Endocrine and Electrolyte Abnormalities
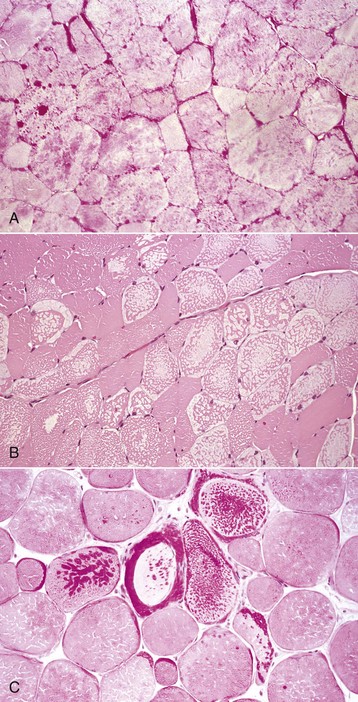
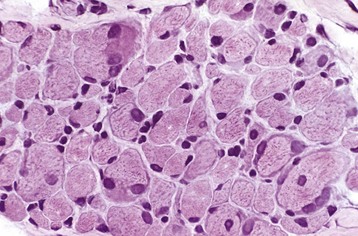
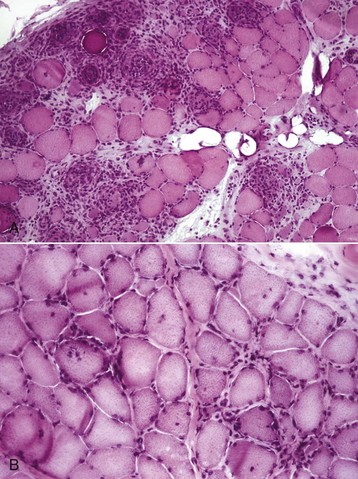
Various endocrinologic abnormalities can result in myopathic conditions (Table 15-9). The most common are hypercortisolism and hypothyroidism in dogs. In horses, pituitary hyperfunction resulting in Cushing’s disease also causes muscle disease. In most cases of endocrine myopathy, the end result is myofiber atrophy, particularly of type 2 fibers. A unique syndrome of muscle hypertrophy and pseudomyotonia occurs in dogs associated with hypercortisolism. Endocrine myopathies can also be complicated by the fact that endocrinopathy can also cause pathologic changes in peripheral nerves, leading to a mixture of myopathic (type 2 fiber atrophy) and neuropathic changes (denervation atrophy and alteration in fiber-type pattern) within muscle. Denervation followed by reinnervation leading to fiber-type grouping can be seen in dogs with chronic hypercortisolism (see Fig. 15-20, B) and hypothyroidism.
TABLE 15-9
Myopathies Caused by Endocrine and Electrolyte Abnormalities
| Disorder | Species Affected |
| Hypothyroidism | Dogs |
| Hypercortisolism | Dogs |
| Hypokalemia | Cattle, cats |
| Hypophosphatemia | Cattle |
| Hypernatremia | Cats |
| Hypocalcemia | Cattle |
| Hypothalamic/pituitary dysfunction | Horses |
Normal electrolyte status is vital to normal skeletal muscle function. Hypocalcemia, hypokalemia, hypernatremia, and hypophosphatemia can cause profound skeletal muscle weakness, sometimes associated with myofiber necrosis, in various species.
Neuropathic and Neuromuscular Junction Disorders
Dysfunction of the lower motor neurons, peripheral nerves, or neuromuscular junction can have profound effects on muscle function.
Neuropathic Disorders: There are many peripheral nerve disorders and a few motor neuron disorders that can lead to denervation atrophy of muscle in animals. These can be inherited or acquired. Long nerves, such as the sciatic and left recurrent laryngeal nerves, appear to be particularly sensitive to development of acquired neuropathy. Many of the peripheral nerve disorders of animals are discussed in Chapter 14. Characteristic features of denervation atrophy are described in the section on Responses of Muscle to Injury.
Neuromuscular Junction Disorders: The neuromuscular junction is a modification of the postsynaptic myofiber membrane. At the neuromuscular junction, the membrane is folded to increase surface area and is studded with specialized ion channels known as acetylcholine receptors. After arrival of an action potential at the distal end of a motor nerve, the terminal axons release acetylcholine, which diffuses across the synaptic space to bind to the acetylcholine receptors. Binding opens these channels, leading to sodium influx, which initiates the skeletal muscle action potential that culminates in muscle contraction. Acetylcholine is rapidly degraded by acetylcholinesterase released from the postsynaptic membrane, which prevents continued stimulation and thus contraction of the muscle fiber.
Disorders that impair the ability of nerve impulses to travel across the neuromuscular junction have profound effects on skeletal muscle function. Technically, however, the myofibers are still innervated, so denervation atrophy does not occur and no light microscopic abnormalities in the muscle or nerve are present. Various neurotoxins (i.e., in snake and spider venom and in curare-containing plants) and drugs can affect the neuromuscular junction, but the most common neuromuscular junction disorders affecting animals are myasthenia gravis, botulism, and tick paralysis.
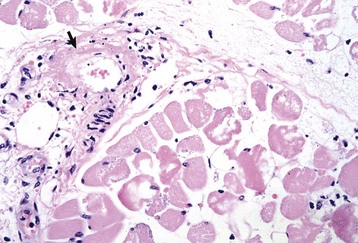
Myasthenia Gravis: Myasthenia gravis can be either acquired or congenital. Acquired myasthenia gravis is an immune-mediated disorder caused by circulating autoantibodies against skeletal muscle acetylcholine receptors (Fig. 15-29). Binding of these antibodies to the acetylcholine receptor on the postsynaptic membrane leads to a severe decrease in the number of functional receptors. The mechanisms by which antibodies damage these receptors are (1) direct damage to the neuromuscular junction, which may be visible with electron microscopy as simplification of the folding of the membrane, and (2) formation of cross-linked antibodies leading to receptor internalization. Sufficient functional acetylcholine receptors are present to initially allow normal neuromuscular transmission, but if there is sustained muscular activity the decrease in the number of available receptors leads to progressive weakness and collapse. Therefore acquired myasthenia gravis results in episodic collapse, and repetitive nerve stimulation causes a characteristic rapid decrease in amplitude of the muscle compound motor action potential. Diagnosis of myasthenia gravis can also be made after intravenous injection of cholinesterase inhibitors such as edrophonium chloride (Tensilon, ICN Pharmaceuticals, Costa Mesa, CA) in collapsed animals. The reduction in cholinesterase activity leads to more active acetylcholine being available within the synapse and rapid, although transient, restoration of skeletal muscle contraction. Detection of autoantibodies to acetylcholine receptors in the blood confirms the diagnosis of acquired myasthenia gravis.
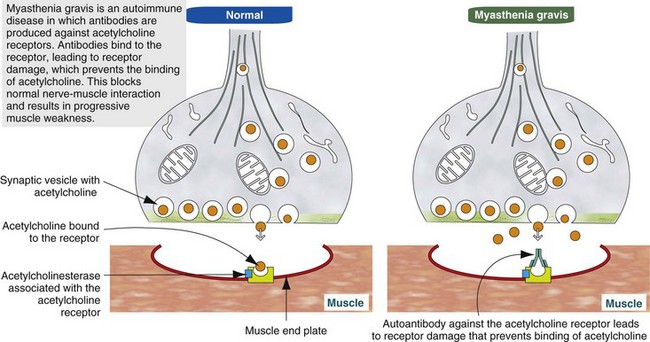
Fig. 15-29 Schematic diagram of the pathogenesis of acquired (autoimmune) myasthenia gravis. (Modified from Kierszenbaum AL: Histology and cell biology: an introduction to pathology, ed 2, St Louis, 2006, Mosby.)
The origin of the autoantibodies causing myasthenia gravis is not always known, but there is a strong link between thymic abnormalities and development of myasthenia gravis in both humans and animals. Specialized cells within the thymic medulla, known as myoid cells, express skeletal muscle proteins, including those of the acetylcholine receptor. It is thought that these cells participate in development of self-tolerance. Abnormalities of the thymus, most commonly thymoma in animals and thymic follicular hyperplasia in humans, can lead to loss of self-tolerance to acetylcholine receptors. In such cases, removal of the abnormal thymus can result in restoration of normal neuromuscular junction activity. When thymic abnormalities are not present, treatment with long-acting anticholinesterase agents and in some cases immunosuppressive agents, such as corticosteroids, is necessary.
Congenital myasthenia gravis is an inherited disorder that is much less common than acquired myasthenia gravis. To date it has been described only in humans, dogs, and cats. Animals with congenital myasthenia gravis are born with defective neuromuscular junctions that often have a decreased membrane surface area, best visualized with electron microscopy, and as a consequence an inherently reduced acetylcholine receptor density. Such animals may be normal at birth because there are sufficient functional acetylcholine receptors to support muscle contraction in a neonate. But, with rapid postnatal growth, clinical signs of profound, sustained, and progressive weakness occur as a consequence of insufficient functional receptors to support the function of growing muscles.
Botulism: Botulism is a neuromuscular disorder caused by the exotoxin of the bacterium Clostridium botulinum. Botulinum toxin is considered one of the deadliest of the known toxins. Botulism is characterized by profound generalized flaccid paralysis. Seven serologically distinct but structurally similar forms of botulinum toxin are designated A, B, C, D, E, F, and G. Sensitivity to these toxin types varies among different species. Dogs are most sensitive to type C toxin, ruminants to types C and D, and horses to types B and C.
Botulinum toxin consists of a light chain and a heavy chain linked by a disulfide bond. Binding of botulinum toxin to receptors on the presynaptic terminals of peripheral nerves is followed by endocytosis of the toxin. Within the endocytotic vesicle of the terminal nerve, the disulfide bond is cleaved, and the released light chain is translocated into the axonal cytoplasm (see Fig. 4-27). Botulinum toxin light chains are metalloproteinases. Numerous proteins are involved in the release of acetylcholine from presynaptic vesicles, and botulinum toxin blocks release of acetylcholine by irreversible enzymatic cleavage of one or more of these proteins. Different forms of botulinum toxin affect different proteins, but the end result is the same. Active neuromuscular junctions are the most sensitive, which has led to the use of low concentrations of locally injected botulinum toxin as a treatment for localized muscular disorders resulting in spasm.
Clostridium botulinum spores are commonly present in the gastrointestinal tract of animals and in the soil. Under favorable anaerobic and alkalinic conditions, these spores become active, with resultant toxin production. Botulism can occur because of ingestion of preformed toxin, such as in feed contaminated by dead rodents or soil-borne organisms, or from toxin produced by Clostridium botulinum organisms within the gastrointestinal tract or superficial wounds (Box 15-9). Dogs and cats are the species most likely to ingest dead rodents containing botulinum toxin and are quite resistant to developing botulism. In veterinary medicine, horses are the most sensitive to botulinum toxin. Death of horses, most often the result of respiratory muscle paralysis, can result from exposure to only very small amounts of botulinum toxin. The damage to presynaptic axon terminals is irreversible, and recovery from botulism occurs only after terminal axon sprouting and reestablishment of new functioning synapses.
Tick Paralysis: Dermacentor and Ixodes ticks can elaborate a toxin that also blocks release of acetylcholine from axon terminals. Tick paralysis is seen most often in dogs and children. Recovery after tick removal can be rapid (within 24 to 48 hours), indicating that the mechanism of toxin action in tick paralysis does not result in irreversible presynaptic damage and thus is different from that of botulinum toxin.
Neoplasia
Neoplasms involving skeletal muscle are most often those that arise within the muscle or its supporting structures or that invade muscle from adjacent tissue. Neoplasms metastatic to muscle are rare.
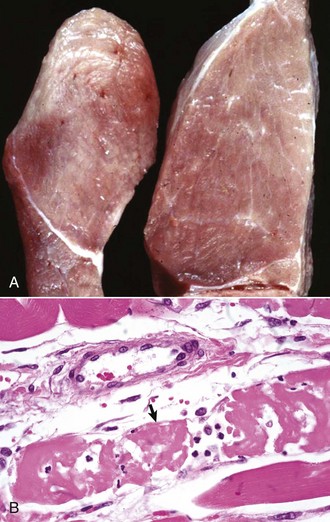
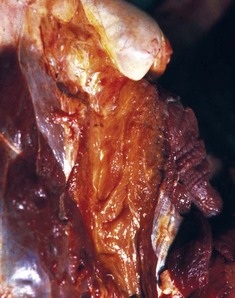
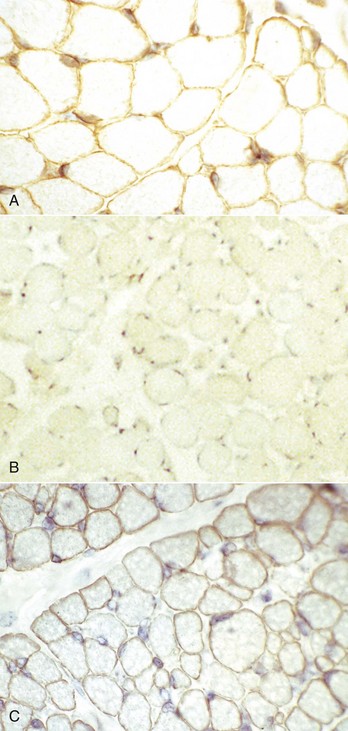
Primary Muscle Tumors: Tumors with striated muscle differentiation are thought to arise from intramuscular pluripotential stem cells rather than from satellite cells. These tumors are uncommon and are either benign (rhabdomyoma) or malignant (rhabdomyosarcoma [Fig. 15-30]). Primary intramuscular tumors can also arise from fibrous tissue, vasculature, or neural elements. The most common tumor to arise from muscle-supporting structures is hemangiosarcoma.
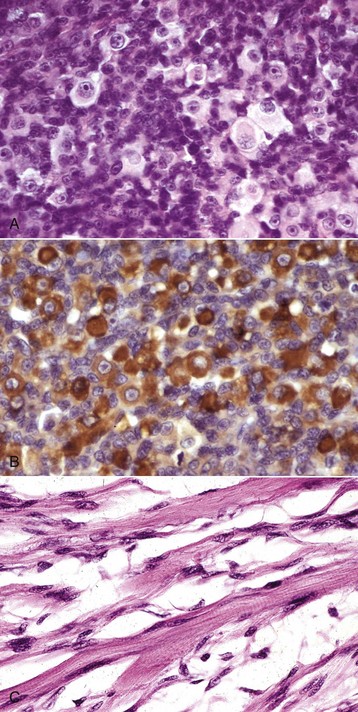
Fig. 15-30 Rhabdomyosarcoma.
A, Skeletal muscle, cat. An admixture of small round basophilic cells with a lesser number of larger round cells with prominent eosinophilic cytoplasm is characteristic of embryonal rhabdomyosarcoma. Nuclei are central and euchromatic, most often with a single large nucleolus. H&E stain. B, Immunostaining reaction of the same rhabdomyosarcoma as depicted in A, showing intense cytoplasmic expression of desmin in many tumor cells, indicative of muscle origin (skeletal, cardiac, or smooth). These cells also express myoglobin and sarcomeric actin (not shown), which differentiates skeletal muscle tumors from smooth muscle tumors. Immunoperoxidase reaction for desmin. C, Botryoid rhabdomyosarcoma, urinary bladder, large breed dog. Cross-striations, characteristic of a well-differentiated rhabdomyosarcoma, are present in the elongated multinucleate tumor cells. H&E stain. (A and B courtesy College of Veterinary Medicine, Cornell University. C courtesy Dr. B.A. Valentine, College of Veterinary Medicine, Oregon State University.)
Rhabdomyoma and Rhabdomyosarcoma: Tumors of striated muscle that occur at sites other than within muscle are rhabdomyomas of the heart or lung and botryoid rhabdomyosarcomas of the urinary bladder; these are not discussed in this section. Rhabdomyoma and rhabdomyosarcoma arising within skeletal muscle are most common in the dog, followed by the horse and cat. Morphologic variants include round cell, spindle cell, and mixed round and spindle cell, reflecting the developmental stages of skeletal muscle. Historically, diagnosis of tumors of skeletal muscle has relied on identification of cross striations indicative of sarcomeric differentiation. Cross striations are most often seen in elongated multinucleate cells known as strap cells (Fig. 15-30, C) and in ovoid cells known as racquet cells. They are most easily recognized after staining with phosphotungstic acid hematoxylin (PTAH) stain, but the search for cross striations can be extremely frustrating and often unrewarding. These days the diagnosis of tumors of skeletal muscle origin relies primarily on results of immunohistochemical examination using antibodies for muscle-specific proteins. Muscle actin and desmin are expressed by smooth and skeletal muscle tumors, but myoglobin, sarcomeric actin, myogenin, and MyoD1 are specific for skeletal muscle. Evidence of muscle differentiation, such as primitive myofilaments and Z-band structures, can also be detected by electron microscopy.
Rhabdomyoma is most often a round cell tumor and occurs most commonly in the larynx of adult dogs. The youngest reported age is 2 years. Tumors are generally smooth and nodular, pink, and unencapsulated. Histologic features are closely packed plump round cells that have central euchromatic nuclei, generally with a single prominent nucleus, and abundant vacuolated to granular eosinophilic cytoplasm. A small number of multinucleate and elongate strap cells can also be seen. Mitoses are rare, and evidence of invasion is uncommon.
Similar to the situation in humans, rhabdomyosarcomas in animals most often occur at a young age and are most common in the neck or oral cavity, especially in the tongue. These tumors are pink and fleshy, and they often have prominent local invasion. The most common and most distinctive form of rhabdomyosarcoma in animals is embryonal rhabdomyosarcoma, composed of primitive round cells with prominent euchromatic nuclei, a single prominent nucleolus, and either indistinct or prominent eosinophilic cytoplasm (“rhabdomyoblasts”; see Fig. 15-30, A and B). Rhabdomyosarcoma can also contain elongate multinucleate strap cells (see Fig. 15-30, C) and ovoid racquet cells. Cellular and nuclear pleomorphism is common, as is mitotic activity. These tumors are locally invasive and frequently metastasize, although too few cases have been studied to document any pattern of metastasis.
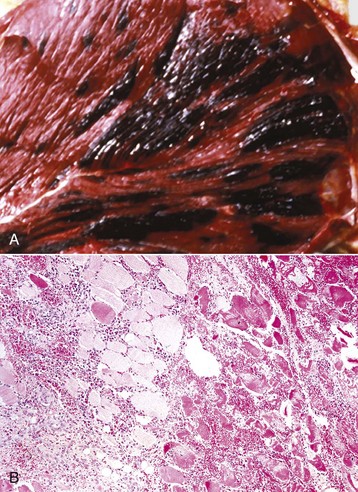
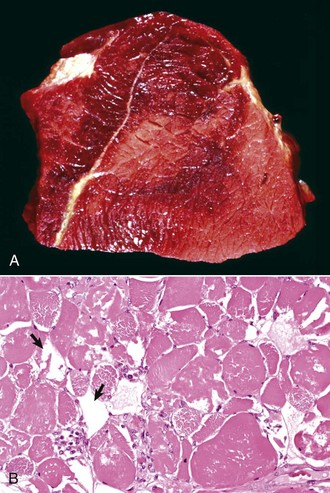
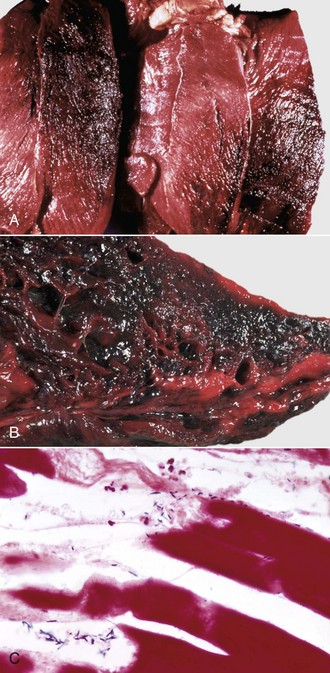
Hemangiosarcoma: Malignant vascular neoplasms (hemangiosarcoma) arising within muscle are most common in the horse and dog (Fig. 15-31). Clinical signs include swelling within a muscle, often with associated lameness. Cytologic preparations frequently reveal only peripheral blood, which is suggestive of a hematoma. Pathologic diagnosis can be difficult if multiple sites within the lesion are not sampled, as the amount of hemorrhage often far exceeds the area composed of proliferating neoplastic endothelial cells. Intramuscular hemangiosarcoma has a high incidence of metastasis, often to the lungs.
Other Tumors Involving Skeletal Muscle: A variant of lipoma, known as infiltrative lipoma, is often located in skeletal muscle. Characteristic gross pathologic and histopathologic findings are mature adipocytes invading skeletal muscle. This tumor is most common in the dog but has also been reported in young horses. Wide excision is the treatment of choice because this tumor recurs as a result of local invasion, but it does not metastasize.
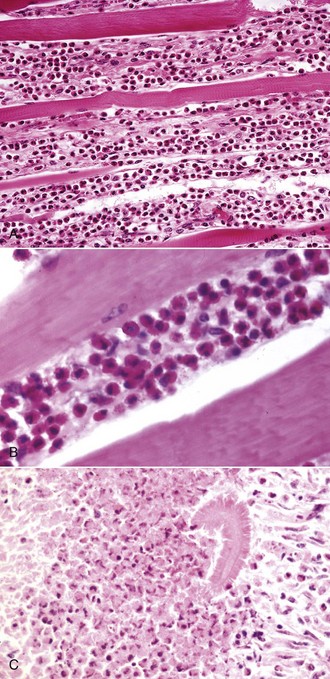
Infiltration of skeletal muscle by neoplastic lymphocytes is not uncommon. Neoplastic lymphocytic infiltrates surround myofibers and can cause myofiber atrophy. These cells do not invade myofibers, however, and myonecrosis is rare. This helps to distinguish intramuscular lymphoma from lymphocytic myositis. Careful examination of infiltrating neoplastic cells typically reveals a relatively monomorphic population of lymphocytes, which may be atypical in appearance. Immunohistochemistry to confirm a single infiltrating cell type is also useful.
Vaccine-associated sarcoma in the muscle of the cat can arise within an intramuscular vaccination site or extend into underlying skeletal muscle from a subcutaneous injection site. Occasionally, mast cell tumors and carcinomas exhibit prominent skeletal muscle invasion. Melanoma arising in the skin of older gray horses often metastasizes to muscle fascia and may exhibit some extension into the muscle itself. Intramuscular metastasis of tumors is rare (see the section on Defense Mechanisms). Intramuscular metastasis of carcinoma, particularly prostatic, can occur in dogs. When carcinomas with areas of sclerosis involve muscle, either by extension or by metastasis, the muscle basement membrane of adjacent myofibers is typically destroyed, often resulting in bizarre multinucleate cells representing attempts at muscle regeneration (see Fig. 15-17). These bizarre cells should not be misidentified as tumor cells.



 years of age, at which time weakness and neurologic deficits are evident. Affected Brahman cattle grow poorly and have muscular weakness and neurologic disease. Electrocardiographic studies reveal abnormalities of cardiac conduction. Serum concentrations of CK and AST can be increased, with notable increases evident in severely weak animals before death.
years of age, at which time weakness and neurologic deficits are evident. Affected Brahman cattle grow poorly and have muscular weakness and neurologic disease. Electrocardiographic studies reveal abnormalities of cardiac conduction. Serum concentrations of CK and AST can be increased, with notable increases evident in severely weak animals before death.