Oral Manifestations of Systemic Diseases
MUCOPOLYSACCHARIDOSIS
The mucopolysaccharidoses are a heterogeneous group of metabolic disorders that are usually inherited in an autosomal recessive fashion. These disorders are all characterized by the lack of any one of several normal enzymes needed to process the important intercellular substances known as glycosaminoglycans. These substances used to be known as mucopolysaccharides, thus the term mucopolysaccharidosis. Examples of glycosaminoglycans include the following:
The type of mucopolysaccharidosis that is seen clinically depends on which of these substrates lacks its particular enzyme. The mucopolysaccharidoses as a group occur with a frequency of approximately 1 in 15,000 to 29,000 live births, although some types are much less common.
CLINICAL AND RADIOGRAPHIC FEATURES: The clinical features of the mucopolysaccharidoses vary, depending on the particular syndrome that is examined (Table 17-1). Furthermore, affected patients with a particular type of this disorder often exhibit a wide range of severity of involvement. Most types of mucopolysaccharidosis display some degree of mental retardation. Often the facial features of affected patients are somewhat coarse, with heavy browridges (Fig. 17-1), and there are other skeletal changes, such as stiff joints. Cloudy degeneration of the corneas, a problem that frequently leads to blindness, is seen in several forms of mucopolysaccharidosis.
Table 17-1
Features of Selected Mucopolysaccharidosis Syndromes
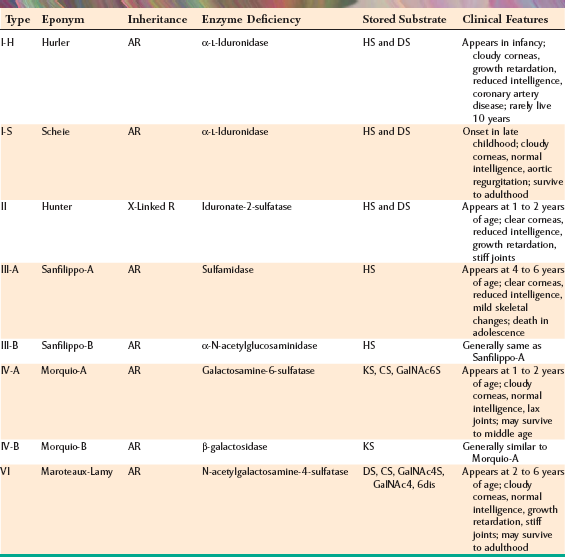
AR, Autosomal recessive; CS, chondroitin sulfate; dis, disulfate; DS, dermatan sulfate; GalNAc, N-acetylgalactosamine; HS, heparan sulfate; KS, keratan sulfate; R, recessive; S, sulfate.
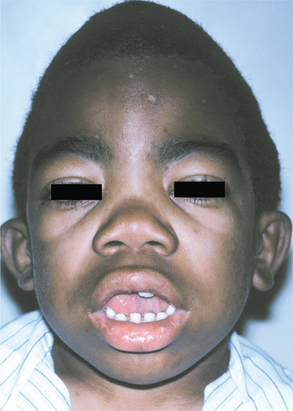
Fig. 17-1 Mucopolysaccharidosis. This patient affected by Hunter syndrome exhibits the characteristic facial features of this disorder.
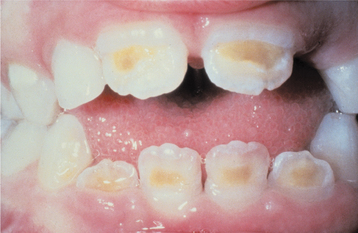
The oral manifestations vary according to the particular type of mucopolysaccharidosis. Most types show some degree of macroglossia. Gingival hyperplasia may be present, particularly in the anterior regions, as a result of the drying and irritating effects of mouth breathing. The dental changes include thin enamel with pointed cusps on the posterior teeth, although this seems to be a feature unique to mucopolysaccharidosis type IVA. Other dental manifestations include numerous impacted teeth with prominent follicular spaces (Fig. 17-2), possibly caused by the accumulation of glycosaminoglycans in the follicular connective tissue. Some investigators have reported the occurrence of multiple impacted teeth that are congregated in a single large follicle, forming a rosette pattern radiographically.

Fig. 17-2 Mucopolysaccharidosis. Radiographic examination of the dentition of a child affected by Hunter syndrome typically shows radiolucencies (arrows) associated with the crowns of unerupted teeth.
Although the clinical findings may suggest that a patient is affected by one of the mucopolysaccharidoses, the diagnosis is confirmed by finding elevated levels of glycosaminoglycans in the urine, as well as deficiencies of the specific enzymes in the patient’s leukocytes and fibroblasts.
TREATMENT AND PROGNOSIS: No satisfactory systemic treatment of the mucopolysaccharidoses exists at this time. Several forms of mucopolysaccharidosis are associated with a markedly reduced life span and with mental retardation. Attempts to improve the survival and quality of life of these patients using allogeneic bone marrow transplantation have met with some success. Unfortunately, not all aspects of the disease are corrected, and the com-plications associated with transplantation must be addressed. Such complications are associated with a 15% to 20% mortality rate. Enzyme replacement therapy currently is available for mucopolysaccharidosis I. Initiation of the enzyme, laronidase, early in the patient’s life appears to improve significantly many of the aspects of the disease, although complete resolution does not occur. Enzyme replacement strategies are also being developed for several of the other forms of this condition. Because of the rarity of these conditions and the expense of developing the treatments, the annual cost for such therapy typically exceeds $340,000. Genetic counseling is indicated for the parents and siblings of a patient affected by one of the mucopolysaccharidosis syndromes. Prenatal diagnosis is available for family planning as well.
Management of the dental problems of these patients is essentially no different from that of other patients. However, several factors may have to be taken into account:
Depending on which of these factors is present and the extent of involvement, dental care may warrant sedation, hospitalization, or general anesthesia of the patient for optimal results. General anesthesia and sedation may be challenging, however, because of excess amounts of pharyngeal tissues that often produce a smaller than normal airway. In severely affected patients, general anesthesia probably should be considered only in life-threatening situations.
LIPID RETICULOENDOTHELIOSES
The lipid reticuloendothelioses are a relatively rare group of inherited disorders. These include the following conditions:
These conditions are seen with increased frequency in patients with Ashkenazi Jewish heritage. Affected patients lack certain enzymes necessary for processing specific lipids, and this results in an accumulation of the lipids within a variety of cells. Because of this accumulation, it appeared that cells were attempting to store these substances; therefore, the term storage disease was commonly used for these disorders.
In Gaucher disease (the most common of the reticuloendothelioses), a lack of glucocerebrosidase results in the accumulation of glucosylceramide, particularly within the lysosomes of cells of the macrophage and monocyte lineage. Three types of Gaucher disease are now recognized: type 1 (nonneuronopathic) is seen primarily in the Ashkenazi Jewish population, and types 2 and 3 (neuronopathic) have a panethnic distribution.
Niemann-Pick disease is characterized by a deficiency of acid sphingomyelinase, resulting in the accumulation of sphingomyelin, also within the lysosomes of macrophages.
Tay-Sachs disease is caused by a lack of b-hexosaminidase A, which results in the accumulation of a ganglioside, principally within the lysosomes of neurons.
All these disorders are inherited as autosomal recessive traits. When the genetic mutation known to cause Gaucher disease was evaluated for the Ashkenazi Jewish population, researchers found that approximately 1 in 10 persons carried the defective gene. Most of the persons identified as having the gene, however, were heterozygous and, therefore, asymptomatic.
CLINICAL AND RADIOGRAPHIC FEATURES:
GAUCHER DISEASE: The clinical features of Gaucher disease are generally the result of the effects of the abnormal storage of glucosylceramide. Macrophages laden with this glucocerebroside are typically rendered relatively nonfunctional, and they tend to accumulate within the bone marrow of the affected patient. This accumulation displaces the normal hematopoietic cells and produces anemia and thrombocytopenia. In addition, these patients are susceptible to bone infarctions. The resulting bone pain is often the presenting complaint. Characteristic Erlenmeyer flask deformities of the long bones, particularly of the femur, are often identified. Accumulations of the macrophages in the spleen and liver result in visceral enlargement. Many affected patients show a significant degree of growth retardation. Neurologic deterioration occurs in patients with the less common types 2 and 3 Gaucher disease. Jaw lesions typically appear as ill-defined radiolucencies that usually affect the mandible without causing devitalization of the teeth or resorption of the lamina dura. Decreased salivary flow has been documented for patients with Gaucher disease compared with an age- and sex-matched population, although this decrease may not be clinically significant.
NIEMANN-PICK DISEASE: Niemann-Pick disease occurs as three different types, each associated with a different clinical expression and prognosis. Types A and B are caused by a deficiency of acid sphingomyelinase, whereas type C is primarily the result of mutation of NPC-1, a gene involved with cholesterol processing. Types A and C have neuronopathic features, characterized by psychomotor retardation, dementia, spasticity, and hepatosplenomegaly, with death occurring during the first or second decade of life. Type B patients normally survive into adulthood and exhibit visceral signs, primarily hepatosplenomegaly, and sometimes pulmonary involvement.
TAY-SACHS DISEASE: Tay-Sachs disease may have a wide clinical range because the condition is genetically heterogeneous. Some forms are mild, with patients surviving into adulthood. In the severe infantile form, however, rapidly progressive neuronal degeneration develops shortly after birth. Signs and symptoms include blindness, developmental retardation, and intractable seizures. Death usually occurs by 3 to 5 years of age.
HISTOPATHOLOGIC FEATURES: Histopathologic examination of an osseous lesion of Gaucher disease shows sheets of lipid-engorged macrophages (Gaucher cells) exhibiting abundant bluish cytoplasm, which has a fine texture resembling wrinkled silk. In Niemann-Pick disease, the characteristic cell seen on examination of a bone marrow aspirate is the “sea blue” histiocyte.
GAUCHER DISEASE: For patients with a mild expression of Gaucher disease, no treatment may be necessary. For more severe forms of Gaucher disease, enzyme replacement therapy with macrophage-targeted glucocerebrosidase (imiglucerase for injection) is used; however, this is quite expensive, often costing more than $200,000 per year for treatment. After 9 to 12 months of therapy, patients exhibit improvement in the status of their anemia, a decrease in plasma glucocerebroside levels, and a decrease in hepatosplenomegaly. Resolution of the radiographic bone changes takes place over a longer period. Children treated with this regimen may show significant gain in height. Unfortunately enzyme replacement therapy has shown minimal effect on the neuronopathic Gaucher disease types 2 and 3. Bone marrow transplantation has also been attempted; however, the problems inherent in graft-versus-host disease (GVHD) are still present with that form of therapy, and thus it is not recommended. A case-control study showed that adults with Gaucher disease have an increased risk for hematologic malignancies, particularly lymphoma and multiple myeloma. Genetic counseling should be provided to all affected patients.
NIEMANN-PICK AND TAY-SACHS DISEASE: The neuronopathic forms of Niemann-Pick disease and the infantile form of Tay-Sachs disease are associated with a poor prognosis. Genetic counseling should be provided for affected families. Molecular markers of these disorders have been developed to identify carriers. Such identification allows earlier intervention in terms of counseling, and targeted population screening for the gene that causes Tay-Sachs disease has resulted in a marked decrease in affected patients during the past 3 decades.
LIPOID PROTEINOSIS (HYALINOSIS CUTIS ET MUCOSAE; URBACH-WIETHE SYNDROME)
A rare condition, lipoid proteinosis is inherited as an autosomal recessive trait. It is characterized by the deposition of a waxy material in the dermis and submucosal connective tissue of affected patients. The earliest thorough description of lipoid proteinosis was by Urbach and Wiethe in 1929, and more than 300 patients, most of whom are of European background, have been reported to date. Mutations of the ECM1 gene, which encodes a glycoprotein known as extracellular matrix protein 1, have recently been identified as the cause for this condition.
CLINICAL FEATURES: The laryngeal mucosa and vocal cords are usually the sites that are initially affected by lipoid proteinosis. Therefore, the first sign of the disease may be one of the following:
The vocal cords become thickened as the accumulation of an amorphous material begins to affect the laryngeal mucosa. This infiltrative mucosal process may also involve the pharynx, esophagus, tonsils, vulva, and rectum. Skin lesions also develop early in life, appearing as thickened, yellowish, waxy papules; plaques; or nodules that often affect the face, particularly the lips and the margins of the eyelids (Fig. 17-3). Some lesions may begin as dark-crusted vesicles that heal as atrophic hyperpigmented patches.
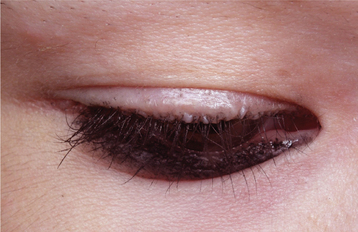
Fig. 17-3 Lipoid proteinosis. Thickened papules are present along the margin of the eyelid. (Courtesy of Dr. Maria Copete.)
Eventually, most patients exhibit a thickened, furrowed appearance of the skin. Other areas of the skin that may be involved include the neck, palms, axillae, elbows, scrotum, knees, and digits. In those areas subjected to chronic trauma, a hyperkeratotic, verrucous surface often develops. In addition to the cutaneous manifestations, symmetrical intracranial calcifications of the medial temporal lobes have been identified in approximately 70% of affected patients. These lesions are usually asymptomatic, although a few patients with such calcifications have been reported to have a seizure disorder.
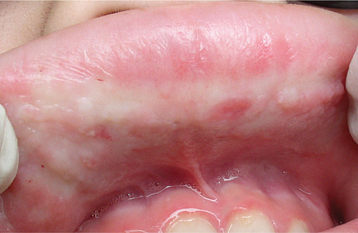
The oral mucosal abnormalities typically become evident in the second decade of life. The tongue, labial mucosa, and buccal mucosa become nodular, diffusely enlarged, and thickened because of infiltration with waxy, yellow-white plaques and nodules (Fig. 17-4). The dorsal tongue papillae are eventually destroyed, and the tongue develops a smooth surface. The accumulation of the amorphous material within the tongue may result in its being bound to the floor of the mouth. Therefore, the patient may not be able to protrude the tongue. Gingival enlargement appears to be an infrequent finding.
HISTOPATHOLOGIC FEATURES: A biopsy specimen of an early lesion of lipoid proteinosis typically reveals the deposition of a lamellar material around the blood vessels, nerves, hair follicles, and sweat glands. This material stains positively with the periodic acid-Schiff (PAS) method and is not digested by diastase. The location of this material, its staining properties, and the presence of increased laminin, type IV collagen, and type V collagen suggest a basement membrane origin.
A biopsy specimen of a lesion in its later stages usually shows not only the lamellar material but also deposition of an amorphous substance within the dermal connective tissue (Fig. 17-5).
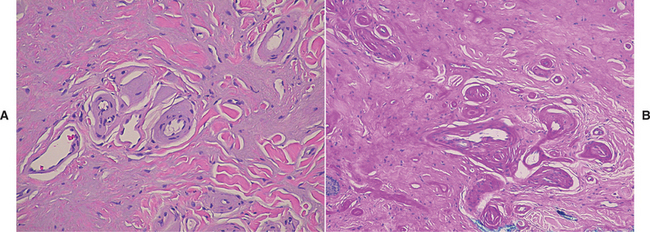
TREATMENT AND PROGNOSIS: Generally, no specific treatment is available for lipoid proteinosis other than genetic counseling. In rare instances, the infiltration of the laryngeal mucosa may produce difficult breathing for some infants, in which case debulking of the mucosal lesions may be necessary. Most patients with lipoid proteinosis have a normal life span. Certainly, however, the vocal hoarseness and the appearance of the skin may influence the quality of life for affected patients.
JAUNDICE (ICTERUS)
Jaundice is a condition characterized by excess bilirubin in the bloodstream. The bilirubin accumulates in the tissues, which results in a yellowish discoloration of the skin and mucosa. To understand jaundice, it is important to know something about the metabolism of bilirubin. Most bilirubin is derived from the breakdown of hemoglobin, the oxygen-carrying pigment of erythrocytes. The average life span of an erythrocyte in the circulation is 120 days. After this time, it undergoes physiologic breakdown. The hemoglobin is degraded and processed by the cells of the reticuloendothelial system, and bilirubin is liberated into the bloodstream in an unconjugated state. In the liver, bilirubin is taken up by the hepatocytes and conjugated with glucuronic acid, which produces conjugated bilirubin, a soluble product that can be excreted in the bile.
There are numerous causes for increased serum levels of bilirubin; some are physiologic, and many are pathologic. Therefore, the presence of jaundice is not a specific sign and generally necessitates physical examination and laboratory studies to determine the precise cause. The basic disturbances associated with increased bilirubin levels include an increased production of bilirubin. This occurs when the red blood cells (RBCs) are being broken down at such a rapid rate that the liver cannot keep pace with processing. This breakdown is seen in such conditions as autoimmune hemolytic anemia or sickle cell anemia.
In addition, the liver may not be functioning correctly, resulting in decreased uptake of the bilirubin from the circulation or decreased conjugation of bilirubin in the liver cells. Jaundice is frequently present at birth as a result of the low level of activity of the enzyme system that conjugates bilirubin. Defects in this enzyme system may also be seen with certain inherited problems, one of the more common of which is Gilbert syndrome. This innocuous condition is often detected on routine examination, and it is estimated to affect up to 5% of people in the United States. Because most of these examples of jaundice occur with impaired processing of bilirubin, laboratory studies usually show unconjugated bilirubin in the serum.
The presence of conjugated bilirubinemia in jaundice can usually be explained by the reduced excretion of bilirubin into the bile ducts. This can be the result of swelling of the hepatocytes (resulting in an occlusion of the bile canaliculi) or hepatocyte necrosis, with disruption of the bile canaliculi and liberation of conjugated bilirubin. Thus liver function may be disturbed because of any one of a variety of infections (e.g., viruses) or toxins (e.g., alcohol). Occlusion of the bile duct from gallstones, stricture, or cancer can also force conjugated bilirubin into the bloodstream.
CLINICAL FEATURES: The patient affected by jaundice exhibits a diffuse, uniform, yellowish discoloration of the skin and mucosa. The color varies in intensity, depending on the serum level of bilirubin and the anatomic site. Because elastin fibers have an affinity for bilirubin, tissues that have a high content of elastin, including the sclera, lingual frenum, and soft palate, are prominently affected. The sclera of the eye is often the first site at which the yellow color is noted (Fig. 17-6). The yellow discoloration caused by hypercarotenemia (resulting from excess ingestion of carotene, a vitamin-A precursor found in yellow vegetables and fruits) may be confused with jaundice, but the sclera is not involved in that condition.
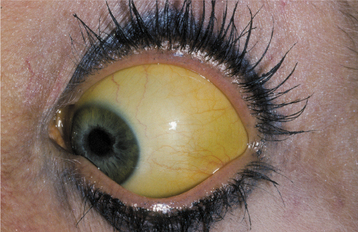
Other signs and symptoms associated with jaundice vary with the underlying cause of the hyperbilirubin-emia. For example, patients with viral hepatitis usually have a fever, abdominal pain, anorexia, and fatigue. The patient with jaundice typically requires a complete medical evaluation to determine the precise cause of the condition so that proper therapy can be instituted.
TREATMENT AND PROGNOSIS: The treatment and prognosis of the patient with jaundice vary with the cause. The jaundice that is commonly noted at birth often resolves spontaneously; however, if the infant is placed under special lights, then the clearing will occur more quickly because conjugation of the bilirubin molecule is triggered by exposure to blue light. If the episode of jaundice is due to significant liver damage, as may be seen with viral hepatitis B or hepatotoxic chemical injury, then the prognosis will vary, depending on the extent of liver damage. The prognosis for patients with jaundice secondary to liver damage associated with metastatic malignancy is poor.
AMYLOIDOSIS
Amyloidosis represents a heterogeneous group of conditions characterized by the deposition of an extracellular proteinaceous substance called amyloid. Virchow coined the term amyloid in the middle of the nineteenth century because he believed it to be a starchlike material (amyl = starch; oid = resembling). We now understand that amyloid can be formed in a variety of settings, each with its own specific type of amyloid protein. Many of these amyloid proteins have been identified precisely with respect to their biochemical composition, and ideally an attempt should be made to categorize the type of amyloid specifically when this diagnosis is made. The various amyloid proteins are designated with an A, to indicate amyloid, followed by an abbreviation for the specific amyloid protein. For example, AL would identify amyloid composed of immunoglobulin light (L) chain molecules. Although amyloid may have several sources, all types of amyloid have the common feature of a b-pleated sheet molecular configuration, which can be seen with x-ray diffraction crystallographic analysis. Because of this similarity of molecular structure, the different types of amyloid have similar staining patterns with special stains.
Amyloidosis can produce a variety of effects, depending on the organ of involvement and the extent to which the amyloid is deposited. With limited cutaneous forms of amyloidosis, virtually no effect on survival is seen. With some forms of systemic amyloidosis, however, death may occur within a few years of the diagnosis as a result of cardiac or renal failure. Furthermore, the presence of amyloid may be associated with other problems, such as multiple myeloma or chronic infections.
CLINICAL FEATURES: Several classifications of amyloidosis have been proposed in the past decade, each evolving as the knowledge of this unusual condition increases. None of the classifications is completely satisfactory, although in recent years, the biochemical makeup of these proteins has figured more prominently in most classifications. This discussion attempts to be as concise and direct as possible. Essentially, amyloidosis may be divided into organ-limited and systemic forms from a clinical standpoint.
ORGAN-LIMITED AMYLOIDOSIS: Although organ-limited amyloidosis may occur in a variety of organs, it has rarely been reported in the oral soft tissues. An example of a limited form of amyloidosis is the amyloid nodule, which appears as a solitary, otherwise asymptomatic, submucosal deposit. Most of the organ-limited forms of amyloidosis consist of aggregates of immunoglobulin light chains, which in some cases are produced by a focal collection of monoclonal plasma cells. By definition, such amyloid deposits are not associated with any systemic alteration.
SYSTEMIC AMYLOIDOSIS: Systemic amyloidosis may occur in several forms:
Primary and Myeloma-Associated Amyloidosis: The primary and myeloma-associated forms of amyloidosis usually affect older adults (average age, 65 years), and a slight male predilection is present. These types of amyloidosis are caused by deposition of light chain molecules (thus the designation AL), with most cases being idiopathic, although approximately 15% to 20% are associated with multiple myeloma. The initial signs and symptoms may be nonspecific, often resulting in a delayed diagnosis. Fatigue, weight loss, paresthesia, hoarseness, edema, and orthostatic hypotension are among the first indications of this disease process. Eventually, carpal tunnel syndrome, mucocutaneous lesions, hepatomegaly, and macroglossia develop as a result of the deposition of the amyloid protein. The skin lesions appear as smooth-surfaced, firm, waxy papules and plaques. These most commonly affect the eyelid region (Fig. 17-7), the retroauricular region, the neck, and the lips. The lesions are often associated with petechiae and ecchymoses. Macroglossia has been reported in 10% to 40% of these patients and may appear as diffuse or nodular enlargement of the tongue (Fig. 17-8). Sometimes oral amyloid nodules show ulceration and submucosal hemorrhage overlying the lesions. Infrequently, patients may complain of dry eyes or dry mouth, which is secondary to amyloid infiltration and destruction of the lacrimal and salivary glands. When significant blood vessel infiltration has occurred, claudication of the jaw musculature may be noticed.
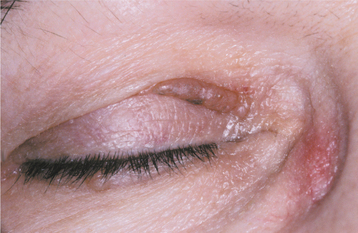
Fig. 17-7 Amyloidosis. This patient exhibits a firm, waxy nodular lesion in the periocular region, a finding that is characteristic of this condition.
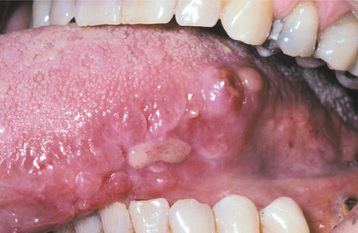
Fig. 17-8 Amyloidosis. Same patient as depicted in Fig. 17-7. Note amyloid nodules of lateral tongue, some of which are ulcerated. The patient’s amyloidosis was the result of previously undiagnosed multiple myeloma.
Secondary Amyloidosis: Secondary amyloidosis is so named because it characteristically develops as a result of a chronic inflammatory process, such as long-standing osteomyelitis, tuberculosis, or sarcoidosis. Cleavage fragments of a circulating acute-phase reactant protein appear to comprise this type of amyloidosis, which is thus designated AA. The heart is usually not affected as in other forms of amyloidosis. Liver, kidney, spleen, and adrenal involvement are typical, however. With the advent of modern antibiotic therapy, this form of amyloidosis has become much less common in the United States.
Hemodialysis-Associated Amyloidosis: Patients who have undergone long-term renal dialysis also are susceptible to amyloidosis, although in this case the amyloid protein has been identified as b2-microglobulin, and this type of amyloidosis is designated as Aβ2M. β2-Microglobulin is a normally occurring protein that is not removed by the dialysis procedure, and it accumulates in the plasma. Eventually, it forms deposits, particularly in the bones and joints. Often, carpal tunnel syndrome occurs, as well as cervical spine pain and dysfunction. Tongue involvement has been reported.
Heredofamilial Amyloidosis: Heredofamilial amyloidosis is an uncommon but significant form of the disease. Several kindred have been identified in Swedish, Portuguese, and Japanese populations, and most types are inherited as autosomal dominant traits. An autosomal recessive form, known as familial Mediterranean fever, has also been described. Several of these conditions appear as polyneuropathies, although other manifestations, such as cardiomyopathy, cardiac arrhythmias, congestive heart failure, and renal failure, eventually develop as the amyloid deposition continues.
HISTOPATHOLOGIC FEATURES: Biopsy of rectal mucosa has classically been used to confirm a diagnosis of primary or myeloma-associated amyloidosis, with up to 80% of such biopsy specimens being positive. Aspiration biopsy of abdominal subcutaneous fat is a simpler procedure, however, and the sensitivity of this technique has been reported to range from 55% to 75%. Alternative tissue sources, however, are the gingiva and labial salivary glands. Histopathologic examination of gingival tissue that has been affected by amyloidosis shows extracellular deposition in the submucosal connective tissue of an amorphous, eosinophilic material, which may be arranged in a perivascular orientation or may be diffusely present throughout the tissue (Fig. 17-9). Relatively low sensitivity has been reported for gingival biopsies, whereas labial salivary gland tissue shows deposition of amyloid in a periductal or perivascular location in more than 80% of the cases.
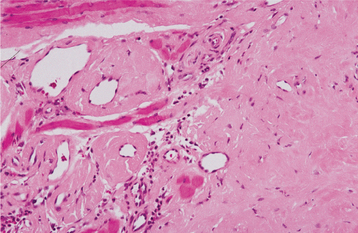
Fig. 17-9 Amyloidosis. This medium-power photomicrograph shows the eosinophilic, acellular deposits that are characteristic of amyloid deposition.
A standard means of identifying amyloid uses the dye, Congo red, which has an affinity for the abnormal protein. In tissue sections stained with Congo red, the amyloid appears red. When the tissue is viewed with polarized light, it exhibits an apple-green birefringence (Fig. 17-10). Microscopic sections stained with crystal violet reveal a characteristic metachromasia; this normally purple dye appears more reddish when it reacts with amyloid. Staining with thioflavine T, a fluorescent dye, also gives positive results if amyloid is present. Ultrastructurally, amyloid is seen as a collection of 7.5- to 10-nm diameter, nonbranching, linear fibrils.
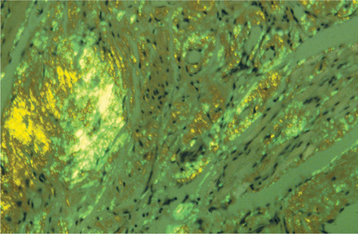
DIAGNOSIS: Once the histopathologic diagnosis of amyloidosis has been made, the patient must be evaluated medically to determine the type of amyloidosis that is present. This often entails a workup that includes serum immunoelectrophoresis to determine whether a monoclonal gammopathy exists so that multiple myeloma can be ruled out. Immunohistochemical studies are proving to be very useful in distinguishing the specific type of amyloid protein. Family history and physical examination findings are also important.
TREATMENT AND PROGNOSIS: In most instances, no effective therapy is available for amyloidosis. Surgical debulking of amyloid deposition in the tongue has met with limited success. Selected forms of amyloidosis may respond to treatment, or at least their progression may be slowed, depending on the underlying cause. In cases of secondary amyloidosis associated with an infectious agent, treatment of the infection and reduction of the inflammation often result in clinical improvement. Renal transplantation may arrest the progression of the bone lesions in hemodialysis-associated amyloidosis, but this procedure apparently does not reverse the process. Liver transplantation can improve the prognosis of several forms of inherited amyloidosis, particularly the transthyretin variant. Familial Mediterranean fever may respond to systemic colchicine therapy. Genetic counseling is also appropriate for patients affected by the inherited forms of amyloidosis. Treatment of primary amyloidosis (AL) with colchicine, prednisone, and melphalan appears to improve the prognosis of patients who do not have cardiac or renal involvement, although the outlook is guarded to poor in most instances. Most patients die of cardiac failure, arrhythmia, or renal disease within months to a few years after the diagnosis.
VITAMIN DEFICIENCY
In the United States today, significant vitamin deficiencies are not common. Patients with malabsorption syndromes or eating disorders, persons who follow “fad diets,” and alcoholics are the groups most commonly affected.
Vitamin A (retinol) is essential for the maintenance of vision, and it also plays a role in growth and tissue differentiation. Vitamin A can be obtained directly from dietary sources, such as organ meats (particularly liver), or the body can synthesize it from b-carotene, which is abundant in red and yellow vegetables.
Vitamin B1 (thiamin) acts as a coenzyme for several metabolic reactions and is thought to maintain the proper functioning of neurons. Thiamin is found in many animal and vegetable food sources.
Vitamin B2 (riboflavin) is necessary for cellular oxidation-reduction reactions. Foods that contain sig-nificant amounts of riboflavin include milk, green vegetables, lean meat, fish, legumes, and eggs.
Vitamin B3 (niacin) acts as a coenzyme for oxidation-reduction reactions. Rich sources include food from animal sources, especially lean meat and liver, milk, eggs, whole grains, peanuts, yeast, and cereal bran or germ.
Vitamin B6 (pyridoxine) serves as a cofactor associated with enzymes that participate in amino acid synthesis. It is found in many animal and vegetable food sources.
Vitamin C (ascorbic acid) is necessary for the proper synthesis of collagen. This vitamin is present in a wide variety of vegetables and fruits, although it is particularly abundant in citrus fruits.
Vitamin D, which is now considered to be a hormone, can be synthesized in adequate amounts within the epidermis if the skin is exposed to a moderate degree of sunlight. Most milk and processed cereal is fortified with vitamin D in the United States today, however. Appropriate levels of vitamin D and its active me-tabolites are necessary for calcium absorption from the gut.
Vitamin E (a-tocopherol) is a fat-soluble vitamin that is widely stored throughout the body. It probably functions as an antioxidant. Vegetable oils, meats, nuts, cereal grains, and fresh greens and vegetables are good sources of vitamin E.
Vitamin K is a fat-soluble vitamin found in a wide variety of green vegetables, as well as milk, butter, and liver; intestinal bacteria also produce it. This vitamin is necessary for the proper synthesis of various proteins, including the clotting factors II, VII, IX, and X.
VITAMIN A: A severe deficiency of vitamin A during infancy may result in blindness. The early changes associated with a lack of this vitamin later in life include an inability of the eye to adapt to reduced light conditions (i.e., night blindness). With more severe, prolonged deficiency, dryness of the skin and conjunctiva develop, and the ocular changes may progress to ulceration of the cornea, leading to blindness.
THIAMIN: A deficiency of thiamin results in a condition called beriberi, a problem that is relatively uncommon in the Western world except in alcoholics or other individuals who do not receive a balanced diet. Thiamin deficiency has also been documented in patients who have had gastric bypass surgery for weight control, presumably because an adequate amount of the vitamin is not obtained in the diet. The condition became prevalent in southeast Asia when the practice of removing the outer husks of the rice grain by machine was introduced. Because these outer husks contained nearly all of the thiamin, people who subsisted on the “polished” rice became deficient in this vitamin. The disorder is manifested by cardiovascular problems (e.g., peripheral vasodilation, heart failure, edema) and neurologic problems (including peripheral neuropathy and Wernicke’s encephalopathy). Patients with Wernicke’s encephalopathy experience vomiting, nystagmus, and progressive mental deterioration, which may lead to coma and death.
RIBOFLAVIN: A diet that is chronically deficient in riboflavin causes a number of oral alterations, including glossitis, angular cheilitis, sore throat, and swelling and erythema of the oral mucosa. A normocytic, normochromic anemia may be present, and seborrheic dermatitis may affect the skin.
NIACIN: A deficiency of niacin causes a condition known as pellagra, a term derived from the Italian words pelle agra, meaning rough skin. This condition may occur in populations that use maize as a principal component of their diets, because corn is a poor source of niacin. Pellagra was once common in the southeastern United States and may still be seen in some parts of the world. The classic systemic signs and symptoms include the triad of dermatitis, dementia, and diarrhea. The dermatitis is distributed symmetrically; sun-exposed areas, such as the face, neck, and forearms, are affected most severely (Fig. 17-11). The oral manifestations have been described as stomatitis and glossitis, with the tongue appearing red, smooth, and raw. Without correction of the niacin deficiency, the disease may evolve and persist over a period of years, eventually leading to death.
PYRIDOXINE: A deficiency of pyridoxine is unusual because of its widespread occurrence in a variety of foods. A number of drugs, such as the antituberculosis drug isoniazid, act as pyridoxine antagonists; therefore, patients who receive these medications may have a deficiency state. Because the vitamin plays a role in neuronal function, patients may show weakness, dizziness, or seizure disorders. Cheilitis and glossitis, reported in people with pellagra, are also reported in patients with pyridoxine deficiency.
VITAMIN C: A deficiency of vitamin C is known as scurvy, and its occurrence in the United States is usually limited to people whose diets lack fresh fruits and vegetables. Commonly affected groups include inner-city infants (whose diets often consist entirely of milk) and older edentulous men, particularly those who live alone.
The clinical signs of scurvy are typically related to inadequate collagen synthesis. For example, weakened vascular walls may result in widespread petechial hemorrhage and ecchymosis. Similarly, wound healing is delayed, and recently healed wounds may break down. In childhood, painful subperiosteal hemorrhages may occur.
The oral manifestations are well documented and include generalized gingival swelling with spontaneous hemorrhage, ulceration, tooth mobility, and increased severity of periodontal infection and periodontal bone loss. The gingival lesions have been termed scorbutic gingivitis (Fig. 17-12). If untreated, scurvy may ultimately lead to death, often as a result of intracranial hemorrhage.
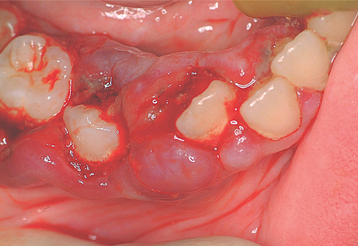
VITAMIN D: A deficiency of vitamin D during infancy results in a condition called rickets; adults who are deficient in this vitamin develop osteomalacia. With the vitamin-D supplementation of milk and cereal, rickets is a relatively uncommon disease today in the United States. In past centuries, however, rickets was often seen, particularly in the temperate zones of the world, which often do not receive adequate sunlight to ensure physiologic levels of vitamin D. Even today in the United States, children who are dark skinned and do not receive adequate sun exposure, as well as solely breast-fed infants, remain at risk for developing rickets. Nutritional rickets remains a problem in many developing countries, although the condition is thought to be associated more with calcium deficiency than vitamin-D deficiency.
Clinical manifestations of rickets include irritability, growth retardation, and prominence of the costochondral junctions (rachitic rosary). As the child ages and begins to put weight on the long bones of the legs, significant bowing results because of the poor mineralization of the skeleton.
A similar pattern of poorly mineralized bone is seen in osteomalacia in adults. Bone normally undergoes continuous remodeling and turnover, and the osteoid that is produced during this process does not have sufficient calcium to mineralize completely. Thus a weak, fragile bone structure results. Patients affected by osteomalacia frequently complain of diffuse skeletal pain, and their bones are susceptible to fracture with relatively minor injury.
VITAMIN E: A deficiency of vitamin E is rare and occurs primarily in children who suffer from chronic cholestatic liver disease. These patients have severe malabsorption of all fat-soluble vitamins, but particularly vitamin E. Multiple neurologic signs develop as a result of abnormalities in the central nervous system (CNS) and peripheral nervous system.
VITAMIN K: A deficiency of vitamin K may be seen in patients with malabsorption syndromes or in those whose intestinal microflora has been eliminated by long-term, broad-spectrum antibiotic use. Oral anticoagulants in the dicumarol family also inhibit the normal enzymatic activity of vitamin K. A deficiency or inhibition of synthesis of vitamin K leads to a coagulopathy because of the inadequate synthesis of prothrombin and other clotting factors. Intraorally, this coagulopathy is most often manifested by gingival bleeding. If the coagulopathy is not corrected, death may result from uncontrolled systemic hemorrhage.
TREATMENT AND PROGNOSIS: Replacement therapy is indicated for vitamin deficiencies. However, such deficiencies are uncommon, except for the situations described earlier. In fact, vitamin excess is perhaps more likely to be encountered in the United States today because so many people self-medicate with unnecessary and potentially harmful vitamin supplements. For example, excess vitamin A may cause abdominal pain, vomiting, headache, joint pain, and exostoses, whereas excess vitamin C may induce the formation of additional kidney stones in individuals with a history of nephrolithiasis.
IRON-DEFICIENCY ANEMIA
Iron-deficiency anemia is the most common cause of anemia in the United States and throughout the world. This form of anemia develops when the amount of iron available to the body cannot keep pace with the need for iron in the production of red blood cells (RBCs). This type of anemia develops under four conditions:
It is estimated that 20% of women of childbearing age in the United States are iron deficient as a result of the chronic blood loss associated with excessive menstrual flow (menorrhagia). Similarly, 2% of adult men are iron deficient because of chronic blood loss, usually associated with gastrointestinal disease, such as peptic ulcer disease, diverticulosis, hiatal hernia, or malignancy.
An increased demand for erythrocyte production occurs during childhood growth spurts and during pregnancy. A decreased intake of iron may be seen during infancy when the diet consists of relatively iron-poor foods, such as cereals and milk. Likewise, the diets of older people may be deficient if their dental condition prohibits them from eating the proper foods or if they cannot afford iron-rich foods, such as meats and vegetables. In the developing world, intestinal parasites (especially hookworms) are a common cause of iron deficiency in children and pregnant women.
Decreased absorption is a much less common problem; however, it can be seen in patients who have had a complete gastrectomy or who have celiac sprue, a condition that results in severe chronic diarrhea because of sensitivity to the plant protein, gluten.
CLINICAL FEATURES: Patients with iron-deficiency anemia that is severe enough to cause symptoms may complain of fatigue, easy tiring, palpitations, lightheadedness, and lack of energy. Oral manifestations include angular cheilitis and atrophic glossitis or generalized oral mucosal atrophy. The glossitis has been described as a diffuse or patchy atrophy of the dorsal tongue papillae, often accompanied by tenderness or a burning sensation. Such findings are also evident in oral candidiasis, and some investigators have suggested that iron deficiency predisposes the patient to candidal infection, which results in the changes seen at the corners of the mouth and on the tongue. Such lesions are rarely seen in the United States, perhaps because the anemia is usually detected relatively early before the oral mucosal changes have had a chance to develop.
LABORATORY FINDINGS: The diagnosis should be established by means of a complete blood count with RBC indices because many other conditions, such as hypothyroidism, other anemias, or chronic depression, may elicit similar systemic clinical complaints. The laboratory evaluation characteristically shows hypochromic microcytic RBCs in addition to reduced numbers of erythrocytes. Additional supporting evidence for iron deficiency includes the findings of low serum iron levels and ferritin concentration together with elevated total iron-binding capacity.
TREATMENT AND PROGNOSIS: Therapy for most cases of iron-deficiency anemia consists of dietary iron supplementation by means of oral ferrous sulfate. For patients with malabsorption problems, parenteral iron may be given periodically. The response to therapy is usually prompt, with red cell parameters returning to normal within 1 to 2 months. The underlying cause of the anemia should be identified so that it may be addressed, if feasible.
PLUMMER-VINSON SYNDROME (PATERSON-KELLY SYNDROME; SIDEROPENIC DYSPHAGIA)
Plummer-Vinson syndrome is a rare condition characterized by iron-deficiency anemia, seen in conjunction with glossitis and dysphagia. Its incidence in developed countries has been declining, probably as a result of the improved nutritional status of the populations. The condition is significant in that it has been associated with a high frequency of both oral and esophageal squamous cell carcinoma; therefore, it is considered a premalignant process.
CLINICAL AND RADIOGRAPHIC FEATURES: Most reported patients with Plummer-Vinson syndrome have been women of Scandinavian or Northern European background, between 30 and 50 years of age. Patients typically complain of a burning sensation associated with the tongue and oral mucosa. Sometimes this discomfort is so severe that dentures cannot be worn. Angular cheilitis is often present and may be severe (Fig. 17-13). Marked atrophy of the lingual papillae, which produces a smooth, red appearance of the dorsal tongue, is seen clinically (Fig. 17-14).
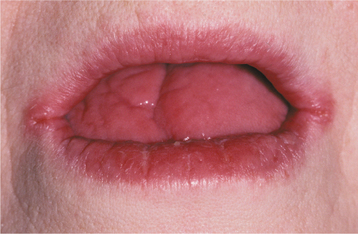
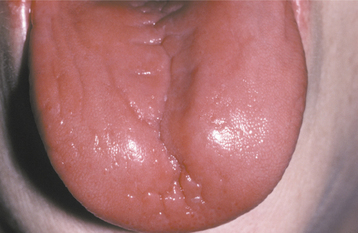
Fig. 17-14 Plummer-Vinson syndrome. The diffuse papillary atrophy of the dorsal tongue is characteristic of the oral changes. (From Neville BW, Damm DD, White DK: Color atlas of clinical oral pathology, ed 2, Philadelphia, 1999, Lippincott Williams & Wilkins.) Lippincott Williams & Wilkins
Patients also frequently complain of difficulty in swallowing (dysphagia) or pain on swallowing. An evaluation with endoscopy or esophageal barium contrast radiographic studies usually shows the presence of abnormal bands of tissue in the esophagus, called esophageal webs. Another sign is an alteration of the growth pattern of the nails, which results in a spoon-shaped configuration (koilonychia). The nails may also be brittle.
Symptoms of anemia may prompt patients with Plummer-Vinson syndrome to seek medical care. Fatigue, shortness of breath, and weakness are characteristic symptoms.
LABORATORY FINDINGS: Hematologic studies show a hypochromic microcytic anemia that is consistent with an iron-deficiency anemia.
HISTOPATHOLOGIC FEATURES: A biopsy specimen of involved mucosa from a patient with Plummer-Vinson syndrome typically shows epithelial atrophy with varying degrees of submucosal chronic inflammation. In advanced cases, evidence of epithelial atypia or dysplasia may be seen.
TREATMENT AND PROGNOSIS: Treatment of Plummer-Vinson syndrome is primarily directed at correcting the iron-deficiency anemia by means of dietary iron supplementation. This therapy usually resolves the anemia, relieves the glossodynia, and may reduce the severity of the esophageal symptoms. Occasionally, esophageal dilation is necessary to help improve the symptoms of dysphagia. Patients with Plummer-Vinson syndrome should be evaluated periodically for oral, hypopharyngeal, and esophageal cancer because a 5% to 50% prevalence of upper aerodigestive tract malignancy has been reported in affected persons.
PERNICIOUS ANEMIA
Pernicious anemia is an uncommon condition that occurs with greatest frequency among older patients of Northern European heritage, although recent studies have identified the disease in black and Hispanic populations as well. The disease is a megaloblastic anemia caused by poor absorption of cobalamin (vitamin B12, extrinsic factor). Intrinsic factor, which is produced by the parietal cells of the stomach lining, is needed for vitamin-B12 absorption. Normally, when cobalamin is ingested, it binds to intrinsic factor in the duodenum. Because the lining cells of the intestine preferentially take up the cobalamin-intrinsic factor complex, significant amounts of the vitamin cannot be absorbed unless both components are present.
In the case of pernicious anemia, most patients lack intrinsic factor because of an autoimmune destruction of the parietal cells of the stomach, and this results in decreased absorption of cobalamin. Antibodies directed against intrinsic factor are also found in the serum of these patients. Vitamin B12 deficiency may occur for other reasons, and although the resulting signs and symptoms may be identical to those of pernicious anemia, these should be considered as distinctly different deficiency disorders. For example, a decreased ability to absorb cobalamin may also occur after gastrointestinal bypass operations. In addition, because cobalamin is primarily derived from animal sources, some strict vegetarians (vegans) may develop vitamin B12 deficiency. In older patients, gastritis associated with Helicobacter pylori infection can result in decreased vitamin B12 absorption.
Because cobalamin is necessary for normal nucleic acid synthesis, anything that disrupts the absorption of the vitamin causes problems, especially for cells that are multiplying rapidly and, therefore, synthesizing large amounts of nucleic acids. The cells that are the most mitotically active are affected to the greatest degree, especially the hematopoietic cells and the gastrointestinal lining epithelial cells.
CLINICAL FEATURES: With respect to systemic complaints, patients with pernicious anemia often report fatigue, weakness, shortness of breath, headache, and feeling faint. Such symptoms are associated with most anemias and probably reflect the reduced oxygen-carrying capacity of the blood. In addition, many patients report paresthesia, tingling, or numbness of the extremities. Difficulty in walking and diminished vibratory and positional sense may be present. Psychiatric symptoms of memory loss, irritability, depression, and dementia have also been described.
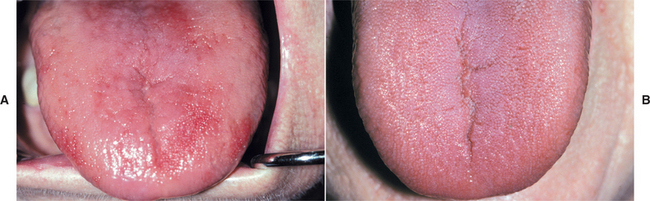
Oral symptoms often consist of a burning sensation of the tongue, lips, buccal mucosa, or other mucosal sites. Clinical examination may show focal patchy areas of oral mucosal erythema and atrophy (Fig. 17-15), or the process may be more diffuse, depending on the severity and duration of the condition. The tongue may be affected in as many as 50% to 60% of patients with pernicious anemia, but it may not show as much involvement as other areas of the oral mucosa in some instances. The atrophy and erythema may be easier to appreciate on the dorsal tongue than at other sites, however.
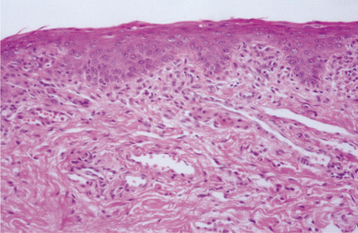
HISTOPATHOLOGIC FEATURES: Histopathologic examination of an erythematous portion of the oral mucosa shows marked epithelial atrophy with loss of rete ridges, an increased nuclear-to-cytoplasmic ratio, and prominent nucleoli (Fig. 17-16). This pattern can be misinterpreted as epithelial dysplasia at times, although the nuclei in pernicious anemia typically are pale staining and show peripheral chromatin clumping. A patchy diffuse chronic inflammatory cell infiltrate is usually noted in the underlying connective tissue.
LABORATORY FINDINGS: Hematologic evaluation of vitamin B12 deficiency shows a macrocytic anemia and reduced serum cobalamin levels. The Schilling test for pernicious anemia has been used to determine the pathogenesis of the cobalamin deficiency by comparing absorption and excretion rates of radiolabeled cobalamin. However, this study is rather complicated to perform, and it appears to be falling out of favor. The presence of serum antibodies directed against intrinsic factor is quite specific for pernicious anemia.
TREATMENT AND PROGNOSIS: Once the diagnosis of pernicious anemia is established, treatment traditionally has consisted of monthly intramuscular injections of cyanocobalamin. The condition responds rapidly once therapy is initiated, with reports of clearing of oral lesions within 5 days. High-dose oral cobalamin therapy has also been shown to be an equally effective treatment, however, with advantages being its cost-effectiveness and the elimination of painful injections. One study has confirmed an increased risk of malignancy, particularly gastric carcinoma, a complication that affects between 1% and 2% of pernicious anemia patients.
PITUITARY DWARFISM
Pituitary dwarfism is a relatively rare condition that results from either the diminished production of growth hormone by the anterior pituitary gland or a reduced capacity of the tissues to respond to growth hormone. Affected patients are typically much shorter than normal, although their body proportions are generally appropriate.
Several conditions may cause short stature, and a careful evaluation of the patient must be performed to rule out other possible causes, such as the following:
1. Intrinsic defects in the patient’s tissues (e.g., certain skeletal dysplasias, chromosomal abnormalities, idiopathic short stature)
2. Alterations in the environment of the growing tissues (e.g., malnutrition, hypothyroidism, diabetes mellitus)
If a lack of growth hormone is detected, the cause should be determined. Sometimes the fault lies with the pituitary gland itself (e.g., aplasia, hypoplasia). In other instances, the problem may be related to destruction of the pituitary or hypothalamus by tumors, therapeutic radiation, or infection.
If the hypothalamus is affected, a deficiency in growth hormone-releasing hormone, which is produced by the hypothalamus, results in a deficiency of growth hormone. Often deficiencies in other hormones, such as thyroid hormone and cortisol, are also detected in patients with primary pituitary or hypothalamic disorders.
Some patients exhibit normal or even elevated levels of growth hormone, yet still show little evidence of growth. These individuals usually have inherited an autosomal recessive trait, resulting in abnormal and reduced growth hormone receptors on the patients’ cells. Thus normal growth cannot proceed.
CLINICAL FEATURES: Perhaps the most striking feature of pituitary dwarfism is the remarkably short stature of the affected patient. Sometimes this is not noticed until the early years of childhood, but a review of the patient’s growth history should show a consistent pattern of failure to achieve the minimal height on the standard growth chart. Often the patient’s height may be as much as three standard deviations below normal for a given age. Unlike the body proportions in many of the dysmorphic syndromes and skeletal dysplasias, the body proportions of patients affected by a lack of growth hormone are usually normal. One possible exception is the size of the skull, which is usually within normal limits. Because the facial skeleton does not keep pace with the skull, however, the face of an affected patient may appear smaller than it should be. Mental status is generally within normal limits.
The maxilla and mandible of affected patients are smaller than normal, and the teeth show a delayed pattern of eruption. The delay ranges from 1 to 3 years for teeth that normally erupt during the first decade of life and from 3 to 10 years for teeth that normally erupt in the second decade of life. Often the shedding of deciduous teeth is delayed by several years, and the development of the roots of the permanent teeth also appears to be delayed. A lack of development of the third molars seems to be a common finding. The size of the teeth is usually reduced in proportion to the other anatomic structures.
LABORATORY FINDINGS: Radioimmunoassay for human growth hormone shows levels that are markedly below normal.
TREATMENT AND PROGNOSIS: Replacement therapy with human growth hormone is the treatment of choice for patients with pituitary dwarfism if the disorder is detected before closure of the epiphyseal growth plates. In the past, growth hormone was extracted from cadaveric pituitary glands; today, genetically engineered human growth hormone is produced with recombinant DNA technology. For patients with a growth hormone deficiency caused by a hypothalamic defect, treatment with growth hormone-releasing hormone is appropriate. If patients are identified and treated at an early age, they can be expected to achieve a relatively normal height. The craniofacial bone structure also assumes a less childlike pattern. Evaluation of a series of patients who had been treated for long periods with growth hormone determined that up to half developed acromegalic features, including larger feet and a larger mandible. For patients who lack growth hormone receptors, no treatment is available.
GIGANTISM
Gigantism is a rare condition caused by an increased production of growth hormone, usually related to a functional pituitary adenoma. The increased production of growth hormone takes place before closure of the epiphyseal plates, and the affected person grows at a much more rapid pace, becoming abnormally tall. Although the average height of the population of the United States has been gradually increasing during the past several decades, individuals who exceed the mean height by more than three standard deviations may be considered candidates for endocrinologic evalua-tion. Familial examples of gigantism have also been described.
CLINICAL AND RADIOGRAPHIC FEATURES: Patients with gigantism usually show markedly accelerated growth during childhood, irrespective of normal growth spurts. Radiographic evaluation of the skull often shows an enlarged sella as a result of the presence of a pituitary adenoma. The adenoma may result in hormonal deficiencies, such as hypothyroidism and hypoadrenocorticism, if the remaining normal pituitary gland tissue is compressed and destroyed. McCune-Albright syndrome (polyostotic fibrous dysplasia and café au lait pigmentation with associated endocrinologic disturbances) (see page 636) may account for as many as 20% of the cases of gigantism.
If the condition remains uncorrected for a prolonged period, extreme height (more than 7 feet tall) will be achieved, and enlargement of the facial soft tissues, the mandible, and the hands and feet will become apparent. These changes often resemble those seen in acromegaly (discussed later). Another oral finding is true generalized macrodontia.
TREATMENT AND PROGNOSIS: Appropriate management of gigantism involves the surgical removal of the functioning pituitary ade-noma, usually by a transsphenoidal approach. Radiation therapy may also be used.
The life span of patients with gigantism is usually markedly reduced. Complications associated with hypertension, peripheral neuropathy, osteoporosis, and pulmonary disease contribute to increased morbidity and mortality.
ACROMEGALY
Acromegaly is an uncommon condition characterized by the excess production of growth hormone after closure of the epiphyseal plates in the affected patient. Usually, this increase in growth hormone is due to a functional pituitary adenoma. The incidence is estimated to be approximately three to five new cases diagnosed per million population per year. The prevalence is believed to be 66 affected patients per million.
CLINICAL AND RADIOGRAPHIC FEATURES: Because most patients with acromegaly have a pituitary adenoma, symptoms related directly to the space-occupying mass of the tumor may be present. These symptoms include headaches, visual disturbances, and other signs of a brain tumor. Sometimes pressure atrophy of the residual normal pituitary by the adenoma results in diminished production of other pituitary hormones and causes other indirect endocrine problems. The direct effects of increased levels of growth hormone include a variety of problems, such as hypertension, heart disease, hyperhidrosis, arthritis, and peripheral neuropathy.
Renewed growth in the small bones of the hands and feet (Fig. 17-17) and in the membranous bones of the skull and jaws is typically observed. Patients may complain of gloves or hats becoming “too small.” The soft tissue is also often affected, producing a coarse facial appearance (Fig. 17-18). Hypertrophy of the soft palatal tissues may cause or accentuate sleep apnea. Because these signs and symptoms are slow to develop and are vague at the onset, an average time of nearly 9 years elapses from the onset of symptoms to the diagnosis of disease. The average age at diagnosis is 42 years, and no sex predilection is seen.
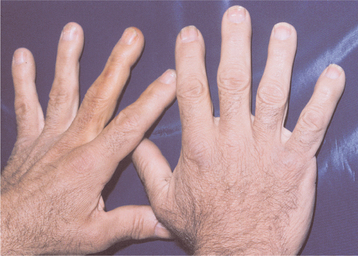
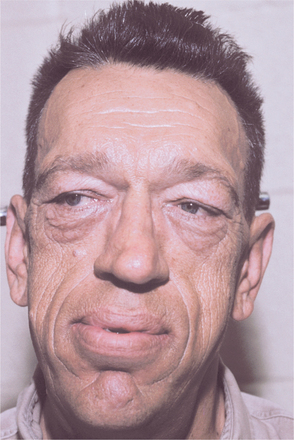
Fig. 17-18 Acromegaly. This patient shows the typical coarse facial features. (Courtesy of Dr. William Bruce.)
From a dental perspective, these patients have mandibular prognathism as a result of the increased growth of the mandible (Fig. 17-19), which may cause apertognathia (anterior open bite). Growth of the jaws also may cause spacing of the teeth, resulting in diastema formation. Soft tissue growth often produces uniform macroglossia in affected patients.
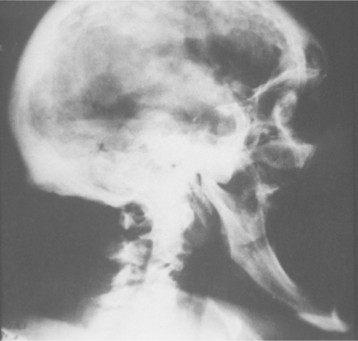
LABORATORY FINDINGS AND DIAGNOSIS: If acromegaly is suspected, measurement of serum growth hormone levels is done after giving the patient a measured quantity of glucose orally. Normally, this glucose challenge will reduce the production of growth hormone, but if the patient has acromegaly, growth hormone will not be suppressed. Usually magnetic resonance imaging (MRI) will identify the pituitary adenoma that is responsible for inappropriate growth hormone secretion.
TREATMENT AND PROGNOSIS: The treatment of a patient with acromegaly is typically directed at the removal of the pituitary tumor mass and the return of the growth hormone levels to normal. The most effective treatment with the least associated morbidity is surgical excision by a transsphenoidal approach. The prognosis for such a procedure is good, although a mortality rate of approximately 1% is still expected. The condition is usually controlled with this procedure, but patients with larger tumors and markedly elevated growth hormone levels are less likely to be controlled.
Radiation therapy may be used in some instances, but the return of the growth hormone levels to normal is not as rapid or as predictable as with surgery. Because some patients also experience hypopituitarism caused by radiation effects on the rest of the gland, some centers may offer radiation therapy as treatment only when surgery fails or is too risky. Pharmacotherapy with one of the somatostatin analogues (e.g., octreotide, lanreotide, vapreotide) helps to control acromegaly if surgical treatment is unsuccessful or if surgery is contraindicated. A growth hormone receptor-blocking agent, pegvisomant, has also been developed and may be used in conjunction with one of the somatostatin analogues or by itself if the patient cannot tolerate the somatostatin analogue. Pegvisomant is injected daily and acts in the peripheral tissues to inhibit the action of growth hormone. These drugs are also used as an adjunct to radiation therapy during the prolonged period that is sometimes necessary for that treatment to take effect.
The prognosis for untreated patients is guarded, with an increased mortality rate compared with that of the general population. Hypertension, diabetes mellitus, coronary artery disease, congestive heart failure, respiratory disease, and colon cancer are seen with increased frequency in acromegalic patients, and each of these contributes to the increased mortality rate. Although treatment of the patient with acromegaly helps to control many of the other complicating problems and improves the prognosis, the life span of these patients still is shortened.
HYPOTHYROIDISM (CRETINISM; MYXEDEMA)
Hypothyroidism is a condition that is characterized by decreased levels of thyroid hormone. When this decrease occurs during infancy, the resulting clinical problem is known as cretinism. If an adult has markedly decreased thyroid hormone levels for a prolonged period, then deposition of a glycosaminoglycan ground substance is seen in the subcutaneous tissues, producing a nonpitting edema. Some call this severe form of hypothyroidism myxedema; others use the terms myxedema and hypothyroidism interchangeably.
Hypothyroidism may be classified as either primary or secondary. In primary hypothyroidism, the thyroid gland itself is in some way abnormal; in secondary hypothyroidism, the pituitary gland does not produce an adequate amount of thyroid-stimulating hormone (TSH), which is necessary for the appropriate release of thyroid hormone. Secondary hypothyroidism, for example, often develops after radiation therapy for brain tumors, resulting in unavoidable radiation damage to the pituitary gland. Most cases, however, represent the primary form of the disease.
Screening for this disorder is routinely carried out at birth, and the prevalence of congenital hypothyroidism in North America is approximately 1 in 4000 births. Usually, this is due to hypoplasia or agenesis of the thyroid gland. In other areas of the world, hypothyroidism in infancy is usually due to a lack of dietary iodine. In adults, hypothyroidism is often caused by autoimmune destruction of the thyroid gland (known as Hashimoto’s thyroiditis) or iatrogenic factors, such as radioactive iodine therapy or surgery for the treatment of hyperthyroidism. Because thyroid hormone is necessary for normal cellular metabolism, many of the clinical signs and symptoms of hypothyroidism can be related to the decreased metabolic rate in these patients.
CLINICAL FEATURES: The most common features of hypothyroidism include such signs and symptoms as lethargy, dry and coarse skin, swelling of the face (Fig. 17-20) and extremities, huskiness of the voice, constipation, weakness, and fatigue. The heart rate is usually slowed (bradycardia). Reduced body temperature (hypothermia) may be present, and the skin often feels cool and dry to the touch. In the infant, these signs may not be readily apparent, and the failure to grow normally may be the first indication of the disease.
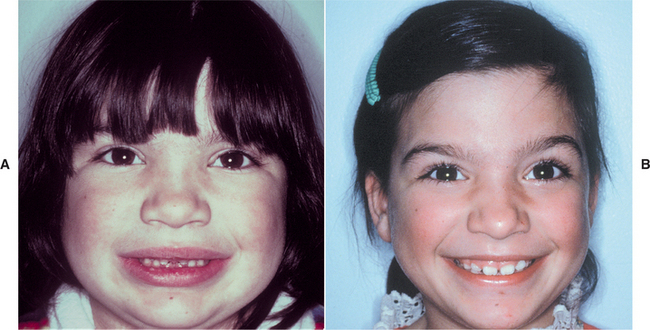
Fig. 17-20 Hypothyroidism. A, The facial appearance of this 9-year-old child is due to the accumulation of tissue edema secondary to severe hypothyroidism. B, Same patient after 1 year of thyroid hormone replacement therapy. Note the eruption of the maxillary permanent teeth.
With respect to the oral findings, the lips may appear thickened because of the accumulation of glycosaminoglycans. Diffuse enlargement of the tongue occurs for the same reason (Fig. 17-21). If the condition develops during childhood, the teeth may fail to erupt, although tooth formation may not be impaired (Figs. 17-22 and 17-23).
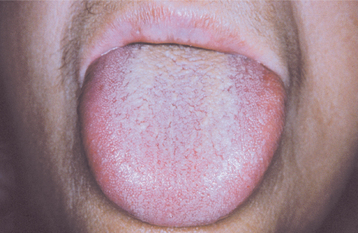
Fig. 17-21 Hypothyroidism. The enlarged tongue (macroglossia) is secondary to edema associated with adult hypothyroidism (myxedema). (Courtesy of Dr. George Blozis.)
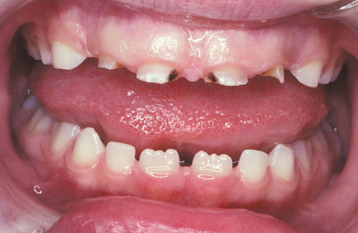
Fig. 17-22 Hypothyroidism. Photograph of the same patient depicted in Fig. 17-20 before hormone replacement therapy. Note the retained deciduous teeth, for which the patient was initially referred.
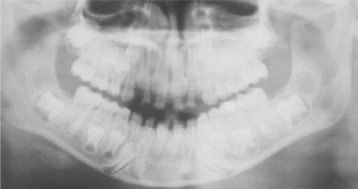
Fig. 17-23 Hypothyroidism. Panoramic radiograph of the same patient in Figs. 17-20 and 17-22. Note the unerupted, yet fully developed permanent dentition.
LABORATORY FINDINGS: The diagnosis is made by assaying the free thyroxine (T4) levels. If these levels are low, then TSH levels are measured to determine whether primary or secondary hypothyroidism is present. With primary thyroid disease, TSH levels are elevated. With secondary disease caused by pituitary dysfunction, TSH levels are normal or borderline.
TREATMENT AND PROGNOSIS: Thyroid replacement therapy, usually with levothyroxine, is indicated for confirmed cases of hypothyroidism. The prognosis is generally good for adult patients. If the condition is recognized within a reasonable time, the prognosis is also good for children. If the condition is not identified in a timely manner, however, permanent damage to the central nervous system may occur, resulting in mental retardation. For affected children, thyroid hormone replacement therapy often results in a dramatic resolution of the condition (see Fig. 17-20).
HYPERTHYROIDISM (THYROTOXICOSIS; GRAVES’ DISEASE)
Hyperthyroidism is a condition caused by excess production of thyroid hormone. This excess production results in a state of markedly increased metabolism in the affected patient. Most cases (60% to 90%) are due to Graves’ disease, a condition that was initially described in the early nineteenth century. It is thought to be triggered by autoantibodies that are directed against receptors for thyroid-stimulating hormone (TSH) on the surface of the thyroid cells. When the autoantibodies bind to these receptors, they seem to stimulate the thyroid cells to release inappropriate thyroid hormone.
Other causes of hyperthyroidism include hyperplastic thyroid tissue and thyroid tumors, both benign and malignant, which secrete inappropriate thyroid hormone. Similarly, a pituitary adenoma may produce TSH, which can then stimulate the thyroid to secrete excess thyroid hormone.
CLINICAL FEATURES: Graves’ disease is five to 10 times more common in women than in men and is seen with some frequency. It affects almost 2% of the adult female population. Graves’disease is most commonly diagnosed in patients during the third and fourth decades of life.
Most patients with Graves’ disease exhibit diffuse thyroid enlargement. Many of the signs and symptoms of hyperthyroidism can be attributed to an increased metabolic rate caused by the excess thyroid hormone. Patients usually complain about nervousness, heart palpitations, heat intolerance, emotional lability, and muscle weakness. The following are often noted during the clinical evaluation:
• Weight loss despite increased appetite
• Widened pulse pressure (increased systolic and decreased diastolic pressures)
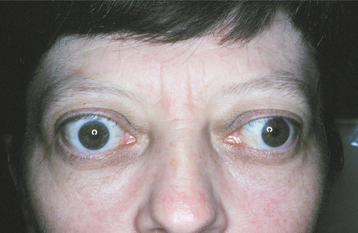
Ocular involvement, which develops in 20% to 40% of affected patients, is perhaps the most striking feature of this disease. In the early stages of hyperthyroidism, patients have a characteristic stare with eyelid retraction and lid lag. With some forms of Graves’ disease, protrusion of the eyes (exophthalmos or proptosis) develops (Fig. 17-24). This bulging of the eyes is due to an accumulation of glycosaminoglycans in the retro-orbital connective tissues.
LABORATORY FINDINGS: The diagnosis of hyperthyroidism is made by assaying free T4 (thyroxine) and TSH levels in the serum. In affected patients, the T4 levels should be elevated and the TSH concentration is typically depressed.
HISTOPATHOLOGIC FEATURES: Diffuse enlargement and hypercellularity of the thy-roid gland are seen in patients with Graves’ disease, typically with hyperplastic thyroid epithelium and little apparent colloid production. Lymphocytic infiltration of the glandular parenchyma is also often noted.
TREATMENT AND PROGNOSIS: In the United States, radioactive iodine (131I) is the most commonly used form of therapy for adult patients with Graves’ disease. The thyroid gland normally takes up iodine from the bloodstream because this element is a critical component of thyroid hormone. When radioactive iodine is given to a patient with Graves’ disease, the thyroid gland quickly removes it from the bloodstream and sequesters the radioactive material within the glandular tissue. The radioactivity then destroys the hyperactive thyroid tissue, bringing the thyroid hormone levels back to normal. Most of the radiation is received during the first few weeks because the half-life of 131I is short.
Other techniques include drug therapy with agents that block the normal use of iodine by the thyroid gland, and this form of therapy is initially favored in most European centers. The two widely used drugs are propylthiouracil and methimazole. At times, they are used before the radioactive iodine therapy. Sometimes they may be administered chronically in the hope that a remission may be induced. In addition, a portion of the thyroid gland may be removed surgically, thereby reducing thyroid hormone production.
Drug therapy alone is often unsuccessful in controlling hyperthyroidism. Unfortunately, with radioactive iodine and surgery, the risk of hypothyroidism is relatively great, although thyroid hormone replacement therapy can be instituted, if needed.
In a patient with uncontrolled hyperthyroidism, a definite risk exists with respect to an inappropriate release of large amounts of thyroid hormone at one time, resulting in a condition called a thyroid storm. A thyroid storm may be precipitated by infection, psychologic trauma, or stress. Clinically, patients may have delirium, convulsions, an elevated tempera-ture (up to 106° F), and tachycardia (sometimes more than 140 beats/minute). Such individuals should be hospitalized immediately because the mortality rate associated with thyroid storm is 20% to 50%. The clinician should be aware of the potential for this problem, and patients with hyperthyroidism should ideally have the condition under control before dental treatment.
HYPOPARATHYROIDISM
Calcium levels in extracellular tissues are normally regulated by parathyroid hormone (PTH) (parathormone) in conjunction with vitamin D. If calcium levels drop below a certain point, then the release of PTH is stimulated. The hormone then acts directly on the kidney and the osteoclasts of the bone to restore the calcium to normal levels. In the kidney, calcium reabsorption is promoted, phosphate excretion is enhanced, and the production of vitamin D is stimulated, which increases the absorption of calcium from the gut. Osteoclasts are activated to resorb bone and thus liberate calcium.
If a reduced amount of PTH is produced, the relatively rare condition known as hypoparathyroidism results. Usually, hypoparathyroidism is due to inadvertent surgical removal of the parathyroid glands when the thyroid gland is excised for other reasons, but sometimes it is the result of autoimmune destruction of the parathyroid tissue. Rare syndromes, such as DiGeorge syndrome and the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome (endocrine-candidiasis syndrome), may be associated with hypoparathyroidism.
CLINICAL FEATURES: With the loss of parathyroid function, the serum levels of calcium drop, resulting in hypocalcemia. Often the patient with chronic hypoparathyroidism adapts to the presence of hypocalcemia and is asymptomatic unless situations that further reduce the calcium levels are encountered. Such situations include metabolic alkalosis, as seen during hyperventilation, when a state of tetany may become evident.
Chvostek’s sign is an oral finding of significance, characterized by a twitching of the upper lip when the facial nerve is tapped just below the zygomatic process. A positive response suggests a latent degree of tetany. If the hypoparathyroidism develops early in life during odontogenesis, then a pitting enamel hypoplasia and failure of tooth eruption may occur (Fig. 17-25). The presence of persistent oral candidiasis in a young patient may signal the onset of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome (see page 219). Hypoparathyroidism may be only one of several endocrine deficiencies associated with this condition.
LABORATORY FINDINGS: PTH can be measured by means of a radioimmunoassay. If serum PTH levels are decreased in conjunction with a decreased serum calcium concentration, elevated serum phosphate level, and normal renal function, then a diagnosis of hypoparathyroidism can be made.
TREATMENT AND PROGNOSIS: Patients with hypoparathyroidism are usually treated with oral doses of a vitamin-D precursor (ergocalciferol, vitamin D2). Additional supplements of dietary calcium may also be necessary to maintain the proper serum calcium levels. With this regimen, patients can often live a fairly normal life. Teriparatide, a recombinant form of the active component of human parathormone, has been developed recently. When given twice daily as subcutaneous injections, this drug has also shown promise as an alternative management strategy for hypoparathyroidism, although it is relatively expensive.
PSEUDOHYPOPARATHYROIDISM (ALBRIGHT HEREDITARY OSTEODYSTROPHY; ACRODYSOSTOSIS)
The rare condition known as pseudohypoparathyroidism represents at least two broad disorders in which normal parathyroid hormone (PTH) is present in adequate amounts but the biochemical pathways responsible for activating the target cells are not functioning properly. The clinical result is a patient who appears to have hypoparathyroidism.
In the case of pseudohypoparathyroidism type I, three subcategories have been defined. For type Ia, a molecular defect of a specific intracellular binding protein known as Gsa seems to prevent the formation of cyclic adenosine monophosphate (cAMP), a critical component in the activation of cell metabolism. Because other hormones also require binding with Gsa to carry out their functions, patients have multiple problems with other endocrine organs and functions. This condition is usually inherited as an autosomal dominant trait.
With respect to pseudohypoparathyroidism type Ib, the problem is thought to be caused by defective receptors for the PTH on the surface of the target cells (the proximal renal tubules). For this reason, no other endocrine tissues or functions are affected. An autosomal dominant mode of inheritance has been suggested for a few families affected by type Ib pseudohypoparathyroidism, but most cases are apparently sporadic. The mechanism of action for pseudohypoparathyroidism type Ic is less clear, but it may involve a defect in adenylate cyclase or a subtle Gsa alteration.
Pseudohypoparathyroidism type II is characterized by the induction of cAMP by PTH in the target cells; however, a functional response by the cells is not invoked. All of the reported cases of this form of the disease appear to be sporadic.
CLINICAL FEATURES: Pseudohypoparathyroidism most commonly appears as type Ia disease. Patients affected by pseudohypoparathyroidism, either type Ia or Ic, have a characteristic array of features that includes mild mental retardation, obesity, round face, short neck, and markedly short stature. Midfacial hypoplasia is also commonly observed. The metacarpals and metatarsals are usually shortened, and the fingers appear short and thick. Subcutaneous calcifications (osteoma cutis) may be identified in some patients. Other endocrine abnormalities that are typically encountered include hypogonadism and hypothyroidism.
Patients with type Ib and II disease clinically appear normal, aside from their symptoms of hypocalcemia.
Dental manifestations of pseudohypoparathyroidism include generalized enamel hypoplasia, widened pulp chambers with intrapulpal calcifications, oligodontia, delayed eruption, and blunting of the apices of the teeth. The pulpal calcifications are often described as “dagger” shaped.
The diagnosis of pseudohypoparathyroidism is made based on elevated serum levels of PTH seen concurrently with hypocalcemia, hyperphosphatemia, and otherwise normal renal function. More sophisticated studies are necessary to delineate the various subtypes.
TREATMENT AND PROGNOSIS: Pseudohypoparathyroidism is managed by the administration of vitamin D and calcium. The serum calcium levels and urinary calcium excretion are carefully monitored. Because of individual patient differences, the medication may need to be carefully adjusted; however, the prognosis is considered to be good.
HYPERPARATHYROIDISM
Excess production of parathyroid hormone (PTH) results in the condition known as hyperparathyroidism. PTH normally is produced by the parathyroid glands in response to a decrease in serum calcium levels.
Primary hyperparathyroidism is the uncontrolled production of PTH, usually as a result of a parathyroid adenoma (80% to 90% of cases) or parathyroid hyperplasia (10% to 15% of cases). Rarely (approximately 1% of cases), a parathyroid carcinoma may be the cause of primary hyperparathyroidism. Infrequently this endocrine disturbance is caused by any one of several inherited syndromes, including multiple endocrine neoplasia type 1 or type 2a, or hyperparathyroidism-jaw tumor syndrome. In the latter condition, affected patients develop multiple jaw lesions that histopathologically are consistent with central cemento-ossifying fibroma. There also appears to be an in-creased risk for these patients to develop parathyroid carcinoma.
Secondary hyperparathyroidism develops when PTH is continuously produced in response to chronic low levels of serum calcium, a situation usually associated with chronic renal disease. The kidney processes vitamin D, which is necessary for calcium absorption from the gut. Therefore, in a patient with chronic renal disease, active vitamin D is not produced and less calcium is absorbed from the gut, resulting in lowered serum calcium levels.
CLINICAL AND RADIOGRAPHIC FEATURES: Most patients with primary hyperparathyroidism are older than 60 years of age. Women have this condition two to four times more often than men do. Typically the condition is identified on routine serologic testing, and the majority of patients are relatively asymptomatic.
Patients with the classic triad of signs and symptoms of hyperparathyroidism are described as having “stones, bones, and abdominal groans.”
Stones refers to the fact that these patients, particularly those with primary hyperparathyroidism, have a marked tendency to develop renal calculi (kidney stones, nephrolithiasis) because of the elevated serum calcium levels. Metastatic calcifications are also seen, frequently involving other soft tissues, such as blood vessel walls, subcutaneous soft tissues, the sclera, the dura, and the regions around the joints.
Bones refers to a variety of osseous changes that may occur in conjunction with hyperparathyroidism. One of the first clinical signs of this disease is seen radiographically as subperiosteal resorption of the phalanges of the index and middle fingers. Generalized loss of the lamina dura surrounding the roots of the teeth is also seen as an early manifestation of the condition (Fig. 17-26). Alterations in trabecular pattern characteristically develop next. A decrease in trabecular density and blurring of the normal trabecular pattern occur; often a “ground glass” appearance results.
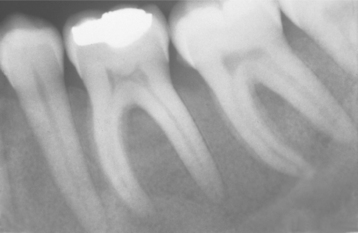
Fig. 17-26 Hyperparathyroidism. This periapical radiograph reveals the “ground glass” appearance of the trabeculae and loss of lamina dura in a patient with secondary hyperparathyroidism. (Courtesy of Dr. Randy Anderson.)
With persistent disease, other osseous lesions develop, such as the so-called brown tumor of hyperparathyroidism. This lesion derives its name from the color of the tissue specimen, which is usually a dark red-brown because of the abundant hemorrhage and hemosiderin deposition within the tumor. These lesions appear radiographically as well-demarcated unilocular or multilocular radiolucencies (Fig. 17-27). They commonly affect the mandible, clavicles, ribs, and pelvis. They may be solitary but are often multiple, and long-standing lesions may produce significant cortical expansion. Typically, the other osseous changes are observable if brown tumors are present. The most severe skeletal manifestation of chronic hyperparathyroidism has been called osteitis fibrosa cystica, a condition that develops from the central degeneration and fibrosis of long-standing brown tumors. In patients with secondary hyperparathyroidism caused by end-stage renal disease (renal osteodystrophy), striking enlargement of the jaws has been known to occur (Fig. 17-28) and produce a “ground-glass” radiographic pattern (see Fig. 17-26).
Fig. 17-27 Hyperparathyroidism. This occlusal radiograph of the edentulous maxillary anterior region shows a multilocular radiolucency characteristic of a brown tumor of primary hyperparathyroidism. (Courtesy of Dr. Brian Blocher.)
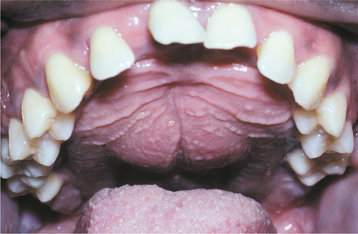
Fig. 17-28 Hyperparathyroidism. Palatal enlargement is characteristic of the renal osteodystrophy associated with secondary hyperparathyroidism.
Abdominal groans refers to the tendency for the development of duodenal ulcers. In addition, changes in mental status are often seen, ranging from lethargy and weakness to confusion or dementia.
HISTOPATHOLOGIC FEATURES: The brown tumor of hyperparathyroidism is histopathologically identical to the central giant cell granuloma of the jaws, a benign tumorlike lesion that usually affects teenagers and young adults (see page 626). Both lesions are characterized by a proliferation of exceedingly vascular granulation tissue, which serves as a background for numerous multinucleated osteoclast-type giant cells (Fig. 17-29). Some lesions may also show a proliferative response characterized by a parallel arrangement of spicules of woven bone set in a cellular fibroblastic background with variable numbers of multinucleated giant cells (Fig. 17-30). This pattern is often associated with secondary hyperparathyroidism related to chronic renal disease (renal osteodystrophy).
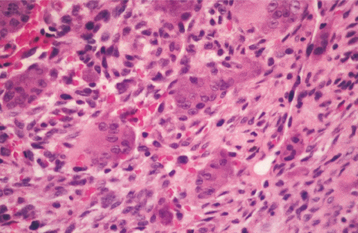
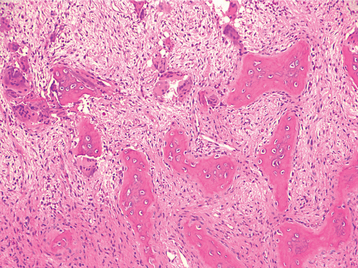
TREATMENT AND PROGNOSIS: In primary hyperparathyroidism, the hyperplastic parathyroid tissue or the functional tumor must be removed surgically to reduce PTH levels to normal.
Secondary hyperparathyroidism may evolve to produce signs and symptoms related to renal calculi or renal osteodystrophy. Restriction of dietary phosphate, use of phosphate-binding agents, and pharmacologic treatment with an active vitamin D metabolite (e.g., calcitriol) may avert problems. Exposure to aluminum salts, which inhibit bone mineralization, should be eliminated also. Patients who do not respond to medical therapy may require parathyroidectomy. Renal transplantation may restore the normal physiologic processing of vitamin D, as well as phosphorus and calcium reabsorption and excretion; however, this does not occur in every case. Cinacalcet is a recently approved medical treatment for managing the overproduction of parathormone associated with secondary hyperparathyroidism. This medication is a calcimimetic agent that sensitizes the calcium receptors of the parathyroid cells to extracellular calcium, causing the cells to reduce their output of parathormone.
HYPERCORTISOLISM (CUSHING’S SYNDROME)
Hypercortisolism is a clinical condition that results from a sustained increase in glucocorticoid levels. In most cases this increase is due to corticosteroid therapy that is prescribed for other medical purposes. If the increase is caused by an endogenous source, such as an adrenal or pituitary (adrenocorticotropic hormone [ACTH]-secreting) tumor, then the condition is known as Cushing’s disease. This latter condition is rather rare and usually affects young adult women.
CLINICAL FEATURES: The signs of Cushing’s syndrome usually develop slowly. The most consistent clinical observation is weight gain, particularly in the central areas of the body. The accumulation of fat in the dorsocervical spine region results in a “buffalo hump” appearance; fatty tissue deposition in the facial area results in the characteristic rounded facial appearance known as moon facies (Fig. 17-31). Other common findings include the following:
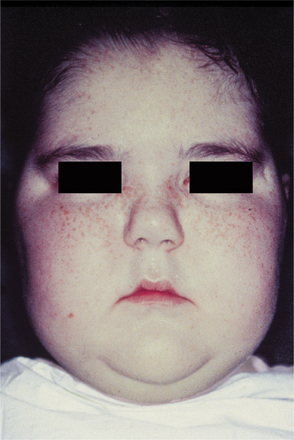
DIAGNOSIS: If the patient has been receiving large amounts of corticosteroids (greater than the equivalent of 20 mg of prednisone) on a daily basis for several months, then the diagnosis is rather obvious, given the classic signs and symptoms described earlier. The diagnosis may be more difficult to establish in patients with a functioning adrenal cortical tumor or an ACTH-secreting pituitary adenoma. Evaluation of these patients should include the measurement of free cortisol in the urine and an assay of the effect of dexamethasone (a potent artificial corticosteroid) on the serum ACTH and cortisol levels. In an unaffected patient, the levels of free cortisol should be within normal limits, and the administration of an exogenous corticosteroid, such as dexamethasone, should suppress the normal level of ACTH, with a concomitant decrease in the cortisol levels. Because functioning tumors do not respond to normal feedback mechanisms, the anticipated decreases in ACTH and cortisol would not be seen in a patient with such a tumor.
TREATMENT AND PROGNOSIS: The clinician should be aware of the signs and symptoms of hypercortisolism to refer affected patients for appropriate endocrinologic evaluation and diagnosis. Once the diagnosis is established and the cause is determined to be an adrenal or pituitary tumor, surgical removal of the lesion is the treatment of choice. Radiation therapy also may be effective, particularly in younger patients. For patients with unresectable tumors, drugs that inhibit cortisol synthesis, such as ketoconazole, metyrapone, or aminoglutethimide, may be used to help control the excess production of cortisol.
Most cases of hypercortisolism, however, are caused by systemic corticosteroid therapy that is given for a variety of immunologic reasons, including treatment of autoimmune diseases and allogeneic transplant recipients. Certain strategies, such as the use of corticosteroid-sparing agents or alternate-day therapy, may minimize the corticosteroid dose needed. The goal should be for patients to use the lowest dose possible to manage immunologic disease.
In normal situations, cortisol is critical to the function of the body, particularly in dealing with stress. As the hormone is metabolized and serum levels drop, feedback to the pituitary gland signals it to produce ACTH, which stimulates the adrenal gland to produce additional cortisol. Unfortunately, therapeutic corticosteroids suppress the production of ACTH by the pituitary gland to the extent that the pituitary gland may not be able to produce ACTH in response to stress, and an acute episode of hypoadrenocorticism (addisonian crisis) may be precipitated. Therefore, the clinician must be aware of the potential side effects of chronic high-dose corticosteroid use and must be able to adapt the treatment of the patient accordingly. For stressful dental and surgical procedures especially, it is often necessary to increase the corticosteroid dose because of the greater need of the body for cortisol. Consultation with the physician who is managing the corticosteroid therapy is advised to determine to what extent the dose should be adjusted.
ADDISON’S DISEASE (HYPOADRENOCORTICISM)
Insufficient production of adrenal corticosteroid hormones caused by the destruction of the adrenal cortex results in the condition known as Addison’s disease, or primary hypoadrenocorticism. The incidence of new cases diagnosed in the Western hemisphere is 110 to 140 per million population per year. The causes are diverse and include the following:
• Infections (e.g., tuberculosis and deep fungal diseases, particularly in patients with acquired immunodeficiency syndrome [AIDS])
• Rarely, metastatic tumors, sarcoidosis, hemochromatosis, or amyloidosis
If the pituitary gland is not functioning properly, secondary hypoadrenocorticism may develop because of decreased production of ACTH, the hormone responsible for maintaining normal levels of serum cortisol.
CLINICAL FEATURES: The clinical features of hypoadrenocorticism do not actually begin to appear until at least 90% of the glandular tissue has been destroyed. With gradual destruction of the adrenal cortex, an insidious onset of fatigue, irritability, depression, weakness, and hypotension is noted over a period of months. A generalized hyperpigmentation of the skin occurs, classically described as bronzing. The hyperpigmentation is generally more prominent on sun-exposed skin and over pressure points, such as the elbows and knees; it is caused by increased levels of beta-lipotropin or ACTH, each of which can stimulate melanocytes. The patient usually complains of gastrointestinal upset with anorexia, nausea, vomiting, diarrhea, weight loss, and a peculiar craving for salt. When hypoadrenocorticism is accompanied by hypoparathyroidism and mucocutaneous candidiasis, the possibility of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome should be considered (see page 219).
The oral manifestations include diffuse or patchy, brown, macular pigmentation of the oral mucosa caused by excess melanin production (Fig. 17-32). Often the oral mucosal changes are the first manifestation of the disease, with the skin hyperpigmentation occurring afterward. Sometimes the oral hypermelanosis may be difficult to distinguish from physiologic racial pigmentation, but a history of a recent onset of oral pigmentation should suggest the possibility of Addison’s disease.
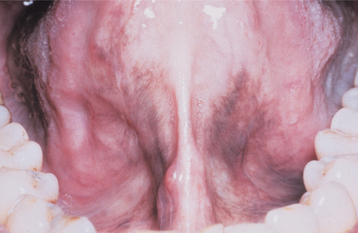
LABORATORY FINDINGS: The diagnosis of hypoadrenocorticism is confirmed by a rapid ACTH stimulation test and measurement of serum cortisol levels and plasma ACTH levels. If serum cortisol levels are below 20 mg/dL, then the patient has adrenal insufficiency. In primary hypoadrenocorticism, the plasma ACTH levels are high (>100 ng/L). In secondary hypoadrenocorticism, the levels are normal (9 to 52 ng/L) or low, as would be expected because the condition results from decreased ACTH production by the pituitary gland.
TREATMENT AND PROGNOSIS: Addison’s disease is managed with corticosteroid replacement therapy. The physiologic dose of corticosteroid is considered to be approximately 5.0 to 7.5 mg of prednisone or its equivalent per day, usually given in divided doses. Because the body’s need for corticosteroid hormones increases during stressful events, the patient must take this into account and increase the dose accordingly. This adjustment is generally not required for dental procedures performed using local anesthesia and lasting less than 1 hour, but an increased dose may be necessary for certain dental and oral surgical procedures that are more lengthy or are done under general anesthesia.
Before the availability of corticosteroids, the prognosis for patients with hypoadrenocorticism was poor, with most patients surviving less than 2 years. Even today, if the condition is not recognized promptly, death may result in a relatively short period of time. With proper diagnosis and management, most affected patients can expect to have a normal life span.
DIABETES MELLITUS
Diabetes mellitus is a common disorder of carbohydrate metabolism that is thought to have several causes, although the basic problem is one of either decreased production of insulin or tissue resistance to the effects of insulin. The net result of this abnormal state is an increase in the blood glucose level (hyperglycemia).
Diabetes mellitus is usually divided into two pre-sentations:
1. Type I—insulin-dependent diabetes mellitus (IDDM) or juvenile-onset diabetes
2. Type II—non-insulin-dependent diabetes mellitus (NIDDM) or adult-onset diabetes
Type I diabetes mellitus is characterized by a lack of insulin production. Patients usually exhibit severe hyperglycemia and ketoacidosis. The disease is typically diagnosed during childhood, and patients require exogenous insulin injections to survive.
Type II diabetes mellitus is sometimes more difficult to diagnose. It usually occurs in older, obese adults. Although hyperglycemia is present, ketoacidosis rarely develops. Furthermore, patients can produce some endogenous insulin. A few patients may take insulin to help control their disease; the insulin injections, however, are usually not necessary for the patient’s survival.
With respect to epidemiology, in the United States diabetes mellitus affects approximately 7% of the population, or 21 million people, although nearly 6 million of these cases remain undiagnosed. More than 1 million new cases are identified each year in the United States. Of these affected patients, most have type II diabetes; only 5% to 10% have type I.
Diabetes is an important disease when we consider the many complications associated with it and the economic effect it has on society. One of the main complications of diabetes is peripheral vascular disease, a problem that results in kidney failure, as well as ischemia and gangrenous involvement of the limbs. By some estimates, 25% of all new cases of kidney failure occur in diabetic patients. Thus diabetes is the leading cause of kidney failure in the United States. Each year more than 50,000 amputations are performed for the gangrenous complications of diabetes. This disease is the leading cause of lower limb amputations in the United States. Retinal involvement often results in blindness; thus the leading cause of new cases of blindness in working-age adults in the United States is diabetes, with more than 12,000 people affected annually. Complications because of diabetes are estimated to contribute to the deaths of more than 200,000 Americans each year.
The cause of diabetes mellitus is essentially un-known, although most cases of type I diabetes appear to be caused by autoimmune destruction of the pancreatic islet cells, and this immunologic attack may be precipitated by a viral infection in a genetically susceptible individual. Type II diabetes does not appear to have an autoimmune cause, however, because no destruction of the islet cells is seen microscopically. Instead, genetic abnormalities have been detected in patients with certain types of type II diabetes, which may explain why the condition occurs so often in families. If one parent is affected by type II diabetes, then the chances of a child having the disorder is about 40%. Similarly, if one identical twin has type II diabetes, then the chances are 90% that the disease will also develop in the other twin.
CLINICAL FEATURES: Although a complete review of the pathophysiology of diabetes mellitus is beyond the scope of this text, the clinical signs and symptoms of a patient with this disease are easier to understand with some basic knowledge of the process. The hormone insulin, produced by the beta cells of the pancreatic islets of Langerhans, is necessary for the uptake of glucose by the cells of the body. When insulin binds to its specific cell surface receptor, a resulting cascade of intracellular molecular events causes the recruitment of intracellular glucose-binding proteins, which facilitate the uptake of glucose by each cell.
TYPE I DIABETES MELLITUS: Because patients with type I diabetes have a deficiency in the amount of insulin, the body’s cells cannot absorb glucose and it remains in the blood. Normal blood glucose levels are between 70 and 120 mg/dL; in diabetic patients, these levels are often between 200 and 400 mg/dL. Above 300 mg/dL, the kidneys can no longer reabsorb the glucose; therefore, it spills over into the urine. Because glucose is the main source of energy for the body, and because none of this energy can be used because glucose cannot be absorbed, the patient feels tired and lethargic. The body begins to use other energy sources, such as fat and protein, resulting in the production of ketones as a by-product of those energy consumption pathways. The patient often loses weight, despite increased food intake (polyphagia). With the hyperglycemia, the osmolarity of the blood and urine increases. The increased osmolarity results in frequent urination (polyuria) and thirst, which leads to increased water intake (polydipsia). Clinically, most patients with type I diabetes are younger (average age at diagnosis being 14 years), and they have a thin body habitus.
TYPE II DIABETES MELLITUS: By contrast, patients with type II diabetes are usually older than 40 years of age at diagnosis, and 80% to 90% of them are obese. In this situation, it is thought that a decrease in the number of insulin receptors or abnormal postbinding molecular events related to glucose uptake results in glucose not being absorbed by the body’s cells. Thus patients are said to show “insulin resistance” because serum insulin levels are usually within normal limits or even elevated. If the hyperglycemia is taken into account, however, the amount of circulating insulin is typically not as much as would be present in a normal person with a similar level of blood glucose. Therefore, many of these patients are described as having a relative lack of insulin.
The symptoms associated with type II diabetes are much more subtle in comparison to those seen with type I. The first sign of type II diabetes is often detected with routine hematologic examination rather than any specific patient complaint. Ketoacidosis is almost never seen in patients with type II diabetes. Nevertheless, many of the other complications of diabetes are still associated with this form of the disease.
COMPLICATIONS: Many complications of diabetes mellitus are directly related to the microangiopathy caused by the disease. The microangiopathy results in occlusion of the small blood vessels, producing peripheral vascular disease. The resultant decrease in tissue perfusion results in ischemia. The ischemia predisposes the patient to infection, particularly severe infections such as gangrene. Another contributing factor is the impairment of neutrophil function, particularly neutrophil chemotaxis.
Amputation of the lower extremity often is necessary because of the lack of tissue perfusion and the patient’s inability to cope with infection. Similar vascular occlusion may affect the coronary arteries (which places the patient at risk for myocardial infarction) or the carotid arteries and their branches (predisposing the patient to cerebrovascular accident, or stroke). When microvascular occlusion affects the retinal vessels, blindness typically results. Kidney failure is the outcome of renal blood vessel involvement. If the ketoacidosis is not corrected in type I diabetes, the patient may lapse into a diabetic coma.
The oral manifestations of diabetes mellitus are generally limited to patients with type I diabetes. Problems include periodontal disease, which occurs more frequently and progresses more rapidly than in normal patients. Healing after surgery may be delayed, and the likelihood of infection is probably increased. Diffuse, nontender, bilateral enlargement of the parotid glands, called diabetic sialadenosis (see page 470), may be seen in patients with either form of diabetes. In uncontrolled or poorly controlled diabetic patients, a striking enlargement and erythema of the attached gingiva has been described (Fig. 17-33). In addition, these patients appear to be more susceptible to oral candidiasis in its various clinical forms (see page 213). Erythematous candidiasis, which appears as central papillary atrophy of the dorsal tongue papillae, is reported in up to 30% of these patients. Zygomycosis (see page 232) may occur in patients with poorly controlled type I diabetes. Some investigators have identified an increased prevalence of benign migratory glossitis (see page 779) in patients with type I diabetes; however, others have not been able to confirm this finding. Xerostomia, a subjective feeling of dryness of the oral mucosa, has been reported as a complaint in one third of diabetic patients. Unfortunately, studies that attempt to confirm an actual decrease in salivary flow rate in diabetic patients have produced conflicting results. Some studies show a decrease in salivary flow; some, no difference from normal; and some, an increased salivary flow rate.
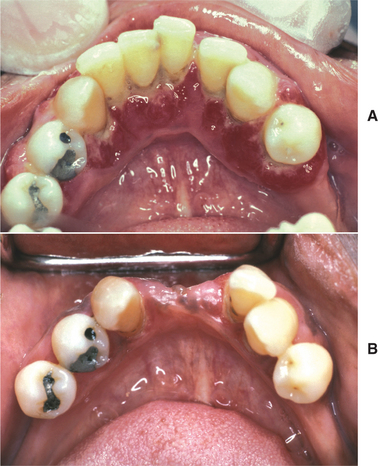
Fig. 17-33 Diabetes mellitus. A, This diffuse, erythematous enlargement of the gingival tissues developed in a diabetic patient who discontinued taking her insulin. B, The gingival tissues have greatly improved after reinstitution of regular insulin injections. Several incisors were extracted because of severe periodontal bone loss.
TREATMENT AND PROGNOSIS: For patients with type II diabetes, dietary modification coupled with exercise may be the only treatment necessary, with the goal being weight loss. The dietary and lifestyle changes may need to be coupled with one or more oral hypoglycemic agents. These drugs are designed to affect different pathophysiologic aspects of the disease. For example, secretagogues increase the insulin supply. These include the second-generation sulfonylurea medications such as glipizide or glyburide. Metformin is a biguanide that increases glucose utilization and decreases insulin resistance and hepatic glucose production. Thiazolidinediones, such as rosiglitazone and pioglitazone, also reduce insulin resistance. Acarbose and miglitol are a-glucosidase inhibitors that reduce the absorption of glucose from the gastrointestinal tract by inhibiting enzymatic degradation of more complex sugars. If these modalities do not control the blood glucose levels, then treatment with insulin is necessary.
For patients with type I diabetes, injections of insulin are required to control blood glucose levels. Different types of insulin are marketed, each type having different degrees of duration and times of peak activity. Insulin was previously extracted primarily from beef and pork pancreata. In some patients, however, antibodies developed to this foreign protein and rendered the insulin useless. To overcome this problem, pharmaceutical companies have developed brands of insulin that have the molecular structure of human insulin. Laboratories produce this human insulin with genetically engineered bacteria using recombinant DNA technology.
The patient’s schedule of insulin injections must be carefully structured and monitored to provide optimal control of blood glucose levels. This schedule is carefully formulated by the patient’s physician and takes into account such factors as the patient’s activity level and the severity of the insulin deficiency. It is imperative that adequate dietary carbohydrates be ingested after the administration of the insulin; otherwise, a condition known as insulin shock may occur. If carbohydrates are not consumed after an insulin injection, then the blood glucose levels may fall to dangerously low levels. The brain is virtually dependent on blood glucose as its energy source. If the blood glucose level drops below 40 mg/dL, the patient may go into shock. This condition can be treated by administration of sublingual dextrose paste, intravenous (IV) infusion of a dextrose solution, or injection of glucagon.
In summary, diabetes mellitus is a common, complex medical problem with many complications. The prognosis is guarded. Studies suggest that strict control of blood glucose levels results in a slowing of the development of the late complications of type I diabetes (e.g., blindness, kidney damage, neuropathy) and reduces the frequency of these complications. Health care practitioners should be aware of the problems these patients may have and should be prepared to deal with them. Consultation with the patient’s physician may be necessary, particularly for patients with type I diabetes who show poor blood glucose control, have active infections, or require extensive oral surgical procedures.
HYPOPHOSPHATASIA
Hypophosphatasia is a rare metabolic bone disease that is characterized by a deficiency of tissue-nonspecific alkaline phosphatase. Approximately 150 distinct mutations of the gene responsible for alkaline phosphatase production have been described. One of the first presenting signs of hypophosphatasia may be the premature loss of the primary teeth, presumably caused by a lack of cementum on the root surfaces. In the homozygous autosomal recessive form, there are rather severe manifestations, and many of these patients are identified in infancy. The milder forms of the disease are inherited in an autosomal dominant or recessive fashion, appearing in childhood or even adulthood, with variable degrees of expression. Generally, the younger the age of onset, the more severe the expression of the disease. The common factors in all types include the following:
• Reduced levels of the bone, liver, and kidney isozyme of alkaline phosphatase
• Increased levels of blood and urinary phos-phoethanolamine
Most authorities believe that the decreased alkaline phosphatase levels probably are responsible for the clinically observed abnormalities. Alkaline phosphatase is thought to play a role in the production of bone, but its precise mechanism of action is unknown.
CLINICAL AND RADIOGRAPHIC FEATURES: Four types of hypophosphatasia are generally recognized, depending on the severity and the age of onset of the symptoms:
PERINATAL HYPOPHOSPHATASIA: The perinatal form has the most severe manifestations. It is usually diagnosed at birth, and the infant rarely survives for more than a few hours. Death is due to respiratory failure. Marked hypocalcification of the skeletal structures is observed.
INFANTILE HYPOPHOSPHATASIA: Babies affected by infantile hypophosphatasia may appear normal up to 6 months of age; after this time, they begin to show a failure to grow. Vomiting and hypotonia may develop as well. Skeletal malformations that suggest rickets are typically observed; these malformations include shortened, bowed limbs. Deformities of the ribs predispose these patients to pneumonia, and skull deformities cause increased intracranial pressure. Nephrocalcinosis and nephrolithiasis also produce problems for these infants. Radiographs show a markedly reduced degree of ossification with a preponderance of hypomineralized osteoid. If these infants survive, premature shedding of the deciduous teeth is often seen.
CHILDHOOD HYPOPHOSPHATASIA: The childhood form is usually detected at a later age and has a wide range of clinical expression. One of the more consistent features is the premature loss of the primary teeth without evidence of a significant inflammatory response (Figs. 17-34 and 17-35). The deciduous incisor teeth are usually affected first and may be the only teeth involved. In some patients, this may be the only expression of the disease. The teeth may show enlarged pulp chambers in some instances, and a significant degree of alveolar bone loss may be seen. More severely affected patients may have open fontanelles with premature fusion of cranial sutures. This early fusion occasionally leads to increased intracranial pressure and subsequent brain damage. Affected patients typically have a short stature, bowed legs, and a waddling gait. The development of motor skills is often delayed.
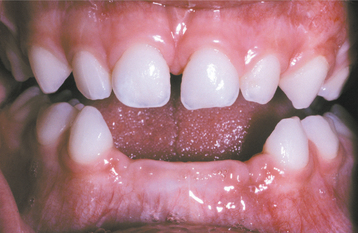
Fig. 17-34 Hypophosphatasia. Premature loss of the mandibular anterior teeth. (Courtesy of Dr. Jackie Banahan.)
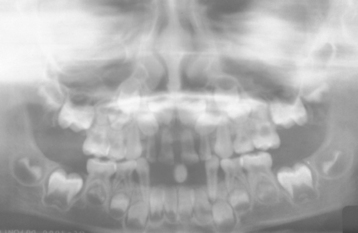
Fig. 17-35 Hypophosphatasia. This panoramic radiograph shows the loss of the mandibular anterior teeth. (Courtesy of Dr. Jackie Banahan.)
Radiographically, the skull has the appearance of “beaten copper,” and it shows uniformly spaced, poorly defined, small radiolucencies. This pattern may be the result of areas of thinning of the inner cortical plate produced by the cerebral gyri.
ADULT HYPOPHOSPHATASIA: The adult form is typically mild. Patients often have a history of premature loss of their primary or permanent dentition, and many of these patients are edentulous. Stress fractures that involve the metatarsal bones of the feet may be a presenting sign of the condition, or an increased number of fractures associated with relatively minor trauma may alert the clinician to this disorder.
DIAGNOSIS: The diagnosis of hypophosphatasia is based on the clinical manifestations and the finding of decreased levels of serum alkaline phosphatase and increased amounts of phosphoethanolamine in both the urine and the blood. Interestingly, as some patients grow older, serum alkaline phosphatase levels may approach normal.
HISTOPATHOLOGIC FEATURES: The histopathologic evaluation of bone sampled from a patient affected with the infantile form of hypophosphatasia shows abundant production of poorly mineralized osteoid. In the childhood or adult form, the bone may appear relatively normal or it may show an increased amount of woven bone, which is a less mature form of osseous tissue.
The histopathologic examination of either a primary or permanent tooth that has been exfoliated from an affected patient often shows an absence or a marked reduction of cementum that covers the root’s surface (Fig. 17-36). This reduced amount of cementum is thought to predispose to tooth loss because of the inability of periodontal ligament fibers to attach to the tooth and to maintain it in its normal position.

TREATMENT AND PROGNOSIS: The treatment of hypophosphatasia is essentially symptomatic because the lack of alkaline phosphatase cannot be corrected. Attempts to treat this condition by infusing alkaline phosphatase have been unsuccessful, presumably because the enzyme functions within the cell rather than in the extracellular environment. Basically, fractures are treated with orthopedic surgery, followed by rehabilitation. Prosthetic appliances are indicated to replace missing teeth, but satisfactory results are not always possible because the alveolar bone is hypoplastic. Because mutational analysis of DNA can identify carriers of the defective gene, patients and their parents should be provided with genetic counseling. As stated earlier, the prognosis varies with the onset of symptoms; the perinatal and infantile types are associated with a rather poor outcome. The childhood and adult forms are usually compatible with a normal life span.
VITAMIN D-RESISTANT RICKETS (HEREDITARY HYPOPHOSPHATEMIA; FAMILIAL HYPOPHOSPHATEMIC RICKETS)
After the use of vitamin D to treat rickets became widespread, it was recognized that some individuals with clinical features characteristic of rickets did not seem to respond to therapeutic doses of the vitamin. For this reason, this condition in these patients was called vitamin D-resistant rickets. Most cases of this rare condition appear to be inherited as an X-linked dominant trait; therefore, males are usually affected more severely than females, who presumably have attenuated features because of lyonization. In the United States, this condition occurs at a frequency of 1 in 20,000 births. In addition to the rachitic changes, these patients are also hypophosphatemic and show a decreased capacity for reabsorption of phosphate from the renal tubules. The disorder is caused by mutations in a zinc metalloproteinase gene known as PHEX (phosphate-regulating gene with endopeptidase activity on the X chromosome). Although the precise mechanisms of action of this gene are unclear, it appears to play a role in vitamin D metabolism.
In contrast, patients affected by the rare autosomal recessive condition known as vitamin D-dependent rickets exhibit hypocalcification of the teeth, unlike those with vitamin D-resistant rickets. Otherwise, the two disorders have similar clinical features. Vitamin D-dependent rickets is caused by a lack of 1a-hydroxylase, the enzyme responsible for converting the relatively inactive vitamin D precursor, 25-hydroxycholecalciferol (calcifediol) to the active metabolite 1,25-dihydroxycholecalciferol (calcitriol) in the kidney. Therefore, these patients respond to replacement therapy with active vitamin D (calcitriol).
CLINICAL FEATURES: Patients with vitamin D-resistant rickets have a short stature. The upper body segment appears more normal, but the lower body segment is shortened. The lower limbs are generally shortened and bowed.
Laboratory investigation reveals hypophosphatemia with diminished renal reabsorption of phosphate and decreased intestinal absorption of calcium. This typically results in rachitic changes that are unresponsive to vitamin D (calciferol). With aging, ankylosis of the spine frequently develops.
From a dental standpoint, the teeth have large pulp chambers, with pulp horns extending almost to the dentinoenamel junction (Figs. 17-37 and 17-38). In some cases the cuspal enamel may be worn down by attrition to the level of the pulp horn, causing pulpal exposure and pulp death. The exposure may be so small that the resulting periapical abscesses and gingival sinus tracts seem to affect what appear to be otherwise normal teeth (Fig. 17-39). Studies have also shown that microclefts may develop in the enamel, giving the oral microflora access to the dentinal tubules and subsequently to the pulp. One study examined a series of affected children and found that 25% of these patients had multiple abscesses involving the primary dentition.
Fig. 17-37 Vitamin D-resistant rickets. This radiograph of an extracted tooth shows a prominent pulp chamber with pulp horns extending out toward the dentinoenamel junction.

Fig. 17-38 Vitamin D-resistant rickets. Ground section of the same tooth depicted in Fig. 17-37. A pulp horn extends to the dentinoenamel junction. (Courtesy of Dr. Carl Witkop.)
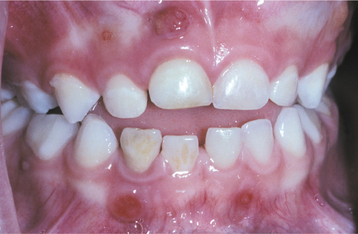
HISTOPATHOLOGIC FEATURES: Microscopic examination of an erupted tooth from a patient with vitamin D-resistant rickets usually shows markedly enlarged pulp horns. The dentin appears abnormal and is characterized by the deposition of globular dentin, which often exhibits clefting. The clefts may extend from the pulp chamber to the dentino-enamel junction. Microclefts are also seen within the enamel. The pulp frequently is nonvital, presumably because of the bacterial contamination associated with both the enamel and the dentinal clefts.
TREATMENT AND PROGNOSIS: For a normal stature to develop, patients with vitamin D-resistant rickets usually need early treatment with calcitriol and multiple daily doses of phosphate. Endodontic therapy is necessary for the pulpally involved teeth. Initiating therapy in early childhood with a synthetic vitamin D compound (1a-hydroxycholecalciferol) appears to reduce dental problems in affected patients when compared with untreated historic controls. Interestingly, the radiographic dental abnormalities do not seem to be improved. Although serum and urine calcium levels must be monitored care-fully to prevent nephrocalcinosis with its potential for kidney damage, patients generally have a normal life span.
CROHN’S DISEASE (REGIONAL ILEITIS; REGIONAL ENTERITIS)
Crohn’s disease is an inflammatory and probably an immunologically mediated condition of unknown cause that primarily affects the distal portion of the small bowel and the proximal colon. It is now well established that the manifestations of Crohn’s disease may be seen anywhere in the gastrointestinal tract, from the mouth to the anus. In addition, other extraintestinal sites of disease involvement, such as the skin, eyes, and joints, have also been identified. The oral lesions are significant because they may precede the gastrointestinal lesions in as many as 30% of the cases that have both oral and gastrointestinal involvement. It is interesting that the prevalence of Crohn’s disease appears to be increasing, but the reasons for this increase have not been determined.
CLINICAL FEATURES: Most patients with Crohn’s disease are teenagers when the disease first becomes evident, although another diagnostic peak of disease activity occurs in patients more than 60 years of age. Gastrointestinal signs and symptoms usually include abdominal cramping and pain, nausea, and diarrhea, occasionally accompanied by fever. Weight loss and malnutrition may develop, which can lead to anemia, decreased growth, and short stature.
A wide range of oral lesions has been clinically reported in Crohn’s disease; however, many of the abnormalities described are relatively nonspecific and may be associated with other conditions that cause orofacial granulomatosis (see page 341). The more prominent findings include diffuse or nodular swelling of the oral and perioral tissues, a cobblestone appearance of the mucosa, and deep, granulomatous-appearing ulcers. The ulcers are often linear and develop in the buccal vestibule (Fig. 17-40). Patchy erythematous macules and plaques involving the attached and un-attached gingivae have been termed mucogingivitis and may represent one of the more common lesions related to Crohn’s disease. Soft tissue swellings that resemble denture-related fibrous hyperplasia may be seen, as well as smaller mucosal tags. Another manifestation that has been reported is aphthouslike oral ulcerations, although the significance of this finding is uncertain because aphthous ulcerations are found rather frequently in the general population, including the same age group that is affected by Crohn’s disease. One large study showed no difference in the prevalence of aphthous ulcers in patients with Crohn’s disease compared with a control population. Fewer than 1% of patients with Crohn’s disease may develop diffuse stomatitis, with some cases apparently caused by Staphylococcus aureus, and others being nonspecific. In at least one instance, recurrent severe buccal space infections resulted in cutaneous salivary fistula formation. Infrequently, pyostomatitis vegetans (see next topic) has been associated with Crohn’s disease.
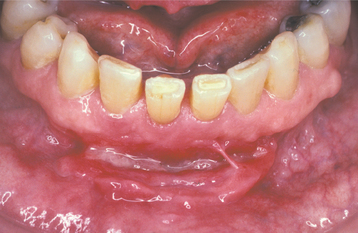
HISTOPATHOLOGIC FEATURES: Microscopic examination of lesional tissue obtained from the intestine or from the oral mucosa should show nonnecrotizing granulomatous inflammation within the submucosal connective tissue (Fig. 17-41). The severity of the granulomatous inflammation may vary tremendously from patient to patient and from various sites in the same patient. Therefore, a negative biopsy result at any one site and time may not necessarily rule out a diagnosis of Crohn’s disease. As with the clinical lesions, the histopathologic pattern is relatively nonspecific, resembling orofacial granulomatosis. Special stains should be performed to rule out the possibility of deep fungal infection, tertiary syphilis, or mycobacterial infection.
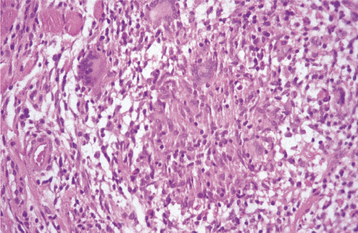
TREATMENT AND PROGNOSIS: Most patients with Crohn’s disease are initially treated medically with a sulfa type of drug (sulfasalazine), and some patients respond well to this medication. Metronidazole may be used if no response is seen with sulfasalazine therapy. With moderate to severe involvement, systemic prednisone may be used and is often effective, particularly when combined with the immunosuppressive drug, azathioprine. Infliximab, a monoclonal antibody directed against tumor necrosis factor-a (TNF-a), has shown promise in refractory cases of Crohn’s disease. Sometimes the disease cannot be maintained in remission by medical therapy, and complications develop that require surgical intervention. Complications may include bowel obstruction or fistula or abscess formation. If a significant segment of the terminal ileum has been removed surgically or is involved with the disease, then periodic injections of vitamin B12 may be necessary to prevent megaloblastic anemia secondary to the lack of ability to absorb the vitamin. Similar supplementation of magnesium, iron, the fat-soluble vitamins, and folate may also be required because of malabsorption.
Oral lesions have been reported to clear with treatment of the gastrointestinal process in many cases. Occasionally persistent oral ulcerations will develop, and these may have to be treated with topical or intralesional corticosteroids. Systemic thalidomide and infliximab have been used successfully to manage refractory oral ulcers of Crohn’s disease.
PYOSTOMATITIS VEGETANS
Pyostomatitis vegetans is a relatively rare condition that has a controversial history. It has been associated in the past with diseases such as pemphigus or pyodermatitis vegetans. Most investigators today, however, believe that pyostomatitis vegetans is an unusual oral expression of inflammatory bowel disease, particularly ulcerative colitis or Crohn’s disease. The pathogenesis of the condition, like that of inflammatory bowel disease, is poorly understood. A few patients with pyostomatitis vegetans have also been noted to have one of several concurrent liver abnormalities.
CLINICAL FEATURES: Patients with pyostomatitis vegetans exhibit characteristic yellowish, slightly elevated, linear, serpentine pustules set on an erythematous oral mucosa. The lesions primarily affect the buccal and labial mucosa, soft palate, and ventral tongue (Figs. 17-42 and 17-43). These lesions have been called “snail track” ulcerations, although in most instances the lesions are probably not truly ulcerated. Oral discomfort is variable but can be surprisingly minimal in some patients. This variation in symptoms may be related to the number of pustules that have ruptured to form ulcerations. The oral lesions may appear concurrently with the bowel symptoms, or they may precede the intestinal involvement.
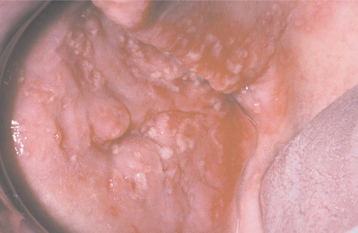
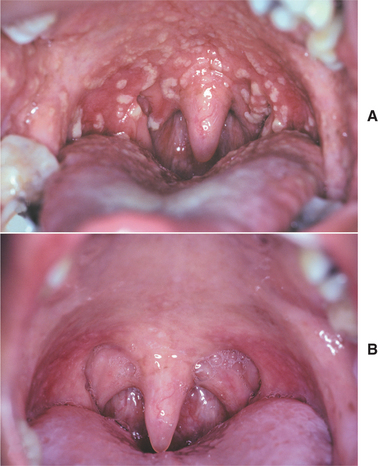
HISTOPATHOLOGIC FEATURES: A biopsy specimen of an oral lesion of pyostomatitis vegetans usually shows marked edema, causing an acantholytic appearance of the involved epithelium. This may be the result of the accumulation of numerous eosinophils within the spinous layer, often forming intraepithelial abscesses (Fig. 17-44). Subepithelial eosinophilic abscesses have been reported in some instances. The underlying connective tissue usually supports a dense mixed infiltrate of inflammatory cells that consists of eosinophils, neutrophils, and lymphocytes. Perivascular inflammation may also be present.
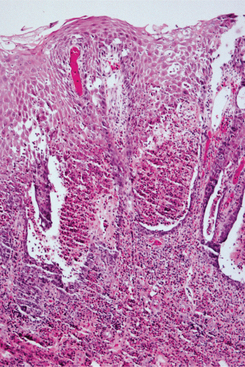
TREATMENT AND PROGNOSIS: Usually, the intestinal signs and symptoms of inflammatory bowel disease are of most concern for patients with pyostomatitis vegetans. Medical management of the bowel disease with sulfasalazine or systemic corticosteroids also produces clearing of the oral lesions (see Fig. 17-40). Often the oral lesions clear within days after systemic corticosteroid therapy is begun, and they may recur if the medication is withdrawn. If the bowel symptoms are relatively mild, then the oral lesions have been reported to respond to topical therapy with some of the more potent corticosteroid preparations.
UREMIC STOMATITIS
Patients who have either acute or chronic renal failure typically show markedly elevated levels of urea and other nitrogenous wastes in the bloodstream. Uremic stomatitis represents a relatively uncommon complication of renal failure. In two series that included 562 patients with renal failure, only eight examples of this oral mucosal condition were documented. Nevertheless, for the patients in whom uremic stomatitis develops, this can be a painful disorder. The cause of the oral lesions is unclear, but some investigators suggest that urease, an enzyme produced by the oral microflora, may degrade urea secreted in the saliva. This degradation results in the liberation of free ammonia, which presumably damages the oral mucosa.
CLINICAL FEATURES: Most cases of uremic stomatitis have been reported in patients with acute renal failure. The onset may be abrupt, with white plaques distributed predominantly on the buccal mucosa, tongue, and floor of the mouth (Fig. 17-45). Patients may complain of unpleasant taste, oral pain, or a burning sensation with the lesions, and the clinician may detect an odor of ammonia or urine on the patient’s breath. The clinical appearance occasionally has been known to mimic oral hairy leukoplakia.
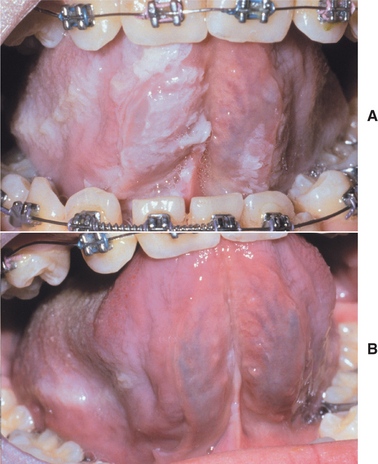
TREATMENT AND PROGNOSIS: In some instances, uremic stomatitis may clear within a few days after renal dialysis, although such resolution may take place over 2 to 3 weeks. In other instances, treatment with a mildly acidic mouth rinse, such as diluted hydrogen peroxide, seems to clear the oral lesions. For control of pain while the lesions heal, patients may be given palliative therapy with ice chips or a topical anesthetic, such as viscous lidocaine or dyclonine hydrochloride. Although renal failure itself is life threatening, at least one example of a uremic plaque that presumably caused a patient’s death has been recorded. This event was thought to have been caused by the dislodging of the plaque with subsequent obstruction of the patient’s airway.
BIBLIOGRAPHY
Alpöz, AR, Çoker, M, Çelen, E, et al. The oral manifestations of Maroteaux-Lamy syndrome (mucopolysaccharidosis VI): a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:632–637.
Brady, RO. Enzyme replacement for lysosomal diseases. Annu Rev Med. 2006;57:283–296.
Danos, O, Heard, J-M. Mucopolysaccharidosis. Mol Cell Biol Hum Dis Ser. 1995;5:350–367.
Downs, AT, Crisp, T, Ferretti, G. Hunter’s syndrome and oral manifestations: a review. Pediatr Dent. 1995;17:98–100.
Fahnehjelm, KT, Törnquist, A-L, Malm, G, et al. Ocular findings in four children with mucopolysaccharidosis I-Hurler (MPS I-H) treated early with haematopoietic stem cell transplantation. Acta Ophthalmol Scand. 2006;84:781–785.
Harmatz, P, Giugliani, R, Schwartz, I, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or RHASB) and follow-on, open-label extension study. J Pediatr. 2006;148:533–539.
Kinirons, MJ, Nelson, J. Dental findings in mucopolysaccharidosis type IV A (Morquio’s disease type A). Oral Surg Oral Med Oral Pathol. 1990;70:176–179.
Meikle, PJ, Hopwood, JJ, Clague, AE, et al. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254.
Mikles, M, Stanton, RP. A review of Morquio syndrome. Am J Orthop. 1997;26:533–540.
Muenzer, J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144:S27–S34.
Nakamura, T, Miwa, K, Kanda, S, et al. Rosette formation of impacted molar teeth in mucopolysaccharidoses and related disorders. Dentomaxillofac Radiol. 1992;21:45–49.
Nelson, J, Crowhurst, J, Carey, B, et al. Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet. 2003;123A:310–313.
Northover, H, Cowie, RA, Wraith, JE. Mucopolysaccharidosis type IVA (Morquio syndrome): a clinical review. J Inherit Metab Dis. 1996;19:357–365.
Nussbaum, BL. Dentistry for the at-risk patient—mucopolysaccharidosis III (Sanfilippo syndrome): a nine-year case study. ASDC J Dent Child. 1990;57:466–469.
Pastores, GM, Arn, P, Beck, M, et al. The MPS I registry: design, methodology, and early findings of a global disease registry for monitoring patients with mucopolysaccharidosis type I. Mol Genet Metab. 2007;91:37–47.
Peters, C, Shapiro, EG, Anderson, J, et al. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLSA-haploidentical related donor bone marrow transplantation in fifty-four children. Blood. 1998;91:2601–2608.
Scott, HS, Bunge, S, Gal, A, et al. Molecular genetics of mucopolysaccharidosis type I: diagnostic, clinical, and biological implications. Hum Mutat. 1995;6:288–302.
Sifuentes, M, Doroshow, R, Hoft, R, et al. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol Genet Metab. 2007;90:171–180.
Simmons, MA, Bruce, IA, Penney, S, et al. Otorhinolarygological manifestations of the mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2005;69:589–595.
Smith, KS, Hallett, KB, Hall, RK, et al. Mucopolysaccharidosis: MPS VI and associated delayed tooth eruption. Int J Oral Maxillofac Surg. 1995;24:176–180.
Staba, SL, Escolar, ML, Poe, M, et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N Engl J Med. 2004;350:1960–1969.
Vellodi, A, Young, EP, Cooper, A, et al. Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch Dis Child. 1997;76:92–99.
Vijay, S, Wraith, JE. Clinical presentation and follow-up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Paediatrica. 2005;94:872–877.
Wraith, JE. The mucopolysaccharidoses: a clinical review and guide to management. Arch Dis Child. 1995;72:263–267.
Wraith, JE, Hopwood, JJ, Fuller, M, et al. Laronidase treatment of mucopolysaccharidosis I. BioDrugs. 2005;19:1–7.
Barranger, JA, Rice, E, Sakallah, SA, et al. Enzymatic and molecular diagnosis of Gaucher disease. Clin Lab Med. 1995;15:899–913.
Beutler, E. Lysosomal storage diseases: natural history and ethical and economic aspects. Mol Genet Metab. 2006;88:208–215.
Beutler, E, Nguyen, NJ, Henneberger, MW, et al. Gaucher disease: gene frequencies in the Ashkenazi Jewish population. Am J Hum Genet. 1993;52:85–88.
Blitzer, MG, McDowell, CA. Tay-Sachs disease as a model for screening inborn errors. Clin Lab Med. 1992;12:463–479.
Dayan, B, Elstein, D, Zimran, A, et al. Decreased salivary output in patients with Gaucher disease. Q J Med. 2003;96:53–56.
Fernandes-Filho, JA, Shapiro, BE. Tay-Sachs disease. Arch Neurol. 2004;61:1466–1468.
Gieselmann, V. Lysosomal storage diseases. Biochim Biophys Acta. 1995;1270:103–136.
Grabowski, GA. Recent clinical progress in Gaucher disease. Curr Opin Pediatr. 2005;17:519–524.
Gravel, RA, Triggs-Raine, BL, Mahuran, DJ. Biochemistry and genetics of Tay-Sachs disease. Can J Neurol Sci. 1991;18:419–423.
Imrie, J, Dasgupta, S, Besley, GTN, et al. The natural history of Niemann-Pick disease type C in the UK. J Inherit Metab Dis. 2007;30:51–59.
Kolodny, EH. Niemann-Pick disease. Curr Opin Hematol. 2000;7:48–52.
Levran, O, Desnick, RJ, Schuchman, EH. Niemann-Pick disease: a frequent missense mutation in the acid sphingomyelinase gene of Ashkenazi Jewish type A and B patients. Proc Natl Acad Sci U S A. 1991;88:3748–3752.
Lustmann, J, Ben-Yehuda, D, Somer, M, et al. Gaucher’s disease affecting the mandible and maxilla: report of a case. Int J Oral Maxillofac Surg. 1991;20:7–8.
McGovern, MM, Aron, A, Brodie, SE, et al. Natural history of type A Niemann-Pick disease. Possible endpoints for therapeutic trials. Neurology. 2006;66:228–232.
Morales, LE. Gaucher’s disease: a review. Ann Pharmacother. 1996;30:381–388.
Morris, JA, Carstea, ED. Niemann-Pick disease: cholesterol handling gone awry. Mol Med Today. 1998;4:525–531.
Rosenthal, DI, Doppelt, SH, Mankin, HJ, et al. Enzyme replacement therapy for Gaucher disease: skeletal responses to macrophage-targeted glucocerebrosidase. Pediatrics. 1995;96:629–637.
Sévin, M, Lesca, G, Baumann, N, et al. The adult form of Niemann-Pick disease type C. Brain. 2007;130:120–133.
Shiran, A, Brenner, B, Laor, A, et al. Increased risk of cancer in patients with Gaucher disease. Cancer. 1993;72:219–224.
Sidransky, E, Tayebi, N, Ginns, EI. Diagnosing Gaucher disease. Early recognition, implications for treatment, and genetic counseling. Clin Pediatr (Phila). 1995;34:365–371.
Starzyk, K, Richards, S, Yee, J, et al. The long-term international safety experience of imiglucerase therapy for Gaucher disease. Mol Genet Metab. 2007;90:157–163.
Tamura, H, Takahashi, T, Ban, N, et al. Niemann-Pick type C disease: Novel NPC1 mutations and characterization of the concomitant acid sphingomyelinase deficiency. Mol Genet Metab. 2006;87:113–121.
Vanier, MT. Prenatal diagnosis of Niemann-Pick diseases types A, B and C. Prenat Diagn. 2002;22:630–632.
Weinstein, LB. Selected genetic disorders affecting Ashkenazi Jewish families. Fam Community Health. 2007;30:50–62.
Aroni, K, Lazaris, AC, Papadimitriou, K, et al. Lipoid proteinosis of the oral mucosa: case report and review of the literature. Pathol Res Pract. 1998;194:855–859.
Bahadir, S, Çobanogglu, Ü, Kapiciogglu, Z, et al. Lipoid proteinosis: a case with ophthalmological and psychiatric findings. J Dermatol. 2006;3:215–218.
Bazopoulou-Kyrkanidou, E, Tosios, KI, Zabelis, G, et al. Hyalinosis cutis et mucosae: gingival involvement. J Oral Pathol Med. 1998;27:233–237.
Botha, P. Oral lipoid proteinosis. SADJ. 1999;54:371–373.
Chaudhary, SJ, Dayal, PK. Hyalinosis cutis et mucosae. Review with a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995;80:168–171.
Hamada, T. Lipoid proteinosis. Clin Exp Dermatol. 2002;27:624–629.
Hamada, T, Wessagowit, V, South, AP, et al. Extracellular matrix protein 1 gene (ECM1) mutations in lipoid proteinosis and genotype-phenotype correlation. J Invest Dermatol. 2003;120:345–350.
Horev, L, Potikha, T, Ayalon, S, et al. A novel splice-site mutation in ECM-1 gene in a consanguineous family with lipoid proteinosis. Exp Dermatol. 2005;14:891–897.
Lupo, I, Cefalu, AB, Bongiorno, MR, et al. A novel mutation of the extracellular matrix protein 1 gene (ECM1) in a patient with lipoid proteinosis (Urbach-Wiethe disease) from Sicily. Br J Dermatol. 2005;153:1019–1022.
Muda, AO, Paradisi, M, Angelo, C, et al. Lipoid proteinosis: clinical, histologic, and ultrastructural investigations. Cutis. 1995;56:220–224.
Staut, CCV, Naidich, TP. Urbach-Wiethe disease (lipoid proteinosis). Pediatr Neurosurg. 1998;28:212–214.
Bansal, V, Schuchert, VD. Jaundice in the intensive care unit. Surg Clin North Am. 2006;86:1495–1502.
Cohen, SM. Jaundice in the full-term newborn. Pediatr Nurs. 2006;32:202–208.
Gordon, SC. Jaundice and cholestasis. Postgrad Med. 1991;90:65–71.
Hass, PL. Differentiation and diagnosis of jaundice. AACN Clin Issues. 1999;10:433–441.
Pratt, DS, Kaplan, MM. Jaundice. In: Kasper DL, Braunwald E, Fauci AS, et al, eds. Harrison’s principles of internal medicine. ed 16. New York: McGraw-Hill; 2005:238–243.
Murtagh, J. Jaundice. Aust Fam Physician. 1991;20:457–466.
Roche, SP, Kobos, R. Jaundice in the adult patient. Am Fam Physician. 2004;69:299–304.
Fahrner, KS, Black, CC, Gosselin, BJ. Localized amyloidosis of the tongue: a review. Am J Otolaryngol. 2004;25:186–189.
Falk, RH, Comenzo, RL, Skinner, M. The systemic amyloidoses. N Engl J Med. 1997;337:898–909.
Gertz, MA, Lacy, MQ, Dispenzieri, A. Amyloidosis: recognition, confirmation, prognosis, and therapy. Mayo Clin Proc. 1999;74:490–494.
Hachulla, E, Grateau, G. Diagnostic tools for amyloidosis. Joint Bone Spine. 2002;69:538–545.
Hachulla, E, Janin, A, Flipo, RM, et al. Labial salivary gland biopsy as a reliable test for the diagnosis of primary and secondary amyloidosis: a prospective clinical and immunohistologic study in 59 patients. Arthritis Rheum. 1993;36:691–697.
Hirschfield, GM. Amyloidosis: a clinico-pathophysiological synopsis. Semin Cell Dev Biol. 2004;15:39–44.
Johansson, I, Ryberg, M, Steen, L, et al. Salivary hypofunction in patients with familial amyloidotic polyneuropathy. Oral Surg Oral Med Oral Pathol. 1992;74:742–748.
Kaplan, B, Martin, BM, Livneh, A, et al. Biochemical subtyping of amyloid in formalin-fixed tissue samples confirms and supplements immunohistologic data. Am J Clin Pathol. 2004;121:794–800.
Lachmann, HJ, Hawkins, PN. Systemic amyloidosis. Curr Opin Pharmacol. 2006;6:214–220.
Mardinger, O, Rotenberg, L, Chaushu, G, et al. Surgical management of macroglossia due to primary amyloidosis. Int J Oral Maxillofac Surg. 1999;28:129–131.
Merlini, G, Westermark, P. The systemic amyloidoses: clearer understanding of the molecular mechanisms offers hope for more effective therapies. J Intern Med. 2004;255:159–178.
Nandapalan, V, Jones, TM, Morar, P, et al. Localized amyloidosis of the parotid gland: a case report and review of the localized amyloidosis of the head and neck. Head Neck. 1998;20:73–78.
Penner, CR, Müller, S. Head and neck amyloidosis: a clinicopathologic study of 15 cases. Oral Oncol. 2006;42:421–429.
Raymond, AK, Sneige, N, Batsakis, JG. Amyloidosis in the upper aerodigestive tract. Ann Otol Rhinol Laryngol. 1992;101:794–796.
Skinner, M, Anderson, JJ, Simms, R, et al. Treatment of 100 patients with primary amyloidosis: a randomized trial of melphalan, prednisone, and colchicine versus colchicine only. Am J Med. 1996;100:290–298.
Stoopler, ET, Alawi, F, Laudenbach, JM, et al. Bullous amyloidosis of the oral cavity: a rare clinical presentation and review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:734–740.
Stoopler, ET, Sollecito, TP, Chen, S-Y. Amyloid deposition in the oral cavity: a retrospective study and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95:674–680.
Westermark, P, Benson, MD, Buxbaum, JN, et al. Amyloid: toward terminology clarification. Report from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid. 2005;12:1–4.
Xavier, SD, Filho, IB, Müller, H. Macroglossia secondary to systemic amyloidosis: case report and literature review. Ear Nose Throat J. 2005;84:358–361.
Blanck, HM, Bowman, BA, Serdula, MK, et al. Angular stomatitis and riboflavin status among adolescent Bhutanese refugees living in southeastern Nepal. Am J Clin Nutr. 2002;76:430–435.
Fairfield, KM, Fletcher, RH. Vitamins for chronic disease prevention in adults. JAMA. 2002;287:3116–3126.
Halligan, TJ, Russell, NG, Dunn, WJ, et al. Identification and treatment of scurvy: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:688–692.
Harper, C. Thiamine (vitamin B1) deficiency and associated brain damage is still common throughout the world and prevention is simple and safe!. Eur J Neurol. 2006;13:1078–1082.
Heath, ML, Sidbury, R. Cutaneous manifestations of nutritional deficiency. Curr Opin Pediatr. 2006;18:417–422.
Hirschmann, JV, Raugi, GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895–906.
Mason, ME, Jalagani, H, Vinik, AI. Metabolic complications of bariatric surgery: diagnosis and management issues. Gastroenterol Clin North Am. 2005;34:25–33.
Nield, LS, Mahajan, P, Joshi, A, et al. Rickets: not a disease of the past. Am Fam Physician. 2006;74:619–626. [629-630].
Olmedo, JM, Yiannias, JA, Windgassen, EB, et al. Scurvy: a disease almost forgotten. Int J Dermatol. 2006;45:909–913.
Powers, HJ. Riboflavin (vitamin B2) and health. Am J Clin Nutr. 2003;77:1352–1360.
Sethuraman, U. Vitamins. Pediatr Rev. 2006;27:44–55.
Thacher, TD, Fischer, PR, Pettifor, JM, et al. A comparison of calcium, vitamin D, or both for nutritional rickets in Nigerian children. N Engl J Med. 1999;341:563–568.
Touyz, LZG. Oral scurvy and periodontal disease. J Can Dent Assoc. 1997;63:837–845.
Russell, RM. Vitamin and trace mineral deficiency and excess. In: Kasper DL, Braunwald E, Fauci AS, et al, eds. Harrison’s principles of internal medicine. ed 16. New York: McGraw-Hill; 2005:403–411.
Wolpowitz, D, Gilchrest, BA. The vitamin D questions: how much do you need and how should you get it? J Am Acad Dermatol. 2006;54:301–317.
Brigden, ML. Iron deficiency anemia: every case is instructive. Postgrad Med. 1993;93:181–192.
Brown, RG. Normocytic and macrocytic anemias. Postgrad Med. 1991;89:125–136.
Cook, JD. Diagnosis and management of iron-deficiency anaemia. Best Pract Res Clin Haematol. 2005;18:319–332.
Killip, S, Bennett, JM, Chambers, MD. Iron deficiency anemia. Am Fam Physician. 2007;75:671–678.
Massey, AC. Microcytic anemia: differential diagnosis and management of iron deficiency anemia. Med Clin North Am. 1992;76:549–566.
Osaki, T, Ueta, E, Arisawa, K, et al. The pathophysiology of glossal pain in patients with iron deficiency anemia. Am J Med Sci. 1999;318:324–329.
Provan, D. Mechanisms and management of iron deficiency anaemia. Br J Haematol. 1999;105(suppl 1):19–26.
Umbreit, J. Iron deficiency: a concise review. Am J Hematol. 2005;78:225–231.
Atmatzidis, K, Papaziogas, B, Pavlidis, T, et al. Plummer-Vinson syndrome. Dis Esophagus. 2003;16:154–157.
Bredenkamp, JK, Castro, DJ, Mickel, RA. Importance of iron repletion in the management of Plummer-Vinson syndrome. Ann Otol Rhinol Laryngol. 1990;99:51–54.
Chen, TSN, Chen, PSY. Rise and fall of the Plummer-Vinson syndrome. J Gastroenterol Hepatol. 1994;9:654–658.
Dantas, RO, Villanova, MG. Esophageal motility impairment in Plummer-Vinson syndrome: correction by iron treatment. Dig Dis Sci. 1993;38:968–971.
Geerlings, SE, Statius van Eps, LW. Pathogenesis and consequences of Plummer-Vinson syndrome. Clin Invest. 1992;70:629–630.
Hoffman, RM, Jaffe, PE. Plummer-Vinson syndrome: a case report and literature review. Arch Intern Med. 1995;155:2008–2011.
Nagai, T, Susami, E, Ebiham, T. Plummer-Vinson syndrome complicated by gastric cancer: a case report. Keio J Med. 1990;39:106–111.
Seitz, ML, Sabatino, D. Plummer-Vinson syndrome in an adolescent. J Adolesc Health. 1991;12:279–281.
Wahlberg, PCG, Andersson, KEH, Biörklund, AT, et al. Carcinoma of the hypopharynx: analysis of incidence and survival in Sweden in over a 30-year period. Head Neck. 1998;20:714–719.
Yukselen, V, Karaoglu, AO, Yasa, MH. Plummer-Vinson syndrome: a report of three cases. Int J Clin Pract. 2003;57:646–648.
Colon-Otero, G, Menke, D, Hook, CC. A practical approach to the differential diagnosis and evaluation of the adult patient with macrocytic anemia. Med Clin North Am. 1992;76:581–596.
Dharmarajan, TS, Adiga, GU, Norkus, EP. Vitamin B12 deficiency: recognizing subtle symptoms in older adults. Geriatrics. 2003;58:30–38.
Drummond, JF, White, DK, Damm, DD. Megaloblastic anemia with oral lesions: a consequence of gastric bypass surgery. Oral Surg Oral Med Oral Pathol. 1985;59:149–153.
Field, EA, Speechley, JA, Rugman, FR, et al. Oral signs and symptoms in patients with undiagnosed vitamin B12 deficiency. J Oral Pathol Med. 1995;24:468–470.
Greenberg, M. Clinical and histologic changes of the oral mucosa in pernicious anemia. Oral Surg Oral Med Oral Pathol. 1981;52:38–42.
Hsing, AW, Hansson, L-E, McLaughlin, JK, et al. Pernicious anemia and subsequent cancer: a population-based cohort study. Cancer. 1993;71:745–750.
Kleinegger, CL, Krolls, SO. Severe pernicious anemia presenting with burning mouth symptoms. Miss Dent Assoc J. 1996;52:12–14.
Lehman, JS, Bruce, AJ, Rogers, RS. Atrophic glossitis from vitamin B12 deficiency: a case misdiagnosed as burning mouth disorder. J Periodontol. 2006;77:2090–2092.
Loffeld, BCAJ, van Spreeuwel, JP. The gastrointestinal tract in pernicious anemia. Dig Dis. 1991;9:70–77.
Oh, RC, Brown, DL. Vitamin B12 deficiency. Am Fam Physician. 2003;67:979–986. [993-994].
Toh, B-H, Alderuccio, F. Pernicious anemia. Autoimmunity. 2004;37:357–361.
Toh, B-H, van Driel, IR, Gleeson, PA. Pernicious anemia. N Engl J Med. 1997;337:1441–1448.
Ayuk, J, Sheppard, MC. Growth hormone and its disorders. Postgrad Med J. 2006;82:24–30.
Buduneli, N, Alpoz, AR, Candan, U, et al. Dental management of isolated growth hormone deficiency: a case report. J Clin Pediatr Dent. 2005;29:263–266.
Carvalho, LR, Justamante de Faria, ME, Farah-Osorio, MG, et al. Acromegalic features in growth hormone (GH)-deficient patients after long-term GH therapy. Clin Endocrinol. 2003;59:788–792.
Funatsu, M, Sato, K, Mitani, H. Effects of growth hormone on craniofacial growth. Angle Orthod. 2006;76:970–977.
Grumbach, MM, Bin-Abbas, BS, Kaplan, SL. The growth hormone cascade: progress and long-term results of growth hormone treatment in growth hormone deficiency. Horm Res. 1998;49(suppl 2):41–57.
Kosowicz, J, Rzymski, K. Abnormalities of tooth development in pituitary dwarfism. Oral Surg Oral Med Oral Pathol. 1977;44:853–863.
Lamberts, SWJ, de Herder, WW, van der Lely, AJ. Pituitary insufficiency. Lancet. 1998;352:127–134.
Laron, Z. Short stature due to genetic defects affecting growth hormone activity. N Engl J Med. 1996;334:463–465.
Daughaday, WH. Pituitary gigantism. Endocrinol Metab Clin North Am. 1992;21:633–647.
Eugster, EA, Pescovitz, OH. Gigantism. J Clin Endocrinol Metab. 1999;84:4379–4384.
Kant, SG, Wit, JM, Breuning, MH. Genetic analysis of tall stature. Horm Res. 2005;64:149–156.
Van Haelst, MM, Hoogeboom, JJM, Baujat, G, et al. Familial gigantism caused by an NSD1 mutation. Am J Med Genet. 2005;139A:40–44.
Ayuk, J, Sheppard, MC. Growth hormone and its disorders. Postgrad Med J. 2006;82:24–30.
Ben-Shlomo, A, Melmed, S. Skin manifestations in acromegaly. Clin Dermatol. 2006;24:256–259.
Burt, MG, Ho, KKY. Newer options in the management of acromegaly. Intern Med J. 2006;36:437–444.
Cohen, RB, Wilcox, CW. A case of acromegaly identified after patient complaint of apertognathia. Oral Surg Oral Med Oral Pathol. 1993;75:583–586.
Farinazzo-Vitral, RW, Motohiro-Tanaka, O, Reis-Fraga, M, et al. Acromegaly in an orthodontic patient. Am J Orthod Dentofacial Orthop. 2006;130:388–390.
Gayle, C, Sonksen, P. The presentation of acromegaly in general practice. Practitioner. 1999;243:110–117.
Guistina, A, Barkan, A, Casanueva, FF, et al. Criteria for cure of acromegaly: a consensus statement. J Clin Endocrinol Metab. 2000;85:526–529.
Laws, ER, Vance, ML, Thapar, K. Pituitary surgery for the management of acromegaly. Horm Res. 2000;53(suppl 3):71–75.
Melmed, S. Acromegaly. N Engl J Med. 2006;355:2558–2573.
Newman, CB. Medical therapy for acromegaly. Endocrinol Metab Clin North Am. 1999;28:171–190.
Ron, E, Gridley, G, Hrubec, Z, et al. Acromegaly and gastrointestinal cancer. Cancer. 1991;68:1673–1677.
Burman, KD, McKinley-Grant, L. Dermatologic aspects of thyroid disease. Clin Dermatol. 2006;24:247–255.
LaFranchi, S. Congenital hypothyroidism: etiologies, diagnosis, and management. Thyroid. 1999;9:735–740.
Lazarus, JH, Obuobie, K. Thyroid disorders—an update. Postgrad Med J. 2000;76:529–536.
Martinez, M, Derksen, D, Kapsner, P. Making sense of hypothyroidism. Postgrad Med. 1993;93:135–145.
Mg’ang’a, PM, Chindia, ML. Dental and skeletal changes in juvenile hypothyroidism following treatment: case report. Odontostomatol Trop. 1990;13:25–27.
Roberts, CGP, Ladenson, PW. Hypothyroidism. Lancet. 2004;363:793–803.
Topliss, DJ, Eastman, CJ. Diagnosis and management of hyperthyroidism and hypothyroidism. Med J Aust. 2004;180:186–193.
Woeber, KA. Update on the management of hyperthyroidism and hypothyroidism. Arch Fam Med. 2000;9:743–747.
Caruso, DR, Mazzaferri, EL. Intervention in Graves’ disease. Postgrad Med. 1992;92:117–134.
Cooper, DS. Hyperthyroidism. Lancet. 2003;362:459–468.
McKeown, NJ, Tews, MC, Gossain, VV, et al. Hyperthyroidism. Emerg Med Clin North Am. 2005;23:669–685.
Pearce, EN. Diagnosis and management of thyrotoxicosis. Br Med J. 2006;332:1369–1373.
Pérusse, R, Goulet, J-P, Turcotte, J-Y. Contraindications to vasoconstrictors in dentistry. Part II: hyperthyroidism, diabetes, sulfite sensitivity, cortico-dependent asthma, and pheochromocytoma. Oral Surg Oral Med Oral Pathol. 1992;74:687–691.
Reid, JR, Wheeler, SF. Hyperthyroidism: diagnosis and treatment. Am Fam Physician. 2005;72:623–630. [635-636].
Singer, PA, Cooper, DS, Levy, EG, et al. Treatment guidelines for patients with hyperthyroidism and hypothyroidism. JAMA. 1995;273:808–812.
Tietgens, ST, Leinung, MC. Thyroid storm. Med Clin North Am. 1995;79:169–184.
Topliss, DJ, Eastman, CJ. Diagnosis and management of hyperthyroidism and hypothyroidism. Med J Aust. 2004;180:186–193.
Yeatts, RP. Graves’ ophthalmopathy. Med Clin North Am. 1995;79:195–209.
Ahonen, P, Myllärniemi, S, Sipilä, I, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829–1836.
Angelopoulos, NG, Goula, A, Tolis, G. Sporadic hypoparathyroidism treated with teriparatide: a case report and literature review. Exp Clin Endocrinol Diabetes. 2007;115:50–54.
Greenberg, MS, Brightman, VJ, Lynch, MA, et al. Idiopathic hypoparathyroidism, chronic candidiasis, and dental hypoplasia. Oral Surg Oral Med Oral Pathol. 1969;28:42–53.
Perheentupa, J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab. 2006;91:2843–2850.
Walls, AWG, Soames, JV. Dental manifestations of autoimmune hypoparathyroidism. Oral Surg Oral Med Oral Pathol. 1993;75:452–454.
Winer, KK, Ko, CW, Reynolds, JC, et al. Long-term treatment of hypoparathyroidism: a randomized controlled study comparing parathyroid hormone-(1-34) versus calcitriol and calcium. J Clin Endocrinol Metab. 2003;88:4214–4220.
Yasuda, T, Niimi, H. Hypoparathyroidism and pseudohypoparathyroidism. Acta Paediatr Jpn. 1997;39:485–490.
Brown, MD, Aaron, G. Pseudohypoparathyroidism: case report. Pediatr Dent. 1991;13:106–109.
Faull, CM, Welbury, RR, Paul, B, et al. Pseudohypoparathyroidism: its phenotypic variability and associated disorders in a large family. Q J Med. 1991;78:251–264.
Gelfand, IM, Eugster, EA, DiMeglio, LA. Presentation and clin-ical progression of pseudohypoparathyroidism with multi-hormone resistance and Albright hereditary osteodystrophy: a case series. J Pediatr. 2006;149:877–880.
Goeteyn, V, De Potter, CR, Naeyaert, JM. Osteoma cutis in pseudohypoparathyroidism. Dermatology. 1999;198:209–211.
Jüppner, H, Linglart, A, Fröhlich, LF, et al. Autosomal-dominant pseudohypoparathyroidism type Ib is caused by different microdeletions within or upstream of the GNAS locus. Ann N Y Acad Sci. 2006;1068:250–255.
Levine, MA. Pseudohypoparathyroidism: from bedside to bench and back. J Bone Miner Res. 1999;14:1255–1260.
Mallette, LE. Pseudohypoparathyroidism. Curr Ther Endocrinol Metab. 1997;6:577–581.
Potts, JT. Diseases of the parathyroid gland and other hyper- and hypocalcemic disorders. In: Kasper DL, Braunwald E, Fauci AS, et al, eds. Harrison’s principles of internal medicine. ed 16. New York: McGraw-Hill; 2005:2249–2268.
Simon, A, Koppeschaar, HP, Roijers, JF, et al. Pseudohypoparathyroidism type Ia, Albright hereditary osteodystrophy: a model for research on G protein-coupled receptors and genomic imprinting. Neth J Med. 2000;56:100–109.
Aggunlu, L, Akpek, S, Coskun, B. Leontiasis ossea in a patient with hyperparathyroidism secondary to chronic renal failure. Pediatr Radiol. 2004;34:630–632.
Antonelli, JR, Hottel, TL. Oral manifestations of renal osteodystrophy: case report and review of the literature. Spec Care Dentist. 2003;23:28–34.
Daniels, JSM. Primary hyperparathyroidism presenting as a palatal brown tumor. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;98:409–413.
Farford, B, Presutti, J, Moraghan, TJ. Nonsurgical management of primary hyperparathyroidism. Mayo Clin Proc. 2007;82:351–355.
Gavaldá, C, Bagán, JV, Scully, C, et al. Renal hemodialysis patients: oral, salivary, dental and periodontal findings in 105 adult cases. Oral Dis. 1999;5:299–302.
Hata, T, Irei, I, Tanaka, K, et al. Macrognathia secondary to dialysis-related renal osteodystrophy treated successfully by parathyroidectomy. Int J Oral Maxillofac Surg. 2006;35:378–382.
Kalyvas, D, Tosios, KI, Leventis, MD, et al. Localized jaw enlargement in renal osteodystrophy: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:68–74.
Khan, A, Bilezikian, J. Primary hyperparathyroidism: pathophysiology and impact on bone. CMAJ. 2000;163:173–175.
Ogata, H, Koiwa, F, Ito, H, et al. Therapeutic strategies for secondary hyperparathyroidism in dialysis patients. Ther Apher Dial. 2006;10:355–363.
Prado, FO, Rosales, AC, Rodrigues, CI, et al. Brown tumor of the mandible associated with secondary hyperparathyroidism: a case report and review of the literature. Gen Dent. 2006;54:341–343.
Sakhaee, K, Gonzalez, GB. Update on renal osteodystrophy: pathogenesis and clinical management. Am J Med Sci. 1999;317:251–260.
Silverman, S, Jr., Gordon, G, Grant, T, et al. The dental structures in primary hyperparathyroidism. Studies in forty-two consecutive patients. Oral Surg Oral Med Oral Pathol. 1962;15:426–436.
Silverman, S, Jr., Ware, WH, Gillooly, C. Dental aspects of hyperparathyroidism. Oral Surg Oral Med Oral Pathol. 1968;26:184–189.
Slatopolsky, E, Brown, A, Dusso, A. Pathogenesis of secondary hyperparathyroidism. Kidney Int Suppl. 1999;73:S14–S19.
Solt, DB. The pathogenesis, oral manifestations, and implications for dentistry of metabolic bone disease. Curr Opin Dent. 1991;1:783–791.
Tejwani, NC, Schachter, AK, Immerman, I, et al. Renal osteodystrophy. J Am Acad Orthop Surg. 2006;14:303–311.
Triantafillidou, K, Zouloumis, L, Karakinaris, G, et al. Brown tumors of the jaws associated with primary or secondary hyperparathyroidism. A clinical study and review of the literature. Am J Otolaryngol. 2006;27:281–286.
Findling, JW, Raff, H. Cushing’s syndrome: important issues in diagnosis and management. J Clin Endocrinol Metab. 2006;91:3746–3753.
Gabrilove, JL. Cushing’s syndrome. Compr Ther. 1992;18:13–16.
Katz, J, Bouloux, P-MG. Cushing’s: how to make the diagnosis. Practitioner. 1999;243:118–124.
Newell-Price, J, Bertagna, X, Grossman, AB, et al. Cushing’s syndrome. Lancet. 2006;367:1605–1617.
Shibli-Rahhal, A, Van Beek, M, Schlechte, JA. Cushing’s syndrome. Clin Dermatol. 2006;24:260–265.
Tsigos, C. Differential diagnosis and management of Cushing’s syndrome. Annu Rev Med. 1996;47:443–461.
Coursin, DB, Wood, KE. Corticosteroid supplementation for adrenal insufficiency. JAMA. 2002;287:236–240.
Jabbour, SA. Cutaneous manifestations of endocrine disorders: a guide for dermatologists. Am J Clin Dermatol. 2003;4:315–331.
Marzotti, S, Falorni, A. Addison’s disease. Autoimmunity. 2004;37:333–336.
Neiman, LK, Chanco-Turner, ML. Addison’s disease. Clin Dermatol. 2006;24:276–280.
Porter, SR, Haria, S, Scully, C, et al. Chronic candidiasis, enamel hypoplasia, and pigmentary anomalies. Oral Surg Oral Med Oral Pathol. 1992;74:312–314.
Shah, SS, Oh, CH, Coffin, SE, et al. Addisonian pigmentation of the oral mucosa. Cutis. 2005;76:97–99.
Werbel, SS, Ober, KP. Acute adrenal insufficiency. Endocrinol Metab Clin North Am. 1993;22:303–328.
Ziccardi, VB, Abubaker, AO, Sotereanos, GC, et al. Precipitation of an addisonian crisis during dental surgery: recognition and management. Compend Contin Educ Dent. 1992;13:518–523.
Belazi, M, Velegraki, A, Fleva, A, et al. Candidal overgrowth in diabetic patients: potential predisposing factors. Mycoses. 2005;48:192–196.
Bergman, SA. Perioperative management of the diabetic patient. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:731–737.
Cianciola, LJ, Park, BH, Bruck, E, et al. Prevalence of periodontal disease in insulin-dependent diabetes mellitus (juvenile diabetes). J Am Dent Assoc. 1982;104:653–660.
Emancipator, K. Laboratory diagnosis and monitoring of diabetes mellitus. Am J Clin Pathol. 1999;112:665–674.
Franco, OH, Steyerberg, EW, Hu, FB, et al. Associations of diabetes mellitus with total life expectancy and life expectancy with and without cardiovascular disease. Arch Intern Med. 2007;167:1145–1151.
Guggenheimer, J, Moore, PA, Rossie, K, et al. Insulin-dependent diabetes mellitus and oral soft tissue pathologies: II. Prevalence and characteristics of non-candidal lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;89:563–569.
Guggenheimer, J, Moore, PA, Rossie, K, et al. Insulin-dependent diabetes mellitus and oral soft tissue pathologies: II. Prevalence and characteristics of Candida and candidal lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;89:570–576.
Manfredi, M, Al-Karaawi, Z, McCullough, MJ, et al. The isolation, identification and molecular analysis of Candida spp. isolated from the oral cavities of patients with diabetes mellitus. Oral Microbiol Immunol. 2002;17:181–185.
Manfredi, M, McCollough, MJ, Vescovi, P, et al. Update on diabetes mellitus and related oral diseases. Oral Dis. 2004;10:187–200.
McKenna, SJ. Dental management of patients with diabetes. Dent Clin North Am. 2006;50:591–606.
Mealey, BL, Oates, TW. Diabetes mellitus and periodontal diseases. J Periodontol. 2006;77:1289–1303.
Miley, DD, Terezhalmy, GT. The patient with diabetes mellitus: etiology, epidemiology, principles of medical management, oral disease burden, and principles of dental management. Quintessence Int. 2005;36:779–795.
Moore, PA, Weyant, RJ, Mongelluzzo, MB, et al. Type 1 diabetes mellitus and oral health: assessment of tooth loss and edentulism. J Public Health Dent. 1998;58:135–142.
Murrah, VA. Diabetes mellitus and associated oral manifestations: a review. J Oral Pathol. 1985;14:271–281.
Newton, CA, Raskin, P. Diabetic ketoacidosis in type 1 and type 2 diabetes mellitus: clinical and biochemical differences. Arch Intern Med. 2004;164:1925–1931.
Seppälä, B, Seppälä, M, Ainamo, J. A longitudinal study on insulin-dependent diabetes mellitus and periodontal disease. J Clin Periodontol. 1993;20:161–165.
Tsai, C, Hayes, C, Taylor, GW. Glycemic control of type 2 diabetes and severe periodontal disease in the US adult population. Community Dent Oral Epidemiol. 2002;30:182–192.
Wang, PH, Lau, J, Chalmers, TC. Meta-analysis of effects of intensive blood-glucose control on late complications of type I diabetes. Lancet. 1993;341:1306–1309.
Winer, N, Sowers, JR. Epidemiology of diabetes. J Clin Pharmacol. 2004;44:397–405.
Wysocki, GP, Daley, T. Benign migratory glossitis in patients with juvenile diabetes. Oral Surg Oral Med Oral Pathol. 1987;63:68–70.
Zachariasen, RD. Diabetes mellitus and xerostomia. Compend Contin Educ Dent. 1992;13:314–322.
Brun-Heath, I, Taillandier, A, Serre, J-L, et al. Characterization of 11 novel mutations in the tissue non-specific alkaline phosphatase gene responsible for hypophosphatasia and genotype-phenotype correlations. Mol Genet Metab. 2005;84:273–277.
Caswell, AM, Whyte, MP, Russell, RGG. Hypophosphatasia and the extracellular metabolism of inorganic pyrophosphate: clinical and laboratory aspects. Crit Rev Clin Lab Sci. 1991;28:175–232.
Fallon, MD, Teitelbaum, SL, Weinstein, RS, et al. Hypophosphatasia: clinicopathologic comparison of the infantile, childhood, and adult forms. Medicine. 1984;63:12–24.
Hu, C-C, King, DL, Thomas, HF, et al. A clinical and research protocol for characterizing patients with hypophosphatasia. Pediatr Dent. 1996;18:17–23.
Mornet, E. Hypophosphatasia: the mutations in the tissue-nonspecific alkaline phosphatase gene. Hum Mutat. 2000;15:309–315.
Olsson, A, Matsson, L, Blomquist, HK, et al. Hypophosphatasia affecting the permanent dentition. J Oral Pathol Med. 1996;25:343–347.
Plagmann, H-H, Kocher, T, Kuhrau, N, et al. Periodontal manifestation of hypophosphatasia. A family case report. J Clin Periodontol. 1994;21:710–716.
Spentchian, M, Merrien, Y, Herasse, M, et al. Severe hypophosphatasia: characterization of fifteen novel mutations in the ALPL gene. Hum Mutat. 2003;22:105–106.
Van den Bos, T, Handoko, G, Niehof, A, et al. Cementum and dentin in hypophosphatasia. J Dent Res. 2005;84:1021–1025.
Archard, HO, Witkop, CJ. Hereditary hypophosphatemia (vitamin D-resistant rickets) presenting primary dental manifestations. Oral Surg Oral Med Oral Pathol. 1966;22:184–193.
Batra, P, Tejani, Z, Mars, M. X-linked hypophosphatemia: dental and histologic findings. J Can Dent Assoc. 2006;72:69–72.
Berndt, M, Ehrich, JHH, Lazovic, D, et al. Clinical course of hypophosphatemic rickets in 23 adults. Clin Nephrol. 1996;45:33–41.
Carpenter, TO. New perspectives on the biology and treatment of X-linked hypophosphatemic rickets. Pediatr Clin North Am. 1997;44:443–466.
Chaussin-Miller, C, Sinding, C, Wolikow, M, et al. Dental abnormalities in patients with familial hypophosphatemic vitamin D-resistant rickets: prevention by early treatment with 1-hydroxyvitamin D. J Pediatr. 2003;142:324–331.
Fadavi, S, Rowold, E. Familial hypophosphatemic vitamin D-resistant rickets: review of the literature and report of case. ASDC J Dent Child. 1990;57:212–215.
Hanna, JD, Niimi, K, Chart, JCM. X-linked hypophosphatemia: genetic and clinical correlates. Am J Dis Child. 1991;145:865–870.
Hillman, G, Geurtsen, W. Pathohistology of undecalcified primary teeth in vitamin D-resistant rickets. Review and report of two cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;82:218–224.
McWhorter, AG, Seale, NS. Prevalence of dental abscess in a population of children with vitamin D-resistant rickets. Pediatr Dent. 1991;13:91–96.
Miller, WL, Portale, AA. Genetic causes of rickets. Curr Opin Pediatr. 1999;11:333–339.
Murayama, T, Iwatsubo, R, Akiyama, S, et al. Familial hypophosphatemic vitamin D-resistant rickets: dental findings and histologic study of teeth. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:310–316.
Pereira, CM, de Andrade, CR, Vargas, PA, et al. Dental alterations associated with X-linked hypophosphatemic rickets. J Endod. 2004;30:241–245.
Scriver, CR, Tenenhouse, HS, Glorieux, HI. X-linked hypophosphatemia: an appreciation of a classic paper and a survey of progress since 1958. Medicine (Baltimore). 1991;70:218–228.
Seow, WK, Needleman, HL, Holm, IA. Effect of familial hypophosphatemic rickets on dental development: a controlled, longitudinal study. Pediatr Dent. 1995;17:346–350.
Shroff, DV, McWhorter, AG, Seale, NS. Evaluation of aggressive pulp therapy in a population of vitamin D-resistant rickets patients: a follow-up of 4 cases. Pediatr Dent. 2002;24:347–349.
Wang, JT, Lin, C-J, Burridge, SM, et al. Genetics of vitamin D 1a-hydroxylase deficiency in 17 families. Am J Hum Genet. 1998;63:1694–1702.
Zambrano, M, Nikitakis, NG, Sanchez-Quevedo, MC, et al. Oral and dental manifestations of vitamin D-dependent rickets type I: report of a pediatric case. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95:705–709.
Brunner, B, Hirschi, C, Weimann, R, et al. Treatment-resistant lingual Crohn’s disease disappears after infliximab. Scand J Gastroenterol. 2005;40:1255–1259.
Challacombe, SJ. Oro-facial granulomatosis and oral Crohn’s disease: are they specific diseases and do they predict systemic Crohn’s disease? Oral Dis. 1997;3:127–129.
Dunlap, CL, Friesen, CA, Shultz, R. Chronic stomatitis: an early sign of Crohn’s disease. J Am Dent Assoc. 1997;128:347–348.
Dupuy, A, Cosnes, J, Revuz, J, et al. Oral Crohn disease: clinical characteristics and long-term follow-up of 9 cases. Arch Dermatol. 1999;135:439–442.
Galbraith, SS, Drolet, BA, Kugathasan, S, et al. Asymptomatic inflammatory bowel disease presenting with mucocutaneous findings. Pediatrics. 2005;116:e439–e444.
Girlich, C, Bogenrieder, T, Palitzsch, K-D, et al. Orofacial granulomatosis as initial manifestation of Crohn’s disease: a report of two cases. Eur J Gastroenterol Hepatol. 2002;14:873–876.
Friedman, S, Blumberg, RS. Inflammatory bowel disease. In: Kasper DL, Braunwald E, Fauci AS, et al, eds. Harrison’s principles of internal medicine. ed 16. New York: McGraw-Hill; 2005:1776–1789.
Harty, S, Fleming, P, Rowland, M, et al. A prospective study of the oral manifestations of Crohn’s disease. Clin Gastroenterol Hepatol. 2005;3:886–891.
Hegarty, A, Hodgson, T, Porter, S. Thalidomide for the treatment of recalcitrant oral Crohn’s disease and orofacial granulomatosis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95:576–585.
Hoffmann, JC, Zeitz, M. Treatment of Crohn’s disease. Hepatogastroenterology. 2000;47:90–100.
Kalmar, JR. Crohn’s disease: orofacial considerations and disease pathogenesis. Periodontol 2000. 1994;6:101–115.
Lisciandrano, D, Ranzi, T, Carrassi, A, et al. Prevalence of oral lesions in inflammatory bowel disease. Am J Gastroenterol. 1996;91:7–10.
Mills, CC, Amin, M, Manisali, M. Salivary duct fistula and recurrent buccal space infection: a complication of Crohn’s disease. J Oral Maxillofac Surg. 2003;61:1485–1487.
Ottaviani, F, Schindler, A, Capaccio, P, et al. New therapy for orolaryngeal manifestations of Crohn’s disease. Ann Otol Rhinol Laryngol. 2003;112:37–39.
Plauth, M, Jenss, H, Meyle, J. Oral manifestations of Crohn’s disease: an analysis of 79 cases. J Clin Gastroenterol. 1991;13:29–37.
Podolsky, DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429.
Rehberger, A, Püspök, A, Stallmeister, T, et al. Crohn’s disease masquerading as aphthous ulcers. Eur J Dermatol. 1998;8:274–276.
Sánchez, AR, Rogers, RS, Sheridan, PJ. Oral ulcerations are associated with the loss of response to infliximab in Crohn’s disease. J Oral Pathol Med. 2005;34:53–55.
Scheper, HJ, Brand, HS. Oral aspects of Crohn’s disease. Int Dent J. 2002;52:163–172.
Scully, C, Cochran, KM, Russell, RI, et al. Crohn’s disease of the mouth: an indicator of intestinal involvement. Gut. 1982;23:198–201.
Van de Scheur, MR, van der Waal, RIF, Völker-Dieben, HJ, et al. Orofacial granulomatosis in a patient with Crohn’s disease. J Am Acad Dermatol. 2003;49:952–954.
Weinstein, TA, Sciubba, JJ, Levine, J. Thalidomide for the treatment of oral aphthous ulcers in Crohn’s disease. J Pediatr Gastroenterol Nutr. 1999;28:214–216.
Ayangco, L, Rogers, RS, Sheridan, PJ. Pyostomatitis vegetans as an early sign of reactivation of Crohn’s disease: a case report. J Periodontol. 2002;73:1512–1516.
Ballo, FS, Camisa, C, Allen, CM. Pyostomatitis vegetans: report of a case and review of the literature. J Am Acad Dermatol. 1989;21:381–387.
Calobrisi, SD, Mutasim, DF, McDonald, JS. Pyostomatitis vegetans associated with ulcerative colitis. Temporary clearance with fluocinonide gel and complete remission after colectomy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995;79:452–454.
Chan, SWY, Scully, C, Prime, SS, et al. Pyostomatitis vegetans: oral manifestation of ulcerative colitis. Oral Surg Oral Med Oral Pathol. 1991;72:689–692.
Chaudhry, SI, Philpot, NS, Odell, EW, et al. Pyostomatitis vegetans associated with asymptomatic ulcerative colitis: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:327–330.
Ficarra, G, Cicchi, P, Amorosi, A, et al. Oral Crohn’s disease and pyostomatitis vegetans: an unusual association. Oral Surg Oral Med Oral Pathol. 1993;75:220–224.
Healy, CM, Farthing, PM, Williams, DM, et al. Pyostomatitis vegetans and associated systemic disease. A review and two case reports. Oral Surg Oral Med Oral Pathol. 1994;78:323–328.
Hegarty, AM, Barrett, AW, Scully, C. Pyostomatitis vegetans. Clin Exp Dermatol. 2004;29:1–7.
Markiewicz, M, Suresh, L, Margarone, J, et al. Pyostomatitis vegetans: a clinical marker of silent ulcerative colitis. J Oral Maxillofac Surg. 2007;65:346–348.
Neville, BW, Laden, SA, Smith, SE, et al. Pyostomatitis vegetans. Am J Dermatopathol. 1985;7:69–77.
Philpot, HC, Elewski, BE, Banwell, JG, et al. Pyostomatitis vegetans and primary sclerosing cholangitis: markers of inflammatory bowel disease. Gastroenterology. 1992;103:668–674.
Thornhill, MH, Zakrzewska, JM, Gilkes, JJH. Pyostomatitis vegetans: report of three cases and review of the literature. J Oral Pathol Med. 1992;21:128–133.
Halazonetis, J, Harley, A. Uremic stomatitis: report of a case. Oral Surg Oral Med Oral Pathol. 1967;23:573–577.
Hovinga, J, Roodvoets, AP, Gailliard, J. Some findings in patients with uraemic stomatitis. J Maxillofac Surg. 1975;3:124–127.
Larato, DC. Uremic stomatitis: report of a case. J Periodontol. 1975;46:731–733.
Leão, JC, Gueiros, LAM, Segundo, AVL, et al. Uremic stomatitis in chronic renal failure. Clinics. 2005;60:259–262.
McCreary, CE, Flint, SR, McCartan, BE, et al. Uremic stomatitis mimicking oral hairy leukoplakia. Report of a case. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:350–353.
Proctor, R, Kumar, N, Stein, A, et al. Oral and dental aspects of chronic renal failure. J Dent Res. 2005;84:199–208.
Ross, WF, Salisbury, PL. Uremic stomatitis associated with undiagnosed renal failure. Gen Dent. 1994;42:410–412.