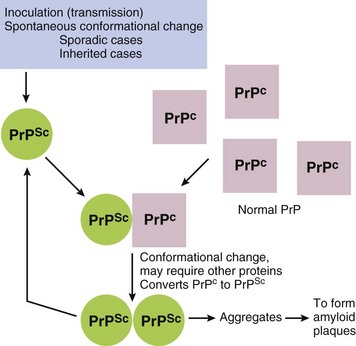
Circulatory Disturbances
Many diseases of the CNS in veterinary medicine result from injury to the circulatory system and vascular endothelium. Vascular diseases of the CNS can result from inflammation/infection, either as a component of a systemic disease process or from extension of inflammatory meningeal or cerebral disease. The incidence of cerebrovascular diseases analogous to those in humans, including trauma, is low in animals, and neurologic manifestations associated with these diseases are uncommon. Arteriosclerosis (“hardening” of arteries) can be categorized as lipid (atherosclerosis) or nonlipid arteriosclerosis; the latter includes arterial fibrosis, mineralization, and amyloid deposition (see Chapter 10).
Atherosclerosis: Atherosclerosis is reported in a variety of animals, including nonhuman primates, pigs, dogs, and several avian species. Older pigs are most commonly and severely affected. Occasional older dogs with chronic hypothyroidism or diabetes mellitus can have severe atherosclerosis. The pathogenesis of atherosclerosis and atherosclerotic plaque formation is most well understood in humans, and the results of experimental studies may have some application to understanding atherosclerosis in domestic animals.
Atherosclerotic plaques arise from a complex and partially understood interaction among endothelium, smooth muscle cells, platelets, T lymphocytes, and monocytes. Endothelial injury induced by oxidized low-density lipoprotein [LDL] cholesterol results in vascular inflammation of the tunica intima. Monocytes migrate into the intima of the vessel wall to phagocytose LDL cholesterol. This process results in the formation of foam cells characteristic of early atherosclerosis (fatty streak). In addition, activated macrophages produce factors that also injure the endothelium. LDL cholesterol concentrations in foam cells and smooth muscle cells often exceed the antioxidant properties of normal endothelium. Oxidized LDL leads to additional metabolic changes that foster a procoagulant microenvironment and enhanced platelet-mediated thrombus formation, as well as initiates a cascade of events leading to the lesions associated with the development of mature atherosclerotic plaques (fibrous plaques with a cap [lipid-laden macrophages walled-off by connective tissue]). The location of atherosclerotic plaques in the circulatory system depends on fluid shear stresses and their interaction with injured vascular endothelium. Atherosclerotic plaques characteristically occur in areas of vessel branching or areas where blood undergoes a sudden change in velocity and/or direction of flow.
Although atherosclerotic plaques can reach sizes large enough to significantly reduce blood flow to regions of the brain, the stability of the plaques determines the seriousness of the disease. A stable plaque is characterized by an excess of smooth muscle cells with few lipid-containing macrophages. An unstable plaque is characterized by a large lipid-rich core with abundant lipid-containing macrophages, thin fibrous cap, and inflammation. Rupture of unstable plaques can lead to vascular thrombosis or thromboembolism and infarction of areas supplied by these vessels in the CNS.
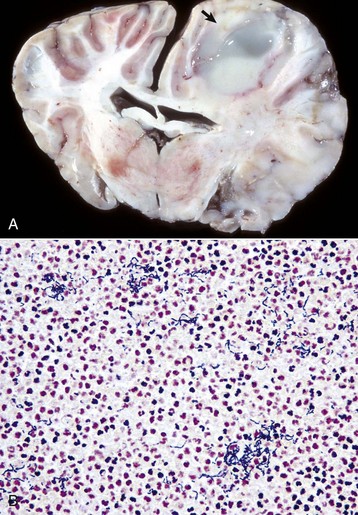
Grossly, vessels that can be involved include the aorta and its major branches, extramural coronary arteries, renal arteries, and cerebral arteries. Affected arteries are rigid, irregularly thickened, and white to yellow-white (atheromatous plaques) (Fig. 14-58, A). Arterial lumina are narrowed or almost obliterated, but there is usually no appreciable ulceration, thrombosis, or hemorrhage (Fig. 14-58, B). Intimal thickening in intracranial arteries contain less lipid and have a greater tendency for fibrosclerosis than other vessels. In arteries within the brain, there is collagenous adventitial or transmural thickening. The arterial lesions can be associated with hemorrhage or infarcts involving basal nuclei, fornix, internal and external capsules, hippocampus, and thalamus.
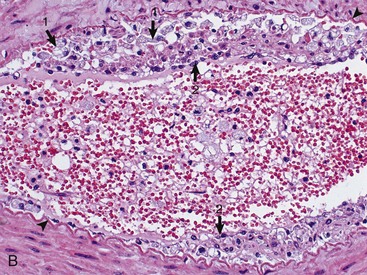
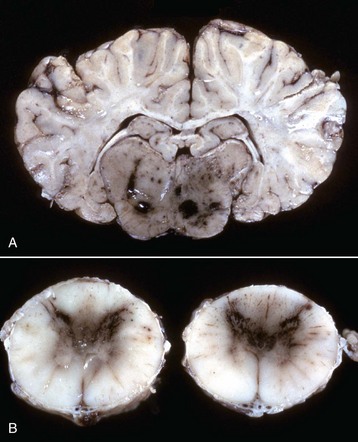
Fig. 14-58 Atherosclerosis, cerebral arteries, brain, ventral surface, dog.
A, The basilar artery, the arteries of the circle of Willis, and the cerebral arteries are segmentally yellow, thickened, and beaded in appearance from atheroma (arrows). This dog had long-standing hypothyroidism. B, The intima of this muscular artery with atherosclerosis contains numerous foamy (lipid laden) macrophages (arrows 1). Arrowheads = internal elastic lamina; arrows 2 = endothelium. H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
In the dog, lesions involve cerebral, coronary, and renal arteries and vessels elsewhere in the body and are most severe in the intima and media. Hemorrhage, ischemia, and infarction of the cerebral cortex are uncommon but can occur.
Cerebral Edema (Permeability Changes): The causes and mechanisms of cerebral edema are presented in the section on vasogenic, cytotoxic, and interstitial edema. Cerebral edema has also been associated with “water intoxication,” which can result from an increased body hydration caused by (1) excessive, faulty intravenous hydration; (2) compulsive drinking caused by abnormal mental function; or (3) altered antidiuretic hormone secretion. The increased body hydration produces a hypotonic (hypo-osmolar) plasma, with subsequent development of an osmotic gradient between the hypotonic plasma and the relatively hypertonic state of the normal cerebral tissue. Fluid moves from the plasma into the brain. In this type of edema, the blood-brain barrier remains intact. If it did not, the change in plasma osmolarity would soon be transmitted to the brain tissue (through vascular leakage) and would abolish the necessary osmotic gradient. Fluid accumulation occurs primarily intracellularly but can also be present extracellularly. In addition, typically a pronounced increase occurs in the rate of formation of CSF originating from the choroid plexus and the extracellular fluid of the brain.
The gross lesions that accompany cerebral edema are the result of enlargement of an organ in an enclosed, limited space; the degree of swelling obviously determines the type and extent of lesions that develop. In evaluating lesions, it is particularly important initially to examine the brain and spinal cord in the fresh state and in situ.
Microscopically, in contrast to some other tissues, such as the lungs, the extracellular fluid associated with vasogenic edema fluid is often not detectable, except in instances of marked vascular injury. When the extracellular space-occupying fluid cannot be identified, only its effects (separation of the cells and their processes causing reduced staining intensity) can be recognized. Additionally, after prolonged vasogenic edema, the lesions include hypertrophy and hyperplasia of astrocytes, activation of microglia, and demyelination. Cytotoxic edema is characterized by cellular swelling, including swelling of astrocytes.
Because of compression against the cranium, an affected brain has flattened gyri and shallow sulci, and it can shift in position. If the edema is confined to one side, the displacement is unilateral, which can be associated with herniation of the cingulated gyrus under the falx cerebri; the extent of the unilateral intracerebral enlargement can be best appreciated after the examination of transverse sections. Diffuse swelling usually causes a caudal shifting that can result in herniation of the brain (parahippocampal gyri of temporal lobes) beneath the tentorium cerebelli (Fig. 14-59) or herniation of the cerebellar vermis through the foramen magnum, resulting in “coning” of the vermis (Fig. 14-60). On a cut surface, the white matter is most often affected (frequently with the vasogenic type of edema, which is the most common). It is swollen and soft, has a damp appearance, and is light yellow in the fresh, unfixed state.
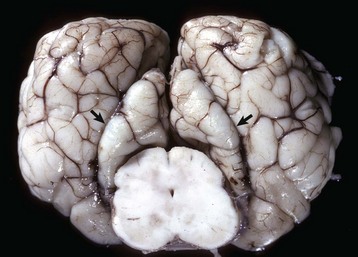
Fig. 14-59 Gyral herniation, parahippocampal gyri, brain, transverse section, caudal face, at level of the rostral colliculi and crus cerebri, horse.
The caudal displacement of the parahippocampal gyri (arrows) was caused by a sudden swelling of the brain (increase in intracranial pressure) from severe cerebral blunt force trauma to the head. The other cerebral gyri are swollen and flattened and sulci are indistinct (cerebral edema). (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
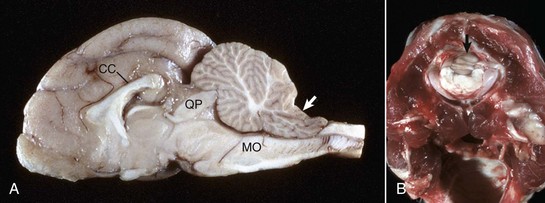
Fig. 14-60 Coning of the cerebellar vermis, brain, cat.
A, Sagittal section. Coning of the cerebellum. The caudal cerebellar vermis has been displaced caudally through the foramen magnum; note the notch on the dorsal surface (arrow). This result has compressed the medulla oblongata (MO), which can cause death from compression of the respiratory center. Note the elevation of the corpus callosum (CC) and focal compression of the rostral cerebellar vermis by the tectum (quadrigeminal plate) (QP). B, Coning of the cerebellum through the foramen magnum, caudal view through the foramen magnum. Note that in this case not only has the cerebellar vermis (arrow) been displaced caudally but also the medulla. The caudal cerebellar peduncles have been displaced caudally as far as the foramen magnum. (A courtesy Dr. D. Cho, College of Veterinary Medicine, Louisiana State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy College of Veterinary Medicine, University of Illinois.)
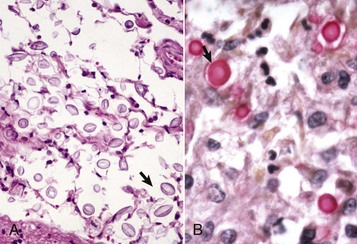
Ischemic Myelopathy (Fibrocartilaginous Embolus Formation): Ischemic myelopathy (necrotizing myelopathy) has been described in the dog, cat, horse, pig, lamb, and turkey. Herniation of degenerative disk material into the vasculature, forming occlusive emboli, is commonly accepted, but the route taken by fibrocartilaginous material into the spinal (or cerebral) vessels is unproved. It has been suggested that trauma to a metaplastic nucleus pulposus causes it to fragment and that the pressure of trauma forces small fragments into damaged veins, venous plexuses, or small arterioles.
The gross lesion is an acute focal infarct involving cervical or lumbar spinal cord most commonly, but any portion can be affected (Fig. 14-61). Microscopically, emboli histochemically identical to the fibrocartilage of the nucleus pulposus of intervertebral disks occlude meningeal or CNS arteries or veins, or both, in affected areas (Fig. 14-62). Clinically, there is a sudden onset of spinal cord deficits, sometimes with cerebral involvement, in certain species. In dogs, larger breeds are more commonly affected. The disease occurs in young and old animals. One study reported that 60% of confirmed cases of canine ischemic myelopathy had a history of trauma or exercise.
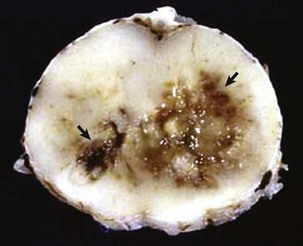
Fig. 14-61 Spinal cord infarction (ischemic necrosis), dog.
The yellow-brown region of necrosis (arrows) in the right lateral and ventral funiculi was the result of fibrocartilaginous emboli that occluded branches of the ventral spinal artery and obstructed blood flow. (Courtesy Dr. J. Edwards, College of Veterinary Medicine, Texas A&M University; and Dr. J. King, College of Veterinary Medicine, Cornell University.)
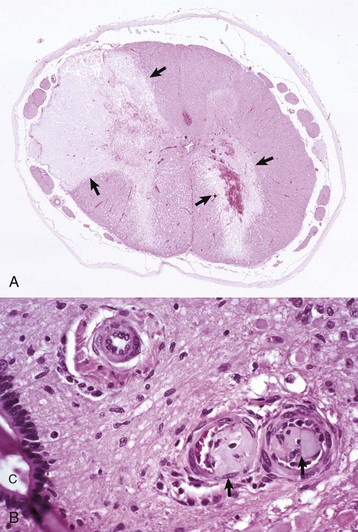
Fig. 14-62 Fibrocartilaginous embolus, spinal cord, dog.
A, Vascular occlusion and infraction. Fibrocartilaginous emboli have obstructed the dorsolateral artery (top left) and branches of the ventral spinal artery to the right ventral gray horn and adjacent white matter, causing infarction (arrows). H&E stain. B, Fibrocartilaginous emboli in arterioles (arrows). C, Central canal. H&E stain. (A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B courtesy Dr. J. Van Vleet, College of Veterinary Medicine, Purdue University.)
Nonlipid Arteriosclerosis: Arterial fibrosis occurs more frequently in older animals and has been described in dogs and horses. In the dog, fibrosis of the intima, media, or adventitia occurs with some frequency in cerebrospinal vessels of all types and caliber. Fibrous thickening of the adventitia of small meningeal and CNS arteries can be accompanied by variable degree of extension of fibrosis into other layers of the vessel wall. A preferential site is the choroid plexus. In old horses, a similar pattern of fibrosis occurs in vessels, and the adventitia may be preferentially affected. Amyloid deposits in meningeal and cerebral vessels are reported in older dogs and other animals. Mineralization (deposition of calcium or iron salts) of cerebral blood vessels occurs in the brains of several species but is especially common in adult horses. Vessels of the internal capsule, globus pallidus, cerebellar dentate nucleus, and infrequently the hippocampus are preferentially affected in horses, cattle, and less commonly, dogs. Meningeal vessels in old cats, old horses, and cattle and vessels of the choroid plexus in old cats are other sites of vascular mineralization. Overt ischemic damage is rarely associated with these nonlipomatous vascular lesions in any species, therefore clinical signs are not seen with this lesion.
Lysosomal Storage Diseases
Dysfunction of lysosome-mediated degradation of products (substrates) of normal cellular metabolism results in diseases referred to as lysosomal storage diseases. These substrates cannot be degraded by lysosomes, and the accumulated substrate eventually results in death of the affected cells.
Cell death is the endpoint of a chronic and progressive process of substrate accumulation that interferes with cellular biochemical processes and transport systems. When neurons or myelinating cells die, they release their accumulated substrate into adjacent tissue. Macrophages are recruited from the bloodstream as monocytes, and they phagocytose cellular debris and unprocessed substrate released from dead cells. Macrophages, however, have the same genetic defect and thus also accumulate substrate in their lysosomes. Although less vulnerable to the effects of substrate accumulation, macrophages eventually die and their released substrate is phagocytosed by additional macrophages recruited from the blood.
Lipid storage diseases, such as globoid cell leukodystrophy, are covered in more detail later. Features of some selected lysosomal storage diseases of animals are provided in Table 14-7.
TABLE 14-7
Classification of Selected Lysosomal Storage Diseases that Involve the CNS of Animals
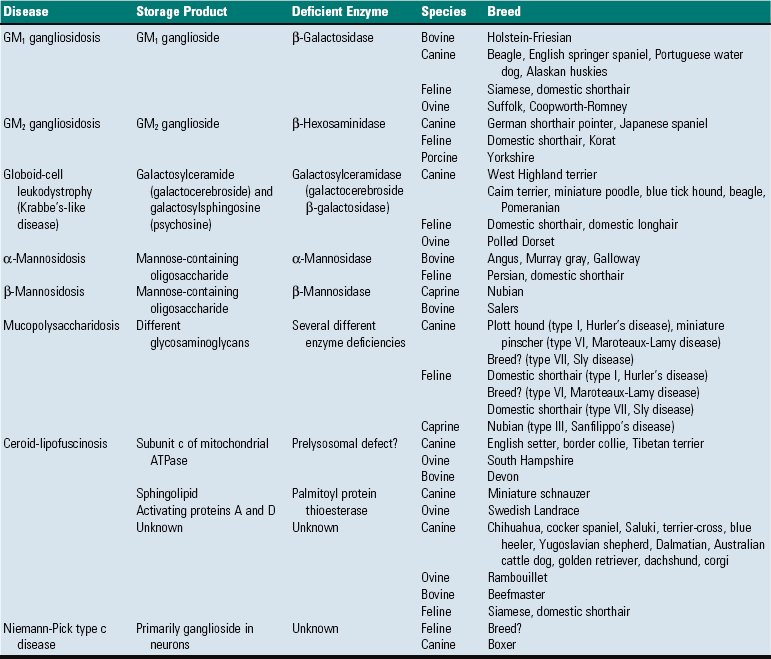
?, Unknown; ATPase, adenosine triphosphatase.
Lysosomal storage diseases were originally thought to develop exclusively because of mutations that result in a reduction in lysosomal enzyme synthesis. More recently, however, it has become clear that there are other defects such as the following:
1. Synthesis of catalytically inactive proteins that resemble normal active enzymes.
2. Defects in posttranslational processing (glycosylation, phosphorylation, addition of fatty acids in the Golgi) of the enzyme, which results in it being misdirected to sites (extracellular) other than to lysosomes.
3. Lack of enzyme activator (an enzyme that normally increases the rate of an enzyme-catalyzed reaction) or protector protein (facilitate repair and refolding of stress-damaged proteins).
4. Lack of substrate activator protein required to assist with the hydrolysis of substrate.
5. Lack of transport protein required for elimination of digested material from lysosomes.
Characterization of lysosomal disorders has therefore been broadened to include involvement of any protein that is essential for normal lysosomal function.
The best-known diseases are characterized by accumulation of the substrate or substrate precursors and sometimes even by the absence of a critical metabolic product for normal lysosomal function. As a general principle, cell swelling and cytoplasmic vacuolation occur because of the accumulation of unprocessed substrate in the lysosomes; therefore differences in the size and appearance of cells (neurons versus hepatocytes) depend on the availability of the substrate (carbohydrate or lipid) in the organ system. Many lipids and glycolipids are unique to the nervous system, thus, when there is a lysosomal defect, neural cells often accumulate in the substrate.
Examples of lysosomal storage diseases that affect humans and animals are the gangliosidoses. With few exceptions, these diseases are inherited in an autosomal recessive pattern. They are also often gene-dose dependent and correspondingly, recessive homozygotes manifest the disease, whereas heterozygotes are phenotypically and functionally normal, but the affected enzyme’s activity is reduced by approximately 50% of normal. The age of onset of clinical signs and the severity of the disease process can vary among the different diseases because the deficiency of the involved enzyme is not always the same. If the gene defect is such that the mutant enzyme is not synthesized at all, there is an early onset of a severe disease. Conversely, if there is some residual synthesis of the deficient enzyme, later onset and a milder form of the disease result because partial catabolism of the accumulated substrate permits a longer period of time before the lysosomes are so distended with substrate that they cause loss of cell function.
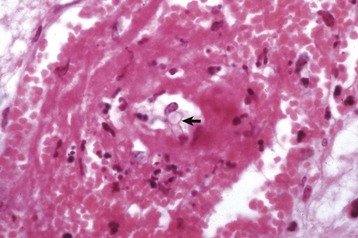
Gross lesions of the CNS vary among the different types of lysosomal storage diseases. Brain atrophy occurs with globoid leukodystrophy in latter stages of the diseases because of the loss of myelin. Brain atrophy can also be seen with ceroid-lipofuscinosis but is not prominent in other lysosomal storage diseases, although brains of animals with gangliosidoses can have a firm, rubbery consistency. Microscopically, affected neurons often have a foamy, finely vacuolated, or granular cytoplasm, which is a reflection of the degree to which the stored material is removed during histologic processing (Fig. 14-63). The specific features of the stored material can be best appreciated by ultrastructural examination.
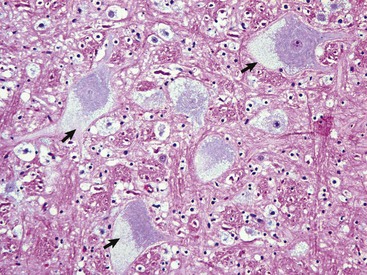
Fig. 14-63 Glycogen/carbohydrate (lysosomal) storage disease, brainstem, neuron cell bodies, cat.
Note the enlargement of the neuron cell bodies, displacement of nuclei, and accumulation of unprocessed substrate in the cytoplasm of the neuronal cell bodies (arrows) giving the appearance of “foamy” cytoplasm. H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Ceroid-lipofuscinosis is a lysosomal storage disease characterized by abnormal sphingolipid (lipopigments) metabolism that occurs in cats, dogs, cattle, and sheep. Its lysosomal dysfunction has not been clearly identified, but experimental studies have shown alterations in the activity of palmitoyl-protein thioesterase and concentration of acid protease. The disease resembles other lysosomal storage diseases in that it can have a recessive mode of inheritance, but it is dissimilar in that it has no gene-dose effect. Brain atrophy occurs with ceroid-lipofuscinosis in later stages of the diseases (in sheep) (see Fig. 14-17). The atrophy, which most frequently involves the cerebral cortex but also sometimes the cerebellum, can result in a 50% reduction in brain weight. The cerebral hemispheres are increased in firmness and often have a tan color, whereas the gyri are thinned and the sulci widened, a clear indication of cerebrocortical atrophy. Microscopically, the cytoplasm of affected neurons has an eosinophilic granular material (with H&E staining) and a decrease in the number of neurons. Reactive astrogliosis is prominent, and microgliosis may also be observed.
Globoid Cell Leukodystrophy: As discussed previously, lysosomal storage generally refers to a cellular alteration in which an increased amount of substrate material, which normally is degraded, accumulates within lysosomes, often eventually resulting in cell death. These diseases have a hereditary basis, occur in young animals, and are transmitted in an autosomal recessive pattern. Features of some selected lysosomal storage diseases of animals are given in Table 14-7.
Globoid cell leukodystrophy, a sphingolipidosis, is a lysosomal storage disease; its principal lesion is primary demyelination involving oligodendrocytes of the CNS and Schwann cells of the PNS. The disease, which has an autosomal recessive inheritance in the Cairn and West Highland white terrier, is generally seen in younger animals, often younger than 1 year old. It has also been described in beagles, miniature poodles, basset hounds, Pomeranians, blue tick hounds, and domestic short- and long-haired cats.
Mechanistically, the proposed sequence of events in this disease includes (1) early “normal” myelination that progresses up to a certain stage; (2) disruption of normal myelin turnover because of deficient galactosylceramidase activity; (3) degeneration and necrosis of myelinating cells because of the accumulation of psychosine; (4) primary demyelination; (5) recruitment of phagocytes, both resident microglia and trafficking blood monocytes; and (6) infiltration of macrophages, which become globoid cells after phagocytosing myelin byproducts into the nervous tissue. The last-named changes occur in response to the demyelination and unmetabolized galactocerebroside.
Affected oligodendroglia and Schwann cells are deficient in the lysosomal hydrolase, galactosylceramide β-galactosidase (GALC), which is responsible for degradation of galactosylsphingosine (psychosine) and galactosylceramide (galactocerebroside). Psychosine is highly toxic and because it is not degraded, it has been hypothesized that it accumulates during the disease and causes direct injury to oligodendrocytes and Schwann cells, possibly through an apoptotic mechanism of cell death, in part, mediated by psychosine-induced production of cytokines and inducible nitric oxide synthase (iNOS).
In globoid cell leukodystrophy, the composition of myelin is not qualitatively abnormal. Galactosylceramide is highly concentrated in myelin but is nearly absent in systemic organs except for the kidney. Peak synthesis and turnover of galactosylceramide coincides with the peak period of myelin formation and turnover during the first year of life. GALC activity also increases in relation to the galactosylceramide peak. Myelination continues at a slower rate as animals mature, and in an adult myelin formation is stable with minimal turnover.
A deficiency in GALC activity results in the accumulation of galactosylceramide, especially during the early phase of myelin maturation and turnover, and in the formation of globoid cells discussed later. Psychosine also accumulates, leading to rapid and massive degeneration of oligodendroglia and Schwann cells, extensive myelinolysis, and reduction in myelination.
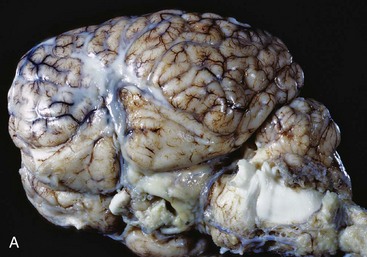
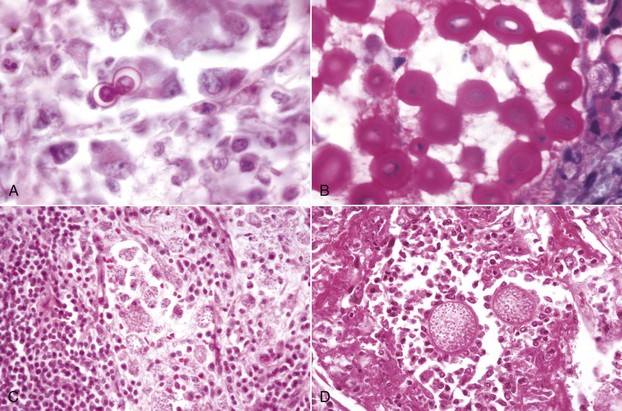
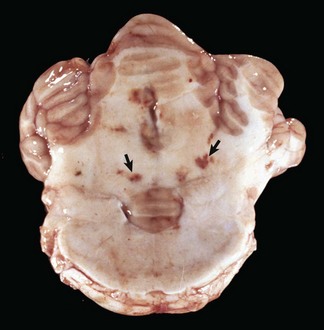
Gross lesions of the CNS are characterized by a gray discoloration of the white matter, especially of the centrum semiovale of the cerebral hemispheres and white matter of the spinal cord (Fig. 14-64). The lesion in the spinal cord tends to start in the peripheral white matter and spread inward. Microscopically, such areas have pronounced loss of myelin (Fig. 14-65, A) and prominent globoid cells containing galactocerebroside that can be demonstrated with a PAS stain (Fig. 14-65, B). Peripheral nerves are also affected and lesions are typified by primary demyelination and secondary axonal degeneration. Small sensory branches of peripheral nerves are useful sites to take biopsies to make diagnoses (see Fig. 14-112).
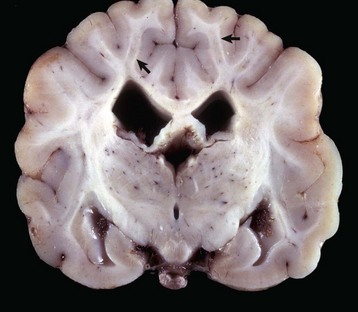
Fig. 14-64 Lipid storage disease, globoid cell leukodystrophy, brain, transverse section at the level of the mammillary body, dog.
The white matter, especially of the gyri, has an off-white to light gray appearance (arrows). Macrophages (globoid cells) derived from blood monocytes (also enzymatically deficient in β-galactocerebrosidase) accumulate in white matter to phagocytose galactocerebroside and oligodendroglial debris secondary to the toxic effects of galactosylsphingosine (psychosine) on oligodendroglia (and in the PNS Schwann cells). There is also bilateral hydrocephalus of the lateral ventricles, presumably hydrocephalus ex vacuo, as a result of the loss of neurons and their axons. (Courtesy Dr. H.B. Gelberg, College of Veterinary Medicine, Oregon State University.)
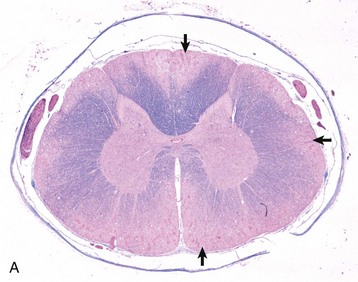
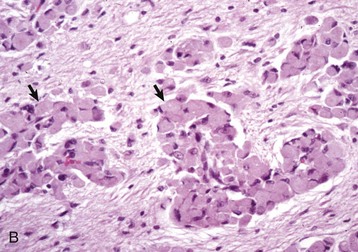
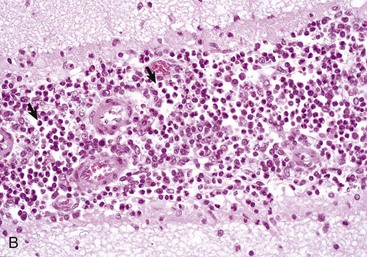
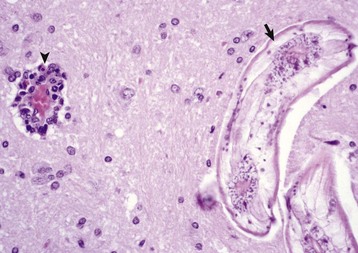
Fig. 14-65 Globoid cell leukodystrophy, dog.
A, Spinal cord. This section of spinal cord has been stained with Luxol fast blue, a histochemical reaction that stains myelin blue. Note the loss of myelin from the periphery of the cord, where axons are heavily myelinated (arrows), the first area to be affected. Luxol fast blue stain with a nuclear fast red counterstain. B, Cerebrum. Globoid cells deficient in β-galactocerebrosidase (arrows), usually located in the perivascular space, increase in size and number because they are unable to degrade galactocerebroside to a less complex substrate. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Clinically, affected animals are ataxic and have limb weakness and tremors that progress to paralysis and muscular atrophy. Poor vision or blindness may also occur.
Traumatic Injury
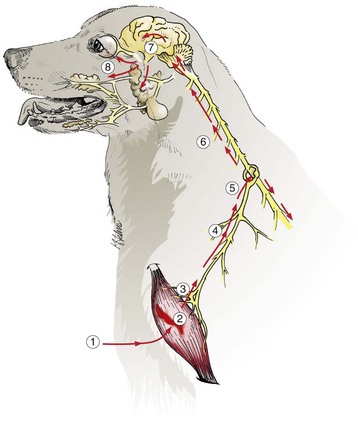
Traumatic injury of the CNS is caused by physical insults such as compression, stretching, and/or laceration of neurons/axons. When the brain and spinal cord collide with the bony ridges lining the cranial vault and the bony wall of the vertebral canal, respectively, or when axial, rotational, and angular forces (Fig. 14-66) are applied to neurons and axons during trauma, the force of impact and sudden acceleration of neurons and axons, both within the CNS and in the adjacent cranial and spinal nerves, can cause them to compress, twist, stretch, and tear. Concurrently, the same type of forces can injure blood vessels in the CNS and leptomeninges and may result in small hemorrhages or hematomas within the parenchyma of the brain and in the leptomeninges (subarachnoid space) (Fig. 14-67).
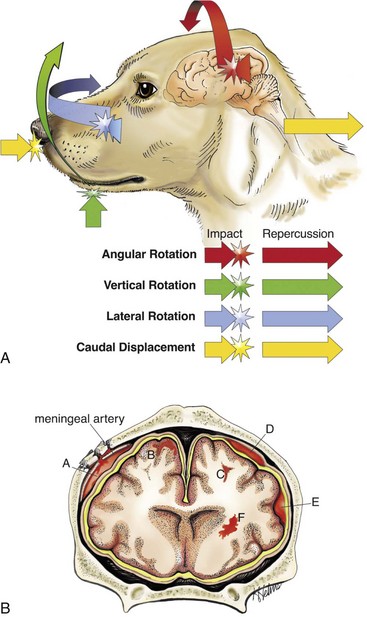
Fig. 14-66 Traumatic CNS injury and hemorrhage.
A, Axial, rotational, and angular energy applied to the brain during trauma determine the severity of shear, tensile, and compressive forces that cause neuronal and vascular injury. B, Locations of hemorrhage, dog, brain. (A) Epidural hemorrhage with laceration of meningeal artery; (B) cortical hemorrhage; (C) hemorrhage in subcortical white matter; (D) subdural hemorrhage secondary to laceration of a bridging vein; (E) subarachnoid hemorrhage; (F) deep intracerebral hemorrhage. (A courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. Redrawn and modified from Leech RW, Shuman RM: Neuropathology: a summary for students, Philadelphia, 1982, Harper & Row.)
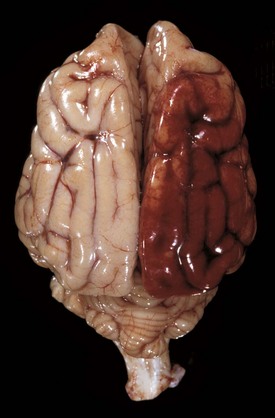
Fig. 14-67 Leptomeningeal (subarachnoid) hemorrhage, brain, right cerebral hemisphere, dog. (Courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University.)
Diffuse brain injury (often present in concussion) is caused by acceleration/deceleration forces applied to many areas of the CNS rather than in one specific location. Diffuse brain injury involves neuronal processes, cell bodies, transmitter mechanisms, and macroglial cells and blood vessels. The most severe injury to axons appears to be at the gray matter–white matter junction. A variant of diffuse brain injury is diffuse axonal injury in which axons of large myelinated nerve fibers are injured by shearing forces. In diffuse brain and axonal injury, mechanical deformation results in physical disruption of cell membranes and cytoskeleton and increased membrane permeability, resulting in major ionic fluxes in and out of the cell. Such changes can lead to excessive release of glutamate, excitotoxicity and cell death, free radical formation, apoptosis, and delayed inflammatory responses. The end result can be Wallerian degeneration.
In general, trauma of the CNS in animals occurs less frequently than in humans. Animals are not exposed as frequently to potentially trauma-causing situations (e.g., automobile travel) as are humans, and there are anatomic differences (see later), including a quadruped posture, which increases stability and helps protect the brains of animals.
Among animals, trauma to the brain is probably most frequently caused in dogs as a result of automobile-induced injury and in cats from falls from significant heights (high-rise apartment buildings, balconies, and roofs). Even after falling from considerable heights, cats often have remarkably minor injury to the CNS. Other examples include fracture of the spinal column or cranium of jumping horses and fractious animals, such as horses and ruminants, during excitement and restraint.
Predisposition to cerebral trauma is also influenced by anatomic differences. The percentage of brain mass in relation to skull size is much less in domestic animals than in primates, and in the bovine and porcine species, the cranial cavity is additionally protected dorsally by prominent frontal sinuses. Birth trauma, which can be important in humans, is essentially insignificant in animals because in the latter, the shoulders and particularly the pelvis, rather than the head, are likely to be compressed in the birth canal. Exceptions to this generalization include brachycephalic breeds of dogs. Several factors also influence the susceptibility of the spinal cord to trauma. The amount of space between the spinal cord and the wall of the vertebral canal is very important in determining the degree of injury after edema or compression with disk herniation. This space is greater in the cervical area of the dog than at the thoracolumbar level. Thus disk herniation at the latter area is more likely to result in severe spinal cord injury.
Functional factors also play an important role in brain injury. The brain of a freely movable head is much more susceptible to injury than one that is fixed in place. The increased susceptibility of the former has been attributed to the ability of the cranium (the bone) and its contents (the brain) to impact each other after nonpenetrating trauma. This interaction occurs because the brain does not completely fill the cranial cavity, thus resulting in a very short distance (or space) between brain and bone.
The type and location of the lesion (contusion and/or hemorrhage) depends on the location of the point of contact and the direction of the blow, relative to the head. If the blow is directly on the back or front of the head, the head and brain will move straightforward or backward, respectively (see Fig. 14-66, axial force). If the blow is horizontal to the top of the head or horizontal to the rostral portion, the head will rotate on the atlanto-occipital axis (angularly and rotationally, respectively). In the case of an axial blow to the back of the head (an animal falling onto the back of its head), the head and thus the cranial vault will accelerate faster than the brain, which will lag behind, and the caudal aspect of the vault may move forward and contact the caudal aspect of the cerebral hemispheres, usually the occipital cortex. In the case of an animal falling onto the back of its head, the head will accelerate abruptly and the momentum will carry the brain caudally, where it may strike the inside of the caudal brain in the cranial vault. A vertical blow delivered directly down onto the dorsal surface of the head will have the same type of result on the dorsal aspect of the cerebral hemispheres as the blow to the back of the head. It is more common, from the same blows described previously, to see hemorrhage, usually subarachnoid, on the opposite side of the point of impact with the brain. For example, a vertical blow to the top of the head causes hemorrhage of the ventral surface of the medulla and cerebral hemispheres. Thus, after an impact on a stationary, freely movable head, the bone of the cranial vault will move on the stationary brain and injure it (coup injury) and on the opposite side the nerves and blood vessels will be stretched, possibly resulting in nerve damage and hemorrhage (contrecoup injury). In addition, the mass and velocity of the object striking the head are important. Trauma after impact of a relatively large blunt object can create notable head movement and a large-impact injury, whereas a small object, such as a bullet moving at a high rate of speed, can cause less head movement and a smaller but deeper area of direct tissue damage. In summary, the basic concept is the transfer of kinetic energy by the striking object to the head. A large blunt object will cause the head to accelerate without deforming it; a smaller object, such as a bullet, will penetrate.
Factors involved in the protection of the brain include the rigidity of the cranium (depending on age), the round shape of the dorsum of the skull, the structure of the parietal, occipital, and temporal cranial bones (two layers of compact bone separated by spongy bone referred to as diploë), cranial sutures, sinuses, ridges in the floor of the cranial cavity, meninges, and CSF. The spinal cord is enclosed and protected by the vertebral column, which is surrounded by soft adipose tissue and muscle. Other structures that help protect the spinal cord by absorbing shock are the intervertebral disks and the cancellous bone of the vertebrae. Vertebral ligaments maintain the alignment of the vertebral column; denticulate ligaments support the spinal cord in the middle of the vertebral canal, and the meninges, particularly the CSF, cushions trauma.
Clinically, animals with CNS trauma have signs referable to the area injured, brain, or spinal cord. With brain trauma, signs can vary widely and range from unconsciousness lasting a few seconds followed by complete recovery and return to normal function to depression, abnormal behaviors such as disorientation and irritability, semiconsciousness with responsiveness only to noxious stimuli, and unconsciousness with no response to any stimulus. With spinal cord trauma, signs vary, depending on the severity of the injury and the rate of onset. Paralysis results from severance of the cord or ruptured disks. Paresis and ataxia result from less severe injury.
Concussion: Concussion is often thought of as a clinical designation of temporary loss of consciousness with recovery after head injury. As in humans, a movable head is much more susceptible to trauma than a fixed, supported one. Application of an appropriate concussive trauma to the mobile head of an animal results in a reversible cerebral dysfunction that lasts for a matter of seconds or at most, a few minutes and is usually reversible, with stronger blows causing more severe injury and even death.
Concussive injuries of the diffuse type also occur in animals, but there are some differences between animals and humans. For example, it is difficult to produce severe concussion in animals because the margin between the force of a stunning blow and one causing fatal injury is very small. The smaller the brain, the less vulnerable it is to rotational forces and the larger are the forces necessary to cause concussion. It should be noted, however, that concussion, particularly when there is rapid recovery from unconsciousness, can occur more frequently than appreciated in animals because the clinical signs may not be recognized.
Diffuse brain injury does not usually cause gross lesions. Microscopic lesions detected in animals include diffuse axonal injury characterized by axonal degeneration, and this may be followed by Wallerian degeneration. Damage to neurons ranges from central chromatolysis to death and neuronal loss. The more severe forms of diffuse brain injury can also have generalized acute brain swelling caused by unregulated vasodilation, which can be followed after some time by cerebral edema.
Spinal concussion is the term applied to the immediate and temporary loss of function that sometimes follows severe direct blows to the spinal column. Loss of function usually affects the long tracts/bundles of nerve fibers (funiculi), but usually there is no demonstrable external change in the vertebrae or spinal cord. As with cerebral concussion, there is often only a temporary functional disability of the cord after injury, but if the trauma is more severe, permanent neurologic deficits can result.
Contusion: Contusion means bruising, which is generally associated with rupture of blood vessels, and in the cerebrum, this injury results in grossly detectable lesions, such as hemorrhage, which can, like concussion, result in unconsciousness and even death. The factors that cause concussion and contusion can occur together in the same animal. Lesions can be superficial (cerebral gyri) or more central (brainstem), and there can be concurrent skull fractures.
Although hemorrhage is the most common lesion, contusion of the brain can also result in tearing of CNS tissue. Tearing results in tissue necrosis and neuronal loss. Two designations are used to identify the location of contusive injury. A coup contusion is located at the impact site, and a contrecoup contusion at a location on the opposite side of the brain. When the two lesions occur together (coup-contrecoup or contrecoup-coup), the first term indicates the site of most severe injury. Box 14-10 summarizes the pathogenesis of coup-contrecoup contusion.
BOX 14-10 Pathogenesis of Coup-Contrecoup Contusion
Conditions Involved in Coup-Contrecoup Contusion
1. Head freely movable.
2. Head accelerated rapidly (by being struck by a broad object, such as an automobile) or decelerated rapidly (head strikes pavement after a fall from a standing position).
3. Because the brain does not fill the cranial vault, it may lag behind the movement of the cranium when the head is accelerated or decelerated rapidly.
4. As a result, the inside of the cranial vault may strike the stationary brain at the point of impact (coup injury), or the lesion may occur on the opposite side (contrecoup), either from the stretching and tearing of vessels at that site or by the brain being struck by the inside of the cranial vault on the opposite side when there is reduced amount of cerebrospinal fluid buffer present.
Many investigations have been made to determine the mechanisms involved in the development of contusive lesions, and the kinetics are complicated and still not completely resolved. Factors considered to be significant include the ability of the head to move freely, the occurrence of a rotational movement of the brain over rough surfaces on the inside of the cranial vault, and the development within the cranial cavity of positive and negative pressures and gravitational forces. The basic results of the different types of blows to the heads of animals have been discussed previously, and it is interesting to compare these with the lesions in humans. Several neuropathologic principles have been generally accepted regarding craniocerebral contusive trauma in humans, as follows:
1. A blow to the stationary (but freely movable) head produces a cerebrocortical coup contusion beneath the point of cranial impact, but with rare exceptions causes no cerebrocortical contrecoup contusion opposite the point of cranial impact. This outcome is not always true of animals, in which a blow to the dorsum of the head from a flat object, such as a spade, causes marked subarachnoid hemorrhage on the ventral surface (contrecoup).
2. An impact of a moving head (moving before impact, as in a fall from a standing position) against a firm or unyielding surface causes a cerebrocortical contrecoup contusion opposite the point of cranial collision (often at the poles and inferior surfaces of the frontal and temporal lobes), but with rare exceptions there is no contusion beneath the point of impact. In contrast, horses that fall backward and land on their backs and strike the occiput often have subarachnoid hemorrhage over the occipital poles of the cerebral hemispheres.
3. Falls from great heights and crushing of the head between a strong external force and unyielding surface are generally not associated with the occurrence of contrecoup lesions.
Dawson and co-workers proposed a mechanism for both contrecoup and coup injury of the human brain that also addressed the specific deficiencies of mechanisms that have been advanced by others. An example explaining the mechanism of contrecoup injury states that when a person falls backward from a standing position because of loss of balance, the gravitational torque acting on the body causes downward acceleration of the head in excess of the acceleration as a result of gravity. Under these circumstances the brain lags toward the trailing anterior surface of the cranium before impact (causing displacement of the protective CSF layer between the brain and skull) and permits compressive stress to develop at this site, although impact occurs at the opposite side of the head. Because dissipation of the CSF at the anterior (contrecoup) site allows compressive stress to be focal at this contrecoup site and because of the shearing stress generated, injury occurs. In addition, a relative rotational gliding motion between the brain and skull is produced when the impact suddenly stops the skull’s motion and rotation, thus creating an additive shearing stress because the fluid lubrication necessary to facilitate gliding of the brain over the cranial surface is reduced. The concentration of this rotational shearing stress is likely to occur beneath the frontal and temporal lobes because of the rough surface of the skull that exists in this location. In contrast to contrecoup injury, coup contusions occur infrequently in this type of fall. In such situations, the brain lags away from the impact site, which results in a thickening of the protective CSF layer between the brain and skull immediately beneath the point of impact, which helps explain the absence of coup injury in primates and humans in typical moving head trauma.
Coup injury in humans can occur when a stationary but freely movable head is impacted. In this type of trauma, there is neither brain lag nor disproportionate distribution of CSF before impact, which accounts for the typical absence of contrecoup contusions. With regard to falls from great heights, the dynamics involving rotation of the body about a fixed point of ground contact associated with a fall from a standing position do not occur. Because gravity produces no torque on a freely falling object, no angular acceleration of the body is produced, therefore such a fall is a true free-fall state that is associated with an absence of brain lag. For this reason, contrecoup lesions occur infrequently with this type of trauma. One point should be emphasized with regard to the evaluation of coup and contrecoup cortical contusions just described. Displacement of bone associated with skull fracture can contuse the subjacent brain, regardless of the resting or moving status of the head, and such fracture-contusions have nothing to do with the coup-contrecoup mechanisms described. Also, even though the basic mechanisms discussed earlier apply to humans, they should also be considered when evaluating cerebral contusions of domestic animals, but the situation in human brains is made far more complex because of the numerous wide bony ridges that project into the cranial vault.
Evaluation of spinal cord trauma should include examination not only of the spinal cord but also of the vertebral column and spinal nerve roots. Injuries to the spinal cord can involve concussion, contusion, hemorrhage, laceration, transection, and compression secondary to vertebral trauma and fracture. Contusion in the spinal cord is characterized by vascular tears, hemorrhage, and necrosis. Tears are generally focal and grossly visible. Contusion can occur without fracture of the vertebral column, with fracture, and with fracture plus dislocation of the spinal column. The latter combination can result in tearing and transection of the spinal cord.
CNS Hemorrhage: Although hemorrhage and hematomas in animal brains can be caused by a wide variety of injuries, trauma to the head is the most common cause. Box 14-11 lists common causes of brain hemorrhage)
BOX 14-11 Common Causes of Hemorrhage in Animal Brains
Vasculitis (such as Histophilus somni [formerly Haemophilus somnus] infection)
Damage to endothelium lining blood vessels (by the virus of canine infectious hepatitis, by septicemia or endotoxemia, by immune complexes, or by parasite larval migration)
Trauma
Contusion
• Coup lesion
• Contrecoup lesion
Penetrating wounds
After trauma to the head, hemorrhages can develop in the epidural, subdural, and subarachnoid space, under the pia mater (subpial), and in the brain (see Fig. 14-66, B). Hemorrhage can be diffuse (see Fig. 14-67) or focal (e.g., hematomas) (Fig. 14-68). Such hemorrhages can result from sliding of the brain over bony ridges (jugae) within the cranium, with resultant stretching and tearing of blood vessels and tissue, after the cutting and penetration of bone fragments from skull fracture. Cerebral epidural hemorrhage, which is not commonly described in animals, has been reported in the horse, especially jumpers, resulting from falls while working. Epidural hemorrhage does not usually occur because the dura is tightly adhered to the inner surface of the calvarium and there is no epidural space. In trauma causing skull fractures, bleeding from local blood vessels can separate the dura from the calvarium, forming a hematoma in the epidural space.
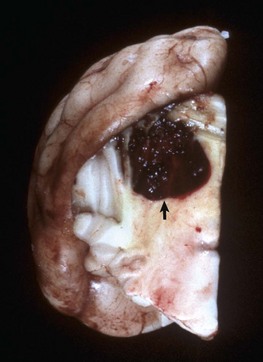
Fig. 14-68 Hematoma (arrow), cerebellum, transverse section, dog.
Traumatic injury to the head resulted in hemorrhage and the formation of a hematoma from shear, tensile, compressive axial, rotational, and angular forces. (Courtesy Dr. H.B. Gelberg, College of Veterinary Medicine, Oregon State University.)
Subdural hemorrhage, which is an extravasation of blood between the dura mater and arachnoid membrane, occurs in dogs and cats. It is rare and usually diffuse and does not form hematomas as seen in humans, where they can be life threatening from compression and herniation of the brain. Subarachnoid and intracerebral hemorrhages are most common in all species after head injury. Hemorrhage can result from injury to the brain with or without a fractured cranium by the mechanisms given previously and also from penetrating objects (bullets and stab wounds).
The same types of hemorrhage that affect the brain (epidural [rare], subdural [rare], leptomeningeal, and parenchymal) also occur in the spinal cord and its meninges. Causes are similar to those for the brain.
Hematomyelia (Hemorrhagic Myelomalacia): Traumatic injury to the spinal cord can cause stretching and tearing of blood vessels, usually arterioles, within the gray matter, resulting in hematomyelia. Hematomyelia is also particularly associated with severe type I disk herniation. If larger vessels are torn, blood pressure can force blood into the gray matter. This outcome results in the formation of a dissecting blood-filled cavity ascending and/or descending initially within the gray matter of the spinal cord (Fig. 14-69). This lesion, which is characterized by a softening to semiliquefaction (myelomalacia) and hemorrhage of the tissue, can develop within 12 to 24 hours after injury and can progress both cranially and caudally from the original site of trauma. As the cavity extends cranially, the hemorrhage at the original site also extends into the white matter and can transect the spinal cord. If this lesion extends to the fifth cervical cord segment, the phrenic nerves to the diaphragm will be denervated and respiratory paralysis will result. Bleeding continues until pressure in the blood-filled cavity is equal to the vascular pressure or until bleeding ceases because of hemostasis in the vessel. Hemorrhage can also result from bleeding in arteriovenous malformations within the spinal cord. Hematomyelia is characterized by neurologic deficits consistent with a sudden onset of ascending or descending flaccid paralysis and sensory abnormalities.
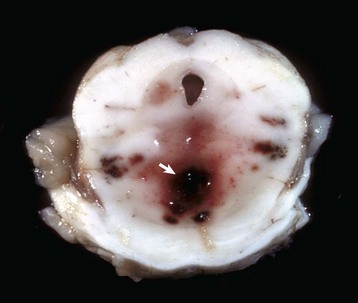
Fig. 14-69 “Duret” hemorrhages, brainstem, transverse section at the level of the pons.
Note the multiple hemorrhages in the periventricular white matter (arrow). These hemorrhages are the result of twisting of the brainstem on a longitudinal axis from rotational and axial forces. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Compressive Injury: Diseases resulting in compressive injury can affect the brain, spinal cord, or both concurrently. In the brain, diseases, such as neoplasia, reticulosis, canine granulomatous meningoencephalitis, and chronic cerebral abscesses, can compress adjacent nervous tissue. In the spinal cord, compression can be intramedullary (within the spinal cord) or extramedullary (outside the spinal cord). Causes of intramedullary compression include hemorrhages, neoplasms such as nephroblastoma of the young dog, and chronic expansile inflammatory diseases. Extramedullary compression can be caused by intervertebral disk herniation in the dog; cervical stenotic myelopathy (wobbler syndrome) in the horse and dog; vertebral fracture and dislocation; neoplasms of the meninges, such as the meningioma; nerve rootlets, such as nerve sheath tumors; or tumor metastasis, such as lymphosarcoma. Finally, developmental anomalies of bone, such as atlanto-occipital malformation, and vertebral deformities with hemivertebrae, such as scoliosis, lordosis, and kyphosis (Fig. 14-70), can result in compression of the spinal cord.
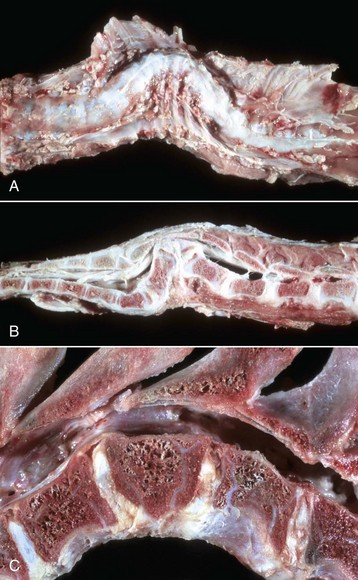
Fig. 14-70 Vertebral abnormalities, vertebral column.
A, Scoliosis, thoracic vertebrae, ventrodorsal view, sheep. Lateral deviation of the spinal column. B, Kyphosis, thoracolumbar vertebrae, lateral view, sheep. Dorsal deviation of several vertebrae and a wedge-shaped vertebra (hemivertebra; Fig. 14-70, C) have resulted in compression of the spinal cord. C, Hemivertebra, lumbar vertebrae, dog. Note that the craniodorsal portion of the body of the hemivertebra has protruded into the vertebral canal. (A and B courtesy College of Veterinary Medicine, University of Illinois. C courtesy Department of Veterinary Biosciences, The Ohio State University.)
Compression of CNS tissue causes neuronal dysfunction by impeding normal anterograde and retrograde axoplasmic flow in axons (see Web Fig. 14-1). In addition, compression of nerves may result in reduced blood flow to nerves and thus also contribute to neuronal dysfunction. Mild compression can result in partial blockage of slow axoplasmic flow and gradual accumulation of neurofilaments and microtubules, which results in mild enlargement of the axon proximal to the compression site and atrophy of the axon distal to the compression. Eventually, with a long period of time of complete blockage, the distal axon is lost.
Brain Displacements: See the discussion of cerebral edema (permeability changes) in the section on Circulatory Disturbances.
Cervical Stenotic Myelopathy: Cervical stenotic myelopathy, or wobbler syndrome, is characterized by stenosis of the cervical vertebral canal, which causes compressive trauma to the cervical spinal cord (Fig. 14-71). This disease occurs primarily in young rapidly growing large breeds of horses and dogs. Reports indicate that the disease is not caused by a straightforward mechanism but apparently involves several factors (multifactorial disease). For example, stallions that have a genetic predisposition for rapid growth and large body size are reported to be at greater risk of developing the disease. Oversupplementation with protein, vitamins, and minerals used to promote rapid growth may also be another environmental factor in developing cervical stenotic myelopathy.
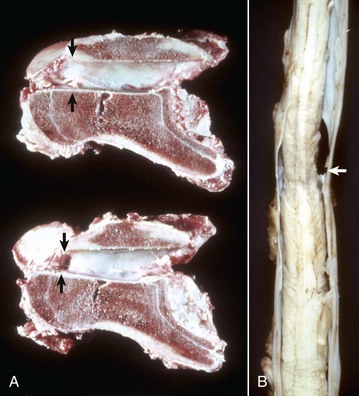
Fig. 14-71 Cervical stenotic myelopathy.
A, Cervical static stenosis, vertebral column, sagittal section, sixth cervical vertebra, horse. The vertebra on the bottom is stenotic (arrows) and will compress the spinal cord. The vertebra on the top is normal (arrows). B, Cervical vertebral instability, spinal cord, fifth cervical segment, dog. Note the narrowing of the spinal cord at the site of compression (arrow). (Courtesy College of Veterinary Medicine, University of Illinois.)
Gross and microscopic lesions of the CNS in cervical stenotic myelopathy are similar to the lesions in intervertebral disk herniation. The severity depends on the speed and degree to which the compression is applied and the specific area of the cord that is involved. Central nervous tissue can tolerate a considerable degree of compression if it is applied slowly. Rapid compression can lead to quickly developing hypoxia-ischemia, necrosis, and direct damage to compressed axons. Lesions detected include a spectrum of changes characterized by axonal injury and disruption of myelin sheaths resulting in Wallerian degeneration and necrosis of the gray or white matter or of both. Lesions can be visible grossly from the exterior but are more commonly seen on cross-section of the spinal cord.
Microscopically, particularly at the site of injury, there is initial swelling of axons followed after several days by loss of architecture of the CNS as a result of necrosis and a beginning accumulation of gitter cells that have phagocytosed the lipid-rich tissue debris. Eventually the necrotic area is cleared and a cystic space is formed, which is surrounded by varying degrees of astrocytosis and astrogliosis, although not usually prominent unless there is severe destruction. Rostral and caudal to this location, the lesion is primarily one of Wallerian degeneration in the white matter, and the pattern of lesion development seen depends on the level of the spinal cord that is examined relative to the site of compression. At the site of injury, all parts of the white and gray matter of the spinal cord are affected and often necrotic, if the compressive force is sufficient. Rostral to this site, white matter degeneration is generally limited to the ascending tracts in the dorsal funiculi and the superficial portions of the dorsolateral part of the lateral funiculi. Caudal to the area of injury, degeneration is limited to the descending tracts in the ventral funiculi, and the more central portions of the lateral funiculi. It should also be noted that (1) a lesion may occur at the point of compression because of ischemia, but a lesion can also occur on the opposite side of the compression also because of ischemia that results from compression of the tissue against bone on that side, and (2) Wallerian degeneration can be observed in distal segments of affected axons far from the point of compression within the spinal cord. In the former case, compressive forces can be transferred through the spinal cord to axons and blood vessels away from the contact point, whereas in the latter example, degeneration of the distal axon segments can extend for a length of centimeters to meters away from the point of contact.
Cervical stenotic myelopathy has been known for many years to affect horses and more recently has been recognized in the large dog breeds. The disease in the horse has been referred to by several designations: the wobbler syndrome, wobbles, equine incoordination, and more recently, cervical stenotic myelopathy. The disease has been described in many horse breeds.
Cervical stenotic myelopathy in the horse has been divided into two syndromes: cervical static stenosis and cervical vertebral instability (dynamic stenosis). Cervical static stenosis commonly affects horses 1 to 4 years of age. The spinal cord is compressed at C5 through C7 as the result of an acquired dorsal or dorsolateral narrowing of the spinal canal (see Fig. 14-71). The stenosis is due to formation of bone that requires time to develop. The compressive effect with this type of stenosis is present regardless of head position. The second form of cervical stenotic myelopathy (cervical vertebral instability [dynamic stenosis]) occurs in horses ranging in age from 8 to 18 months and is characterized by a narrowing of the spinal canal during flexion of the neck, primarily at C3 through C5 vertebrae.
A disease process with many similarities to that in the horse also affects the dog. It has been known as wobbler syndrome, vertebral instability, vertebral subluxation, and cervical spondylolisthesis. The disease has been most frequently described in the Great Dane and Doberman pinscher breeds but has also been reported in the Saint Bernard, Irish setter, fox terrier, basset hound, Rhodesian ridgeback, and Old English sheepdog. Dogs can have signs develop between 8 months and 1 year of age, with a range of 1 month to 9 years. Great Danes tend to have lesions develop at a young age (8 months to 1 year), whereas Doberman pinschers are generally older, often more than 1 year of age. The vertebral and associated spinal cord lesions in the dog have been reported to most often involve the caudal cervical area from C5 through C7 vertebrae. An exception is the basset hound in which C3 is affected.
Tumors: In the brain, diseases, such as neoplasia, cause compression of adjacent nervous tissue. Compression of CNS tissue causes neuronal dysfunction by impeding normal anterograde and retrograde axoplasmic flow in axons (see the later discussion of specific types of CNS tumors).
Leptomeningeal Hemorrhage: Physical trauma to the CNS compresses, twists, and stretches blood vessels until they are torn. Such injury results in bleeding and leptomeningeal (subarachnoid) hemorrhage (see Fig. 14-67). In the spinal cord, laceration of a large artery or vein can result in prolonged bleeding that ascends or descends the leptomeninges, causing neurologic dysfunction on examination compatible with ascending or descending neurologic deficits in spinal nerve roots.
Tumors
Neoplasms of the CNS of animals are not as rare as was once believed. In fact, neoplasms occur with a frequency and variety, at least in the dog, similar to those in humans. The majority of the neoplasms described have been in the dog and cat, and a large portion of these tumors occur in the older population. The intention in discussing CNS neoplasms in this chapter is to present a brief overview of the more common or better-known neoplasms that occur in animals and is not meant to be all inclusive. The location and characteristics of the primary and common secondary (metastatic) tumors of the nervous system are summarized in Table 14-9.
TABLE 14-9
Primary Tumors of the Nervous System
WM&GM, White matter and gray matter.
*Other names for this tumor type include schwannoma, neurofibroma, and neurilemmoma.
Embryonal or Primitive Neoplasms: Considering the complexities of brain development and the fact that astrocytes and oligodendrocytes arise from common stem cells early in development, it should not be surprising that some neoplasms arising in the CNS have multiple lines of differentiation as indicated by immunohistochemistry. One such neoplasm is the rare primitive neuroectodermal tumor. This neoplasm has recently been recognized in a young calf. An additional report in a primate has been made. Peripheral neuroectodermal tumors are tumors composed of poorly differentiated embryonal cells, tend to occur in young age groups, and are aggressive. Immunohistochemical staining for specific markers confirms multiple lines of differentiation such as neuronal, astrocytic, and oligodendroglial. Typically, one cell type dominates, most commonly neuronal.
Another neoplasm of uncertain histogenesis is the rhabdoid tumor. In humans, this neoplasm most commonly arises in the kidneys and brain and is believed to develop from embryonal stem cells. As is true of other embryonal neoplasms, young individuals are commonly affected. A novel concept is the proposal that these neoplasms arise from primordial stem cells that adopt the phenotypic characteristics of cells in the tissue of origin.
Rhabdoid tumors arising in the brain have cell markers of neuronal or glial differentiation. Neoplasms in the kidneys typically lack these markers. Despite their primary location, cell morphology is similar. The cells are large and round to polyhedral, and cytoplasm is eosinophilic and abundant and contains large inclusions composed of skeins of intermediate filaments. A rhabdoid tumor has been reported in the brain of an 18-month-old dog. Immunohistochemical staining demonstrated that cells distributed throughout the tumor stained for vimentin; however, only scattered cells stained for neuron-specific enolase and GFAP. No cells in the tumor stained for S-100.
Medulloblastoma: Medulloblastoma has characteristics similar to the less frequently occurring neuroblastoma, and both are considered to arise from cells of the neuronal lineage. The cell of origin for the medulloblastoma has not been definitely determined, but it has been proposed that the neoplasm can arise from primitive cells originating in the neuroepithelial roof of the fourth ventricle that give rise to the external granular cell layer.
In animals, medulloblastomas have been reported in dogs, cats, cows, and pigs. There is a predilection for these tumors in young animals. The neoplasm chiefly occurs in the cerebellum of puppies and calves and sometimes also in adult dogs. The neoplasm is well circumscribed, soft, gray to pink, and usually does not have hemorrhages, cysts, or necrosis. The growth can compress the fourth ventricle and cause an obstructive hydrocephalus and can infiltrate adjacent structures, including the leptomeninges. It can metastasize through the CSF in the ventricles or subarachnoid space. Microscopically, the neoplasm is highly cellular and consists of round-to-elongated nuclei that have prominent chromatin in an ill-defined cytoplasm. The cells are arranged in sheets or broad bands and also can form pseudorosettes. Mitoses can be numerous.
Astrocytomas: Astrocytomas have been morphologically classified based on their degree of differentiation (histologic features in H&E stained sections) and include the following three types: diffuse astrocytomas, anaplastic astrocytomas, and glioblastoma multiforme. The degree of differentiation refers to how closely astrocytes forming the tumor resemble normal astrocytes within the CNS. Diffuse astrocytomas tend to have the most well-differentiated astrocytes, whereas glioblastoma multiforme has the most poorly differentiated astrocytes. All of these tumors are malignant; however, the degree of malignancy is often inversely related to the degree of differentiation.
Astrocytomas have been reported in dogs, cats, and cattle but are most frequently diagnosed in dogs (10% incidence) and uncommon in the cat. Brachycephalic breeds, such as Boston terriers and boxers, and dogs 5 to 11 years of age are most commonly affected. Common sites include the cerebral hemispheres, especially the temporal and pyriform lobes, thalamus-hypothalamus, midbrain, and less frequently, the cerebellum and spinal cord.
Astrocytomas often displace normal tissue; however, their gross appearance often depends on their rate of growth and degree of differentiation (Fig. 14-72). Slow-growing, well-differentiated astrocytomas (less malignant) are usually difficult to distinguish from normal tissue and are rather solid or firm and gray-white. Rapidly growing, poorly differentiated astrocytomas are more malignant and easier to discern because they have areas of necrosis, hemorrhage, cavitation, and edema.
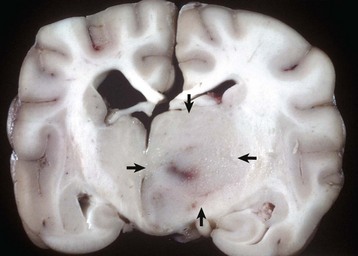
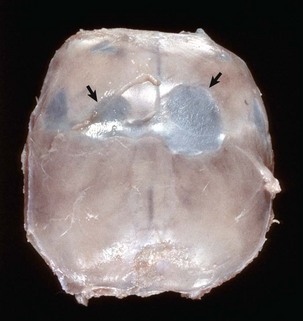
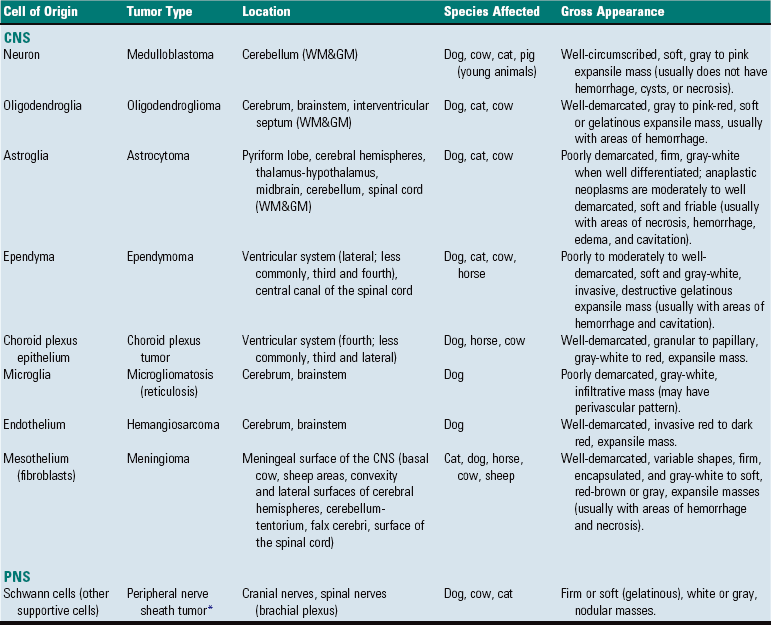
Fig. 14-72 Astrocytoma, brain, transverse section at the level of the thalamus, dog.
The deep ventromedial area (thalamus/hypothalamus) of the right hemisphere (arrows) contains a poorly demarcated, nonencapsulated, expansile mass, which is a space-occupying lesion that has displaced the midline to the left and compressed the right lateral ventricle. The left lateral ventricle is mildly dilated, most likely from compression of the interventricular foramen. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Microscopically, the more well-differentiated diffuse astrocytomas consist of a rather uniform cell type loosely organized. Cell size varies, and distinct ramifying cytoplasmic processes can be observed. Nuclei vary in size and shape and contain more chromatin than normal astrocytes. Cells tend to be arranged around and along blood vessels. The boundary between neoplastic and normal tissue is indistinct. Anaplastic astrocytomas and glioblastoma multiforme have extreme cellular pleomorphism, often with giant cell formation.
Increasing usage of immunohistochemical markers for glial cells and indicators of cellular proliferation can undoubtedly enhance the specificity and prognostic significance of the diagnosis of different glial cell neoplasms in animals. The most reliable marker for astrocytomas is GFAP, although vimentin has proved useful in some cases.
Clinical signs in animals with astrocytomas vary, depending on the location of the tumor in the CNS but can include behavioral changes, ataxia, tetraparesis, seizures, circling, and abnormal cranial nerve and proprioceptive reflexes.
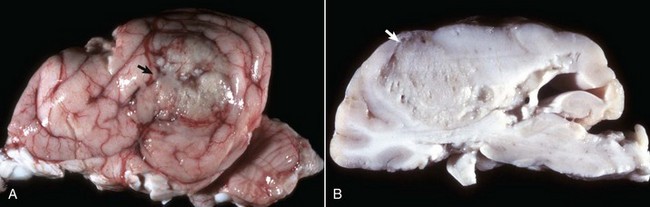
Choroid Plexus Papillomas and Carcinomas: Choroid plexus papilloma occurs most commonly in the dog but has been reported in the horse and in cattle. There is no breed predilection in dogs. In the dog, the neoplasm occurs most frequently in the fourth ventricle, but it also can be located in the third and lateral ventricles.
Grossly, the neoplasm is a well-defined, expansive, granular-to-papillary growth located within the ventricular system that is gray-white to red and compresses the adjacent nervous tissue (Fig. 14-73). Noncommunicating hydrocephalus may result from obstruction of CSF flow within the ventricular system. Microscopically, these neoplasms generally resemble the choroid plexus and are characterized by an arborizing vascular connective tissue stroma that is covered with a cuboidal to columnar epithelial layer. Mitoses are not present in the benign form. A more malignant variety, choroid plexus carcinoma, is characterized by invasiveness, presence of mitoses, additional occurrence of solid tumor growth, and a tendency to metastasize within the ventricular system or into the subarachnoid space (through the lateral apertures), where implantation in the ependyma or meninges, respectively, occurs. Currently, no reliable immunohistochemical markers for these tumors exist.
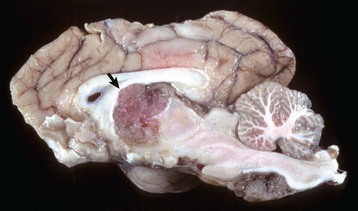
Fig. 14-73 Choroid plexus tumor (carcinoma), brain, sagittal section, dog.
The third ventricle contains an expansile mass (arrow) that has invaded the normal tissue ventral to it. The mass ventral to the medulla (right) may be a metastasis arising from tumor cells that entered the third ventricle and then spread in the cerebrospinal fluid caudally, through the mesencephalic duct, into the fourth ventricle, and out through a lateral aperture into the subarachnoid space. (Courtesy Dr. Y. Niyo, College of Veterinary Medicine, Iowa State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
The age range of dogs with choroid plexus tumors in one study was 5 to 13 years, except for one dog that was 2 years of age. Clinical signs in animals with choroid plexus tumors vary, depending on the location of the tumor in the CNS but can include behavioral changes, ataxia, paresis, seizures, circling, and abnormal cranial nerve and proprioceptive reflexes.
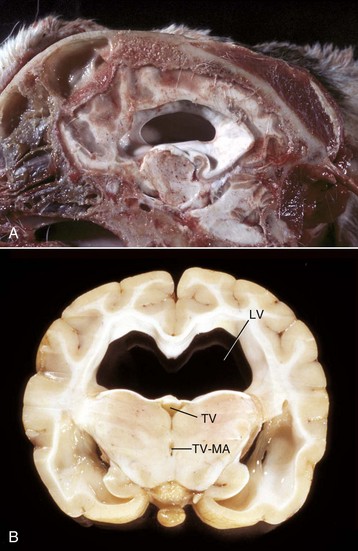
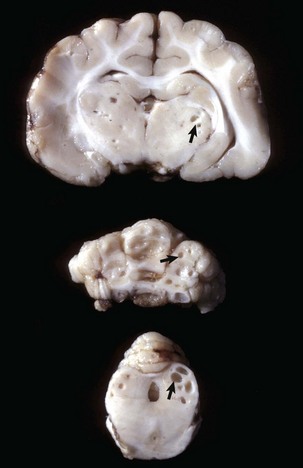
Ependymomas: Ependymomas are one of the less frequently occurring neoplasms in dogs, cats, cattle, and horses. Some reports on the dog indicate a higher frequency in brachycephalic breeds. Ependymomas usually involve the lateral or less commonly, third and fourth ventricles. They also occur in the central canal of the spinal cord. The neoplasm can be observed within the ventricular system and subarachnoid space, likely attributable to local metastasis via the CSF. Noncommunicating hydrocephalus may result from obstruction of CSF flow within the ventricular system.
Grossly, ependymomas are usually large expansile intraventricular masses with generally well-demarcated margins (Fig. 14-74). The neoplasm is soft and gray-white to red, depending on blood content, and has a smooth cut surface in dogs. In cats, the cut surface can have a granular texture. In some ependymomas, the cut surface may have gelatinous consistency and be cavitated. More aggressive tumors show invasion into the normal tissue at its margins. Microscopically, ependymomas are highly cellular and well vascularized. Cells have hyperchromic, round-to-oval nuclei with scant or undetectable cytoplasm. Cells form perivascular rosettes (pseudorosettes) with nuclear polarity away from the vessel wall. Cells are also arranged in sheets and bands. The mitotic rate is variable. Hemorrhage, mucinous and cystic degeneration, and capillary proliferation occur. Malignancy is indicated by invasive growth, frequent mitoses, and anaplasia. Currently, no reliable immunohistochemical markers for ependymomas exist.
Fig. 14-74 Ependymoma, brain, transverse section at the level of the hippocampus, dog.
The third ventricle contains a moderately well-demarcated expansile mass (arrows) that has invaded normal tissue ventral to it. Moderate hydrocephalus is present in both lateral ventricles from blockage of the third ventricle. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
The average age of affected dogs ranges from 6 to 12 years. Occurrences in a cat as young as 18 months of age and in a calf 5 months of age have been reported. Clinical signs in animals with ependymomas vary, depending on the location of the tumor in the CNS but can include behavioral changes, ataxia, paresis, seizures, circling, and abnormal cranial nerve and proprioceptive reflexes.
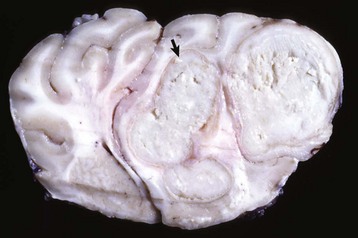
Oligodendrogliomas: Neoplasms composed of oligodendrocytes occur most commonly in the dog, but cases have been reported in cats and cattle. The reported incidence varies; some reports indicate oligodendrogliomas as the most common neuroectodermal neoplasm (5% to 12% incidence), whereas others place it second to astroglial neoplasms. As with astrocytomas in dogs, there is a predilection for brachycephalic breeds (Boston terriers, boxers, and bulldogs), and the age range is the same as for astrocytomas (5 to 11 years of age). Neoplasms occur in all areas of the white matter of the cerebrum, brainstem, and interventricular septum (Fig. 14-75). Neoplasms tend to extend to meningeal and ventricular surfaces, but dissemination in the CSF is uncommon.
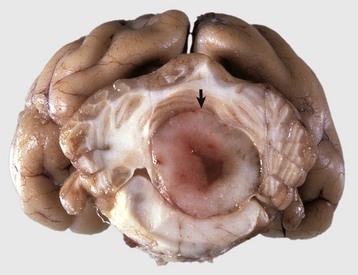
Fig. 14-75 Oligodendroglioma, brain, cerebellum, transverse section caudal to the pons, dog.
The tumor is relatively well demarcated from the surrounding cerebellum. Oligodendrogliomas may be well demarcated or merge with adjacent normal brain. They vary in size, are gray to pink-red, and are soft or gelatinous with areas of hemorrhage (arrow). (Courtesy Dr. W. Crowell, College of Veterinary Medicine, University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
Grossly, most oligodendrogliomas can be relatively well demarcated from surrounding tissue or their margins can be indistinct and blend in with adjacent normal white matter. The neoplasms vary in size, can grow quite large, are gray to pink-red, and are soft or gelatinous with areas of hemorrhage. In larger tumors, the central area may be cystic. Microscopically, the neoplasms are composed of densely packed cells. Nuclei are centrally located, hyperchromic, and surrounded by pale or nonstaining cytoplasm creating a perinuclear halo. Other patterns include cells arranged in rows, especially peripherally, or in semicircles. Mitoses are generally infrequent. Regressive changes include mucoid degeneration, edema, cavitation, and rarely mineralization. Extensive necrosis is uncommon. Currently, no reliable immunohistochemical markers for oligodendrogliomas exist.
Meningiomas: Meningioma is the most common mesodermal neoplasm of the CNS of animals, especially in cats. Sites of occurrence in the dog include the basal area of the brain, the area over the convexity of the cerebral hemispheres, the cerebellum-tentorium area, the lateral surface of the brain, the falx cerebri, and the surface of the spinal cord. Retrobulbar involvement (originating from the optic nerve sheath) also occurs. In the cat, the neoplasm uniquely occurs in the tela choroidea of the third ventricle but also occurs over the cerebral hemispheres, along the falx cerebri, over the cerebellum and tentorium, and rarely at the base of the brain. Occurrence in the meninges of the spinal cord is not common. Meningiomas have been reported to arise from arachnoid “cap cells,” which are on the external surface of the arachnoid membrane. These cells cover the surface of the arachnoid layer that oppose the surface layer of the dura mater, and thus these tumors project into the subdural space and often into the CNS parenchyma.
Grossly, neoplasms in the dog are solitary and vary in size. The neoplasms are well defined, spherical, lobulated, lenticular, or plaquelike in shape; firm; encapsulated; and gray-white (Fig. 14-76). Sometimes on the cut surface there are soft, red, brown, or gray areas of hemorrhage and necrosis. Because these neoplasms grow slowly, they cause pressure atrophy of the adjacent nervous tissue. Meningiomas can be invasive, and sometimes there is hyperostosis of the overlying bone. In the cat, meningiomas vary in size from barely detectable to 2 cm in diameter. Cats and occasionally cattle can have more than one neoplasm. Other characteristics are comparable with those described in the dog.
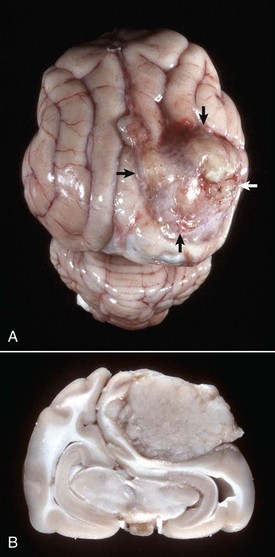
Fig. 14-76 Meningioma, brain, cat.
A, On the surface of the right parietal cortex is a mass (arrows) that has compressed and distorted the adjacent parenchyma. It is a space-occupying lesion that has displaced the midline (cerebral longitudinal fissure) to the left. B, Transverse section at the level of the hippocampus of the brain depicted in A. The tumor has compressed the right cerebral hemisphere and this has resulted in the midline being displaced to the left with compression of the left cerebral hemisphere. The meningioma does not invade the brain and can be “shelled” out at necropsy or surgery. (Courtesy College of Veterinary Medicine, University of Illinois.)
Microscopically, several patterns of neoplastic cells can occur, and more than one can be present in a given neoplasm. Based on their cytomorphologic features, these tumors are grouped into one of six categories: (1) epithelioid/meningotheliomatous; (2) fibroblastic; (3) transitional (features of both epithelioid and fibroblastic); (4) psammomatous; (5) angioblastic; and (6) anaplastic/malignant. Cytomorphologic features are beyond the scope of this chapter; however, the more common pattern is characterized by the formation of nests, islands, or laminated whorls of cells. The cells have large cell bodies with abundant cytoplasm, ill-defined cell boundaries, and elongated, oval, open nuclei with peripherally located chromatin. The number of cells making up a whorl can vary from a few to many, with mineralized material (referred to as psammoma bodies) in the center of such structures. A second pattern is characterized by strands or streams of elongated cells with a rather irregular or parallel orientation. Regressive changes include hemorrhage and cavernous vascular formations. Invasive growth occurs but is less common than growth by expansion. Currently, vimentin and pancytokeratin may be useful immunohistochemical markers for these tumors.
Meningiomas occur most frequently in the dog and cat but have been reported in other species of domestic animals, including horses, cattle, and sheep. In the dog, this neoplasm occurs in several breeds, with dolichocephalic animals being frequently represented. The majority of meningiomas are in dogs between 7 and 14 years of age and in cats 10 years old or older.
Other benign neoplasms of mesenchymal origin, such as neurofibromas of spinal cord nerve roots, occur in the meninges. Spindle cell sarcomas, such as neurofibrosarcomas and dural osteosarcomas, have also been reported in the meninges.
Microgliomatosis: Microgliomatosis, which has been classified as a proliferative neoplastic disease, has some features that are quite distinct from the inflammatory and neoplastic forms of reticulosis described in a later section (see the section on Reticulosis/Granulomatous Meningoencephalitis). Gross lesions are usually not present. The cells, which infiltrate the CNS without topographic perivascular arrangement, resemble microglia in that their nuclei, which vary in size and shape and have prominent chromatin, are the only visible cellular component after conventional staining. Also, mitoses can be common, and there is no accompanying reticulin fiber formation such as occurs in the inflammatory and neoplastic reticuloses. Microgliomatosis occurs in older dogs.
Hemangiosarcoma: Primary hemangiosarcoma of the CNS is a rare neoplasm arising from endothelial cells. The disease is most common in dogs but can occur in all domestic species. The neoplasm is a solitary expansile red-to-dark red mass within the cerebral cortex. Its color and bloody consistency are helpful in differentiating it from a primary or metastatic melanoma. Most hemangiosarcomas found in the CNS are from metastases.
Metastatic Tumors: Hematogenously metastasizing neoplasms occur and affect the brain more often than the spinal cord. The species in which metastasis has been most commonly reported is the dog; the next most frequent is the cat. Of the metastasizing carcinomas, mammary gland carcinoma in the dog has been reported to occur most frequently, although others have been described. Hemangiosarcoma of the heart, liver, and spleen is one of the most common metastasizing sarcomas in the dog; others are the mesenchymal component of the malignant mixed mammary gland tumor, lymphosarcoma, fibrosarcoma, and malignant melanoma. In the CNS, metastatic hemangiosarcomas appear to have predilection for the gray matter–white matter interface (Fig. 14-77). In the cat, the neoplasms that metastasize to the CNS include the mammary gland carcinoma (Fig. 14-78) and lymphosarcoma. Primary CNS lymphomas have been reported.
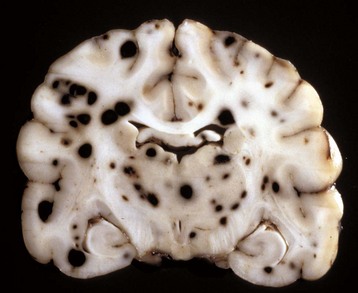
Fig. 14-77 Hemangiosarcoma, metastatic, brain, formalin-fixed, transverse section at the level of the thalamus, dog.
Note the prominent hematogenous metastases, which appear as black nodules of various sizes distributed throughout the brain, sometimes at the gray matter–white matter interface. In an unfixed (fresh) specimen, the nodules would be red to dark red from erythrocytes. Black nodules in a fresh specimen would be consistent with metastatic melanoma. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
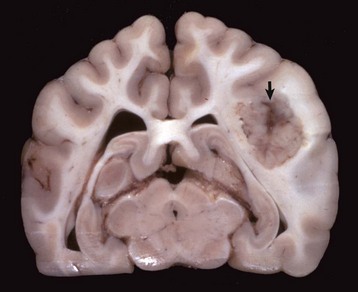
Fig. 14-78 Mammary carcinoma, metastatic, brain, transverse section at the level of the hippocampus, dog.
The right cerebral hemisphere contains a well-demarcated mass (arrow), which has caused enlargement of the right cerebral hemisphere and compression of the right lateral ventricle. The left lateral ventricle is slightly dilated, probably because of pressure on the interventricular foramen. (Courtesy Dr. F. Moore and Dr. J. Carpenter, Angell Memorial Animal Hospital; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)