Dermatologic Diseases
ECTODERMAL DYSPLASIA
Ectodermal dysplasia represents a group of inherited conditions in which two or more ectodermally derived anatomic structures fail to develop. Thus depending on the type of ectodermal dysplasia, hypoplasia or aplasia of tissues (e.g., skin, hair, nails, teeth, sweat glands) may be seen. The various types of this disorder may be inherited in any one of several genetic patterns, including autosomal dominant, autosomal recessive, and X-linked patterns. Even though by some accounts more than 170 different subtypes of ectodermal dysplasia can be defined, these disorders are considered to be relatively rare, with an estimated frequency of seven cases occurring in every 10,000 births. For fewer than 20% of these conditions, the specific genetic mutations and their chromosomal locations have been identified. Systematically classifying these conditions can be challenging because of their wide-ranging clinical features; however, some investigators have suggested that a classification scheme based on the molecular genetic alteration associated with each type might be appropriate. Thus groups of ectodermal dysplasia syndromes could be categorized as being caused by mutations in genes encoding cell-cell signals, genes encoding adhesion molecules, or genes regulating transcription.
CLINICAL FEATURES: Perhaps the best known of the ectodermal dysplasia syndromes is hypohidrotic ectodermal dysplasia. In most instances, this disorder seems to show an X-linked inheritance pattern, with the gene mapping to Xq12-q13.1; therefore, a male predominance is usually seen. However, a few families have been identified that show autosomal recessive or autosomal dominant patterns of inheritance.
Affected individuals typically display heat intolerance because of a reduced number of eccrine sweat glands. Sometimes the diagnosis is made during infancy because the baby appears to have a fever of undetermined origin; however, the infant simply cannot regulate body temperature appropriately because of the decreased number of sweat glands. Uncommonly, death results from the markedly elevated body temperature, although this generally happens only when the condition is not identified. Sometimes, as a diagnostic aid, a special impression can be made of the patient’s fingertips and then examined microscopically to count the density of the sweat glands. Such findings should be interpreted in conjunction with appropriate age-matched controls.
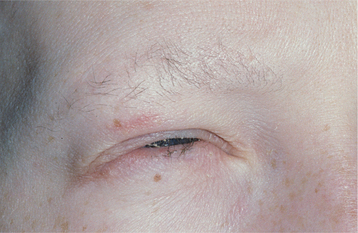
Other signs of this disorder include fine, sparse hair, including a reduced density of eyebrow and eyelash hair (Fig. 16-1). The periocular skin may show a fine wrinkling with hyperpigmentation (Fig. 16-2), and midface hypoplasia is frequently observed, often resulting in protuberant lips. Because the salivary glands are ectodermally derived, these glands may be hypoplastic or absent, and patients may exhibit varying degrees of xerostomia. The nails may also appear dystrophic and brittle.
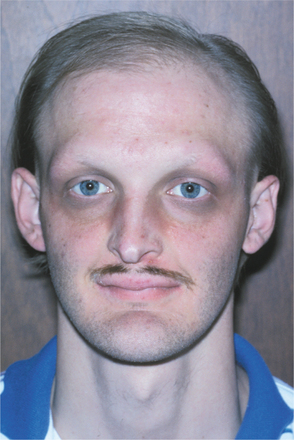
Fig. 16-1 Ectodermal dysplasia. The sparse hair, periocular hyperpigmentation, and mild midfacial hypoplasia are characteristic features evident in this affected patient.
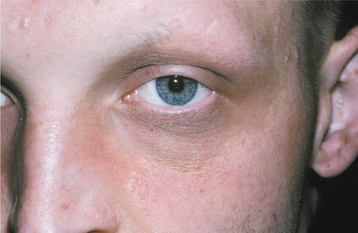
Fig. 16-2 Ectodermal dysplasia. Closer view of the same patient depicted in Fig. 16-1. Fine periocular wrinkling, as well as sparse eyelash and eyebrow hair, can be observed.
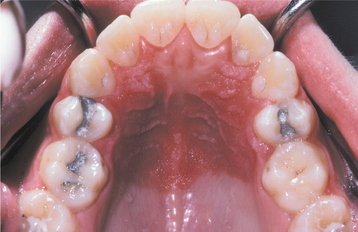
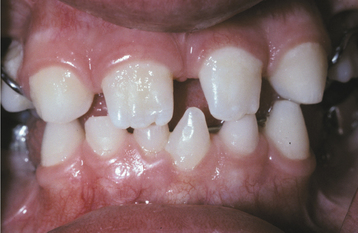
The teeth are usually markedly reduced in number (oligodontia or hypodontia), and their crown shapes are characteristically abnormal (Fig. 16-3). The incisor crowns usually appear tapered, conical, or pointed, and the molar crowns are reduced in diameter. Complete lack of tooth development (anodontia) has also been reported, but this appears to be uncommon.
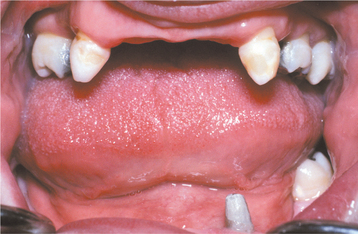
Fig. 16-3 Ectodermal dysplasia. Oligodontia and conical crown forms are typical oral manifestations. (Courtesy of Dr. Charles Hook and Dr. Bob Gellin.)
Female patients may show partial expression of the abnormal gene; that is, their teeth may be reduced in number or may have mild structural changes. This incomplete presentation can be explained by the Lyon hypothesis, with half of the female patient’s X chro mosomes expressing the normal gene, and the other half expressing the defective gene.
HISTOPATHOLOGIC FEATURES: Histopathologic examination of the skin from a patient with hypohidrotic ectodermal dysplasia shows a decreased number of sweat glands and hair follicles. The adnexal structures that are present are hypoplastic and malformed.
TREATMENT AND PROGNOSIS: Management of hypohidrotic ectodermal dysplasia warrants genetic counseling for the parents and the patient. The dental problems are best managed by prosthetic replacement of the dentition with complete dentures, overdentures, or fixed appliances, depending on the number and location of the remaining teeth. With careful site selection, endosseous dental implants may be considered for facilitating prosthetic management of patients older than 5 years of age.
WHITE SPONGE NEVUS (CANNON’S DISEASE; FAMILIAL WHITE FOLDED DYSPLASIA)
White sponge nevus is a relatively rare genodermatosis (a genetically determined skin disorder) that is inherited as an autosomal dominant trait displaying a high degree of penetrance and variable expressivity. This condition is due to a defect in the normal keratinization of the oral mucosa. In the 30-member family of keratin filaments, the pair of keratins known as keratin 4 and keratin 13 is specifically expressed in the spinous cell layer of mucosal epithelium. Mutations in either of these keratin genes have been shown to be responsible for the clinical manifestations of white sponge nevus.
CLINICAL FEATURES: The lesions of white sponge nevus usually appear at birth or in early childhood, but sometimes the condition develops during adolescence. Symmetrical, thickened, white, corrugated or velvety, diffuse plaques affect the buccal mucosa bilaterally in most instances (Fig. 16-4). Other common intraoral sites of involvement include the ventral tongue, labial mucosa, soft palate, alveolar mucosa, and floor of the mouth, although the extent of involvement can vary from patient to patient. Extraoral mucosal sites, such as the nasal, esophageal, laryngeal, and anogenital mucosa, appear to be less commonly affected. Patients are usually asymptomatic.
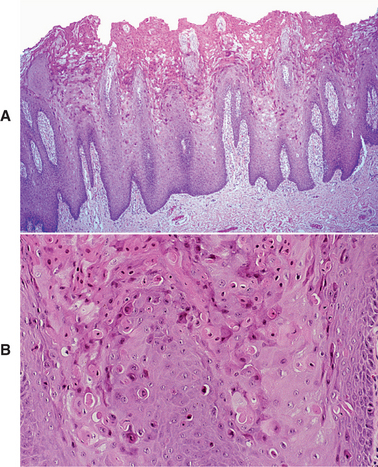
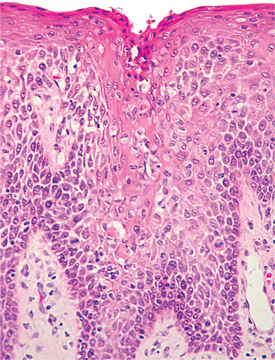
HISTOPATHOLOGIC FEATURES: The microscopic features of white sponge nevus are characteristic but not necessarily pathognomonic. Prominent hyperparakeratosis and marked acanthosis with clearing of the cytoplasm of the cells in the spinous layer are common features (Figs. 16-5 and 16-6); however, similar microscopic findings may be associated with leukoedema and hereditary benign intra-epithelial dyskeratosis (HBID). In some instances, an eosinophilic condensation is noted in the perinuclear region of the cells in the superficial layers of the epithelium, a feature that is unique to white sponge nevus. Ultrastructurally, this condensed material can be identified as tangled masses of keratin tonofilaments.
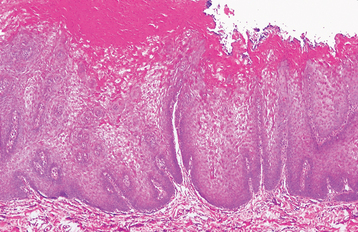
Fig. 16-5 White sponge nevus. This low-power photomicrograph shows prominent hyperparakeratosis, marked thickening (acanthosis), and vacuolation of the spinous cell layer.
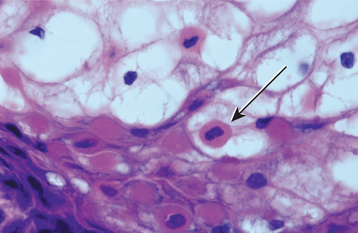
Fig. 16-6 White sponge nevus. This high-power photomicrograph shows vacuolation of the cytoplasm of the cells of the spinous layer, with no evidence of epithelial atypia. Perinuclear condensation of keratin tonofilaments can also be observed in some cells.
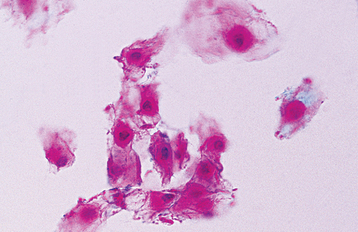
Exfoliative cytologic studies may provide more definitive diagnostic information. A cytologic preparation stained with the Papanicolaou method often shows the eosinophilic perinuclear condensation of the epithelial cell cytoplasm to a greater extent than does the histopathologic section (Fig. 16-7).
HEREDITARY BENIGN INTRAEPITHELIAL DYSKERATOSIS (WITKOP-VON SALLMANN SYNDROME)
Hereditary benign intraepithelial dyskeratosis (HBID) is a rare autosomal dominant genodermatosis primarily affecting descendants of a triracial isolate (Native American, black, and white) of people who originally lived in North Carolina. Examples of HBID have sporadically been reported from other areas of the United States because of migration of affected individuals, and descriptions of affected patients with no apparent connection to North Carolina have also appeared in the literature.
CLINICAL FEATURES: The lesions of HBID usually develop during childhood, in most instances affecting the oral and conjunctival mucosa. The oral lesions are similar to those of white sponge nevus, with both conditions showing thick, corrugated white plaques involving the buccal and labial mucosa (Fig. 16-8). Milder cases may exhibit the opalescent appearance of leukoedema. Other oral mucosal sites, such as the floor of the mouth and lateral tongue, may also be affected. These oral lesions may exhibit a superimposed candidal infection as well.
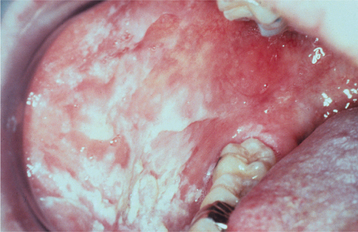
Fig. 16-8 Hereditary benign intraepithelial dyskeratosis (HBID). Oral lesions appear as corrugated white plaques of the buccal mucosa. (Courtesy of Dr. John McDonald.)
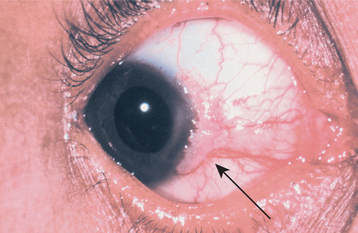
The most interesting feature of HBID is the ocular lesions, which begin to develop very early in life. These appear as thick, opaque, gelatinous plaques affecting the bulbar conjunctiva adjacent to the cornea (Fig. 16-9) and sometimes involving the cornea itself. When the lesions are active, patients may experience tearing, photophobia, and itching of the eyes. In many patients, the plaques are most prominent in the spring and tend to regress during the summer or autumn. Sometimes blindness may result from the induction of vascularity of the cornea secondary to the shedding process.
HISTOPATHOLOGIC FEATURES: The histopathologic features of HBID include prominent parakeratin production in addition to marked acanthosis. A peculiar dyskeratotic process, similar to that of Darier’s disease, is scattered throughout the upper spinous layer of the surface oral epithelium (Fig. 16-10). With this dyskeratotic process, an epithelial cell appears to be surrounded or engulfed by an adjacent epithelial cell, resulting in the so-called cell-within-a-cell phenomenon.
TREATMENT AND PROGNOSIS: Because HBID is a benign condition, no treatment is generally required or indicated for the oral lesions. If superimposed candidiasis develops, then an antifungal medication can be used. Patients with symptomatic ocular lesions should be referred to an ophthalmologist. Typically, the plaques that obscure vision must be surgically excised. This procedure, however, is recognized as a temporary measure because the lesions often recur.
PACHYONYCHIA CONGENITA (JADASSOHN-LEWANDOWSKY TYPE; JACKSON-LAWLER TYPE)
Pachyonychia congenita is a group of rare genodermatoses that are usually inherited as an autosomal dominant trait. The nails are dramatically affected in most patients, but oral lesions are seen only in patients affected by the Jadassohn-Lewandowsky form of the disease (pachyonychia congenita type 1). Fewer than 500 cases have been reported. Specific mutations in either the keratin 6a or keratin 16 gene have been detected for the Jadassohn-Lewandowsky type of pachyonychia congenita, whereas mutations of either the keratin 6b or keratin 17 gene are associated with the Jackson-Lawler form (pachyonychia congenita type 2).
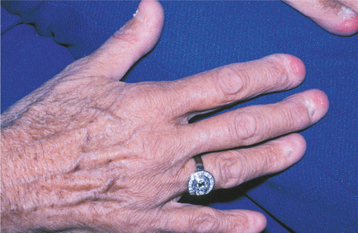
CLINICAL FEATURES: Virtually all patients with pachyonychia congenita exhibit characteristic nail changes, either at birth or in the early neonatal period. The free margins of the nails are lifted up because of an accumulation of keratinaceous material in the nail beds. This results in a pinched, tubular configuration. Ultimately, nail loss may occur (Fig. 16-11).
Other skin changes that may occur include marked hyperkeratosis of the palmar and plantar surfaces, producing thick, callouslike lesions (Fig. 16-12). Hyperhidrosis of the palms and soles is also commonly present. The rest of the skin shows punctate papules, representing an abnormal accumulation of keratin in the hair follicles. One disabling feature of the syndrome is the formation of painful blisters on the soles of the feet after a few minutes of walking during warm weather.
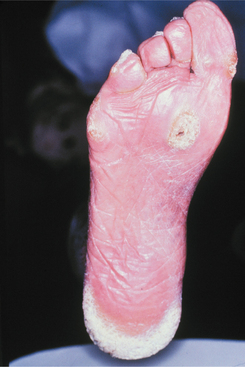
Fig. 16-12 Pachyonychia congenita. The soles of the feet of affected patients typically show marked calluslike thickenings. (Courtesy of Dr. Lou Young.)
The oral lesions seen in the Jadassohn-Lewandowsky form consist of thickened white plaques that involve the lateral margins and dorsal surface of the tongue. Other oral mucosal regions that are frequently exposed to mild trauma, such as the palate, buccal mucosa, and alveolar mucosa, may also be affected (Fig. 16-13). Neonatal teeth have been reported in patients affected by the Jackson-Lawler form, but these individuals do not have oral white lesions. Hoarseness and dyspnea have been described in some patients as a result of laryngeal mucosal involvement.
HISTOPATHOLOGIC FEATURES: Microscopic examination of lesional oral mucosa shows marked hyperparakeratosis and acanthosis with perinuclear clearing of the epithelial cells.
TREATMENT AND PROGNOSIS: Because the oral lesions of pachyonychia congenita show no apparent tendency for malignant transformation, no treatment is required. The nails are often lost or may need to be surgically removed because of the deformity. In addition, the keratin accumulation on the palms and soles can be quite uncomfortable and distressing to many of the affected individuals. Most patients have to pay continuous attention to removal of the excess keratin, and issues related to quality of life often arise. Patients should receive genetic counseling, as an aid in family planning. Identification of the various keratin mutations associated with these disorders is now possible, using molecular techniques to evaluate material obtained from chorionic villus sampling, thereby allowing prenatal diagnosis.
DYSKERATOSIS CONGENITA (COLE-ENGMAN SYNDROME; ZINSSER-COLE-ENGMAN SYNDROME)
Dyskeratosis congenita is a rare genodermatosis that is usually inherited as an X-linked recessive trait, resulting in a striking male predilection. Autosomal dominant and autosomal recessive forms, although less common, have been reported. Mutations in the DKC1 gene have been determined to cause the X-linked form of dyskeratosis congenita. The mutated gene appears to disrupt the normal maintenance of telomerase, an enzyme that is critical in determining normal cellular longevity. The clinician should be aware of the condition because the oral lesions may undergo malignant transformation, and patients are susceptible to aplastic anemia.
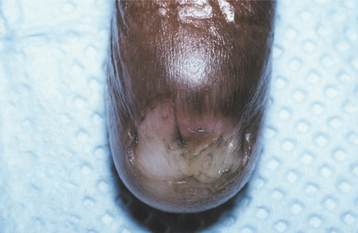
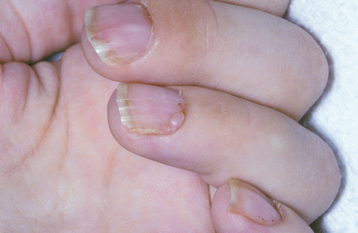
CLINICAL FEATURES: Dyskeratosis congenita usually becomes evident during the first 10 years of life. A reticular pattern of skin hyperpigmentation develops, affecting the face, neck, and upper chest. In addition, abnormal, dysplastic changes of the nails are evident at this time (Fig. 16-14).
Intraorally, the tongue and buccal mucosa develop bullae; these are followed by erosions and, eventually, leukoplakic lesions (Fig. 16-15). The leukoplakic lesions are considered to be premalignant, and approximately one third of them become malignant in a 10- to 30-year period. The actual rate of transformation may be higher, but this may not be appreciated because of the shortened life span of these patients. Rapidly progressive periodontal disease has been reported sporadically.
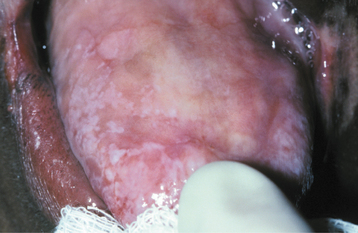
Fig. 16-15 Dyskeratosis congenita. Atrophy and hyperkeratosis of the dorsal tongue mucosa are visible.
Thrombocytopenia is usually the first hematologic problem that develops, typically during the second decade of life, followed by anemia. Ultimately, aplastic anemia develops in approximately 70% of these patients (see page 580). Mild to moderate mental retardation may also be present. Generally, the autosomal recessive and X-linked recessive forms show a more severe pattern of disease expression.
HISTOPATHOLOGIC FEATURES: Biopsy specimens of the early oral mucosal lesions show hyperorthokeratosis with epithelial atrophy. As the lesions progress, epithelial dysplasia develops until frank squamous cell carcinoma evolves.
TREATMENT AND PROGNOSIS: The discomfort of the oral lesions is managed symptomatically, and careful periodic oral mucosal ex-aminations are performed to check for evidence of malignant transformation. Routine medical evaluation is warranted to monitor the patient for the development of aplastic anemia. Selected patients may be considered for allogeneic bone marrow transplantation once the aplastic anemia is identified.
As a result of these potentially life-threatening complications, the prognosis is guarded. The average life span for the more severely affected patients is 32 years of age. The parents and the patient should receive genetic counseling, but identification of the DKC1 gene should allow for accurate confirmation of carriers of the gene and for prenatal diagnosis.
XERODERMA PIGMENTOSUM
Xeroderma pigmentosum is a rare genodermatosis in which numerous cutaneous malignancies develop at a very early age. The prevalence of the condition in the United States is estimated to be 1 in 250,000 to 500,000. The condition is inherited as an autosomal recessive trait and is caused by one of several defects in the excision repair and/or postreplication repair mechanism of DNA. As a result of the inability of the epithelial cells to repair ultraviolet (UV) light-induced damage, mutations in the epithelial cells occur, leading to the development of skin cancer at a rate 1000 to 4000 times what would normally be expected in people younger than 20 years of age.
CLINICAL FEATURES: During the first few years of life, patients affected by xeroderma pigmentosum show a markedly increased tendency to sunburn. Skin changes, such as atrophy, freckled pigmentation, and patchy depigmentation, soon follow (Fig. 16-16). In early childhood, actinic keratoses begin developing, a process that normally does not take place before 40 years of age. These lesions quickly progress to squamous cell carcinoma, with basal cell carcinoma also appearing; consequently, in most patients a nonmelanoma skin cancer develops during the first decade of life. Melanoma develops in about 5% of patients with xeroderma pigmentosum, but it evolves at a slightly later time. As a consequence of sun exposure, the head and neck region is the site most frequently affected by these cutaneous malignancies. Neurologic manifestations include subnormal intelligence in 80% of affected individuals.
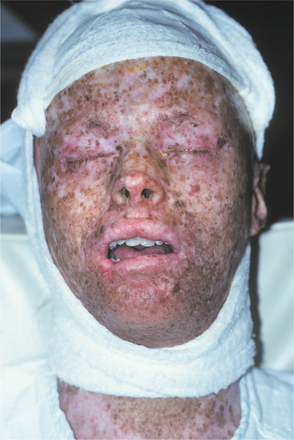
Fig. 16-16 Xeroderma pigmentosum. The atrophic changes and pigmentation disturbances shown are characteristic of xeroderma pigmentosum.
Oral manifestations, which often occur before 20 years of age, include development of squamous cell carcinoma of the lower lip and the tip of the tongue. This latter site is most unusual for oral cancer, and its involvement is again undoubtedly related to the increased sun exposure, however minimal, which this area receives in contrast to the rest of the oral mucosa.
The diagnosis of xeroderma pigmentosum is usually made when the patient is evaluated for the cutaneous lesions, because it is highly unusual for a very young person to have skin cancer. Because xeroderma pigmentosum is an autosomal recessive trait, a family history of the disorder is not likely to be present, but the possibility of a consanguineous relationship of the affected child’s parents should be investigated.
HISTOPATHOLOGIC FEATURES: The histopathologic features of xeroderma pigmentosum are relatively nonspecific, in that the cutaneous premalignant lesions and malignancies that occur are microscopically indistinguishable from those observed in unaffected patients.
TREATMENT AND PROGNOSIS: Treatment of xeroderma pigmentosum is challenging because in most instances significant sun damage has already occurred by the time of diagnosis. Patients are advised to avoid sunlight and unfiltered fluorescent light and to wear appropriate protective clothing and sunscreens if they cannot avoid sun exposure. Before receiving dental treatment, a calibrated UV light meter should be used to evaluate the amount of UV light being emitted from various sources in the dental office, such as the examination light, the radiograph viewbox, computer screens in the area, fiber-optic lights, or lights that are used for curing composite restorations. Some authors have suggested that any reading greater than 0 nm/cm2 would be unacceptable. A dermatologist should evaluate the patient every 3 months to monitor the development of cutaneous lesions.
Topical chemotherapeutic agents (e.g., 5-fluorouracil) may be used to treat actinic keratoses. Nonmelanoma skin cancers should be excised conservatively, preferably with microscopically controlled excision (Mohs surgery) to preserve as much normal tissue as possible. Patients should also receive genetic counseling, because a high number of consanguineous marriages have been reported in some series.
The prognosis is still poor. Most patients die 30 years earlier than the normal population, either directly from cutaneous malignancy or from complications associated with the treatment of the cancer.
HEREDITARY MUCOEPITHELIAL DYSPLASIA
Hereditary mucoepithelial dysplasia is a rare disorder that may occur sporadically or may be inherited as an autosomal dominant trait. Approximately 30 cases have been reported. For reasons that are not entirely understood, the mucosal epithelial cells do not develop in a normal fashion, and for this reason the designation of dysplasia has been given. However, in this situation, no increased risk of malignant transformation is seen. When a cervical exfoliative cytologic preparation (“Pap smear”) is done, the epithelial cells that are harvested may be interpreted as appearing cytologically unusual or atypical; in the past, some female patients have been advised to undergo hysterectomy because of this misinterpretation. Consequently, accurate identification of this disorder is extremely important for these patients.
CLINICAL FEATURES: Hereditary mucoepithelial dysplasia is characterized by both cutaneous and mucosal abnormalities. Patients typically have sparse, coarse hair with nonscarring alopecia. Eyelashes and eyebrows are generally affected (Fig. 16-17). Severe photophobia develops at an early age, and most of these patients will have evidence of cataracts beginning in childhood or early adult life. Corneal vascularization, keratitis, and nystagmus are also commonly described. As would be expected, vision is usually markedly impaired for these patients. Other skin manifestations include a prominent perineal rash that appears during infancy, as well as a widespread rough, dry texture because of follicular keratosis.
Pulmonary complications related to mucoepithelial dysplasia can range in severity, presumably because of the degree of gene expression. In one family, cavitary bullae were reported to form within the lung parenchyma, and these led to recurrent bouts of pneumonia, often resulting in life-threatening complications.
The oral manifestations of hereditary mucoepithelial dysplasia are usually quite striking, appearing as demarcated fiery-red erythema of the hard palate (Fig. 16-18), with generally less involvement of the attached gingivae and tongue mucosa. These mucosal alterations are typically asymptomatic, despite their remarkable clinical appearance. The nasal, conjunctival, vaginal, cervical, urethral, and bladder mucosa may have the same unusual erythematous features.
HISTOPATHOLOGIC FEATURES: Biopsies of the mucosal lesions of hereditary mucoepithelial dysplasia show epithelium with minimal keratinization and a disorganized maturation pattern. The squamous epithelial cells may have a relatively high nuclear/cytoplasmic ratio, but significant nuclear or cellular pleomorphism is not observed. Cytoplasmic vacuoles have been described and may appear as grayish inclusions (Fig. 16-19). These vacuoles also may be observed in exfoliative cytology samples. Ultrastructurally, the lesional cells have been described as having reduced numbers of desmosomes and internalized gap junctions.
INCONTINENTIA PIGMENTI (BLOCH-SULZBERGER SYNDROME)
Incontinentia pigmenti is a relatively rare inherited disorder, with approximately 800 cases reported worldwide. It typically evolves in several stages, primarily affecting the skin, eyes, and central nervous system (CNS), as well as oral structures. There is a marked female predilection, with a 37:1 female-to-male ratio reported. The condition is inherited as an X-linked dominant trait, with the single unpaired gene on the X chromosome being lethal for most males. The mutated gene responsible for producing the phenotypic features of incontinentia pigmenti maps to the Xq28 locus, where the genetic information related to nuclear factor-kB essential modulator (NEMO) is found. Of the few males who survive, a small percentage have Klinefelter syndrome (XXY karyotype), whereas the rest usually show mosaicism for the NEMO gene, suggesting a postzygotic mutation.
CLINICAL FEATURES: The clinical manifestations of incontinentia pigmenti usually begin in the first few weeks of infancy. There are four stages:
1. Vesicular stage—Vesiculobullous lesions appear on the skin of the trunk and limbs. Spontaneous resolution occurs within 4 months.
2. Verrucous stage—Verrucous cutaneous plaques develop, affecting the limbs. These clear by 6 months of age, evolving into the third stage.
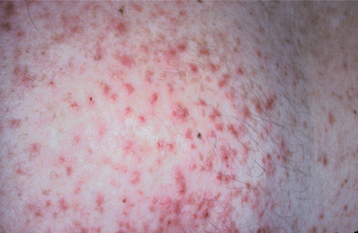
3. Hyperpigmentation stage—Macular, brown skin lesions appear, characterized by a strange swirling pattern (Fig. 16-20), although these tend to fade around the time of puberty.
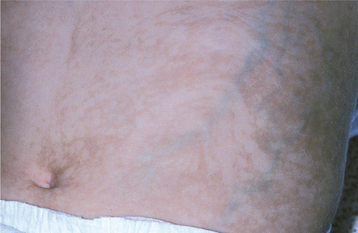
4. Atrophy and depigmentation stage—Atrophy and depigmentation of the skin ultimately occur.
CNS abnormalities occur in approximately 30% of affected patients. The most common problems are mental retardation, seizure disorders, and motor difficulties. Ocular problems (e.g., strabismus, nystagmus, cataracts, retinal vascular abnormalities, optic nerve atrophy) may also be identified in nearly 35% of these patients.
The oral manifestations of incontinentia pigmenti, noted in 70% to 95% of the cases, include oligodontia (hypodontia), delayed eruption, and hypoplasia of the teeth (Fig. 16-21). The teeth are small and cone shaped; both the primary and permanent dentitions are affected.
HISTOPATHOLOGIC FEATURES: The microscopic findings in incontinentia pigmenti vary, depending on when a biopsy of the skin lesions is performed.
In the initial vesicular stage, intraepithelial clefts filled with eosinophils are observed. During the verrucous stage, hyperkeratosis, acanthosis, and papillomatosis are noted. The hyperpigmentation stage shows numerous melanin-containing macrophages (melanin incontinence) in the subepithelial connective tissue, the feature from which the disorder derives its name.
TREATMENT AND PROGNOSIS: Treatment of incontinentia pigmenti is directed toward the various abnormalities. Dental management includes appropriate prosthodontic and restorative care, although this is sometimes difficult if CNS problems are severe. Prenatal genetic testing can be performed, but currently this is not widely available.
DARIER’S DISEASE (KERATOSIS FOLLICULARIS; DYSKERATOSIS FOLLICULARIS; DARIER-WHITE DISEASE)
Darier’s disease is an uncommon genodermatosis with rather striking skin involvement and relatively subtle oral mucosal lesions. The condition is inherited as an autosomal dominant trait, having a high degree of penetrance and variable expressivity. A lack of cohesion among the surface epithelial cells characterizes this disease, and mutation of a gene that encodes an intracellular calcium pump has been identified as the cause for abnormal desmosomal organization in the affected epithelial cells. Estimates of the prevalence of Darier’s disease in Northern European populations range from 1 in 36,000 to 1 in 100,000.
CLINICAL FEATURES: Patients with Darier’s disease have numerous erythematous, often pruritic, papules on the skin of the trunk and the scalp that develop during the first or second decade of life (Fig. 16-22). An accumulation of keratin, producing a rough texture, may be seen in association with the lesions, and a foul odor may be present as a result of bacterial degradation of the keratin. The process generally becomes worse during the summer months, either because of sensitivity of some patients to UV light or because increased heat results in sweating, which induces more epithelial clefting. The palms and soles often exhibit pits and keratoses. The nails show longitudinal lines, ridges, or painful splits.
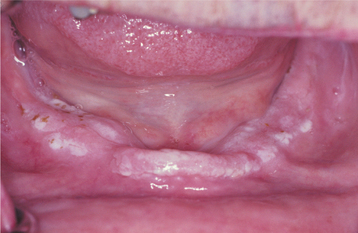
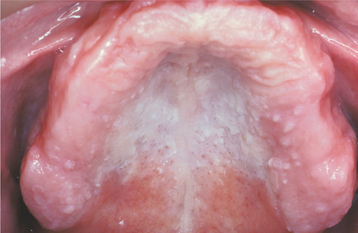
The oral lesions are typically asymptomatic and are discovered on routine examination. The frequency of occurrence of oral lesions ranges from 15% to 50%. They consist of multiple, normal-colored or white, flat-topped papules that, if numerous enough to be confluent, result in a cobblestone mucosal appearance (Fig. 16-23). These lesions affect the hard palate and alveolar mucosa primarily, although the buccal mucosa or tongue may be occasionally involved. If the palatal lesions are prominent, then the condition may resemble inflammatory papillary hyperplasia or nicotine stomatitis. Some patients with this condition also experience recurrent obstructive parotid swelling secondary to duct abnormalities.
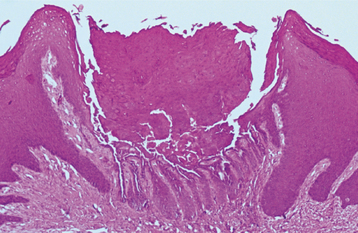
HISTOPATHOLOGIC FEATURES: Microscopic examination of the cutaneous or mucosal lesions shows a dyskeratotic process characterized by a central keratin plug that overlies epithelium exhibiting a suprabasilar cleft (Fig. 16-24). This intraepithelial clefting phenomenon, also known as acantholysis, is not unique to Darier’s disease and may be seen in conditions such as pemphigus vulgaris (see page 765). In addition, the epithelial rete ridges associated with the lesions appear narrow, elongated, and “test tube”-shaped. Closer inspection of the epithelium reveals varying numbers of two types of dyskeratotic cells, called corps ronds (round bodies) or grains (because they resemble cereal grains).
TREATMENT AND PROGNOSIS: Treatment of Darier’s disease depends on the severity of involvement. Photosensitive patients should use a sunscreen, and all patients should minimize unnecessary exposure to hot environments. For relatively mild cases, keratolytic agents or emollients may be the only treatment required. For more severely affected patients, systemic retinoids are often beneficial, but the side effects of such medications are often quite bothersome to the patient and can be significant; therefore, the physician should carefully monitor their use. Although the condition is not premalignant or otherwise life threatening, genetic counseling is appropriate.
WARTY DYSKERATOMA (ISOLATED DARIER’S DISEASE; ISOLATED DYSKERATOSIS FOLLICULARIS; FOCAL ACANTHOLYTIC DYSKERATOSIS; FOLLICULAR DYSKERATOMA)
The warty dyskeratoma is a distinctly uncommon solitary lesion that can occur on skin or oral mucosa. It is histopathologically identical to Darier’s disease. For this reason the lesion has been termed isolated Darier’s disease. The lesion is not otherwise related to Darier’s disease, however, and its cause remains unknown.
CLINICAL FEATURES: The cutaneous warty dyskeratoma typically appears as a solitary, asymptomatic, umbilicated papule on the skin of the head or neck of an older adult. The intraoral lesion also develops in patients older than age 40, and a slight male predilection has been identified. The intraoral warty dyskeratoma appears as a pink or white, umbilicated papule located on the keratinized mucosa, especially the hard palate and the alveolar ridge (Fig. 16-25). A warty or roughened surface is noted in some lesions. Most warty dyskeratomas are smaller than 0.5 cm in diameter.
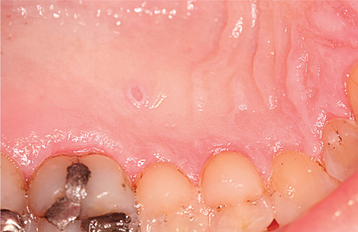
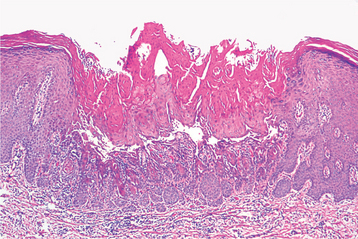
HISTOPATHOLOGIC FEATURES: Histopathologically, the warty dyskeratoma appears very similar to keratosis follicularis. Both conditions display dyskeratosis, basilar hyperplasia, and a suprabasilar cleft (Fig. 16-26). The warty dyskeratoma is a solitary lesion, however, and the formation of corps ronds and grains is not a prominent feature.
TREATMENT AND PROGNOSIS: Treatment of the warty dyskeratoma consists of conservative excision. The prognosis is excellent; these lesions have not been reported to recur, and they have no apparent malignant potential. Careful histopathologic evaluation of the tissue should be performed, because some epithelial dysplasias may show a marked lack of cellular cohesiveness, resulting in a similar acantholytic appearance microscopically.
PEUTZ-JEGHERS SYNDROME
Peutz-Jeghers syndrome is a relatively rare but well-recognized condition, having a prevalence of approximately 1 in 100,000 to 200,000 births. It is characteri-zed by freckle-like lesions of the hands, perioral skin, and oral mucosa, in conjunction with intestinal polyposis and predisposition for affected patients to develop cancer. The syndrome is generally inherited as an autosomal dominant trait, although as many as 35% of cases represent new mutations. Mutation of the STK11 gene (also known as LKB1) on chromosome 19p13.3, which encodes for a serine/threonine kinase, is responsible for most cases of Peutz-Jeghers syndrome.
CLINICAL FEATURES: The skin lesions of Peutz-Jeghers syndrome usually develop early in childhood and involve the periorificial areas (e.g., mouth, nose, anus, genital region). The skin of the extremities is affected in about 50% of patients (Fig. 16-27). The lesions resemble freckles, but they do not wax and wane with sun exposure, as do true freckles.
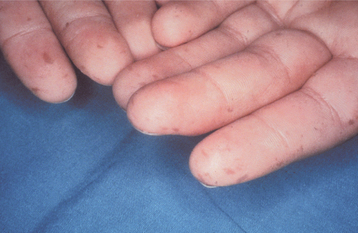
Fig. 16-27 Peutz-Jeghers syndrome. Cutaneous lesions appear as brown, macular, frecklelike areas, often concentrated around the mouth or on the hands. (Courtesy of Dr. Ahmed Uthman.)
The intestinal polyps, generally considered to be hamartomatous growths, are scattered throughout the mucus-producing areas of the gastrointestinal tract. The jejunum and ileum are most commonly affected. Patients often have problems with intestinal obstruction because of intussusception (“telescoping” of a proximal segment of the bowel into a distal portion), a problem that usually becomes evident during the third decade of life. Most of these episodes are self-correcting, but surgical intervention is sometimes necessary to prevent ischemic necrosis of the bowel, with subsequent peritonitis. Gastrointestinal adenocarcinoma develops in a significant percentage of affected patients, although the polyps themselves do not appear to be premalignant. In one large series of cases, 9% of the patients had developed gastrointestinal malignancy by 40 years of age and 33% by 60 years of age. This compares to 0.1% and 1.0%, respectively, in the general population. Other tumors affecting the pancreas, male and female genital tract, breast, and ovary may also develop. In women, the risk of developing breast cancer approaches 50% by 60 years of age. The increased frequency of malignancy in these patients overall is estimated to be approximately 18 times greater than normal.
The oral lesions essentially represent an extension of the perioral freckling. These 1- to 4-mm brown to blue-gray macules primarily affect the vermilion zone, the labial and buccal mucosa, and the tongue; they are seen in more than 90% of these patients (Fig. 16-28). The number of lesions and the extent of involvement can vary markedly from patient to patient. Some degree of fading of the pigmented lesions may be noted during adolescence.
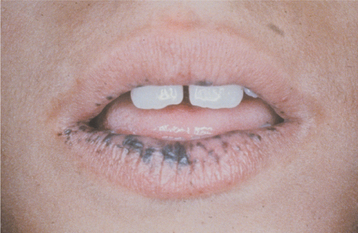
HISTOPATHOLOGIC FEATURES: The gastrointestinal polyps of Peutz-Jeghers syndrome histopathologically represent benign overgrowths of intestinal glandular epithelium supported by a core of smooth muscle. Epithelial atypia is not usually a prominent feature, unlike the polyps of Gardner syndrome (see page 651).
Microscopic evaluation of the pigmented cutaneous lesions shows slight acanthosis of the epithelium with elongation of the rete ridges. No apparent increase in melanocyte number is detected by electron microscopy, but the dendritic processes of the melanocytes are elongated. Furthermore, the melanin pigment appears to be retained in the melanocytes rather than being transferred to adjacent keratinocytes.
HEREDITARY HEMORRHAGIC TELANGIECTASIA (OSLER-WEBER-RENDU SYNDROME)
Hereditary hemorrhagic telangiectasia (HHT) is an uncommon mucocutaneous disorder that is inherited as an autosomal dominant trait, and recent epidemiologic studies suggest a prevalence of at least 1 in 10,000 people. Mutation of either one of two different genes at two separate loci is responsible for the condition. HHT1 is caused by a mutation of the ENG (endoglin) gene on chromosome 9, whereas mutation of ALK1 (activin receptor-like kinase-1; ACVRL1), a gene located on chromosome 12, produces HHT2. The proteins produced by these genes may play a role in blood vessel wall integrity. With both types of HHT, numerous vascular hamartomas develop, affecting the skin and mucosa; however, other vascular problems, such as arteriovenous fistulas, may also be seen. Patients affected with HHT1 tend to have more pulmonary involvement, whereas those with HHT2 generally have a later onset of their telangiectasias and a greater degree of hepatic involvement. The clinician should be familiar with HHT because the oral lesions are often the most dramatic and most easily identified component of this syndrome.
CLINICAL FEATURES: Patients with HHT are often diagnosed initially because of frequent episodes of epistaxis. On further examination, the nasal and oropharyngeal mucosae exhibit numerous scattered red papules, 1 to 2 mm in size, which blanch when diascopy is used. This blanching indicates that the red color is due to blood contained within blood vessels (in this case, small collections of dilated capillaries [telangiectasias] that are close to the surface of the mucosa). These telangiectatic vessels are most frequently found on the vermilion zone of the lips, tongue, and buccal mucosa, although any oral mucosal site may be affected (Figs. 16-29 and 16-30). With aging, the telangiectasias tend to become more numerous and slightly larger.
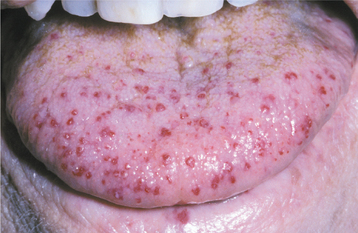
Fig. 16-29 Hereditary hemorrhagic telangiectasia (HHT). The tongue of this patient shows multiple red papules, which represent superficial collections of dilated capillary spaces.
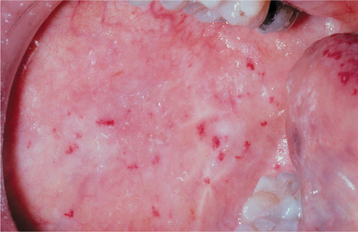
Fig. 16-30 Hereditary hemorrhagic telangiectasia (HHT). Red macules similar to the tongue lesions are observed on the buccal mucosa.
In many patients, telangiectasias are seen on the hands and feet. The lesions are often distributed throughout the gastrointestinal mucosa, the genitourinary mucosa, and the conjunctival mucosa. The gastrointestinal telangiectasias have a tendency to rupture, which may cause significant blood loss. Chronic iron-deficiency anemia is often a problem for such individu als. Significantly, arteriovenous fistulas may develop in the lungs (30% of HHT patients), liver (30%), or brain (10% to 20%). The brain lesions seem to predispose these patients to the development of brain abscesses. In at least one instance, periodontal vascular malformations were felt to be the cause of septic pulmonary emboli that resolved only after several teeth with periodontal abscesses were extracted.
A diagnosis of HHT can be made if a patient has three of the following four criteria:
1. Recurrent spontaneous epistaxis
2. Telangiectasias of the mucosa and skin
3. Arteriovenous malformation involving the lungs, liver, or CNS
In some instances, CREST syndrome (Calcinosis cutis, Raynaud’s phenomenon, Esophageal dysfunction, Sclerodactyly, and Telangiectasia) (see page 801) must be considered in the differential diagnosis. In these cases, serologic studies for anticentromere autoantibodies often help to distinguish between the two conditions because these antibodies typically would be present only in CREST syndrome.
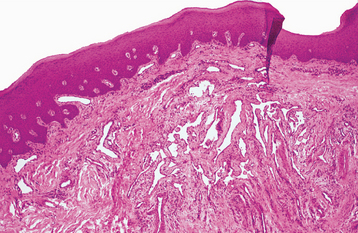
HISTOPATHOLOGIC FEATURES: If one of the telangiectasias is submitted for biopsy, the microscopic features essentially show a superficially located collection of thin-walled vascular spaces that contain erythrocytes (Fig. 16-31).
TREATMENT AND PROGNOSIS: For mild cases of HHT, no treatment may be required. Moderate cases may be managed by selective cryosurgery or electrocautery of the most bothersome of the telangiectatic vessels. Laser ablation of the telangiectatic lesions has also been used, although this approach appears to be most successful for patients with mild to moderate disease. More severely affected patients, particularly those troubled by repeated episodes of epistaxis, may require a surgical procedure of the nasal septum (septal dermoplasty). The involved nasal mucosa is removed and replaced by a skin graft; however, some long-term follow-up studies suggest that the grafts eventually become revascularized, resulting in recurrence of the problem. Nasal closure is another surgical technique that has been performed for patients with severe epistaxis in whom other methods have failed.
Combined progesterone and estrogen therapy may benefit some patients, but because of the potentially serious side effects, this should be limited to the most severely affected individuals. Iron replacement therapy is indicated for the iron-deficient patient, and occasionally blood transfusions may be necessary to compensate for blood loss.
From a dental standpoint, some authors recommend the use of prophylactic antibiotics before dental procedures that might cause bacteremia in patients with HHT and evidence of a pulmonary arteriovenous malformation. For patients with a history of HHT, such antibiotics are advocated until a pulmonary arteriovenous malformation is ruled out because of the 1% prevalence of brain abscesses in affected individuals. Researchers believe that antibiotic coverage, similar to that for endocarditis prophylaxis, may prevent this serious complication. Patients with a history of HHT should be screened for arteriovenous malformations, which can be eliminated by embolization or other vasodestructive techniques using interventional radiologic methods.
The prognosis is generally good, although a 1% to 2% mortality rate is reported from complications related to blood loss. For patients with brain abscesses, the mortality rate is 10%, even with early diagnosis and appropriate therapy.
EHLERS-DANLOS SYNDROMES
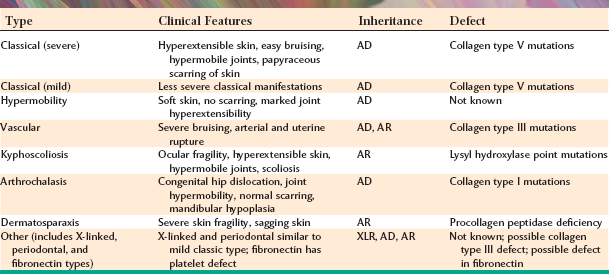
The Ehlers-Danlos syndromes, a group of inherited connective tissue disorders, are relatively heterogeneous. At least 10 types have been described over the years, but recent clinical and molecular evidence suggests that seven categories of this disease may be more appropriate. The patient exhibits problems that are usually attributed to the production of abnormal collagen, the protein that is the main structural component of the connective tissue. Because the production of collagen necessitates many biochemical steps that are controlled by several genes, the potential exists for any one of these genes to mutate, producing selective defects in collagen synthesis. The various forms of abnormal collagen result in many overlapping clinical features for each of the types of the Ehlers-Danlos syndrome (Table 16-1). This discussion concentrates on the most common and significant forms of this group of conditions.
Typical clinical findings include hypermobility of the joints, easy bruisability, and marked elasticity of the skin. Some patients have worked in circus sideshows as the “rubber” man and the “contortionist” as a result of their pronounced joint mobility and ability to stretch the skin.
CLINICAL FEATURES: The pattern of inheritance and the clinical manifestations vary with the type of Ehlers-Danlos syndrome being examined. About 80% of patients have the classical type in either the mild or severe form. Classical Ehlers-Danlos syndrome is inherited as an autosomal dominant trait, and defects of type I collagen have been reported in some families, whereas problems with type V collagen have been identified in others, suggesting genetic heterogeneity. Hyperelasticity of the skin (Fig. 16-32) and cutaneous fragility can be observed. An unusual healing response that often occurs with rela tively minor injury to the skin is termed papyraceous scarring because it resembles crumpled cigarette paper (Fig. 16-33).
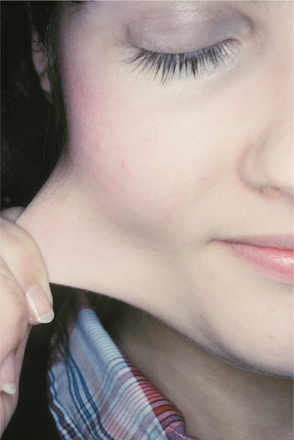
Fig. 16-32 Ehlers-Danlos syndrome. The hyperelasticity of the skin is evident in this patient affected by the mild form of classical Ehlers-Danlos syndrome.
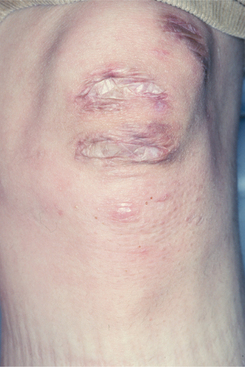
Fig. 16-33 Ehlers-Danlos syndrome. Scarring that resembles crumpled cigarette paper (papyraceous scarring) is associated with minimal trauma in patients with Ehlers-Danlos syndromes. These lesions involve the skin of the knee.
Patients with the hypermobility type of Ehlers-Danlos syndrome exhibit remarkable joint hypermobility but no evidence of scarring.
The vascular type of Ehlers-Danlos used to be known as the ecchymotic type because of the extensive bruising that occurs with everyday trauma. Defects in type III collagen have been identified in this disorder. This form is inherited in an autosomal dominant pattern, and the patient may be mistaken for a victim of child abuse. The life expectancy of these patients is often greatly reduced because of the tendency for aortic aneurysm formation and rupture.
One rare form of Ehlers-Danlos syndrome (originally reported as type VIII) was described as having dental manifestations as a hallmark feature, with patients showing marked periodontal disease activity at a relatively early age. Although these patients may have overlapping features with either the classical or vascular forms of the disease, recent studies examining five affected families in Sweden have suggested that this form of Ehlers-Danlos syndrome is linked to a specific mutation of a gene that has been mapped to chromosome 12p13.
The oral manifestations of Ehlers-Danlos syndrome include the ability of 50% of these patients to touch the tip of their nose with their tongue (Gorlin sign), a feat that can be achieved by less than 10% of the general population. Some authors have noted easy bruising and bleeding during minor manipulations of the oral mucosa; others state that oral mucosal friability is present. A tendency for recurrent subluxation of the temporomandibular joint (TMJ) and the development of other TMJ disorders has also been reported.
Most patients with Ehlers-Danlos syndrome have normal teeth. A variety of dental abnormalities have been described, however, including malformed, stunted tooth roots, large pulp stones, and hypoplastic enamel. Although most of these findings have not been consistently correlated with any particular type of the syndrome, pulp stones seem to occur more commonly in patients affected by classical Ehlers-Danlos syndrome.
TREATMENT AND PROGNOSIS: The prognosis for the patient with Ehlers-Danlos syndrome depends on the type. Some forms, such as the vascular type, can be very serious, with sudden death occurring from rupture of the aorta secondary to the weakened, abnormal collagen that constitutes the vessel wall. The mild classical type is generally compatible with a normal life span, although affected women may have problems with placental tearing and hemorrhage during gestation.
Accurate diagnosis is important because it affects the prognosis heavily. Similarly, because the various types of this syndrome show a variety of inheritance patterns, an accurate diagnosis is required so that appropriate genetic counseling can be provided.
TUBEROUS SCLEROSIS (EPILOIA; BOURNEVILLE-PRINGLE SYNDROME)
Tuberous sclerosis is an uncommon syndrome that is classically characterized by mental retardation, seizure disorders, and angiofibromas of the skin. The condition is often inherited as an autosomal dominant trait, but two thirds of the cases are sporadic and appear to represent new mutations. These mutations involve either one of two genes: TSC1 (found on chromosome 9) or, more commonly, TSC2 (found on chromosome 16). Both of these gene products are believed to contribute to the same intracellular biochemical pathway that seems to have a tumor suppressor function. The multiple hamartomatous growths that are seen in this disorder are thought to arise from disruption of the normal tumor suppressor function of these genes. Tuberous sclerosis has a wide range of clinical severity, although patients who have the TSC2 mutation have a more severe expression of the disease than patients who have the TSC1 mutation. Milder forms of tuberous sclerosis may be difficult to diagnose.
The prevalence is at least 1 in 10,000 in the general population, although in some long-term care facilities tuberous sclerosis accounts for as high as 1% of the mentally retarded patients.
CLINICAL FEATURES: Several clinical features characterize tuberous sclerosis. The first of these, facial angiofibromas, used to be called adenoma sebaceum. Because these lesions are neither adenomas nor sebaceous, the use of that term should be discontinued. Facial angiofibromas appear as multiple, smooth-surfaced papules and occur primarily in the nasolabial fold area (Fig. 16-34). Similar lesions, called ungual or periungual fibromas, are seen around or under the margins of the nails (Fig. 16-35).
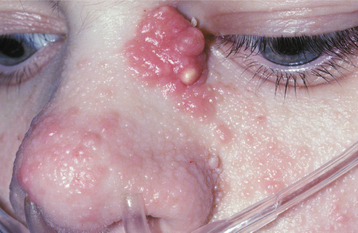
Fig. 16-34 Tuberous sclerosis. Patients typically have multiple papular facial lesions that microscopically are angiofibromas.
Two other characteristic skin lesions are connective tissue hamartomas called shagreen patches and ovoid areas of hypopigmentation called ash-leaf spots. Even though approximately 5% of the general population may have an ash-leaf spot, studies have reported that 60% to 97% of children with tuberous sclerosis display these lesions. The shagreen patches, so named because of their resemblance to sharkskin-derived shagreen cloth, affect the skin of the trunk. The ash-leaf spots may appear on any cutaneous surface and may be best visualized using UV (Wood’s lamp) illumination.
CNS manifestations include seizure disorders in 70% to 80% of affected patients and mental retardation in 33% to 45%. In addition, hamartomatous proliferations in the CNS develop into the potato-like growths (“tubers”) seen at autopsy, from which the term tuberous sclerosis is derived (Fig. 16-36). The tuberous hamartomas can best be visualized using T2-weighted magnetic resonance imaging (MRI) and are present in 80% to 95% of these patients.
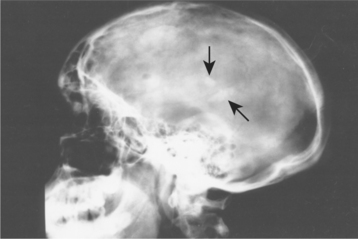
Fig. 16-36 Tuberous sclerosis. Patchy calcifications (arrows) associated with intracranial hamartoma formation are seen on this lateral skull radiograph. (Courtesy of Dr. Reg Munden.)
A relatively rare tumor of the heart muscle, called cardiac rhabdomyoma, is also typically associated with this syndrome. This lesion, which probably represents a hamartoma rather than a true neoplasm, occurs in approximately 30% to 50% of affected patients and is typically identified in childhood. Problems with myocardial function often develop as a result of this process, but many of these tumors undergo spontaneous regression.
Another hamartomatous type of growth related to this disorder is the angiomyolipoma. This is a benign neoplasm composed of vascular smooth muscle and adipose tissue and occurs primarily in the kidney.
Oral manifestations of tuberous sclerosis include developmental enamel pitting on the facial aspect of the anterior permanent dentition in 50% to 100% of patients. These pits are more readily appreciated after applying a dental plaque-disclosing solution to the teeth. Multiple fibrous papules affect 11% to 56% of patients. The fibrous papules are seen predominantly on the anterior gingival mucosa (Fig. 16-37), although the lips, buccal mucosa, palate, and tongue may be involved. Diffuse fibrous gingival enlargement is reported in affected patients who are not taking phenytoin; however, most cases of gingival hyperplasia in these individuals are probably related to medication taken to control seizures. Some patients with tuberous sclerosis may also exhibit radiolucencies of the jaws that represent dense fibrous connective tissue proliferations (Fig. 16-38).
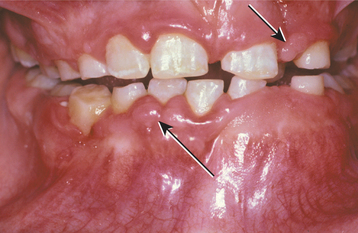
Fig. 16-37 Tuberous sclerosis. Patients often exhibit gingival hyperplasia, which may be secondary to phenytoin medications used to control seizures in some cases. Fibrous papules of the gingiva (arrows) may also be present.
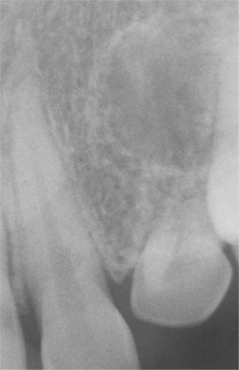
Fig. 16-38 Tuberous sclerosis. Periapical radiograph exhibiting a well-defined radiolucency apical to the maxillary left lateral incisor. Biopsy revealed an intraosseous fibrous proliferation.
The diagnosis of tuberous sclerosis can be based on finding at least two of the following major features:
The presence of one major and two minor features may also confirm the diagnosis. The minor features include the following:
HISTOPATHOLOGIC FEATURES: Microscopic examination of the fibrous papules of the oral mucosa or the enlarged gingivae shows a nonspecific fibrous hyperplasia. Similarly, the radiolucent jaw lesions consist of dense fibrous connective tissue that resembles desmoplastic fibroma or the simple type of central odontogenic fibroma. The angiofibroma of the skin is a benign aggregation of delicate fibrous connective tissue characterized by plump, uniformly spaced fibroblasts with numerous interspersed thin-walled vascular channels.
TREATMENT AND PROGNOSIS: For patients with tuberous sclerosis, most of the treatment is directed toward the management of the seizure disorder with anticonvulsant agents. Periodic MRI of the head may be done to screen for intracranial lesions, whereas ultrasound evaluation is performed for evaluation of kidney involvement. Mentally retarded patients may have problems with oral hygiene procedures, and poor oral hygiene contributes to phenytoin-induced gingival hyperplasia. Patients affected by this condition have a slightly reduced life span compared with the general population, with death usually related to CNS or kidney disease. Genetic counseling is also appropriate for affected patients, and genetic testing is available for both TSC1 and TSC2 mutations if prenatal or preimplantation family planning is desired.
MULTIPLE HAMARTOMA SYNDROME (COWDEN SYNDROME; PTEN HAMARTOMA-TUMOR SYNDROME)
Multiple hamartoma syndrome is a rare condition that has important implications for the affected patient, because malignancies, in addition to the benign hamartomatous growths, develop in a high percentage of these individuals. Usually, the syndrome is inherited as an autosomal dominant trait showing a high degree of penetrance and a range of expressivity. The gene responsible for this disorder has been mapped to chromosome 10, and a mutation of the PTEN (phosphatase and tensin homolog deleted on chromosome 10) gene has been implicated in its pathogenesis. The estimated prevalence of this condition is approximately 1 in 200,000, and more than 300 affected patients have been described in the literature. In recent years, overlapping clinical features of multiple hamartoma syndrome with Lhermitte-Duclos disease, Bannayan-Riley-Ruvalcaba syndrome, and Proteus-like syndrome have been noted, and all of these disorders have demonstrated mutations of the PTEN gene.
CLINICAL FEATURES: Cutaneous manifestations are present in almost all patients with multiple hamartoma syndrome, usually developing during the second decade of life. The majority of the skin lesions appear as multiple, small (less than 1 mm) papules, primarily on the facial skin, especially around the mouth, nose, and ears (Fig. 16-39). Microscopically, most of these papules represent hair follicle hamartomas called trichilemmomas. Other commonly noted skin lesions are acral keratosis, a warty-appearing growth that develops on the dorsal surface of the hand, and palmoplantar keratosis, a prominent calluslike lesion on the palms or soles. Cutaneous hemangiomas, neuromas, xanthomas, and lipomas have also been described.
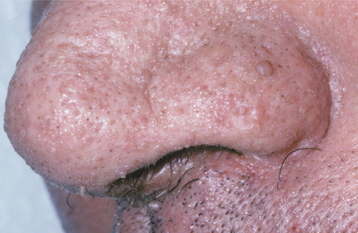
Fig. 16-39 Multiple hamartoma syndrome. These tiny cutaneous facial papules represent hair follicle hamartomas (trichilemmomas).
Other problems can appear in these patients as well. Thyroid disease usually appears as either a goiter or a thyroid adenoma, but follicular adenocarcinoma may develop. In women, fibrocystic disease of the breast is frequently observed. Unfortunately, breast cancer occurs with a relatively high frequency (25% to 50%) in these patients. The mean age at diagnosis of breast malignancy is 40 years, which is much younger than usual. In the gastrointestinal tract, multiple benign hamartomatous polyps may be present. In addition, several types of benign and malignant tumors of the female genitourinary tract occur more often than in the normal population.
The oral lesions vary in severity from patient to patient and usually consist of multiple papules affecting the gingivae, dorsal tongue, and buccal mucosa (Figs. 16-40 and 16-41). These lesions have been reported in more than 80% of affected patients and generally produce no symptoms. Other possible oral findings include a high-arched palate, periodontitis, and extensive dental caries, although it is unclear whether the latter two conditions are significantly related to the syndrome.
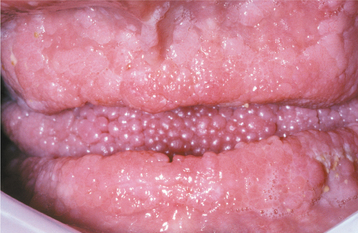
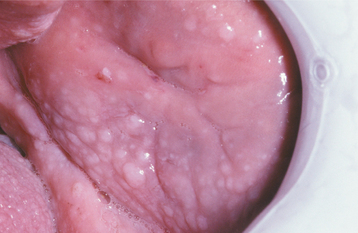
HISTOPATHOLOGIC FEATURES: The histopathologic features of the oral lesions are rather nonspecific, essentially representing fibroepithelial hyperplasia. Other lesions associated with this syndrome have their own characteristic histopathologic findings, depending on the hamartomatous or neoplastic tissue origin.
DIAGNOSIS: The diagnosis can be based on the finding of two of the following three pathognomonic signs:
A variety of other major and minor diagnostic criteria, as well as a positive family history, are also helpful in confirming the diagnosis. Genetic testing for mutations of the PTEN gene are clinically available, but 20% of patients who otherwise have characteristic multiple hamartoma syndrome will not demonstrate a genetic abnormality; therefore, a negative test does not necessarily preclude the diagnosis of multiple hamartoma syndrome.
TREATMENT AND PROGNOSIS: Treatment of multiple hamartoma syndrome is con-troversial. Although most of the tumors that develop are benign, the prevalence of malignancy is higher than in the general population; therefore, annual physical examinations that focus specifically on anatomic sites of increased tumor prevalence are appropriate. Some investigators recommend bilateral prophylactic mastectomies as early as the third decade of life for female patients because of the associated increased risk of breast cancer.
EPIDERMOLYSIS BULLOSA
The term epidermolysis bullosa describes a heterogeneous group of inherited blistering mucocutaneous disorders. Each has a specific defect in the attachment mechanisms of the epithelial cells, either to each other or to the underlying connective tissue. Recent advances in the understanding of the clinical, epidemiologic, and molecular genetic abnormalities of these conditions have led to the identification of approximately 25 different forms. Depending on the defective mechanism of cellular cohesion, there are four broad categories:
Each category consists of several forms of the disorder. A variety of inheritance patterns may be seen, depending on the particular form. The degree of severity can range from relatively mild, annoying forms, such as the simplex types, through a spectrum that includes severe, fatal disease. For example, many cases of junctional epidermolysis bullosa result in death at birth because of the significant sloughing of the skin during passage through the birth canal. Specific mutations in the genes encoding keratin 5 and keratin 14 have been identified as being responsible for most of the simplex types, whereas mutations in the genetic codes for the a3, b3, and g2 subunits of laminin have been documented for the junctional types. Most of the dystrophic types appear to be caused by mutations in the genes responsible for type VII collagen production, with nearly 200 distinctly different mutations identified to date. The hemidesmosomal type is the most recently characterized pattern of this group of disorders, and mutations of genes associated with various hemidesmosomal attachment proteins, such as plectin, type XVII collagen (BP180), and a6b4 integrin, have been established.
A few representative examples of the types of epidermolysis bullosa are summarized in Table 16-2. Because oral lesions are most commonly observed in the dystrophic forms, this discussion centers on these forms. Dental abnormalities, such as anodontia, enamel hypoplasia, pitting of the enamel, neonatal teeth, and severe dental caries, have been variably associated with several of the different types of epidermolysis bullosa, although studies have suggested that the prevalence of dental abnormalities is significantly increased only with the junctional type. A disorder termed epidermolysis bullosa acquisita is mentioned because of the similarity of its name; however, this appears to be an unrelated condition, having an autoimmune (rather than a genetic) origin (see page 744).
Table 16-2
Examples of Epidermolysis Bullosa
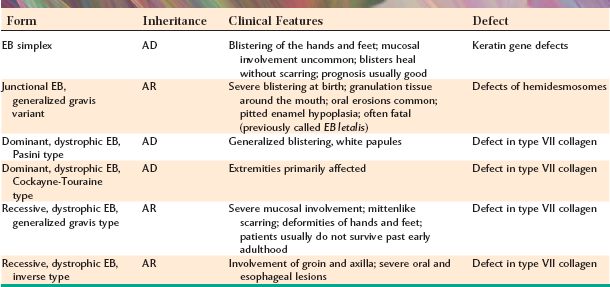
EB, Epidermolysis bullosa; AD, autosomal dominant; AR, autosomal recessive.
DOMINANT DYSTROPHIC TYPES: The dystrophic forms of epidermolysis bullosa that are inherited in an autosomal dominant fashion are not usually life threatening, although they may certainly be disfiguring and pose many problems. The initial lesions are vesicles or bullae, which are seen early in life and develop on areas exposed to low-grade, chronic trauma, such as the knuckles or knees (Fig. 16-42). The bullae rupture, resulting in erosions or ulcerations that ultimately heal with scarring. In the process, appendages such as fingernails may be lost.
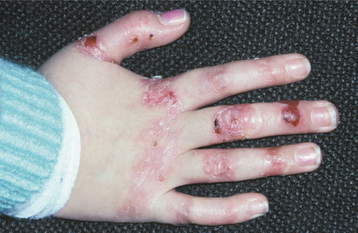
Fig. 16-42 Epidermolysis bullosa. A young girl, affected by the dominant dystrophic form of epidermolysis bullosa, shows the characteristic hemorrhagic bullae, scarring, and erosion associated with minimal trauma to the hands.
The oral manifestations are typically mild, with some gingival erythema and tenderness. Gingival recession and reduction in the depth of the buccal vestibule may be observed (Fig. 16-43).
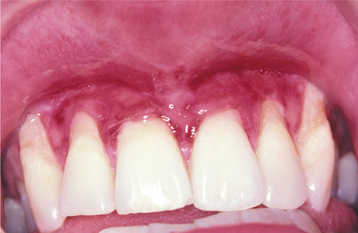
RECESSIVE DYSTROPHIC TYPES: Generalized recessive dystrophic epidermolysis bullosa represents one of the more debilitating forms of the disease. Vesicles and bullae form with even minor trauma. Secondary infections are often a problem because of the large surface areas that may be involved. If the patient manages to survive into the second decade, then hand function is often greatly diminished because of the repeated episodes of cutaneous break down and healing with scarring, resulting in fusion of the fingers into a mittenlike deformity (Fig. 16-44).
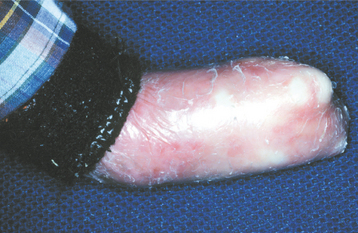
Fig. 16-44 Epidermolysis bullosa. A 19-year-old man, affected by recessive dystrophic epidermolysis bullosa, shows the typical mittenlike deformity of the hand caused by scarring of the tissue after damage associated with normal activity.
The oral problems are no less severe. Bulla and vesicle formation is induced by virtually any food having some degree of texture. Even with a soft diet, the repeated cycles of scarring often result in microstomia (Fig. 16-45) and ankyloglossia. Similar mucosal injury and scarring may cause severe stricture of the esophagus. Because a soft diet is usually highly cariogenic, carious destruction of the dentition at an early age is common.
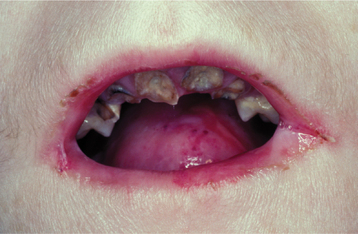
Fig. 16-45 Epidermolysis bullosa. Same patient as depicted in Fig. 16-44. Microstomia has been caused by repeated trauma and healing with scarring. Note the severe dental caries activity associated with a soft cariogenic diet.
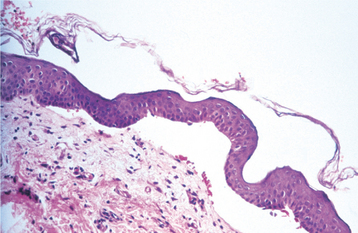
HISTOPATHOLOGIC FEATURES: The histopathologic features of epidermolysis bullosa vary with the type being examined. The simplex form shows intraepithelial clefting by light microscopy. Junctional, dystrophic, and hemidesmosomal forms show subepithelial clefting (Fig. 16-46). Electron microscopic examination, which is still considered the diagnostic “gold standard,” reveals clefting at the level of the lamina lucida of the basement membrane in the junctional forms and below the lamina densa of the basement membrane in the dystrophic forms. In contrast, the hemidesmosomal form shows clefting just below the basal cell layer, at its interface with the lamina lucida. Immunohistochemical evaluation of perilesional tissue may help to identify specific defects to classify and subtype the condition further. Molecular genetic analysis is now being used to confirm the diagnosis in some instances.
TREATMENT AND PROGNOSIS: Treatment of epidermolysis bullosa varies with the type. For milder cases, no treatment other than local wound care may be needed. Sterile drainage of larger blisters and the use of topical antibiotics are often indicated in these situations. For the more severe cases, intensive management with oral antibiotics may be necessary if cellulitis develops; despite intensive medical care, some patients die as a result of infectious complications.
The “mitten” deformity of the hands, seen in recessive dystrophic epidermolysis bullosa, can be corrected with plastic surgery, but the problem usually recurs after a period of time, and surgical intervention is required every 2 years on average. With esophageal involvement, dysphagia may be a significant problem, resulting in malnutrition and weight loss. Placement of a gastrostomy tube may be necessary at times. Patients with the recessive dystrophic forms are also predisposed to development of cutaneous squamous cell carcinoma. This malignancy often develops in areas of chronic ulceration during the second through third decades of life and represents a significant cause of death for these patients. Infrequently, the lingual mucosa of affected patients has been reported to undergo malignant transformation as well.
Management of the oral manifestations also depends on the type of the disease. For patients who are susceptible to mucosal bulla formation, dental manipulation should be kept to a minimum. To achieve this, topical 1% neutral sodium fluoride solution should be administered daily to prevent dental caries. A soft diet that is as noncariogenic as possible, as well as atraumatic oral hygiene procedures, should be encouraged. Maintaining adequate nutrition for affected patients is critical to ensure optimal wound healing.
If dental restorative care is required, the lips should be lubricated to minimize trauma. Injections for local anesthesia can usually be accomplished by depositing the anesthetic slowly and deeply within the tissues. For extensive dental care, endotracheal anesthesia may be performed without significant problems in most cases.
Unfortunately, because of the genetic nature of these diseases, no cure exists. Genetic counseling of affected families is indicated. Both prenatal diagnosis and preimplantation diagnosis are available as adjuncts to family planning.
Immune-Mediated Diseases and Their Evaluation
Several conditions discussed in this chapter are the result of inappropriate production of antibodies by the patient (autoantibodies). These autoantibodies are directed against various constituents of the molecular apparatus that hold epithelial cells together or that bind the surface epithelium to the underlying connective tissue. The ensuing damage produced by the interaction of these autoantibodies with the host tissue is seen clinically as a disease process, often termed an immunobullous disease. Because each disease is characterized by production of specific types of autoantibodies, identification of the antibodies and the tissues against which they are targeted is important diagnostically. The two techniques that are widely used to investigate the immunobullous diseases are (1) direct immunofluorescence and (2) indirect immunofluorescence studies. Following is a brief overview of how they work.
Direct immunofluorescence is used to detect autoantibodies that are bound to the patient’s tissue. Before testing can take place, several procedures must occur. Inoculating human immunoglobulins into a goat creates antibodies directed against these human immunoglobulins. The antibodies raised in response to the human immunoglobulins are harvested from the animal and tagged with fluorescein, a dye that glows when viewed with ultraviolet (UV) light. As illustrated on the left side of Fig. 16-47, a frozen section of the patient’s tissue is placed on a slide, and this is incubated with fluorescein-conjugated goat antihuman antibodies. These antibodies bind to the tissue at any site where human immunoglobulin is present. The excess antibody suspension is washed off, and the section is then viewed with a microscope having a UV light source.
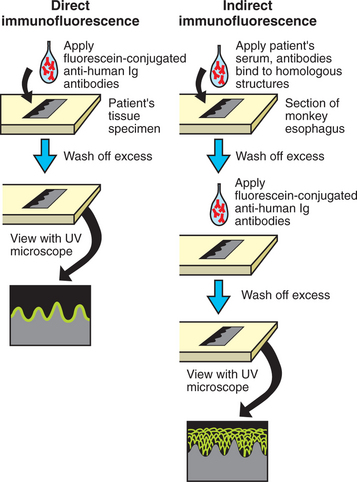
Fig. 16-47 Immunofluorescence techniques. Comparison of the techniques for direct and indirect immunofluorescence. The left side depicts the direct immunofluorescent findings in cicatricial pemphigoid, a disease that has autoantibodies directed toward the basement zone. The right side shows the indirect immunofluorescent findings for pemphigus vulgaris, a disease that has autoantibodies directed toward the intercellular areas between the spinous cells of the epithelium. Ig, Immunoglobulin; UV, ultraviolet.
With indirect immunofluorescence studies, the patient is being evaluated for presence of antibodies that are circulating in the blood. As shown on the right side of Fig. 16-47, a frozen section of tissue that is similar to human oral mucosa (e.g., Old World monkey esophagus) is placed on a slide and incubated with the patient’s serum. If there are autoantibodies directed against epithelial attachment structures in the patient’s serum, then they will attach to the homologous structures on the monkey esophagus. The excess serum is washed off, and fluorescein-conjugated goat antihuman antibody is incubated with the section. The excess is washed off, and the section is examined with UV light to detect the presence of autoantibodies that might have been in the serum.
Examples of the molecular sites of attack of the autoantibodies are seen diagrammatically in Fig. 16-48. Each site is distinctive for a particular disease; however, the complexities of the epithelial attachment mechanisms are still being elucidated, and more precise mapping may be possible in the future. A summary of the clinical, microscopic, and immunopathologic features of the more important immune-mediated mucocutaneous diseases is found in Table 16-3.
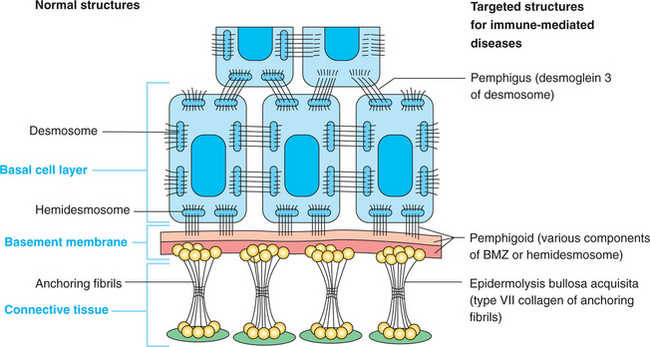
Fig. 16-48 Epithelial attachment apparatus. Schematic diagram demonstrating targeted structures in several immune-mediated diseases. BMZ, Basement membrane zone.
PEMPHIGUS
The condition known as pemphigus represents four related diseases of an autoimmune origin:
Only the first two of these affect the oral mucosa, and the discussion is limited to pemphigus vulgaris. Pemphigus vegetans is rare; most authorities now feel it represents simply a variant of pemphigus vulgaris.
Pemphigus vulgaris is the most common of these disorders (vulgaris is Latin for common). Even so, it is not seen very often. The estimated incidence is one to five cases per million people diagnosed each year in the general population. Nevertheless, pemphigus vulgaris is an important condition because, if untreated, it often results in the patient’s death. Furthermore, the oral lesions are often the first sign of the disease, and they are the most difficult to resolve with therapy. This has prompted the description of the oral lesions as “the first to show, and the last to go.”
The blistering that typifies this disease is due to an abnormal production, for unknown reasons, of autoantibodies that are directed against the epidermal cell surface glycoproteins, desmoglein 3 and desmoglein 1. These desmogleins are components of desmosomes (structures that bond epithelial cells to each other), and the autoantibodies attach to these desmosomal components, effectively inhibiting the molecular interaction that is responsible for adherence. As a result of this immunologic attack on the desmosomes, a split develops within the epithelium, causing a blister to form. Desmoglein 3 is preferentially expressed in the parabasal region of the epidermis and oral epithelium, whereas desmoglein 1 is found primarily in the superficial portion of the epidermis, with minimal expression in oral epithelium. Patients who have developed autoantibodies directed against desmoglein 3, with or without desmoglein 1, will histopathologically show intraepithelial clefting just above the basal layer, and clinically oral mucosal blisters of pemphigus vulgaris will form. Patients who develop autoantibodies directed against only desmoglein 1 will histopathologically show superficial intraepithelial clefting of the epidermis, but oral mucosa will not be affected. Clinically, the fine scaly red lesions of pemphigus foliaceous or pemphigus erythematosus will be evident.
Occasionally, a pemphigus-like oral and cutaneous eruption may occur in patients taking certain medications (e.g., penicillamine, angiotensin-converting enzyme [ACE] inhibitors, nonsteroidal antiinflammatory drugs [NSAIDs]) or in patients with malignancy, especially lymphoreticular malignancies (so-called paraneoplastic pemphigus) (see page 769). Similarly, a variety of other conditions may produce chronic vesiculoulcerative or erosive lesions of the oral mucosa, and these often need to be considered in the differential diagnosis (see Table 16-3). In addition, a rare genetic condition termed chronic benign familial pemphigus or Hailey-Hailey disease may have erosive cutaneous lesions, but oral involvement in that process appears to be uncommon.
CLINICAL FEATURES: The initial manifestations of pemphigus vulgaris often involve the oral mucosa, typically in adults. The average age at diagnosis is 50 years, although rare cases may be seen in childhood. No sex predilection is observed, and the condition seems to be more common in persons of Mediterranean, South Asian, or Jewish heritage.
Patients usually complain of oral soreness, and examination shows superficial, ragged erosions and ulcerations distributed haphazardly on the oral mucosa (Figs. 16-49 to 16-52). Such lesions may affect virtually any oral mucosal location, although the palate, labial mucosa, buccal mucosa, ventral tongue, and gingivae are often involved. Patients rarely report vesicle or bulla formation intraorally, and such lesions can seldom be identified by the examining clinician, probably because of early rupture of the thin, friable roof of the blisters. More than 50% of the patients have oral mucosal lesions before the onset of cutaneous lesions, sometimes by as much as 1 year or more. Eventually, however, nearly all patients have intraoral involvement. The skin lesions appear as flaccid vesicles and bullae (Fig. 16-53) that rupture quickly, usually within hours to a few days, leaving an erythematous, denuded surface. Infrequently ocular involvement may be seen, usually appearing as bilateral conjunctivitis. Unlike cicatricial pemphigoid, the ocular lesions of pemphigus do not tend to produce scarring and symblepharon formation (see page 772).
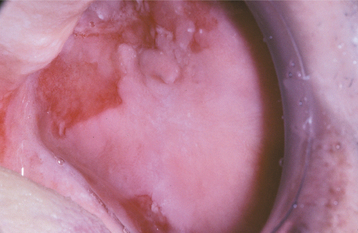
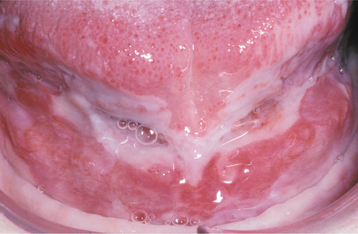
Fig. 16-50 Pemphigus vulgaris. Large, irregularly shaped ulcerations involving the floor of the mouth and ventral tongue.
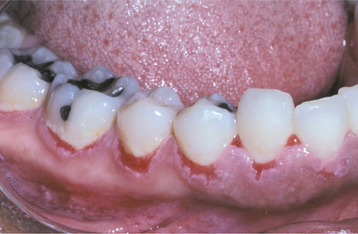
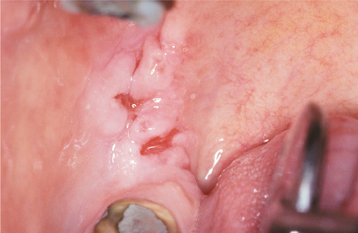
Fig. 16-52 Pemphigus vulgaris. The patient, with a known diagnosis of pemphigus vulgaris, had been treated with immunosuppressive therapy. The oral erosions shown here were the only persistent manifestation of her disease.
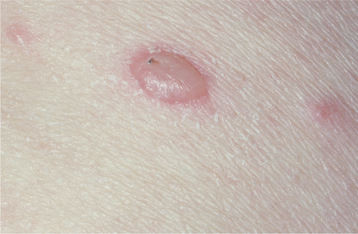
Without proper treatment, the oral and cutaneous lesions tend to persist and progressively involve more surface area. A characteristic feature of pemphigus vulgaris is that a bulla can be induced on normal-appearing skin if firm lateral pressure is exerted. This is called a positive Nikolsky sign.
HISTOPATHOLOGIC FEATURES: Biopsy specimens of perilesional tissue show characteristic intraepithelial separation, which occurs just above the basal cell layer of the epithelium (Fig. 16-54). Sometimes the entire superficial layers of the epithelium are stripped away, leaving only the basal cells, which have been described as resembling a “row of tombstones.” The cells of the spinous layer of the surface epithelium typically appear to fall apart, a feature that has been termed acantholysis, and the loose cells tend to assume a rounded shape (Fig. 16-55). This feature of pemphigus vulgaris can be used in making a diagnosis based on the identification of these rounded cells (Tzanck cells) in an exfoliative cytologic preparation. A mild-to-moderate chronic inflammatory cell infiltrate is usually seen in the underlying connective tissue.
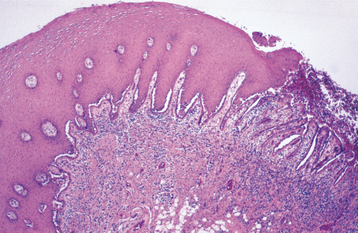
Fig. 16-54 Pemphigus vulgaris. Low-power photomicrograph of perilesional mucosa affected by pemphigus vulgaris. An intraepithelial cleft is located just above the basal cell layer.
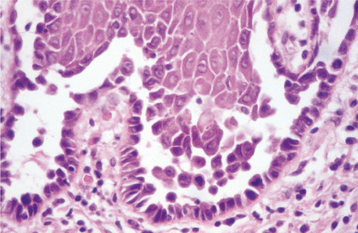
Fig. 16-55 Pemphigus vulgaris. High-power photomicrograph showing rounded, acantholytic epithelial cells sitting within the intraepithelial cleft.
The diagnosis of pemphigus vulgaris should be confirmed by direct immunofluorescence examination of fresh perilesional tissue or tissue submitted in Michel’s solution. With this procedure, antibodies (usually IgG or IgM) and complement components (usually C3) can be demonstrated in the intercellular spaces between the epithelial cells (Fig. 16-56) in almost all patients with this disease. Indirect immunofluorescence is also typically positive in 80% to 90% of cases, demonstrating the presence of circulating autoantibodies in the patient’s serum. Enzyme-linked immunosorbent assays (ELISAs) have been developed to detect circulating autoantibodies as well.
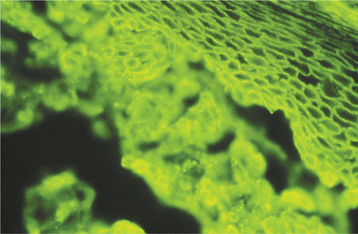
Fig. 16-56 Pemphigus vulgaris. Photomicrograph depicting the direct immunofluorescence pattern of pemphigus vulgaris. Immunoreactants are deposited in the intercellular areas between the surface epithelial cells.
It is critical that perilesional tissue be obtained for both light microscopy and direct immunofluorescence to maximize the probability of a diagnostic sample. If ulcerated mucosa is submitted for testing, then the results are often inconclusive because of either a lack of an intact interface between the epithelium and connective tissue or a great deal of nonspecific inflammation.
TREATMENT AND PROGNOSIS: A diagnosis of pemphigus vulgaris should be made as early in its course as possible because control is generally easier to achieve. Pemphigus is a systemic disease; therefore, treatment consists primarily of systemic corticosteroids (usually prednisone), often in combination with other immunosuppressive drugs (so-called steroid-sparing agents), such as azathioprine. Although some clinicians have advocated the use of topical corticosteroids in the management of oral lesions, the observed improvement is undoubtedly because of the absorption of the topical agents, resulting in a greater systemic dose. The potential side effects associated with the long-term use of systemic corticosteroids are significant and include the following:
Ideally, a physician with expertise in immunosuppressive therapy should manage the patient. The most common approach is to use relatively high doses of systemic corticosteroids initially to clear the lesions, then attempt to maintain the patient on as low a dose of corticosteroids as is necessary to control the condition. Often the clinician can monitor the success of therapy by measuring the titers of circulating autoantibodies using indirect immunofluorescence, because disease activity frequently correlates with the abnormal antibody levels. Pemphigus may undergo complete resolution, although remissions and exacerbations are common. One study suggested that up to 75% of patients will have disease resolution after 10 years of treatment.
Before the development of corticosteroid therapy, as many as 60% to 90% of these patients died, primarily as a result of infections and electrolyte imbalances. Even today, the mortality rate associated with pemphigus vulgaris is in the range of 5% to 10%, usually because of the complications of long-term systemic corticosteroid use.
PARANEOPLASTIC PEMPHIGUS (NEOPLASIA-INDUCED PEMPHIGUS; PARANEOPLASTIC AUTOIMMUNE MULTIORGAN SYNDROME)
Paraneoplastic pemphigus is a rare vesiculobullous disorder that affects patients who have a neoplasm, usually lymphoma or chronic lymphocytic leukemia. Approximately 150 cases have been documented. Although the precise pathogenetic mechanisms are unknown, some evidence suggests abnormal levels of the cytokine, interleukin 6 (IL-6), could be produced by host lymphocytes in response to the patient’s tumor. IL-6 may then be responsible for stimulating the abnormal production of antibodies directed against antigens associated with the desmosomal complex and the basement membrane zone of the epithelium. In addition to a variety of different antibodies that attack these epithelial adherence structures, recent reports have described cutaneous and mucosal damage that appears to be mediated by cytotoxic T lymphocytes in some cases of paraneoplastic pemphigus. As a result of this multifaceted immunologic attack, the disease manifests in an array of clinical features, histopathologic findings, and immunopathologic findings that may be perplexing if the clinician is unfamiliar with this condition.
CLINICAL FEATURES: Patients typically have a history of a malignant lymphoreticular neoplasm, or less commonly, a benign lymphoproliferative disorder such as angiofollicular lymph node hyperplasia (Castleman’s disease) or thymoma. In approximately one third of reported cases, paraneoplastic pemphigus developed before a malignancy was identified, thus signaling the presence of a tumor. The neoplastic disease may or may not be under control at the time of onset of the paraneoplastic condition. Signs and symptoms of paraneoplastic pemphigus usually begin suddenly and may appear polymorphous. In some instances, multiple vesiculobullous lesions affect the skin (Fig. 16-57) and oral mucosa. Palmar or plantar bullae may be evident, a feature that is uncommon in pemphigus vulgaris. For other patients, skin lesions can appear more papular and pruritic, similar to cutaneous lichen planus. The lips often show hemorrhagic crusting similar to that of erythema multiforme (Fig. 16-58). The oral mucosa shows multiple areas of erythema and diffuse, irregular ulceration (Fig. 16-59), affecting virtually any oral mucosal surface. If the lesions remain untreated, then they persist and worsen. Some patients may develop only oropharyngeal lesions, without cutaneous involvement.
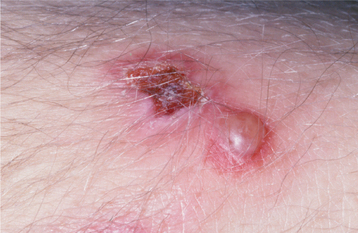
Fig. 16-57 Paraneoplastic pemphigus. The bulla and crusted ulcerations on this patient’s arm are representative of the polymorphous cutaneous lesions.
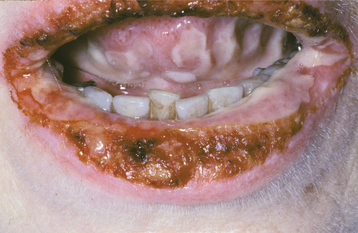
Fig. 16-58 Paraneoplastic pemphigus. Crusted, hemorrhagic lip lesions may be mistaken for erythema multiforme or herpes simplex infection.
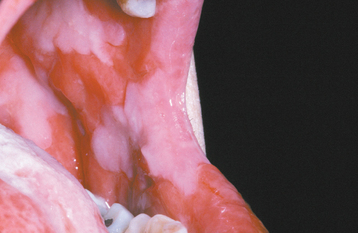
Other mucosal surfaces are also commonly affected, with 70% of patients having involvement of the conjunctival mucosa. In this area, a cicatrizing (scarring) conjunctivitis develops, similar to that seen with cicatricial pemphigoid (Fig. 16-60). The vaginal mucosa and mucosa of the respiratory tract may be involved.
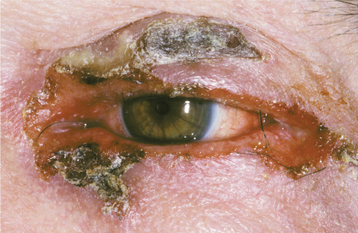
HISTOPATHOLOGIC FEATURES: The features of paraneoplastic pemphigus on light microscopic examination may be as diverse as the clinical features. In most cases, a lichenoid mucositis is seen, usually with subepithelial clefting (like pemphigoid) or intraepithelial clefting (like pemphigus) (Fig. 16-61).
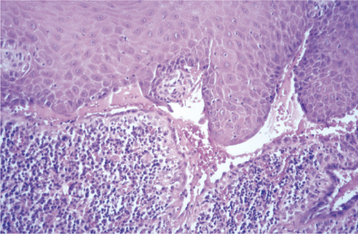
Fig. 16-61 Paraneoplastic pemphigus. This medium-power photomicrograph shows both intraepithelial and subepithelial clefting.
Direct immunofluorescence studies may show a weakly positive deposition of immunoreactants (IgG and complement) in the intercellular zones of the epithelium and/or a linear deposition of immunoreactants at the basement membrane zone. Indirect immunofluorescence should be conducted using a transitional type of epithelium (e.g., rat urinary bladder mucosa) as the substrate. This shows a fairly specific pattern of antibody localization to the intercellular areas of the epithelium. Immunoprecipitation studies remain the gold standard for the diagnosis of paraneoplastic pemphigus, however, because the various antibodies that characterize this condition can be identified with a considerable degree of specificity. Antibodies directed against desmoplakin I and II, major bullous pemphigoid antigen, envoplakin, and periplakin, in addition to desmoglein 1 and 3, are typically detected. Examples of paraneoplastic pemphigus that show only a lichen oid reaction with no demonstrable autoantibody production have infrequently been described.
TREATMENT AND PROGNOSIS: Paraneoplastic pemphigus is often a very serious condition with a high morbidity and mortality rate. For the infrequent cases associated with a benign lymphoproliferative condition, surgical removal of the tumor may result in regression of the paraneoplastic pemphigus. For those cases associated with malignancy, treatment essentially consists of systemic prednisone, typically combined with another immunosuppressive agent, such as azathioprine, methotrexate, or cyclophosphamide. As with pemphigus vulgaris, the skin lesions usually respond more quickly to treatment than the oral lesions. Unfortunately, although the immunosuppressive therapy often manages to control the autoimmune disease, this immunosuppression often seems to trigger a reactivation of the malignant neoplasm. Thus a high mortality rate is seen, with patients succumbing to complications of the vesiculobullous lesions, complications of immune suppressive therapy, or progression of malignant disease. Occasionally, long-term survivors are reported, but these seem to be in the minority. As more of these patients are identified, therapeutic strategies can be better evaluated and modified for optimal care in the future.
MUCOUS MEMBRANE PEMPHIGOID (CICATRICIAL PEMPHIGOID; BENIGN MUCOUS MEMBRANE PEMPHIGOID)
Evidence has accumulated to suggest that mucous membrane pemphigoid represents a group of chronic, blistering, mucocutaneous autoimmune diseases in which tissue-bound autoantibodies are directed against one or more components of the basement membrane. As such, this condition has a heterogeneous origin, with autoantibodies being produced against any one of a variety of basement membrane components, all of which produce similar clinical manifestations. The precise incidence is unknown, but most authors believe that it is at least twice as common as pemphigus vulgaris.
The term pemphigoid is used because clinically it often appears similar (the meaning of the -oid suffix) to pemphigus. The prognosis and microscopic features of pemphigoid, however, are very different.
Although a variety of terms have been used over the decades to designate this condition, a group of experts from both medicine and dentistry met in 1999 and came to an agreement that mucous membrane pemphigoid would be the most appropriate name for the disease. Cicatricial pemphigoid, another commonly used name for this process, is derived from the word cicatrix, meaning scar. When the conjunctival mucosa is affected, the scarring that results is the most significant aspect of this disorder because it invariably results in blindness unless the condition is recognized and treated. Interestingly, the oral lesions seldom exhibit this tendency for scar formation.
CLINICAL FEATURES: Mucous membrane pemphigoid usually affects older adults, with an average age of 50 to 60 years at the onset of disease. Females are affected more frequently than males by a 2:1 ratio. Oral lesions are seen in most patients, but other sites, such as conjunctival, nasal, esophageal, laryngeal, and vaginal mucosa, as well as the skin (Fig. 16-62), may be involved.
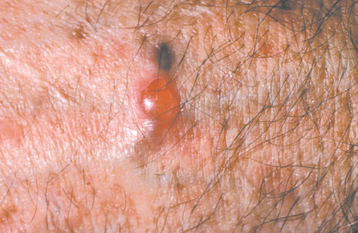
Fig. 16-62 Mucous membrane pemphigoid. Although cutaneous lesions are not common, tense bullae such as these may develop on the skin of 20% of affected patients. (Courtesy of Dr. Charles Camisa.)
The oral lesions of pemphigoid begin as either vesicles or bullae that may occasionally be identified clinically (Fig. 16-63). In contrast, patients with pemphigus rarely display such blisters. The most likely explanation for this difference is that the pemphigoid blister forms in a subepithelial location, producing a thicker, strong-er roof than the intraepithelial, acantholytic pemphigus blister. Eventually, the oral blisters rupture, leaving large, superficial, ulcerated, and denuded areas of mucosa (Fig. 16-64). The ulcerated lesions are usually painful and persist for weeks to months if untreated.
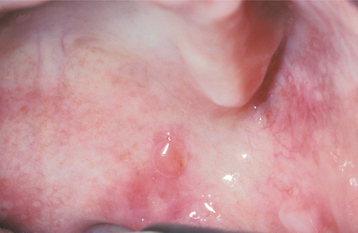
Fig. 16-63 Mucous membrane pemphigoid. One or more intraoral vesicles, as seen on the soft palate, may be detected in patients with cicatricial pemphigoid. Usually, ulcerations of the oral mucosa are also present.
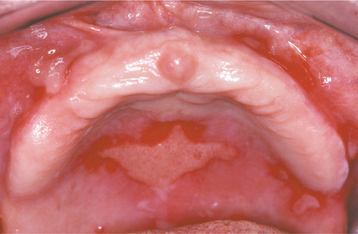
Fig. 16-64 Mucous membrane pemphigoid. Large, irregular oral ulcerations characterize the lesions after the initial bullae rupture.
Often this process is seen diffusely throughout the mouth, but it may be limited to certain areas, especially the gingiva (Fig. 16-65). Gingival involvement produces a clinical reaction pattern termed desquamative gingivitis (see page 162). This pattern may also be seen in other conditions, such as erosive lichen planus or, much less frequently, pemphigus vulgaris.
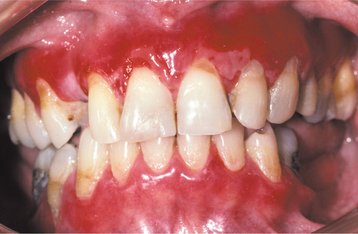
Fig. 16-65 Mucous membrane pemphigoid. Often the gingival tissues are the only affected site, resulting in a clinical pattern known as desquamative gingivitis. Such a pattern may also be seen with lichen planus and pemphigus vulgaris.
The most significant complication of mucous membrane pemphigoid, however, is ocular involvement. Although exact figures are not available, up to 25% of patients with oral lesions may eventually develop ocular disease. One eye may be affected before the other. The earliest change is subconjunctival fibrosis, which usually can be detected by an ophthalmologist using slit-lamp examination. As the disease progresses, the conjunctiva becomes inflamed and eroded. Attempts at healing lead to scarring between the bulbar (lining the globe of the eye) and palpebral (lining the inner surface of the eyelid) conjunctivae. Adhesions called symblepharons result (Fig. 16-66). Without treatment the inflammatory changes become more severe, although conjunctival vesicle formation is rarely seen (Fig. 16-67). Scarring can ultimately cause the eyelids to turn inward (entropion). This causes the eyelashes to rub against the cornea and globe (trichiasis) (Fig. 16-68). The scarring closes off the openings of the lacrimal glands as well, and with the loss of tears, the eye becomes extremely dry. The cornea then produces keratin as a protective mechanism; however, keratin is an opaque material, and blindness ensues. End-stage ocular involvement may also be characterized by adhesions between the upper and lower eyelids themselves (Fig. 16-69).
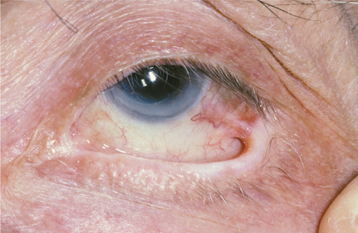
Fig. 16-66 Mucous membrane pemphigoid. Although the earliest ocular changes are difficult to identify, patients with ocular involvement may show adhesions (symblepharons) between the bulbar and palpebral conjunctivae before severe ocular damage occurs.
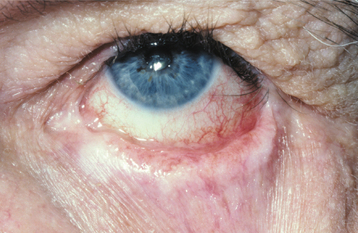
Fig. 16-67 Mucous membrane pemphigoid. The disease has caused the upper eyelid of this patient to turn inward (entropion), resulting in the eyelashes rubbing against the eye itself (trichiasis). Also note the obliteration of the lower fornix of the eye.
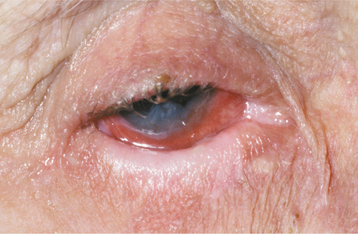
Fig. 16-68 Mucous membrane pemphigoid. A patient with ocular involvement shows severe conjunctival inflammation. An ophthalmologist removed the lower eyelashes because of trichiasis associated with entropion.
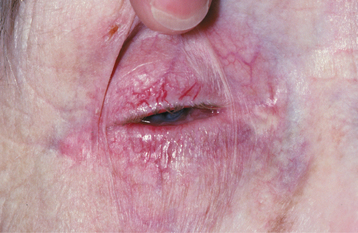
Fig. 16-69 Mucous membrane pemphigoid. In this patient, the ocular involvement has resulted in nearly complete scarring between the conjunctival mucosa and the eyelids themselves, producing blindness.
Other mucosal sites may also be involved and cause considerable difficulty for the patient. In females, the vaginal mucosal lesions may cause considerable pain during attempts at intercourse (dyspareunia).
Laryngeal lesions, which are fairly uncommon, may be especially significant because of the possibility of airway obstruction by the bullae that are formed. Patients who experience a sudden change in vocaliza tion or who have difficulty breathing should undergo examination with laryngoscopy.
HISTOPATHOLOGIC FEATURES: Biopsy of perilesional mucosa shows a split between the surface epithelium and the underlying connective tissue in the region of the basement membrane (Fig. 16-70). A mild chronic inflammatory cell infiltrate is present in the superficial submucosa.
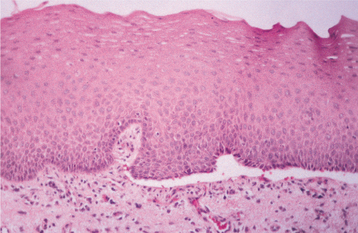
Fig. 16-70 Mucous membrane pemphigoid. Medium-power photomicrograph of perilesional tissue shows characteristic subepithelial clefting.
Direct immunofluorescence studies of perilesional mucosa show a continuous linear band of immunoreactants at the basement membrane zone in nearly 90% of affected patients (Fig. 16-71). The immune deposits consist primarily of IgG and C3, although IgA and IgM may also be identified. One study has suggested that, when IgG and IgA deposits are found in the same patient, the disease may be more severe. All of these immunoreactants may play a role in the pathogenesis of the subepithelial vesicle formation by weakening the attachment of the basement membrane through a variety of mechanisms, including complement activation with recruitment of inflammatory cells, particularly neutrophils.
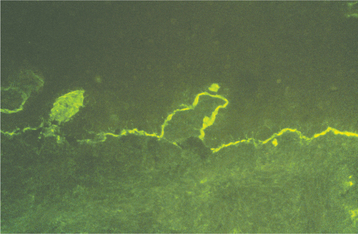
Fig. 16-71 Mucous membrane pemphigoid. Direct immunofluorescence studies show a deposition of immunoreactants at the basement membrane zone of the epithelium. (Courtesy of Dr. Ronald Grimwood.)
Indirect immunofluorescence is positive in only 5% to 25% of these patients, indicating a relatively consistent lack of readily detectable circulating autoanti-bodies. One type of mucous membrane pemphigoid produces low levels of circulating autoantibodies to epiligrin (laminin-5), a component of the basement membrane. Antiepiligrin mucous membrane pemphigoid seems to have more widespread involvement, affecting oral, nasal, ocular, and laryngeal mucosa, compared with other forms of mucous membrane pemphigoid. In contrast, another group of investigators has shown that pemphigoid patients with only oral mucosal involvement have circulating autoantibodies to a6 integrin, a component of the hemidesmosome.
For an accurate diagnosis, perilesional tissue—rather than the ulcerated lesion itself—should be obtained. Often the epithelium in the area of the lesion is so loose that it strips off as the clinician attempts to perform the biopsy. Such tissue is not usually adequate for diagnostic purposes because the interface between the epithelium and connective tissue is no longer intact (although some investigators have shown positive immunofluorescence with this tissue).
Other relatively rare conditions can mimic pemphigoid histopathologically. These include linear IgA bullous dermatosis, angina bullosa hemorrhagica, and epidermolysis bullosa acquisita.
LINEAR IgA BULLOUS DERMATOSIS: Linear IgA bullous dermatosis, as the name indicates, is characterized by the linear deposition of only IgA along the basement membrane zone. Even though some cases of mucous membrane pemphigoid may have IgA antibodies, linear IgA bullous dermatosis predominantly affects the skin and, therefore, can usually be distinguished from mucous membrane pemphigoid on a clinical basis.
ANGINA BULLOSA HEMORRHAGICA: Angina bullosa hemorrhagica is a rare, poorly characterized oral mucosal disorder that exhibits variably painful, blood-filled vesicles or bullae, usually affecting the soft palate of middle-aged or older adults. The blisters typically rupture spontaneously and heal without scarring. A subepithelial cleft is noted microscopically. No hematologic or immunopathologic abnormalities have been detected, and although the cause is unknown, many patients have a history of trauma or corticosteroid inhaler use.
EPIDERMOLYSIS BULLOSA ACQUISITA: Epidermolysis bullosa acquisita is an immunologically mediated condition characterized by autoantibodies directed against type VII collagen, the principal component of the anchoring fibrils. The anchoring fibrils play an important role in bonding the epithelium to the underlying connective tissue. As a result, their immunologic destruction leads to the formation of bullous lesions of the skin and mucosa with minimal trauma.
Oral lesions are present in nearly 50% of the cases, although such lesions are uncommon in the absence of cutaneous lesions. To distinguish epidermolysis bullosa acquisita from other immunobullous diseases with subepithelial clefting, a special technique is performed. A sample of the patient’s perilesional skin is incubated in a concentrated salt solution; this causes the epithelium to separate from the connective tissue, forming an artificially induced bulla. Immunohistochemical evaluation shows deposition of IgG autoantibodies on the floor (connective tissue side) of the bulla where type VII collagen resides. This finding is in contrast to that of most forms of mucous membrane pemphigoid, in which the autoantibodies are usually localized to the roof of the induced blister.
TREATMENT AND PROGNOSIS: Once the diagnosis of mucous membrane pemphigoid has been established by light microscopy and direct immunofluorescence, the patient should be referred to an ophthalmologist who is familiar with the ocular lesions of this condition for a baseline examination of the conjunctivae. This should be done whether or not the patient is experiencing ocular complaints. In addition, if the patient is experiencing symptoms at other anatomic sites, then the appropriate specialist should be consulted.
Because this condition is characterized by heterogeneous pathogenetic mechanisms, it is not surprising that treatments advocated over the years have been varied. In fact, there is no single good therapy for every patient; treatment must be individualized, depending on lesional distribution, disease activity, and therapeutic response. Perhaps as the various forms of pemphigoid are better defined immunopathologically, more specific, directed therapy can be devised.
TOPICAL AGENTS: If only oral lesions are present, sometimes the disease can be controlled with application of one of the more potent topical corticosteroids to the lesions several times each day. Once control is achieved, the applications can be discontinued, although the lesions are certain to flare up again. Sometimes alternate-day application prevents such exacerbations of disease activity.
Patients with only gingival lesions often benefit from good oral hygiene measures, which can help to decrease the severity of the lesions and reduce the amount of topical corticosteroids required. As an additional aid in treating gingival lesions, a flexible mouth guard may be fabricated to use as a carrier for the corticosteroid medication.
SYSTEMIC AGENTS: If topical corticosteroids are unsuccessful, systemic corticosteroids plus other immunosuppressive agents (particularly cyclophosphamide) may be used if the patient has no medical contraindications. This type of aggressive treatment is absolutely indicated in the presence of advancing ocular disease. Recent studies have suggested that treatment with intravenous (IV) human immunoglobulin may be more effective in managing ocular lesions of pemphigoid than systemic corticosteroid therapy. Attempts at surgical correction of any symblepharons that might have formed must be done when the disease is under control or quiescent; otherwise, the manipulation often induces an acute flare of the ocular lesions.
For patients with mild-to-moderate involvement by mucous membrane pemphigoid, an alternative systemic therapy that may produce fewer serious side effects is the use of dapsone, a sulfa drug derivative. Some centers report good results with dapsone, but others observe that a minority of patients respond adequately. Contraindications to its use include glucose-6-phosphate dehydrogenase deficiency or allergy to sulfa drugs.
Another alternative systemic therapy that may be used for patients with less severe disease is tetracycline or minocycline and niacinamide (nicotinamide). Systemic daily divided doses of 0.5 to 2.0 g of each drug have been reported (in open-label trials) to be effective in controlling mucous membrane pemphigoid. Double-blind, placebo-controlled studies on larger groups of patients should be done to confirm this form of therapy, however.
BULLOUS PEMPHIGOID
Bullous pemphigoid is the most common of the autoimmune blistering conditions, occurring at an estimated rate of 10 cases per million population per year. The disease is characterized by the production of autoantibodies directed against components of the basement membrane. In many respects, bullous pemphigoid resembles mucous membrane pemphigoid, but most investigators note that there are enough differences to consider these diseases as distinct but related entities. One significant difference is that the clinical course in patients with bullous pemphigoid is usually limited, whereas the course in patients with cicatricial pemphigoid is usually protracted and progressive.
CLINICAL FEATURES: Bullous pemphigoid typically develops in older people; most patients are between 60 and 80 years of age. No sex or racial predilection is generally reported, although one group of investigators noted that men are overrepresented in this disease by a 2:1 margin when one corrects for the skewing of the aging population toward the female gender. Pruritus may be an early symptom. This is followed by the development of multiple, tense bullae on either normal or erythematous skin (Fig. 16-72). These lesions eventually rupture after several days, causing a superficial crust to form. Eventually, healing takes place without scarring.
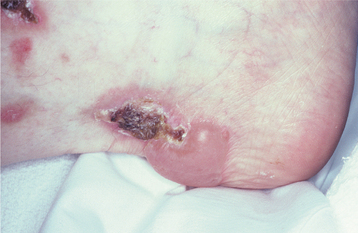
Fig. 16-72 Bullous pemphigoid. Cutaneous vesiculobullous lesions of the heel. The bullae eventually rupture, leaving hemorrhagic crusted areas.
Oral mucosal involvement is uncommon, although the reported prevalence in several series of cases has ranged from 8% to 39%. Referral bias may explain the discrepancy in prevalence rates. The oral lesions, like the skin lesions, begin as bullae, but they tend to rupture sooner, probably as a result of the constant low-grade trauma to which the oral mucosa is subjected. Large, shallow ulcerations with smooth, distinct margins are present after the bullae rupture (Fig. 16-73).
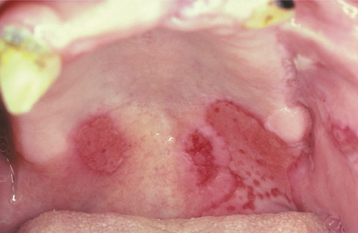
HISTOPATHOLOGIC FEATURES: Microscopic examination of tissue obtained from the perilesional margin of a bulla shows separation of the epithelium from the connective tissue at the basement membrane zone, resulting in a subepithelial separation. Modest numbers of both acute and chronic inflammatory cells are typically seen in the lesional area, and the presence of eosinophils within the bulla itself is characteristic.
Direct immunofluorescence studies show a continuous linear band of immunoreactants, usually IgG and C3, localized to the basement membrane zone in 90% to 100% of affected patients. These antibodies may bind to proteins associated with hemidesmosomes, structures that bind the basal cell layer of the epithelium to the basement membrane and the underlying connective tissue. These proteins have been designated as bullous pemphigoid antigens (BP180 and BP230), and immunoelectron microscopy has demonstrated the localization of BP180 to the upper portion of the lamina lucida of the basement membrane.
In addition to the tissue-bound autoantibodies, 50% to 90% of the patients also have circulating autoantibodies in the serum, producing an indirect immunofluorescent pattern that is identical to that of the direct immunofluorescence. Unlike pemphigus vulgaris, the antibody titers seen in bullous pemphigoid do not appear to correlate with disease activity. The antibodies alone do not appear to be capable of inducing bullae in this disease. Instead, binding of the antibodies to the basement membrane initiates the complement cascade, which in turn results in degranulation of mast cells, with recruitment of neutrophils and eosinophils to the area. The damage to the basement membrane is thought to be mediated by elastases and matrix metalloproteinases released by these inflammatory cells.
TREATMENT AND PROGNOSIS: Management of the patient with bullous pemphigoid consists of systemic immunosuppressive therapy. Moderate daily doses of systemic prednisone usually control the condition, after which alternate-day therapy may be given to reduce the risk of corticosteroid complications. If the lesions do not respond to prednisone alone, then another immunosuppressive agent, such as azathioprine, may be added to the regimen. Dapsone, a sulfa derivative, may be used as an alternative therapeutic agent, and tetracycline and niacinamide therapy is reported to be effective for some patients. The more severe, resistant cases require prednisone combined with cyclophosphamide; however, this regimen has the potential for significant side effects.
The prognosis is generally good, with many patients experiencing spontaneous remission after 2 to 5 years. Problems may develop with immunosuppressive therapy in this older adult population, however, and mortality rates of up to 27% have been reported in some series.
ERYTHEMA MULTIFORME
Erythema multiforme is a blistering, ulcerative mucocutaneous condition of uncertain etiopathogenesis. This is probably an immunologically mediated process, although the cause is poorly understood. In about 50% of the cases, the clinician can identify either a preced-ing infection, such as herpes simplex or Mycoplasma pneumoniae, or exposure to any one of a variety of drugs and medications, particularly antibiotics or analgesics. These agents may trigger the immunologic derangement that produces the disease. Sophisticated techniques in molecular biology have demonstrated the presence of herpes simplex DNA in patients with recurrent erythema multiforme, thus supporting the concept of an immunologic precipitating event. Interestingly, direct and indirect immunofluorescence studies are nonspecific and are not really very useful diagnostically except to rule out other vesiculobullous diseases.
A spectrum of severity of this disease has been recognized for many years, ranging from erythema multiforme minor through erythema multiforme major (traditionally thought to be synonymous with Stevens-Johnson syndrome) and toxic epidermal necrolysis (Lyell’s disease). Recent publications have suggested that erythema multiforme minor and major may represent a distinctly different process from the latter two conditions. However, considerable overlap in the clinical features of erythema multiforme major and Stevens-Johnson syndrome is seen, and this classification scheme remains controversial.
CLINICAL FEATURES: Erythema multiforme usually has an acute onset and may display a wide spectrum of clinical disease. On the mild end of the spectrum, ulcerations develop, affect ing the oral mucosa primarily. In its most severe form, diffuse sloughing and ulceration of the entire skin and mucosal surfaces may be seen (toxic epidermal necrolysis or Lyell’s disease).
Patients affected by erythema multiforme are usually young adults in their 20s or 30s. Men are affected more frequently than women.
Prodromal symptoms include fever, malaise, headache, cough, and sore throat, occurring approximately 1 week before onset. Although the disease is self-limiting, usually lasting 2 to 6 weeks, about 20% of patients experience recurrent episodes, usually in the spring and autumn.
Erythematous skin lesions develop in about 50% of cases. A variety of appearances (multiforme means many forms) may be present. Typically, early lesions appear on the extremities and are flat, round, and dusky-red in hue. These become slightly elevated and may evolve into bullae with necrotic centers. Sometimes particular skin lesions develop that are highly characteristic for the disease. These lesions appear as concentric circular erythematous rings resembling a target or bull’s-eye (target lesions) (Fig. 16-74).
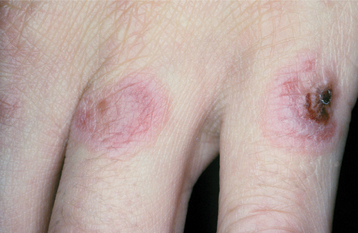
Fig. 16-74 Erythema multiforme. The concentric erythematous pattern of the cutaneous lesions on the fingers resembles a target or bull’s-eye.
The oral lesions begin as erythematous patches that undergo epithelial necrosis and evolve into large, shallow erosions and ulcerations with irregular borders (Fig. 16-75). Hemorrhagic crusting of the vermilion zone of the lips is common (Fig. 16-76). These oral lesions, like the skin lesions, emerge quickly and are uncomfortable. Sometimes patients are dehydrated because they are unable to ingest liquids as a result of mouth pain. The ulcerations often have a diffuse distribution. The lips, labial mucosa, buccal mucosa, tongue, floor of the mouth, and soft palate are the most common sites of involvement. Usually, the gingivae and hard palate are relatively spared.
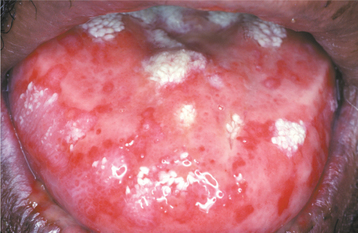
Fig. 16-75 Erythema multiforme. Diffuse ulcerations and erosions involving the dorsal surface of this patient’s tongue.
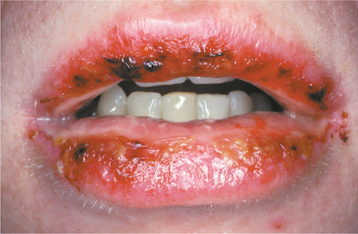
Fig. 16-76 Erythema multiforme. Ulceration of the labial mucosa, with hemorrhagic crusting of the vermilion zone of the lips.
ERYTHEMA MULTIFORME MAJOR: A more severe form of the disease, known as erythema multiforme major or Stevens-Johnson syndrome, is usually triggered by a drug rather than infection. For such a diagnosis to be made, either the ocular (Fig. 16-77) or genital (Fig. 16-78) mucosae should be affected in conjunction with the oral and skin lesions. With severe ocular involvement, scarring (symblepharon formation) may occur, similar to that in cicatricial pemphigoid (see page 772).
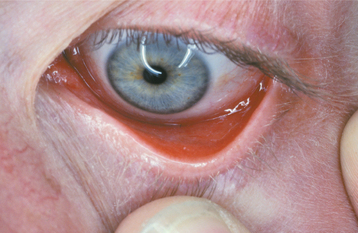
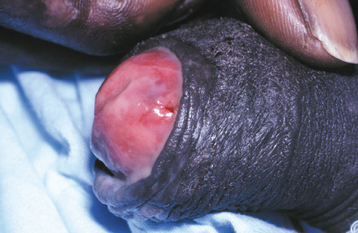
TOXIC EPIDERMAL NECROLYSIS: Many dermatologists consider toxic epidermal necrolysis to represent the most severe form of erythema multiforme. It is almost always triggered by drug exposure, with more than 200 different medications having been implicated. Recent studies have shown that the damage to the epithelium is due to increased apoptosis of the epithelial cells. Diffuse sloughing of a significant proportion of the skin and mucosal surfaces makes it appear as if the patient had been badly scalded (Figs. 16-79 and 16-80). In contrast to erythema multiforme major, toxic epidermal necrolysis tends to occur in older people. A female predilection is observed. If the patient survives, then the cutaneous process resolves in 2 to 4 weeks; however, oral lesions may take longer to heal, and significant residual ocular damage is evident in half the patients. These more severe presentations of erythema multiforme are rare. Erythema multiforme major occurs at an average rate of five cases per million population per year, and toxic epidermal necrolysis occurs at a rate of about one case per million per year.
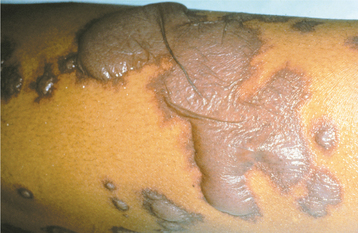
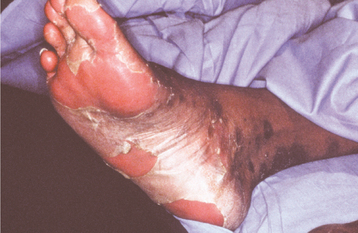
HISTOPATHOLOGIC FEATURES: Histopathologic examination of the perilesional mucosa in erythema multiforme reveals a pattern that is characteristic but not pathognomonic. Subepithelial or intraepithelial vesiculation may be seen in association with necrotic basal keratinocytes (Fig. 16-81). A mixed inflammatory infiltrate is present, consisting of lymphocytes, neutrophils, and often eosinophils. Sometimes these cells are arranged in a perivascular orientation (Fig. 16-82). Because the immunopathologic features are also nonspecific, the diagnosis is often based on the clinical presentation and the exclusion of other vesiculobullous disorders.
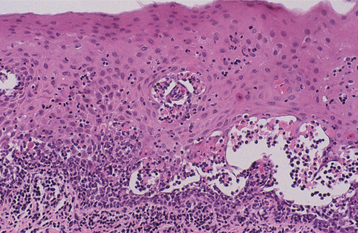
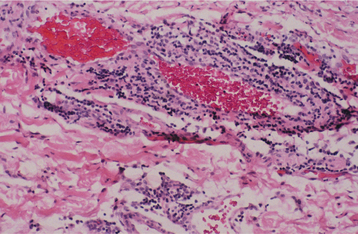
TREATMENT AND PROGNOSIS: Management of erythema multiforme, in many respects, remains controversial. In the past, the use of systemic or topical corticosteroids was often advocated, especially in the early stages of the disease. There is little good clinical evidence that such treatment alters the course of this disease, however. If a causative drug is identified or suspected, then it should be discontinued immediately.
If the patient is dehydrated as a result of an inability to eat because of oral pain, then intravenous rehydration may be necessary along with topical anesthetic agents to decrease discomfort.
If recurrent episodes of erythema multiforme are a problem, then an initiating factor, such as recurrent herpesvirus infection or drug exposure, should be sought. If disease is triggered by herpes simplex, then continuous oral acyclovir or valacyclovir therapy can prevent recurrences.
Generally, erythema multiforme is not life threatening except in its most severe forms. The mortality rate in patients with toxic epidermal necrolysis historically has been approximately 34%; the rate in those with Stevens-Johnson syndrome is 2% to 10%. Corticosteroids should be avoided in the management of toxic epidermal necrolysis because some investigators have found that such drugs may be detrimental. Intravenous administration of pooled human immunoglobulins has been shown in several open-label trials to produce remarkable resolution of toxic epidermal necrolysis, presumably because of blockade of Fas ligand, which is believed to be responsible for inducing epithelial cell apoptosis. Because the lesions of toxic epidermal necrolysis are analogous to those suffered by burn patients, management of these patients in the burn unit of the hospital is recommended.
ERYTHEMA MIGRANS (GEOGRAPHIC TONGUE; BENIGN MIGRATORY GLOSSITIS; WANDERING RASH OF THE TONGUE; ERYTHEMA AREATA MIGRANS; STOMATITIS AREATA MIGRANS)
Erythema migrans is a common benign condition that primarily affects the tongue. It is often detected on routine examination of the oral mucosa. The lesion occurs in 1% to 3% of the population. Females are affected more frequently than males by a 2:1 ratio. Patients may occasionally consult a health care professional if they happen to notice the unusual appearance of their tongue or if the lingual mucosa becomes sensitive to hot or spicy foods as a result of the process.
Even though erythema migrans has been documented for many years, the etiopathogenesis is still unknown. Some investigators have suggested that erythema migrans occurs with increased frequency in atopic individuals; however, one recent large epidemiologic study in the United States found no statistically significant association between erythema migrans and a variety of conditions that had previously been postulated either to cause or influence this process. Erythema migrans was not seen as frequently in cigarette smokers, while there seemed to be no significant differences in frequency related to age, sex, oral contraceptive use, presence of allergies, diabetes mellitus, or psychological or dermatologic conditions.
CLINICAL FEATURES: The characteristic lesions of erythema migrans are seen on the anterior two thirds of the dorsal tongue mucosa. They appear as multiple, well-demarcated zones of erythema (Figs. 16-83 and 16-84), concentrated at the tip and lateral borders of the tongue. This erythema is due to atrophy of the filiform papillae, and these atrophic areas are typically surrounded at least partially by a slightly elevated, yellow-white, serpentine or scalloped border (Fig. 16-85). The patient who is aware of the process is often able to describe the lesions as appearing quickly in one area, healing within a few days or weeks, then developing in a very different area. Frequently, the lesion begins as a small white patch, which then develops a central erythematous atrophic zone and enlarges centrifugally. Often patients with fissured tongue (see page 13) are affected with erythema migrans as well. Some patients may have only a solitary lesion, but this is uncommon. The lesions are usually asymptomatic, although a burning sensation or sensitivity to hot or spicy foods may be noted when the lesions are active. Only rarely is the burning sensation more constant and severe.
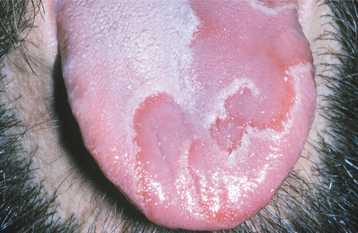
Fig. 16-83 Erythema migrans. The erythematous, well-demarcated areas of papillary atrophy are characteristic of erythema migrans affecting the tongue (benign migratory glossitis). Note the asymmetrical distribution and the tendency to involve the lateral aspects of the tongue.
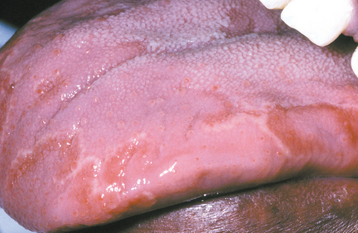
Fig. 16-84 Erythema migrans. Lingual mucosa of a different patient than the one in Fig. 16-83. The lateral distribution of the lesions is shown.
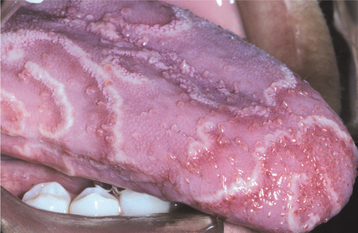
Very infrequently, erythema migrans may occur on oral mucosal sites other than the tongue. In these instances, the tongue is almost always affected; however, other lesions develop on the buccal mucosa, on the labial mucosa, and (less frequently) on the soft palate (Figs. 16-86 and 16-87). These lesions typically produce no symptoms and can be identified by a yellow-white serpentine or scalloped border that surrounds an erythematous zone. These features should prevent confusion with such conditions as candidiasis or erythroplakia.
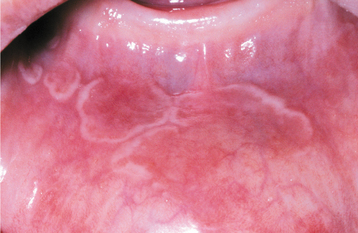
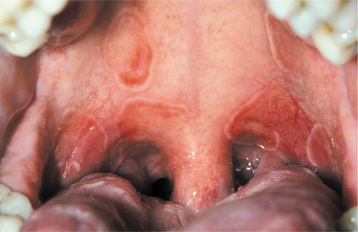
HISTOPATHOLOGIC FEATURES: If a biopsy specimen of the peripheral region of erythema migrans is examined, a characteristic histopathologic pattern is observed. Hyperparakeratosis, spongiosis, acanthosis, and elongation of the epithelial rete ridges are seen (Fig. 16-88). In addition, collections of neutrophils (Munro abscesses) are observed within the epithelium (Fig. 16-89); lymphocytes and neutrophils involve the lamina propria. The intense neutrophilic infiltrate may be responsible for the destruction of the superficial portion of the epithelium, thus producing an atrophic, reddened mucosa as the lesion progresses. Because these histopathologic features are reminiscent of psoriasis, this is called a psoriasiform mucositis. Despite the apparent lack of association between dermatologic conditions and erythema migrans in one recent report, another case-control study of psoriatic patients showed that erythema migrans occurred at a rate of about 10%; only 2.5% of an age-matched and sex-matched population were affected. A Brazilian study determined that both patients with psoriasis and those with benign migratory glossitis were more likely to have the same human leukocyte antigen (HLA) group, namely HLA-Cw6. Whether these findings mean that erythema migrans represents oral psoriasis or that patients with psoriasis are just more susceptible to erythema migrans is open to debate.
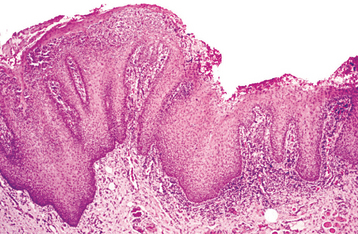
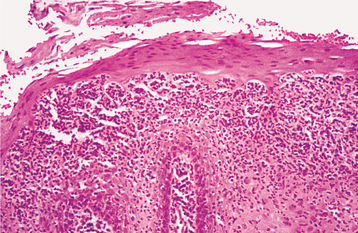
TREATMENT AND PROGNOSIS: Generally no treatment is indicated for patients with erythema migrans. Reassuring the patient that the condition is completely benign is often all that is necessary. Infrequently, patients may complain of tenderness or a burning sensation that is so severe that it disrupts their lifestyle. In such cases, topical corticosteroids, such as fluocinonide or betamethasone gel, may provide relief when applied as a thin film several times a day to the lesional areas.
REACTIVE ARTHRITIS (REITER’S SYNDROME)
Reactive arthritis represents a group of uncommon diseases that most likely have an immunologically mediated cause. Current evidence suggests that these disorders may be triggered by any one of several infectious agents in a genetically susceptible person. In some instances, the arthritis will be accompanied by mucocutaneous findings, including oral lesions. A classic triad of signs has been described:
However, most patients do not exhibit all three of these signs. Although reactive arthritis with a mucocutaneous component is also known as Reiter syndrome, some authors have advocated removing the Reiter eponym because of Hans Reiter’s Nazi criminal activities during World War II and he was not the first to describe this syndrome.
It is interesting that reactive arthritis has been reported with some frequency in patients infected with the human immunodeficiency virus (HIV).
CLINICAL FEATURES: Reactive arthritis is particularly prevalent in young adult men. According to most series, there is a male-to-female ratio of 9:1. The majority (60% to 90%) of these patients are positive for HLA-B27, a haplotype present in only 4% to 8% of the population. The syndrome usually develops 1 to 4 weeks after an episode of dysentery or venereal disease; in fact, two French physicians published a description of this entity affecting four postdysenteric soldiers 1 week before Reiter’s paper appeared.
Urethritis is often the first sign and is seen in both affected males and females. Females may also have inflammation of the uterine cervix. Conjunctivitis usually appears concurrently with the urethritis, and after several days, arthritis ensues. The arthritis usually affects the joints of the lower extremities, although TMJ involvement has been identified in one third of these patients, typically as erosion of the condylar head. Skin lesions often take the form of a characteristic lesion of the glans penis (balanitis circinata). These lesions develop in about 20% to 30% of patients with reactive arthritis, and they appear as well-circumscribed erythematous erosions with a scalloped, whitish linear boundary.
The oral lesions, which occur in slightly less than 20% of patients with this disorder, are described in various ways. Some reports mention painless erythematous papules distributed on the buccal mucosa and palate; other reports describe shallow, painless ulcers that affect the tongue, buccal mucosa, palate, and gingiva. Some authors have even implied that geographic tongue may be a component of reactive arthritis, probably because geographic tongue bears a superficial resemblance to the lesions of balanitis circinata.
The American Rheumatism Association has defined reactive arthritis based on the clinical findings of a peripheral arthritis that lasts longer than 1 month in conjunction with urethritis, cervicitis, or both.
HISTOPATHOLOGIC FEATURES: The histopathologic findings of the cutaneous lesions in patients with reactive arthritis are frequently similar to those found in patients with psoriasis, particularly with respect to the presence of microabscesses within the superficial layers of the surface epithelium. Other features in common with psoriasis include hyperparakeratosis with elongated, thin rete ridges.
TREATMENT AND PROGNOSIS: Some patients with reactive arthritis experience spontaneous resolution of their disease after 3 to 12 months, but many others have chronic symptoms that may wax and wane. Treatment may not be necessary for the milder cases. Nonsteroidal antiinflammatory drugs (NSAIDs) are initially used for managing arthritis, and sulfasalazine may be helpful in resolving cases that do not respond. Immunosuppressive agents, including corticosteroids, azathioprine, and methotrexate, are reserved for the most resistant cases if they are not associated with HIV infection.
Physical therapy probably helps to reduce joint fibrosis associated with arthritis. About 10% to 25% of patients with this disorder have severe disability, usually from arthritis.
LICHEN PLANUS
Lichen planus is a relatively common, chronic dermatologic disease that often affects the oral mucosa. The strange name of the condition was provided by the British physician Erasmus Wilson, who first described it in 1869. Lichens are primitive plants composed of symbiotic algae and fungi. The term planus is Latin for flat. Wilson probably thought that the skin lesions looked similar enough to the lichens growing on rocks to merit this designation. Even though the term lichen planus suggests a flat, fungal condition, current evidence indicates that this is an immunologically mediated mucocutaneous disorder.
A variety of medications may induce lesions that can appear clinically very similar to the idiopathic form of the condition; however, the term lichenoid mucositis (or lichenoid dermatitis, depending on the site involved) is probably a better name for the drug-related alterations (see page 347). Similarly, foreign material that becomes inadvertently em-bedded in the gingiva may elicit a host response that is termed lichenoid foreign body gingivitis (see page 160). Reports of hepatitis C infection associated with oral lichen planus occasionally have appeared in the literature, usually from the Mediterranean countries, but this does not appear to be a significant association in the United States or Great Britain. More recent, carefully controlled epidemiologic studies do not appear to support an association of oral lichen planus with hepatitis C. However, genetic influences presumably may have an effect on the expression of lichen planus in select populations.
The relationship of stress or anxiety to the development of lichen planus is controversial, and most cited cases appear to be anecdotal or lack appropriate controls. Those studies that have applied psychologic questionnaires often find increased levels of anxiety in these patients; however, many patients who have been told that they have lichen planus are aware that anxiety has been linked to the disorder. Whether this awareness may influence the manner in which they answer the psychologic questionnaires could be debated. In one study that used psychologic questionnaires to attempt to resolve this question, patients with oral lichen planus had no greater degree of stress in their lives than did age-matched and sex-matched control patients. It might be that stress has no bearing on the pathogenesis of lichen planus; however, an alternative explanation might be that those patients who have lichen planus simply respond in this fashion to levels of stress that do not induce lesions in other people.
CLINICAL FEATURES: Most patients with lichen planus are middle-aged adults. It is rare for children to be affected. Women predominate in most series of cases, usually by a 3:2 ratio over men. Approximately 1% of the population may have cutaneous lichen planus. The prevalence of oral lichen planus is between 0.1% and 2.2%.
The skin lesions of lichen planus have been classically described as purple, pruritic, polygonal papules (Fig. 16-90). These usually affect the flexor surfaces of the extremities. Excoriations may not be visible, despite the fact that the lesions itch, because it hurts the patient when he or she scratches them.
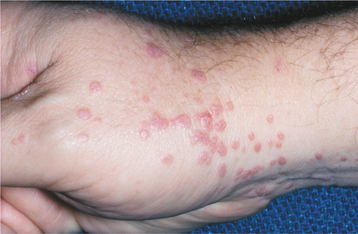
Fig. 16-90 Lichen planus. The cutaneous lesions on the wrist appear as purple, polygonal papules. Careful examination shows a network of fine white lines (Wickham’s striae) on the surface of the papules.
Careful examination of the surface of the skin papules reveals a fine, lacelike network of white lines (Wickham’s striae). Other sites of extraoral involvement include the glans penis, the vulvar mucosa, and the nails (Fig. 16-91). Essentially there are two forms of oral lesions: reticular and erosive.
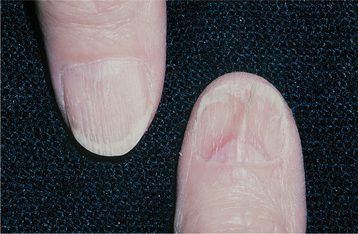
RETICULAR LICHEN PLANUS: Reticular lichen planus is much more common than the erosive form, but the erosive form predominates in several studies. This is probably because of referral bias (because the erosive form is symptomatic and, therefore, the patient is more likely to be referred to an academic center for evaluation). The reticular form usually causes no symptoms and involves the posterior buccal mucosa bilaterally (Figs. 16-92 and 16-93). Other oral mucosal surfaces may also be involved concurrently, such as the lateral and dorsal tongue, the gingivae, the palate, and vermilion border (Fig. 16-94).
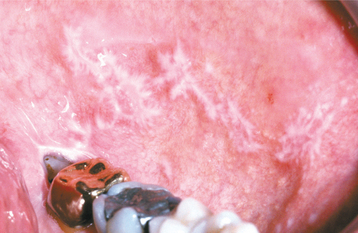
Fig. 16-92 Lichen planus. The interlacing white lines are typical of reticular lichen planus involving the posterior buccal mucosa, the most common site of oral involvement.
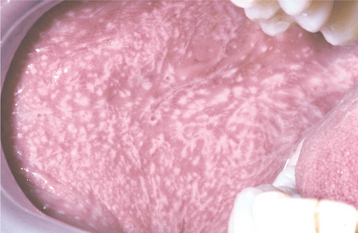
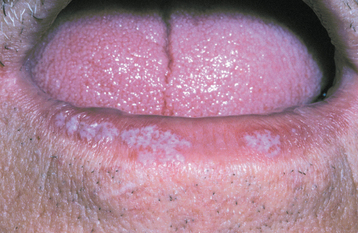
Reticular lichen planus is thus named because of its characteristic pattern of interlacing white lines (also referred to as Wickham’s striae); however, the white lesions may appear as papules in some instances. These lesions are typically not static but wax and wane over weeks or months (Fig. 16-95). The reticular pattern may not be as evident in some sites, such as the dorsal tongue, where the lesions appear more as keratotic plaques with atrophy of the papillae (Fig. 16-96). In addition, superficial mucoceles may develop within, or adjacent to, mucosal areas that are involved by lichen planus.
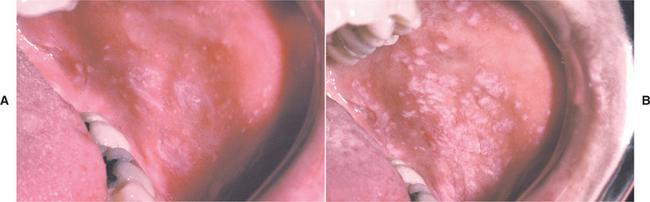
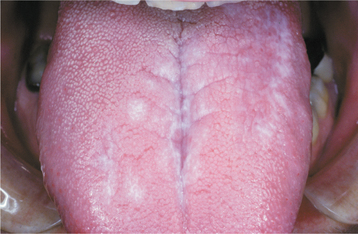
EROSIVE LICHEN PLANUS: Erosive lichen planus, although not as common as the reticular form, is more significant for the patient because the lesions are usually symptomatic. Clinically, there are atrophic, erythematous areas with central ulceration of varying degrees. The periphery of the atrophic regions is usually bordered by fine, white radiating striae (Figs. 16-97 and 16-98). Sometimes the atrophy and ulceration are confined to the gingival mucosa, producing the reaction pattern called desquamative gingivitis (see page 162) (Fig. 16-99). In such cases, biopsy specimens should be obtained for light microscopic and immunofluorescent studies of perilesional tissue, because mucous membrane pemphigoid (see page 771) and pemphigus vulgaris (see page 765) may appear in a similar fashion.
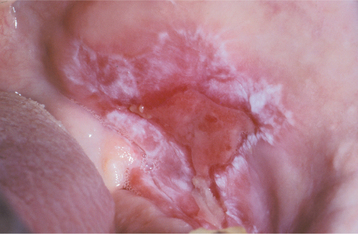
Fig. 16-97 Lichen planus. Ulceration of the buccal mucosa shows peripheral radiating keratotic striae, characteristic of oral erosive lichen planus.
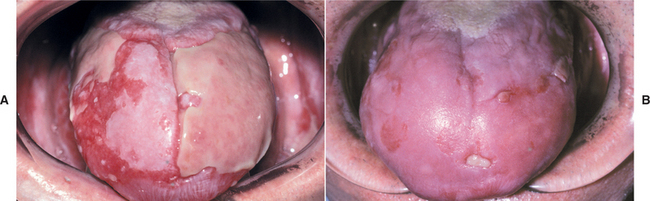
Fig. 16-98 Lichen planus. A, The dorsal surface of the tongue shows extensive ulceration caused by erosive lichen planus. Note the fine white streaks at the periphery of the ulcerations. B, Same patient after systemic corticosteroid therapy. Much of the mucosa has reepithelialized, with only focal ulcerations remaining.
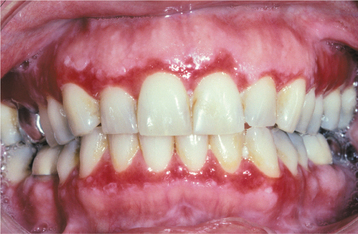
Fig. 16-99 Lichen planus. Erosive lichen planus often appears as a desquamative gingivitis, producing gingival erythema and tenderness.
If the erosive component is severe, epithelial separation from the underlying connective tissue may occur. This results in the relatively rare presentation of bullous lichen planus.
HISTOPATHOLOGIC FEATURES: The histopathologic features of lichen planus are characteristic but may not be specific, because other conditions, such as lichenoid drug reaction, lichenoid amalgam reaction, oral graft-versus-host disease (GVHD), lupus erythematosus (LE), chronic ulcerative stomatitis, and oral mucosal cinnamon reaction may also show a similar histopathologic pattern. Varying degrees of orthokeratosis and parakeratosis may be present on the surface of the epithelium, depending on whether the biopsy specimen is taken from an erosive or reticular lesion.
The thickness of the spinous layer can also vary. The rete ridges may be absent or hyperplastic, but they classically have a pointed or “saw-toothed” shape (Fig. 16-100).
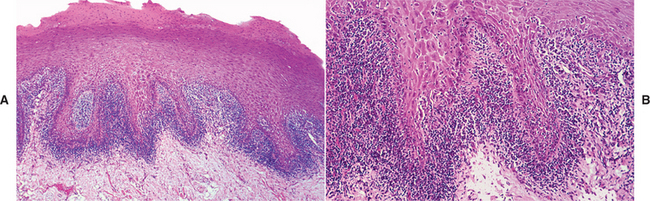
Fig. 16-100 Lichen planus. A, This low-power photomicrograph of an oral lesion shows hyperkeratosis, saw-toothed rete ridges, and a bandlike infiltrate of lymphocytes immediately subjacent to the epithelium. B, Higher-power view showing migration of lymphocytes into the lower epithelium with interface degeneration of the basal cell layer.
Destruction of the basal cell layer of the epithelium (hydropic degeneration) is also evident. This is accompanied by an intense, bandlike infiltrate of predominantly T lymphocytes immediately subjacent to the epithelium (Fig. 16-101). Degenerating keratinocytes may be seen in the area of the epithelium and connective tissue interface and have been termed colloid, cytoid, hyaline, or Civatte bodies. No significant degree of epithelial atypia is expected in oral lichen planus, although lesions having a superimposed candidal infection may appear worrisome. These should be reevaluated histopathologically after the candidal infection is treated. On occasion, the chronic inflammatory host response to the atypical cells of epithelial dysplasia can appear virtually indistinguishable histopathologically from lichen planus, particularly in milder cases of epithelial dysplasia. Such ambiguity may contribute to the controversy related to the malignant transformation potential of lichen planus.
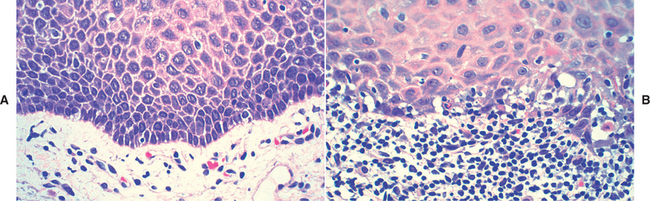
Fig. 16-101 Lichen planus. A, High-power photomicrograph of normal epithelium showing an intact basal cell layer and no inflammation. B, High-power photomicrograph of lichen planus showing degeneration of the basal epithelial layer and an intense lymphocytic infiltrate in the superficial lamina propria.
The immunopathologic features of lichen planus are nonspecific. Most lesions show the deposition of a shaggy band of fibrinogen at the basement membrane zone.
DIAGNOSIS: The diagnosis of reticular lichen planus can often be made based on the clinical findings alone. The interlacing white striae appearing bilaterally on the posterior buccal mucosa are virtually pathognomonic. Difficulties in diagnosis may arise if candidiasis is superimposed on the lesions because the organism may disturb the characteristic reticular pattern of the lichen planus (Fig. 16-102).
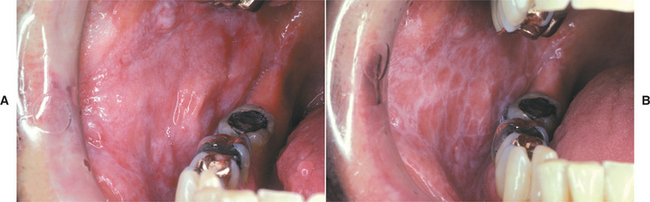
Fig. 16-102 Lichen planus. A, These relatively nondescript white lesions affected the buccal mucosa of a patient who had complained of a burning sensation. Histopathologic evaluation of the lesion showed a lichenoid mucositis with superimposed candidiasis. B, Same patient 2 weeks after antifungal therapy. Once the mucosal reaction to the candidal organism was eliminated, the characteristic white striae of reticular lichen planus were identified.
Erosive lichen planus is sometimes more challenging to diagnose (based on clinical features alone) than the reticular form. If the typical radiating white striae and erythematous, atrophic mucosa are present at the periphery of well-demarcated ulcerations on the posterior buccal mucosa bilaterally, then the diagnosis can sometimes be rendered without the support of histopathologic findings. However, a biopsy is often necessary to rule out other ulcerative or erosive diseases, such as lupus erythematosus or chronic ulcerative stomatitis.
Specimens of isolated erosive lichenoid lesions, particularly those of the soft palate, the lateral and ventral tongue, or the floor of the mouth, should be obtained for biopsy to rule out premalignant changes or malignancy. Another condition that may mimic an isolated lesion of lichen planus, both clinically and histopathologically, is a lichenoid reaction to dental amalgam (see page 354).
TREATMENT AND PROGNOSIS: Reticular lichen planus typically produces no symptoms, and no treatment is needed. Occasionally, affected patients may have superimposed candidiasis, in which case they may complain of a burning sensation of the oral mucosa. Antifungal therapy is necessary in such a case. Some investigators recommend annual reevaluation of the reticular lesions of oral lichen planus.
Erosive lichen planus is often bothersome because of the open sores in the mouth. Because it is an immunologically mediated condition, corticosteroids are recommended. The lesions respond to systemic corticosteroids, but such drastic therapy is usually not necessary. One of the stronger topical corticosteroids (e.g., fluocinonide, betamethasone, clobetasol gel) applied several times per day to the most symptomatic areas is usually sufficient to induce healing within 1 or 2 weeks. Some investigators have recommended compounding corticosteroid ointments with an adhesive methylcellulose base, but patient compliance may be reduced because this material is difficult to apply. The patient should be warned that the condition will undoubtedly flare up again, in which case the corticosteroids should be reapplied. In addition, the possibility of iatrogenic candidiasis associated with corticosteroid use should be monitored (Fig. 16-103). Although the use of agents such as topical retinoids, tacrolimus, or cyclosporine has occasionally been advocated for recalcitrant cases of erosive lichen planus, reports of their efficacy have been contradictory. Furthermore, their side effects can be significant, and in the case of cyclosporine, the cost of the drug may be prohibitive. Some investigators suggest that patients with oral erosive lichen planus be evaluated every 3 to 6 months, particularly if the lesions are not typical.
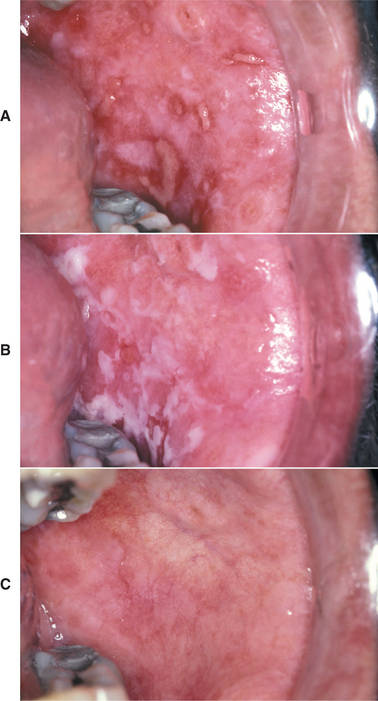
Fig. 16-103 Lichen planus. A, This patient was diagnosed with erosive lichen planus affecting the buccal mucosa and was treated with topical corticosteroids. B, Same patient 2 weeks later. The creamy-white plaques of pseudomembranous candidiasis have developed as a result of the corticosteroid therapy. C, Same patient after antifungal therapy. At this point, he was asymptomatic.
The question of the malignant potential of lichen planus, particularly the erosive form, is yet to be resolved. Most cases of reported malignant transformation are rather poorly documented. Some of these reported cases may not have been true lichen planus, but rather they may have actually been dysplastic leukoplakias with a secondary lichenoid inflammatory infiltrate that mimicked lichen planus (“lichenoid dysplasia”). In addition, the argument can be made that because both lichen planus and squamous cell carcinoma are not rare, some people may have both problems simultaneously, and the two processes may be unrelated to one another. Conversely, some investigators say that the atrophic epithelium of lichen planus may be more susceptible to the action of carcinogens, resulting in an increased risk of malignant transformation. One study examined the molecular characteristics of classic reticular lichen planus, comparing the loss of heterozygosity at purported tumor suppressor gene loci in these lesions with that of varying grades of oral epithelial dysplasia, squamous cell carcinoma, normal oral mucosa, and oral reactive lesions. The molecular profile of oral lichen planus more closely resembled that of normal or reactive oral mucosa, a finding that provides less support for the concept of lichen planus being precancerous. Another study evaluated the malignant transformation rate of typical oral lichen planus compared with oral “lichenoid” lesions. The lichenoid lesions had some features of lichen planus, but were not completely representative, either clinically or histopathologically, of that disease. These investigators found that there was no transformation of characteristic lichen planus, although several of the lichenoid lesions developed into squamous cell carcinoma. Additional prospective clinical studies with strict clinical and histopathologic criteria for the definition of oral lichen planus will need to be performed to resolve this question. If the potential for malignant transformation exists, then it appears to be small. Most of the reported cases have been confined to patients with either the erosive or so-called plaque-type form of lichen planus.