Astrocytosis: Increased numbers of astrocytes.
Astrogliosis: Reactive astrocytic response with increased number (variable), length, and complexity of cell processes. In the CNS, reparative processes after injury, such as inflammation and necrosis, are facilitated by astrogliosis.
Axonopathy, distal, of the PNS: A neuropathy with degeneration of the terminal and preterminal axon of peripheral nerves.
Axonopathy, distal of the CNS and PNS: Degeneration of axons involving distal portions of peripheral nerves, and distal portions of long axons in the CNS (spinal cord).
Axonotmesis: Axonal injury of a peripheral nerve in which there is degeneration of the part distal to the site of trauma, leaving the supporting framework intact and allowing for improved potential for regeneration and effective reinnervation.
Blood-brain barrier of the CNS: A barrier to free movement of certain substances from cerebral capillaries into CNS tissue. Relies on tight junctions between capillary endothelial cells and selective transport systems in these cells. Endothelial cell basement membrane and foot processes of astrocytes abutting the basement membrane may play role in barrier function.
Blood-CSF barrier of the CNS: A barrier that consists of tight junctions located between epithelial cells of the choroid plexus and the cells of the arachnoid membrane that respectively separate fenestrated blood vessels of the choroid plexus stroma and dura mater from the CSF.
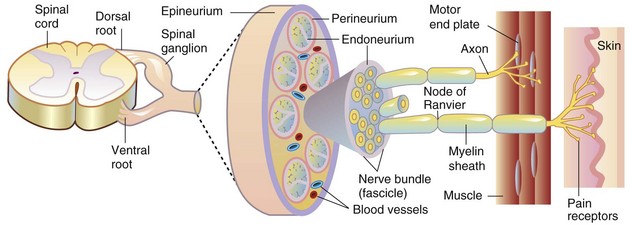
Blood-nerve barrier: A barrier to free movement of certain substances from the blood to the endoneurium of peripheral nerves. Barrier properties are conferred by tight junctions between capillary endothelial cells of the endoneurium and between perineurial cells and selective transport systems in the endothelial cells.
Brain edema: Increase in tissue water within the brain that results in an increase in brain volume. The fluid may be present in the intracellular or extracellular compartments or both. The term also is used to include the accumulation of plasma, especially in association with severe injury to the vasculature.
Brain swelling: Marked, rapidly developing, sometimes unexplained, increase in cerebral blood volume and brain volume because of relaxation (dilation) of the arterioles that occurs after brain injury.
Büngner, cell bands of: A column of proliferating Schwann cells that forms within the space previously occupied by an axon following wallerian degeneration. The proliferating column of cells is surrounded by the persisting basement membrane of the original Schwann cells.
Central chromatolysis: Dissolution of cytoplasmic Nissl substance (arrays of rough endoplasmic reticulum and polysomes) in the central part of the neuronal cell body that results from injury to the neuron (often involving the axon). The cell body is swollen, and the nucleus frequently is displaced peripherally to the cell membrane. These structural changes functionally represent a response to injury that can be found (if the cell survives) by axonal regeneration with protein synthesis to produce components of the axon required for fast and slow axonal transport.
Cranium bifidum: A dorsal midline cranial defect through which meninges alone or meninges and brain tissue may protrude into a sac (-cele), covered by skin.
Demyelination: A disease process in which demyelination (destruction of the myelin sheath) is the primary lesion, although some degree of axonal injury may occur. Primary demyelination is caused by injury to myelin sheaths and/or myelinating cells and their cell processes. Secondary demyelination occurs with axonal injury, as in wallerian degeneration.
Dysraphism: Dysraphia, which literally means an abnormal seam, refers to a defective closure of the neural tube during development. This defect, which may occur at any point along the neural tube, is exemplified by anencephaly, prosencephalic hypoplasia, cranium bifidum, spina bifida, and myeloschisis.
Encephalitis: Inflammation of the brain.
Encephalo-: A combining form that refers to the brain.
Encephalopathy: A degenerative disease process of the brain.
Ganglionitis: Inflammation of peripheral (sensory or autonomic or both) ganglia.
Gemistocyte: Reactive, hypertrophied astrocyte that develops in response to injury of the CNS. The cell body and processes of gemistocytes become visible with conventional staining (e.g., H&E stain). The cell bodies and processes of normal astrocytes are not visible with H&E staining.
Gitter cell: Macrophage that accumulates in areas of necrosis of CNS tissue. The cytoplasm is typically distended, with abundant lipid-containing material derived from the lipid-rich nervous tissue. Gitter cell nuclei are often displaced peripherally to the cell membrane. These cells are often referred to as “foamy” macrophages.
Hydranencephaly: A large, fluid-filled cavity in the area normally occupied by CNS tissue of the cerebral hemispheres resulting from abnormal development. The nervous tissue may be so reduced in thickness that the meninges form the outer part of a thin-walled sac. The lateral ventricles are variably enlarged because of their expansion into the area normally occupied by tissue.
Hydrocephalus: Accumulation of excess CSF resulting from obstruction within the ventricular system (noncommunicating form) associated with enlargement of any or all of the following: lateral ventricles, third ventricle, mesencephalic aqueduct, and fourth ventricle. Hydrocephalus can also occur with communication of the CSF between the ventricular system and the subarachnoid space (communicating form). Hydrocephalus ex vacuo (compensatory hydrocephalus) is characterized by an expansion of the lateral ventricle (or ventricles) that follows loss of brain tissue.
Leuko-: Combining form referring to white matter of the brain or spinal cord.
Leukoencephalitis: Inflammation of the white matter of the brain.
Macroglia: A collective term referring to astrocytes and oligodendrocytes. Has also been variously used to refer solely to astrocytes or to astrocytes, oligodendrocytes, and ependymal cells of the CNS, and Müller cells of the retina.
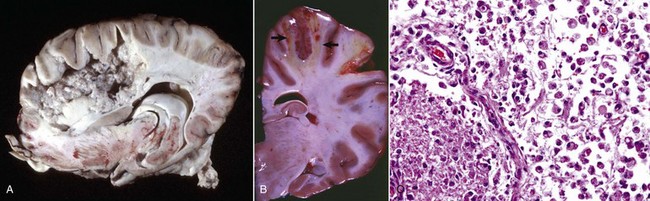
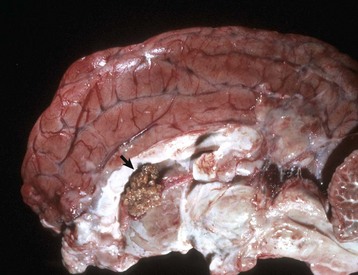
Malacia: Grossly detectable (macroscopic lesion) softening of CNS tissue, usually the result of necrosis.
Meningo-: Combining form referring to meninges.
Meningomyelocele: A form of spina bifida in which meninges and spinal cord herniate through a defect in the vertebral column into a sac (-cele) covered by skin.
Microglia: Resident cells of the CNS believed to arise from monocytes that populate the brain during embryonic development.
Motor neuron, lower: Large multipolar neurons in the brainstem and ventral horns of the spinal cord with axons extending into the PNS.
Motor neuron, upper: Motor neurons with axons residing solely in the CNS that control lower motor neurons.
Myelitis: Inflammation of the spinal cord.
Myelo-: Combining form referring to spinal cord.
Myelopathy: A degenerative disease process of the spinal cord.
Myeloschisis: Similar to spina bifida, except in its severe form is characterized by complete failure of the spinal neural tube to close, therefore a lack of development of the entire dorsal vertebral column.
Neuroglial cells: Astrocytes, oligodendroglia, ependymal cells, and microglia of the CNS.
Neuronophagia: Accumulation of microglial cells around a dead neuron.
Neuropil: The gray matter feltwork that consists of intermingled and interconnected processes of neurons (axons and dendrites) and their synaptic junctions, plus processes of oligodendroglia, astrocytes, and microglia.
Neurapraxia: Traumatic injury to a peripheral nerve with temporary conduction block but with no permanent axonal damage.
Neurotmesis: Complete transection of a nerve and supporting framework with little potential for normal reinnervation.
Onion bulb: Concentric arrays of Schwann cell cytoplasm around an axon signifying multiple episodes of demyelination and remyelination.
Polio-: Combining form referring to gray matter of the CNS.
Polioencephalomalacia: Softening (usually the result of necrosis) of the gray matter of the brain.
Polioencephalomyelitis: Inflammation of the gray matter of the brain and spinal cord.
Poliomyelomalacia: Softening (usually the result of necrosis) of the gray matter of the spinal cord.
Porencephaly: A cleft or cystic defect in the cerebral hemisphere that communicates with the subarachnoid space and also may communicate with the ventricular system. The defect may contain CSF.
Radiculoneuritis (polyradiculoneuritis): Inflammation of a spinal nerve rootlet (or rootlets).
Rarefaction: Reduction in density of CNS tissue that may result from edema, necrosis, and the like. This lesion is usually observed microscopically.
Satellitosis: An accumulation of oligodendroglia around neuronal cell bodies. Although this feature can be seen in normal brains, some consider that it may also be associated with neuronal injury.
Sclerosis: Literally means induration or hardening and, when used in describing lesions of the CNS, often refers to induration or hardening of the brain or spinal cord resulting from astrogliosis (astrocytic scar formation).
Spina bifida: A dorsal midline defect involving one to several vertebrae of the spinal column caused by failure of the neural tube to close, permitting exposure of the underlying meninges and spinal cord. The lesion may be associated with herniation of meninges alone or meninges and spinal cord tissue into a sac (-cele) covered by skin, or there may be no herniation (spina bifida occulta).
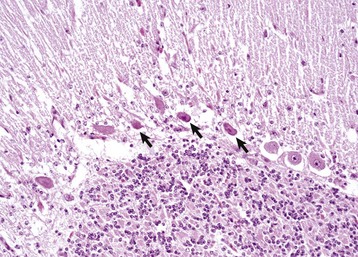
Status spongiosus: An encompassing term meaning the presence of small focal, ovoid to round “clear (unstained or poorly stained [H&E stain])” spaces in the CNS. The lesion can result from several different tissue alterations, which include splitting of the myelin sheath, accumulation of extracellular fluid, swelling of cellular (e.g., astrocytic and neuronal) processes, and axonal injury (wallerian degeneration) when swollen axons are no longer detectable within distended spaces.
Syringomyelia: A tubular cavitation (syrinx) in the spinal cord that is not lined by ependyma and may extend over several segments.
Wallerian degeneration: Degeneration of the distal component of an injured (compressed or severed) axon. Although the term originally referred to injury of axons in the PNS, current usage also includes the CNS. This process also results in functional and structural alterations in the cell body (central chromatolysis) and proximal internode segment of the axon, and in secondary demyelination.
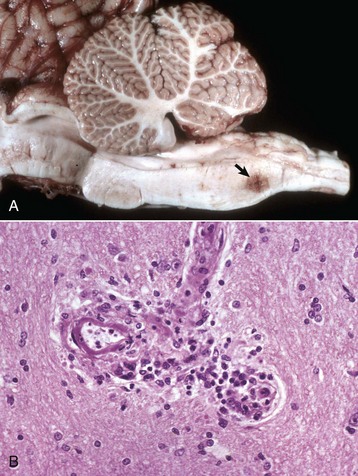
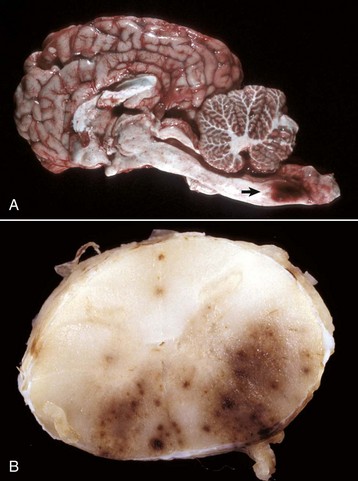
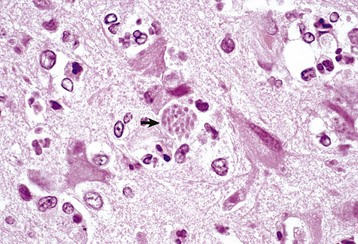
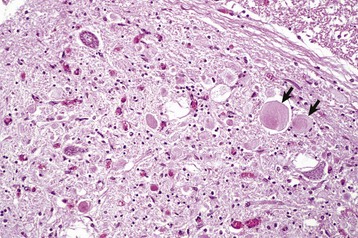
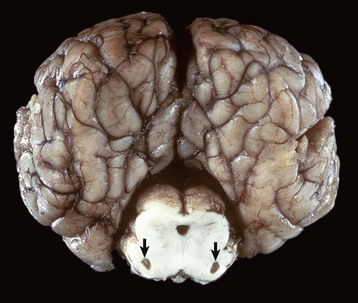
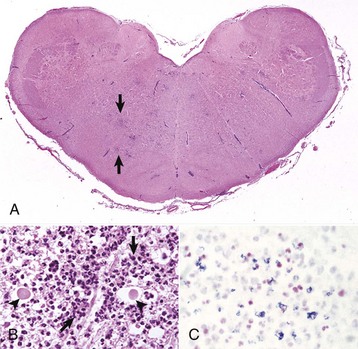
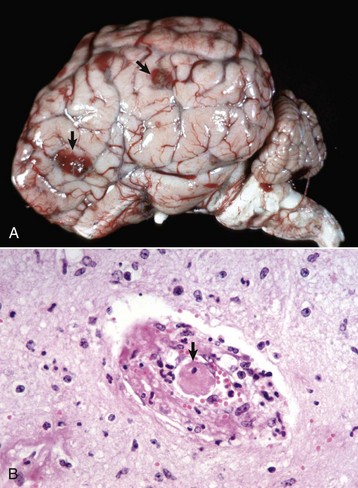
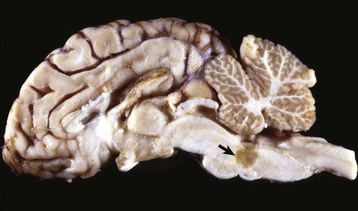
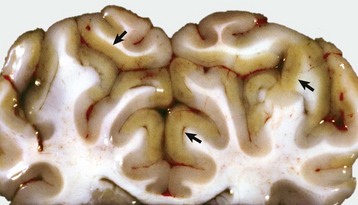
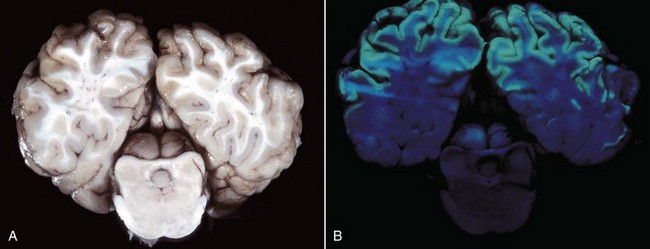
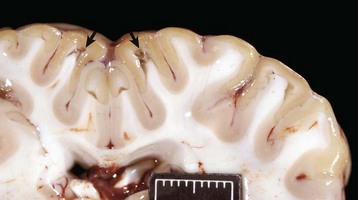
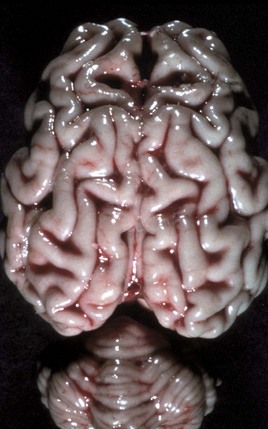
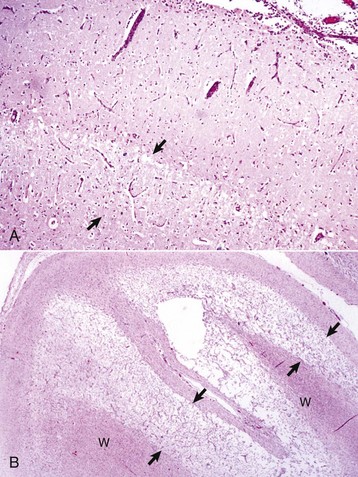
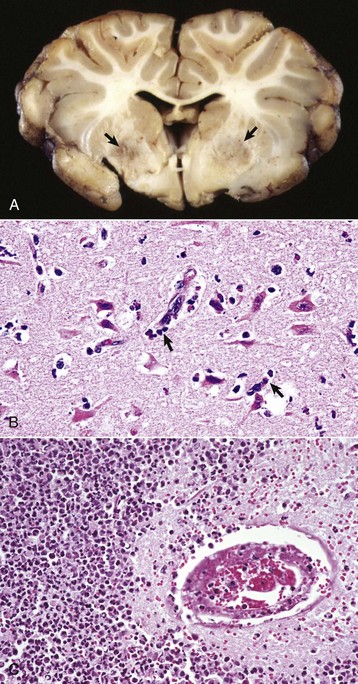
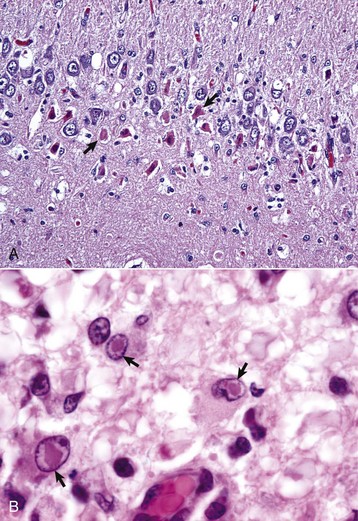
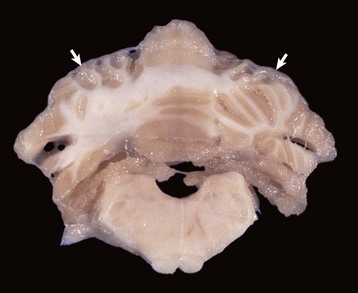
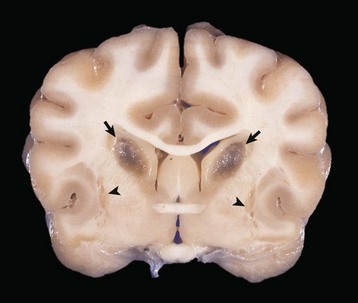
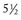 months of age, is characterized clinically by rear limb ataxia, intention tremors, hypermetria of front and rear limbs, and atrophy of appendicular and epaxial muscles, presumably from disuse.
months of age, is characterized clinically by rear limb ataxia, intention tremors, hypermetria of front and rear limbs, and atrophy of appendicular and epaxial muscles, presumably from disuse.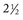 and 5 months of age, and there is onset of progressive cerebellar ataxia, spastic paresis, and collapse.
and 5 months of age, and there is onset of progressive cerebellar ataxia, spastic paresis, and collapse.