Degenerative Diseases of the Central Nervous System
Degenerative diseases of the central nervous system (CNS) can affect grey matter, white matter, or both. The neurodegenerative disorders are characterized by loss of functionally related groups of neurons. The pattern of neuronal loss is selective, affecting one or more groups of neurons while leaving others intact. The cause of the neuronal loss is unknown but is clearly multifactorial. The diseases appear to arise without any clear inciting event in individuals without previous neurologic deficits. The clinical symptoms produced depend on which neuronal populations are lost. Degenerative changes in grey matter diseases interfere with the function of the neuronal cell bodies and synapses. The diseases can affect the cortex, the grey matter of the spinal cord, or both. The most common factor in this group of diseases is the slow deterioration of body functions controlled by the brain and spinal cord.
The neuropathologic findings observed in the degenerative diseases reflect changes in different components. In some disorders there are intracellular abnormalities such as Lewy bodies and neurofibrillary tangles, while in others, there is primary loss of neurons.29 Some degenerative diseases have prominent involvement of the cerebral cortex, such as Alzheimer’s disease; others are more restricted to subcortical areas and may present with movement disorders such as tremors and dyskinesias, such as Parkinson’s disease. Demyelination has the major impact on other disorders such as multiple sclerosis. Through genetic and molecular studies of these diseases it is becoming clearer that there are many shared features across the disorders.
Cellular stress is a major component of neurodegenerative disease. When the cell is stressed, the proteins that form filaments and microtubules creating the cytoskeleton can collapse and form perinuclear bundles or clumps of protein aggregates. If the stress experienced by the cell is not lethal, the cell adapts and manufactures several proteins that may restore functional activity of partially denatured proteins. If the proteins cannot be restored, then a process begins to destroy the proteins. If the proteins do not fully degrade, they become clumped together to form intracellular inclusions. The inclusions in neurodegenerative disorders are examples of such aggregates. The aggregated proteins are generally cytotoxic, but the mechanisms by which protein aggregation is linked to cell death may be different in these various diseases. The histologic characteristics of the inclusions often form the diagnostic hallmarks of these different diseases.
Disorders of movement associated with grey matter destruction are reflected in functional loss and decreased fractionation of movement. Dementia can be present and always represents a pathologic process; dementia, despite popular belief, is not part of normal aging. The majority of the degenerative diseases that affect the basal ganglia are associated with involuntary movements. Disruption of smooth coordination of muscles can be seen in diseases affecting the cerebellum and brainstem. Many of the disorders appear later in life and mimic the normal deterioration of the nervous system that comes with aging.
The cost of care for people with degenerative neurologic disease is significant due to the protracted time of disability before death and the extent of the disability. Although medical science has made tremendous progress in the past few years, degenerative disorders continue to be a challenge to health care providers and a scourge to modern society.
AMYOTROPHIC LATERAL SCLEROSIS
Overview and Definition
Amyotrophic lateral sclerosis (ALS) is a disorder that is generally recognized as an adult-onset progressive motor neuron disease but is also a complex disease process underlying a multisystem illness. It is the most physically devastating of the neurodegenerative diseases. ALS is a progressive disease of unknown cause, characterized by degeneration and scarring of the motor neurons in the lateral aspect of the spinal cord, brainstem, and cerebral cortex, giving rise to the terms lateral and sclerosis in identifying the disease. Peripheral nerve changes result in muscle fiber atrophy or amyotrophy. The resulting weakness causes profound limitation of movement.125 Executive dysfunction, characterized by deficiencies in attention, language comprehension, planning, and abstract reasoning, represents cortical involvement.
Incidence
The incidence of ALS is about 2 per 100,000, with 5000 people in the United States diagnosed each year. In the western Pacific the incidence increases to 14 to 55 per 100,000.102 Electrodiagnostic equipment for testing may not be available in an area, so there may be greater numbers than are currently identified.
Etiologic and Risk Factors
Approximately 90% of cases of ALS occur sporadically and are clinically manifest in the fifth decade or later. The cause is unknown. There have been clusters of ALS noted, where there have been three or four individuals living or working in close proximity or individuals participating in the same sport, and military service appears to hold a possible risk for ALS. Chronic intoxication with heavy metals, such as lead or mercury, has been suggested as an etiologic agent, but there still does not seem to be a clear cause. ALS occurs predominantly in men, although bulbar onset occurs more often in women. There may be an increased incidence in white males, with a lower rate reported in Mexico, Poland, and Italy.125 There appears to be little evidence of active poliovirus in persons with ALS. In fact, there are very few postpolio individuals who develop ALS, and it is postulated that polio may protect against developing ALS. There is an increased incidence of cancer in individuals with motor neuron disease.26
Familial ALS (FALS) is an inherited autosomal trait. It occurs in 5% to 10% of all ALS cases. The identification of at least one additional family member with ALS in successive generations is essential for the diagnosis of FALS. Most FALS is inherited in an autosomal dominant pattern and is characterized by an early onset. Family linkage may be missed if there was a death of a family member before the usual age of onset. Copper and zinc superoxide dismutase (SOD1) gene mutation may account for about 15% of cases of FALS. SOD1 genetic testing can be used for genetic counseling in families in which SOD1 mutations are already established.
Pathogenesis
The pathologic features are primarily degeneration of motor cells in the spinal cord, brainstem, and to a lesser extent the cerebral cortex, with secondary degeneration of pyramidal tracts. Eighty percent of FALS cases have degeneration of the spinocerebellar tract. Dementia may be due to changes in the frontotemporal cortices or in the substantia nigra (basal ganglia). Destruction of large motor neurons of the anterior horn cells is greatest in the cervical and lumbar regions of the cord and between the internal capsule and the bulbar pyramids. Critical neurons are sparse, and the dendrites are shortened, fragmented, and disorganized. Microscopic examination demonstrates a reduction in the number of anterior horn neurons throughout the length of the spinal cord, with associated reactive gliosis and loss of anterior root myelinated fibers. There are similar findings in the cranial nerve nuclei. Diffuse and patchy loss of myelin appears in all areas of the spinal cord except the posterior columns, allowing for preservation of sensation.132
Some histologic changes seen in ALS are consistent with those in other lower motor neuron diseases, but there are some changes that may be unique to ALS. Ribonucleic acid (RNA) content is reduced in the damaged and normal neurons in the area. Excessive accumulation of the pigmented lipid (lipofuscin) develops that normally is not seen until advanced age. The production of free radicals may be responsible for the changes in the lipid molecules, eventually causing cell death.21 Spheroids, the axonal swelling containing packed neurofilaments found in the dendrites and axons distant from the cell body, are found more frequently in the early cases with shorter clinical courses. Spheroids are not found in FALS. The spheroids may represent the slowing of transport in the axon and the abnormal processing of neurofilaments. Inclusion bodies (see Chapter 28) of several types are found in the motor neurons and axons. The skeinlike inclusion consisting of threadlike linear or tubular structures of filaments, similar to Lewy body inclusions, may be specific to ALS. Lymphocytes indicating immunoreaction have been found in the more rapidly advancing cases. Antibodies have been found in some cases of ALS that may indicate involvement of hormones in the immune process; however, there appears to be a poor response to immunologic treatment.12
Glutamate, the principal excitatory neurotransmitter in the human motor system, can cause excitotoxic damage when the extracellular glutamate concentration increases. Plasma glutamate levels in individuals with motor neuron disease can be twice that of normal. This may be related to a defect in the transport and breakdown of excitatory amino acids predisposing the person to neurotoxicity, especially noted in males. Environmental toxins may act as excitotoxins.20,153
Oxidative damage appears to play a role in the damage to nerve cells in ALS. Calcium-mediated excitotoxicity can generate free radicals. In FALS, the mutated SOD1 appears to cause increased reactivity to hydrogen peroxide and lead to an increase in free radicals. Copper ions in a reduced state will cause this process to become harmful to the neuron. Oxidative damage and glutamate toxicity may interact or potentiate each other. They may also contribute to other mechanisms of motor neuron degeneration, including axon transport abnormalities and apoptosis.125
The death of the peripheral motor neuron in the brainstem and spinal cord leads to denervation and atrophy of the corresponding muscle fibers. In the early phases of the illness, denervated muscle may be reinnervated by sprouting of preserved nearby distal motor axon terminals, although reinnervation in this disease is less extensive than in other chronic neurologic disorders.26
There is remarkable selectivity of neuronal cell death, involving motor neurons of the brainstem and spinal cord with relative sparing of the oculomotor nuclei. There is eventual spread into the prefrontal, parietal, and temporal areas, as well as into the subthalamic nuclei and reticular formation. In persons kept breathing with ventilatory support, there may eventually be sensory system changes.
Clinical Manifestations
Cognitive impairments are noted in up to 50% of individuals with ALS. With careful assessment, these deficiencies can be noted early on. Executive function deficits can be found in visual attention, working memory, cognitive flexibility, problem solving, and visual-perceptual skills. Verbal fluency declines before dysarthria develops. The cognitive deficits are due to changes in frontal lobe function and may be related to frontotemporal dementia. Bulbar onset is more predictive of cognitive impairment than limb onset. Pseudobulbar affect, resulting in emotional lability, emotional outbursts, and pathologic laughing or crying, is not related to a psychologic or psychiatric condition and is not a part of the frontotemporal dementia.
The motor control manifestations of ALS vary depending on whether upper or lower motor neurons are predominantly involved. With lower motor neuron cell death and early denervation, the first evidence of the disease typically is insidiously developing asymmetric weakness, usually of the distal aspect of one limb progressing to weakness of the contiguous muscles. Extensor muscles become weaker than flexor muscles, especially in the hands. Cervical extensor weakness develops and can lead to drooping of the head and pain associated with overstretched muscles. Increased lumbar lordosis occurs as part of the compensatory strategy to attempt to right the head and bring the eyes level.
The neurons innervating muscles controlling articulation, chewing, and swallowing originate in the medulla, or the “bulb,” and any weaknesses in the muscles are considered bulbar signs. In the lower motor or flaccid component, facial muscles are affected. Inability to hold the eye closed against pressure is a standard test. Weakness around the mouth develops, and air leaks out. The movement of the tongue is decreased, and atrophy is present. Fasciculations in the tongue are present with lower motor neuron dysfunction. Dysarthria associated with lower motor neuron involvement is reflected by inability to shout or sing, a hoarse or whispering quality of the voice, and nasal tone. Manipulating food inside the mouth becomes difficult. Eventually, weak swallowing may trigger reflex coughing.
Individuals with ALS complain of drooling because of the absence of automatic swallowing, which is made worse by the forward head position. Breathing becomes difficult, and accessory breathing replaces diaphragmatic breathing. Respiratory distress can occur when sleeping, especially on the back.
Deformities of the extremities are common, especially since weakness causes shortening of the extensor muscles. Clawhand develops as the weakness of lumbricals and interossei hinders metacarpal flexion and tenodesis flexes the distal joints. Fig. 31-1 shows the hands of an individual with ALS.
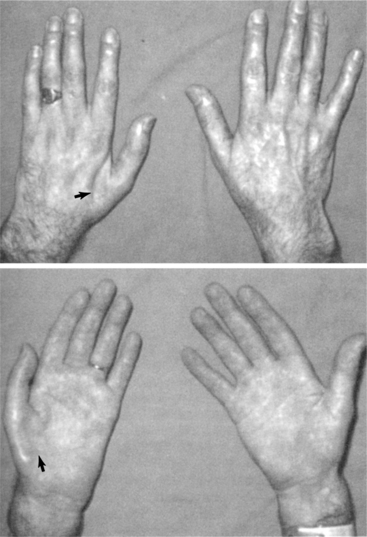
Figure 31-1 Wasting of hand muscles in amyotrophic lateral sclerosis. (From Parsons M: Color atlas of clinical neurology, London, 1993, Wolfe.) Wolfe
Weakness caused by denervation is associated with progressive wasting and atrophy of muscles. Cramping with volitional movement in the early morning is often reported, with complaints of stiffness. Muscle cramps indicate lower motor neuron dysfunction. It may be related to hyperexcitability of distal motor axons. Early in the disease there are fasciculations, or spontaneous twitching of muscle fibers. Fasciculations are the result of spontaneous contractions of a group of muscle fibers belonging to a single motor unit. The impulse for the fasciculation appears to arise from hyperexcitable distal motor axons. This is random in time and in muscles affected. It should be noted that both muscle cramping and fasciculations are found in healthy adults and should never be taken alone as a concern for the development of ALS.
Upper motor neuron symptoms are characterized by loss of inhibition and the resulting lack of dexterity and spasticity. Muscle strength is decreased along an upper motor neuron pattern. Extensor muscles of the upper extremity and flexor muscles of the lower extremity are weakened, since spasticity develops as the result of loss of brainstem control of the vestibulospinal and reticular formation control. As in other upper motor lesions, spasticity can limit the ability to accurately assess muscle strength.
Spastic bulbar palsy occurs when upper motor neurons and the corticobulbar fibers controlling speech, mastication, and swallowing are affected. This is termed pseudobulbar palsy and differs from the palsy associated with lower motor neuron loss in the brainstem. Pseudobulbar affect may manifest as inappropriate laughter, irritability, anger, and tearfulness.
The tendon, or muscle stretch, reflexes become hyperactive based on the loss of the Ia inhibitory reflex. This also extends to the development of clonus, in which manual quick stretch of a muscle induces repeated rhythmic muscle contraction. Babinski’s response is positive, characterized by extension of the great toe, often accompanied by fanning of the other toes in response to stroking the outer edge of the ipsilateral sole upward from the heel with a blunt object. If there is enough wasting of the dorsiflexors, the response may appear to be flexor despite upper motor neuron involvement.
It is characteristic of ALS that, regardless of whether the initial disease involves upper or lower motor neurons, both categories are eventually implicated. In most persons with ALS, Babinski’s and Hoffmann’s signs are present or the tendon jerks are disproportionately active.156 Throughout the course of the disease, eye movements and sensory, bowel, and bladder functions are preserved.
ALS is characterized by differing areas of CNS involvement and has been categorized in terms of four major groups of symptoms listed below.67,110 Fig. 31-2 shows the levels of dysfunction associated with the terms that describe them.
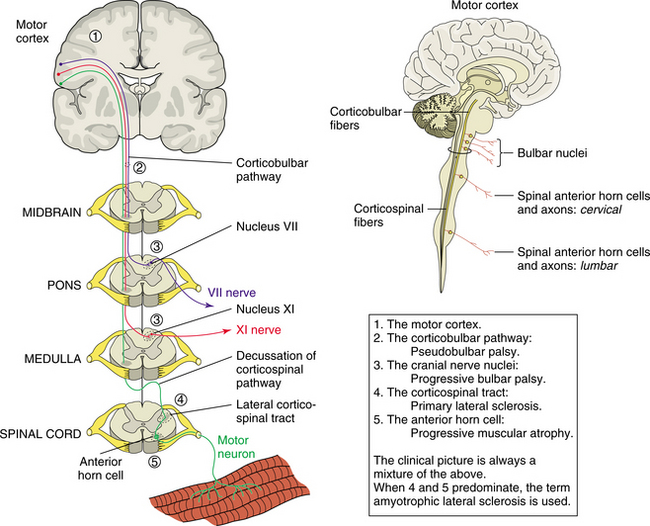
Figure 31-2 Areas of damage in the central and peripheral nervous system as a result of amyotrophic lateral sclerosis. (From Lindsay KW, Bone I, Callander R: Neurology and neurosurgery illustrated, New York, 1986, Churchill Livingstone. Insert from Noble J: Textbook of primary care medicine, ed 3, Copyright 2001, Mosby, Inc., and borrowed from Pryse-Phillips WM, Murray TJ: Essentials of neurology: a concise textbook, New York, 1992, Medical Examination Publisher, p 660.) Medical Examination Publisher
1. Pseudobulbar palsy reflects damage in the corticobulbar tract.
2. Progressive bulbar palsy is a result of cranial nerve nuclei involvement. There is weakness of the muscles involved in swallowing, chewing, and facial gestures. Fasciculations of the tongue are usually prominent. With early bulbar involvement, there can be difficulty with respiration before weakness of the limbs. Dysarthria and exaggeration of the expression of emotion, or pseudobulbar affect, indicate involvement of the corticobulbar tract. The oculomotor system is usually not involved, and eye movement remains normal.
3. Primary lateral sclerosis results in neuronal loss in the cortex. Signs of corticospinal tract involvement include hyperactivity of tendon reflexes with spasticity causing difficulty with active movement. Weakness and spasticity of specific muscles represent the level and progression of the disease along the corticospinal tracts. There is no muscle atrophy, and fasciculations are not present. This form of ALS is rare.
4. In progressive spinal muscular atrophy there is progressive loss of motor neurons in the anterior horns of the spinal cord, often beginning in the cervical area. There is progressive weakness, wasting, and fasciculations involving the small muscles of the hands. Other levels of the spinal cord can be the site of the initial disease process, with symptoms reflecting the level involved. These areas of weakness can be present without evidence of higher-level corticospinal involvement, such as spasticity.
ALS with probable upper motor neuron signs is a condition in which there are no overt upper motor neuron signs, but involvement of the corticospinal tracts is indicated by the incongruous presence of active tendon reflexes in limbs with weak, wasted, and twitching muscles. Upper and lower limbs are usually affected first, with progression to facial symptoms and respiratory failure.
MEDICAL MANAGEMENT
Diagnosis is predominantly made by the combination of clinical presentation and electromyogram (EMG). The time to diagnosis differs, typically according to the first presenting symptoms. With upper limb onset the time to diagnosis is approximately 15 months, with lower extremity onset it is 21 months, and with bulbar involvement as the first sign it is approximately 17 months.25 Box 31-1 describes diagnostic criteria.
The symptoms are generally first reported to a primary care physician, and it appears that there is a greater delay in reaching the diagnosis in such cases than when the initial symptoms are reported to a neurologist. Often in the early cases and those that are progressing slowly, there may be minimal abnormality on the first EMG, and the changes that lead to diagnosis may not appear for 6 to 12 months. In electrodiagnosis, rapidly progressive ALS shows different changes on the EMG compared with slowly progressive ALS. It is believed that some of these differences comes from the adaptation and sprouting that occur early in the process. This adaptation cannot be sustained as the disease progresses.100
EMG studies include the muscles of the extremities and trunk and are selected based on the propensity for weakness in ALS. EMG criteria for the diagnosis of ALS include the presence of fibrillations, positive waveforms, fasciculations, and motor unit potential changes in multiple nerve root distributions in at least three limbs and the paraspinal muscles. These changes occur without change in sensory response.126
In 1990 the World Federation of Neurology El Escorial criteria for the diagnosis of ALS were established, and four categories of ALS were outlined (Box 31-2). Suspected ALS is characterized by lower motor neuron signs alone in two or more regions, to which might be added upper motor neuron signs on the basis of the clinical examination. Possible ALS is defined as upper and/or lower motor neuron signs in only one region, possibly with a grouping of upper or lower motor neuron signs in other regions. Exclusion of structural lesions would be attempted. Probable ALS is considered if there are upper and lower motor neuron signs in two regions, and the upper motor neuron signs are above the lower motor neuron signs. Structural lesions must definitely be ruled out by neuronal imaging studies. Definite ALS requires lower motor neuron signs to be present in addition to upper motor neuron signs in three CNS regions concomitantly with upper or lower motor neuron signs in other regions with structural lesions excluded. Definite EMG signs of lower motor neuron degeneration require the presence of evidence of acute denervation with fibrillations or positive sharp waves and chronic denervation represented by large-amplitude and long-duration motor unit potentials as well as reduced recruitment in each muscle.25
Box 31-2 WORLD FEDERATION OF NEUROLOGY EL ESCORIAL CRITERIA FOR DIAGNOSIS OF AMYOTROPHIC LATERAL SCLEROSIS (ALS)

EMG, Electromyography; NCV, nerve conduction velocity; LMN, lower motor neuron; UMN, upper motor neuron; (), proposal to add this category to diagnostic criteria for ALS, WFN El Escorial Revisited; LAUS, laboratory abnormalities of uncertain significance.
From Brooks BR: Introduction: defining optimal management in ALS: from first symptoms to announcement, Neurology 53(suppl 5):S1-S3, 1999.
There are several disorders that resemble ALS that are treatable. ALS must be differentiated from other conditions that produce a combination of upper and lower motor neuron lesions. Lymphoma and Lyme disease can cause diffuse lower motor axonal neuropathy. Disorders of the cervical cord, such as skull base deformities, syringomyelia, cord tumors, and cervical spondylosis, must be ruled out.12 Box 31-3 describes disorders that can mimic ALS.
In such cases, however, there should not be any evidence of anterior horn cell involvement in the legs or trunk, but only in the upper limbs. Cervical myelopathy can look like ALS if cord compression is combined with root involvement. The lower motor neuron findings are only in the arms, an important diagnostic feature, and this situation can be confirmed by imaging of the cervical cord and use of EMG to determine if there are fasciculations in the legs. Any signs of disease due to a lesion above the foramen magnum, such as bulbar signs or cranial nerve V or VII nerve involvement, would rule out a cervical cause. Lower motor neuron lesions may be predominant with spinal arachnoiditis (usually syphilitic) and radiculitis, cervical ribs, and peripheral nerve lesions, including the postpolio syndrome. Weakness and wasting are typical of all forms of hereditary motor neuropathy, some of which occur first in adult life, and in hereditary motor and sensory neuropathy. The same findings, although without fasciculations, are also seen in primary muscle disease, rheumatoid arthritis, and myotonic dystrophy. If there is doubt about evidence of anterior horn cell disease in the trunk or legs, EMG should be able to demonstrate that which cannot be seen clinically.
Weight loss may suggest carcinoma, and investigations should be undertaken to rule out underlying malignancy if there is any atypical feature on examination or investigation, such as marked slowing of motor nerve conduction velocities. Most other mimics can be excluded by history, such as hereditary neuropathy, prior gastrectomy, polio, or electrical injury. Dyspnea may suggest chronic obstructive pulmonary disease or heart failure. Thorough examination will reveal hyperthyroidism or acromegaly. Laboratory tests will reveal lead or other metal poisoning. ALS symptoms may mimic nonneurologic diseases, and neurologic signs may be missed.9 Involvement of the sensory system or conduction block with evoked potential testing may indicate other neurodegenerative disease processes.
TREATMENT.
Multiple pathways have been implicated in the pathogenesis of ALS. A medication or combination of medications that targets more than one pathogenic pathway may slow disease progression in an additive or synergistic fashion. Such combination therapy has been successful in oncology, although there are multiple drug interactions and increased incidence of drug side effects. Drug resistance can develop with prolonged periods of medication. Specific targets for therapy are not well defined, and issues of dose, duration, and bioavailability in the diseased state are unknown. Furthermore, the timing of drug initiation is an important factor in determining the response to therapy, which is based on the ratio of reversible and irreversible injury at any time point in the disease
Riluzole remains the only Food and Drug Administration (FDA)–approved drug for ALS based on the 3-month improvement in survival observed in two large clinical trials. Riluzole has a broad range of pharmacologic effects, including inhibition of glutamate release, postsynaptic glutamate receptor activation, and voltage-sensitive sodium channel inactivation. Riluzole appears to be neuroprotective and slows the disease course by approximately 10% to 15%, but it is not curative. There is controversy over the best time to begin therapy with riluzole.113 Neuroprotective effects would be more extensive when there are more motor units intact to preserve, and this argument supports early treatment.38 Asthenia and gastrointestinal side effects are common, and the long-term neurotoxic effects are unknown.
Neuroinflammation occurs in the brainstem and spinal cord of individuals with ALS, suggesting that antiinflammatory agents may be effective in treating this disease. Celastrol is a potent antiinflammatory and antioxidant that suppresses nitric oxide production. Tamoxifen may be neuroprotective in ALS because of its ability to inhibit protein kinase C, which mediates inflammation in spinal cords of individuals with ALS. The drug penetrates the CNS and is generally well tolerated.
Talampanel is a noncompetitive modulator of ionotropic glutamate receptors primarily under development as an antiepileptic agent. The most common side effects are ataxia and sedation. The antiepileptic properties of talampanel indicate that the drug crosses the blood-brain barrier.
Agents that decrease aggregation have been hypothesized to be neuroprotective.180 Scriptaid was identified in a screen for small molecules that disrupt in vitro aggresome formation in cultured COS cells transfected with mutant SOD1-GFP. ONO-2506 is similar to valproate and works to restore normal astrocyte functions after brain damage. This agent has additional antiglutamate and antiinflammatory cyclooxygenase-2 (COX-2) inhibitor properties.
The role of apoptosis in motor neuron degeneration is increasingly recognized. Minocycline is a second-generation tetracycline antibiotic that prevents microglial activation. Typical side effects include gastrointestinal upset, vertigo, and cumulative dose-dependent photosensitivity. Therapeutic manipulation of the programmed cell death pathway could represent one way of improving the disease course. However, concern has been expressed that inhibition of apoptosis could preserve viability of motor neurons that are severely injured and dysfunctional. The best approach might therefore be to combine treatment inhibiting apoptosis with other approaches that target upstream cellular pathways of motor neuron injury.
Memantine is an amino adamantine derivative licensed as a neuroprotective agent for Alzheimer’s disease. The drug penetrates the CNS and is well tolerated by individuals with Alzheimer’s disease; there is limited study of its use in individuals with ALS.
Myotrophin (insulin-like growth factor I [IGF-I]) appears to affect motor dysfunction by promoting the survival of motor neurons and regeneration of motor nerves.76
The high metabolic load of motor neurons and the consequent dependence of these cells on oxidative phosphorylation may make them particularly vulnerable to the loss of mitochondrial function. Coenzyme Q10 is an antioxidant and an essential mitochondrial cofactor facilitating electron transfer in the respiratory chain. This commonly used nutraceutical is being tested in neurodegenerative conditions in which mitochondrial dysfunction has been implicated, including ALS. Dosages up to 3000 mg/day are safe and well tolerated in individuals with ALS. Coenzyme Q10 is lipophilic and effectively crosses the blood-brain barrier. Use of vitamin E as an antioxidant early in the course has been advocated. The impetus for studying and treating individuals with antioxidant therapy is its role in protecting against motor neuron injury.166
A ketogenic diet similar to the one employed to control epilepsy may be of some effect as ketones have the ability to alter mitochondrial function and have a positive effect in ALS based on animal studies. However, the diet has yet to be tested in human beings. Researchers have begun studies in individuals with ALS to determine whether similar findings can be demonstrated in human beings. With the possible advent of ketone esters for oral administration, clinical trials of dietary supplements in ALS might be possible in the near future.167
Oxidative stress appears to put people at risk for ALS comparable to that found in those with Alzheimer’s disease. People at risk for ALS might be identified by use of bioassays showing increased oxidative damage comparable to that in individuals with diagnosed disease. These assays could lead to early intervention and primary prevention.
Although no medication can stop the disease, much can be done in the form of symptomatic therapy. Health care providers should emphasize the value of maintaining the highest level of function throughout the course of the disease, providing education and support to prepare for the rapid decline in function. Symptomatic measures may include the use of anticholinergic drugs to control drooling and baclofen or diazepam to control spasticity. Dextromethorphan, a drug long used for cough suppression, has been effective in controlling the tearfulness that comes with pseudobulbar involvement.168
Maintenance of nutrition is a significant problem because of the difficulty chewing and swallowing. Weakness of jaw movement, loss of tongue mobility, and difficulty in lip closure, in addition to impairment of the swallowing reflex, are common. This may lead to respiratory complications from aspiration. By modifying the consistency and texture of food and fluids, the risk of aspiration is reduced.6
There is a shift in the care of individuals with ALS toward the use of multidisciplinary ALS clinics to provide coordinated care. Survival has been found to be longer for individuals with bulbar symptoms, the use of aids and appliances was greater, and the mental quality of life was better for the individuals with ALS treated at the multidisciplinary clinics than for individuals who do not receive specialty clinic care. With focused care, up to 80% of individual with ALS can die at home.54
PROGNOSIS.
The course of ALS is relentlessly progressive. It appears that earlier onset (less than 50 years of age) has a longer course. Death from the adult-onset sporadic type usually occurs within 2 to 5 years, resulting mainly from pneumonia caused by respiratory compromise. In general, those with bulbar palsy have a more rapid course than those with primary lateral sclerosis, in whom the prognosis is markedly better. Respiratory failure and inability to eat are part of the final stages of ALS. Nasogastric tube feeding and use of a respirator may be options to prolong the life of the client. Individual and family wishes concerning these procedures should be discussed as early as possible in the course of the disease, since some clients may experience a rapid decline in function at any time.
31-1 SPECIAL IMPLICATIONS FOR THE THERAPIST
Impaired Motor Function and Sensory Integrity Associated with Progressive Disorders of the Central Nervous System
The ALS-Specific Quality of Life Instrument (ALSSQOL) is based on the McGill Quality of Life Questionnaire (MQOL), modified by changes in format and by adding questions on religiousness and spirituality. A 59-item tool with a completion time averaging 15 minutes, it is a practical tool for the assessment of overall quality of life in individuals with ALS and appears to be valid and useful across large samples. Validation studies of a shortened version are now under way.164
The ALS Functional Rating Scale (ALSFRS-R), which can be found at and downloaded from http://www.alsconnection.com/ALSFRS.asp, is a functional scale that can be used to follow the progression of ALS. Six months are needed to detect changes in the ALSFRS-R score because of variability, due principally to differing rates of progression among patients.66
The relationship between verbal associative fluency, verbal abstract reasoning, and judgment in ALS can be evaluated using a 20-minute screening evaluation. Deficiencies in these measures were found in 20% to 35% of patients with limb-onset ALS and in 37% to 60% of patients with bulbar-onset ALS. This simple screen identifies deficits that affect discussions of treatment interventions and end-of-life issues.
Muscle strength declines in an overall linear progression throughout the course of the disease. Staging of ALS helps the therapist to determine the most effective intervention based on the current functional status and on the predicted progression of the disease. Table 31-1 describes interventions associated with the various stages of the disease.
Table 31-1
Exercise and Rehabilitation Programs for Clients with Amyotrophic Lateral Sclerosis
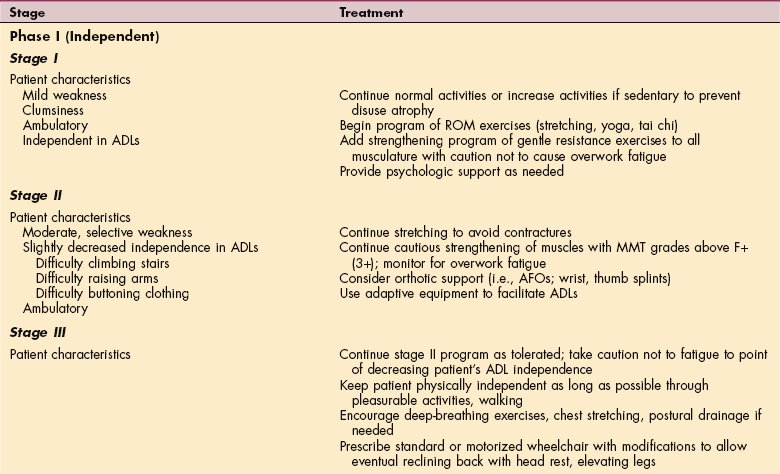

ADLs, Activities of daily living; ROM, range of motion; MMT, manual muscle test; AFOs, ankle-foot orthoses; HMV, home mechanical ventilation.
Modified from Sinaki M: Exercise and rehabilitation measures in amyotrophic lateral sclerosis. In Yase Y, Tsubaki T, eds: Amyotrophic lateral sclerosis: recent advances in research and treatment, Amsterdam, 1988, Elsevier.
The rate of loss is stable within a broad range after the first year, but during the first year there is fluctuation of strength that may be due to the potential for adaptation within the CNS. At this point, the goal of therapy is to maintain general physical activity and muscular tone. Regular exercise in moderation can help alleviate fatigue and have a beneficial effect on the client’s general well-being.17 Complaints of diffuse pain will start in the early stages, related to joint stiffness and decreases in muscle control in the limbs or trunk.
Spasticity contributes to complaints of weakness. Consistent slow stretching that decreases tone may be of benefit. The Ashworth scale can be used to measure the degree of spasticity. Cramps, which can be a source of pain, also respond to a daily stretching routine.
Changes in gait are significant, and gait analysis is necessary to assess the need for assistive devices. Falls caused by weakness are a major problem. Ankle dorsiflexion is lost before loss of strength in plantar flexion. Hamstring strength appears to correlate with walking, and the decrease parallels the loss of walking ability. Isometric muscle strength as a percentage of normal shows a dramatic decrease late in the course of the disease when fewer muscle fibers are available. This is when the greatest functional losses are noted. A surprisingly small amount of muscle activity is necessary across the joints to allow normal function of a joint. Some movement and joint stability are maintained until the degeneration causes atrophy of muscle activity to less than 20% of normal. The weakness and wasting often produce painful subluxation of the scapulohumeral joint, and the arm should be supported.3 Contractures should be routinely stretched, taking care to support the joints, since there is minimal control of muscle activity in the late stages. Complaints of pain may begin when the client is unable to shift weight in bed or in sitting. Changing the reclining angle of the bed or wheelchair or the position of the legs will give some relief. Caregivers need to be educated in this aspect of care.
Braces, other assistive devices, and motorized scooters or wheelchairs help to maintain mobility and freedom. Many upper extremity devices are available to maintain joint alignment at rest, to make daily activities easier to perform, and to support mobility when it is lost. Braces for the lower extremity can extend the time of upright walking, and braces for the back and neck can assist with head and trunk control. Pain is often a complaint brought to the therapist. Thermal modalities and transcutaneous electrical nerve stimulation can help the pain associated with muscle shortening, joint stiffness, and muscle cramping.
Evaluation of the home environment, providing rails, hoists, or supports; eliminating stairs where possible; and advising on helpful devices for feeding, shaving, dressing is essential as the individual becomes limited to household mobility. Posture for activities of daily living (ADLs) may be improved with a collar, a brace, or spring-loaded splints. Special beds can be leased or borrowed. Family and friends may organize a roster of people to sleep over, sparing the spouse from waking every 3 hours to turn the patient. The legs should be elevated and elastic stockings used if leg swelling is a problem. Avoid using diuretics for leg swelling.
Frustration and boredom are common. Family and volunteers can be mobilized from neighborhood groups, such as church or social groups, to visit to talk, listen, play cards, turn pages or read, or just to be there for a while.
Sexual frustration is common and is not often discussed. The clinician should do so freely and without embarrassment with both the patient and spouse; the problems are mainly matters of method. The partner may need counseling to understand that he or she needs to take the active role and to learn effective techniques that overcome weakness and muscle spasms.
Respiratory changes cause the most disability and eventually lead to death. Respiratory distress is a mechanical problem because of lack of muscle support. From the earliest stages of care of the client with ALS, prevention of respiratory complications should be emphasized. Early evidence of respiratory involvement may be shortness of breath, poor cough reflex, and headache. Some clients can be taught to use their abdominal muscles to increase inspiration and expiration when the muscles of the diaphragm and intercostal muscles become weak. Swallowing becomes difficult and should be evaluated by a speech pathologist. Pseudobulbar affect causing uncontrolled laughing or crying decreases the capacity to regulate breathing and increases risk of shortness of breath. Aspiration is common, and techniques to control this can be taught. Mechanical ventilation is an option to prolong the ability to breathe. Individuals with a relatively slow disease progression, and those with spinal onset, might benefit more from treatment with noninvasive ventilation than patients with rapid disease progression or bulbar onset. Noninvasive positive pressure ventilation (NIPPV) has been shown to improve patient quality of life, despite progression of ALS, and without increasing the caregiver burden or stress.75 In addition, suction, intermittent positive pressure breathing, and postural drainage appear to be useful in maintaining bronchial hygiene. Only 5% of individuals choose long-term, invasive ventilation because of the restriction of activity, caregiver involvement, and overall cost.99 Communication becomes limited, again because of loss of oral muscle control and breath support. Communication strategies can be taught, and augmentative equipment is available. In all cases the individual becomes dependent over time. In the terminal stages, the comfort of the patient is the therapeutic goal.
As patients with ALS weaken, the decisions facing the patient progress from issues of morbidity to mortality. Traditionally, the neurologist and other therapeutic support staff have deferred to the wishes of the patient and family members in determining level of support in response to progressive physical decline. Impairments in judgment have potentially significant clinical implications that should be considered by health care providers and caregivers when discussing treatment interventions and end-of-life issues with these patients.60
All patients with ALS and their families have to come to grips with the many end-of-life decisions that confront them. These include the need to get the many events in life in order, come to terms with relationships, and decide how forthcoming disabilities will be handled. Decisions regarding care at home versus in a nursing facility should be made as early as possible. Information about advance directives, living wills, and power of attorney should be available. Patients may raise the question of suicide or assisted suicide, and the caregivers should be comfortable not only talking about these issues but also calling on others who may have more expertise and experience in discussing these issues. It makes things more difficult for the patient and family if the caregivers avoid these sensitive areas and talk only about the disease and medical management. Psychologic and emotional support for the individual and the family is critical. A direct and informative approach is appreciated; giving false hope should be avoided.
ALZHEIMER’S DISEASE, ALZHEIMER’S DEMENTIA, AND VARIANTS
Overview and Definition
The two terms Alzheimer’s disease (AD) and Alzheimer’s dementia are related but not synonymous. AD is the disease process that ultimately results in Alzheimer’s dementia. Alzheimer’s dementia has a characteristic cognitive pattern. In some individuals, early in the disease course AD may cause memory loss of insufficient severity to warrant the designation of dementia. In other individuals, AD may follow an atypical course with progressive aphasia or progressive apraxia rather than a typical Alzheimer’s dementia. Most of the time AD causes Alzheimer’s dementia.
Dementia is a term for a decline in intellectual functioning severe enough to interfere with a person’s relationships and ability to carry out daily activities. A significant decline in memory is a hallmark of dementia but is not the only characteristic. Age-associated memory impairment, or benign senescent forgetfulness, is a decline in short-term memory that does not progress to other mental or intellectual impairments. Other causes of dementia must be carefully ruled out, and there are syndromes that mimic AD in relationship to the dementia but have different neurologic causes. Listed here are a few of the other dementias that cause change in cognitive status.
Pick’s Disease.: Much less common and sometime clinically indistinguishable from AD, Pick’s disease is characterized by cortical atrophy involving predominantly the frontal and temporal regions with sparing of the posterior two thirds. Loss of frontal inhibition of socially unacceptable and previously suppressed behavior emerges early in the disease, often overshadowing the memory disturbance. The inclusions are known as Pick bodies. The neurons balloon in the area of involved tissue, but there are not the plaques or tangles seen in AD.
Lewy Body Dementia.: This disorder exhibits highly variable clinical and neuropathologic overlap with AD and Parkinson’s disease. It is characterized by initial parkinsonism unresponsive to standard medications, progressing to deterioration of cognition. Cellular changes include presence of the Lewy bodies found in Parkinson’s disease and neurofibrillary tangles, senile plaques, and granulovacuolar degeneration similar to those in AD.
Corticobasal ganglionic degeneration is characterized by striking asymmetrical gait and speed apraxia, “alien hand” syndrome, rigidity, myoclonus, and cortical sensory loss. Dementia is usually a late manifestation of the disease.
Frontotemporal Dementia.: This term is used to describe the various progressive disorders that have a predilection for the frontal lobes. The cellular neuropathology is variable, and in some cases it seems to be the frontal lobe manifestations of AD, Pick’s disease, and Lewy body dementia.
AD is the most common cause of dementia overall. It is one of the principal causes of disability and decreased quality of life among older adults.3 Progress in clinical knowledge of AD has led to more reliable diagnostic criteria and accuracy; the earliest manifestations and even the presymptomatic phases of the disease may soon be identifiable.
Incidence and Etiologic and Risk Factors
There are approximately 4 to 4.5 million people with AD in the United States and 8 million affected around the world. Barring a cure, by 2050 this number will increase by almost threefold, to 13.2 million. The prevalence of AD rises with each decade of age. The known prevalence is 6% in people over 65 years of age, 20% in people over 80 years of age, and more than 95% in those 95 years of age.193 Because life expectancy continues to rise, so does the potential for more individuals to be afflicted. It is believed that many individuals with the symptoms go undiagnosed and untreated. The cause of AD remains unknown, but there appears to be a relationship among genetic predisposition; the abnormal processing of a normal cellular substance, amyloid; and advanced age.87 Lifetime risk of developing AD is estimated to be between 12% and 17%. Twin studies show evidence that in identical twins, one may develop AD while the other remains dementia free. People with a family history of the disease are at higher than average risk for AD. Researchers are identifying important genetic factors, notably the apolipoprotein E ε4 (ApoE4) gene.
The ApoE ε2 allele (ApoE2) may be protective, and the ApoE ε4 allele (ApoE4) is associated with increased risk. The amyloid precursor protein (APP) gene is located on chromosome 21. Studies have reported the greatest deposits of β-amyloid in people with ApoE4, which is now believed to be a major risk factor for late-onset AD. Some evidence suggests that the ApoE protein removes β-amyloid but that the ApoE4 variant does so less efficiently than other ApoE types.
People inherit a copy of one type of allele from each parent, but AD is not inevitable, even in people with two copies of the ApoE4 allele. People without ApoE4 have an estimated risk of between 9% and 20% for developing AD by age 85. In people with one copy of the gene, the risk is between 25% and 60%. In people with two copies, the risk ranges from 50% to 90%. But only 2% of the population carries two copies of the ApoE4 allele.
Genetic mutations in the genes that control APP are also being targeted as causes of early-onset AD. In the genetic disease Down syndrome, for example, β-APP, the source of β-amyloid, is overproduced, which almost always leads to early AD.
Mutations in genes known as presenilin 1 (PS1) and presenelin 2 (PS2) account for most cases of early-onset inherited AD. The defective genes appear to accelerate β-amyloid plaque formation and apoptosis, a natural process by which cells self-destruct.
Mutations of these and other genes have been identified and provide strong support for the “amyloid cascade hypothesis” of AD (Fig. 31-3). Although the amyloid cascade is currently considered by many researchers as a key contributor to the pathogenesis of AD, some researchers have challenged this assertion and have proposed that β-amyloid occurs secondary to neuron stress and func tions as a protective adaptation to the disease rather than causing cell death.
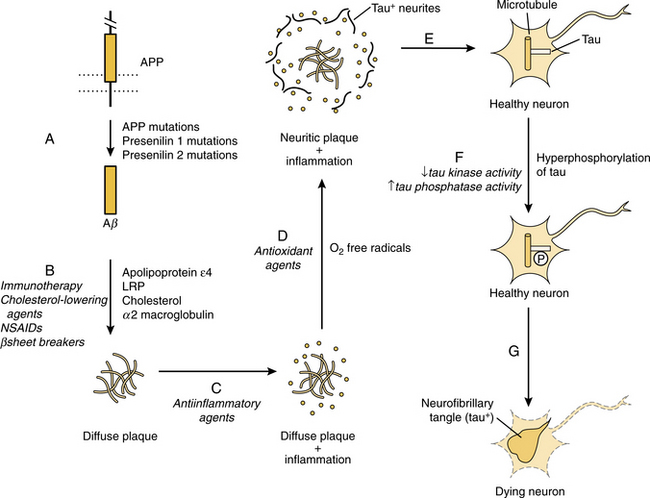
Figure 31-3 The amyloid cascade hypothesis of Alzheimer’s disease pathogenesis and potential therapeutic targets. (In Goetz CG: Textbook of clinical neurology, ed 3, Philadelphia, 2007, Saunders.)
The same genes may have different effects depending on the ethnic population. Dietary and other cultural factors that increase the risk for hypertension and unhealthy cholesterol levels may also play a role. For example, a study of Japanese men showed that their risk increased if they emigrated to America. And the disease is much less common in West Africa than in African Americans, whose risk is the same as or higher than that of Caucasians Americans.
Some studies have reported an association between AD and systolic hypertension (the higher and first number in blood pressure measurement). Furthermore, some studies report a lower risk for AD in individuals whose blood pressure was reduced. Nevertheless, although hypertension is strongly linked to memory and mental difficulties, stronger evidence is needed to prove any causal relationship between hypertension and AD.
There has been research suggesting an association between high cholesterol levels and AD in some people. A number of recent studies support the link between AD and cholesterol by suggesting that certain cholesterol-lowing drugs known as statins may be protective against AD. The ApoE genotype is linked with both atherosclerosis and AD. The ApoE4 genotype reflects abnormal cholesterol transport.191
Box 31-4 outlines some of the key risk factors as well as possible protective factors related to AD.
Pathogenesis
Like other degenerative conditions, AD has no single identified cause. The loss of neurons is thought to be due to the breakdown of several processes necessary for sustaining brain cells. There are several neuropathologic hallmarks of AD. There is progressive accumulation of insoluble fibrous material, amyloid. Senile plaques consisting of extracellular amyloid are found in higher concentrations in the brains of individuals with AD than in normal aging brains. The amyloid deposition appears to have a relationship to β-amyloid protein, a natural substance that is required to maintain fibroblasts and cell function. Components of this protein occur typically as a by-product of neuron function. Normally, the β-amyloid dissolves and is reabsorbed by the brain tissue. When it remains in the fluid surrounding the neurons, the β-amyloid protein may deform its shape by folding in on itself. This abnormal protein then sticks together with other β-amyloid material, forming a sheet of connected proteins; the result is a plaque. The amyloid plaque also includes fragmented axons, altered glial cells, and cellular debris. This plaque triggers an inflammatory response, resulting in increased free radicals that cause damage to the nervous system. Other inflammatory factors of specific interest in Alzheimer’s research are the enzyme COX and its products, called prostaglandins.
APP is a large nerve-protecting protein that is the source of β-amyloid. In AD certain enzymes, particularly those called γ-secretases, snip APP into β-amyloid pieces. This process is controlled by presenilin proteins. Genetic abnormalities that affect either APP or presenilin proteins occur in some inherited cases of early-onset AD. Another important protein in the areas of the brain affected by AD is endoplasmic reticulum–associated binding protein (ERAB), and it appears to combine with β-amyloid, which in turn attracts new β-amyloid from outside the cells. High amounts of ERAB may also enhance the toxicity of β-amyloid.
It appears that the amyloid plaque, when it comes in contact with a neuron, causes chemical changes that may lead to the destruction and destabilization of microtubules, the structural components of the neural cells. A protein molecule called tau normally responsible for holding the microtubules together detaches and causes the microtubule to disintegrate. This process may be due to an enzyme that escapes its normal restraints and breaks down the tau. As the microtubule disintegrates, neurofibrillary tangles form and remain in the system. The overall effects are decreased cell division and loss of axonal transport of neurotransmitters. Fig. 31-4 shows a typical neuritic plaque and neurofibrillary tangle.
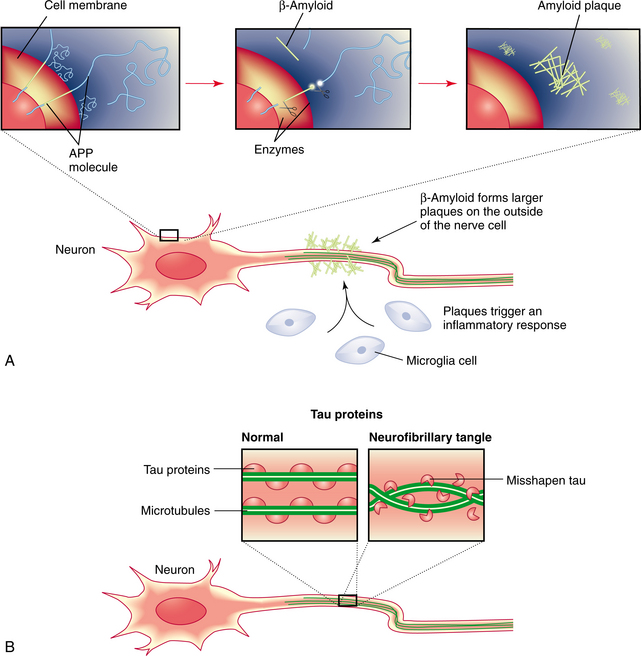
Figure 31-4 Current etiologic theories for the development of Alzheimer’s disease. A, Abnormal amounts of β-amyloid are cleaved from the amyloid precursor protein (APP) and released into the circulation. The β-amyloid fragments come together in clumps to form plaques that attach to the neuron. Microglia react to the plaque, and an inflammatory response results. B, Tau proteins provide structural support for the neuron microtubules. Chemical changes in the neuron produce structural changes in tau proteins. This results in twisting and tangling (neurofibrillary tangles). (From Lewis SM: Medicalsurgical nursing (single volume): assessment and management of clinical problems, ed 7, St. Louis, 2007, Mosby Elsevier Health Science.)
AD is characterized by disruptions in multiple major neurotransmitters, of which cholinergic abnormalities are the most prominent. Acetylcholine is an important neurotransmitter in areas of the brain involved in memory formation, and loss of acetylcholine activity correlates with the severity of AD. The reduction in the number of acetylcholine receptors precedes other pathologic changes, and these receptors are reduced significantly in late AD, particularly in the basal forebrain. There is selective loss of nicotinic receptor subtypes in the hippocampus and cortex. Presynaptic nicotinic receptors control the release of neurotransmitters important for memory and mood, such as acetylcholine, glutamate, serotonin, and norepinephrine. There is still some question whether this plays a primary role in the disease or is a secondary reaction.
Synaptic loss is the best pathologic correlate of cognitive decline, and synaptic dysfunction is evident long before synapses and neurons are lost. Once synaptic function stops, despite the number of surviving neurons, there may be little chance of changing the disease process.
Glutaminergic neurons appear to be prone to formation of neurofibrillary tangles. It appears that the most vulnerable group of cortical neurons is the pyramidal cells with corticocortical and hippocampal projections. Other subgroups of neurons are resistant to the degenerative process, such as the projections from primary sensory to adjacent secondary sensory areas. Increased excitotoxicity due to increased glutamatergic stimulation of N-methyl-D-aspartate (NMDA) receptors results in abnormally high levels of intracellular calcium and may ultimately lead to cell death.
There appears to be a hierarchy of cortical connection systems that are affected differently during the course of AD. A progression of the neurofibrillary tangles seems to move from the entorhinal cortex and hippocampus to the limbic system to the cortex, including the frontal, temporal parietal, and occipital cortices. One study suggests that the effect of AD on hippocampal volume equals the effect of roughly 17 years of aging.189 This may correlate with the changes seen in memory, behavior, and motor skills as the disease progresses. The distribution of the lesions in the cerebral cortex may be different in AD compared with that in other disease processes that cause dementia.
Cerebral amyloid angiopathy (CAA) may predispose an individual to develop AD. CAA is an important feature of senile dementia and AD along with senile plaques, neurofibrillary tangles, neutrophil threads, and synapse loss. Amyloid gradually causes atrophy of the medial smooth muscle cells of the arteries of the brain that weakens them, causing predisposition to hemorrhage.135 There appears to be a relationship between strokes and AD. (See Chapter 32.) Abnormalities have been reported in fibroblasts, red and white blood cells, and platelets. Alterations in blood proteins have been observed. The role of these peripheral changes is not clear.107
Clinical Manifestations
The early symptoms of AD may be overlooked because they resemble signs of natural aging. Still, older adults who begin to notice a persistent mild memory loss for recent events may have a condition called mild cognitive impairment (MCI) MCI is now believed to be a significant sign of early-stage AD in older people. Studies now suggest that older individuals who experience such mild memory abnormalities convert to AD at a rate of about 10% to 15% per year.
Disorders of function are found in the person with AD that correlate with the level of damage in the various components of the cortex as described earlier. Visuospatial deficits are an early clinical finding. Navigating the environment, cooking, and fixing or manipulating mechanical objects in the home are all visuospatial tasks that often are impaired in the first stages of AD. Drawing is abnormal; the ability to draw a three-dimensional object is often lost. The loss of ability to solve mathematical problems and handle money is typical in the early stages of AD. Judgment is impaired, and safety in driving is diminished.
Subtle personality changes occur in AD, such as indifference, egocentricity, impulsivity, and irritability. People with AD become withdrawn and anxious. Memory is affected, and this is seen as inability to recall current events. Studies show that particular memory subsystems are relatively more or less vulnerable to diffuse cortical pathologies.31 People with AD seem to retain higher capacity in implicit memory than was originally thought. AD causes loss of older memories, and recall of events from early life disappears. Language declines in a characteristic progression. Word-finding difficulty is first, followed by inability to remember names (anomia), and finally diminished comprehension. Social situations become difficult, and mood swings are common.119 Between 40% and 60% of individuals with late-onset AD suffer from psychotic symptoms, which may include hallucinations, delusions, and dramatic verbal, emotional or physical outbursts. This is a severe form of AD, with a genetic basis, that has a more rapid and aggressive course. Table 31-2 describes the difference between normal aging and AD.
Table 31-2
Differences Between Normal Signs of Aging and Dementia
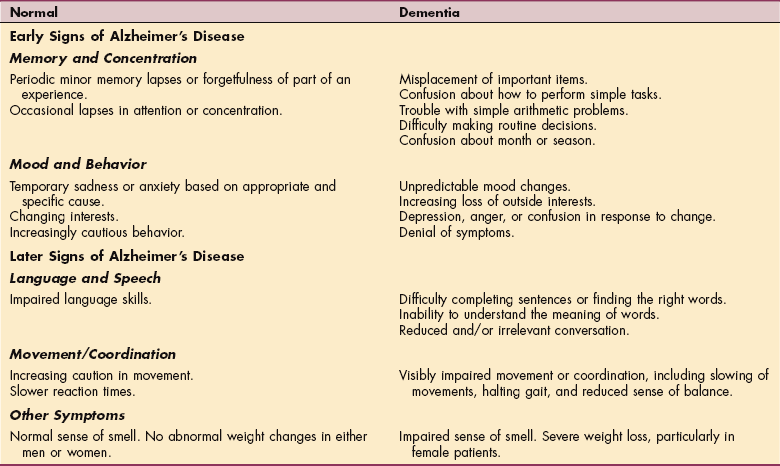
Data from Junior League of the City of New York: Alzheimer’s disease: early warning signs and diagnostic resources, New York, 1988, The League.
Major depression is uncommon, but many persons with AD have periods of depressed mood associated with feelings of inadequacy and hopelessness. AD-associated depression is often more modifiable by environmental manipulation than depressions not associated with AD. As AD progresses, delusions, agitation, and even violence may occur.
Abnormal motor signs are common, related to the area of the brain that is involved, and perhaps due to the type of neurotransmitter dysfunction. A relationship between the motor impairments and levels of function can be seen. Presence of tremor appears to be associated with increased risk for cognitive decline, presence of bradykinesia with increased risk for functional decline, and presence of postural-gait impairments with increased risk for institutionalization and death. This may reflect need for assistance with mobility and number of falls. See Special Implications for the Therapist: Alzheimer’s Disease later in the chapter.
Disorders of sleep, eating, and sexual behavior are common. The electroencephalogram (EEG) shows more awake time in bed, longer latencies to rapid eye movement sleep, and losses in slow-wave sleep.121
MEDICAL MANAGEMENT
The most important diagnostic step in evaluating dementias is to determine whether a chronic encephalopathy results from a potentially reversible cause. Interaction of multiple medications can also trigger dementia and should be assessed.
A decline from previous levels of functioning and impairment in multiple cognitive domains beyond memory are critical in establishing dementia. Determining the rate of change is useful, since abrupt changes are not consistent with AD.18 The progression is usually continuous and does not fluctuate or improve. Information obtained from family members or caregivers can provide data when there seems to be lack of insight from the client. The Functional Activities Questionnaire is a useful informant-based measure.
Clinical screening tests, such as the Short Test of Mental Status, the Mini-Mental State Examination (MMSE), and Mattis Dementia Rating Scale provide a baseline for monitoring the course of cognitive impairment over time and document multiple cognitive impairments.3
A clock drawing test is also a good test for AD. The individual is given a piece of paper with a circle on it and is first asked to write the numbers in the face of a clock and then to show “10 minutes after 11.” The score is based on spacing between the numbers and the positions of the hands.
Neuropsychologic tests can accurately predict the probability of conversion to incident AD after 5 or 10 years.55,177 Clues on physical examination include a variety of findings that may be common in elderly individuals but are not part of the typical picture of AD, such as ataxia, hyperreflexia, and tremulousness.
Depression can be difficult to distinguish from dementia, and it can coexist with dementia. Changes in memory, attention, and the ability to make and carry out plans suggest depression, the most common psychiatric illness in older persons. Marked visuospatial or language impairment suggests a dementing process. Depression scales can be used to determine levels of depression.
Ruling out a partially or completely reversible dementia by performing a blood count, chest radiography, and general neurologic examination is critical in the diagnostic evaluation of a person with suspected AD. Autoimmune and paraneoplastic serologic studies may be helpful in such individuals as well.
Use of neuroimaging can be beneficial in the diagnosis of AD. Both magnetic resonance imaging (MRI) and computed tomography (CT) can identify the changes in brain size that are associated with AD. Diagnostic criteria are based on the measurement of medial temporal lobe atrophy or on the volumetric measurement of the entorhinal cortex and hippocampus.15 The brain demonstrates atrophy with normal aging, so this is not the only diagnostic test.
Single-photon emission computed tomography (SPECT) can be used to determine brain activity, especially in areas where information is processed for memory functions. This may be used in the future to predict potential for development of AD.
Researchers are looking at different components of the human brain cell to identify molecular changes in deoxyribonucleic acid (DNA) and RNA seen in individuals with dementia and AD. Approaches are widespread, since it is clear that the disease is multifactorial. The National Institute on Aging (NIA) held a consensus conference in 1998 with the creation of an NIA Reagan profile that requires that neuropathologic assessment include assessment of both plaques and neurofibrillary tangles.
TREATMENT.
There is currently no cure for AD. Current treatment focuses on establishing an early accurate clinical diagnosis, early institution of cholinesterase inhibitors and/or NMDA receptor–targeted therapy. Treating medical comorbidities and dementia-related complications, ensuring that appropriate services are provided, addressing the long-term well-being of caregivers, and treating behavioral and psychologic symptoms with appropriate nonpharmacologic and pharmacologic interventions also are important.109
Current drugs approved for treatment of AD have modest symptomatic benefits but do not have profound disease-modifying effects. Disease-modification approaches including neuroprotection are now the most active area of investigation, with focus on antiamyloid treatment. Oxidative stress and cell cycle–related abnormalities are early events in AD, occurring before any cytopathology can be identified, and together may create disease pathogenesis. Therefore, antioxidants are an AD prevention strategy under investigation. Inflammation and activation of microglia is a relatively early pathogenic event that precedes the process of neuron destruction in AD. Therefore, despite the early negative results of clinical trials with nonsteroidal antiinflammatory drugs (NSAIDs) for the treatment of AD, these and other antiinflammatory agents may still have a role in reducing the risk for AD. Modulation of cardiovascular risk factors may also reduce the risk for AD. Although hormone replacement therapy with estrogen showed no benefit and even a potential deleterious effect in individuals with AD, estrogen may still have a role in reducing the risk for AD if given early in menopause and when neurons are in a healthy state. Other neurodegenerative processes, such as excitotoxicity and apoptosis, may also have a patho- physiologic role in AD and are now under study. Medications currently in use are outlined in Box 31-5.
Treatment oriented at preventing the breakdown of tau, or the formation of plaques, is being tested now and shows promise. The treatment of those persons identified as at high risk may someday be protective gene therapy. Future drug therapies may be targeted at specific cognitive modules.
Management of the client with AD is a challenge to health providers and to the family who become caregivers. Manipulation of the environment can be effective. It is difficult to manage aggressive behavior in the home, and long-term care in a facility with a special Alzheimer’s unit is often the most appropriate place for that client.
A management model for AD that incorporates a diagnostic protocol to identify and assess people with possible dementia and care management addressing individual function, caregiver support, medical treatment, psychosocial needs, nutritional needs, and advance directives planning is critical. To improve end-of-life care for people with AD, any treatment model should also incorporate patient-centered care and palliative care from the initial diagnosis of AD though its terminal stages. Short-term intensive counseling can significantly reduce the long-term risk for depression among those who care for spouses or partners with AD.50
In many individuals with AD, treating comorbid conditions such as depression, hearing or vision impairment, congestive heart failure, symptomatic urinary tract infection, or hypothyroidism may produce a greater benefit than focusing treatment only on AD. Cardiovascular disease may influence the expression and clinical manifestations of the disease.
There is compelling evidence for the important role of regular physical activity.115 Exercise training combined with behavioral management techniques can improve physical health and depression in individuals with AD. Leisure-time physical activity at midlife is associated with a decreased risk of dementia and AD later in life. Regular physical activity may reduce the risk or delay the onset of dementia and AD, especially among genetically susceptible individuals.155
Diet in midlife shows potential for neuroprotection, and findings can be generalized to a combination of the following: consumption of a diet low in fat, high in omega-3 oils, and high in dark vegetables and fruits; use of soy (for women only); supplementation with vitamin C, coenzyme Q10, and folate; and moderate alcohol intake. It appears that no single item creates the protection, but the foods and supplements may work together to lower risk.
There is growing evidence indicating that oxidative damage caused by the β-amyloid peptide in the pathogenesis of AD may be hydrogen peroxide (H2O2) mediated. Polyphenols from apple and citrus juices, such as quercetin, are able to cross the blood-brain barrier and show neuroprotection against H2O2. The effect of polyphenols from citrus is similar to that of vitamin C, but quercetin from apple juice gives stronger neuroprotection than vitamin C. In addition to their antioxidant properties, many polyphenols, such as quercetin, have potent antiinflammatory properties. In addition to antioxidant vitamins and polyphenols, fruit and vegetable juices also may possess other protective components, such as folate and minerals.45
The mutations in APP, presenilin 1, and presenilin 2 allow genetic screening to be used in suspected cases of familial AD with early onset and for appropriate genetic counseling and support. Although preimplantation genetic diagnosis (PGD) of the embryo, prenatal diagnosis, preimplantation embryo selection, and presymptomatic testing have been offered to families of individuals who have early-onset familial AD, complex legal and ethical issues surrounding these interventions must be addressed before these interventions can be routinely recommended.
PROGNOSIS.
AD is the fourth leading cause of death in adults. The period from onset to death typically is 7 to 11 years. Initially, deficits in higher cortical function are the most noticeable. Motor signs may reflect higher burden or different type or more biologically detrimental localization of neuropathology. The association of different aspects of motor signs with different outcomes may reflect varying underlying neurotransmitter systems being affected. For example, in Parkinson’s disease, tremor and bradykinesia have been viewed as representing more purely dopaminergic manifestations, while posture, balance, and gait disorders may be mediated by other neurotransmitter systems in addition to dopamine. Changes caused by the dementia may advance relentlessly over many years, creating not only deep emotional and psychologic distress but also practical problems related to caregiving that can overwhelm affected families. During the middle stages of the disease, the client often develops behavioral and motor problems. Finally, the client becomes mute and unable to comprehend. Death is often secondary to dehydration or infection.
DYSTONIA
Definition and Overview
Dystonia is a neurologic syndrome dominated by involuntary, sustained muscle contractions frequently causing twisting and repetitive movements. These abnormal postures are often exacerbated when the person performs active voluntary movements.
Traditionally, dystonia was classified according to type, including primary (idiopathic or of unknown cause) or secondary (occurring as a result of injury or other brain illness). These classifications are still reported in the literature, but the current classification scheme for dystonia now describes the disorder in each person according to three separate categories: age of onset, distribution of symptoms, and etiology. A second type of classification is by body involvement. In focal dystonia one body area is affected, in segmental dystonia two or more body areas are involved, and generalized dystonia is wider spread. There has been some confusion in the literature regarding the name cervical dystonia. This neurologically based movement disorder affecting the head and neck is a separate entity from spasmodic torticollis. Torticollis is a musculoskeletal phenomenon treated as an orthopedic condition (see discussion of torticollis in Chapter 23).
Incidence
An estimated 250,000 persons are afflicted with dystonia in North America. About 1.1 in 100,000 persons per year develop dystonia, with a female to male ratio of 1.6: 1.53 The Mayo Clinic in Rochester found 3.4 cases of primary generalized dystonia and 29.5 cases of focal dystonia per 100,000.133 Focal dystonia is estimated to be six times more common than other well-known neuromuscular disorders such as muscular dystrophy, Huntington’s disease, and ALS.
The average age of onset of idiopathic dystonia is 8 years. For focal dystonias, the age of onset is between 30 and 50 years.
Etiologic Factors
Idiopathic, or primary, dystonia is the most common diagnosis, accounting for two thirds of all cases. A genetic basis on the DYT1 gene locus is responsible for causing primary torsion dystonia.22 It appears that persons with generalized dystonia carry a different gene than those with focal dystonias.56 Another inherited dystonia is dopa-responsive dystonia, or Segawa’s dystonia.
Secondary dystonia is the result of small areas of brain damage or scarring of the CNS. The changes have been attributed to drugs, infections, tumors, and demyelinating processes as well as acute trauma, such as caused by auto accidents. Box 31-7 lists the major causes of secondary dystonia.
Focal dystonias involving hand function are particularly common among those in certain occupational groups, such as keyboard operators and musicians. Writer’s cramp is also a focal dystonia. Focal dystonia related to occupational cramps may be a result of abnormal or repetitive biomechanics. Focal dystonia involving the hand may also occur as part of a peripheral nerve disorder.
Drug-induced extrapyramidal symptoms may include dystonia as a common side effect associated with antipsychotic drugs (neuroleptics). This results in various acute and chronic manifestations of neuroleptic-induced dystonia (e.g., blepharospasm [difficulty in opening the eyelids], torticollis or retrocollis [involuntary extension of the neck]).83 The fact that β-blocking agents are effective in reducing symptoms in these cases points to the possibility that neuroleptic drugs increase the activity of β-adrenergic transmitters.
Pathogenesis
Descending pathways involving reciprocal inhibition of motor neurons have been identified as possible sites of pathogenesis in idiopathic dystonia. Nerve conduction velocity studies have shown that there is a failure of neural activities preparing for movement. Defective retrieval of specific motor programs in response to sensory stimuli results in co-contraction of both agonist and antagonist muscles around a joint.154,186 In focal dystonia of the hand, there is somatosensory degradation in the involved hand, with graphesthesia and astereognosis.34
Inherited dystonia mapped to chromosome 9 is probably the result of a protein that affects the function of certain nerve cells. The DYT1 and DYT6 genes have been associated with dystonia.57 The exact malfunction remains unknown, but it may be that a protein is missing or is produced in excess.
The cause of symptomatic, or secondary, dystonia is thought to be chemical dysfunction or scarring within the striatum (caudate nucleus and putamen).69 Overactivity of the direct pathway within the basal ganglia loop (cortex–striatum–internal globus pallidus–thalamus–premotor/motor cortex) is speculated to result in an overflow of motor cortex activity, thus creating the dystonic movements. A defect in the body’s ability to process inhibitory neurotransmitters such as γ-aminobutyric acid (GABA), dopamine, acetylcholine, norepinephrine, and serotonin may contribute to poor inhibition of motor control. Dysfunction of the lenticulothalamic neuronal circuit seems to be related to the development of dystonia following head trauma.108
Recent research suggests that the somatosensory cortex may also function abnormally, contributing to the altered motor output.33,69,90,91,178 Both somatosensory cortex and somatotopic representation at the thalamus degrade in individuals with dystonia.111
The cerebellum has also been implicated using functional MRI. Theoretically, increased activity in the cerebellum results in the excessive movement and dystonic posturing.68,86
Clinical Manifestations
Cervical dystonia is the most common focal dystonia and is characterized by rotation of the neck, lateral flexion, and flexion and extension occurring in various combinations. The condition is usually painful, is disruptive to functional activity, and leads to osteoarthritis and hypertrophy of the sternocleidomastoid muscle if remission does not occur. Dystonia-induced cervical fracture has been reported.
Writer’s or occupational cramp is a form of dystonia that can be particularly disabling, resulting in deterioration of handwriting or fine motor control. The fingers and wrist flex excessively, causing the hand to grasp a pen tightly and press unnecessarily hard on the paper. Another type of cramp results in extension of the fingers, making it difficult to hold a pen. Tremor or myoclonic jerks may occur while writing or trying to play a musical instrument. Lower extremity dystonia is common, including dystonic movements in the foot and toes.
Blepharospasm is uncontrolled blinking or closure of the eyelids for seconds to hours. In oromandibular dystonia, face and jaw muscles contract, causing grimaces or facial distortions, and dysphonia affects the speech muscles of the throat, causing strained, forced, or breathy speech.
Involvement of the respiratory muscles has been considered unusual but may in fact be underestimated, either because it is not conspicuous or because the problem is improperly attributed to another cause. Clinical manifestations of respiratory involvement may include involuntary deep and loud inspirations combined with dystonia, breathing arrests, or broken speech caused by deep inspirations when speaking or reading aloud.105
Dystonia usually is present continually throughout the day whenever the affected body part is used. In more severe cases, the dystonia can appear at rest. The symptoms may begin in one area only with a particular movement. For example, it may be apparent when walking forward but not when walking backward or the foot may turn under after walking or other exercise, causing the person to walk on the lateral border of the foot.
MEDICAL MANAGEMENT
Dystonia is a clinical diagnosis except for those cases that have a genetic basis. Testing for genetic forms of dystonia appears to be most appropriate for people under the age of 26.93 Otherwise, there is no definitive test for dystonia, and the diagnosis of idiopathic dystonia is often delayed 1 year or more. The clinical presentation of dystonic movements, such as head deviation or neck pain, may be the first diagnostic sign. The person usually has a normal perinatal and developmental history. EMG studies show sustained simultaneous contractions of agonists and antagonists. Determining that there is no evidence for symptomatic, or secondary, dystonia is essential in the diagnosis of idiopathic dystonia (see Box 31-7).
TREATMENT.
Treatment remains symptomatic and includes drug therapy, including botulinum toxin type A (BTX) injections, physical and occupational therapy, and sometimes surgery.
Anticholinergics such as trihexyphenidyl (Artane) have been the most widely used medications to decrease acetylcholine and correct a cholinergic imbalance in the basal ganglia. Side effects of these drugs vary, with blurred vision, dry mouth, confusion, voiding, sleeping difficulties, and personality changes observed.
Baclofen and other muscles relaxants are used occasionally for relief.
BTX, injected intramuscularly, has emerged as a safe and effective symptomatic treatment for a number of conditions associated with excessive muscle activity, par ticularly focal dystonias involving a limited number of muscles. These injections are effective in improving postural deviation and pain in about 80% to 90% of people with cervical dystonias.84,85 Injection directly into the actively contracting muscles blocks the neuromuscular junction by acting presynaptically to reduce the release of acetylcholine, producing a chemical denervation. Muscle weakness can result from this treatment. Response occurs in 3 to 7 days and lasts 3 to 4 months. Dysphagia is the most serious side effect but can be decreased in incidence and severity by injecting lower doses, particularly into the sternocleidomastoid. The need to continue indefinitely with repeat injections approximately every 3 months is a major drawback.
Surgery may be considered only when other treatments are no longer effective, although surgical intervention may also lose its effect over time, providing only temporary symptomatic relief. Surgeries to interrupt the pathways or foci responsible for the abnormal movements can be effective. Thalamotomy is the destruction of a portion of the thalamus. Pallidotomy is a destructive operation on the globus pallidus. Pallidus stimulation is achieved by placing an electric stimulator in the globus pallidus. Rhizotomy involves the surgical resection of the anterior cervical spinal nerve roots and is used along with selective peripheral denervation, or removal of the nerves at the point where they enter the contracting muscles.
PROGNOSIS.
Age of onset is the best predictor of prognosis. If dystonia starts in childhood and affects other members of the family, it tends to get progressively worse over the years. If the condition starts in childhood and is secondary to cerebral palsy or other brain injury close to the time of birth, the dystonia tends to remain static for many years. In one third of cases of adult-onset focal dystonia there is progression to segmental dystonia, although there is less than a 20% chance that the disease will progress to generalized dystonia.
Spontaneous remission occurs in 30% of cases within the first year, but the majority of clients show steady progression of their focal dystonia, with maximal disability occurring after 5 years. Neck pain, occurring in 70% to 80% of clients, contributes significantly to disability. Cervical dystonia has important psychosocial consequences, since many people with this condition withdraw from their jobs and social activities.190
HUNTINGTON’S DISEASE
Overview and Definition
Huntington’s disease (HD) is a progressive hereditary disorder characterized by abnormalities of movement, personality disturbances, and dementia. Known also as Huntington’s chorea, it is most often associated with choreic movement that is brief, purposeless, involuntary, and random. However, the disease course involves more than just a movement disorder, and hence the name Huntington’s disease. HD is a disorder of the CNS and is classified as a neurologic disorder, but because it is a condition with effects that are complex, management requires a multidisciplinary approach.71,198
Incidence and Etiologic and Risk Factors
The prevalence of HD in North America ranges from 4 to 8 per 100,000. It is estimated that there are 25,000 cases in the United States. HD may begin at any time after infancy but usually starts in middle age. Twenty-five percent of persons with HD have disease of late onset, which is defined as onset of motor symptoms after age 50 years. There is almost always a history of an affected parent. There is a 50% risk in each child of an affected adult.6 Transmission of the juvenile form of HD (onset before age 20 years) appears to be primarily from the father. With adult onset there is more equal transmission from both parents.
HD is an autosomal dominant disease with the IT15 or HD genetic marker found on the tip of chromosome 4. In a subset of cases, the Junctophilin 3 gene is responsible for the HD genotype. All the people who inherit the gene will develop symptoms of the disease if they do not die prematurely. Because there is no cure for HD, there is an ethical dilemma associated with testing. Studies are under way to determine the psychiatric and social problems that may result from the knowledge that one will develop HD.61
Pathogenesis
Although the cause remains unknown, pathologic findings show a consistent pattern of tissue changes in the brain. The ventricles are enlarged as a result of atrophy of the adjacent basal ganglia, specifically the caudate nucleus and putamen (collectively the striatum) (Fig. 31-5). This is due to extensive loss of small and medium-sized neurons. The volume of the brain can decrease by as much as 20%. Caudate atrophy correlates with a measured decline in Mini-Mental State Examination scores but not with the severity or duration of neurologic symptoms. It is the atrophy of the putamen that correlates with neurologic symptoms. The atrophy of the cortices appears to occur at the same rate as that of the striatum. White matter degeneration in the frontal cortex appears to be associated with the course of the disease. Slower disease progression appears to be correlated with more white matter changes. The more aggressive progressive disease is related to less white matter and more striatal damage. Other subtle changes occur in the cortex and cerebellum, including both loss of neurons and production of glial cells that inhibit neural transmission.70
Figure 31-5 Atrophy seen in the caudate and putamen in a person with Huntington’s disease. As the disease progresses there is a change in the caudate at the interface with the ventricle. The outline becomes more and more concave, representing the progressive atrophy.
As with other progressive diseases, there is selective vulnerability of neurons in a particular region with preservation of others. In the early and middle stages of HD, neurons projecting from the striatum to the substantia nigra are depleted. This reduces the amount of neurotransmitters, including GABA, acetylcholine, and metenkephalin. This leaves relatively higher concentrations of other neurotransmitters, such as dopamine and norepinephrine. The normal balance of inhibition and excitation responses in the complex organization of the basal ganglia and thalamus that allows for smooth, controlled movement is disrupted. The result is an excess of dopamine and excessive excitation of the thalamocortical pathway. This may explain the excessive abnormal involuntary movements described as chorea.9,147,198
In the later stages of HD, there is a loss of the direct inhibitory substance that causes more inhibition of the thalamocortical output with resultant rigidity and bradykinesia, or slowness of movement. By the late stages of HD, virtually all the caudate nucleus projection neurons are affected. The mechanism of neuronal loss is not known. One hypothesis is that an excitotoxin causes the cell death noted in the basal ganglia.146
Clinical Manifestations
Movement Disorders.: Many individuals with suspected early HD will show almost no neurologic abnormality on routine examination other than minor choreic movements. The movements may be suppressed during the examination since they can often be integrated into a purposeful movement, such as raising the hand to the head as if to smooth the hair. Early in the course the involuntary movements may appear to be no more than an exaggeration of normal restlessness, usually involving the upper limbs and face. The chorea is increased by mental concentration, emotional stimuli, performance of complex motor tasks, and walking. Problems with voluntary movement may be detected by asking for rapid tongue movements or finger-to-thumb tapping, or testing for dysdiadochokinesia, the inability to make rapid alternating movements.
Assessment of muscle strength will usually be normal in early cases but may be affected by any significant bradykinesia or general motor disturbance. Tone will usually be normal initially, but rigidity will become part of the clinical picture in many cases as the disease progresses. The tendon reflexes are usually normal.
Abnormalities in eye movement are common in HD. The ability to execute a saccade, a rapid movement of the eyes from one target to another in order to move the visual focus rapidly to different objects, is disturbed. There is often a decrease in the velocity of eye movement, an undershooting of the target, or latency in initiation of movement. Gaze fixation abnormalities have been noted; that is, inability to fix on a light source without the intrusion of small saccadic movements. Smooth pursuit, or tracking of the eye to follow a moving object, is interrupted by the same small, jerky saccadic movements. There is often an inability to suppress reflex saccades to a visual stimulus, which leads to visual distractibility.
The term chorea is derived from the Greek word for dance, and gait abnormalities are common in HD. When chorea is a predominant sign, persons walk with a wide-based, staggering gait. Those persons with bradykinesia and hypertonicity may walk with a slow, stiff, unsteady gait.
Dysarthria, reflected as a decrease in the rate and rhythm of speech, may be mild in the early course with an increase to the point where speech may be unintelligible. In addition to the mechanical problems, neuron loss disrupts linguistic abilities, resulting in reduced vocabulary and syntactic errors. Some persons become mute at a stage before motor disability is severe.
Abnormalities of swallowing, or dysphagia, can cause choking and asphyxia. Dysphagia may involve multiple abnormalities of ingestion, including inappropriate food choices, abnormal rate of eating, poor bolus formation, and inadequate respiratory control.
Cachexia, or the wasting of muscle with weight loss, is found despite an adequate diet. This appears to be independent of the hyperkinesia and is found in persons with rigidity as well.
Sleep disorders become a progressive problem throughout the course of HD. An increased latent period before sleep and increased periods of wakefulness are common in moderately affected persons. Sleep reversal—daytime somnolence and nighttime restlessness—is seen in severely affected persons and is probably related to the dementia. Choreic movements are reduced during the deepest part of sleep.
Urinary incontinence is often a problem. This could be related to dementia, depression, decreased mobility, or hyperreflexia of the muscles that control urine output. There can be a concomitant increase in the incidence of urinary tract infections.
Neuropsychologic and Psychiatric Disorders.: Early mental disturbances in persons with HD include personality and behavioral changes, such as irritability, apathy, depression, decreased work performance, violence, impulsivity, and emotional lability.129 Intellectual decline usually follows the personality changes. The neuropsychologic profile characteristically includes a type of memory disturbance that suggests an impairment of information retrieval. Individuals often have difficulty recalling information on command but are able to give the correct answer in a multiple-choice format. There is difficulty with organization, planning, and sequencing, even when all the information is provided. Other prominent abnormalities include visuospatial deficits, impaired judgment, and ideomotor apraxia, the inability to perform previously learned tasks despite intact elementary motor function.
More than one third of persons with HD will develop an affective disorder. Depression is the most common psychiatric condition and does not appear to be simply a reaction to a fatal illness. Evidence for this is the fact that mood disorders are not randomly distributed but occur in subsets of families with HD.61
MEDICAL MANAGEMENT
The clinical diagnosis of HD depends on recognition of patterns of symptoms given in the client’s history and clinical signs, and the family history. Difficulties in diagnosis arise when the family history appears negative. Some families deny the presence of cognitive or psychiatric disease. Understanding of the clinical signs must take into account the fact that signs change during the course of the illness. Different patterns may be observed depending on the age of onset.71
MRI demonstrates atrophy of the striatum that is most easily appreciated as enlargement of the frontal horn of the lateral ventricles. Fig. 31-6 shows this change in brain structure. This is not of great diagnostic value unless it is very pronounced, given the normal reduction in brain mass with age and the occurrence of atrophy in other disorders that might be confused with HD. Positron emission tomography (PET) will also show atrophy, but its value as a diagnostic tool has the same limitations as MRI.
Figure 31-6 Magnetic resonance scan showing the degeneration of the caudate in a person with Huntington’s disease (HD). The dotted lines show where the tissue would be in a person without HD. (From Ramsey R: Neuroradiology, Philadelphia, 1994, Saunders.)
In addition to genetic linkage analysis, which requires testing of family members, it is now possible to evaluate the DNA of an individual to identify specific components that are diagnostic for HD. This eliminates the need to compare DNA of affected family members, but there are still problems with this method, because there is a small percentage of affected individuals who do not display the characteristics on the specific gene and there are nonaffected individuals who carry the gene.
Recognition of HD in older persons is critical to establish the genetic link for future generations. Often the diagnosis is overlooked in favor of the label of senile chorea, because there are minimal changes in behavior and cognition. The differential diagnosis of HD in the older population includes various degenerative, systemic, and drug-related conditions. An individual treated with neuroleptics for a psychiatric presentation of HD, for example, may go on to develop movement disorders, and these may be confused with the typical side effects of the medication.131
TREATMENT.
Management of HD requires a team approach, including medical and social services. Education of clients and their families about the implications of the disease is important. Genetic, psychologic, and social counseling are started as soon as the diagnosis is confirmed. Organizations designed to help families with HD are often of great help.
Medical treatment is symptomatic. The most useful drugs for the symptomatic relief of chorea are anticonvulsants or antipsychotic agents that block dopamine neurotransmission. They can also help with the emotional outbursts, paranoia, psychosis, and irritability seen in HD. Drug therapy for chorea should be held in reserve if the abnormal movements are slight. There is a high incidence of side effects with the drugs, including acute dystonias, pseudoparkinsonism, and akathisia, which is characterized by uncontrollable physical restlessness. The most serious effect is chronic tardive dyskinesia resulting in involuntary movement of the face, tongue, and lips. Another adverse reaction is neuroleptic malignant hyperpyrexic syndrome, characterized by fever and rigidity.
Surgical procedures to remove the medial globus pallidus, thought to be overexcited by neuronal loss in the striatum, have been tried with mixed results. Implantation of adrenal medullary grafts has not been encouraging; the improvement appears to be transient.
PROGNOSIS.
It is characteristic of the disease that younger people, with onset of symptoms at age 15 to 40 years, will experience a more severe form of the disease than older people, with onset in their fifties and sixties. The advance of the disease is slow, with death occurring on average 15 to 20 years after onset. Survival into the eighties is not uncommon, and persons living to past 90 years old have been recorded. Age at onset and age at death frequently show a familial correlation.
Increasing disability from involuntary movements and mental changes often results in death from intercurrent infection. Suicide accounts for approximately 6% of deaths, and 25% of persons with HD attempt suicide at least once.
MULTIPLE SCLEROSIS
Overview and Definition
Multiple sclerosis (MS) is a major cause of disability in young adults. The name is descriptive of the sclerotic plaques disseminated throughout the CNS that are the hallmark of the disease. There are multiple lesions found throughout the brain and spinal cord. These lesions slow or block neural transmission, resulting in weakness, sensory loss, visual dysfunction, and other symptoms. The course of MS is highly variable. Complications of MS may affect multiple body systems and require profound adjustments in lifestyle and expectations for clients and their families; therefore, a multidisciplinary approach is necessary to optimize clinical care.
MS is a chronic illness that may be manifested in multiple forms and courses. There are four generally recognized subtypes. Relapsing-remitting MS is characterized by relapses or attacks, which are periods of neurologic dysfunction lasting days to months and followed by full or partial recovery. By convention, new symptoms must last at least 24 hours and be separated from other symptoms by at least 30 days to qualify as a new attack. The hallmark is that there is a stable course between relapses. This is the most common pattern, seen in about 85% of newly diagnosed individuals. Secondary progressive MS describes an initial pattern of relapse and remission that changes into a steadily progressive pattern over time in more than 50% of the relapsing individuals. There sometimes are continued relapses during this phase. This conversion generally occurs 5 to 10 years after the initial onset of relapsing symptoms. Primary progressive MS is a steady decline in neurologic function from the outset with episodes of minimal recovery. The most common clinical presentation of primary progressive MS is myelopathy, a gradual, progressive weakening and wasting of muscles, which is typically seen in persons with onset past the age of 40 years. Progressive-relapsing MS is progressive disease from the onset with clear exacerbations. This is considered the rarest form of MS.
Incidence
Marked differences in the prevalence of MS exist among different populations and ethnic groups. MS is a disease of temperate climates. Highest known prevalence occurs in the Orkney Islands, off Scotland. MS is also common in Scandinavia and elsewhere in northern Europe. There are more than 2 million persons affected worldwide; the incidence is 12 per 100,000 persons. In the United States it is estimated that 450,000 people are affected, with about 10,000 new cases per year. Caucasians of northern European descent have significantly higher rates of the disease than other racial groups. MS is extremely rare in Japan and virtually unknown in black Africa, but Japanese Americans and African Americans show an increased prevalence.151 African Americans with MS have a greater likelihood of developing opticospinal MS and transverse myelitis and have a more aggressive disease course than Caucasian Americans.73 Fig. 31-7 shows the incidence rates worldwide.
Figure 31-7 World map showing the relative incidence of multiple sclerosis. Areas of greatest risk are in the northern latitudes. (Adapted from National Institute of Neurological and Communicative Disorders and Stroke: Multiple sclerosis: hope through research. Publication no. 79-75, Washington, DC, 1981, National Institutes of Health. In Umphred DA: Neurological rehabilitation, ed 5, St Louis, 2007, Mosby.)
MS rarely will begin before adolescence; it rises steadily in incidence from the teens to age 35 years and declines gradually thereafter. Similar to many other autoimmune diseases, MS has a predilection for women, with a female to male ratio of 2.5: 1. Men have a slightly later age of onset and more severe clinical outcomes than women. Men with MS transmit the disease to their children (independently of the child’s sex) more often than do women who have MS.39 Relapse rates may decline 70% in the third trimester of pregnancy, most likely due to circulating levels of estriol. These facts suggest a complex hormonal modulation of the immune system.
Etiologic and Risk Factors
There is a clear genetic component in the risk of developing MS.40 When one parent is affected, and especially if that parent developed MS at an early age, before 20 years, a child has a fivefold higher risk of developing MS.158 The human leukocyte antigen (HLA) region on chromosome 6 has been identified as one genetic determinant for MS, but this contributes only a small fraction to the genetic basis of MS.44 Refinement of the genetic linkage map is in progress. The linkages are complex and may involve multiple weak links that are difficult to identify using current research methods. In Scotland, where risk of developing MS is high, there is an increase in the incidence of HLA-DR2 allele in the population that has a proven correlation to MS. The presence of HLA-DR2 is associated with a nearly twofold increase in the likelihood of having a second attack of MS within 5 years. The severity and course of MS may also be associated with different genes. A variety of genes have been associated with the disease, including interleukin-1b receptor, interleukin-1 receptor antagonist, and immunoglobulin Fc receptor.
Coexisting autoimmune disorders are seen in a majority of individuals, such as Hashimoto’s thyroiditis, psoriasis, inflammatory bowel disease, and rheumatoid arthritis. Families with a history of MS have autoimmune disorders in greater than 65% of first-degree relatives. Hashimoto’s thyroiditis, psoriasis, and inflammatory bowel disease are the most common disorders also occurring in family members. The presence of various immune disorders in families with several members with MS suggests that the disease might arise on a background of a generalized susceptibility to autoimmune disorders. A distinct MS phenotype, defined by its association with other autoimmune diseases, segregates with specific genotypes that could underlie the common susceptibility.
Viral infection often precipitates an attack of MS. In fact, it is the only natural event that has been shown unequivocally to increase the risk of a new attack of MS. Less than 10% of infections are followed by relapses, but more than one third of the relapses are proceeded by infection.44 The possibility that infections, especially viral infections, actually may cause MS has been investigated for many years. Numerous candidate viruses and a smaller number of bacteria have been proposed and then rejected. Most recently, human herpesvirus 6 (HHV-6) and the bacterium Chlamydia pneumoniae have been touted as environmental triggers of MS. Research attempting to clarify these issues continues. There appears to be a protective effect of vitamin D intake on risk of developing MS.
Pathogenesis
MS is the classic example of a primary demyelinating disorder, but demyelination alone does not account for the persistent functional disturbances that characterize the disease as it progresses. After myelin is damaged, nerve conduction properties, initially abnormal, can recover, in part related to redistribution of sodium channels. Axonal injury in MS, known from the work of Charcot in the mid-nineteenth century but subsequently associated only with late disease, was recently rediscovered as an important early pathology of considerable importance in MS.72
Myelin loss occurs initially around the small veins and venules. The spinal cord may show similar changes. There are focal abnormalities referred to as plaques disseminated throughout the cerebral hemispheres in the white matter, especially in the white matter surrounding the ventricles, the corpus callosum, the optic nerves, and the brainstem.
The pathologic conditions that occur in MS include inflammation, demyelination, and axon loss. MS is predominantly a T cell–mediated inflammatory disorder with overproduction of proinflammatory cytokines. Demyelination can result from either direct damage to neuronal myelin by inflammatory cells or indirectly because of the extracellular environment. This demyelination can cause neurons to be more susceptible to apoptosis. Although demyelination may produce the relapses that occur during the disease, long-term disability is due primarily to irreversible axon loss and cell death.62
The initial event in MS may be priming of autoreactive, peripheral T cells against myelin antigens by an infectious agent. It is believed that preexisting autoreactive T cells are activated outside the CNS by foreign microbes, self-proteins, or microbial superantigens. Activated T cells cross the blood-brain barrier through a multistep process. In this trafficking process, lymphocytes attached to endothelial cells proceed to pass through the endothelial cell cytoplasm. Chemokines, like cytokines, are secreted polypeptides that can induce increased production of integrin molecules by lymphocytes and switch these integrin molecules into a higher affinity state that allows them to transmigrate more effectively across CNS endothelium. The activated T cells secrete cytokines that stimulate microglial cells and astrocytes, recruit additional inflammatory cells, and induce antibody production by plasma cells. Antimyelin antibodies, activated macrophages or microglial cells, and tumor necrosis factor are believed to cooperate in producing demyelination. In the neurodegenerative phase of the disease, excessive amounts of glutamate are released by lymphocytes, microglia, and macrophages. The glutamate activates various glutamate receptors, causing the influx of calcium through ion channels associated with toxic damage to oligodendrocytes and axons. The oligodendrocyte is a nonneural component of the CNS that is directly related to the cell body or axon. The oligodendrocyte produces the myelin that wraps around the axon of the nerve cell. The myelin sheath increases the speed of the action potential and is critical for smooth and rapid movement.
Both necrosis and apoptosis play a role in the progression of MS. When necrosis occurs in MS lesions, it produces enhancing lesions caused by the substantial breakdown of the blood-brain barrier. In MS lesions, apoptosis occurs in neurons, oligodendrocytes, and leukocytes Apoptosis occurs pathologically in MS, in part because the loss of myelin causes neurons to be much more susceptible to binding of agents that induce apoptosis rather than necrosis. Apoptosis is probably at least partially responsible for the progression that leads to permanent disability and explains why lesion activity on MRI scans can be quiescent at a time when the disability of the individual continues to increase. Apoptosis of oligodendrocytes may represent a very early stage in the evolution of lesions that underlie acute relapses of MS. Therefore, myelin destruction in MS lesions may be caused by processes other than those mediated by macrophages.
Brain atrophy has emerged as a clinically relevant component in MS and begins early in the disease course. Progressive loss of brain tissue bulk can be identified by MRI. The caudate (part of the basal ganglia) can be smaller in volume and also have changes in shape. The demyelinated cortex contains apoptotic neurons. Cortical demyelination may influence dendrite and axon function and decrease neuronal viability, which may further activate disease progression. Axonotmesis as a result of inflammation in MS lesions can lead to retrograde neuronal degeneration and apoptosis.142 In chronic lesions, demyelinated axons undergo a slow, disseminating wallerian degeneration along neural tracts away from the initial site of injury, contributing to long-term disability.
Quantitative MRI indicates that cortical atrophy occurs early in the disease course and is related to physical disability and cognitive impairment. Besides demyelination, the cerebral cortex of individuals with MS may also be affected by tissue loss and atrophy, particularly in areas adjacent to severe white matter pathology. Neurons in such lesions may show signs of retrograde damage, including central chromatolysis. Furthermore, degeneration of cortical neurons could contribute to the depletion of neuronal metabolites in white matter that may not show MRI image changes.
New lesions frequently appear at sites of previous activity or within or at the edges of previously stable lesions. New lesions can form within or overlap shadow plaques (areas of thin remyelination) and may contribute to failed remyelination or the conversion of shadow plaques into classic demyelinated plaques.144 Thus some lesions may appear to be quite large.
The plaques may be acute or chronic lesions. The acute plaques are small, circumscribed areas of hypocellularity, demyelination, and axonal loss. Astrogliosis, resulting in scar tissue formation, is more severe in MS lesions than in most other neuropathologic conditions. Periventricular infiltrates consisting of macrophages, T cells, immunoglobulins, and microglia fill the plaque.188 Axons passing through may be spared, but there seems to be more and more evidence of axon loss within the plaques. New active plaques are pink with faint borders that exhibit an intense inflammatory response. Old inactive plaques are grey and firm, with sharp edges with a gliotic background crossed by axons but lacking oligodendrocytes.134 Abnormal immune function in the blood and cerebrospinal fluid (CSF) is a common finding.74
A variety of pathogenic mechanisms may be involved in the development of MS plaques, which occur as a result of diversity in the sequence of events that causes the condition to arise. The features of the lesions, including the amount and nature of T cell–, macrophage-, and immunoglobulin-related damage, seem to vary from individual to individual, but there is much less variation among the active lesions within an individual. For example, the survival of oligodendrocytes varies among individuals.44 In some individuals, it appears that oligo dendrocyte dystrophy and apoptosis are the primary cause of demyelination. In others, the presence of immunoglobulins suggests that demyelinating antibodies have a pathogenic role.104,112 As with demyelination, axonal destruction could result from direct immunologic attack or inflammatory mediators or from secondary effects of chronic demyelination.182
Clinical Manifestations
In most individuals, MS is characterized by progressive disability over time, but the amount of accumulated disability varies widely. A benign course may affect up to 20% of individuals and is characterized by an abrupt onset with one or a few relapses followed by complete or nearly complete remitting periods. These individuals experience little or no permanent disability and remain relatively symptom free. This is a designation that can only be made with certainty in retrospect, since there are no perfect prognostic markers. More recent MRI scan data suggest that even in individuals with benign MS there is likely significant progression of lesions.
Each individual’s CNS appears to have a different threshold for producing symptoms and signs reflecting the affected regions of the CNS. This threshold, or the capacity of the individual’s brain to adapt to the lesions, will determine the severity of the clinical manifestations.
Optic neuritis is often the first manifestation of MS. The optic nerve is an extension of the cerebral cortex, virtually a tract of the CNS, and is therefore subject to the effects of demyelination with the syndrome of optic neuritis. Optic neuritis typically presents as a unilateral, painful decrease or loss of vision. It is commonly associated with visual field defects, decreased color vision, and reduced clarity of vision.63 Individuals with optic neuritis must be carefully evaluated for an ocular mobility abnormality to determine the possibility of a second anatomically distinct lesion, because the symptoms of blurring may be caused by an eye movement disorder.94
Early-onset MS is associated with lesions within the spinal cord. The spinal cord may be abnormal when the brain MRI is normal. However, an abnormal spinal cord is found more than half the time when there are nine or more brain lesions. Approximately 80% to 85% of individuals present with a relapsing-remitting course, with symptoms and signs evolving over days and typically improving over weeks.165
Sensory changes are most often the initial complaint. This is often a paresthesia or dysesthesia noted in one extremity or in the head and face. Visual blurring, diplopia, weakness, and balance problems also may be early signs. Often these symptoms are transient and not even reported. It is usually when there is a pattern or the symptoms are unchanging that the person seeks medical attention. Dorsal column symptoms include paresthesias (tingling, pricking) and hypoesthesia (diminished sensitivity). These may begin in an extremity and ascend over hours or several days to include the rest of the leg or arm, the perineum, the trunk, and perhaps other extremities. Other sensory complaints include a feeling of swelling, of wetness, or that the body part is tightly wrapped. Involvement of a cord level is diagnostically helpful in distinguishing this attack from a peripheral neuropathologic incident. Other positive dorsal column signs are loss of vibration, position, and two-point discrimination. Sensory complaints are often not substantiated by objective findings, especially if the symptoms are mild or the remission has already begun before the individual is examined. For example, the feeling of numbness may not result in a loss of response to pinprick.
The single most common and most disabling symptom of MS is fatigue.64 Fatigue is typically present in midafternoon and may take the form of increased motor weakness with effort, mental fatigue, and sleepiness. MS-related fatigue presents as an overwhelming feeling of tiredness in those who have done little and are not depressed. People who are depressed, whether they have or do not have MS, often do not sleep properly, eat properly, or feel well. They may describe this as fatigue, but the treatment revolves around the treatment of depression. Deconditioning and lack of endurance may lead to fatigue.
Neuromuscular or short-circuiting fatigue is common. The demyelinated nerve fires again and again until it shorts when it is called upon to do a repetitive task. Thus, progressive resistive exercises often result in a feeling of increased fatigue and weakness rather than a feeling of increased strength. Short breaks with energy conservation can make this fatigue less prominent.
Spasticity, velocity-dependent stiffness about a joint, is an extremely common problem with MS, occurring in 90% of all cases. It can vary from nonexistent to severe, even in the same person, and from moment to moment, making its management challenging. It seems to be the result of disequilibrium in the ascending and descending excitatory and inhibitory pathways in the brain and spinal cord. GABA and other neurotransmitters are involved. Often those who have significant spasticity use their spasticity to walk, transfer, and manage their daily living. Treatment becomes an issue only if the spasticity is causing discomfort, pain, or problems with daily living. Pain, either from an injury or from a bladder infection, exacerbates spasticity. Thus, the management of spasticity begins with the removal of noxious stimuli. In some individuals, spasticity can be intractable. The high doses of medications necessitated by this circumstance sedate and cause disability on their own. Spasms may accompany the spasticity and usually are more severe and frequent at night. They often interrupt the sleep cycle, even in those who do not recognize their awakenings. This may lead to severe fatigue the next day; thus, minimizing nocturnal spasms is important. Associated signs of upper motor neuron syndrome may include clonus, spontaneous extensor or flexor spasms, positive Babinski’s sign, and loss of precise autonomic control.117,123,163
Weakness in MS usually is a result of decreased neuromuscular impulses secondary to demyelination and axonal loss. As discussed previously, progressive resistive exercises may contribute to fatigue and give the appearance of increasing weakness. Signs of muscle weakness secondary to damage of the motor cortex and tracts reflect the loss of orderly recruitment and rate modulation of motor neurons. Muscle activation patterns and agonist-antagonist relationships are disturbed.
Heat, either from increased ambient temperature or from fever, often increases weakness. This may be the result of a conduction block. Cooling often allows more efficient conduction and improved strength if there is appropriate innervation.
Involvement at the level of the brainstem reflects lesions of cranial nerves III through XII at the root, nuclear, or bulbar level. Trigeminal neuralgia (also called tic douloureux) is a shocklike pain in the face. Although not a common finding, it is highly characteristic of MS in a young person. Spasm or weakness of facial muscles can also be seen. Dysarthria, abnormal speech resulting from poor control of the muscles of speech, and dysphagia, including signs of gurgling, coughing, weight loss, pneumonia, choking, or a weak voice, can present in brainstem involvement.
There can also be gaze palsies, the loss of active control of eye movement, and nystagmus, involuntary rhythmic tremor of the eye. Intranuclear ophthalmoplegia is the most common gaze palsy, resulting in lateral gaze paralysis, and is caused by demyelination of the pontine medial longitudinal fasciculus, an area of the brain’s white matter involved in the control of eye movement. Other lesions in the brainstem and reticular formation can cause other palsies, resulting in difficulty with conjugate gaze and ipsilateral gaze palsy. Idiopathic nystagmus that improves over time also can be caused by lesions in the vestibular nuclei or cerebellum.
Other abnormalities of ocular mobility, such as instability of fixation or inability to suppress the vestibuloocular response, are related to lesions involving brainstem nuclei and tracts. Vertigo, the sensation of spinning, may appear suddenly and in dramatic fashion with gait unsteadiness and vomiting. In MS this reflects a brainstem rather than end-organ vestibular disorder; a careful look at associated brainstem symptoms will help to distinguish the cause (see Chapter 38).
Coordination (ataxia) problems often accompany tremor and are among the most difficult symptoms to manage. Compensatory techniques taught via exercises can be helpful but rarely enough to satisfy. Cerebellar syndrome deficits may be symmetric, with all four limbs involved, or asymmetric, with only one side affected. Manifestations include ataxia, hypotonia, and truncal weakness, causing postural and movement disorders. Dysarthria of cerebellar origin (scanning speech, producing abnormalities in the rhythm of speech) is common. Cerebellar signs are often associated with the progressive phases of the illness.181
Pain is surprisingly common in MS, occurring in 50% of individuals. The pain usually is a burning neuropathic type. It may be disturbing, and pain medicine offers little if any relief. The distribution of the pain may not follow any recognizable neurologic distribution. It can be paroxysmal in nature but often is fairly constant, with nocturnal worsening when the body is at rest in bed. Another clinical symptom is Lhermitte’s sign, a momentary electricity-like sensation evoked by neck flexion or cough.
Depression is a primary symptom of MS occurring because of actual changes in the brain and its chemistry. Depression can occur as a direct result of the MS plaques or as a reaction to the diagnosis or disability. Depression may lead to greater disability than that caused by the level of neurologic impairment. Medication use for other symptoms may contribute to the cognitive problems with sedation and confusion; thus, these must be reviewed regularly in those who have dulled cognition. Depression may be an additive culprit and is treatable if recognized. There is no question that reactive (exogenous) depression occurs frequently in MS and is amenable to counseling and other nonpharmacologic techniques, but the brain disease seen in MS clearly leads to chemical (endogenous) depression in many. Cognitive decline is of significance in up to 50% of persons with MS.162 Memory loss is the most common cognitive problem, and there commonly are a variety of slowed response types. The suicide rate for MS seems greater than that for many other neurologic diseases, some of which have a worse perceived prognosis.
Bladder and bowel symptoms are common and usually occur when the spinal cord is involved. Bladder urgency, frequency, and incontinence associated with an overactive bladder are often seen early in the disease and generally precede incontinence. Bladder issues are prominent in those who have MS. The bladder can present itself with frequency, urgency, hesitancy, and incontinence with differing mechanisms. Most frequent is the small, failure-to-store bladder characterized by a low postvoid residual. This can be measured by catheterization or ultrasound. It often has uncontrollable contractions.
More problematic from a management point of view is the large, failure-to-empty bladder. It overfills with residuals from 200 to 900 ml and presents with the same symptomatology despite looking different anatomically and physiologically. Frequently, there is a dyssynergia between the bladder and the urinary sphincter, causing similar symptoms from yet another mechanism. Residual urine after emptying, with subsequent overflow incontinence and heightened risk of urinary tract infections, is a problem for 50% of persons with MS, especially later in the course of the disease. This may require intermittent or chronic catheterization, or use of the Credé method (manual pressure on the bladder to express urine).14 Neurogenic disorders may also impair bowel function, resulting in incontinence or constipation.
Bowel function is affected less than bladder function but can be problematic. Irritable bowel is a common associated problem, and regulating the bowel often means changing treatments, depending on the circumstances. It is better to be slightly constipated than slightly loose.163
Sexual expression may require special attention in MS. Relationships remain of utmost importance and need to be stressed, without diminishing the importance of the actual sexual performance in the face of disability. In men, erectile dysfunction is frequent. Occasionally, penile prostheses may be placed. This is less common today because of alternative treatment options. Female sexual expression may be diminished by vaginal dryness or decreased or altered sensation in the vaginal area.
Unique symptoms in MS are called paroxysmal because they come in bouts. Sometimes they are sensory in nature and may be painful. Trigeminal neuralgia is a common example of this type of painful, paroxysmal symptom. This lancinating, electrical sensation along the distribution of a branch of the trigeminal nerve can be terribly disabling. It may be a stand-alone problem in some individuals but also is a symptom associated with MS. Periodic electrical sensation down the spine with the bending of the neck is called Lhermitte’s sign, another paroxysmal sensory symptom. These types of sensations occasionally may be felt in other parts of the body. Often the paroxysmal symptoms are motor in nature. They may be twitching of an eyelid or myokymia in the facial muscles. They may manifest as a true spasm of an arm in an extended or, occasionally, flexed position lasting for seconds. They can occur frequently, several times an hour. The spasm may occur with speech, a paroxysmal dysarthria or speech arrest. All of these can be disconcerting to the individual and the clinician.
The aggressive use of immune-modulating agents to slow down the actual disease process has contributed to the development of a whole new set of symptoms that the clinician must recognize and manage. Interferon initiation often brings about a fever reaction, which may be disabling to the person who has MS. It often is recommended that interferon be administered in the evening before going to bed. This allows for the impact of the fever during the night, when it may be less disabling to activities of living. Antipyretic medications (ibuprofen or acetaminophen) may be administered 4 hours before the injection, at the time of injection, and then, if necessary, 4 hours after the injection.
Subcutaneous injections can lead to immune reactions within the skin, with inflammation, redness, and significant irritation. Good injection technique is essential. This may involve icing the area before injecting. A common error is injecting too shallowly. Injection devices use improved techniques and have decreased many of these side effects but may contribute to the problem if they are set to deliver the medication too shallowly within the skin rather than beneath it. It is important to allow the air bubble in the syringe to remain to push the medication through the skin and it is recommended that the needle be free of lingering medication as it pierces the skin. Rotation of the injection sites is an absolute necessity to preserve as much skin area as possible and allow healing between injections. For some individuals, the addition of a cortisone cream or a local anesthetic cream may be helpful. With some treatments, pain can linger after the injection. Applying cooling can offer symptomatic relief. Understanding the issues often is helpful in the development of compensatory strategies.
Glatiramer acetate occasionally induces a systemic reaction. This rare, but real, symptom complex presents with a feeling of panic, chest discomfort, shortness of breath, and a feeling of doom and gloom. It usually lasts for 20 minutes and then clears rapidly, but if panic sets in and an emergency is called, the issue may be magnified and complicated. The treatment is to realize that the problem is associated with the medication and requires resting for the duration of the symptoms to allow them to abate on their own. This usually is not a recurrent problem but occasionally can repeat.