Nervous System*
Central Nervous System (CNS)
The CNS consists of neurons, glia, ependyma, endothelial cells and pericytes of blood vessels, and the meninges (Fig. 14-1 and Box 14-1). Neurons vary in size, shape, and function, and their cell bodies are organized into functional groups such as nuclei, gray columns, and cerebral lamina. Neuronal processes called axons and dendrites traverse through the brain and spinal cord, the former often as organized bundles (tracts, fasciculi) forming synapses on cell bodies, dendrites, and axons of other functionally related neurons. It is estimated that there are 1 × 1011 neurons in the human brain. Each neuron makes approximately 10,000 synapses with other neurons; therefore there are about 1 × 1015 synapses in the human brain.
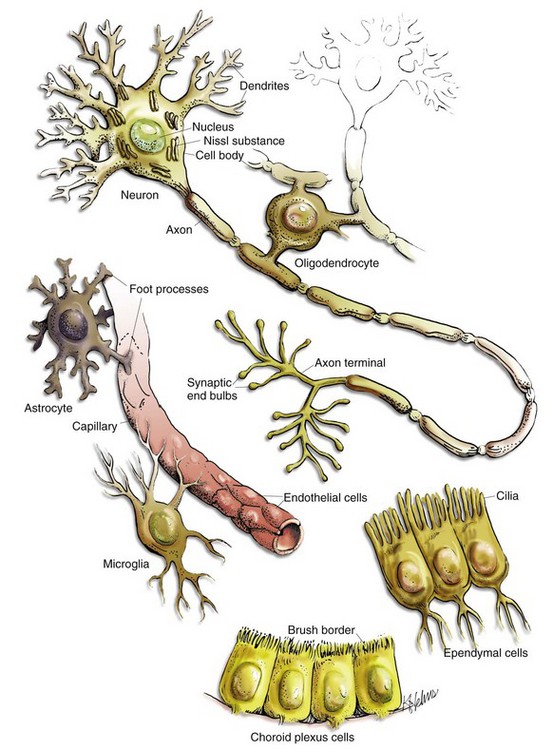
Fig. 14-1 Cell types in the CNS include neurons, astrocytes, oligodendroglia, microglia, ependymal cells, choroid plexus epithelial cells, and vascular endothelial cells. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Exactly which cells are classified as glia has varied over the last few decades. Originally, histologists included astrocytes (astroglia), oligodendrocytes (oligodendroglia), ependymal cells (ependymocytes), and microglia as glial cells; however, they currently recognize astrocytes, oligodendrocytes, and microglia as glial cells. Some classification schemes list astrocytes and oligodendrocytes as macroglia. Astrocytes, oligodendrocytes, and ependymal cells are derived from neuroectoderm; whereas microglia, part of the monocyte-macrophage system, are derived from mesoderm (bone marrow). In the mammalian CNS, glia outnumber neurons 10 to 1. Ependymal cells line the ventricular system, whereas choroid plexus epithelial cells form the outer covering of the choroid plexuses.
The CNS is arranged to form two basic parts: the gray and white matter (Figs. 14-2 and 14-3). In the CNS, gray matter is found in the cerebral cortex, in the cerebellar cortex and cerebellar roof nuclei, around the base of the cerebral hemispheres (basal nuclei [often called basal ganglia]: caudate nucleus, lentiform nucleus [putamen, globus pallidus], amygdaloid nucleus, claustrum), and throughout the brainstem, often in nuclei. The gray matter is typified by numerous neuronal cell bodies, plus a feltwork of intermingled thinly myelinated axons and dendrites, their synaptic junctions, and processes of oligodendroglia, astrocytes, and microglia. This network of processes and synapses in the gray matter is referred to as the neuropil. The white matter consists of well-myelinated axons that arise from neuronal cell bodies in the gray matter and terminate distally in synapses or myoneural junctions, plus oligodendroglia, astrocytes, and microglia. In the cerebral hemispheres, white matter is located centrally; whereas in the brainstem, white matter is intermingled with gray matter (nuclei). In the spinal cord, white matter is located peripherally surrounding the gray matter.
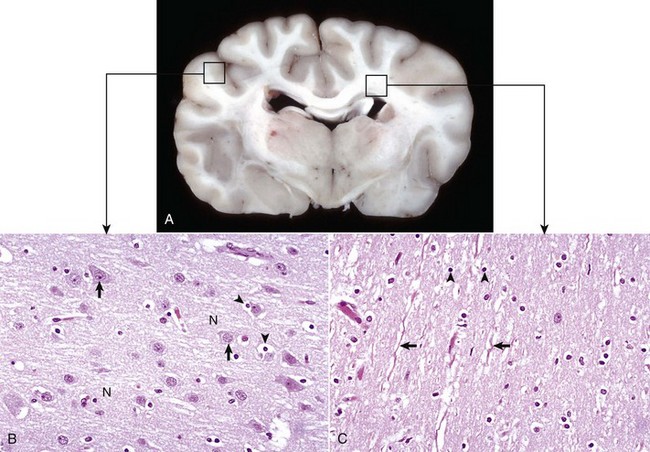
Fig. 14-2 Organization of the brain, gray matter, and white matter.
A, Transverse section at the level of the thalamus, dog. Gray matter (darker areas) of the cerebral cortex lies beneath the leptomeninges on the external surface of the brain, whereas in the thalamus there tends to be a mixture of gray and white matter. Major white matter areas (light areas) include corona radiata, centrum semiovale, and corpus callosum of the cerebrum, and internal capsule and optic tracts bordering the lateral and ventral surfaces of the thalamus, respectively. B, Gray matter consists primarily of the cell bodies of neurons (arrows) and a network of intermingled thinly myelinated axons, dendrites, and glial cell processes. This network is referred to as the neuropil (N). Other components include oligodendroglia (perineuronal satellite cells) (arrowheads), protoplasmic astrocytes, and microglia. H&E stain. C, White matter primarily consists of well-myelinated axons (arrows) plus oligodendroglia (arrowheads) and fibrous astrocytes. The clear spaces surrounding large axons are artifacts formed when the lipid components of myelin lamellae are dissolved away by solvents in the process of embedding tissue in paraffin for sectioning. H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
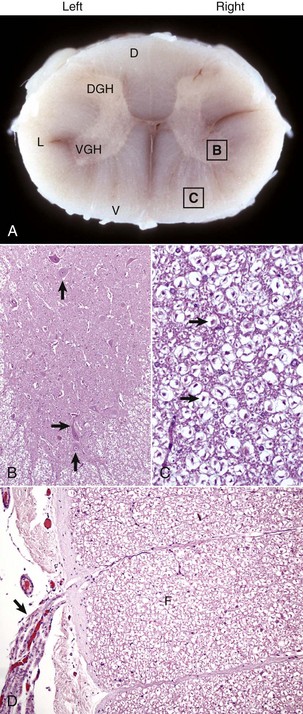
Fig. 14-3 Organization of the spinal cord, gray matter, and white matter.
A, White matter in the spinal cord is located peripherally and divided into dorsal, lateral, and ventral funiculi. As a general rule, dorsal funiculi (D) consist of ascending sensory axons, lateral funiculi (L) have a mixture of sensory and motor axons, and ventral funiculi consist of descending motor axons (V). DGH, Dorsal gray horn; VGH, ventral gray horn. Histologically, the right side is a mirror image of the left side. The areas labeled B and C and contained within the boxes correspond to the areas illustrated in B and C. B, Transverse section of spinal cord, ventral gray horn, horse. The cell bodies of large motor neurons (arrows) are those of lower motor neurons and their axons extend in peripheral nerves to myoneural junctions that innervate skeletal muscle. H&E stain. C, Transverse section of spinal cord, ventral funiculus, horse. Because most axons course up and down the length of the spinal cord, in a transverse section, axons (arrows) are cut in cross section. They are surrounded by myelin sheaths whose lipid components are dissolved out during the preparation of paraffin embedded sections, resulting in clear spaces that are an artifact. H&E stain. D, Efferent spinal nerve (longitudinal section shown here), transverse section of spinal cord, ventral funiculus, dog. Axons of lower motor neurons leave funiculi (F) and assemble as nerve rootlets (arrow) eventually forming peripheral nerves that innervate skeletal muscle. H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The exterior of the CNS is covered by the meninges. The meninges consist of three layers named, from outermost to inner most layers, the dura mater, arachnoid, and pia mater. The arachnoid and pia enclose the subarachnoid space.
Cells of the CNS
Neurons: The structure and basic cellular biology of neurons is similar to that of other cells (Fig. 14-4); however, there are, as discussed later, some notable differences. The neuron consists of three structural components: dendrites, a cell body, and a single axon. The length of the axon varies, depending on the function of the neuron. The length of axons of motor or sensory neurons can be 10,000 to 15,000 times the diameter of the neuronal cell body, which results in these axons being several meters in length. The axon terminates in synaptic processes or neuromuscular junctions.
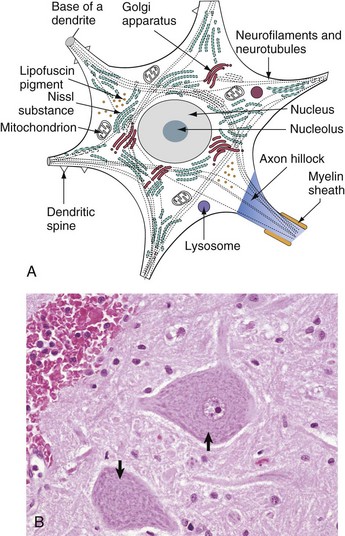
Fig. 14-4 Neuron structure.
A, Basic cell biology and structure of neurons are similar to other cells in the body. Additionally, neurons have dendritic arborizations and an axon, specializations for the initiation, propagation, and transmission of impulses that underlie the basic function of these cells. B, The cytoplasm of the neuronal cell body has blue (basophilic [H&E stain]) granular material (rough endoplasmic reticulum) called Nissl substance (arrows). Nissl substance synthesizes proteins, including precursor neurotransmitter proteins and the structural proteins (neurofilaments), active in maintaining the integrity (length and diameter) of the axon. H&E stain. (A modified from Kierszenbaum AL: Histology and cell biology, St Louis, 2002, Mosby. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Neuronal cell bodies vary considerably in size and shape, from the large neurons of the lateral vestibular nucleus, Purkinje cell layer of the cerebellum, and the ventral gray matter of the spinal cord to the very small lymphocyte-like granule cells of the cerebellar cortex (Fig. 14-5). Neuronal nuclei tend to be vesicular to spherical in shape, tend to be usually centrally located, and often, particularly in large neurons, tend to contain a prominent central nucleolus. Neurons contain focal arrays of rough endoplasmic reticulum and polysomes, termed Nissl substance, that are responsible for the synthesis of proteins involved in many of the neuron’s vital cellular processes such as axonal transport. Nissl substance is present in all neurons, regardless of the size of the cell body, but tends to be more prominent in those cells with voluminous cytoplasm such as motor neurons.
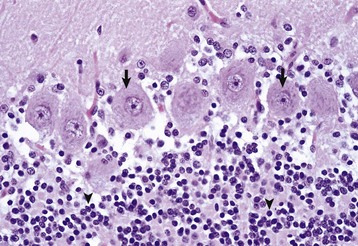
Fig. 14-5 Variations in neuronal morphology, cerebellum, granule cells, and Purkinje neurons, normal animal.
The granule cell neurons of the cerebellar cortex (arrowheads) are very small lymphocyte-like cells that have relatively little demonstrable Nissl substance when compared with Purkinje neurons (arrows) and large motor neurons (depicted in Fig. 14-4, B). H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Axonal Transport: In most cells of the body, proteins and other molecules are distributed throughout the cell by simple diffusion. In neurons, simple diffusion alone is inefficient because synapses are a considerable distance away from the cell body of the neuron. As a result, molecules cannot diffuse the length of the axon; they must be transported the length of the axon to the synapse. In addition, there are no systems in axons or synapses to catabolize molecules resulting from normal metabolic processes in these structures. Thus these molecules need to be returned to the cell body for processing. These processes are facilitated in the axon by retrograde (toward the cell body) and anterograde (toward the synapse) axonal transport systems.
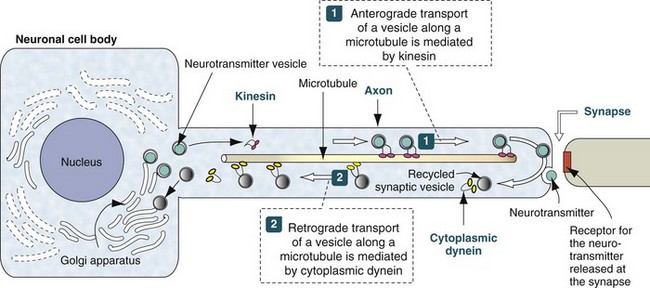
Web Fig. 14-1 Axonal transport systems.
Neurotransmitter vesicles and neurofilament proteins, synthesized in the rough endoplasmic reticulum and packaged in the Golgi apparatus are transported through the length of the axon and to synapses by kinesin. Kinesin is a microtubule motor protein that uses chemical energy from adenosine triphosphate hydrolysis to generate mechanical force and thus bind to and move attached to microtubules. Used vesicles and effete neurofilament proteins are returned along a microtubule (recycled) to the neuron cell body by cytoplasmic dynein, another microtubule motor protein. These transport systems are used by some pathogens (rabies virus, Listeria monocytogenes) to enter and spread within the CNS. (Modified from Kierszenbaum AL: Histology and cell biology, St Louis, 2002, Mosby.)
As a result of these structural differences between neurons and other cells, neurons have developed axonal transport systems to efficiently move molecules and cellular organelles from the cell body through the axon to the synapses and their degradation products back to the cell body (Web Fig. 14-1). Axons can be longer than a meter in length, especially in an animal such as a giraffe. Lower motor neurons, whose cell bodies lie in the ventral gray horn of the spinal cord, and lumbar dorsal root ganglia, whose axons extend to the distal limb and to the caudal medulla, have the longest axons in the body. The neuron expends considerable energy and materials to move biologic materials up and down the axon. Alterations in the function of these transport systems can lead to neuronal dysfunction.
These transport systems are divided into “fast axonal transport” and “slow axonal transport.” The fast axonal transport system has an anterograde component (toward the synapse) and a retrograde component (toward the cell body). The slow axonal transport system has only an anterograde component (toward the synapse).
Fast anterograde axonal transport (up to 400 mm per day) moves materials not intended for use in the cytoplasm of the neuron cell body. These materials formed from the Golgi apparatus are principally membrane-bound vesicles. They include mitochondria and membranous vesicles that contain peptide neurotransmitters, small transmitter molecules, and the enzymes necessary for their activation. These materials are moved down the axon on microtubules by specialized protein motors composed of kinesin and kinesin-related proteins using adenosine triphosphate (ATP) as an energy source.
Fast retrograde axonal transport (200 to 300 mm per day) returns endosomes, mitochondria, and catabolized proteins to the cell body of the neuron for degradation in lysosomes and reuse. This transported material is returned on microtubules, by dynein and microtubule-associated adenosine triphosphatase (ATPase) in the axon. This system will also transport certain toxins, such as tetanus toxin, and viruses, such as rabies virus, from the periphery via the peripheral nervous system (PNS) into the CNS.
Slow anterograde axonal transport (0.2 to 5 mm per day) transfers throughout the axon via microtubules, the major cytoskeletal proteins, such as microtubule and neurofilament proteins, that are necessary to maintain the structural integrity and transport systems within the axon.
Diseases of the axon that result directly or indirectly from alterations in axonal transport systems are discussed later. The character of the histologic lesions affecting injured nerve fibers can often be related to alterations in specific transport systems. Neurofilament proteins are synthesized in the neuronal cell body and are assembled and transported into axons. If neurofilaments accumulate in neuronal cell bodies and proximal axons, this lesion is called an axonopathy and is characterized by alterations in slow transport systems, which results in axonal swelling or atrophy and perikaryal neurofibrillary accumulations. Axonal injury and alterations in neurofilament transport can also cause secondary demyelination.
Membrane Potentials and Transmitter/Receptor Systems: A fundamental activity of neurons is to modulate and effectively transmit chemical and electric signals from one neuron to another via synapses in the CNS or from one neuron to a muscle cell via junctional complexes, myoneural junctions, or motor end-plates in the PNS. The process of nerve impulse conduction is made possible by the establishment and maintenance of an electric potential across the cell membrane of the neuron/axon.
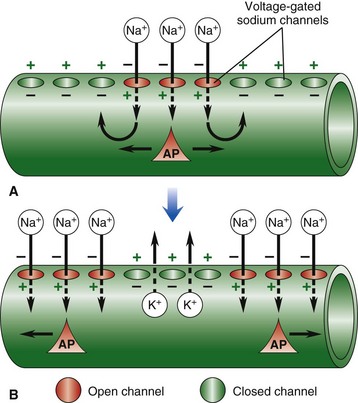
Web Fig. 14-2 Resting and action potentials.
Nerve impulse conduction is made possible by the establishment and maintenance of an electric potential across the cell membrane of the neuron/axon. Resting membrane potential is established and maintained by differences in concentrations of potassium ions inside and sodium outside of the cell membrane. Sodium and potassium ions will leak across the cell membrane, and therefore concentration gradients are maintained by a sodium-potassium pump in the cell membrane. When an event depolarizes the cell membrane to a threshold level of approximately −50 mV, an action potential will occur. A, An action potential is initiated by an event that opens sodium channels, and the action potential is propagated along the cell membrane by the sequential opening of voltage-gated sodium channels in adjacent sections of the membrane. B, The action potential is regenerated in adjacent sections of the cell membrane as additional sodium channels open. Depolarized segments repolarize as sodium channels close and potassium ions move out of the cell. AP, Action potential. (From Copstead LC, Banasik JL: Pathophysiology: biological and behavioral perspectives, ed 2, Philadelphia, 2000, Saunders.)
A fundamental activity of neurons is to modulate and effectively transmit chemical and electric signals from one neuron to another via synapses in the CNS or from one neuron to a muscle cell via junctional complexes, myoneural junctions, or motor end-plates in the PNS. The process of nerve impulse conduction is made possible by the establishment and maintenance of an electric potential across the cell membrane of the neuron/axon. Membrane potential is the difference in voltage between the inside and outside of the neuronal/axonal cell membrane and is called the resting potential. This potential is established and maintained by a membrane sodium ion (Na+)/potassium ion (K+)-ATPase (Na+/K+-ATPase) pump. The pump keeps the concentration of sodium ions outside the cell approximately 10 times greater than inside the cell, and the concentration of potassium ions inside the cell 20 times greater than outside the cell. The differences in concentrations of sodium ions outside and potassium ions inside of the cell membrane keep the membrane resting potential at approximately −70 mV. Thus the inside of the neuron/axon is 70 mV less than the outside. Sodium and potassium ions will leak across the cell membrane, and therefore concentration gradients are maintained by the Na+/K+-ATPase pump in the cell membrane. This established equilibrium and the membrane potential places the neuron in a “resting” condition, ready to generate an action potential.
An action potential arises when a neuron transmits information down an axon, away from the neuronal cell body. An action potential is initiated by an event that depolarizes the cell membrane and causes the resting potential to move toward 0 mV. When depolarization reaches a threshold level of approximately −50 mV, an action potential will occur. Once initiated, the strength of an action potential is always the same because the action potential is an intrinsic property of the neuron cell body and its axon.
Action potentials are caused by the movement of sodium and potassium ions across the neuron cell body/axon cell membrane. With an initiating event, sodium channels are first to open, and large concentrations of sodium ions enter the intracellular microenvironment (Web Fig. 14-2). Because sodium ions are positively charged, the polarity becomes more positive (−70 mV to −50 mV) and the neuron/axon becomes depolarized. Potassium channels open later in the depolarization process, concurrently with the closing of sodium channels. Potassium ions leave the cell and enter the extracellular fluid. These events cause repolarization of the neuron/axon and a return to a resting potential (−70 mV) via the membrane Na+/K+-ATPase pump. Alterations in these ion channels have been correlated with epilepsy in humans and will likely be discovered in animals.
Action potentials are most commonly initiated by neurotransmitters, such as acetylcholine, acting through synapses, but they also occur as a result of mechanical stimuli, such as stretching and sound waves. There are two main classes of synapses: inhibitory and excitatory. Stimulation of inhibitory synapses results in inhibitory postsynaptic potentials that cause hyperpolarization of dendrites and cell bodies. Hyperpolarization decreases the membrane potential (more negative, −80 mV), thus making the neuron less likely to reach the threshold for an action potential. Inhibitory neurotransmitters include γ-aminobutyric acid (GABA), glycine, dopamine, serotonin, norepinephrine (in the CNS), and acetylcholine (in heart muscle).
Stimulation of excitatory synapses results in excitatory postsynaptic potentials that cause depolarization of the dendrites and cell bodies. Depolarization increases the membrane potential (more positive, −50 mV), thus making the neuron more likely to reach the threshold for an action potential. Excitatory neurotransmitters include glutamate, norepinephrine in the PNS, and acetylcholine in skeletal muscle.
The generation of an action potential is a complicated process requiring depolarization of the cell membrane (−50 mV). Inhibitory and excitatory synapses and their inhibitory and excitatory postsynaptic potentials, respectively, are “summed” through processes, termed spatial and temporal summation, occurring in the dendritic network of the neuron. Spatial summation reflects additive input from different parts of the dendritic network, whereas temporal summation reflects additive input from stimuli that occur closely in time. This summation process is a graded potential and ultimately determines if the threshold for an action potential will occur.
The action potential is a flow of depolarization that travels down the axon to synapses at the distal axon. When the axon lacks myelin, the flow of depolarization down the axon is called continuous conduction. When the axon is myelinated, the speed of conduction is determined by the degree of myelination of the axon and is called saltatory conduction. The diameter of unmyelinated axons can range from 0.2 to 1 mm with action potential velocities ranging from 0.2 to 2 m/sec, whereas the diameter of myelinated axons can range from 2 to 20 mm with action potential velocities ranging from 12 to 120 m/sec. The greater the degree of myelination, the faster the speed of impulse conduction down the axon. In unmyelinated axons, action potentials are conducted at a relatively “slower” velocity by the process of ion exchange (continuous conduction). In myelinated axons, action potentials are conducted at a relatively “faster” velocity by a mechanism called saltatory conduction. In this process, action potentials move down the myelinated axon using cable properties, like electric current flow in insulated copper wires. This method is fast, efficient, and requires less energy than ion exchange. However, the action potential would decay if axons were myelinated continuously along their length and likely would not reach synapses at full strength or at all. This decay is caused by loss of current across the cell membrane and capacitance properties of the cell membrane as the action potential travels down the axon. To minimize the decay of action potentials, axons are myelinated in segments called internodes. A gap, called the node of Ranvier, is formed between consecutive internodes and measures between 0.2 and 2 mm in length. At this gap, the action potential is restored to full strength by ion exchange. The node of Ranvier is highly enriched in sodium channels, and these channels are essential for impulse propagation via rapid action potential current restoration. Disease processes that disrupt myelination of axons will interfere with saltatory conduction, slow the action potential, and result in clinical dysfunction of the nervous system (see Fig. 14-21).
The axon can be a very long extension of the neuron cell body extending, for example, up to 2 m from the lumbar dorsal root ganglion in a giraffe. At its distal end, the axon splits into several branches that end as specialized structures called axon terminals/terminal buttons/synaptic bulbs. Synapses present at these axon terminals are functional, and structural points of contact between “networked” neurons and these synapses convert the action potential into chemical signals that stimulate the next neuron in the conduction pathway. The cell membrane that releases chemical neurotransmitters is called the presynaptic membrane, and the cell membrane that has neurotransmitter receptors for the chemical neurotransmitters is called the postsynaptic membrane. These membranes are found on dendrites and cell bodies of the next neuron in the neural conduction pathway. The gap between the presynaptic and postsynaptic membranes that chemical neurotransmitters must cross is called the synaptic cleft. The mechanism of diseases, such as tetanus and botulism, is manifested through presynaptic and postsynaptic membrane receptors.
When an action potential reaches the axon terminal, it causes the release of chemical neurotransmitters from the presynaptic membrane by opening voltage-gated calcium channels, leading to membrane depolarization. The amount of chemical neurotransmitter released into the synaptic cleft is determined by the number of action potentials that reach the axon terminal over time. Chemical neurotransmitters traverse the synaptic cleft and bind to neurotransmitter receptors on dendrites and cell bodies of a new neuron in the neural conduction pathway.
There are two types of chemical neurotransmitter receptors, ionotropic and metabotropic, on the membrane of postsynaptic neurons. Functionally, these receptor types differ in latency and duration of action. Ionotropic receptors have a fast response and short duration of effect, whereas metabotropic receptors have a slower response and a longer duration of effect. In addition, ionotropic receptors are localized to specific sites on the postsynaptic membrane, whereas metabotropic receptors are distributed diffusely and at random.
Chemical neurotransmitter stimulation of ionotropic receptors results in the opening of ion gates or channels, resulting in depolarization of the postsynaptic membrane. Excitatory neurotransmitters, such as glutamate, open postsynaptic membrane sodium channels. Inhibitory neurotransmitters, such as GABA, open postsynaptic membrane chloride channels.
Chemical neurotransmitter stimulation of metabotropic receptors results in the generation of a second messenger such as in the cyclic adenosine monophosphate (cAMP) pathway, which initiates a sequence of metabolic changes in the neuron. Metabotropic receptors are composed of protein subunits that span the postsynaptic cell membrane. An extracellular component of this protein has a high affinity for neurotransmitters and functions as a binding site. After binding the neurotransmitter, the receptor undergoes a configurational change that directly or indirectly activates a cell membrane enzyme, such as intracellular G proteins, leading to the formation of the second messenger. cAMP can activate protein kinase A–induced phosphorylation, leading to functional changes in ion channels and protein transcription. Dopamine is an example of a chemical neurotransmitter that uses metabotropic receptor pathways.
Astrocytes: The functions of astrocytes in the CNS are regulation, repair, and support, as depicted in Fig. 14-6. Mature astrocytes differentiate from pluripotential progenitor cells during the development of the CNS. Astrocytes are the most numerous cell type in the CNS and have traditionally been classified into two types based on morphology. Protoplasmic astrocytes are located primarily in gray matter, whereas fibrous astrocytes occur chiefly in white matter. Microscopically, astrocytes have relatively large vesicular nuclei, indistinct or inapparent nucleoli, and no discernible cytoplasm with routine hematoxylin and eosin (H&E) staining (Fig. 14-7). With suitable histochemical stains, metallic impregnation, or immunohistochemical staining for glial fibrillary acidic protein (GFAP [the major intermediate filament in astrocytes]), the cell body and the extensive arborization and interconnections of astrocytic processes can be demonstrated. Processes vary from short and brushlike to long branching processes in protoplasmic and fibrous astrocytes, respectively (Fig. 14-8). These morphologic features and their corresponding histochemical and immunohistochemical staining reactions serve as important criteria for the classification of tumors of astrocyte origin.
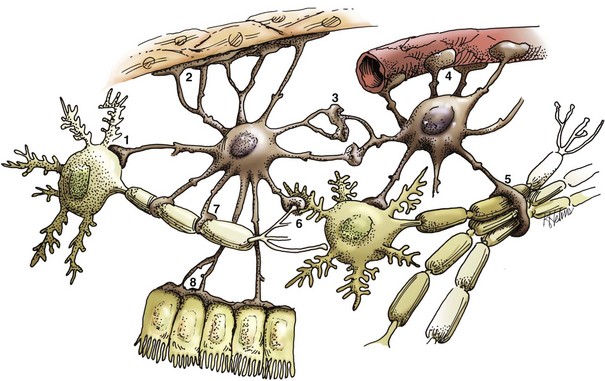
Fig. 14-6 Functions of astrocytes.
Astrocytes provide structural integrity and regulatory oversight, as depicted in this diagram. They: 1, monitor and regulate fluid and electrolyte balances within neurons and surrounding extracellular space; 2, form the glial limitans at the base of the pia mater; 3, interconnect with other astrocytes to provide a system to monitor and regulate fluid and electrolyte balances throughout the CNS; 4, possibly participate in the formation and functions of the blood-brain barrier; 5, participate in the support of axon tracts of functionally related neurons; 6, monitor for and remove excessive release of neurotransmitters in synapses; 7, protect and insulate nodes of Ranvier; and 8, participate in the cerebrospinal fluid–brain barrier. In addition, astrocytes are a reparative (healing) cell after CNS injury with loss of tissue because nervous tissue, per se, is devoid of fibroblasts. Fibroblasts exist in the pia mater and other meninges. Everywhere else, healing depends on the astrocyte, which responds by increased length, branching, and complexity of cellular processes (astrogliosis). The astrocyte has many functions in the nervous system; one of them is to act in healing to produce a scar in attempts to isolate cavities and abscesses. Fibroblasts may also contribute to the formation of a scar, if this cell type is present, as it is in the leptomeninges. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
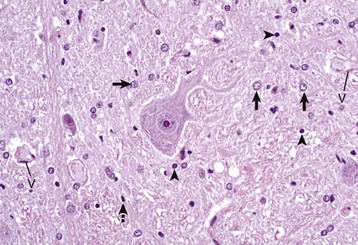
Fig. 14-7 Histologic features of glial cells, ventral gray horn, spinal cord, horse.
A neuronal cell body and its processes are in the center of the illustration. To the inexperienced, identifying specific types of glial cells in H&E stained histologic sections can be challenging. Astrocytes (arrows) have larger vesicular nuclei (dispersed chromatin) and the cell membrane and cytoplasm are rarely seen in nondiseased conditions. Thus these nuclei just seem to “sit” in the midst of the neuropil. The majority of nuclei in the neuropil here are astrocytic. Oligodendroglial cells (arrowheads) have smaller and dense round nuclei (condensed chromatin) often surrounded by a clear zone indicative of cell cytoplasm and a cell membrane. Oligodendroglial cells in gray matter are called perineuronal satellite cells; those in white matter are called interfascicular oligodendrocytes. Microglial cells are difficult to identify in H&E stained sections of the CNS, but they often appear as “rod cells,” which have small, dense elongated nuclei (dashed arrow). The light pink homogeneous tissue distributed in large quantities between these cell types is the neuropil. V, Blood vessels. H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
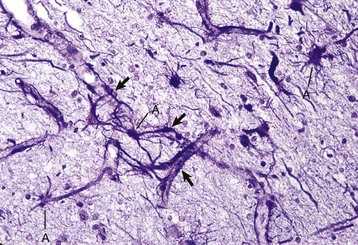
Fig. 14-8 Astrocytic processes, brain, cerebral cortex, normal animal.
Processes of astrocytes arborize extensively throughout the CNS (structures stained purple). Note that some of the processes are on the outside of blood capillaries (end feet) (arrows). A, Cell body of astrocyte. Holzer’s stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Regulation of the microenvironment: The microenvironment of the CNS must be under strict control to maintain normal function. Astrocytes are involved in homeostasis of the CNS and regulate ionic and water balance, antioxidant concentrations, uptake and metabolism of neurotransmitters, and metabolism or sequestration of potential neurotoxins, including ammonia, heavy metals, and excitatory amino acid neurotransmitters such as glutamate and aspartate. Interactions between astrocytes, microglia, and neurons orchestrate immune reactions in the brain. In this regard, astrocytes can express major histocompatibility complex (MHC) class I and II antigens, a variety of cytokines and chemokines, and adhesion molecules that modulate inflammatory events in the CNS. Astrocytes also secrete growth factors and extracellular matrix molecules that play a role not only in development but also in repair of the CNS.
Repair of injured nervous tissue: In the CNS, reparative processes that occur after injury, such as inflammation and necrosis, are chiefly the responsibility of astrocytes. In these reparative processes, astrocytes are analogous to fibroblasts in the rest of the body. Astrocytes do not synthesize collagen fibers, as do fibroblasts. Instead, repair is accomplished by astrocytic swelling and division, and abundant proliferation of astrocytic cell processes containing intermediate filaments composed of GFAP, a process called astrogliosis. As an example, neuronal necrosis occurs in some viral diseases of the CNS. When neurons die, the spaces left by the loss of the neuronal cell bodies are filled and such spaces (<1 mm in diameter) are filled by processes of astrocytes. Larger spaces that form after injury, such as an infarct, are often too large to be filled and therefore exist in the CNS as fluid-filled spaces (cysts) surrounded by a capsule of astrocytic processes. Astrocytes will also attempt to wall off abscesses, but they are not as effective as fibroblasts and the capsule can be incomplete or weak (Fig. 14-9). In the case of direct extension of bacteria from the meninges or meningeal blood vessels, which contain or are surrounded by fibroblasts, respectively, fibroblasts play a larger role in isolating the inflammatory process.
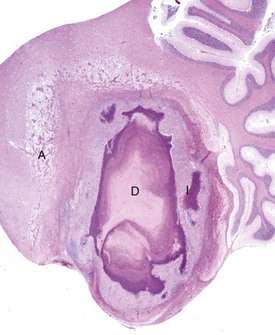
Fig. 14-9 Astrocytic repair, bacterial abscess, brainstem, sheep.
The abscess has a central core of necrotic debris (D) surrounded by a layer of inflammatory cells (I) and a less dense pink-staining zone representing an attempt by astrocytes and fibroblasts to form a capsule (A). This capsule is formed by fibrous tissue on the ventral and right sides, those sides closest to the pia, which contains fibroblasts. A fibrous capsule is absent from the dorsal and left sides of the abscess, adjacent to brain parenchyma. Here, there is no population of resident fibroblasts and the capsule is formed by astrocytes and their processes, which are often delicate and do not form an effective capsule (A). H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Structural support of the CNS: Structurally, astrocytic processes provide support for other cellular elements and ensheathe and insulate synapses. Astrocytes also provide guidance and support of neuronal migration during development; thus, tracts and fasciculi of axons with similar functions are arranged and structurally supported by astrocytic processes. Processes of astrocytes (foot processes) also terminate on blood vessels throughout the CNS, forming a component of the blood-brain barrier. Astrocytes influence the induction of tight junctions between endothelial cells that serve as the structural basis for the blood-brain barrier. A dense meshwork of astrocytic processes also forms the glia limitans beneath the pia mater and is variably prominent in subependymal areas. During CNS development, cells termed radial glia provide a scaffold and guidance for migrating neurons. When development is completed, radial glia mature into astrocytes.
Oligodendroglia: There are two types of oligodendroglia: (1) interfascicular oligodendrocytes and (2) satellite oligodendrocytes (satellite cells). The function of interfascicular oligodendroglia is myelination of axons, whereas the function of satellite oligodendroglia is thought to be regulation of the perineuronal microenvironment. Oligodendroglia have been compared with neurons with regard to their total cell size in that their processes occupy much more space than the cell body. Neurons have very long axons, which account for their size; oligodendroglia have extensive myelin sheaths, which account for their size. In H&E stained sections, oligodendroglia are often confused with lymphocytes because of the similarity of the morphology of their nuclei and cytoplasmic volume. Interfascicular oligodendroglia and perineuronal satellite oligodendroglia are located primarily in white and gray matter of the CNS, respectively (Fig. 14-10); however, interfascicular oligodendroglia can also be found along axons that traverse through the gray matter. The mature, small oligodendrocyte has a spherical, hyperchromatic nucleus (see Figs. 14-7 and 14-10). As with astrocytes, the cell body and processes of this cell do not stain with conventional H&E staining methods and can only be demonstrated following special procedures that include metallic (silver) impregnation and immunohistochemical methods.
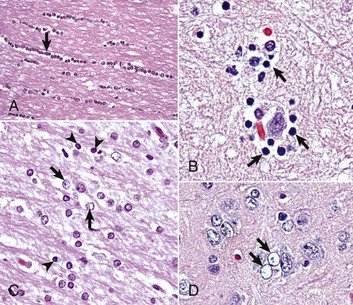
Fig. 14-10 Responses of glial cells to injury in H&E stained CNS sections.
A, White matter. In nondiseased states, oligodendroglia in white matter are often arranged linearly (interfascicular oligodendroglia) (arrow) and are responsible for the formation of myelin around axons. In gray matter (not shown; see Fig. 14-17), oligodendroglia are dispersed as individual cells around neuronal cell bodies as perineuronal satellite cells (Fig. 14-10, B). H&E stain. B, Gray matter. When neurons are injured or there exists some perturbation of the perineuronal microenvironment, a long-held belief was that oligodendroglia around neurons hypertrophy and proliferate in a process referred to as satellitosis. Currently, there is no uniform agreement that these cells respond to neuronal injury in this manner. Perineuronal satellite oligodendroglia (arrows) surround a small degenerate neuron with condensed chromatin and little cytoplasm. H&E stain. C, White matter. Astrocytes (arrows) and oligodendroglia (arrowheads) have a limited repertoire of responses to injury in the CNS. Astrocytic proliferation can occur but is very difficult to determine in sections stained with H&E. Here, astrocyte nuclei are somewhat enlarged and appear more numerous than expected. H&E stain. D, Gray matter. Astrocytes respond to injury in hyperammonemia, such as occurs with hepatic encephalopathy, by forming astrocytes with enlarged, markedly vesicular (“watery”), often elongated nuclei called Alzheimer’s type II astrocytes (arrows). This type of astrocyte may occur in pairs that are surrounded by a clear space indicative of cellular swelling. H&E stain. (A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B to D courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Most interfascicular oligodendroglia (see Fig. 14-10) are aligned in rows parallel to myelinated axons and are responsible for the formation and maintenance of segments (internodes) of myelin sheaths. One oligodendroglial cell can form as many as 50 different internodes of myelin, each of which can be located on many different axons (Fig. 14-11). Altered function of oligodendroglial cells, as occurs in infectious canine distemper virus (CDV) infection, can cause primary demyelination of these segments, resulting in severe neurologic dysfunction. Oligodendroglia also influence maturation and maintenance of axons and inhibit regeneration of established myelinated axons.
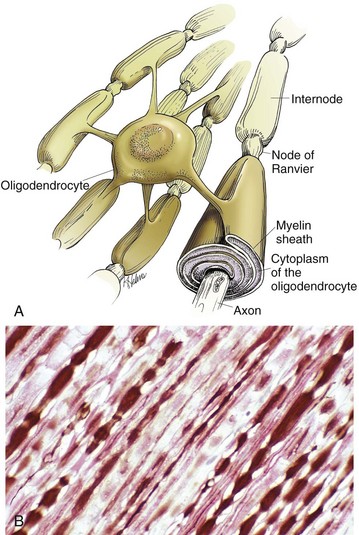
Fig. 14-11 CNS myelin.
Oligodendroglia myelinate axons within the CNS (also see Fig. 14-1). A, As depicted in this illustration, each oligodendrocyte sends out numerous cytoplasmic processes that repetitively encircle (myelinate) the portion of an axon between two nodes of Ranvier (internode) on the same and several different axons. Direct or indirect injury to an oligodendrocyte can result in “demyelination” of those internodes myelinated by that oligodendrocyte. This injury will slow the rate of conduction of an action potential, and depending on the site of the lesion may lead to clinical signs of neural dysfunction (ataxia, proprioception deficits). B, CNS nerves, longitudinal section. Axons and their neurofilaments (brown stain) and myelin (red stain) are well demonstrated by this immunohistochemical stain for neurofilaments and myelin basic protein. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Perineuronal satellite oligodendroglia (see Fig. 14-10) are adjacent to neuronal cell bodies and are also located around blood vessels in the gray matter. They are thought by some investigators to regulate the perineuronal microenvironment and respond to perturbation by proliferation. When the perineuronal microenvironment is altered or neuron cells bodies are injured, perineuronal satellite oligodendroglia, in an attempt to regulate the environmental perturbation, hypertrophy and proliferate in a process referred to as satellitosis. Similarly, alterations in the microenvironment of gray and white matter away from areas surrounding neuron cell bodies results in hypertrophy of oligodendroglia (see Fig. 14-10). Finally, satellitosis can be quite prominent in normal CNS in various areas of the gray matter.
Microglia: The basic functions of microglia are immunosurveillance, immunoregulation, and reparative (phagocytic) activities after neural cell injury and death. The origin of microglia in the CNS has been debated for years. The current consensus is that the cells originate from circulating monocytes (mesoderm-derived) that enter and populate the CNS during embryonic development and early postnatal life, analogous to the formation of the monocyte-macrophage system in other organs. After entry into the CNS, the cells become amoeboid microglia, phagocytosing dead cells and cellular debris during remodeling and maturation of the CNS. Amoeboid cells then enter a quiescent stage and transform into ramified microglia. Ramified microglia constitute up to 20% of the glial cells and are present throughout the mature CNS, serving as sentinels of brain injury. Ramified microglia, also called resting cells, are most numerous in perineuronal and perivascular areas and in interfascicular locations in white matter. Evidence of pinocytosis in ramified cells suggests some role in maintaining the neural microenvironment. The principal function of microglia is phagocytosis, the initiation of and participation in the innate and adaptive immune responses, and in degenerative and inflammatory diseases of the CNS.
Microscopically, ramified microglia have small, hyperchromatic ovoid-, rod-, or comma-shaped nuclei and no appreciable cytoplasm with routine H&E staining, thus the term rod cell is sometimes used to describe them (see Fig. 14-7). With special labeling techniques or metallic impregnation, ramified cells have a few delicate branching processes. The small hyperchromatic nuclei and nuclear shape distinguish microglia from astrocytes and oligodendroglia. However, microglia are often difficult to identify in H&E stained sections without some expertise in neuropathology.
Activated microglial cells are not the major source of active macrophages in inflammation of the CNS. Blood monocytes recruited from the circulation account for up to 70% of the macrophages in inflammatory and degenerative diseases of the CNS. These macrophages differentiate from blood monocytes involved in normal “leukocytic trafficking” through the CNS and can be involved in immunologic and phagocytic responses (gitter cells) to disease processes and infectious organisms. They are found mainly in the leptomeninges, choroid plexus, and perivascular areas.
Ependyma (Including Choroid Plexus Epithelial Cells): The basic functions of ependymal cells, which line the ventricular system, are to move cerebrospinal fluid (CSF) through the ventricular system via movement of their cilia and to regulate the flow of materials between the CNS and the CSF. The ependyma is a single-layered, cuboidal to columnar, epithelium that lines the ventricles and mesencephalic aqueduct of the brain, and central canal of the spinal cord (Fig. 14-12). This layer of cells is therefore situated between the CSF and nervous tissue. Ependymal cells have cilia that project into the CSF and beat in a coordinated manner in the direction of CSF flow. Other structures, referred to as circumventricular organs, which include the choroid plexuses, are covered by highly specialized ependymal cells. The surface of ependymal cells that form the choroid plexus have microvilli (microvillus border) and cilia that occur singly or more often in groups of three or more. The choroid plexus epithelial cells also have specialized tight junctions (zonulae occludentes) that are a functional part of the blood-CSF barrier. In contrast to the choroid plexus, junctions between the conventional ependymal cells include gap junctions (transmembrane proteins form a pore, allowing communication between adjacent cells) and zonulae and fasciae adherentes, which permit movement of materials, such as proteins from the CSF, into the extracellular space of the brain. This cellular lining, however, is not a static membrane in that it regulates several processes that involve interaction between the CSF and brain. The functions include regulation of fluid homeostasis between the ventricular cavities and the brain, secretion and absorption of CSF, endocytosis, phagocytosis, and metabolism of substances such as iron resulting from the lysis of erythrocytes after hemorrhage into the ventricular system. Finally, ependymal cells have the structural and enzymatic characteristics necessary for scavenging and detoxifying a wide variety of substances in the CSF.
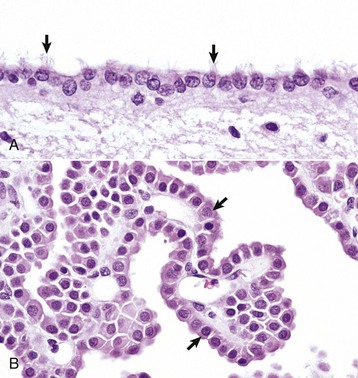
Fig. 14-12 Ependymal and choroid plexus epithelial cells.
A, Ependymal cells are ciliated (arrows) and assist with the flow of cerebrospinal fluid (CSF) through the ventricular system. H&E stain. B, Choroid plexus epithelial cells (arrows) secrete CSF from a brush-border (microvilli) on the luminal surface. The surface of the choroid plexus also has cilia that occur singly or more often in groups of three or more on a single cell. H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
During embryonic development, the medial wall of the lateral ventricle (choroid fissure), the roof of the third ventricle, and the rostral part of the roof of the fourth ventricle consist of a single layer of neuroectoderm that is adherent on its outer surface to the pia mater. This neuroectoderm-pia union forms the tela choroidea, providing an anchor for the choroid plexuses, which is formed by an invagination of this bilayer membrane into the ventricular spaces.
Choroid plexus epithelial cells are modified ependymal cells. The choroid plexus epithelium is a single-layered, cuboidal to columnar, epithelium with a microvillus border (see Fig. 14-12). CSF is secreted from the microvillus border. Choroid plexus epithelial cells, along with capillaries and the pia mater, form the choroid plexuses that project into the lateral, third, and fourth ventricles. The basic function of choroid plexuses is to produce the CSF that fills the ventricular system and the subarachnoid space. CSF has two important functions: (1) to act as a “shock absorber” to mitigate the effects of trauma to the brain and spinal cord and (2) to deliver nutrients to and remove wastes from the CNS.
The normal flow pattern of CSF is regulated by an intraventricular biologic pressure gradient in which the pressure created by secretion of CSF exceeds the pressure created by its absorption in arachnoid villi (arachnoid granulations). Arachnoid villi are focal extensions of the arachnoid and subarachnoid space that extend into the dorsal sagittal venous sinus of the brain. CSF is secreted by the choroid plexuses in the lateral, third, and fourth ventricles. It should be noted, however, that fluid from other sources, such as secretion by the ependyma, interstitial fluid of the brain, and ultrafiltrate of the blood, has also been reported to contribute to the formation of CSF. It moves from the lateral ventricles into the third ventricle, from the third ventricle through the mesencephalic aqueduct (aqueduct of Sylvius in humans), and then to the fourth ventricle. Once in the fourth ventricle, the CSF exits through the two lateral apertures of the fourth ventricle to enter the subarachnoid space. Lateral apertures are the two openings in the caudal medullary velum that forms the roof of the fourth ventricle into the subarachnoid space, one at each side of the cerebellopontine angle. Although the central canal of the spinal cord is connected to the ventricular system at the caudal end of the fourth ventricle, there apparently is little active movement of CSF within the central canal. CSF in the subarachnoid space is reabsorbed by the arachnoid villi in the brain. Recent evidence indicates that other routes of CSF drainage, in addition to arachnoid granulations, also exist and vary in different species. Venous sinuses, lymphatic drainage, and the cribriform plate appear to play important roles in CSF drainage and the maintenance of normal interventricular CSF pressure. In fact, experimental evidence suggests that the cribriform plate route may be the most important of the four. In humans, the entire volume of CSF is circulated approximately four times a day; however, with aging, the entire volume of CSF circulates less than two times a day.
Meninges: The meninges, which enclose the CNS, consist of three layers: the dura mater (outermost layer [pachymeninges]), the arachnoid membrane (mater), and the pia mater (innermost layer) (Fig. 14-13). Together, the arachnoid membrane and pia mater are frequently referred to as the leptomeninges, pia-arachnoid layer, or pia-arachnoid. The arachnoid membrane and pia mater are held together by bands of fibrous tissue called arachnoid trabeculae. This arrangement forms a compartment called the subarachnoid space in which CSF flows and which also contains blood vessels and nerves. There is also limited evidence based on studies in humans with neuro–acquired immunodeficiency syndrome that the brain has a primitive lymphatic system. The leptomeninges form a protective covering for the CNS and provide an external envelope filled with CSF that provides additional protection.
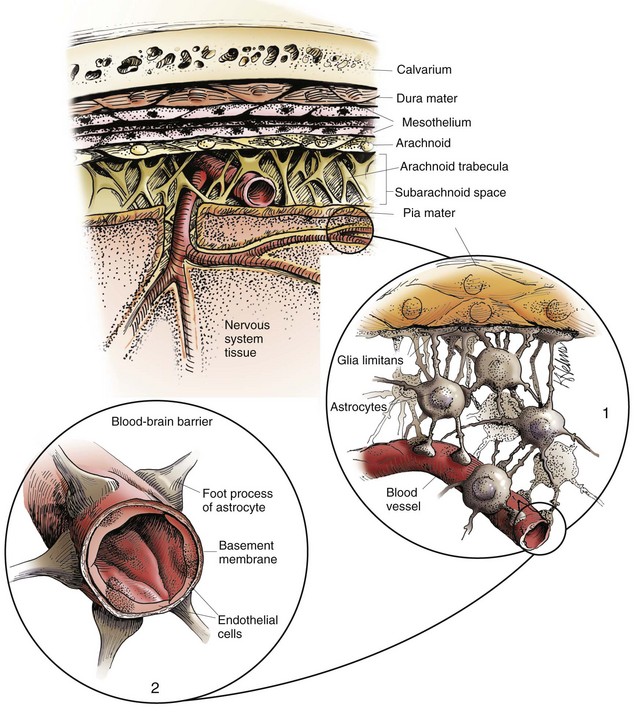
Fig. 14-13 Organization of the meninges.
The meninges, from outside to inside, are the dura mater, arachnoid mater, and pia mater as illustrated in the diagram. The arachnoid mater and the pia mater form the leptomeninges. These two layers of the leptomeninges also enclose the subarachnoid space, which contains the arteries, veins, and nerves and is filled with cerebrospinal fluid. The pia mater is attached to the surface of the CNS. Astrocytes and their foot processes underlie the pia mater and form the glia limitans (inset 1) and surround the endothelial cells that form the blood-brain barrier. As arterioles penetrate the cortex to supply the tissue with blood, they carry the pia and glia limitans with them for 1 to 3 mm until the arteriole structurally becomes a capillary. At this transition site within the cortex, the capillary penetrates the pia and is surrounded by the glia limitans, and the end feet of the astrocytes become part of the blood-brain barrier (inset 2). Components of the blood-brain barrier are capillary endothelial cells, basement membrane, and astrocytic foot processes, but the barrier is formed structurally by tight junctions between endothelial cells and functionally by specialized transport systems in these cells. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The dura mater, once referred to as the pachymeninx (thick meninges), is a strong and dense collagenous membrane (Fig. 14-14). In the cranium, the dura consists of two layers that are fused with each other. The outer layer serves as the periosteum of the cranial bone, except in the areas of the venous sinuses (surrounded by dura) and falx cerebri, which is the longitudinal layer that extends ventrally between the two cerebral hemispheres. At the level of the foramen magnum, the two layers become separated; the outer layer continues to function as the periosteum of the vertebral (spinal) canal, and the inner layer forms the free dural membrane that surrounds the spinal cord. The inner aspect of dura mater is lined by elongated, flattened mesothelial-like cells. Except in neonates, there is no epidural (extradural) space in the cranial vault as there is in the spinal cord. There can be a “potential” epidural or extradural space in mature animals from hemorrhage caused by trauma.
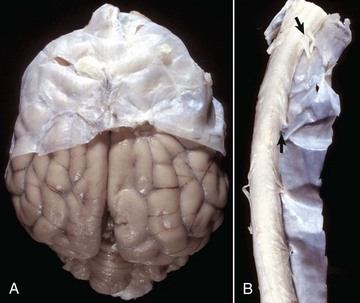
Fig. 14-14 Layers of the meninges.
A, Brain, dog. The dura matter is a thick opaque layer. Here it covers the rostral (cranial) half of the brain and has been dissected away from the caudal half of the brain to expose the underlying leptomeninges. In old animals, the dura mater often fuses with the periosteum of the calvarium, and at necropsy to expose the brain, it is usually removed attached to the calvarium. The leptomeninges are present, but because they are so transparent, in this photograph they are barely visible on the surface of the caudal half of the brain between gyri. B, Spinal cord, horse. The dura mater is the thick opaque layer dissected from and lying to the right of the spinal cord. The leptomeninges (pia-arachnoid layer) are present (but not readily visible in this photograph) on the exposed surface of the spinal cord. Arrows indicate spinal nerve roots. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The arachnoid consists of both the multilayered membrane composed of cells that overlap one another and the trabeculae that join it to the pia. The arachnoid has tight junctions between its cells, although other junctions have also been described. It contains no blood vessels and has an outer smooth surface formed by mesothelial-like cells that abut similar cells in the dura mater. The mesothelium-like surfaces of the dura and arachnoid oppose and slide over each other, analogous to the parietal and visceral surfaces of other serous membranes.
The pia mater is closely adherent to the surface of the brain and spinal cord and is penetrated by a large number of blood vessels that supply the underlying nervous tissue (Fig. 14-15). The pia mater consists of flat, thin, overlapping connective tissue cells (fibroblasts) that are separated from the underlying neural tissue by variable amounts of loose collagen fibers and the glia limitans. In many areas the pia, which lacks a basal lamina, is only one-cell-layer thick and has fenestrations, so that the glia limitans is exposed directly to the subarachnoid space. Pial and arachnoid cells also ensheathe blood vessels, collagen bundles, and nerves that are within or cross the subarachnoid space and also are around arteries that penetrate into the CNS up to 1 to 2 mm in depth. Macrophages also are present throughout the leptomeninges.
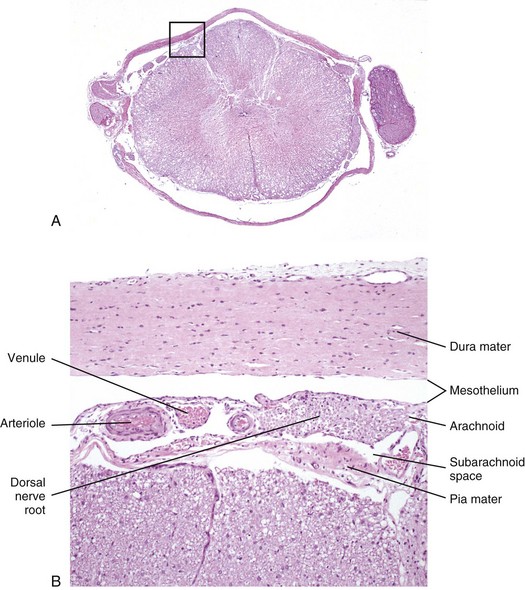
Fig. 14-15 Histologic section of spinal cord and meninges.
A, Low magnification of a cross-section of the spinal cord and meninges with spinal nerve rootlets and a dorsal root ganglion from which B was selected (box). H&E stain. B, The inner surface of the dura mater and the outer surface of the arachnoid mater are covered with mesothelial cells, and the space between them is the subdural space. Blood vessels and nerves of the dorsal and ventral roots traverse in the subarachnoid space. H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Endothelium: The basic functions of endothelium in the CNS are to line luminal surfaces of blood vessels; form the blood-brain barrier; regulate thrombosis, thrombolysis, and platelet adherence; and maintain a nonthrombogenic boundary between coagulation cascade molecules and luminal surfaces of endothelial cells. Additionally, endothelial cells function as regulatory barriers to small and large molecules crossing the endothelium, and they control the adherence of leukocytes to their luminal surfaces. The endothelial cells of the blood-brain barrier actively transport those molecules that the brain consumes rapidly and in large quantities such as glucose, amino acids, lactate, and ribonucleosides.
Responses to Injury
Ground Rules for Understanding Injury in the CNS
Before the responses of the CNS to injury are discussed, some fundamental concepts are reviewed in Box 14-2.
Neurons
Neurons are the most vulnerable cells in the nervous system and probably within the body. They have large requirements for energy to maintain normal metabolism, transport systems, and the formation of cytoskeleton proteins in the axon, which can extend over long distances (>1 m), but neurons lack adequate intracellular glucose reserves. Therefore they are completely dependent for survival on an adequate blood supply to provide glucose. Additionally, neurons are vulnerable to free radical oxidative stresses and have a limited ability to buffer shifts of calcium ions into the cell, which can interfere with oxidative phosphorylation and ATP production, such as occurs with ischemia.
Neurons are especially sensitive to excessive stimulation with excitatory amino acid neurotransmitters called excitotoxins (e.g., glutamate and aspartate). These neurotransmitters are also released in a wide variety of neuronal injuries, especially in neuronal ischemia. Under normal conditions, astrocytic processes surrounding synapses have efficient uptake systems to remove excitotoxins, and neurons are not injured. In excessive quantities, persistent binding of excitotoxins to receptors can lead to neuronal degeneration and death.
The microscopic appearance of the neuronal cell body can vary according to the injury. Characteristic changes of the neuronal cell body are reviewed in Box 14-3.
Neuronal Cell Death: Neurons can die after injury as a result of one of two mechanisms: apoptotic cell death and necrotic cell death. These mechanisms are summarized next and discussed in greater detail in Chapter 1. Both apoptotic and necrotic neuronal cell death can occur concurrently or in temporal or spatial sequences within the nervous system. Although apoptotic and necrotic neuronal death represent different responses of neurons to injury, identical receptors, messenger systems, and mechanisms of cytotoxicity are likely involved in both apoptotic and necrotic cell death. Factors that determine whether the apoptotic or necrotic pathway is activated are unclear but appear to depend on the character on the initiating ligand or injury, type of cell membrane receptors activated, and caspases expressed in response to injury.
Apoptotic Cell Death (Programmed Cell Death): Apoptosis is a single cell-initiated, gene-directed cellular, and self-destructive regulatory mechanism that leads to “programmed” cell death. This mechanism is used (1) during the development of the nervous system to ensure proper migration and orientation of cell layers and removal of excess embryonic cells, (2) to remove “aged” cells (i.e., cell turnover) in organs, and (3) to maintain cell number homeostasis in organ systems that have regenerative capacity (endocrine glands).
Apoptotic neuronal death is characterized by a sequence of cellular degenerative steps that can be identified biochemically and morphologically. After appropriate signals are recognized and interpreted by cell membrane receptors (Fas, tumor necrosis factor [TNF] receptor-1, TNF-related apoptosis-inducing ligand receptors), a family of proteins known as caspases are activated. Caspases cleave cellular substrates that are required for cellular function and include cytoskeleton proteins and nuclear proteins such as DNA repair enzymes. Caspases also activate other degradative enzymes, such as DNAases, which cleave nuclear DNA.
The role of apoptotic neuronal death in specific neurologic diseases is discussed in greater detail in subsequent sections. As examples, some viral infections that occur in utero produce developmental anomalies by initiating apoptosis that leads to faulty differentiation of embryonic granule and Purkinje cell layers such as occurs in experimental Borna disease. Mild ischemia, excitotoxins, hormones, corticosteroids, and proinflammatory cytokines can induce apoptotic cell death. Rabies virus has been linked experimentally to apoptotic neuronal death.
Apoptosis results in characteristic morphologic changes in cells such as shrinkage, cytoplasmic condensation and blebbing, and chromatin clumping and fragmentation (see Figs. 1-30 through 1-34). As cells continue to shrink, nuclear chromatin is cleaved into smaller units and along with condensed cytoplasm is packaged for removal by macrophages. Inflammation is not induced by apoptotic cell death.
Necrotic Cell Death: Necrosis is a process that usually affects groups of cells in contrast to single isolated cells as observed in apoptosis. Necrosis is characterized by the following sequence: hydropic degeneration, swelling of mitochondria, pyknosis and fragmentation of the nucleus, and eventual cell lysis caused by cell membrane damage and the inability of the plasma membrane to control ion and fluid gradients (see Figs 1-11 through 1-17). Cellular debris associated with necrotic neuronal death elicits an inflammatory response in contrast to apoptotic neuronal death.
Acute neuronal necrosis (ischemic cell change): Acute neuronal necrosis is a common response to a variety of CNS injuries, such as cerebral ischemia caused by blood loss and hypovolemic shock, vascular thrombosis, and cardiac failure; inflammatory mediators; bacterial toxins; thermal injury; heavy metals; and nutritional deficiencies, such as thiamine deficiency; and trauma. Additionally, conditions that reduce ATP generation through oxidative phosphorylation also lead to neuronal degeneration and death. Such conditions include (1) interference with cytochrome oxidase activity in mitochondria caused by cyanide poisoning, (2) competitive inhibition of oxygen uptake in carbon monoxide poisoning, and (3) inadequate availability of glucose for neuronal metabolism in hypoglycemia.
The susceptibility of cells and tissue structures of the CNS to ischemia in decreasing order of susceptibility are neurons, oligodendroglia, astrocytes, microglia, and blood vessels. However, within groups of neurons, some neurons are more sensitive to injury than others. This phenomenon is called selective neuronal vulnerability. Purkinje cells; some striatal neurons; neurons of the third, fifth, and sixth cerebral cortical lamina; and hippocampal pyramidal cells have the highest vulnerability. A regional vulnerability of neurons has also been reported (cerebral cortex and striatum > thalamus > brainstem > spinal cord). It is hypothesized that the most vulnerable neurons likely produce the most excitotoxins, such as glutamate, and are the most sensitive to them. Because of the microanatomic arrangement of the cerebral cortex, ischemic neurons often occur in a laminar pattern within the cerebrocortical gray matter. This microanatomic pattern accounts for the laminar lesions observed in thiamine deficiency–induced polioencephalomalacia in ruminants and in other diseases such as salt poisoning in pigs and lead poisoning in ruminants.
After the various types of CNS injury, there is an early increase in ATP-dependent release of normally sequestered intracellular calcium ions from altered mitochondria and endoplasmic reticulum. Also during this time, neuronal depolarization potentiates the release of the neuroexcitatory neurotransmitter glutamate. Persistent activation of glutamate receptors of target cells results in a disturbance referred to as excitotoxicity. This altered activity leads to a notable influx of extracellular calcium into cells, causing further impairment of mitochondrial function and the generation of reactive oxygen species, such as superoxide, hydrogen peroxide, hydroxyl radicals, and nitric oxide. These reactive oxygen species, exerting their effects especially on lipid-rich cell membranes, can enhance the existing excitotoxicity, cause further influx of calcium into cells as a result of membrane damage, and ultimately result in neuronal dysfunction and death. Additionally, reperfusion of ischemic tissue after the initial ischemic injury can enhance the generation of reactive oxygen metabolites, thus amplifying the tissue damage. Other influencing factors include the temperature of the brain at the time of ischemia, with lower temperatures (as little as 2° C decrease) having a sparing effect and elevated temperatures having an enhanced effect on neuronal injury following ischemia.
Neurons depend on a continuous supply of oxygen to remain viable, and if the supply is interrupted for several minutes, the vulnerable neurons as described previously degenerate. Ischemic cell change can also result from metabolic disturbances other than ischemia, such as in thiamine deficiency and cyanide toxicosis, which interferes with oxygen use. In H&E stained sections, the cytoplasm of the neuronal cell body is shrunken, deeply eosinophilic, and frequently sharply angular to triangular in shape (Fig. 14-16). The nucleus is reduced in size, is often triangular, and is pyknotic. The nucleolus and Nissl substance are usually not detectable. Ischemia neurons die and are removed either by a process called neuronophagia, which is phagocytosis by microglial cells and macrophages or by lysis (see Fig. 14-16). After neuronal necrosis, there is swelling of perineuronal and perivascular astrocytic processes.
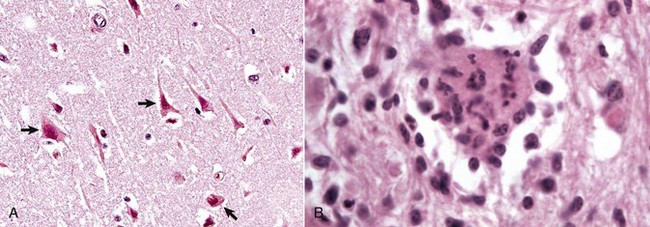
Fig. 14-16 Neuronal necrosis (acute), so-called ischemic cell change, cerebrum, dog.
A, Neuronal ischemia. Neuronal cell bodies of cerebral cortical laminae are red, angular, and shrunken (arrows) and their nuclei are contracted and dense. This lesion can be caused by neuronal ischemia. H&E stain. B, Neuronophagia. This necrotic neuron cell body (center of figure) is surrounded by macrophages that will phagocytose the cell debris. H&E stain. (A courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Chronic neuronal necrosis (brain atrophy): Neuronal death and loss of neurons can occur as a result of progressive disease processes of long duration in the CNS. This loss, termed simple neuronal atrophy, is seen with slowly progressive neurologic diseases, such as cerebral cortical atrophy of aging, ceroid-lipofuscinosis, and primary and multisystem and cerebellar neuronal degeneration. Gross lesions are usually not visible, but when cerebrocortical neurons die, there can be atrophy of cerebral gyri, which results in widening of the sulci (Fig. 14-17). Microscopic lesions indicative of an earlier loss of neurons are diminished numbers of neurons and astrogliosis and atrophy and loss of neurons in functionally related systems. Loss of neurons over time results in progressively worsening neurologic dysfunction.
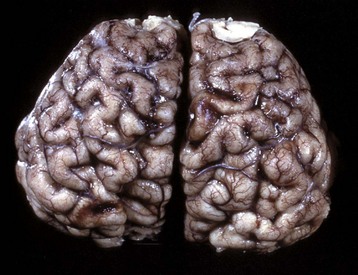
Fig. 14-17 Cerebral cortical atrophy, horse.
Atrophy is seen with a variety of slowly progressive neurologic diseases in which there is a progressive loss of neurons. These diseases include cerebral cortical atrophy of aging and ceroid-lipofuscinosis. The characteristic gross lesions are narrowing of the cerebral gyri with a consequent widening of the sulci. (Courtesy the Department of Veterinary Biosciences, The Ohio State University.)
Wallerian Degeneration and Central Chromatolysis: Injury to axons of the CNS and PNS can result from a variety of causes such as (1) traumatic transection leading to Wallerian degeneration, (2) compression and crushing, (3) therapeutic neurectomies, (4) nerve stretching injury, and (4) intoxication.
Wallerian Degeneration: In 1850, Dr. Augustus Volney Waller described the pattern of microscopic lesions (necrosis) in axons and myelin sheaths after transection. These changes are characteristic of Wallerian degeneration. Although Waller described this process in peripheral nerves, the term Wallerian degeneration is also used to describe necrosis that occurs in nerve fibers in the CNS after axons are injured (compressed or severed). These reactions include swelling of the neuronal cell body, dispersion of centrally located Nissl substance, and peripheral displacement of the nucleus termed central chromatolysis (Fig. 14-18) and swelling of the axon and myelin distal to the site of injury are to swell and break, and the rate is proportional to the diameter of the fiber (Fig. 14-19). Thus the larger the diameter of the axon, the faster the rate of Wallerian degeneration.
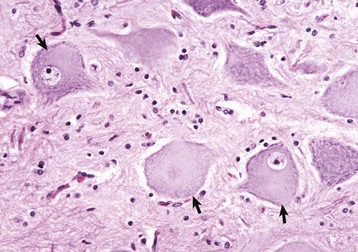
Fig. 14-18 Central chromatolysis, neuron cell body, dog.
Compare with Figs. 14-4, B, and 14-7. Affected neurons have eccentric nuclei and pale central cytoplasm with dispersed Nissl substance (arrows). (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
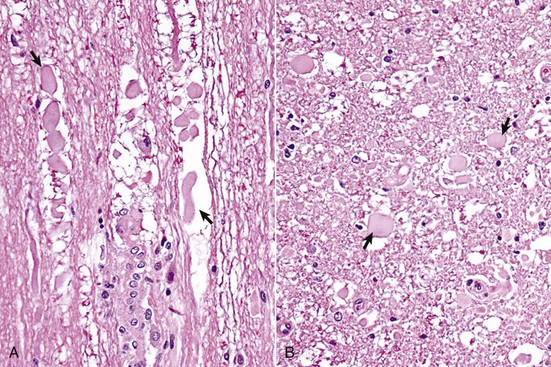
Fig. 14-19 Wallerian degeneration, transverse section of spinal cord, dog.
A, Longitudinal section. Arrows show swollen axons. H&E stain. B, Transverse section. H&E stain. Laceration and/or severe compression of myelinated nerves cause a specific sequence of structural and functional changes in the axon and the myelin (distal from the point of injury), referred to as Wallerian degeneration (see Web Fig. 14-3). Axons are initially swollen (arrows) and are eventually removed by phagocytosis to leave clear spaces, which were once the sites of nerve fibers. The cell bodies of affected neurons usually have central chromatolysis, but are metabolically active in an attempt to regenerate the lost portion of the axon (not shown; see Fig. 14-18). (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
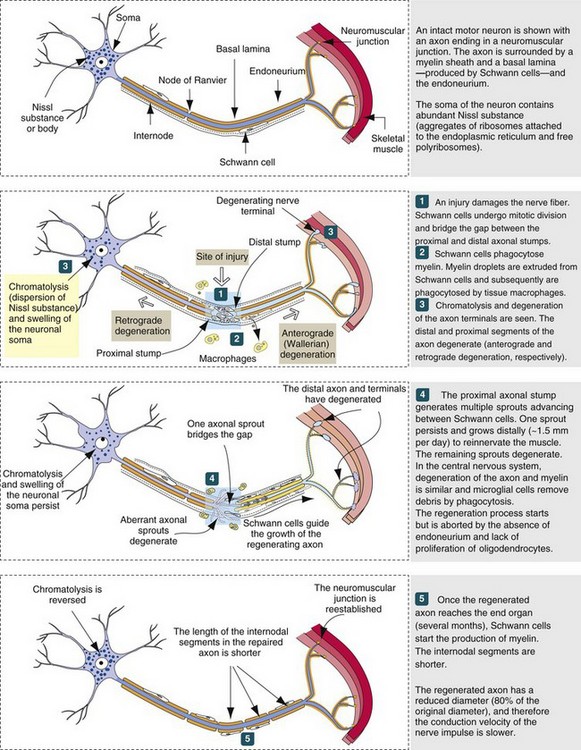
Web Fig. 14-3 Peripheral nerve degeneration and regeneration (also applicable to neurons within the CNS). (Modified from Kierszenbaum AL: Histology and cell biology, St Louis, 2002, Mosby.)
Wallerian degeneration in the CNS follows the same sequence of events as in peripheral nerve fibers, but the speed of degeneration and phagocytosis is slower and there is little or no regeneration (Web Fig. 14-3). In addition, an axon of the PNS has the advantage of (1) efficient phagocytosis with removal of debris, (2) Schwann cells to remyelinate the regenerated axon, and (3) an endoneurial tube to guide the axon as it extends into the distal segment. On the other hand, an axon of the CNS has (1) few microglia (sparse in the white matter) to remove myelin debris and (2) oligodendrocytes with a more limited capability to remyelinate axons. In the CNS, severed axons have a very limited ability to regenerate and successfully reinnervate their appropriate sensory or motor structures. In the PNS, if the severed nerves have a large distance between the cut ends, fibrous tissue scarring can prevent the axons from the proximal segment from entering the distal endoneurial tubes and thus prevent the reparative response, resulting in the formation of a neuroma. If the severed ends of nerves are sewn together but the endoneurial tubes are malaligned, which usually occurs, the sensory and motor nerves regenerate but reinnervate inappropriate sensory and motor structures.
The sequence of Wallerian degeneration in the PNS (similar in CNS except for significant regeneration; see previously) includes the following:
1. Degeneration and fragmentation of axon and myelin within several days. Proximal segment degenerates back to the next node of Ranvier, but all the distal segment dies.
2. Removal of axonal and myelin debris by phagocytosis. Some phagocytes are from the blood and some phagocytosis is by Schwann cells. All of the debris is cleared out of the endoneurial tube within a few weeks.
3. Regeneration of axon if the endoneurium is intact to allow the axon of the proximal segment to enter and slide down the tube.
Microscopically, another early lesion in Wallerian degeneration is central chromatolysis, characterized by swelling of the neuronal cell body, dispersion of centrally located Nissl substance, and peripheral displacement of the nucleus (see Fig. 14-18). The time of onset depends on how much of the axon has been lost, and thus on how close to the neuronal cell body the axon has been transected. Onset can begin within 24 to 48 hours and reach its maximum in about 18 days after axonal injury. The dispersal of Nissl substance (chromatolysis) is indicative of enhanced synthesis of transport and structural proteins required for regeneration of the axon and reestablishment of fast and slow axonal transport systems. The extent to which chromatolysis develops is related to the degree and location of axonal injury. It is more prominent and can even be followed by neuronal death, the more severe the loss of the volume of the axon and the closer the axonal injury is to the cell body. The time required for recovery of cell bodies can be several months and in most cases will vary from 3 to 6 months, depending on the severity of the axonal injury and the length of axon regenerated.
The change in the axon distal to the point of injury is first evident within 24 hours of injury. Wallerian degeneration is initially characterized by irregularity of the axonal diameter, which is followed after 48 to 72 hours by fragmentation of the axon and myelin along its length (see Fig. 14-19). This axonal alteration is followed by disintegration, and usually there is no evidence of the axon remaining by the second week after the injury.
Changes in the myelin sheath surrounding myelinated axons are evident by 28 to 96 hours after injury when axonal disintegration is well advanced. Initially, there are irregularities in the sheath accompanied by folding, lamellar splitting, fracturing, and fragmentation (secondary demyelination). The fragmented myelin can form droplets (termed ellipsoids), which surround and enclose isolated fragments and debris of the former axon. Both axonal and myelin debris are then removed by macrophages through phagocytosis. Degeneration of myelin is usually completed by the end of the second week, although evidence of myelin debris can be detected up to 1 to 3 months after axonal injury (the time required for macrophages in the PNS to phagocytose and clear the debris). Myelin debris can be detected in the CNS (when compared with the PNS) for a much longer time after injury.
If the neuronal cell body survives the injury to its axon, regeneration from the proximal stump in the PNS can occur. The degree of axonal regeneration depends on the status of the endoneurial tube (sheath) distal to the original point of injury (see Web Fig. 14-3). The normal endoneurial tube (sheath) and its contents consist (from outside inward) of (1) a connective tissue investment referred to as the endoneurium, (2) the basement membrane that surrounds the Schwann cells plus the Schwann cell cytoplasm, (3) the myelin sheath of myelinated axons, and (4) the axon. Approximately 24 to 72 hours after axonal injury, the endoneurial tube (sheath), formed by persisting basement membrane and endoneurium, contains degenerating remnants of the previously existing axon along with Schwann cells. Schwann cells begin to proliferate and eventually form a longitudinal column of cells referred to as the bands of Büngner.
If the endoneurial tube (sheath) remains intact, as can occur following a compression injury to a peripheral nerve, neural regeneration through the formation of axonal sprouts can occur. A regenerating sprout from the proximal axonal stump can enter the column of Schwann cells and regenerate uninterrupted along its original pathway to the periphery, reestablishing innervation with an end-organ (skeletal muscle). Such axons then become remyelinated and regain their physiologic function of impulse transmission. Because of axoplasmic flow, a regenerating neuron lengthens at a rate of approximately 1 to 4 mm/day. Although the time required for axonal regeneration can vary, depending on the length of axon to be regenerated, examples of times required for morphologic and functional recovery after crush injury of a peripheral nerve are 250 to 300 and 456 to 486 days, respectively. If, however, the integrity of the endoneurial tube (sheath) is destroyed, as would occur after complete severance of a peripheral nerve, regeneration might not occur, because the proximal axonal stump might be prevented from reaching the distal endoneurial tube (sheath) by proliferated fibrous connective tissue (scar formation) at the site of axonal severance. Regenerating axons may also enter inappropriate endoneurial tubes (sheaths), resulting in improper impulse transmission, such as a sensory neuron axonal sprout entering an endoneurial tube intended for a nerve innervating a muscle.
The microscopic lesions for Wallerian degeneration within the CNS are similar to those described for the PNS, except that in the CNS, degenerated axons and myelin sheaths can remain for months to years before complete removal. With injury to the CNS, some cell bodies have central chromatolysis; others initially have central chromatolysis followed by atrophy and death. Affected axons and their myelin sheaths undergo a rather characteristic series of changes as they degenerate. Initially, axons form linear and bulbous swellings at and some distance from the site of injury. These enlargements are termed axonal spheroids. Spheroids consist of neurofilaments, microtubules, and cellular organelles. Because injury to the axon results in dysfunction of axoplasmic flow, any disruption of anterograde and retrograde axonal transport results in the accumulation of neurofilaments, microtubules, cellular organelles, and recycled molecules at or near the point of injury. This process occurs in peripheral nerves as well.
The axonal enlargements can be seen as early as a few hours at the site of injury and remain prominent, particularly for the first week or so (see Fig. 14-19). The surrounding myelin sheath is usually distended to create a space between the sheath and the axonal swelling. Progressively, such affected axons and myelin sheaths fragment along their length, forming ellipsoids as in the PNS. Eventually ellipsoids are removed through degeneration and phagocytosis, leaving an empty space, or one still containing myelin debris and macrophages (gitter cells). The latter is termed a digestion chamber. With time and continued lysis and phagocytosis of the debris, most of the lesion will consist of enlarged empty spaces. The absence of swollen axons in such dilated spaces, especially in the early stages after CNS trauma, does not necessarily mean that the entire axon has degenerated and been removed, which can require several months. It might instead be the site of separation of an enlarged axon from the adjacent axon at the level of the section being examined.
Macroglia
Astrocytes: Common astrocytic reactions in CNS injury are swelling, hypertrophy, division, and the laying down of intermediate filaments in cell processes. The term astrocytosis means that astrocytes have increased in size and number in response to injury, whereas the term astrogliosis (somewhat synonymous with hypertrophy) implies synthesis of intermediate filaments and an increased length, complexity, and branching of the astrocytic processes. The recognition of these differences is based on histopathologic evaluation.
Swelling is an acute response and is reversible, or it may progress with time to hypertrophy. Swollen astrocytes have clear-staining or vacuolated cytoplasm. Astrocytes swell after ischemia because of the increased uptake of sodium, chloride, and potassium ions and water in an effort to maintain homeostasis in the extracellular microenvironment. It is important to remember that such swelling depends on the astrocyte being viable and still having a semipermeable plasma membrane, even though its function may be altered. With progression and if the degree and duration of ischemia are sufficiently severe to result in cell death, the plasma membrane becomes fully permeable, and the cell does not swell but becomes shriveled or shrunken and undergoes disintegration, as described for the ischemic cell change of neurons.
If injury is severe, astrocytic processes fragment and disappear followed by lysis of the cell body. Hypertrophied astrocytes, often referred to as reactive, represent a response to a milder and more protracted injury to the CNS. Because of increases in intermediate filaments, mainly GFAP, the cytoplasm becomes apparent along with increased length and branching of the processes with H&E staining. The increase of intermediate filaments and consequently the intensity of GFAP immunohistochemical staining in these cells are so dramatic that some have defined reactive astrocytes on the basis of this change. In protracted degenerative conditions, astrocytes termed gemistocytes can be observed (Fig. 14-20). These cells have eccentric nuclei and abundant pink homogeneous cytoplasm, in contrast to the lack of visible cytoplasm in normal astrocytes, with routine H&E staining. Animals with hepatic encephalopathy can have a unique microscopic lesion in the brain affecting astrocytes of the cerebral cortices. Astrocytic nuclei tend to be in pairs occasionally with prominent central nucleoli and surrounded by a clear space, which is the edematous cytoplasm. They are called Alzheimer’s type II astrocytes (see Fig. 14-10, D).
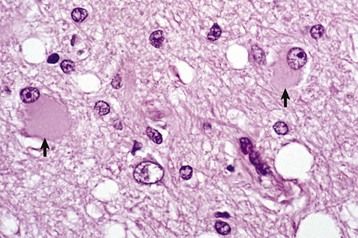
Fig. 14-20 Gemistocytes (gemistocytic astrocytes), cerebrum, dog.
When astrocytes react to injury, initially by hypertrophy and later by the synthesis of increased glial filaments (astrogliosis), the nuclei enlarge and often the cell body, which is not normally visible in H&E stained sections, will become visible. These astrocytes are called gemistocytes (plump astrocytes) (arrows). They occur in diseases in which there is alteration of intracellular and extracellular fluid balances or injury to the parenchyma, where healing will be by glial scarring (astrogliosis, e.g., to encapsulate a deep abscess or fill in a small area of dead space). H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Astrocytic proliferation can occur in CNS injury, but in most instances, proliferative capacity is limited. When it occurs, the most dramatic examples are associated with attempts by reactive astrocytes (astrogliosis) to “wall off” abscesses and neoplasms or to fill in cavitated areas that result after lysis of necrotic neurons with the processes of astrocytes. This laying down of glial fibers is referred to as a glial scar. It is formed by a network of interlaced astrocytic processes and provides a loose barrier that separates the injured brain from the more normal adjacent tissue. This astroglia-fibroblast interface in injured CNS attempts to reform the glia limitans, restore the blood-brain barrier, and reestablish fluid and electrolyte balances.
Oligodendrocytes: Oligodendroglia react to injury by cell swelling, hypertrophy, and degeneration. Both perineuronal and interfascicular oligodendroglia can swell, hypertrophy, and degenerate; however, only oligodendroglia precursor cells can proliferate to replace degenerate cells. The role that perineuronal or satellite oligodendroglia play in normal neuronal function and neuronal injury has not been definitively clarified. Microscopically, these cells swell and hypertrophy around injured neurons; this response to injury has been called satellitosis (see Fig. 14-10, B).
Degeneration of interfascicular oligodendroglia caused by ischemia, certain viruses, lead toxicity, and autoimmunity can result in selective degeneration of myelin sheaths referred to as primary demyelination. Primary demyelination is the loss of myelin around an intact axon and results in the alteration of the conduction velocity of an action potential down the axon leading to clinical dysfunction (Fig. 14-21). Mechanisms of primary demyelination are summarized in Box 14-4. Protracted or repetitive injury to myelinating cells and their myelin sheaths can lead to irreversible neuronal atrophy. Oligodendroglia precursor cells located in the subventricular zone of the CNS can mature into interfascicular oligodendroglia and can also proliferate in response to noncytocidal injury and become involved in remyelination after primary demyelination.
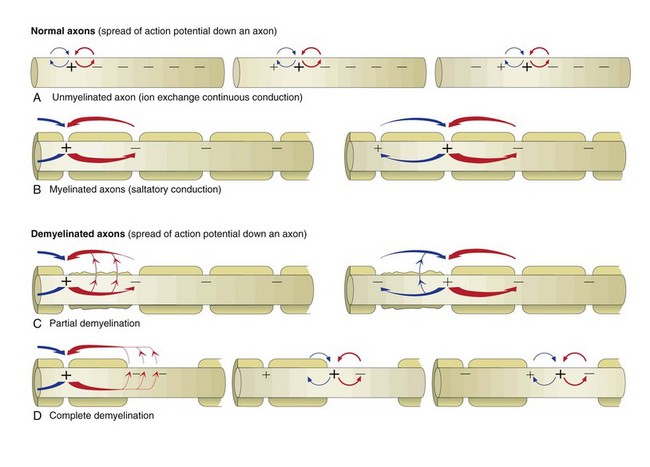
Fig. 14-21 Axonal action potential conduction and the effect of demyelination.
The speed of the conduction process is determined by the diameter of the axon and the degree of myelination. As axons increase in diameter, the resistance to ion flow decreases, allowing the action potential to flow faster. In addition, the degree of myelination is directly proportional to the diameter of the axon. Thus the concept that the more myelin the faster the speed of the impulse is true up to the point in which the myelin is normal in thickness. For an axon whose myelin is reduced, conduction of the action potential is slower. Under normal conditions, locomotion is a well-coordinated event that requires precise timing (speed) of impulse conduction to get coordinated movements. If the speed of the action potential is altered by disease, especially demyelination, then the conduction of the action potential will be delayed and what are normally coordinated movements become uncoordinated. A, In unmyelinated axons, action potentials are conducted at a relatively “slower” velocity by the process of ion exchange continuous conduction (Web Fig. 14-2). B, In myelinated axons, action potentials are conducted at a relatively “faster” velocity by a mechanism called saltatory conduction. Optimal function of saltatory conduction is dependent on having the proper degree of myelination of the axon (as determined by axonal diameter) throughout the full length of the axon. C, In axons that have lost some but not all of their myelin lamellae from one or more internodes so that there is a “thinner” covering of myelin, the speed of saltatory conduction is reduced because of leakage of the action potential across this thinner myelin sheath, resulting in clinical dysfunction of the nervous system. D, In axons that have lost all of their myelin from one or more internodes (complete primary demyelination of the internode), the speed of saltatory conduction is reduced because of the conversion from saltatory conduction to ion exchange continuous conduction in the areas where internodes have lost their myelin. Thus the speed and timing of the action potential is substantially reduced, leading to clinical dysfunction of the nervous system. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
CNS or PNS injury can also lead to loss of myelin secondary to injury of the axon and its cell body or to death of the neuron. When axons are injured, myelin lamellae forming the internodes are retracted and removed by phagocytosis. In some instances, oligodendroglia or Schwann cells, the myelin-forming cells in the PNS, also degenerate. This form of myelin degeneration is termed secondary demyelination and is secondary to axon degeneration or loss (resembles Wallerian degeneration).
Ependymal Cells: Ependymal and choroid plexus epithelial cell responses to injury include atrophy, degeneration, and necrosis. Atrophy usually occurs in response to enlargement of the ventricles as occurs with hydrocephalus. The cilia and microvilli of affected cells are reduced in number, and there is also a reduction in their cellular organelles such as endoplasmic reticulum and mitochondria. An additional lesion that accompanies ventricular enlargement is stretching and tearing of the ependymal lining. In such instances, the resulting areas of ependymal discontinuity result in the subependymal CNS being directly exposed to the CSF. Unfortunately, mammalian ependymal cells do not regenerate and therefore do not repair the denuded areas. After 1 to 2 weeks, astrogliosis, which varies greatly in degree and uniformity, occurs in the exposed areas. Astrogliosis can extend into the ventricular space or be minimal in extent and confined to the periventricular area. Periventricular interstitial edema, myelin loss, and axon loss can ensue.
Inflammation of the ependyma, called ependymitis, can also occur, with infection being the most common cause. Microbes most commonly gain entrance to the ependyma via the circulation by lodging in the choroid plexuses, by direct contamination from a rupture of a cerebral abscess into the ventricular system, and by retrograde reflux through the lateral apertures of infected CSF from the subarachnoid space in cases of leptomeningitis. In the case of bacterial infection, the suppurative exudate that forms in the CSF can cause obstructive hydrocephalus, although the development of hydrocephalus cannot always be explained on the basis of obstruction.
Microglia
Microglia are often the first cells in the CNS to react to injury, and the magnitude of the response is graded to correlate with the severity of damage. The responses of microglia to injury include hypertrophy, hyperplasia, phagocytosis of cellular and myelin debris, and neuronophagia, which is the removal of dead neuron cell bodies. After injury, microglia progress through a stage of activation, becoming fully immunocompetent reactive cells. These reactive cells readily proliferate, either focally, forming glial nodules (Fig. 14-22), or more diffusely, depending on the nature of the injury. As mentioned, in concert with astrocytes and neurons, microglia help coordinate inflammatory events in the CNS. Resident microglia and blood-derived macrophages express MHC class I and II antigens, serve as antigen-presenting cells, and possess a broad armament of adhesion molecules, cytokines, and chemokines. Once activated, these cells can also produce nitric oxide, reactive oxygen intermediates, and other chemical mediators of inflammation that can damage the CNS if not under strict control. When tissue necrosis occurs, macrophages derived from blood monocytes phagocytose the lipid-laden debris of dead neurons and glia, and they become foamy macrophages termed gitter cells (Fig. 14-23) and accumulate in the damaged CNS.
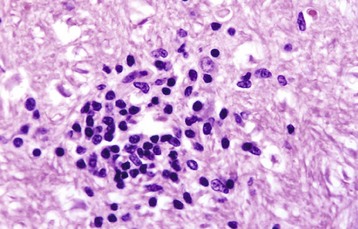
Fig. 14-22 Glial nodule, brainstem, dog.
These nodules (center of figure), formed by reactive microglial cells and macrophages, occur most frequently in viral encephalitides. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
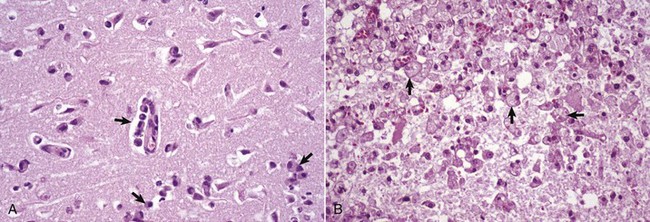
Fig. 14-23 Gitter cells, cerebrum.
A, Early polioencephalomalacia, cow. Note the angular, eosinophilic neurons with pyknotic nuclei (ischemic cell change). Monocytes (arrows) in the perivascular space have been recruited from the vasculature. These cells will become macrophages and phagocytose cellular debris from the necrotic neurons and the myelin from the nerve fibers undergoing degeneration after the death of their neurons. Microglia also participate in this phagocytic response. Macrophages, which have ingested degenerate myelin or other cellular debris, have foamy cytoplasm and are termed gitter cells. H&E stain. B, Old necrotic area, dog. The normal brain parenchyma has liquefied and the debris has been ingested by macrophages (arrows), which has resulted in the cytoplasm of these cells becoming foamy. They are now designated as gitter cells or, simply, foamy macrophages. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Meninges
Pathologic processes that initially involve the meninges, most commonly the leptomeninges, can secondarily invade the CNS because of the close apposition between the two tissues. Conversely, processes that primarily affect the CNS can secondarily affect the meninges, most commonly the leptomeninges.
Meningitis refers to inflammation of the meninges. In common usage, the term generally refers to inflammation of the leptomeninges in contrast to inflammation of the dura mater, which is referred to as pachymeningitis. Leptomeningitis can be acute, subacute, or chronic and depending on the cause, suppurative, nonsuppurative, or granulomatous, and the exudate and inflammatory cells are chiefly in the subarachnoid space. Besides retrograde axonal transport, as occurs with, for example, Listeria monocytogenes, infectious agents spread to the meninges hematogenously by direct extension or by leukocytic trafficking.
Other meningeal lesions include (1) inflammation of the external periosteal dura after osteomyelitis, formation of extradural abscesses, and skull fracture and involve the inner dura as an extension of leptomeningitis and (2) proliferation of the inner dural mesothelial cells, arachnoid cells, fibroblasts, and cells of the pia mater in response to irritation. Additional lesions likely related to aging or degeneration include formation of cellular nests of mesothelial-like cells on the outer surface of the arachnoid membrane, mineralization of the arachnoid membrane in humans, and mineralization plus ossification of the dura mater of the spinal cord in both humans and dogs. Dural ossification in the dog, which tends to affect the ventral, cervical, and lumbar dura mater, is most commonly encountered in large breeds, although smaller breeds can be affected. The clinical significance of this lesion has been debated but not decided.
Circulatory System
Endothelial Cell (and Blood Vessel) Responses to Injury: Because many of the infectious and neoplastic disease processes demonstrated in this book are spread through the body via the circulatory system, endothelial cells lining blood vessels, especially capillaries, are subject to a variety of injuries. Bacterial hematogenous CNS diseases occur at the interface between the white and gray matter in the cerebral hemispheres. This phenomenon is thought to result from abrupt changes in vascular flow or luminal diameter of vessels at the interface. These changes may make endothelial cells more susceptible to injury, vasculitis, and thrombosis or predispose the vessels to entrapment of tumor or bacterial emboli.
Endothelial injury can be reversible or nonreversible, resulting in necrosis. Injury resulting in endothelial dysfunction can include the activation and release of vasoactive mediators, such as histamine, leading to local and/or systemic changes in vascular flow, pressure, and permeability. Bacterial products and elicited inflammatory cytokines can directly or indirectly cause vascular inflammation (vasculitis) leading to thrombosis and disseminated intravascular coagulation. Thrombotic meningoencephalitis of cattle caused by the bacterium Histophilus somni (formerly Haemophilus somnus) is an example of this type of injury (see Fig. 14-89). Certain herpesviruses and protozoa can also infect endothelial cells and cause endothelial necrosis with vasculitis, hemorrhage, and thrombosis. Finally, some pathogens, such as angioinvasive Mucor spp., directly invade blood vessels, resulting in necrosis of the endothelium. Vasculitis resulting in thrombosis can cause tissue ischemia, infarction, and vasogenic edema of the affected area of the CNS. A review of endothelial injury can be found in Chapter 2.
Infarction: Infarction means necrosis of a tissue after obstruction (ischemia) of its arterial blood supply. The rate at which ischemia occurs in the CNS determines the degree of injury that follows. The more rapid the onset of ischemia, the more severe the lesion. However, if the obstruction is sudden, as caused by an embolus formed as an example by tumor cells, many of the neurons can die within minutes and other components within hours (Fig. 14-24). This outcome also applies to compressive injuries to the CNS that produce a sudden reduction in blood flow, such as can happen with sudden compression in rapidly occurring Hansen type I disk herniation in the dog. If the blood flow through an artery is gradually reduced, for example, because of arteriosclerosis, there is often sufficient time for anastomotic vessels to dilate and compensate. Anastomoses of the arteries that penetrate from the ventral and cortical surfaces of the brain are insufficient to prevent infarction after sudden occlusion of one or more of these arteries. If the compression is slow—such as is caused by a slowly developing Hansen type II disk herniation in a dog or by a slowly growing neoplasm from the exterior, such as meningioma in a cat—adjacent neural tissue will atrophy to accommodate the mass.
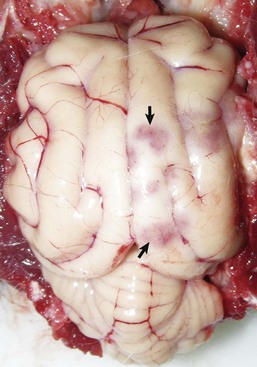
Fig. 14-24 Malacia, vascular occlusion, ischemia, infarction, cerebrum, cat.
Several red-pink foci (arrows) are areas of ischemic necrosis secondary to vascular occlusion caused by cerebral metastasis of a bronchoalveolar carcinoma. (Courtesy Drs. C.A. Lichtensteiger and R.A. Doty, College of Veterinary Medicine, University of Illinois.)
Cerebral necrosis, comparable to infarction after vascular occlusion, can also result from other causes, including cessation of cerebral circulation caused by cardiac arrest, sudden hypotension caused by reduced cardiac output, and reduced or absent oxygen in inspired air. Additional causes include altered function of hemoglobin as a result of carbon monoxide poisoning, inhibition of tissue respiration after cyanide poisoning, ingesting toxic substances and poisons, and nutritional deficiencies.
When an artery supplying the CNS is suddenly occluded, blood supply to cells at the center of the infarcted area is rapidly stopped, and if maintained for a sufficient period, all cells die. Neurons at the border of this area continue to receive some blood from unobstructed vessels. It is proposed that the axonal terminals of degenerated ischemic neurons in the center of the infarct release excessive amounts of the neurotransmitter glutamate, causing injury to still-viable neurons in the borders, which increases the extent of the infarct. This process begins after the binding of the neurotransmitter glutamate to receptors on viable neurons in the borders, inducing an abnormal movement of calcium ions into the recipient cells followed by an increase in intracellular calcium ion concentration. This buildup of calcium ions contributes to a multifunctional cascade that leads to neuronal death. When there is hemorrhage with the infarct, the mechanical injury from the pressure, plus tissue displacement by the hemorrhage, can cause additional damage. See Table 14-1 for the reparative responses associated with the resolution of infarcts.
TABLE 14-1
Chronologic Sequence of Changes within Infarcted Tissue (in the Living Animal) after an Ischemic Event
| Time Following Ischemic Event | Tissue Change |
| Immediate (seconds) | Cessation of blood flow (ischemia) and accumulation of waste products |
| Few minutes | Cellular injury and death; coagulation necrosis and edema; hemorrhage (especially in gray matter) |
| 20 minutes | First microscopic evidence of neuronal injury (perfusion-fixation) |
| 1-2 hours | First microscopic evidence of neuronal injury (immersion fixation) |
| 2 hours | Pale staining of infarct microscopically (white matter); swelling of capillary endothelium; increase in size of astrocytic nuclei |
| 3-5 hours | Ischemic cell change in most neurons; swelling of oligodendroglia and astroglia; beginning clasmatodendrosis of astrocytes |
| 6-24 hours | Beginning neutrophilic infiltration; alteration of myelin (pale staining), 8-24 hours; degeneration and decrease of oligodendroglia, 8-24 hours; astrocytic swelling and retraction and fragmentation of processes (clasmatodendrosis), and degeneration*; cytoplasm of astrocytes visible, 8-24 hours*; vascular degeneration and fibrin deposition, 8-24 hours; thrombosis,† 6-24 hours; beginning endothelial proliferation at margin of infarct, 9 hours |
| 8-24 (up to 48) hours | Initial gross detection of infarct unless hemorrhagic; infarct edematous (swollen), soft, pale, or hemorrhagic and demarcated |
| 1-2 days | Swelling of axons and myelin sheaths; prominent neutrophilic infiltration |
| 2 days | Prominent loss of neuroectodermal cells; continued proliferation of endothelial cells; reduced number of neutrophils; beginning increase in mononuclear cells (gitter cells) |
| 3-5 days | Prominent number of mononuclear cells (gitter cells); disappearance of neutrophils; continued endothelial cell proliferation; number of capillaries appear increased; beginning of astrocytic proliferation (often at margin of infarct) |
| 5-7 days | Grossly, swelling of infarct reaches maximum |
| 8-10 days | Reduction in gross swelling of infarct; liquefaction necrosis; prominent number of mononuclear cells (gitter cells); continued endothelial cell proliferation; beginning fibroblastic activity with collagen formation, variable but most prominent in CNS tissue adjacent to the meninges; beginning increase of astroglial fiber production, 5-13 days |
| 3 weeks-6 months | Mononuclear cells decreased; astroglial fiber density increased (especially at margin); astrocytic proliferation reduced; astrocytes return to original appearance; cystic stage of infarct, 2-4 months; vascular network may be present within cyst; endothelial cell proliferation reduced |
*The degree of astrocytic injury depends on location (e.g., central or peripheral) of the cells within the infarct.
†Obviously, thrombosis may occur earlier than 6 hours. This is the time when it may initially be prominent.
Although they occur through the same mechanisms, areas of cerebral infarction differ somewhat in gross appearance from infarcts in other tissues (Fig. 14-25). The abundance of lipids and enzymes, plus the relative lack of fibrous connective tissue stroma in the brain and spinal cord, results in the affected areas eventually becoming soft because of liquefaction necrosis. The gross appearance of infarction may also differ according to location. Lesions affecting the gray matter tend to be hemorrhagic, whereas infarction of the white matter is often pale. This difference is probably due in part to the less-dense capillary meshwork in the white matter and in part to the fact that the vessels supplying the white matter have fewer anastomoses than those of the gray matter. Infarcted tissue goes through a characteristic sequence of changes that can permit a relatively accurate determination of the age of the infarct. An outline of the chronologic events that occur after an ischemic episode that lasts more than 5 to 6 minutes and is followed by resuscitation of an animal is given in Table 14-1. As can be seen, the tissue changes listed in Table 14-1 take different periods of time to develop in the living resuscitated animal after ischemia occurs. Variation in the times that specific lesions occur depends on the extent and duration of the initial ischemic event. Following removal of cellular and myelin debris, the infarct is repaired by astrocytes. If the infarct is small (<1 mm), it is filled via astrogliosis; if the infarct is larger, it is encapsulated to form a cyst.
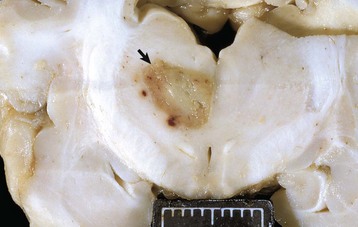
CNS Swelling and Edema
Congestive Brain Swelling: Congestive brain swelling, as distinguished from cerebral edema, is thought to represent, at least in part, an unregulated vasodilation after trauma, and it can cause serious brain damage (even more severe than the primary injury) if not properly controlled. This lesion therefore represents an enlargement of the brain resulting in elevated intracranial pressure caused by the increased diameter of the blood-containing vasculature, whereas edema results in an increased pressure following accumulation of fluid in the interstitium or intracellularly outside the circulation. Acute brain swelling can be localized (usually of lesser significance) when associated with focal lesions or generalized (often serious) when caused by diffuse brain injury. Although rarely seen in domestic animals, one exception to the importance of focal lesions is extracerebral hemorrhage (acute subdural hematoma in humans), which—though principally involving the surface of one hemisphere—can cause more mass effect (brain swelling) in the underlying cerebral hemisphere than the hematoma itself. In such circumstances (acute subdural hematoma), the small amount of blood in the subarachnoid space is not the sole reason for the patient’s neurologic state. If the hematoma is removed, the acute brain swelling can progress so rapidly that the brain protrudes (herniates) through the craniotomy site.
The more serious forms of diffuse brain injury are associated with generalized acute brain swelling. It is sometimes difficult to determine the relative importance of swelling in affected individuals because initial acute swelling (detectable as soon as 30 minutes after injury in humans) can be followed after several hours to days by true cerebral edema (as a result of increased vascular permeability), which can be the actual deleterious lesion. Peroxidative injury to blood vessels has been one proposed cause of pathologic vasodilation in the posttraumatic CNS.
Cerebral Edema: The basis of our current understanding of cerebral edema was advanced by Klatzo in 1967 when he proposed two distinct types: (1) cytotoxic edema, or cell swelling, caused by increased intracellular fluid with normal vascular permeability and (2) vasogenic edema, or tissue swelling, caused by increased extracellular fluid resulting from increased vascular permeability (Fig. 14-26). Other types of cerebral edema have been identified as hydrostatic (or interstitial) edema associated with increased hydrostatic pressure of the CSF (resulting from obstructive internal hydrocephalus) and hypo-osmotic edema, which is dependent on development of an abnormal osmotic gradient between the blood and nervous tissue. The types of edema in the CNS are summarized in Table 14-2. It should be emphasized that, depending on the nature of the injury, multiple mechanisms can contribute to edema in the CNS and these distinctions are not always clearly defined or distinct. For continuity, spongiform change and status spongiosus will also be discussed in this section.
TABLE 14-2
| Type of Edema | Cause | Outcome |
| Cytotoxic | Altered cellular metabolism (often due to ischemia) | Intracellular accumulation of fluid (neurons, glial cells, endothelial cells) |
| Vasogenic | Vascular injury with breakdown of the blood-brain barrier | Extracellular accumulation of fluid (cerebrocortical white matter) |
| Hydrostatic (interstitial) | Elevated ventricular hydrostatic pressure (hydrocephalus) | Extracellular accumulation of fluid (periventricular white matter) |
| Hypo-osmotic | Osmotic imbalances (blood plasma versus extracellular and intracellular microenvironments of the CNS) | Extracellular and intracellular accumulation of fluid (cerebrocortical gray and white matter) |
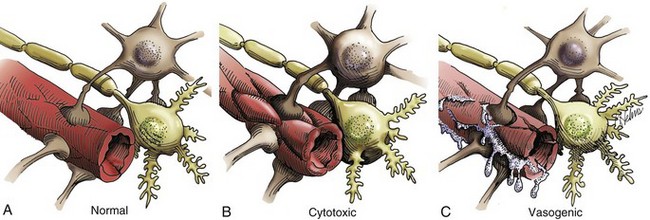
Fig. 14-26 Types of cerebral edema.
A, Normal blood-brain barrier. Endothelial cells are red; astrocytes are beige; neurons are light yellow. B, Cytotoxic edema. Cytotoxic edema is characterized by the accumulation of fluid intracellularly (in neurons, astrocytes, oligodendroglia, and endothelial cells) as a result of altered cellular metabolism, often caused by ischemia. The gray and white matter are both affected. The fluid taken up by swollen cells is primarily derived from the extracellular space, which becomes reduced in size and has an increased concentration of extracellular solutes. C, Vasogenic edema. This type of edema is seen in acute inflammation, and its basic mechanism is an increase in vascular permeability from the breakdown of the blood-brain barrier. This breakdown allows movement of plasma constituents such as water, ions, and plasma proteins into the extracellular space, particularly that of the white matter. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. Based on an illustration from Leech RW, Shuman RM: Neuropathology: a summary for students, Philadelphia, 1982, Harper & Row.)
Vasogenic Edema: In animals, vasogenic edema is the most common type of edema in the CNS. It occurs following vascular injury often adjacent to inflammatory foci, hematomas, contusions, infarcts, cerebral hypertension, and neoplasms. The underlying mechanism of vasogenic cerebral edema is a breakdown of the blood-brain barrier that results in movement of plasma constituents, such as water, ions, and organic osmolytes, and proteins into the perivascular extracellular space, particularly that of the white matter (Fig. 14-27). In addition to extracellular accumulation of fluid, vasogenic edema can also be accompanied by some cellular swelling involving astrocytes. Vasogenic edema with the resulting accumulation of extracellular fluid can cause an increase in intracranial pressure within the CNS. This pressure can also be so severe as to cause neurologic dysfunction and caudal displacement of brain structures such as the parahippocampal gyri and cerebellar vermis (see Figs. 14-59 and 14-60).
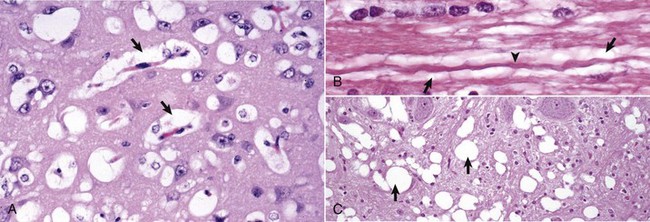
Fig. 14-27 Edema.
A, Vasogenic edema. The perivascular spaces are wide as a result of fluid leakage through the blood-brain barrier (arrows) (see Fig. 14-26). A similar change can be seen around neurons. These fluid-filled spaces are often very difficult to differentiate from artifactual spaces caused by shrinkage from fixation and dehydration in the preparation of the paraffin-embedded sections. H&E stain. B, Intramyelinic edema. Note the accumulation of edema fluid (arrows) between myelin lamellae that surrounds the axon (arrowhead). This lesion was caused by hexachlorophene added to a medicated shampoo. Such products are no longer available for use in veterinary practice. H&E stain. C, Edema (spongy change, status spongiosus), hepatic encephalopathy, dog. This lesion is characterized by variably sized fluid-filled spaces within the white matter (arrows). It can develop by several different mechanisms, which include splitting of myelin sheaths, accumulation of extracellular fluid, and swelling of astrocytic and neuronal cellular processes. Such changes can reflect osmotic imbalances as well as a direct toxic effect on cells (cytotoxic edema). H&E stain. (A and C courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Cytotoxic Edema: Cytotoxic edema is characterized by the accumulation of fluid intracellularly in neurons, astrocytes, oligodendroglia, and endothelial cells (called hydropic degeneration in other cells of the body) as a result of altered cellular metabolism, often caused by ischemia. Although not all of the cells listed previously may be involved in all cases of cytotoxic edema, affected cells swell within seconds of injury. The mechanism is thought to involve an energy deficit that interferes with normal function of the cell’s Na+/K+-ATPase pump. Thus the cell cannot maintain homeostasis, which requires the secretion of intracellular sodium, and the elevated concentration of intracellular sodium and presumably other ions, as well as organic osmolytes, are followed by an increased influx of water. The gray and white matter of the brain are both affected, the brain swells, and the sulci and gyri become indistinct and flattened (Fig. 14-28), respectively. The fluid taken up by the swollen cells is primarily derived from the extracellular space, which becomes reduced in size and has an increased concentration of extracellular solutes.
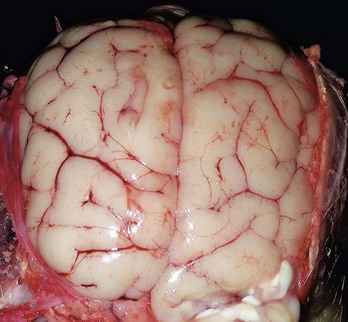
Fig. 14-28 Cerebral edema, dog.
On the dorsal surface, gyri are swollen and flattened and sulci have become less distinct. Accumulation of extracellular fluid has caused the brain to swell, and because space within the cranial vault is limited, the brain has been pressed against the calvarium. In extreme cases, notable brain swelling can cause caudal displacement of the vermis of the cerebellum and the parahippocampal gyri (see Figs. 14-59 and 14-60). (Courtesy Drs. C.A. Lichtensteiger and A. Gal, College of Veterinary Medicine, University of Illinois.)
However, for this lesion to be described accurately as cerebral edema, there must be additional fluid movement into the brain and not merely a change of existing fluid from extracellular to intracellular compartments. In practical terms, this is not always the case and can be difficult to determine. The term cytotoxic edema has been used rather loosely to refer simply to cellular swelling in many cases. Additional fluid may originate from the circulation by way of the transcapillary fluid exchange or possibly from the CSF, which has extensive diffusional communication with the extracellular fluid of the brain. The blood-brain barrier remains intact during development of this type of edema, so fluid does not enter the brain by a disturbance in vascular permeability. Specific causes of this lesion include hypoxia-ischemia, particularly in the early stages; intoxication with metabolic inhibitors, such as 2,4,dinitrophenol, 6-aminonicotinamide, and ouabain; and severe hypothermia.
Interstitial (Hydrostatic) Edema: Interstitial edema is characterized by the accumulation of fluid in the extracellular space of the brain because of elevated ventricular hydrostatic pressure that accompanies hydrocephalus. Fluid moves across the ependyma of the ventricular wall and accumulates extracellularly in the periventricular white matter. Unlike the other forms of cerebral edema that cause swelling of affected CNS tissue, hydrostatic edema causes variable degeneration and loss of the periventricular white matter mostly through primary demyelination accompanied by loss of axons. As the periventricular white matter is reduced in volume, the ventricle expands to fill the void by displacement, thereby exacerbating the hydrocephalus. The blood-brain barrier remains intact in hydrostatic edema.
Hypo-Osmotic Edema: Hypo-osmotic edema occurs after overconsumption of water (water intoxication), leading to dilution of the osmolality of the plasma. Under normal conditions the osmolality of CSF and extracellular fluid in the CNS is slightly greater than that of plasma. When the osmolality of plasma is further decreased, water moves from the vasculature into the brain after the osmotic gradient, resulting in osmotic edema. This form of edema accounts for the clinical signs and lesions in osmotic demyelination syndrome and salt poisoning, as discussed later.
Spongiform Change and Status Spongiosus: Spongiform change is a term whose exact meaning varies in different scientific disciplines and experimental situations. In this chapter, wherever possible, spongiform change is used to describe morphologic changes in H&E stained sections that occur primarily in gray matter. These changes are characterized by small clear vacuoles of varied sizes that form in the cytoplasm of neuron cell bodies and proximal dendrites in diseases such as the transmissible spongiform encephalopathies (TSEs) and rabies encephalitis and in the processes of astrocytes that are spatially related to the affected neurons.
Status spongiosus (spongy degeneration) is also a phrase whose exact meaning varies. It is defined as multiple fluid-filled clear spaces in the white matter of H&E stained sections of the CNS and may be extracellular or intracellular (see Fig. 14-27, C). This lesion results from the accumulation of edema fluid in the white matter secondary to a variety of causes, including cytotoxic edema, vasogenic edema, intramyelinic edema, Wallerian degeneration, and other hypoxic, toxic, and metabolic diseases.
In some instances, the term spongy degeneration or spongy change has been used in the veterinary literature to describe microscopic lesions in a group of diseases of young dogs, cats, and cows characterized by fluid accumulation in white matter. These diseases are discussed in later sections.
Defense Mechanisms
The CNS has several unique structural and functional barrier systems that serve to protect it from diseases affecting the vascular and ventricular systems and to actively facilitate transfer of necessary molecules, such as glucose, to cells within the CNS.
Blood-Brain Barrier: The blood-brain barrier, formed by vascular endothelial cells, endothelial-derived basement membrane, and foot processes of astrocytes, exists in the capillaries of the CNS (see Fig. 14-13). The most important structural component of the blood-brain barrier is the tight junctions between endothelial cells of cerebral capillaries. By means of the blood-brain barrier, the CNS can selectively regulate its extracellular compartment and isolate itself from sudden biochemical changes that may occur in the systemic circulation. In the majority of the CNS, endothelial cells are nonfenestrated and are held together by intercellular tight junctions. These tight junctions actively prevent the movement of protein, hydrophilic molecules, and ions from capillary lumina into the intercellular compartment of the CNS. Endothelial cells also have a transmembrane lipophilic pathway for the diffusion of small lipid molecules and numerous highly selective polarized receptor-mediated transport systems for molecules such as insulin, transferrin, glucose, purines, and amino acids. Finally, endothelial cells express a net negative charge on their abluminal side and at the basement membrane, providing an additional selective mechanism that impedes movement of anionic molecules such as chloride ions across the barrier. Foot processes of astrocytes cover more than 90% percent of the abluminal surface of capillary endothelial cells. Experimental evidence suggests that secretion of growth factors from astrocytes promotes the formation and maintenance of the blood-brain barrier.
Capillaries in the area postrema, median eminence, neurohypophysis, pineal body, subfornical organ, commissural organ, and supraoptic crest lack tight junctions and are fenestrated; thus the blood-brain barrier is absent at these sites.
Glia Limitans: The CNS is separated from the subarachnoid CSF by the pia mater and the glia limitans (see Fig. 14-13). The glia limitans, which covers the outer surface of the brain and spinal cord and is situated immediately subjacent to the pia mater, consists of astrocytic fibers with many foot processes that form a distinct layer that lies subjacent to the pia mater. In many areas the pia is only one-cell-layer thick and has fenestrations, so that the glia limitans is exposed directly to the subarachnoid space. As arterioles penetrate the cerebral cortex to supply the CNS with blood, they carry the pia mater and surrounding glia limitans with them until the arteriole structurally and functionally becomes a capillary. At the capillary level, the pia mater disappears, but the layer of pericapillary astrocytic foot processes remains and serves as a component of the blood-brain barrier. This transition zone occurs at a depth approximately 1 to 3 mm within the cerebral cortex.
Blood–Cerebrospinal Fluid Barrier: The blood-CSF barrier is formed by the choroid plexus and the arachnoid. This barrier is formed by tight junctions between apposing surfaces of choroid plexus epithelial cells that cover the choroid plexuses. As noted previously, blood vessels of the choroid plexus are fenestrated. The barrier formed by tight junctions between choroid plexus epithelial cells restricts the movement of molecules that leak from fenestrated capillaries into the extracellular compartment of the choroid plexus and then into the CSF. Similarly, the arachnoid membrane also has tight junctions that prevent the movement of molecules from the blood into the CSF. The arachnoid membrane is generally impermeable to hydrophilic molecules but lacks specialized transport systems, and its role in forming the blood-CSF barrier is largely passive.
Cerebrospinal Fluid–Brain Barrier (Ependymal Barrier): The CNS is separated from ventricular CSF by ependymal epithelial cells and foot processes of astrocytes. Although the ependymal lining does form a cellular barrier of sorts, materials within the ventricular system can without too much difficulty penetrate into the brain. The CSF-brain barrier is far more permeable than the blood-brain barrier.
Innate and Adaptive Immune Responses
Although infectious agents have developed unique approaches to gain entry into the CNS, the body has also evolved a strong suite of defense mechanisms to protect the CNS against infectious pathogens and disease processes. The skin and mucous membranes of the alimentary, respiratory, and urinary systems provide structural and functional barriers against disease. The inflammatory response, immune system, and monocyte-macrophage system provide a strong local and systemic defense against pathogen replication and disease spread. Finally, barrier systems in the CNS, reviewed in an earlier section, represent structural and functional protection against a wide range of pathogens and toxic injuries. These defense mechanisms are summarized in Box 14-5.
Inflammation of the CNS: Inflammation of the CNS is different from inflammation in other organs because of the presence of the blood-brain barrier. Under normal conditions, this barrier provides limited isolation of the CNS from circulating cellular and humoral elements of the immune system. Macrophages (monocytes) and T lymphocytes can penetrate an intact blood-brain barrier and enter the perivascular and subarachnoid spaces, transit these spaces, and return to the circulation in a role of protective immunologic surveillance of the CNS.
It is important to remember that inflammation in the CNS is regulated by a complex system of recognition and adhesion molecules, cytokines, chemokines, and their corresponding receptors (see Chapters 3 and 5). In particular within the CNS, chemokines and their receptors regulate physiologic and pathologic leukocyte trafficking and cellular migration events.
When pathogens use one of the four portals of entry to gain access to the CNS, the inflammatory process that ensues disrupts the blood-brain barrier. Thus, in addition to inflammation, edema and hemorrhage can result. Selectins and integrins in cooperation with chemokines are active in initiating and regulating the acute inflammatory response and the movement of neutrophils across the blood-brain barrier in response to a variety of pathogens. Migration of inflammatory cells within the CNS is poorly understood. Chemotactic gradients are likely established by chemokines that diffuse from sites of production within foci of inflammation. Activated glial cells, including astrocytes and resident microglia, form chemokine networks in areas of inflammation in response to cytokines produced by T lymphocytes that recognize foreign antigens.
Depending on the type of antigen and the pathogenicity of the infectious agent, the inflammatory response resolves (heals) or progresses to a chronic or granulomatous phase with attempts at resolution and clearance of the infectious agent. In the CNS, the type of inflammatory response can vary with the cause. A rather simplistic guideline, to which there are always exceptions, that compares the type of inflammation with different causal agents is as follows:
1. Serous to suppurative or purulent responses can be the result of several species of bacteria.
2. Eosinophil responses occur in salt poisoning of pigs and with parasitic larval migration.
3. Lymphocytic, monocytic/macrophage, nonsuppurative, lymphomonocytic, and lymphohistiocytic responses can be caused by viruses and certain protozoa.
4. Granulomatous response can be the result of fungi, certain protozoa, and some higher-order bacteria such as the Mycobacterium spp.
Portals of Entry
Disease processes of the CNS enter the brain and spinal cord through one of four principal portals (Box 14-6). These portals include (1) direct extension, (2) hematogenous entry, (3) leukocytic trafficking, and (4) retrograde axonal transport.
Direct Extension
Direct extension is a common portal of entry and includes a wide range of disease processes. Penetrating trauma through the calvarium or vertebrae as a result of a gunshot wound or other forms of trauma can provide a direct portal into the CNS. Disease processes can also extend into the brain and/or spinal cord as a result of (1) a middle and/or inner ear infection (Fig. 14-29), (2) nasal cavity/sinus infection or neoplasia through the cribriform plate or calvarium (Fig. 14-30), or (3) bacterial osteomyelitis or neoplasia of vertebral bodies with extension through the vertebrae into the vertebral canal.
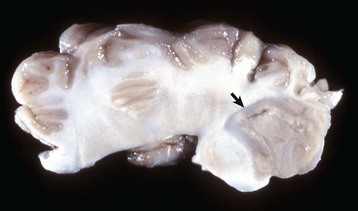
Fig. 14-29 Chronic bacterial abscess, leptomeninges of the cerebellum, sheep.
The abscess (arrow) resulted from direct extension of infection from an inner ear infection and compresses and distorts the cerebellum. It is walled off from the adjacent cerebellum by a distinct fibrous capsule synthesized by fibroblasts from the adjacent leptomeninges. This abscess likely arose in the leptomeninges of the cerebellum and grew to compress adjacent cerebellum. (Courtesy College of Veterinary Medicine, University of Illinois.)
Fig. 14-30 Osteochondrosarcoma, calvarium, dog.
The neoplasm has destroyed and penetrated the calvarium and compressed the cerebral hemispheres (arrows). There is also invasion of frontal or nasal sinuses and nasal cavity. (Courtesy Dr. K. Bailey, College of Veterinary Medicine, University of Illinois.)
Benign growths of the calvarium and vertebrae, such as osteomas, chondromas, and osteochondromas, often extend into and compress the brain and spinal cord. One specific neoplasm, the multilobular osteoma, has been described as originating from the periosteum of the canine skull. Also, malignant neoplasms adjacent to the cranium or spinal column can cause injury by direct invasion. Some examples include extracranial osteosarcoma and fibrosarcoma in the dog. Also, malignant melanoma of the soft palate in the dog and melanoma involving the paravertebral lymph nodes in the horse can invade adjacent CNS tissue. Other examples of direct extension include lymphosarcoma affecting the spinal cord in the bovine species and less frequently involving the spinal cord or brain in the dog and cat and carcinomas of the ethmoidal area and nasal cavity.
Hematogenous Entry
The most common portal of entry into the CNS is the bloodstream. In neonates, infectious agents, such as Escherichia coli, can enter the blood through the umbilical vein or through the venous system after surgical procedures such as castration. A CNS disease in cows called thrombotic meningoencephalitis is caused by Histophilus somni (formerly Haemophilus somnus) bacteremia with localization of bacteria in blood vessels of the brain, which leads to vasculitis, hemorrhage, and thrombosis (see Fig. 14-89). In adult animals, sites of chronic inflammation, such as abscesses, bacterial skin disease, and ear infections, can also serve as sustained sources of bacteria, which can enter the venous system and spread to distant sites through the bloodstream hematogenously.
Capillary beds of the meninges, neuropil, and choroid plexuses are common sites for localization of specific infectious agents. Such localization patterns may be attributable to receptor-mediated phenomena or vascular flow patterns related to the size of the infectious pathogen. Additionally the bloodstream is also a portal of entry into the CNS for metastasizing tumors such as hemangiosarcoma and a variety of carcinomas.
Leukocyte Trafficking
As part of the systemic immunologic surveillance system, macrophages (monocytes) and lymphoid cells continually move in and out of capillary beds in the CNS and thus serve as sentinel cells monitoring for the presence of disease processes within the brain and spinal cord (see Fig. 4-11). As examples, retroviruses, such as feline leukemia virus, and mycotic agents, such as Blastomyces dermatitidis, have stages of their life cycles within the cytoplasm of lymphocytes or macrophages. During the movement of lymphocytes and macrophages in and out of the CNS, cells infected with such agents are activated to release their infectious contents and infect cells of the CNS.
Retrograde Axonal Transport
Retrograde axonal transport provides a unique portal of entry for viruses such as rabies and the bacterium Listeria monocytogenes. These pathogens replicate in tissues richly innervated with receptors and motor end plates from sensory and motor neurons, respectively, which provide a connection between peripheral infection and the CNS. Retrograde axoplasmic flow is then used to gain entry into the CNS (see Web Fig. 14-1).
Disorders of Domestic Animals
Disorders that occur in many or all animal species are discussed in this section. Disorders of individual animal species are discussed in later sections covering disorders unique to that species.
Malformations
Neural Tube Closure Defects (Dysraphia): Dysraphia literally means an abnormal seam, and these anomalies appear to result from defective interaction of neuroepithelium with adjacent notochordal and mesenchymal cells during closure of the neural tube in the early stages of development. Neuroepithelium is the progenitor cell for neurons and astrocytes, oligodendrocytes, and ependymal cells.
Experimental studies of closure of the neural tube show that it occurs at four distinct locations called closure initiation sites in the embryo, and disruption of this process at these sites leads to site-specific dysraphic anomalies. Closure site I contributes to the posterior neuropore (the opening at the posterior end of the embryonic neural canal), whereas closure sites II to IV contribute to the anterior neuropore (the opening at the anterior end of the embryonic neural canal). Anencephaly is caused by a failure of closure sites II or IV; spina bifida is caused by a failure of closure site I. Genes possibly involved in neural tube closure defects include those of the folate metabolic pathway and folate transport.
Dysraphic anomalies, also called neural tube closure defects, in animals are typified by anencephaly and prosencephalic hypoplasia, cranium bifidum, and spina bifida.
Anencephaly and Prosencephalic Hypoplasia: Anencephaly literally means an absence of the brain, but in many instances of so-called anencephaly only the rostral part of the brain (cerebral hemispheres) is absent, or very rudimentary, and to varying degrees the brainstem is preserved. Thus this abnormality is best designated prosencephalic hypoplasia. Such anomalies result from an abnormal development of the rostral aspect of the neural tube. Although the cause for these anomalies is largely unknown, anencephaly has been reported to be associated with anomalies in other body systems in calves. Additionally, anencephaly—after initial cranium bifidum and exencephaly (protrusion of brain not covered by skin or meninges)—has been reported to occur in rat fetuses after exposure of the pregnant dam to excessive concentrations of vitamin A and cyclophosphamide.
Meningoencephalocele and Cranium Bifidum: Cranium bifidum is characterized by a dorsal midline cranial defect through which meningeal and brain tissue can protrude. The protruded material, which forms a sac (-cele), is covered by skin and can be lined by meninges (meningocele) or meninges accompanied by a part of the brain (meningoencephalocele) (Fig. 14-31). These malformations are hereditary in pigs and cats and are also caused by griseofulvin treatment in pregnant cats during the first week of gestation.
Fig. 14-31 Meningocele (M), brain, calf.
A defect in the caudodorsal portion of the skull has allowed the meninges to herniate into a large external pouch covered by skin. The pouch contains fluid and is lined by arachnoid and dura, which are continuous with those surrounding the brain. The cerebellum is small, and the occipital cortex truncated. Scale bar = 5 cm. (Courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University.)
Meningomyelocele and Spina Bifida: Spina bifida is the vertebral counterpart of cranium bifidum. This lesion, which frequently tends to affect the caudal spine, is characterized by a dorsal defect in the closure of one to several vertebral arches that form the dorsal spinal column covering the spinal cord. The lesion results from a failure of the neural tube and developing vertebral arches to close properly, which may result in herniation of either meninges (meningocele) or meninges and spinal cord (meningomyelocele) through the defect, forming a sac covered with skin. In some cases there is no herniation of the meninges or spinal cord through the defect, and this variation is termed spina bifida occulta (Fig. 14-32). In this variation, there is an absence of skin over the affected vertebral arches, vertebral musculature is visible, and the dura mater and spinal cord can be seen in the spinal canal.
Fig. 14-32 Spina bifida occulta, calf.
There is a cleft in several vertebrae of the dorsal spinal column resulting from defective closure of the neural tube. Although not always the case, note the lack of herniation of the meninges or spinal cord through the defect. The spinal cord is not visible (i.e., occulta) because it is located in the vertebral canal at the deepest ventral extent of the cleft and is covered by edematous muscle. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Spina bifida has been reported in several species, including horses, calves, sheep, dogs (especially English bulldogs), and cats, particularly the Manx breed, in which it is inherited as an autosomal dominant trait. An additional lesion, myeloschisis, is similar to spina bifida except in its severe form, it results from failure of the entire spinal neural tube to close. This lesion is therefore characterized by lack of development of the entire dorsal vertebral column.
Hydromyelia: Congenital hydromyelia is an abnormal dilation of the central canal of the spinal cord (Fig. 14-33) that leads to the formation of a cavity in which CSF may accumulate. In animals, this disorder likely results from infectious or genetic injury that results in damage to ependymal cells lining the canal and the subsequent disruption of the normal flow of CSF and the formation of abnormal CSF pressure gradients within the central canal. As CSF accumulates in the enlarging space, the increased pericanalicular pressure placed on the spinal cord compresses the white and gray matter, leading to loss of white matter and possibly neurons in gray matter. Acquired hydromyelia is rare and is caused by obstruction of the central canal CSF flow. Causes of obstruction include infection, inflammation, and neoplasia.
Fig. 14-33 End-stage hydromyelia, spinal cord, dog.
The white and gray matter of the spinal cord are missing as a result of compression atrophy from a space-occupying, fluid-filled central canal. The only recognizable remnants of nervous tissue is the dura (arrows). In less severely affected animals, there would be variable dilatation of the central canal of the spinal cord with much less severe compression atrophy. (Courtesy College of Veterinary Medicine, University of Tennessee.)
Clinical signs in young animals with congenital hydromyelia vary, depending on the location and size of the dilation of the central canal in the spinal cord. Signs may include ataxia, urinary incontinence, respiratory difficulty, muscle weakness in front and/or hind limbs, and abnormal proprioceptive reflexes.
Lissencephaly: Lissencephaly (agyria) and a similar change called pachygyria (large gyria) are developmental anomalies that result in part of or the entire cerebrum having smooth surfaces lacking normal gyri and sulci (Fig. 14-34). The cortex is thicker than normal on a transverse section, and the normal laminar pattern of neurons is disrupted.
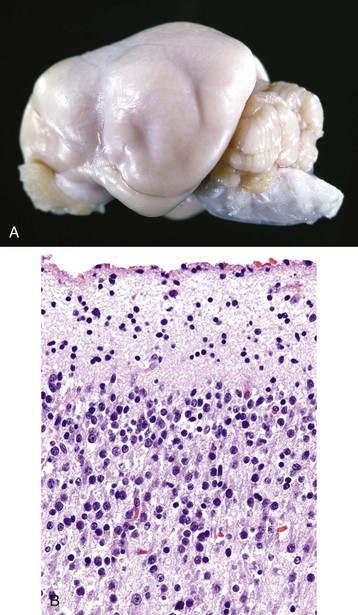
Fig. 14-34 Lissencephaly, brain, dog.
A, Note the smooth surfaces of the cerebral hemispheres, which are without gyri and sulci. Gyri and sulci fail to form, possibly from failure of neuronal development and migration. Lissencephaly is an abnormality in domestic animals, but is normal in mice, rats, rabbits, and birds. B, The development and migration of neurons have been disrupted such that the cortical gray matter lacks normal lamina formed by neuronal cell bodies. H&E stain. (A courtesy Dr. L. Roth, College of Veterinary Medicine, Cornell University. B courtesy Dr. R. Mantene, College of Veterinary Medicine, University of Illinois.)
This lesion is thought to have a genetic basis and results from an arrest of or defect in neuronal migration during development. Recent experimental studies suggest that this migrational disorder is linked to mutations and/or deletions in the doublecortin, filamin-1, LIS1, and reelin genes. These genes control the spatial and temporal expression of proteins in the extracellular microenvironment that subsequently bind to receptors on migrating cells. Patterns of cell membrane binding signals are interpreted by migrating cells and are reflected in their movements by changes in intracellular cytoskeletal reorganization. This process allows cells to migrate to their final destinations within the CNS. Thus alterations in signaling pathways lead to abnormal neuronal migration and CNS anomalies.
The brains of some laboratory animals, such as rabbits, rats, and mice, lack gyri and sulci; therefore agyria is normal in these species and has no functional significance.
Porencephaly and Hydranencephaly: The formation of fluid-filled cavities in the brain, termed porencephaly (small cavities) and hydranencephaly (large cavities), usually occurs in utero during gestation. Porencephaly refers to a cleft or cyst in the wall of the cerebral hemisphere that typically communicates with the subarachnoid space, but it can also communicate with a lateral ventricle. The cavitation results from destruction of immature neuroblasts whose loss prevents normal development as a result of faulty or aberrant neuroblast migration. Hydranencephaly is considered a severe form of porencephaly and is characterized by cavitation in areas normally occupied by the white matter of the cerebral hemispheres and results from improper development of this part of the cerebrum. Hydranencephaly is often quite severe, with very little tissue present between the dilated lateral ventricles and the leptomeninges.
Type I and type II porencephaly have been described in human infants, and cases of porencephaly reported in animals can also be categorized using this scheme. Type I porencephaly is caused by vascular injury or vasculitis. Injury, resulting in infarction in the area of the subependymal germinal matrix, results in the formation of a cyst within the focus of dead cells and effete erythrocytes. The germinal matrix is very sensitive to ischemia because of sparse stroma, delicate vasculature, and high metabolism. The initial focus of hemorrhage can grow by centripetal expansion, depending on the severity of hemorrhage and hypoxia, into a cyst of considerable size. Type II porencephaly is caused by injury of neuroblasts in the germinal matrix and the failure of these neuroblasts to migrate within the matrix to form the cerebral cortex. A cyst results from the expansion of the subarachnoid space into the void left by the absence of the cortex.
Type II porencephaly appears to be the form of porencephaly that occurs in domestic animals. Viruses, such as those that cause Akabane disease, bovine virus diarrhea, blue tongue, border disease, Rift Valley fever, and Wesselsbron disease, infect and destroy differentiating neuroblasts and neuroglial cells in the developing fetus in utero. Although neuroblasts appear to be the primary target for viral infection in these diseases, additional experimental studies need to be conducted to clarify whether endothelial cells are also infected.
Grossly, porencephaly/hydranencephaly appears as thin-walled fluid-filled cysts of varied sizes in the cerebral hemispheres. Because of the lack of brain substance, the ventricles expand into this space (hydrocephalus ex vacuo), and the ependymal lining remains relatively preserved or may have scattered defects characterized by absent ependyma. The cranium and meninges are generally unaltered. In some cases, cerebellar hypoplasia (all or part of the cerebellum) and hypoplasia of the spinal cord may also occur. Microscopically, necrosis of undifferentiated cells, including potential neuroblasts and neuroglia, surrounding a fluid-filled cavity is present in the subventricular zone of the cerebral hemispheres. Degeneration and loss of motor neurons of the ventral horns of the spinal cord may also be observed. This lesion may result in denervation atrophy of limb muscles with a resultant lack of joint movement and arthrogryposis, a persistent congenital flexure or contraction of a joint. Nonsuppurative encephalitis, typified by the accumulation of macrophages, lymphocytes, and plasma cells, also occurs.
Malformations of the Cerebellum:
Cerebellar Hypoplasia: In animals, the most common causes of cerebellar hypoplasia are parvoviruses (kittens: panleukopenia virus [Fig. 14-35] and puppies: canine parvovirus) and pestiviruses (calves: bovine viral diarrhea virus [Fig. 14-36] and piglets: classic swine fever virus). These viruses infect and destroy mitotic cells, primarily the cells of the external granular layer of the cerebellum that are still dividing during the late gestational and early neonatal periods. Necrosis of these cells means they are not available to migrate to form the internal granular layer, and thus the cerebellum is hypoplastic. In calves the cerebellar lesion (cerebellar hypoplasia/atrophy), which follows infection at 150 days of gestation (midtrimester), is considered to involve two processes. One process is typified by early necrosis of the undifferentiated cells in the external granular layer. A second process involves viral-induced vasculitis and ischemia of cerebellar folial white matter.
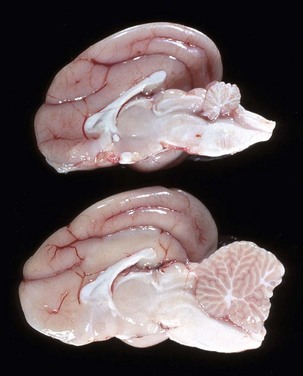
Fig. 14-35 Cerebellar hypoplasia, brain, cat.
In the cat, cerebellar hypoplasia (cerebellar hypoplasia, top specimen; normal cat, bottom specimen) most commonly is the result of in utero infection with feline panleukopenia virus (parvovirus). The virus infects and causes lysis of dividing cells in the external granular layer (on the outside of the cerebellum in the fetus). Because these cells are no longer available to migrate to form the (internal) granular layer, the cerebellum remains small. (Courtesy Dr. Y. Niyo, College of Veterinary Medicine, Iowa State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
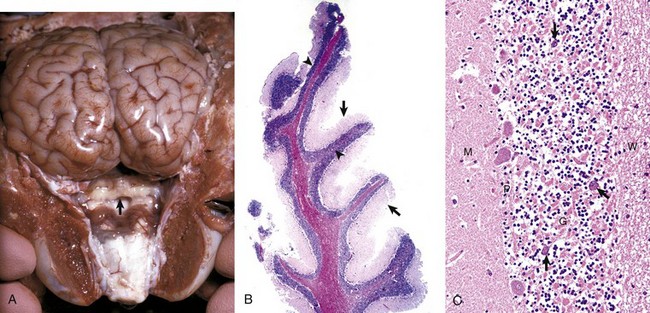
Fig. 14-36 Cerebellum, calf.
In the normal neonatal calf, cells of the external granular layer of the cerebellum migrate to form the (internal) granular layer (not shown). Bovine viral diarrhea virus infects and kills mitotic cells of the external granular layer of the cerebellum. These cells are still dividing during the late gestational and early neonatal periods in the cat and between 100 to 180 days of gestation in the calf. Necrosis of these cells means they are not available to migrate to form the internal granular layer, and thus the cerebellum does not obtain full size. Depending on the stage of gestation, injury can also alter development of cells in others ways including altered patterns of migration, resulting in various other lesions termed dysplasia. A, Cerebellar hypoplasia (arrow). In utero infection with bovine viral diarrhea virus (pestivirus) results in cytolysis of dividing germinal cells of the external granular layer and vascular impairment secondary to vasculitis of the cerebellum during organogenesis. The severity of the lesion involving the granule cells is at its greatest if dividing cells are infected during the earliest stages of cellular differentiation, and occurs between 100 to 180 days of gestation. B, Note the folia of the cerebellum are hypoplastic with a reduced thickness of the molecular layer (arrows) and haphazardly organized and thinned granular cell layer (arrowheads). C. The molecular layer (M) of the cerebellum is reduced in thickness and lacks the normal number of neuronal nuclei. The Purkinje cell layer (P) has large gaps between adjacent cells as the result of the loss of neuron cell bodies or the failure of neurons to migrate properly to form this layer. Note the retention of Purkinje cells (arrows) in the granular cell layer (G). The granular cell layer has significantly reduced numbers of neurons as shown by the lack of nuclei. White matter (W). H&E stain. (A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B and C courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Grossly, the size of the cerebellum is reduced; the reduction in size varies in severity, depending on the age and developmental stage of the brain when the fetus or neonate is infected.
Microscopically, there is necrosis and loss of the external granular layer and degeneration and loss of Purkinje cells that are postmitotic but immature. Reasons for degeneration of Purkinje cells might include infection by the virus or lack of normal development of the cerebellar cortex. The Purkinje cells can also be malpositioned and located in the molecular layer as a result of the viral-induced alteration in development of the cerebellar cortex. In calves, edema of the folial white matter with focal hemorrhage in the cortex, followed by focal cavitation of the white matter and atrophy, may also be present. These latter lesions are due to ischemia resulting from vasculitis. Leptomeningitis, characterized by accumulation of lymphocytes and plasma cells and occasionally fibroplasia, may cause adhesions between adjacent cerebellar folia and focal obliteration of the subarachnoid space.
Malformations of the Spinal Cord:
Syringomyelia: Syringomyelia (congenital and acquired forms) is a disorder in which a cyst forms in the spinal cord. The cyst, a tubular cavitation called a syrinx, is not lined by ependyma and is separate from the central canal. The syrinx can extend over several spinal cord segments. The lesion is well known in humans and has also been described in the dog (Weimaraner breed) and calves. The syrinx can communicate with the central canal but should not be confused with hydromyelia, which means dilatation of the central canal. The cavity contains fluid and is unlined, except for varying degrees of mural astrocytosis, which is usually very mild in the Weimaraner. Proposed causes include presence of an anomalous vascular pattern that results in low-grade ischemia, leading to infarction or failure of cells destined for this area to develop in utero trauma in humans or an infection that causes degeneration and cavitation. A rare acquired form of syringomyelia is similar to congenital syringomyelia; however, it occurs in older animals. Proposed causes include injury after trauma to the central canal or its vascular supply caused by trauma, infection, or neoplasia that result in degeneration and cavitation of the spinal cord.
Although the central canal of the spinal cord is connected to the ventricular system via the fourth ventricle, there apparently is little active movement of CSF within the central canal. Recently, it has been hypothesized that there may be alteration of “normal” CSF flow (see the discussion on ependyma in the section on Cells of the CNS) with redirection of the flow along a pressure gradient into the central canal and into the syrinx. It has also been suggested that pressure differences in the vertebral column cause CSF to continually move into the cyst, resulting in enlargement of the syrinx and additional compressive damage to the spinal cord.
Clinical signs in young dogs and calves with syringomyelia vary, depending on the location and size of the cyst in the spinal cord. Signs may include ataxia, urinary incontinence, respiratory difficulty, muscle weakness in front and/or hind limbs, and abnormal proprioceptive reflexes.