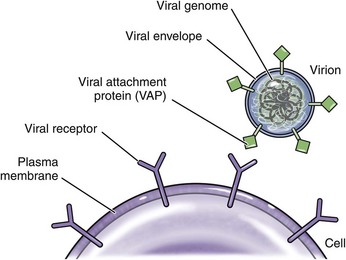
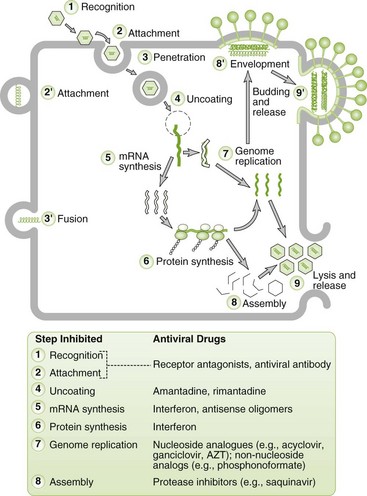
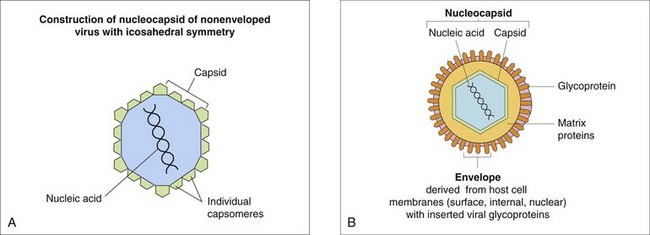
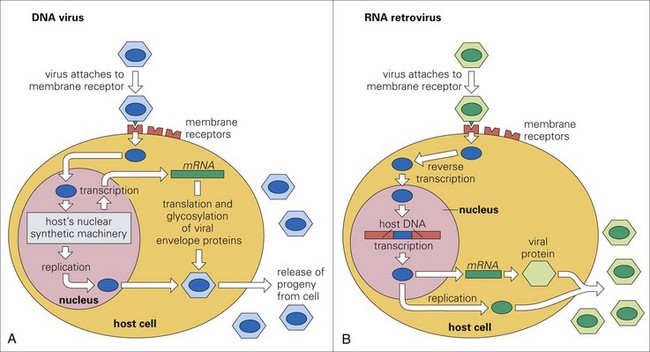
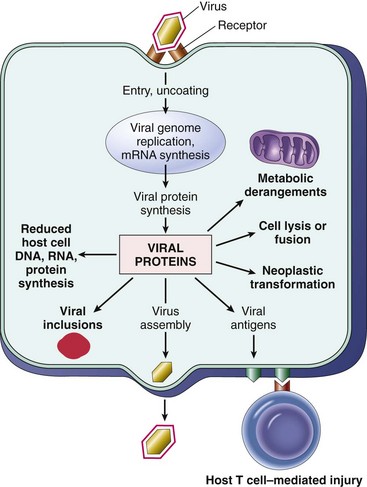
Cardiovascular System and Lymphatic Vessels
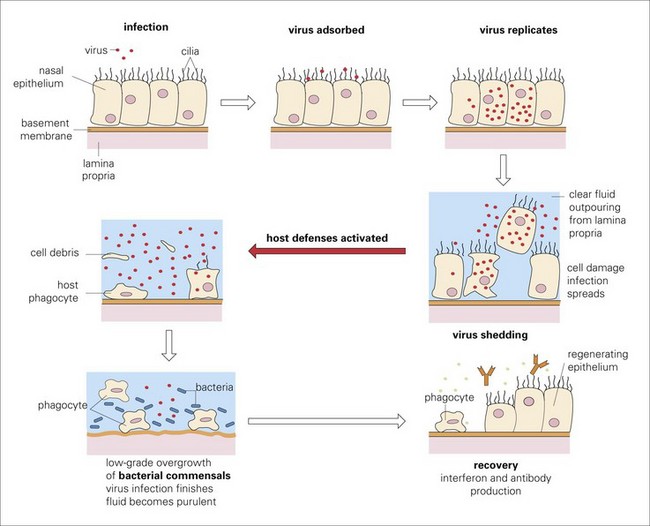
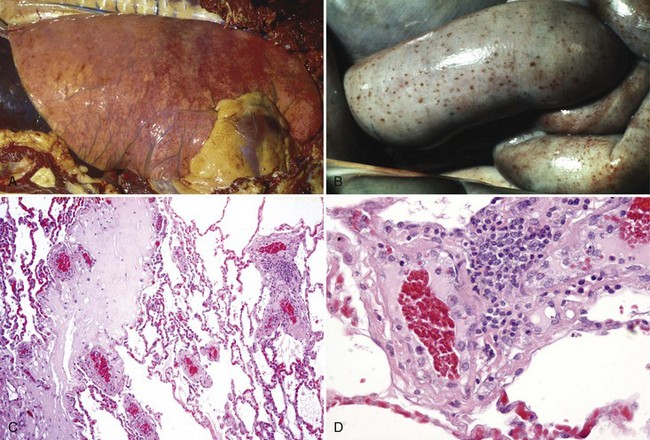
Edema Disease (Escherichia coli): The pathogenesis of edema disease begins as an alimentary enterotoxemia and ends as a fibrinoid arteriopathy/arteriolopathy of the vascular system, especially of the brain, leading to ischemia and malacia. The enterotoxemia phase is discussed in the section on the Alimentary System; the nervous system phase is discussed in the section on the Nervous System. The mechanism of injury is injury and death (coagulative necrosis) of endothelial and smooth muscle cells of arteries and arterioles caused by Shiga toxin 2e (also known as verotoxin 2e) produced by hemolytic strains of E. coli. After colonization of the intestinal mucosae, the toxin is absorbed from the alimentary system and circulates in the blood vascular system. Cells susceptible to the affects of this toxin include endothelial and smooth muscle cells of arteries and arterioles that express receptors for the toxin such as globotetraosylceramide, galactosylgloboside, and globotriaosylceramide. Toxin acts to disrupt protein synthesis leading to vascular permeability changes and cell death and thus edema of affected organs, most notably the eyelids, ventral neck (jowls), the gastric and colonic mesenteries, and the nervous system (see Figs. 7-126 and 7-127). Additionally, endothelial injury caused by this toxin may lead to hemorrhage, intravascular coagulation, microthrombosis, and infarction.
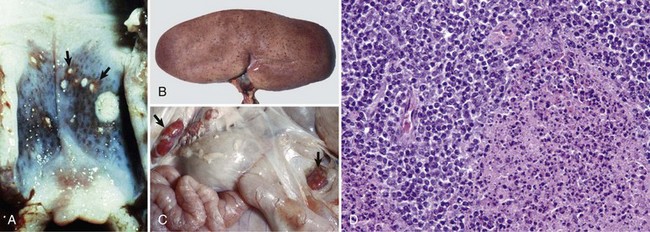
Embolic Vasculopathy/Vasculitis (Actinobacillus equuli, Escherichia coli, Staphylococcus spp., Streptococcus spp., Fusobacterium necrophorum): This section covers a variety of diseases in which the key component of the underlying pathogenesis is embolization through the blood vascular system, leading to vasculitis and potentially thrombosis and ischemia. Such embolic diseases most commonly begin in the skin/subcutis or mucosae but end in a wide variety of highly vascularized organ systems. Examples of embolic diseases include white spotted kidney disease (E. coli), embolic nephritis (foal shigellosis [Actinobacillus equuli]), milk spots in the liver (E. coli), bacterial endocarditis (E. coli), and bacterial hepatitis (Fusobacterium necrophorum). Embolization also occurs in diseases caused by angioinvasive fungi, and they are covered in the section on fungal diseases. The mechanism of injury in embolic vasculopathy/vasculitis is cell death, probably acute coagulative necrosis, caused by bacterial toxins and inflammation and its mediators and degradative enzymes. Gross lesions include gray-white foci of necrosis and inflammation distributed at random (vascular embolization pattern) in tissue such as occur in renal actinobacillosis of foals (see Fig. 11-42).
Bacteria are able to enter and spread in the vascular system by two mechanisms: (1) direct entry into a blood vessel or (2) establishment of a local infection followed by invasion of the vascular system. The former category usually results from bacteria gaining direct access to blood vessels secondary to penetrating trauma, bite wounds, or lacerations, whereas the latter category usually is caused by similar traumatic injury that leads to local inflammation and often abscess formation. In the direct entry mechanism, access to the vascular system, embolization, and entrapment in capillary beds are likely physical interactions based on the anatomy of vascular distribution patterns, physiology of vascular flow and pressures, and pathobiology of endothelial cell surface molecules. As an example in the cerebral cortices, lesions caused by bacterial emboli tend to be observed at the interface between gray and white matter. Anatomically, at this location, capillaries penetrate through the gray matter from the overlying meninges and as they run into the white matter they make abrupt turns (90-degree), so capillaries can run parallel to fiber tracts in the white matter. This flow change causes vascular turbulence and endothelial cell surface perturbations, and under the proper conditions, activation of Virchow’s triad can result in the formation of vascular endothelial surfaces that may be sticky or have attached fibrin that can entrap bacteria. Many of the bacterial virulence determinates discussed throughout this chapter, as well as ligand-receptor interactions, probably are involved to some extent in the origination and entrapment and growth of bacterial emboli in the direct entry mechanism.
In the establishment of a local infection mechanism, contamination of the umbilicus at birth and the skin/subcutis through management practices, such as tail docking, castration, and ear notching, are the common means of establishing local infections. Injury of mucosae, such as occurs in the abomasum from lactic acidosis in grain overload, also provides opportunities for bacteria to enter the portal blood vascular system and then embolize to and colonize the liver. Finally, bacteria that induce biofilms or cause irresolvable inflammatory processes, such as in a dermatitis, otitis, cellulitis, periodontal disease, arthritis, or abscesses, can serve as a site for intermittent bacteremia and embolization. Many of the bacterial virulence determinates discussed throughout this chapter, as well as ligand-receptor interactions, probably are involved to some extent in the origination and entrapment and growth of bacterial emboli in the local infection mechanism.
Septicemic Anthrax (Bacillus anthracis): The sections in this chapter on alimentary and inhalation anthrax should be reviewed for background information pertinent to understanding septicemic anthrax (see Fig. 7-135). Once vegetative forms of the bacteria enter the circulatory system from the respiratory or alimentary systems, septicemia ensues and vascular collapse occurs, resulting from massive release of toxins into the blood plasma. Septicemic anthrax is characterized by animals found dead unexpectedly, often in a classic sawhorse stance and with hemorrhage (unclotted) from body orifices (Fig. 4-23). If inadvertently opened, the spleen will be enlarged and unclotted blood will exit from cut surfaces; lymph nodes will be enlarged, edematous, and hemorrhagic; and body tissues and serosal surfaces will be edematous and hemorrhagic (see Fig. 4-23). Necropsies should not be performed on animals that are suspected of dying from anthrax because the vegetative form proliferates in large numbers in blood; when they are released from lacerated blood vessels onto the ground, they form endospores, which contaminate the area long-term.
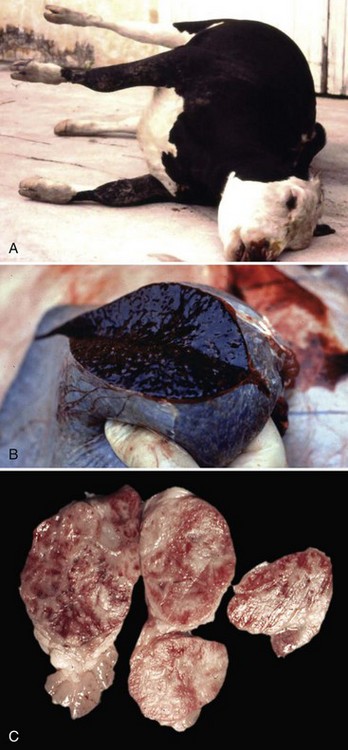
Fig. 4-23 Anthrax, ox.
A, Because of the high fever, cadavers of cattle dying of anthrax decompose rapidly with the usual result of excessive gas formation in the GI tract, abdominal distention and resultant “saw horse” position of the legs. B, The spleen is enlarged and bloody (splenomegaly, bloody spleen). A postmortem examination should not be performed on an animal suspected of dying from anthrax. Air-dried impression smears of blood from external orifices or from an ear vein can be stained and the bacterium identified (see Fig. 7-135). C, Lymph nodes are also enlarged and bloody as a result of anthrax toxins that destroy vascular endothelial cells (see Fig. 13-53). Anthrax toxin can also cause severe injury to the intestines (see Fig. 7-135) and lungs. (A courtesy Dr. D. Driemeier, Federal University of Rio Grande do Sul, Brazil. B and C courtesy Dr. J. King, College of Veterinary Medicine, Cornell University.)
In the circulatory system, vegetative forms proliferate in large numbers and are arranged in long chains in the capillary beds of many organ systems, including the spleen (see Fig. 13-53). Large quantities of edema toxin and lethal toxin are released into the blood, causing dysfunction and cell death of endothelial cells and their barrier systems, thus the toxins increase the permeability of the capillary wall, leading to edema, vasodilation, and hemorrhage in infected organ systems. Anthrax toxins also disrupt the clotting cascade likely through massive activation of DIC and consumption of clotting factors, resulting in unclotted blood at body orifices and within tissues and organs.
Vascular Leptospirosis (Leptospira spp.): The mechanism of injury in vascular leptospirosis is cell death caused by (1) physical properties (penetrating movements) of bacteria that disrupt functions of endothelial cells and (2) bacterial toxins that act directly on membranes of endothelial cells of small blood vessels, including capillaries of the systemic vasculature in all organ systems, leading to coagulative necrosis of affected cells. Gross lesions include acute vasculitis (endothelial cell necrosis) with systemic petechial and ecchymotic hemorrhages, edema, and DIC affecting all organ systems and serosal surfaces (see Fig. 2-18).
Animals encounter Leptospira spp. through direct contact of their oral or conjunctival mucous membranes or skin with leptospira-infected urine or with water from reservoirs or ponds into which infected urine drains. Ingestion may also serve as a portal of entry if water contaminated with leptospira is consumed and the bacteria encounter mucosae of the intestine. During mastication and swallowing, it is likely that the mucosae of the oral pharynx trap the bacteria in its mucus layer. After swallowing and through intestinal peristalsis, the bacteria are moved into contact with villi and crypts, where they are probably trapped in the mucus layer and encounter enterocytes. In the conjunctiva, it is also likely that the mucosae trap the bacteria in a mucus layer. For infection to occur, it has been suggested that the skin and mucosae must have small cuts or abrasions that allow the bacteria to penetrate into the vascularized submucosal or subcutaneous connective tissues and gain access to capillaries and/or postcapillary venules. However, the bacteria are motile and likely able to penetrate mucus layers and invade mucosae by moving directly through mucosal epithelial cells or between the cells through intracellular junctional complexes. In all three of these portals of entry, the goal is for the bacteria to reach well-vascularized ECM tissues. As a group, these spirochetes are highly motile and invasive and using its invasive motility (virulence determinate), it is able to penetrate the vascular wall and endothelial cells of capillaries and postcapillary venules to gain access to the circulatory system. Leptospira spp. may also invade lymphatic vessels and through this system and the thoracic duct eventually gain access to the circulatory system. Leptospira spp. are able to grow and replicate in the circulatory system and then spread systemically to all organ systems. To infect other organs such as the kidney and liver, it appears that the bacteria must first attach to endothelial cell membranes via adhesins before invading these cells and their underlying vascularized tissues and then interact with renal tubular epithelial cells and hepatocytes.
Leptospira spp. initially spread through the vascular system to all tissues of the body and do not appear to specifically target the kidney or liver via a tropism (attraction to a specific cell type or tissue) mechanism. However, once epithelial cells are infected, the reason for dominance of lesions in these organs is unclear and may be related to some essential trophism (nourishment of tissues) provided by these cells to the bacteria for colonization and proliferation.
In the context of interacting with capillary endothelial cells and probably renal epithelium, surface-associated proteins (outer membrane leptospiral protein) appear to be involved in ligand-receptor interactions that facilitate adhesion to host cell receptors like cell-adhesion molecules and ECM proteins (Len protein family). Adhesion to cells also appears to cause an increased expression of adhesion receptors such as E-selectin on endothelial cells resulting in additional adhesion of bacteria, platelets, and neutrophils (acute inflammatory response). This response may be attributable to bacteria wall LPS, peptidoglycans, and outer membrane proteins, thus promoting inflammation in capillaries leading to vasculitis and hemorrhage. Bacterial LPS likely activates cells by binding to TLRs on host cell membranes. Leptospira spp. also produce pore-forming hemolysins, proteases, sphingomyelinases, and collagenases that may assist in this process, but their role in causing endothelial cell injury remains to be determined.
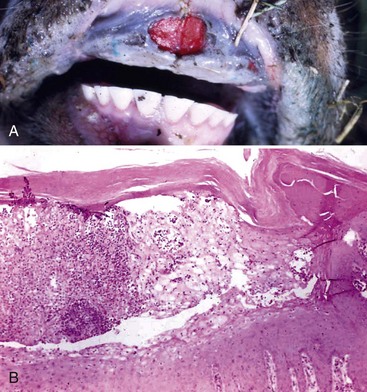
Glanders (Burkholderia mallei; Farcy, Malleus, Droes): The mechanism of injury in glanders is cell death caused by pyogranulomatous inflammation and its mediators and degradative enzymes. It is a disease of lymphatic vessels (and surrounding integument) and of the respiratory system. Gross lesions include ulcers, pustules, and nodules that can affect any part of the body but most frequently involve the skin and lymphatic vessels of the legs and flanks (Fig. 4-24). Nodules typically occur along the course of lymphatic vessels resulting in a raised beaded appearance of the skin. These nodules often rupture because of trauma to the skin or from pressure necrosis caused by the expanding volume of exudate within the nodules. This process results in craterlike ulcers of the skin that discharge a thick yellowish-white viscid and sticky purulent material containing abundant bacteria (see Fig. 4-24), and the outcome has been called farcy pipes. In the respiratory system, pyogranulomas can occur in mucosae of the nasal cavity and in all lobes of the lungs (random pattern).
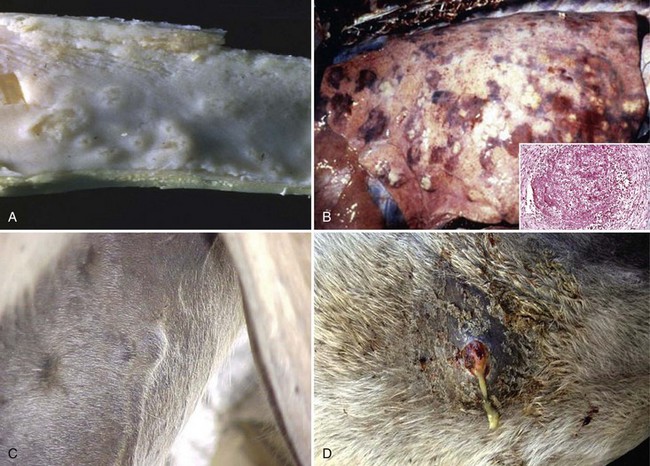
Fig. 4-24 Glanders disease.
A, Mucosa, nasal turbinates, multiple nasal ulcers, and granulomas. Burkholderia mallei colonizes the nasal turbinates resulting in pyogranulomatous inflammation, necrosis, and ulceration of the mucosa. B, When the bacterium colonizes the mucosae of the conductive system of the lung, it spreads into pulmonary parenchyma resulting in the formation of pyogranulomas (inset) throughout the lung. Inset, H&E stain. C, When the bacterium spreads to the skin, it colonizes subcutaneous lymphatic vessels resulting in the formation of pyogranulomatous nodules that typically occur along the course of lymphatic vessels (pyogranulomatous lymphangitis) resulting in a raised beaded appearance of the skin. D, These nodules frequently rupture because of trauma to the skin or from pressure necrosis due to the expanding volume of exudate within the nodules. This process results in crater-like ulcers of the skin that discharge a thick yellowish-white viscid and sticky purulent material containing abundant bacteria. (A courtesy Dr. D.D. Harrington, School of Veterinary Medicine, Purdue University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy United States Animal Health Association, St. Joseph, MO. B inset courtesy Dr. Tyler, College of Veterinary Medicine, University of Georgia and Noah’s Arkive, College of Veterinary Medicine, University of Georgia. C courtesy Dr. D. Driemeier, Federal University of Rio Grande do Sul, Brazil. D courtesy Dr. R. Mota, Universidad Federal Rural de Pernambuco, Recife, Brazil and Dr. M. Brito, Universidad Federal Rural do Rio de Janeiro, Brazil.)
Horses, mules and donkeys encounter Burkholderia mallei, a facultative intracellular bacterium that resides in the soil, via fomites that (1) penetrate the skin by trauma or foreign objects or (2) are inhaled and/or ingested. In either case, these fomites are contaminated with bacteria-laden exudate derived from other infected animals with cutaneous lesions or nasal discharge. It appears that cutaneous glanders can occur through two mechanisms: (1) direct penetration of the skin or (2) through systemic spread to the skin via a systemic bacteremia or leukocytic trafficking from an initial infection of the respiratory or intestinal mucosae. For direct penetration, the skin apparently must have small cuts or abrasions that allow the bacterium to penetrate the subcutaneous connective tissues, gain access to lymphatic vessels, and cause infection and inflammation. Target cells for infection in the cutaneous lymphatic vessels and skin are unknown. Additionally, the role of ligand-receptor interactions in this process has not been determined. It appears that lesions characteristic of glanders probably result from inflammatory responses directed against a variety of molecules within the bacterium, resulting in pyogranulomatous inflammation.
For systemic spread to the skin, the bacterium appears to encounter mucosae of the nasal cavity and conductive component of the respiratory system. It is likely trapped in the mucus layer and then colonizes the mucosae. The development of a biofilm (virulence determinate) may be important in the colonization process. Once mucosae are colonized, the bacterium could then be phagocytosed by mucosal macrophages and/or dendritic cells and carried through mucosae to MALTs such as BALT. It is also possible, but unknown, that the bacterium has a mechanism to penetrate the mucus layer, enter the apical surfaces of mucosal epithelial cells via endocytosis or transcytosis, and exit the lateral-basal surfaces to gain access to mucosal-associated lymphoid tissues. In these lymphoid tissues, the target cells for infection and the role of ligand-receptor interactions have not been determined but appear to result in the nasal and pulmonary pyogranulomas characteristic of the disease. Cells of the monocyte-macrophage system and possibly mucosal dendritic cells probably play a role in the infection, replication, and spread of the bacterium. Once in MALTs, the bacterium could spread to regional lymph nodes in lymphatic vessels (1) as a cell-free bacteremia or (2) via leukocytic trafficking in macrophages, and after infection of and replication in lymphoid tissues, the bacterium could then spread systemically via lymphatic vessels and the thoracic duct to the circulatory system. Via the circulatory system, the bacterium ultimately arrives (cell free or in macrophages) at capillary beds of the skin, passes through the endothelial cells (leukocytic trafficking, endocytosis, or transcytosis), enters the subcutaneous tissues, encounters cutaneous lymphatic vessels, and elicits a pyogranulomatous inflammatory response in these tissues. Pyogranulomas have also been described in the spleen and liver.
Lesions in the respiratory system probably arise from an extension of the processes involved in colonizing mucosae as previously described, with secondary spread into adjacent interstitial tissues and alveoli. Additionally, it is possible that pyogranulomas in the lung could arise from spread of the bacterium through the circulatory system as a cell-free bacteremia or in macrophages as described. The existence of this mechanism of spread has not been determined.
Through ingestion and gastrointestinal peristalsis, Burkholderia mallei can also potentially reach the alimentary system. Herein, they could encounter the mucus layer of intestinal mucosae or M cells overlying Peyer’s patches. How the bacterium could penetrate the mucus layer is unknown, but it could involve mucosae-associated macrophages as discussed previously in respiratory system mucosae. Bacteria could also infect M cells via endocytosis, undergo endocytosis or transcytosis, and exit the basal surfaces via exocytosis to gain access to macrophages within Peyer’s patches. For either of these two pathways, macrophages within Peyer’s patches could then be infected with bacteria and be used to spread bacteria systemically. The likely goal of these types of mucosal encounters is to provide the bacterium with the opportunity to infect the local lymphoid tissues and then gain access to local, regional, and systemic lymph nodules and the circulatory system directly or via macrophages.
How and if the bacterium evades killing by phagosome-lysosome fusion, replicates, and spreads to cutaneous lymphatic vessels via the circulatory system are unknown. Type III and IV secretion systems (virulence determinates) may be possible mechanisms for the invasion, escape from lysosomes or phagolysosomes, and survival in target cells. The bacterium is surrounded by a type I O-antigenic polysaccharide (capsular) antigen, a virulence determinate, that may also block phagocytosis or phagosome-lysosome fusion. LPS, which likely contains a lipid A component, may play a role in tissue injury. Additionally, an interaction of the bacterium with cell membranes is a prerequisite for infection to occur. A type IV pilin-like protein may be involved in the adherence of the bacterium to target cells.
Urinary System
Pulpy Kidney (Overeating) Disease (Clostridium perfringens): The pathogenesis of pulpy kidney disease begins as an enterotoxemia caused by Clostridium perfringens, and this disease in the section on the alimentary system should be reviewed. Because ε-toxin is a permease that alters cell permeability, the vascular beds in affected intestinal tissues readily absorb toxins into the circulatory system. It appears that the sequence of events leading to pulpy kidney disease occur in the first phase or early in the second phase of alimentary enterotoxemia before toxin-induced massive necrosis of the intestine occurs. The mechanism of injury in pulpy kidney disease is cell death caused by ε-toxin that acts directly on renal endothelial and tubular epithelial cell membranes leading to vascular permeability changes and acute coagulative necrosis of tubular cells. Microthrombosis and ischemia from capillary endothelial injury are plausible but unproved mechanisms of tubular cell death. Gross lesions include soft pliable kidneys with hemorrhages; however, the lesions are often attributed to postmortem change. Experimental data suggest that vascular endothelial cells, such as those in the renal cortex supplying epithelial cells of renal tubules, express receptors (ligand-receptor interactions) for ε-toxin. Because ε-toxin is an angiotoxic permease, it increases the permeability of targeted endothelial cells, allowing plasma containing ε-toxin to leak into the ECM surrounding renal tubules. Renal tubular epithelial cells also express receptors for ε-toxin, and toxin binding may lead to membrane-mediated cytotoxicity and cell death.
Necrohemorrhagic Urocystitis (Escherichia coli, Corynebacterium renale, Pseudomonas spp., Proteus vulgaris, or Klebsiella pneumoniae): Necrohemorrhagic urocystitis is a term used to group bacteria whose virulence determinates can cause acute inflammation and hemorrhage of the mucosae of the urinary bladder, especially affecting transitional epithelial cells and the lamina propria and its capillary beds. Because more is known about virulence determinates for uropathogenic E. coli, it is discussed in greater detail; however, the other bacteria listed in this group likely use similar or related mechanisms to cause disease. The mechanism of injury is probably cell death (coagulative necrosis) caused by bacterial toxins that act directly on mucosal epithelial cells and capillaries in the lamina propria of the bladder and acute and chronic inflammation and their effector molecules and degradative enzymes. Gross lesions of the bladder include mucosal edema and mucosae that are rough and granular, red to dark red, and covered with white-gray flecks of fibrin mixed with cellular debris (see Fig. 11-64). Blood vessels in the wall and serosa of the bladder are prominent; this change is due to active hyperemia of the fluidic vascular phase of acute inflammation.
Animals encounter these bacteria through contact with them in fomites or fluid droplets of urinary or fecal origin. They commonly become commensal organisms that reside in the mucous membranes of the vagina and prepuce. Physical changes in pressure across the tubular components of the urinary and reproductive systems caused by parturition and breeding appear to force these commensal bacteria via reflux mechanisms into the urethra and urinary bladder. The length of the urethra, in part, appears to determine why females have cystitis more commonly than males. Environmental stressors, such as peak lactation, traumatic mucosal injury, and a high-protein diet that increases the pH of the urine, make mucosae more susceptible to colonization and alter the commensal relationship, allowing the bacteria to replicate in sufficient numbers to colonize the mucosae of the urinary and reproductive systems and spread bacteria to other animals. Once in the lumen of the urinary bladder, bacteria gain access to mucosal surfaces via random movement of the urine. The urinary mucosae lacks goblet cells, thus there is no mucus layer to penetrate. These bacteria encounter the apical surface of transitional epithelial cells, and using ligand-receptor interactions characteristic of other bacterial diseases, begin the process of adherence, binding, and colonization of the mucosae. Uropathogenic E. coli expresses adhesins, such as type 1 fimbriae, P fimbriae, and S fimbriae, that are involved in this process. These fimbriae (also known as pili) bind to hexagonal arrays of mannosyl-glycoprotein receptors known as uroplakins. They are expressed on the apical (and luminal) surfaces of specialized transitional epithelial cells called umbrella cells. The tips of the type 1 fimbriae express a ligand called FimH adhesin that binds to these uroplakin receptors.
Once bound, the bacteria begin a complicated process of entering and colonizing the cells and mucosae, respectively, through a series of conformational changes in the apical surface of the umbrella cells, leading to cytoskeletal rearrangements and cell entry by a zipper mechanism. Bacterial flagella may also have a role in this mechanism. This process results in colonization of the mucosae and the development of a biofilm-like arrangement affecting the mucosae (also known as biomasses of bacteria or intracellular bacterial communities). After formation of intracellular bacterial communities, bacteria kill infected umbrella cells via hemolysins that produce pores in membranes, thus releasing bacteria into the lumen of the bladder where they colonize new umbrella cells and repeat the infective process or are released into the environment during urination. The edema, hemorrhage, and necrosis characteristic of necrohemorrhagic urocystitis appear to be caused by acute inflammation and a variety of virulence determinates in highly pathogenic strains of uropathogenic E. coli and likely the other bacteria listed previously. Acute inflammation is probably induced via TLRs that recruit neutrophils from the vascular system into the lamina propria and mucosae and in response to cell necrosis, loss of the mucosal barrier, and interaction of the vascularized lamina propria with bacterial toxins. In umbrella cells infected by bacteria, bacterial toxins, such as LT and ST toxins, Shiga-like toxin, cytotoxins, and endotoxin, likely diffuse through the mucosae and cause membrane injury, leading to cell death (necrosis) and loss of the mucosal barrier system. These toxins may also stimulate apoptotic cell death, leading to release of bacteria into the urine. Once dead, these cells slough into the urine and endotoxins and other toxic molecules can be absorbed into the highly vascularized lamina propria, resulting in injury to the capillaries and acute vasculitis with active hyperemia.
Other virulence determinates that contribute to the pathogenesis of necrohemorrhagic urocystitis include bacterial surface molecules such as capsular K antigens and LPS that block phagocytosis and killing of the bacteria by neutrophils and macrophages. Uropathogenic E. coli usually produces siderophores that play a role in iron acquisition for the bacteria during and after colonization. The lytic actions of hemolysins also increase the availability of iron and other nutrients for bacterial growth in colonized mucosae. Hemolysins also can kill lymphocytes and block phagocytosis and chemotaxis by phagocytic cells. Some strains of uropathogenic E. coli have a virulence determinate for the production of urease, which hydrolyzes ammonia in urine into urea, resulting in an alkaline urine that causes additional injury to mucosae. Finally, these bacteria can readily exchange genetic information with less virulent bacterial strains by transduction and conjugation using drug resistance, toxin, and other virulence plasmids. These factors are a few of the reasons why it is often difficult to treat and resolve certain types of acute and chronic bladder infections.
Contagious Bovine Pyelonephritis (Corynebacterium renale or Escherichia coli): Contagious bovine pyelonephritis is most commonly caused by Corynebacterium renale, but E. coli may also cause this disease. These bacteria are likely commensal organisms that reside in the mucous membranes of the vagina and prepuce. Mechanisms that contribute to the occurrence of cystitis that preceded pyelonephritis are discussed in the previous section. Information about the mechanisms used by Corynebacterium renale or E. coli to cause contagious bovine pyelonephritis in cattle is limited. Thus portions of this section are speculative and based on (1) what is known mechanistically about other diseases of the respiratory system caused by Corynebacterium spp. or E. coli and (2) a reasonable probability that inflammation, responses to injury, and lesions that have been described in contagious bovine pyelonephritis are the result of underlying and known pathobiologic mechanisms. The mechanism of injury in contagious bovine pyelonephritis is probably cell death (acute coagulative necrosis) caused by (1) bacterial toxins that act directly on transitional mucosal and tubular epithelial cells of the urinary system possibly by inducing tubular cell and collecting duct cell apoptosis and (2) acute and chronic inflammation and their effector molecules and degradative enzymes. Gross lesions include white-tan streaks often mixed with narrow red streaks (hemorrhage) that radiated from the pelvis, through the medulla, often extending to the cortical medullary junction or deeper into the cortex (see Fig. 11-53). In many ways, these lesions resemble inverted renal cortical infarcts with their bases against the pelvis and their apices extending into the medulla.
Cattle (and probably sheep and goats) encounter these bacteria through contact with them in fomites or fluid droplets of urinary or fecal origin. Environmental stressors such as parturition, peak lactation, traumatic mucosal injury, and a high-protein diet that increases the pH of the urine, making the mucosae more suitable to colonization, could alter the commensal relationship, allowing the bacteria to replicate in sufficient numbers to colonize the mucosae of the urinary and reproductive systems and spread the bacterium to other animals. It has been proposed that contagious bovine pyelonephritis occurs secondary to a chronic and often insidious cystitis that ascends in the lower urinary tract and reaches the renal pelvis via the ureters to then spread though the mucosal barrier formed by transitional epithelium of the renal pelvis into the interstitium of the renal medulla. The mechanism of ascension may be caused by the reflux of infected urine from the ureters and urinary bladder into the renal pelvis. In the pelvis, it may occur via spread of bacteria through the mucosal barrier formed by the transitional epithelium of the pelvis and into vascularized ECM connective tissues supporting the tubules. How each of the events occurs has not been specifically determined; however, ligand-receptor interactions characteristic of other bacterial diseases and their interaction with mucosae likely occur in contagious bovine pyelonephritis.
Although Corynebacterium renale is a nonmotile bacterium, more pathogenic strains of E. coli are motile and this virulence determinate may help in ascension of the bacterium up the ureter to the kidney. It has been shown that pili are required for Corynebacterium renale to adhere to transitional epithelium of the urinary system and to attach to and colonize mucosae of the reproductive system. Additionally, pili may serve to disrupt phagocytosis of the bacteria by neutrophils and macrophages. Binding is strongest to the mucosal epithelial cells of the vulva and vagina. The receptors used to bind to mucosae have not been determined; however, once bound, colonization of the mucosae begins. Once bacteria replicate to sufficient numbers, they then spread by ascending in the lower urinary tract to encounter and colonize the mucosae of the urethra and bladder and then by vesicoureteral reflux to ascend to the ureters and renal pelvis. The development of a chronic insidious urocystitis is often an intervening stage in the disease that serves to produce large numbers of bacteria. After colonization, it is not known how these bacteria cross mucosae to gain access to the medullary interstitium at the pelvis. Degradative enzymes and inflammatory mediators combined with bacterial virulence determinates, such as Renalin, which is an extracellular cytolytic protein produced by Corynebacterium renale, may facilitate spread across mucosal barriers and inflammation and cell death within the medulla. It has been suggested that lesions in the medulla (resembling inverted renal cortical infarcts) may actually begin as a vasculitis from inflammation resulting in thrombosis, ischemia, and necrosis. It has been shown that toxins from E. coli may stimulate apoptotic cell death in renal tubular cells in pyelonephritis.
Renal Leptospirosis (Leptospira spp.): The pathogenesis of renal leptospirosis begins as vascular leptospirosis (see the section on the Cardiovascular System and Lymphatic Vessels) caused by Leptospira spp. The mechanism of injury in renal leptospirosis is cell death caused by (1) physical properties (penetrating movements) of bacteria that disrupt functions of endothelial cells, (2) bacterial toxins that act directly on membranes of tubular epithelial cells, and (3) acute and chronic inflammation and their effector molecules and degradative enzymes. Gross lesions include discrete and coalescing, often linear to radiating white to gray foci of tubular cortical necrosis and acute inflammation intermixed with hemorrhage (see Fig. 11-73). In chronic renal leptospirosis, lesions include discrete and coalescing, often linear to radiating white to gray foci of chronic inflammation and fibrosis (see Fig. 11-20). In the kidney, primary target cells for infection appear to be epithelial cells of the proximal convoluted tubules (cortex) (Fig. 4-25, A) and then later, epithelial cells of the loops of Henley (medulla) (Fig. 4-25, B). Once Leptospira spp. gain access to the circulatory system, they disseminate in glomerular capillaries and then intertubular capillaries of proximal convoluted tubules. Bacteria could access proximal tubular cells via their apical or basolateral surfaces by two routes, vascular by glomerular capillaries and migration into the lumen of the urinary space (apical) or vascular via intertubular capillaries and migration into the interstitium (basolateral). Because glomerular changes are usually unremarkable and bacteria and inflammation are observed in the interstitium, it appears that epithelial cells of the proximal convoluted tubules are infected via the basal-lateral surfaces of the cells via migration through intertubular capillaries.
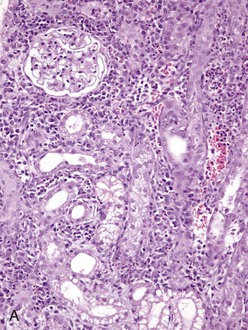
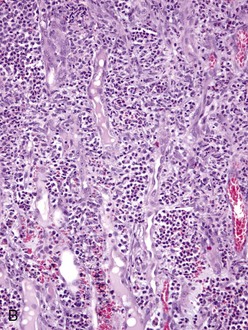
Fig. 4-25 Renal leptospirosis.
A, Kidney, outer cortex. Note the infiltration of mononuclear cells, chiefly macrophages, lymphocytes and plasma cells in the interstitium between the proximal convoluted tubules, the result of the leptospires infecting the proximal tubule cells after exiting the intertubular capillaries. B, Kidney (same as A), inner cortex. Numerous neutrophils distend the interstitium between the loops of Henle. This acute inflammatory response further down the nephron from the area in A, supports the concept that the cells of the loop of Henle are infected later than those in the proximal convoluted tubule. H&E stain. (A and B courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
The bacteria likely attach to endothelial cell membranes of intertubular capillaries via adhesins then penetrate the vessel wall by moving directly through the cells or through their junctional complexes to gain access to the interstitium. In reality, the distance in the interstitium between capillaries and proximal tubular epithelial cells is probably no more than 100 µm and it is likely that the flagella of these motile bacteria propel them to the epithelial cells. It is unclear why the bacteria target proximal tubular epithelial cells for infection. Although undetermined, such specificity could be attributed to ligand-receptor interactions or to a chemical gradient, such as an iron concentration, that could be required for bacterial growth and replication. The bacteria are present in the cytoplasm of these cells; endocytosis and phagosome-lysosome fusion are not involved in cell entry. It appears that the bacteria are able to directly enter these cells via their motility.
The cause of death of proximal tubular cells is probably multifactorial, involving vasculitis and ischemia, trauma from physical injury caused by bacterial motility, inflammatory mediators and degradative enzymes, and bacterial toxins. The inflammatory cells in this lesion progress from neutrophils (suppurative) to lymphocytes, macrophages, and plasma cells (chronic). Epithelial cells lining the loop of Henle could also be infected by an intertubular capillary-interstitial route. This mechanism has not been confirmed. Additionally and based on inflammatory cell responses, it is unclear why cells of the proximal tubules appear to be infected at an earlier point in the disease than those of the loop of Henle. When proximal tubular cells die, they release bacteria into the urinary lumen where they are carried in urine and spread into the environment via urination. During this luminal transit, the bacteria also encounter the apical surfaces of epithelial cells lining the loop of Henle. It is plausible that Leptospira spp. infect epithelial cells of the loop of Henle via their apical surfaces projecting into the urinary lumen, using mechanisms similar to those previously described. Infection appears to result in the same cascade of cell alterations and inflammatory responses as those described for proximal tubular cells. Biofilms (virulence determinate) formed by Leptospira spp. may also play a role in tubular injury. These outcomes in both proximal tubular and loop of Henle cells serve as the basis for characterizing this disease as tubulointerstitial nephritis.
Bone Marrow, Blood Cells, and Lymphatic System
Strangles (Streptococcus equi ssp. equi): The mechanism of injury in strangles is death (coagulative necrosis) of cells of lymphatic vessels, lymph nodes, and the monocyte-macrophage system attributable to acute suppurative inflammation and its mediators and degradative enzymes. Gross lesions include the formation of acute abscesses within regional lymph nodes resulting in enlarged firm lymph nodes that on a cut surface have discrete and coalescing areas of yellow-white suppurative exudate infiltrating and compressing contiguous parenchyma (see Fig. 13-74). Affected retropharyngeal and mandibular lymph nodes may also have draining fistulous tracts between affected nodes and the surface of the skin, the guttural pouches, and the nasal cavities and sinuses, resulting in release of bacteria into the environment. This outcome occurs because degradative enzymes released from dead neutrophils in abscesses digest the capsule of the lymph node and the structures of all contiguous tissues until a fistulous tract is formed.
Foals encounter Streptococcus equi ssp. equi through inhalation or ingestion of fomites or body fluids contaminated with the bacterium. It is deposited on mucosae of the nasal (centrifugal turbulence) and oral pharynx and trapped in the mucus layer. The bacterium is nonmotile and it has not been clearly shown how it penetrates the mucus layer and gains access to mucosal epithelial cells. Mucosal epithelial cells of the tonsils and tonsillar crypts appear to be important target cells for adherence and binding, and this specificity may be determined by unique ligand-receptor interactions. Once in contact with cell membranes, several bacterial cell wall surface proteins, such as M-like proteins (SeM, SzPSe), may act as adhesins and attach to receptors expressed on the membranes of these cells. The characteristics of these receptors are unknown. It has not been determined if the bacterium needs to first colonize the mucosal surface before spreading into subjacent local lymphoid tissues. Additionally, it has not been determined how the bacterium crosses the mucosal barrier of the tonsil. Mucosal macrophages could phagocytose the bacteria in the mucus layer and carry it via leukocyte trafficking through the mucosal barrier into the local lymphoid tissues, or once bound to host cell receptors, it could be transported through the cell via transcytosis and released via exocytosis from the basal membranes of the cells into local lymphoid tissues of the tonsil. Dendritic cells could also be involved in the spread of bacteria across the mucosal barrier and into local and regional lymphoid tissues and lymph nodes. In local lymphoid tissues of the tonsil, the bacterium is able to replicate extracellularly and then spread to regional nodes such as the mandibular or retropharyngeal. Although unknown, the bacterium could spread by means of lymphatic vessels to these regional nodes in macrophages via leukocyte trafficking or through cell-free migration. The bacterium multiplies extracellularly in the lymph tissues and nodes, and the exudate contains large numbers of viable bacteria.
Mechanistically, several bacterial virulence determinates contribute to the character of this exudate (suppurative) and the large number of viable bacteria. Bacterial virulence determinates that act as chemoattractants for neutrophils and that disrupt phagocytosis and killing by neutrophils appear to explain the abundance of exudate and viable bacteria, respectively. Early in the sequence of events when bacteria encounter mucosal and tissue macrophages in local and regional lymphoid tissues and nodes, a bacterial cell wall protein, SeeH, interacts with these cells, resulting in the release of proinflammatory cytokines, increased vascular permeability, and edema. The occurrence of acute suppurative inflammation is facilitated by several virulence determinates. When peptidoglycan of the bacterial cell wall interacts with C3 of complement in the edema fluid via the alternative complement pathway, it produces complement-derived chemotactic factors that attract large numbers of neutrophils from capillary beds into local vascularized connective tissues. Furthermore, bacterial streptokinase interacts with plasminogen in the edema fluid to form active plasmin, which hydrolyzes fibrin. This process appears to increase the spread and dispersion of bacteria in tissue. Normally, fibrin confines bacteria by isolating them within its polymerized fibrillar meshwork so they can be phagocytosed and killed by neutrophils and macrophages, but when fibrin is hydrolyzed, large numbers of bacteria can accumulate in the exudate of an abscess and be readily available for release into the environment (see later). The outcome of these processes also contributes to the initiation of the cellular (leukocytic) phase of acute inflammation.
The surface of Streptococcus equi ssp. equi is coated with numerous protein virulence determinates, such as hyaluronic acid, SeM, and Se18.9 proteins that disrupt phagocytosis and killing. The bacterium also secretes leukocidal toxin and streptolysin S, a cell membrane pore-forming toxin, which kills leukocytes and disrupts phagocytosis. These processes lead to the accumulation of large numbers of viable bacteria in the exudate of lymphoid tissues and nodes and in abscess formation. Additionally, hyaluronic acid in the capsule appears to block interactions between bacteria and neutrophils by increasing the negative charge and hydrophobicity of the bacterial surface and by producing a localized oxygen-reduced environment that protects the activity of oxygen-labile proteases and toxins such as streptolysin S. It is likely but undetermined that bastard strangles occurs because of spread of the bacterium to systemic lymph nodes and organ systems by leukocyte trafficking or cell-free migration via efferent lymphatic vessels and/or capillaries or postcapillary venules in lymph nodes to gain access to the systemic circulatory system.
Caseous Lymphadenitis (Corynebacterium pseudotuberculosis): The mechanism of injury in caseous lymphadenitis is cell death attributable to inflammation and its mediators and degradative enzymes affecting cells of the monocyte-macrophage system and cell populations in lymph nodes and other organ systems. Gross lesions include chronic active pyogranulomatous lymphadenitis (see Figs. 11-75 and 11-76) with enlarged firm lymph nodes that on a cut surface have discrete and coalescing areas of yellow-white caseous exudate infiltrating and compressing contiguous parenchyma and abundant connective tissue. In other organs, such as the lung, abscesses encapsulated by dense bands of fibrous connective tissue and containing yellow-white caseous exudate are common findings.
Sheep and goats encounter Corynebacterium pseudotuberculosis through penetrating wounds and potentially ingestion. The bacterium is a common contaminate of the environment usually from animals that have the cutaneous form of caseous lymphadenitis leading to fistulous tracts from draining cutaneous lymph nodes. Penetrating wounds most commonly involve the skin and mucous membranes of the oral cavity. Management practices, such as shearing, may cause skin abrasions, whereas objects like wire, sticks, and protruding barn or fence nails may puncture the skin. Similar types of injury may occur in the oral cavity by similar objects and mechanisms. Once the bacterium reaches the submucosa or subcutis, it is phagocytosed by neutrophils and macrophages and spread via leukocyte trafficking to regional lymph nodes via lymphatic vessels. The bacterium replicates in the lymph nodes and the inflammatory response results in multiple pyogranulomas (abscesses) that grow in size and with time, notably enlarge and affect the entire lymph node. Macrophages infected with bacteria leave the node via leukocyte trafficking and spread via lymphatic vessels and probably the thoracic duct to the systemic circulation system or spread by entering the capillary or venous circulation within the node to gain access to the systemic circulatory system. Via leukocyte trafficking, macrophages then spread the bacterium to other visceral lymph nodes, especially the mediastinal and bronchial, and to tissues in a wide variety of organ systems, especially the lung. Because the bacterium is able to replicate in large numbers in macrophages and neutrophils, the death of these cells attributable to mycolic acid or cell aging results in the release of bacteria into vascularized ECM tissues. This process activates integrins and adhesins in vascular endothelium and causes massive recruitment of additional neutrophils and macrophages into the tissues as part of a chronic active inflammatory response, thus repeating the inflammatory process of forming pyogranulomas (abscesses). The mechanisms used by Corynebacterium pseudotuberculosis to gain access to lymph nodes via ingestion (if it occurs) are unknown. Two potential pathways could be used and both focus on macrophages and leukocyte trafficking. First, bacteria could interact with the mucus layer and mucosae of the oral pharynx, be phagocytized by macrophages and carried to the tonsils, then to regional lymph nodes, and then systemically. Second, bacteria could be swallowed and via alimentary peristalsis encounter M cells of small intestinal crypts, spread via M cells to macrophages in contiguous Peyer’s patches, then to regional lymph nodes, and then systemically. It is likely that the mechanisms and responses to injury described for the penetrating wounds portal of entry would also apply to these two scenarios.
Corynebacterium pseudotuberculosis has two known bacterial virulence determinates, phospholipase D and mycolic acid, that allow it to colonize tissues and produce pyogranulomas. Phospholipase D increases vascular permeability, which is thought to assist macrophages in migrating in and out of tissues infected with the bacterium, thus favoring the systemic spread of the bacterium. As a potent exotoxin, it also injures cell membranes leading to macrophage and neutrophil dysfunction, disruption, and death and interferes with neutrophil chemotaxis. Corynebacterium pseudotuberculosis does not have a protective capsule but has a waxy mycolic acid coat on the cell wall surface. Mycolic acid induces acute inflammation, has a role in the formation of granulomas, is toxic for macrophages, and prevents killing of the bacterium with phagosome-lysosome fusion likely by protecting against hydrolytic enzymes present within lysosomes.
Brucellosis (Brucella spp.): The mechanism of injury in brucellosis is cell death caused by inflammation and its mediators and degradative enzymes. Brucella spp. do not have virulence determinates for exotoxins or endotoxins that cause direct injury to cells. Gross lesions include chronic active pyogranulomatous lymphadenitis with enlarged firm lymph nodes that on a cut surface have discrete and coalescing areas of yellow-white exudate infiltrating and compressing contiguous parenchyma.
Animals encounter Brucella spp. through inhalation or ingestion of bacteria in fomites contaminated with infected exudates from other body systems such as the female reproductive tract. The bacterium encounters mucosae and their mucus layers through centrifugal turbulence and entrapment in the mucus layer of the nasal pharynx and through mastication, gravity, and entrapment in the mucus layer of the oral pharynx. Bacteria are phagocytosed by macrophages or dendritic cells migrating through, on, or in the mucosae and spread to local lymphoid tissues via leukocyte trafficking by lymphatic vessels, to regional lymph nodes via lymphatic vessels, and then systemically to superficial and visceral lymph nodes and other organs such as spleen, liver, bone marrow, mammary glands, and reproductive organs. Brucella spp. can also cross the mucosal barrier via endocytosis, exit the basal surface of mucosal epithelial cells through exocytosis, and spread cell free in lymphatic vessels to local and regional lymph nodules and nodes where they are phagocytosed by macrophages and then spread systemically as discussed above. Through ingestion and peristalsis, Brucella spp. can also reach the alimentary system where they encounter M cells. Bacteria infect M cells via endocytosis, undergo transcytosis, and exit the basal surfaces via exocytosis to gain access to macrophages within Peyer’s patches. Macrophages within Peyer’s patches are then infected with the bacteria and used to spread bacteria systemically. The goals of these three types of mucosal encounters are to provide Brucella spp. with ample opportunity to infect macrophages and subsequently gain access to local, regional, and systemic lymph nodules and nodes.
In lymph nodes, bacteria-infected macrophages are killed by the bacterium or die through aging and the bacteria are released into vascularized ECM. These bacteria elicit an acute inflammatory response that is quickly replaced by a pyogranulomatous inflammation because of LPS in the bacterial cell wall. The material is not readily degradable, and macrophages (as monocytes) are recruited from the systemic circulation to phagocytose and degrade such material and kill the bacteria. Brucella spp. are able to evade the killing mechanisms when phagocytosed by neutrophils and macrophages. Additionally, they are able to grow and replicate in macrophages and dendritic cells. Once Brucella spp. encounter cell membranes of macrophages, they use ligand-receptor interactions to attach to and enter cells; however, the details remain unclear. Outer membrane protein of the bacterial cell wall and class A scavenger receptors on host target cells are likely involved but not necessarily with each other. TLRs are also likely involved in attachment and entry into macrophages. Entry occurs via endocytosis through a phagosome, but phagosome-lysosome fusion does not occur because bacteria are able to block fusion through rapid acidification of the phagosome. LPS (a PAMP]), a type IV secretion system, and a long list of other putative virulence determinates such as cyclic β-1,2-glucan and heat shock proteins may also be involved in blocking phagosome-lysosome fusion and promoting bacterial growth and replication. The virulence of Brucella spp. strains appears related to the LPS composition of its capsule, with the encapsulated smooth phenotypes generally being more virulent. Additionally, the presence of a smooth capsule enhances bacterial growth and replication in phagosomes.
When Brucella spp. spread via leukocyte trafficking systemically in macrophages, they are able to gain access to tissues in the male and female reproductive systems and the mammary gland (Fig. 4-26; see also Fig. 19-18). In summary, infected macrophages interact with and infect placental trophoblasts in placentomas and epithelial cells of other reproductive tissues. Once such cells are infected, the bacteria likely infect fetal macrophage-like cells that serve to spread the bacteria through the fetus and to other lymphoid tissues in reproductive organs (see Fig. 4-26, C and D). Brucella spp. also survive in macrophages of these tissues by inhibiting the phagosome-lysosome fusion. Bacterial growth and replication with concurrent death of bacteria-infected macrophages results in pyogranulomatous inflammation of these tissues and organ systems.
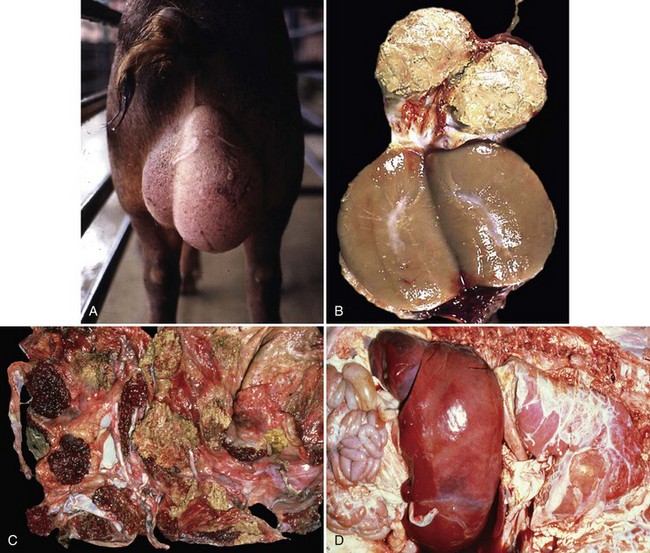
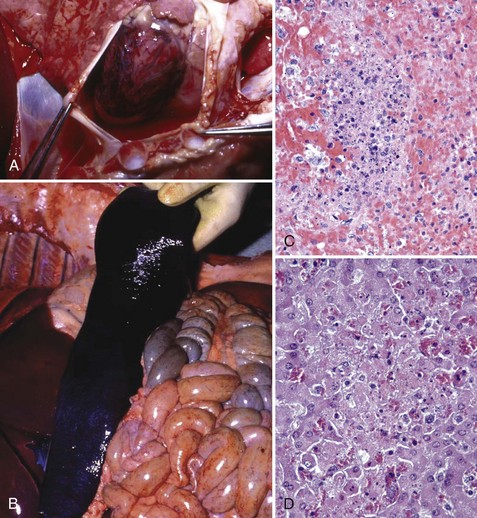
Fig. 4-26 Brucellosis.
Brucellosis is a disease in which the bacterium initially targets lymphocytes and macrophages in mucosae-associated lymphoid tissues, regional lymph nodes, systemic lymph nodes, and the spleen to replicate and grow in number. It uses macrophages to spread to and through these tissues and then systemically to infect cells and tissues in the placenta, sex organs of males and females, and fetuses. Therefore brucellosis is initially and long term characterized by a chronic active pyogranulomatous lymphadenitis with sequelae that affect the reproductive systems. A, Boar, swollen testis. The testis in enlarged due to chronic active pyogranulomatous inflammation. Brucella spp. spread in macrophages via leukocyte trafficking from lymphoid tissues systemically to the testis. B, The epididymis can be filled with pyogranulomatous exudate, which obstructs the flow of spermatozoa and causes infertility. Infected animals can also serve as carriers and spread the bacterium via sexual contact (also see Fig. 19-18). C, Fetal cotyledons. Note the roughened granular yellow-brown surface of cotyledons (arrows) infected with the bacterium. This lesion is caused by pyogranulomatous inflammation leading to severe necrosis of the affected cotyledons. Normal cotyledons are dark red and have a smooth shiny surface. D, Fetal brucellosis, hepatomegaly and fibrinous polyserositis. The bacterium is thought to be spread from infected cotyledons to fetal organs via leukocytic trafficking in fetal macrophage-like cells. The bacterium causes extensive injury of the vascular system and organs through inflammatory responses induced in the fetus. (A courtesy Dr. C. Wallace, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. K. McEntee, Reproductive Pathology Collection, University of Illinois; and Dr. J. King, College of Veterinary Medicine, Cornell University. C and D courtesy Dr. K. McEntee, Reproductive Pathology Collection, University of Illinois.)
Rhodococcal Mesenteric Lymphadenitis (Rhodococcus equi): The pathogenesis of rhodococcal mesenteric lymphadenitis begins as an infection of the respiratory system (see section on the Respiratory System, Mediastinum, and Pleurae), followed by infection of the alimentary system (see section on the Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity). The mechanism of injury in rhodococcal mesenteric lymphadenitis is death of cells of the monocyte-macrophage system and of all cell populations in the lymph node secondary to inflammation and its mediators and degradative enzymes. Gross lesions include chronic active pyogranulomatous lymphadenitis (see Fig. 7-137) with enlarged firm lymph nodes that on a cut surface have discrete and coalescing areas of yellow-white exudate infiltrating and compressing contiguous parenchyma. Rhodococcus equi enters the alimentary system through M cells and is released into Peyer’s patches where it is phagocytosed by tissue macrophages. Bacteria-infected tissue macrophages spread via leukocyte trafficking in lymphatic vessels within the intestinal mesentery to mesenteric lymph nodes, leading to pyogranulomatous lymphadenitis, and then systemically via the thoracic duct and the blood vascular system to additional lymph nodes and lymphoid tissues such as the spleen. The pathogenesis of pyogranulomatous lymphadenitis appears to progress much like that which occurs in the lung.
Nervous System
Edema Disease (Escherichia coli): The pathogenesis of edema disease begins as an enterotoxemia of the alimentary system (see section on the Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity). This disease is caused by a specific strain of hemolytic E. coli having virulence determinates for a bacterial toxin called Shiga toxin 2e (also known as verotoxin 2e). It was initially called edema disease principle. The mechanism of injury is death of endothelial and smooth muscle cells of arterioles (fibrinoid arteriopathy/arteriolopathy), thus the toxin biologically behaves as an angiotoxin. In the brain, vascular lesions are followed by secondary ischemia and necrosis of neural cells, particularly neurons in brainstem nuclei. Gross lesions include symmetric areas of yellow-gray malacia in the brainstem overlying affected nuclei (see Web Fig. 10-24). Pigs encounter E. coli through ingestion and colonization of the intestinal mucosae. The enterotoxemia phase is discussed in the section on the Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity. Shiga toxin 2e is absorbed in the alimentary system and circulates systemically in the blood vascular system. Endothelial and smooth muscle cells of arteritis and arterioles express receptors for this toxin. This toxin causes vascular permeability changes and edema followed by endothelial injury and death leading to hemorrhage, intravascular coagulation, microthrombosis, and infarction (grossly malacia). Shiga toxin 2e acts to disrupt protein synthesis in affected cells leading to cell death.
Focal Symmetric Encephalomalacia (Clostridium perfringens): The pathogenesis of focal symmetric encephalomalacia begins as an enterotoxemia caused by Clostridium perfringens. Because ε-toxin is a permease that alters cell permeability, the vascular beds in affected intestinal tissues readily absorb toxins into the circulatory system. It appears that the sequence of events leading to focal symmetric encephalomalacia occurs in the first phase or early in the second phase of enterotoxemia before toxin-induced massive necrosis of the intestine occurs. The mechanism of injury is cell death caused by bacterial toxins that act directly on endothelial cell membranes causing permeability changes leading to acute coagulative necrosis of affected cells, including endothelial cells and neurons. Gross lesions include bilaterally symmetric malacia (acute coagulative necrosis of neuron cell bodies) and liquefactive necrosis of the basal ganglia, internal capsule, thalamus, and substantia nigra with edema and hemorrhage. Cerebral edema causes indistinct sulci and flattened gyri and in severe cases, coning of the cerebellar vermis through the foramen magnum. Because ε-toxin is a permease that alters cell permeability of the microvascular system, in the nervous system, it behaves as an angiotoxin. The vascular beds in affected intestinal tissues with enterotoxemia readily absorb toxins into the circulatory system. They are then carried to the brain via the blood vascular system where they act to increase the permeability of capillary beds leading to the release of blood plasma containing toxins into the neuropil, resulting in severe generalized vasogenic cerebral edema. Circulating ε-toxin accumulates preferentially in the brain via ligand-receptor interactions. It is likely that receptors expressed on different populations of endothelial cells in the body and within the brain determine in part the specificity for certain neurons and nuclear groups within the nervous system. The cell membrane of cerebral endothelial cells is the probable site of toxin binding, and it appears that toxin-induced injury of endothelial cells leads to the expression of more receptors for circulating ε-toxin. Injury to the endothelium disrupts the integrity of the blood-brain barrier, leading to increased vascular permeability, vasogenic edema, and the diffusion of toxin into the neuropil where it encounters neuron cell bodies. Acute coagulative necrosis of neuron cell bodies has been attributed to toxin-induced microthrombosis of capillaries, resulting in neuronal ischemia and by direct cytotoxic action on neurons and other neural cells. The selective nature of neuronal death caused by ε-toxin may be explained by ligand-receptor interactions, selective metabolic vulnerability of specific populations of neurons, or the concentration of ε-toxin.
Botulism and Tetanus (Clostridium botulinum, Clostridium tetani): The mechanism of injury in botulism is disruption of neurotransmitter vesicle exocytosis at myoneural (flaccid paralysis) junctions (i.e., synapses) by botulinum (Clostridium botulinum) neurotoxin. The mechanism of injury in tetanus is disruption of neurotransmitter vesicle exocytosis at neural-neural (spastic paralysis) junctions (i.e., synapses) by tetanus (Clostridium tetani) neurotoxin (tetanospasmin). These neurotoxins are produced in anaerobic microenvironments (a lowered oxidation-reduction [redox] potential) such as in necrotic tissue occurring in traumatic wounds (e.g., nail penetrating sole of the hoof, gastric ulcers in foals, muscle necrosis). Gross or microscopic lesions are not observed in the nervous system with these diseases.
Animals encounter these bacteria through contact with bacterial endospores present in soil and on environmental objects. Spores are carried into wounds and germinate to vegetative forms, and when this latter form dies, neurotoxins are released into dead tissue. Examples of such wounds include penetration of the skin or sole of the hoof or gastric ulcers. In addition, botulinum neurotoxin can also be released from lysed vegetative forms in the anaerobic environment of decaying vegetable matter (e.g., spoiled silage, hay, grain) and decomposing carcasses and absorbed into the circulatory system from the alimentary system with their ingestion. From a wound (or the alimentary system), neurotoxins access myoneural (botulinum neurotoxin) and neural-neural (tetanus neurotoxin) junctions by two routes, either hematogenously (botulinum neurotoxin) or via retrograde axonal transport (tetanus neurotoxin).
Botulinum neurotoxin enters the blood from (1) wounds as it diffuses via a concentration gradient to the periphery of the wound to areas with adequate circulation in which it is absorbed into the blood via capillaries and (2) absorption through intestinal villi and transfer to capillary beds within the lamina propria of the villi. Botulinum neurotoxin gains access to myoneural junctions via capillary beds that supply muscular tissues. On its release from capillaries, the neurotoxin diffuses in interstitial fluids until contacting the cell membrane of peripheral nerves (e.g., lower motor neuron), where it enters the cytoplasm of the neuron through the formation of endocytotic vesicles.
In contrast, tetanus neurotoxin (tetanospasmin) enters the nervous system and gains access to neural-neural junctions by initially entering the cytoplasm of distal processes of neurons through the formation of endocytotic vesicles in viable nerve endings located in tissue surrounding the site of the wound. The endocytotic vesicles are transported into the central nervous system (CNS) by retrograde axonal transport, and tetanus neurotoxin is released into the interstitial fluid of the neural-neural junctions by exocytosis. Free tetanus neurotoxin binds to the cell membrane of inhibitory interneurons of the spinal cord, is internalized via endocytosis, and acts to disrupt the release of inhibitory neurotransmitters via the same mechanism as used by botulinum toxin: disruption of the synaptic fusion complex. Presynaptic neurons (upper motor neurons) excite postsynaptic neurons (lower motor neurons) on a nearly continuous basis. Inhibitory interneurons acting on lower motor neurons serve to counterbalance and smooth the excitatory effects of acetylcholine released from presynaptic neurons (upper motor neurons) to excite the same lower motor neurons. Thus skeletal muscle groups (opposing flexor and extensor muscles) are given time to relax; as a result, skeletal muscle contractions initiated by lower motor neuron are well regulated and coordinated. Failure to have adequate inhibitory interneuron regulation of lower motor neurons leads to the spastic paralysis observed in tetanus.
Although botulinum and tetanus toxins gain access to targets in the nervous system by different mechanisms, from this point forward in the pathogenesis, both botulinum and tetanus neurotoxins share a common mechanism of injury, the disruption of neurotransmitter vesicle exocytosis by disrupting the synaptic fusion complex. The mechanisms are demonstrated in Figs. 4-27 and 4-28 for botulinum toxin and tetanus toxin, respectively. Thus diseases (clinical signs) that occur are the direct result of disruption of the function of myoneural (flaccid paralysis) and neural-neural (spastic paralysis) junctions. Botulinum and tetanus toxins have heavy and light chains and behave as typical A-B toxins (i.e., diphtheria, cholera, pertussis, and shigellae toxins) composed of two units: a binding B-domain (heavy chain) that mediates transport via endocytosis and exocytosis and an enzymatically active A-domain (light chain) that serves to cleave proteins within the target cell. The heavy chain binds to the neuronal membrane of myoneural junctions (botulinum toxin) and nerve endings (tetanus toxin), and the entire toxin molecule enters the neuron via receptor-mediated endocytosis. The A-domain is cleaved from the B-domain within the target cell endocytotic vesicle and then released into the cytoplasm where it is active. The A-domain (light chain), a zinc-containing endopeptidase, leaves the endocytotic vesicle and enters the cytoplasm of the neuron and acts to cleave proteins that form the synaptic fusion complex. This complex, formed by fusion of synaptic vesicle proteins with presynaptic plasma membrane proteins, serves to bring neurotransmitter vesicles into contact with the neuronal cell membrane at the myoneural (botulinum toxin) and neural-neural (tetanus toxin) junctions and facilitates membrane fusion and release of excitatory (acetylcholine) and inhibitory neurotransmitters (glycine and γ-aminobutyric acid [GABA]), respectively.
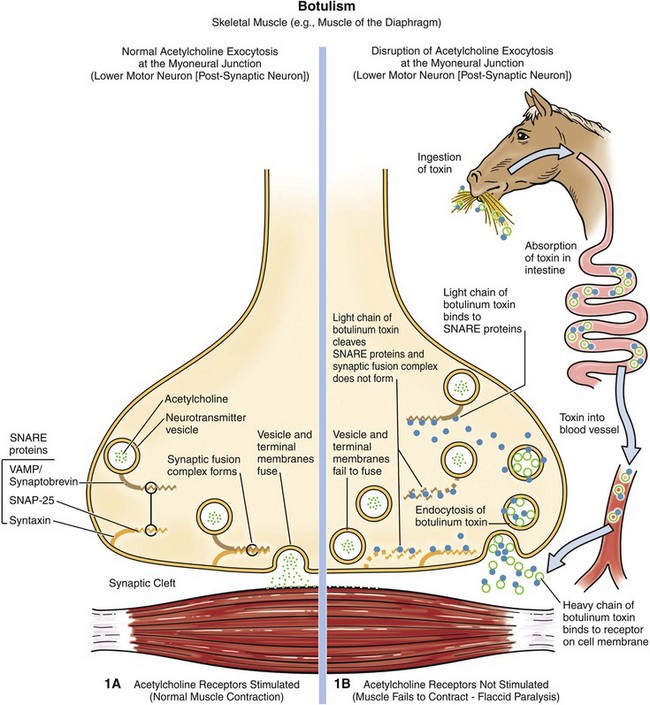
Fig. 4-27 Mechanism of myoneural junction dysfunction in botulism.
Note that botulinum toxin reaches the myoneural junction via the circulatory system. (Courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
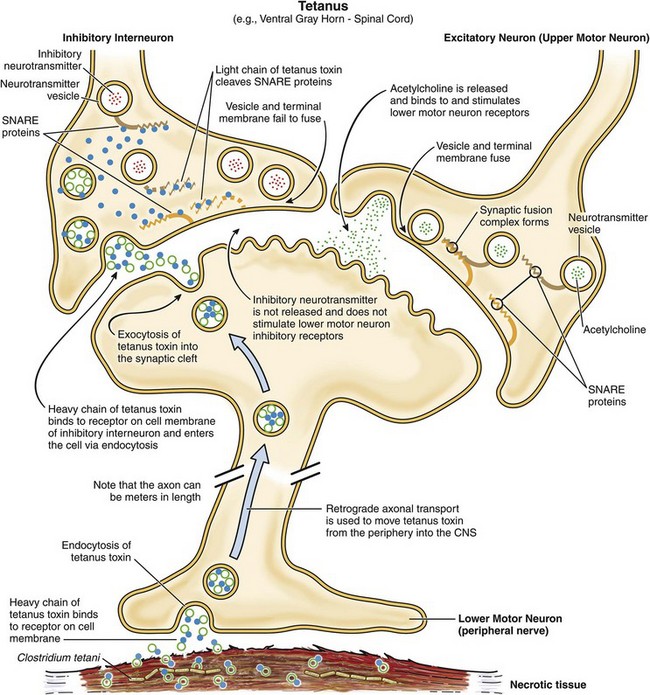
Fig. 4-28 Mechanism of neural-neural junction dysfunction in tetanus.
Note that tetanus toxin reaches the neural-neural junction via retrograde axonal transport. The selectivity of tetanus toxin for inhibitory interneurons is likely mediated by the expression of different glycosylphosphatidylinositol-anchored protein(s) on different types of neurons. The B-domain of tetanus toxin appears to bind only to the type of glycosylphosphatidylinositol-anchored protein expressed on inhibitory interneurons. (Courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
Different types of glycosylphosphatidylinositol-anchored protein(s) may be expressed on different types of neurons, which may explain why B-domain of tetanus toxin only binds to inhibitory interneurons and not other types motor neurons. Disruption of the synaptic fusion complex prevents the neurotransmitter vesicles from fusing with the membrane, which in turn prevents release of neurotransmitters into the synaptic cleft. Proteins that form the synaptic fusion complex (SNARE proteins) include neurotransmitter vesicle proteins (such as vesicle-associated membrane proteins [VAMPs]/synaptobrevin) and presynaptic plasma membrane proteins (syntaxin, synaptosomal-associated protein [SNAP-25]). Different types of Clostridium botulinum produce different types of toxins (toxin type A to G), and these types target and cleave specific types of SNARE proteins, synaptobrevin (cleaved by toxin types B, D, F, and G), syntaxin (cleaved by toxin type C), and synaptosomal-associated protein (cleaved by toxin types A, C, and E). Botulinum toxin seemingly does not cross the blood-brain barrier; therefore neural-neural junction functions in the CNS remain intact. Notwithstanding the profound neurologic signs of spastic paralysis and flaccid paralysis that occur with tetanus (Clostridium tetani) and botulism (Clostridium botulinum), respectively, macroscopic or microscopic lesions are not observed in the nervous system.
Listeriosis (Listeria monocytogenes): The mechanism of injury in listeriosis is cell death that is uniquely localized to the brainstem caused by acute inflammation and its mediators and degradative enzymes. Gross lesions are often not observed, but when present consist of nodules and linear bands of gray-yellow exudate (perivascular microabscesses formed by neutrophils) mixed with active hyperemia and/or hemorrhage commonly arranged in a perivascular pattern (see Fig. 14-88).
Cattle, sheep, and goats encounter Listeria monocytogenes in soil, animal feed, water, and feces; however, the greatest risk of contracting the disease occurs when ruminants are fed improperly stored silage in which the pH is not acidic enough to prevent overgrowth of the bacterium. Consumption of Listeria monocytogenes–contaminated silage is not sufficient to cause CNS disease, unless it occurs with a penetrating injury of the oral cavity caused by a stick or other sharp object (nail) that carries the bacterium in the silage into the submucosal connective tissue of the oral cavity or tongue. At this point, the bacterium colonizes oral tissues, enters nerve endings in the oral cavity, and ascends into the CNS via retrograde axonal transport in cranial nerves. The oral cavity is primarily innervated by the trigeminal and other cranial nerves that terminate in the brainstem. Thus Listeria monocytogenes ultimately localizes to the brainstem (i.e., pons, medulla oblongata, and proximal cervical spinal cord). The mechanism of entry into nerve endings is unknown; however, it has been shown experimentally in cell culture systems that Listeria. monocytogenes gains entry into typically nonphagocytic cells through endocytosis and endocytic vesicles. Bacterial internalization, the entry process, is mediated by internalins (type A and B) that utilize host cell receptor E-cadherin, a transmembrane glycoprotein. Because Listeria monocytogenes resides intracellularly within cell bodies of neurons when it arrives in the brainstem, it initially does not disrupt the blood-brain barrier and thus does not activate defense mechanisms provided by the innate (inflammation) and adaptive immune responses. The cytoplasm of infected neuron cell bodies appears to be permissive and allows free proliferation of the bacterium. This permissive environment also appears to be promoted by a bacterial virulence determinate called listeriolysin O that inhibits immune responses and allows infected cells to hide from defense mechanisms.
Once free in the cytoplasm, the bacterium replicates to sufficient numbers and then begins the process of infecting other cells. In the cytoplasm, the bacterium has a doubling time of approximately 1 hour. When the number of bacterium in the cytoplasm reaches a level sufficient to facilitate infection of adjacent cells, bacteria move themselves, facilitated by a virulence determinate in the cytoplasm, to the inner side of the cell membrane via polymerization and depolymerization of host cell actin filaments. Once near the cell membrane, aggregates of bacteria use a bacterial surface protein called surface protein actA to propel themselves via actin polymerization (listerial actin-based motility) and a pseudopod into cell membranes of adjacent cells, forming invaginations of the membrane that ultimately result in double-membrane endocytotic phagocytic vesicles (Fig. 4-29). This process is random, so it does not appear to target specific cells in the nervous system only neighboring cells. These double-membrane endocytotic phagocytic vesicles are lysed by listeriolysin O, phospholipase C, and lecithinase, and the bacteria are released into the cytoplasm of newly infected cells. Experimentally, Listeria monocytogenes has been shown to infect neutrophils, macrophages, fibroblasts, endothelial cells, and various types of nerve cells, including neurons and microglial cells. It appears that infection of and injury to endothelial cells of capillaries initiates the inflammatory process. Once initiated, the blood-brain barrier is disrupted and activation of the entire inflammatory cascade ensues. Neutrophils are the primary effector cells utilized by animal defense mechanisms to kill the bacterium. Experimentally, Listeria monocytogenes infected endothelial cells express exuberant endothelial adhesion molecules (P- and E-selectin, intercellular adhesion molecule-1 [ICAM-1], and vascular cell-adhesion molecule-1 [VCAM-1]) resulting in activation of the neutrophil adhesion cascade and neutrophil binding, both components of the acute inflammation. The bacterium can also spread from macrophages to endothelial cells.
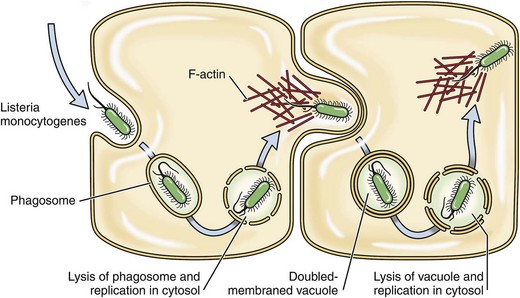
Fig. 4-29 Mechanism of infection in listeriosis.
Listeria monocytogenes propels itself via actin polymerization (Listeria actin-based motility) within a membrane pseudopod into cell membranes of adjacent neural cells forming invaginations of the membrane that ultimately result in double-membrane endocytotic phagocytic vesicles.
Thrombotic Meningoencephalitis (Histophilus somni): The pathogenesis of thrombotic meningoencephalitis (TME) begins as pulmonary histophilosis (see the section on Respiratory System, Mediastinum, and Pleurae). The mechanism of injury in the nervous system is infarction secondary to occlusive ischemia by bacterial-induced arteritis (vasculitis) and subsequent thrombosis caused by acute inflammation and its mediators and degradative enzymes. Gross lesions are red hemorrhagic infarcts of varied sizes distributed at random throughout nervous tissue, especially in the cerebral cortices (see Fig. 14-89).
Cattle encounter Histophilus somni (formerly Haemophilus somnus) via inhalation of fomites or water droplets contaminated with the bacterium. It likely exists in nasal or oral biofilms as a commensal bacterium of mucosae. Environmental stressors, such as overcrowding, combined with other factors, such as poor ventilation and humidity or abrupt changes in ambient air temperature, could alter the commensal relationship, allowing the bacteria to replicate in sufficient numbers to colonize mucosae and spread the bacterium to other animals. After colonization of the respiratory mucosae, the bacterium spreads to the lung (see the section on Pulmonary Histophilosis, Respiratory System, Mediastinum, and Pleurae) and then gains access to the vascular system in areas of inflammation, embolizes to the CNS (septicemia), and colonizes and infects small arterioles likely via ligand-receptor interactions. The first encounter with endothelial cells of arterioles occurs at anatomic sites in the brain in which there are abrupt changes in the laminar flow of blood resulting in turbulence, such as occurs at the interface between gray and white matter in the cerebral cortex. Turbulence favors perturbation of the endothelium and contact of the bacterium with platelets and the endothelium. Experimentally, Histophilus somni and its membrane LOS (a truncated form of LPS) have been shown to activate bovine platelets and increase the expression of adhesion molecules, such as ICAM-1 and E-selectin and tissue factor (factor III) on endothelial cells. Tissue factor is a protein necessary for activation of blood coagulation cascades. Thus, because strains of Histophilus somni have virulence determinates that enhance the adherence of the bacterium to endothelial cells, such areas are prone to endothelial injury, exposure of collagen, platelet aggregation and activation, activation of clotting cascades, arterial thrombosis and obstruction, and infarction (the lesion of TME).
Meningitis (Escherichia coli and Other Bacterial Species): The pathogenesis of meningitis shares many of the mechanisms discussed for porcine polyserositis (see the section on the Respiratory System, Mediastinum, and Pleurae) and embolic vasculopathy/vasculitis (see the section on the Cardiovascular System and Lymphatic Vessels).
Muscle
Blackleg (Clostridium chauvoei): The mechanism of injury in blackleg is necrosis (acute gangrenous myositis) of muscle, connective, and nervous tissues caused by α- and β-toxins released from vegetative forms of Clostridium chauvoei. Gross lesions occur in large striated muscle groups and include dark red to black muscle that appears dry and contains gas bubbles (see Fig. 15-38). Affected muscle may have a rancid (spoiled butter) smell. Cattle, sheep, and goats encounter spores through ingestion of plant matter and topsoil contaminated with spores often after disturbances (excavations) of the soil and pasture bed. Spores are carried by swallowing and peristalsis through the oral pharynx, esophagus, abomasum, and rumen to their final destination, the small intestine. It appears that spores can remain dormant in the small intestine or germinate into vegetative forms and become normal inhabitants of the alimentary system. How spores interact with mucosae and gain access to epithelial cells and mucosal macrophages is unknown. Other similar pathogens use M cells to enter Peyer’s patches, so it is plausible that spores may gain access to and infect macrophages in Peyer’s patches via this mechanism. Although suggested but unproved, spores are likely the form of the bacterium that spreads systemically to muscle. However, it is possible that vegetative forms of the bacterium spread to muscle and produce spores in dendritic cells and macrophages of muscle after their phagocytosis.
Spread to muscle could also occur via leukocyte trafficking followed by interaction with and infection of endothelial cells and then dendritic cells and macrophages of muscle. If it occurs, tropism for dendritic cells and macrophages of muscle is probably mediated by ligand-receptor interactions. Blackleg often follows some form of traumatic injury to muscle. It is thought that injury creates a microenvironment suitable with lowering of the oxidation-reduction (redox) potential (anaerobic environment) required for germination of spores. Such a hypothesis also assumes that the injury has damaged the dendritic cells and macrophages of muscle, allowing them access to this microenvironment. Spores germinate into vegetative forms of the bacterium and produce large quantities of a variety of α- and β-toxins such as oxygen-stable hemolysin, deoxyribonuclease (DNase), hyaluronidase, oxygen-labile hemolysin, and neuraminidase. These toxins diffuse out from the site of bacterial replication and coagulate muscle tissue and its vascular supply, resulting in acute gangrenous myositis.
Malignant Edema (Clostridium septicum): The pathogenesis of malignant edema is similar to that of blackleg (see previous section) regarding bacterial replication, the production of toxins, tissue injury, and gross lesions affecting striated muscle and blood vessels. However, the mechanism of spread to muscle is different. Cattle, sheep, and goats encounter spores through wounds caused by penetrating objects, such as a wire, which carries spores into the wound. Wounds caused by castration, tail docking, unsanitary vaccination, and other management practices can also be infected with spores. The injury must be sufficient to create an anaerobic microenvironment in the wound with lowering of the oxidation-reduction (redox) potential suitable for germination of spores. Once spores germinate, vegetative forms release toxins that injure and coagulate muscles and vascular tissues resulting in necrosis and edema much like in blackleg.
Big Head and Black Disease (Clostridium novyi): The pathogeneses of big head and black disease are very similar to malignant edema and blackleg, respectively. In big head of sheep, penetrating wounds of the skin of the head caused by horns during fighting establish the initial anaerobic microenvironment for spores to germinate. The resulting pathogenesis is similar to that which occurs in malignant edema. Black disease of cattle and sheep occurs because of fluke migration (Fasciola hepatica) through the liver (see Figs. 8-55 and 8-56) that causes hepatocellular necrosis and establishes an anaerobic microenvironment suitable for germination of spores. Kupffer cells likely contain dormant spores. As in blackleg, spores are likely ingested, enter through the mucosae of the small intestine, become phagocytosed by cells of the monocyte-macrophage system, and spread via leukocyte trafficking to Kupffer cells of the liver.
Bone, Joints, Tendons, and Ligaments
Lumpy Jaw (Actinomyces bovis): The mechanism of injury in lumpy jaw is cell death attributable to pyogranulomatous inflammation and its mediators. Gross lesions include enlarged and misshapen bones of the mandible and/or maxilla attributable to abscesses, fibrosis, and fistulous tracts in the bone with secondary remodeling of the bone (i.e., pyogranulomatous alveolar osteomyelitis). The cut surface has numerous randomly distributed discrete and coalescing yellow-white granulomas surrounded by remodeled bone intermixed with bands of fibrous connective tissue (see Fig. 16-58). Actinomyces bovis is a commensal bacterium of mucosae of the oral cavity of cattle and sheep, likely existing in a biofilm. The bacterium can infect bone by several routes: (1) genetic or developmental defects of the tooth root and/or socket that provide access to bone, (2) injury to a tooth and its socket opening a pathway into the bone, and (3) penetrating wounds that give the bacterium access to the bone and its periosteum. During chewing, the bacterium is carried by direct extension through the mucosae into submucosal connective tissues via penetrating wounds such as those caused by sharp foreign bodies like sticks or wires. The object may penetrate the periosteum and the bone, giving the bacterium direct access to these tissues. The bacterium colonizes submucosal connective tissue, and LPS of the cell wall, in part, likely plays a role in the type of inflammatory response that occurs. Little is known about virulence determinates, ligand-receptor interactions, target cells, toxins, capsule antiphagocytic molecules, or other factors that may contribute to the pathogenicity of this bacterium. Actinomyces bovis can spread via lymphatic vessels to regional lymph nodes and cause a similar inflammatory response in these tissues.
Integumentary System
Greasy Pig Disease (Staphylococcus hyicus): The mechanism of injury in greasy pig disease is cell death and exfoliation of cells of the skin secondary to inflammation and its mediators and degradative enzymes. Gross lesions include areas of patchy red skin (active hyperemia of acute inflammation) followed by thickening of the reddened skin and the formation of reddish brown macules, vesicles, and pustules first around the eyes, nose, lips, and ears and then the flanks and abdomen (see Fig. 17-47). Affected skin, mostly through inflammation, exudes large quantities of a greasy exudate consisting of serum and sebum mixed with inflammatory cells, degradative enzymes, and cell debris. This exudate is the basis for naming the disease.
Pigs encounter Staphylococcus hyicus through fomites and body fluids contaminated with the bacterium. This bacterium is likely a commensal organism that resides in the skin and hair follicles of healthy pigs. Environmental stressors, such as skin trauma caused by overcrowding combined with other factors such as poor ventilation and humidity or abrupt changes in ambient air temperature, could alter the commensal relationship, allowing bacteria to replicate in sufficient numbers to colonize the skin, spread the bacterium to other animals, and cause disease. Infected droplets are deposited on the surface of the skin, but the bacterium under most conditions is not able to infect and colonize intact skin. It appears that skin trauma is usually a prerequisite for colonization because abrasions on the feet and legs or lacerations on the body precede the onset of the disease.
The role of virulence determinates, ligand-receptor interactions, and cells of the monocyte-macrophage system, such as Langerhans’ cells, are poorly understood in the pathogenesis of the disease. An exfoliative toxin that induces separation of epithelial cells of the stratum corneum and spinosum aids in the invasion of the bacterium into the skin. It serves to expose vascularized ECM tissues in traumatized skin. Fibronectin-binding proteins expressed on the surface of the bacteria appear to act as adhesins, allowing the bacteria to bind to the fibronectin present in collagen, fibrin, and heparin sulfate proteoglycans of traumatized skin. Fibronectin is a glycoprotein of vascularized ECM tissues and is produced by cells such as fibroblasts. Once the skin is colonized, the infection appears to spread to the hair follicles leading to suppurative inflammation and sebaceous gland hyperplasia and hypersecretion (i.e., greasy pig). It also appears that acute inflammation and its effector cells, such as neutrophils, play a central role in the onset and progression of the skin lesions. Capsule polysaccharides and protein A in the bacterial wall appear to block phagocytosis of the bacterium by neutrophils and increase the ability of bacteria to survive and replicate in vascularized ECM tissues of the skin.
Canine Pyoderma (Staphylococcus intermedius): The pathogenesis of canine pyoderma appears to be similar to that of greasy pig disease (see previous section). Skin trauma arising from pruritus and scratching or from existing skin disease leads to the exposure of vascularized ECM tissues and its colonization by bacteria. Although incompletely characterized, it is likely that a variety of virulence determinates are involved in canine pyoderma, including surface proteins (colonization of host tissues); invasins such as leukocidin, kinases, hyaluronidase (promote bacterial spread in tissues); surface factors such as capsule polysaccharides and protein A (inhibit phagocytosis); and exotoxins and exfoliative toxins such as hemolysins, leukotoxin, and leukocidin (cause cell death).
Diamond Skin Disease (Erysipelothrix rhusiopathiae): The mechanism of injury in diamond skin disease is cell death and infarction of skin secondary to cutaneous vasculitis. Gross lesions include active hyperemia and red-purple skin affecting the ears, ventral abdomen, and legs followed by thrombosis, ischemia, and infarction resulting in rhomboidal (diamond) red-purple areas of skin (cutaneous infarcts) (see Figs. 17-51 and 10-81).
Pigs encounter Erysipelothrix rhusiopathiae through ingestion of fomites and body fluids contaminated with the bacterium. This bacterium is likely a commensal organism that resides in a biofilm of the mucosae of the pharynx and tonsillar epithelia of healthy pigs. Environmental stressors, such as overcrowding combined with other factors such as poor ventilation and humidity or abrupt changes in ambient air temperature, can alter the commensal relationship, allowing the bacteria to replicate in sufficient numbers to colonize mucosae and spread the bacterium to other animals. Infected droplets are deposited on pharyngeal mucosae, where bacteria encounter the mucus layer and mucosal epithelial cells. It is unclear how this nonmotile bacterium is able to penetrate the mucus layer and gain direct access to the luminal membrane of epithelial cells. Additionally, it is unclear if and how the bacterium colonizes the mucus layer and mucosae. It appears that neuraminidase may be a virulence determinate for Erysipelothrix rhusiopathiae potentially involved in initial interactions with and invasion of the mucus layer of pharyngeal mucosae. It acts to remove sialic acid from glycoproteins, glycolipids, and oligosaccharides expressed on host cells, potentially exposing new receptors for the bacterium. It is also probably important later in the disease process in the attachment, colonization, and invasion of endothelial cells by the bacterium leading to vasculitis, thrombosis, infarction, and DIC.
Other virulence determinates involved in mucosal colonization and systemic spread of the bacterium include capsular polysaccharides (antiphagocytic properties), surface proteins (adhesins, antiphagocytic properties, biofilm formation), invasins such as hyaluronidase (invade ECM tissues), and enzymes such as superoxide dismutase and catalase (block the effects of the respiratory burst of phagocytosis and oxygen free radicals). Transcytosis could move bacteria through mucosal cells to the basal surface of mucosal epithelial cells to encounter local macrophages and lymphoid cells in the tonsils. Alternatively, mucosal macrophages could phagocytose bacteria in the mucus layer, migrate through the mucosal barrier, and spread them via leukocyte trafficking to the same cells.
Macrophages within the tonsil are likely used by the bacterium for replication and growth and then to spread them via leukocyte trafficking in lymphatic vessels to regional lymph nodes to infect additional macrophages. Ligand-receptor interactions are likely involved and bacterial surface proteins appear to serve as adhesins to macrophage and endothelial cell membrane receptors. Specific bacterial adhesins and host cell receptors for them on macrophages and endothelial cells have not been identified. Once bound to cell membrane, the bacterium is phagocytosed and retained in a phagosome within the cell cytoplasm. Erysipelothrix rhusiopathiae grows and replicates intracellularly in phagosomes and phagolysosomes. Capsular polysaccharides are able to inhibit phagocytosis of the bacterium by neutrophils and to a limited extent by macrophages. However, macrophages are used by the bacterium to isolate it from host innate and adaptive immune responses. Although it appears that phagosome-lysosome fusion occurs, capsular polysaccharides appear to block the oxidative burst and prevent killing of the bacterium by molecules present in the lysosome. Although undetermined, bacteria-infected macrophages in regional lymph nodes could spread the bacterium systemically via leukocyte trafficking using lymphatic vessels and the thoracic duct or postcapillary venules and the venous system to the systemic circulatory system and then to capillary beds within the skin. Cutaneous infarcts (also vascular-related lesions in other organs such as the kidney) suggest these bacteria may have tropisms for vascular endothelial cells. It is unclear why this occurs but is likely linked to the expression of bacterial virulence determinates and ligand-receptor interactions with host endothelial cells. In addition to cell death attributable to direct infection of endothelial cells by the bacterium, bacterial neuraminidase may also activate the alternative complement pathway and induce thrombocytopenia, producing complement-derived chemotactic factors that could contribute to injury of capillary beds in local vascularized connective tissues. These mechanisms may contribute, in part, to the development of vegetative valvular endocarditis and arthritis that occurs in the chronic septicemic form of this disease.
Female Reproductive System
Brucellosis (Brucella spp.): The pathogenesis of Brucellosis begins as an infection of the regional and systemic lymph nodes (see the section on Bone Marrow, Blood Cells, and Lymphatic System) facilitated by entry through mucosae of the respiratory and alimentary systems. The mechanism of injury is cell death caused by pyogranulomatous inflammation and its mediators and degradative enzymes. Gross lesions include aborted fetuses; necrosis, inflammation, and fibrinoid exudation of uterine caruncles and fetal cotyledons (see Fig. 4-26); and a yellow-white uterine exudate. Bacteria spread in macrophages via leukocyte trafficking from regional lymph nodes to the caruncular side of placentomas when they likely leave the vascular system to migrate into and through these tissues. Although it is unclear what additional cells in the placentoma are infected during transplacental spread to the fetus, trophoblasts are infected with bacteria. Other cell types may be involved. Bacteria then could spread in fetal macrophage-like cells within the fetal circulatory system to the fetus via the umbilical cord or within the allantoic and amniotic membranes and infect the fetus via contact with fetal mucosae of the respiratory or alimentary systems, but if this occurs or what cells facilitate this spread are unclear.
Bovine Mastitis (Staphylococcus aureus, Streptococcus agalactiae, Streptococcus dysgalactiae, and Escherichia coli): The mechanism of injury in bovine mastitis is death of all cell populations in the mammary gland from (1) bacterial toxins, (2) inflammation and its mediators and degradative enzymes, and (3) induced reparative responses such as fibrosis. Gross lesions in acute mastitis include firm, swollen, edematous, and occasionally hemorrhagic glands and ectatic ducts and sinuses containing yellow-white exudate (see Figs. 18-53 to 18-55, 18-57, and 18-58). In chronic mastitis, tissues are firm and consist of large zones of fibrous connective tissue that have replaced and displaced remaining normal glands (see Fig. 18-59). Inflammatory exudate is difficult to observe unless abscesses have formed. Ducts and sinuses may be ectatic.
Animals encounter these bacteria through physical contact in fomites or fluid droplets from mammary gland, uterine, or fecal origin on milking equipment and human hands. They commonly become commensal organisms that reside in biofilms of the mucous membranes of the teat canal and mammary duct and sinuses. Trauma to mucosae in the gland induced by pressure changes acting on the duct system caused by milking likely makes the mucosae more suitable for colonization and alters the commensal relationship, allowing the bacteria to replicate in sufficient numbers to spread the bacterium within the gland and to other animals mechanically during milking. Mastitis is an ascending infection, and milk in canals and sinuses is a suitable culture media for initial growth of bacteria. This environment is not suitable in the long-term for survival of the bacteria, thus they attempt to colonize mucosae to sustain the infection. Ligand-receptor interactions are likely involved in the adherence of these bacteria to receptors on mucosal epithelial cells; however, specific bacterial adhesins and host cell receptors have not been clearly identified. Once mucosae are colonized, bacteria employ mechanisms to sustain the infection. For example, Staphylococcus aureus produces toxins, such as superantigens, leukocidins, hemolysins, coagulase, and likely α-, β-, and δ-toxins (virulence determinates) that result in cell membrane injury and cell death and the activation of mucosal macrophages. The severity of this lesion and its progression to gangrenous mastitis in the peracute and acute forms are dependent on the type and quantity of toxins secreted by the bacterium as determined by its virulence determinates. Additionally, activated mucosal macrophages secrete proinflammatory cytokines resulting in the recruitment of neutrophils from the systemic circulation, through the mucosae, and ultimately into the milk, thus increasing the somatic cell count. With the focus of inflammation on the mucosae, epithelial cells and subjacent basement membrane are injured, killed, and sloughed, providing bacteria with access to vascularized ECM tissues of the gland. Using bacterial surface proteins, they are able to adhere to and colonize ECM tissues likely using receptors expressed on molecules such as fibronectin, vitronectin, laminin, and collagen in the matrix. This process allows bacteria to evade many of the harmful actions of the innate and adaptive immune responses. Additionally, capsular polysaccharides block phagocytosis by neutrophils and macrophages. As a result, acute inflammation progresses with time to chronic inflammation with fibrosis (see Chapter 3), which is a common manifestation of mastitis caused by Staphylococcus aureus.
Chronic mastitis is often linked with the formation of mucosal biofilms. In mastitis caused by Streptococcus agalactiae and Streptococcus dysgalactiae, the bacteria use most of the mechanisms employed by Staphylococcus aureus with one important exception. They lack virulence determinates that injure the mucosae and allow the bacterium to invade vascularized ECM tissues and colonize this area. Thus the bacterium is limited to colonizing the mucosae and causing inflammation at the mucosal barrier. The outcome of this process is loss of mucosal epithelial cells lining glands, collapse of the glands, and replacement of the glands with fibrous connective tissue. In mastitis caused by E. coli and other coliforms, the bacterium uses most of the mechanisms as discussed previously. However, in the initial phases of colonizing the mucosae, endotoxins (LPS) and other toxic molecules released from Gram-negative bacteria cause tissue injury and cell death, affecting the mucosae, submucosae, and capillary beds. The concurrent acute inflammatory response with neutrophils and their degradative enzymes exacerbate the severity of the injury. This outcome leads to tissue necrosis, edema, and hemorrhage. Endotoxin is also absorbed by the capillaries and can cause endotoxic shock of the circulatory system and death of affected animals (see Chapters 2 and 3).
Male Reproductive System
Brucellosis (Brucella spp.): The pathogenesis of Brucellosis begins as an infection of regional and systemic lymph nodes (see the sections on Bone Marrow, Blood Cells, and Lymphatic Systems and Female Reproductive System) facilitated by entry through the mucosae of the respiratory and alimentary systems. The mechanism of injury is cell death caused by pyogranulomatous inflammation and its mediators and degradative enzymes. Brucella spp. spread in macrophages via leukocyte trafficking from regional lymph nodes to the testes, epididymides, and other male reproductive tissues. Gross lesions include enlarged and deformed testes and epididymides attributable to a yellow-white pyogranulomatous exudate against the bacteria in the tissues (see Fig. 4-26).