Dilation or dissection of the descending thoracic or abdominal aorta (<50 years of age) |
| Pulmonary | None | Spontaneous pneumothorax Apical blebs (ascertained by chest x-ray) |
| Skin | None | Stretch marks not associated with marked weight changes, pregnancy, or repetitive stress Recurrent incisional hernias |
| Dura | Lumbosacral dural ectasia by CT or MRI | None |
| Family genetic history | Having a parent, child, or sibling who meets these diagnostic criteria independently Presence of a haplotype around FBN1, inherited by descent, known to be associated with unequivocally diagnosed Marfan syndrome in the family |
None |
CT, computed tomography; MRI, magnetic resonance imaging.
* If there is an affected family member, only one major criterion is necessary for diagnosis. If no family history, two major criteria (organ systems) and one minor criterion (involvement of a third organ system) are necessary.
New mutations in FBN1 account for 25% of cases of MS. The gene is large and complex; more than 200 mutations have been identified in affected individuals, making molecular diagnosis difficult.
Treatment had previously focused on providing beta-blockers to lower preload and decrease wear-and-tear on the aorta and, when the aortic root reaches 5.5 cm in diameter, to surgically place a composite graft to replace the aortic valve and ascending aorta. It is now understood that the cardiovascular effects of MS are caused not by the defect in the fibrillin protein itself but rather by an excess in transforming growth factor-beta (TGF-β), a protein usually bound by fibrillin. Losartan, an angiotensin II receptor antagonist that also lowers levels of TGF-β, has shown great efficacy in preventing aneurysms in animal models of MS.
Autosomal Recessive Disorders
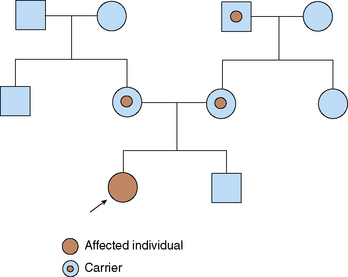
Disorders that are inherited in an AR manner manifest only when both copies of a gene pair have a mutation (Table 47-6). Affected children usually are born to unaffected parents, each of whom carries one copy of the mutation. If both members of a couple are carriers (or heterozygotes) for this mutation, each of their offspring has a 25% chance of being affected (Fig. 47-3).
TABLE 47-6 Rules of Autosomal Recessive Inheritance
Trait appears in siblings, not in their parents or their offspring
On average, 25% of siblings of the proband are affected (at the time of conception, each sibling has a 25% chance of being affected)
A normal sibling of an affected individual has a two thirds chance of being a carrier (heterozygote)
Males and females are likely to be affected equally
Rare traits are likely to be associated with parental consanguinity
Traits generally involve mutations in genes that code for enzymes (e.g., phenylalanine hydroxylase–deficient in PKU) and are associated with serious illness and shortened life span
PKU, phenylketonuria.
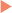
X-Linked Disorders
More than 500 genes have been identified on the X chromosome, whereas only about 50 are believed to be present on the Y chromosome. Females, whose cells have two copies of an X chromosome, possess two copies of each gene on the X chromosome, whereas males, who have one X chromosome and a Y chromosome, have only one copy of these genes. Early in female embryonic development, one X chromosome is randomly inactivated in each cell. There are many X-linked disorders (colorblindness, Duchenne muscular dystrophy, hemophilia A) in which heterozygous females show some manifestations of the disorder. Selective (skewed) inactivation of the normal gene produces symptoms in carrier women.
X-Linked Recessive Inheritance
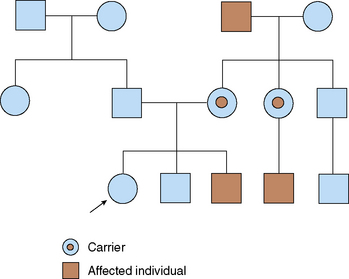
Most disorders involving the X chromosome are recessive. With only one copy of the X chromosome, males are more likely to manifest these diseases than females. Each son born to a female carrier of an X-linked recessive trait has a 50% chance of inheriting the trait, but none of this woman's daughters would be affected (each daughter has a 50% chance of being a carrier). An affected father transmits the mutation to all of his daughters, who are carriers, but not to his sons; having received their father's Y chromosome, they would not be affected (as such, there is no male-to-male transmission) (Table 47-7 and Fig. 47-4).
TABLE 47-7 Rules of X-Linked Recessive Inheritance
Incidence of the trait is higher in males than in females
Trait is passed from carrier females, who may show mild expression of the gene, to half of their sons, who are more severely affected
Each son of a carrier female has a one in two chance of being affected
Trait is transmitted from affected males to all of their daughters; it is never transmitted father to son
Because the trait can be passed through multiple carrier females, it may skip generations
X-Linked Dominant Inheritance
Only a few X-linked dominant disorders have been described. It might be expected that males and females would be equally affected, with females being less severely affected than males, the result of X inactivation. This is the case for X-linked vitamin D–resistant rickets (hypophosphatemic rickets), a disorder in which the kidney's ability to reabsorb phosphate is impaired. Phosphate levels and resulting rickets are not as severe in females as in males.
However, another group of X-linked dominant disorders are lethal in males. Affected mothers can have affected or normal daughters and normal sons. Affected sons die in utero. Incontinentia pigmenti involves a characteristic swirling skin pattern of hyperpigmentation that develops after a perinatal skin rash with blistering. Affected females also have variable involvement of the central nervous system, hair, nails, teeth, and eyes. In Rett syndrome, females are normal at birth, but later in the first year of life, they develop microcephaly and developmental regression and plateau. About 50% of patients develop seizures. Girls often are diagnosed with autism and, by 2 years of age, adopt a handwashing posture that causes them to lose all purposeful hand movements.
OTHER TYPES OF GENETIC DISORDERS
Multifactorial Disorders
Often termed polygenic inheritance, multifactorially inherited disorders result from the interplay of genetic and environmental factors. In addition to 20% of congenital malformations, including cleft lip and palate and spina bifida, most common disorders of childhood and adult life, such as asthma, atherosclerosis, diabetes, and cancer, result from an interaction between genes and the environment. These disorders do not follow simple mendelian modes of inheritance; rather, affected individuals tend to cluster in families. The disorders occur more often in first- and second-degree relatives than would be expected by chance, and they are more likely to be concordant (although not 100%) in monozygotic twins than in dizygotic twins.
Hypertrophic Pyloric Stenosis
Occurring in about 1 in 300 children, hypertrophic pyloric stenosis (HPS) is five times more likely to occur in males than in females. When a child with HPS is born, the recurrence risk in future progeny is 5% to 10% for males and 1.5% to 2% for females. In adulthood, the risk of an affected male having an affected child is markedly increased over the general population: 4% of sons and 1% of daughters of such men would be likely to be affected. Even more striking is the risk to children born to affected females: 17% to 20% of sons and 7% of daughters are affected.
The thickness of the pyloric muscle may be distributed across a bell-shaped curve; the position on the bell-shaped curve is determined by many factors, including the expression of multiple, unknown genes. HPS may result when an individual's genetic and environmental influences cause him or her to fall to an extreme position on this curve, past a certain point, which is called a threshold. In HPS, this threshold is farther to the left for males than it is for females.
Neural Tube Defects
Prior to 1998, myelomeningocele affected 1 in 1000 liveborn infants in the United States. Anencephaly occurred with a similar frequency, although most infants were either stillborn or died in the neonatal period. Since 1998, because of the supplementation of food staples with folic acid, both of these conditions have become far less frequent. Multiple genetic and nongenetic factors dictate the speed with which the neural tube closes, as follows:
Disorders with Unusual Patterns of Inheritance
Mitochondrial Inheritance
Human cells contain non-nuclear DNA; a single chromosome is present in each mitochondrion and mutations within this DNA are associated with a group of diseases. Mitochondrial DNA (mtDNA), which is circular and 16.5 kb in length, replicates independently of nuclear DNA. Involved in energy production used to run the cell, mtDNA codes for a few respiratory chain proteins (most mitochondrial proteins are coded on nuclear DNA) and for a set of transfer RNAs unique to mitochondrial protein synthesis. Virtually all mitochondria are supplied by the oocyte, which means that mtDNA is maternally derived. A woman with a mutation in mtDNA passes this mutation to all of her children. More than one population of mitochondria may be present in the oocyte, a phenomenon called heteroplasmy. The mtDNA mutation may be present in a few or many mitochondria. When the fertilized egg divides, mitochondria are distributed randomly. The presence of symptoms in the offspring, and their severity depends on the ratio of mutant to wild-type mtDNA present in that tissue. If an abundance of mutant mitochondria exists in tissue that has high energy requirements (brain, muscle, and liver), clinical symptoms occur. If fewer mutant mitochondria are present, few clinical symptoms may be seen.
MELAS (mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes) is an example of a mitochondrial disorder. Normal in early childhood, individuals affected with MELAS develop episodic vomiting, seizures, and recurrent cerebral insults that resemble strokes between 5 and 10 years of age. In 80% of cases, analysis of the mtDNA reveals a specific mutation (A3243G) in the MTTL1 gene (a gene that codes for a mitochondrial transfer RNA).
In families in which MELAS occurs, a range of symptoms is seen in first-degree relatives, including progressive external ophthalmoplegia, hearing loss, cardiomyopathy, and diabetes mellitus. Although all offspring of a woman who carries a mutation would be affected, because of heteroplasmy, the severity of disease varies, depending on the percentage of mitochondria bearing the mutation that are present.
Uniparental Disomy
Evaluation of a child with uniparental disomy (UPD) reveals a normal karyotype. However, chromosomal markers for one particular chromosome are identical to the markers found on the chromosomes of the patient's mother or father (but not both). In UPD, the patient inherits two copies of one parent's chromosome and no copy from the other parent.
UPD probably occurs through a few mechanisms, but the most common results from a spontaneous rescue mechanism. At the time of conception, through nondisjunction, the fertilized egg is trisomic for a particular chromosome, with two copies of one parent's chromosome and one copy of the other parent's chromosome; conceptuses with trisomy often miscarry early in development. Patients with UPD survive because they spontaneously lose one of three copies of the affected chromosome. If the single chromosome from one parent is lost, the patient has UPD.
An alternate explanation involves monosomy for a chromosome rather than trisomy. Had the patient, at the time of conception, inherited only a single copy of a chromosome, spontaneous duplication of the single chromosome would lead to UPD.
Prader-Willi and Angelman Syndromes
Prader-Willi syndrome (PWS), which occurs in approximately 1 in 10,000 infants, is characterized by hypotonia of prenatal onset; postnatal growth delay; a characteristic appearance, including almond-shaped eyes and small hands and feet; developmental disability; hypogonadotropic hypogonadism; and obesity after infancy. Early in life, affected infants are so hypotonic that they cannot take in enough calories to maintain their weight. Nasogastric feeding is invariably necessary, and failure to thrive is common. During the first year of life, muscle tone improves and children develop a voracious appetite. Some 60% to 70% of patients with PWS have a small deletion of chromosome 15 (15q11). In patients without a deletion, 20% have UPD of chromosome 15.
Angelman syndrome (AS) is a condition with moderate to severe mental retardation, absence of speech, ataxic movements of the arms and legs, a characteristic craniofacial appearance, and a seizure disorder that is characterized by inappropriate laughter. AS is also characterized by a deletion in the 15q11 region in 70% of affected individuals; UPD for chromosome 15 can be demonstrated in approximately 10% of AS patients.
If the deletion occurs in paternal chromosome 15, the affected individual has PWS, whereas AS results from a deletion occurring only in the maternal chromosome 15. When UPD is responsible, maternal UPD results in PWS, whereas paternal UPD results in AS. To summarize, if the patient lacks a copy of paternal chromosome 15q11.2, PWS will occur; if maternal chromosome 15q11.2 is lacking, AS results.
This phenomenon is explained by genomic imprinting. Imprinting is an epigenetic phenomenon, a nonheritable change in the DNA that causes an alteration in gene expression based on parental origin of the gene. PWS is caused by deficiency of the protein product of the gene SNRPN (small nuclear ribonucleoprotein). Although SNRPN is present on both maternally derived and paternally derived chromosome 15, it is expressed only in the paternally derived chromosome. Expression is normally blocked in the maternal chromosome because the bases of the open reading frame are methylated; this physical change in the DNA prevents gene expression. PWS results whenever a paternal chromosome 15 is missing, either through deletion or through UPD.
AS results from a lack of expression of ubiquitin-protein ligase E3A (UBE3A), a second gene in the chromosome 15q11.2 region. UBE3A is normally expressed only in the maternally derived chromosome 15. Although the gene is present in paternal chromosome 15, UBE3A is methylated, and gene expression is blocked. Therefore, if either the critical region of maternal chromosome 15 is deleted, or paternal UPD occurs, the individual will manifest symptoms of AS.
Expansion of a Trinucleotide Repeat
More than 50% of human DNA appears as repeat sequences, two or three bases repeated over and over again. Disorders caused by expansion of trinucleotide repeats include fragile X syndrome, Huntington disease, myotonic dystrophy, Friedreich ataxia, and the spinocerebellar ataxias. Although an increase in the number of the three repeated bases is at the heart of each disorder, the molecular mechanism differs.
Fragile X syndrome (FRAX), which occurs with a frequency of approximately 1 in 2000 children, is the most common cause of inherited mental retardation. Features include characteristic craniofacial findings (large head; prominent forehead, jaw, and ears); macro-orchidism with testicular volume twice normal in adulthood; a mild connective tissue disorder, including joint laxity, patulous eustachian tubes, and mitral valve prolapse; and a characteristic neurobehavioral profile, including mental retardation (ranging from mild to profound), autism spectrum disorder, and pervasive developmental disorder.
Positional cloning in the Xq27 region has identified a triplet repeat region composed of one cytosine and two guanine residues (CGG). Occurring in the CpG island, a part of the promoter region of a gene that has been called FMR-1, unaffected individuals who have no family history of FRAX have 0 to 45 CGG repeats (most have 25 to 35). In individuals with FRAX, the number of repeats is greater than 200; such people are said to have a full mutation. Between these two categories, a third group has 56 to 200 repeats; always phenotypically normal, these individuals are premutation carriers.
FRAX results from a failure to express the FMRP, the protein product of the FMR-1 gene. Fragile X mental retardation protein (FMRP) is a carrier protein that shuttles mRNA between the nucleus and cytoplasm in the central nervous system and other areas (e.g., the developing testis) during early embryonic development. Although FMRP is produced in unaffected individuals and premutation carriers, in those with the full mutation, FMRP transcription of the protein is blocked because the large number of CGG repeats in the CPG island become methylated (an epigenetic phenomenon). Thus, FRAX occurs as a consequence of a loss-of-function mutation—the failure of expression of FMRP because of methylation of the promoter sequence.
In female premutation carriers, an expansion in the number of repeats from the premutation to the full mutation range may occur during gametogenesis. The cause of this expansion is not understood. Although these females do not have symptoms and signs of FRAX, premutation carriers may manifest ovarian failure and/or a neurologic condition known as fragile X tremor/ataxia syndrome later in life.
TERATOGENIC AGENTS
Approximately 6.5% of all birth defects are attributed to teratogens, which are chemical, physical, or biologic agents that have the potential to damage embryonic tissue and result in one or more congenital malformations. Agents known to be teratogenic include drugs (prescription and nonprescription); intrauterine infections (rubella); maternal diseases, such as diabetes mellitus; and environmental substances, such as heavy metals. Knowledge of teratogenic agents and their effect on the developing fetus is important, because limiting exposure to these agents is an effective way to prevent birth defects (see Chapters 58, 59, and 60).
Maternal Infections
Rubella was the first maternal infection known to cause a pattern of malformations in fetuses affected in utero. Cytomegalovirus, Toxoplasma gondii, herpes simplex, and varicella are additional potentially teratogenic in utero infections (see Chapter 66).
Maternal Disease
Maternal diabetes mellitus and maternal phenylketonuria can result in congenital anomalies in the fetus. Strict control of these disorders before and during pregnancy protects the developing child (see Chapter 59).
Medications and Chemicals
Fetal alcohol spectrum disorder, which occurs in 10 to 20 per 1000 children, may be the most common teratogenic syndrome. Features include prenatal and postnatal growth deficiency, developmental disabilities, microcephaly, skeletal and cardiac anomalies, and a characteristic facial appearance. To cause the full-blown fetal alcohol syndrome, pregnant women must drink at least 6 ounces of alcohol each day during the pregnancy. Lesser consumption during all or part of the gestation will lead to milder symptoms. Warfarin, retinoic acid, and phenytoin are additional teratogenic agents (see Chapter 59).
Radiation
High-dose radiation exposure during pregnancy in Hiroshima, Japan, and Nagasaki, Japan, was shown to increase the rate of spontaneous abortion and result in children born with microcephaly, mental retardation, and skeletal malformations. Estimates of exposure to cause these effects were approximately 25 rad. The dose from routine radiologic diagnostic examinations is in the millirad range.
Methyl Mercury
An inadvertent spill of methyl mercury into the water supply of Minamata, Japan, led to an outbreak of congenital malformations. Children were born with neurologic abnormalities, including mental retardation, cerebral palsy–like movement disorders, and blindness. This incident raised concern about exposure of fetuses to heavy metals (especially mercury) from contaminated fish in the diets of pregnant women.