CHAPTER 35 Clinical Manifestations of Hepatobiliary Disease
GENERAL CONSIDERATIONS
Clinical signs of hepatobiliary disease in cats and dogs can be extremely variable, ranging from anorexia and weight loss to abdominal effusion, jaundice, and hepatic coma (Box 35-1). However, none of these signs are pathognomonic for hepatobiliary disease, and they must be distinguished from identical signs caused by disease of other organ systems. The severity of the clinical sign does not necessarily correlate with the prognosis or with the degree of liver injury, although several of these signs are often seen together in dogs and cats with end-stage hepatic disease (e.g., ascites, metabolic encephalopathy from hepatocellular dysfunction, and acquired portosystemic venous shunting with gastrointestinal bleeding); however, ascites has recently been shown to be a significant negative prognostic indicator in dogs with chronic hepatitis. At the opposite end of the spectrum of hepatobiliary disease, because of the tremendous reserve capacity of the liver, there may be no clues for the presence of a hepatic disorder except for abnormal screening blood test results obtained before an elective anesthetic procedure.
 BOX 35-1
BOX 35-1ABDOMINAL ENLARGEMENT
ORGANOMEGALY
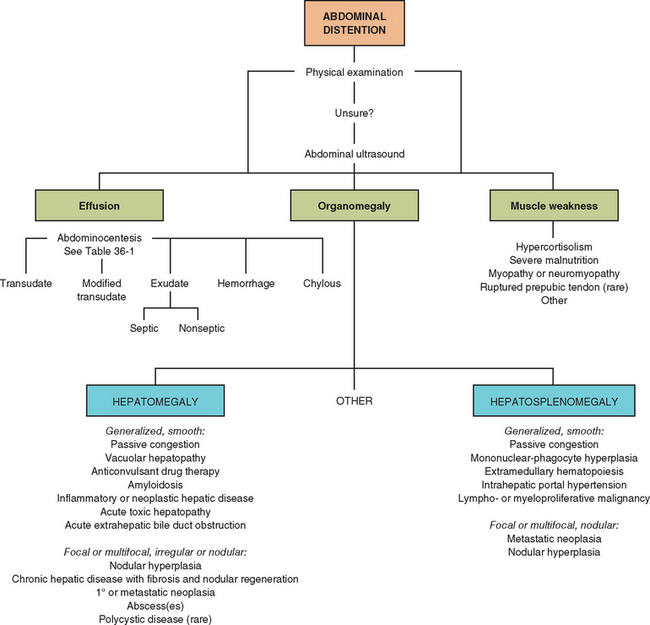
Abdominal enlargement may be the presenting complaint of owners of cats and dogs with hepatobiliary disease, or it may be noted during physical examination. Organomegaly, fluid expansion of the peritoneal space, or poor abdominal muscle tone is usually the cause of this abnormality.
Enlarged organs that most often account for increased abdominal size are the liver, the spleen (see Chapter 88), and occasionally the kidneys (see Chapter 41). Normally, the liver is palpable just caudal to the costal arch along the ventral body wall in the cat and dog, but it may not be palpable at all. Inability to palpate the liver, especially in dogs, does not automatically mean that the liver is abnormally small. In lean cats it is usually possible to palpate the diaphragmatic surface of the liver. In cats or dogs with pleural effusion or other diseases that expand thoracic volume, the liver may be displaced caudally and erroneously appear to be enlarged.
Liver enlargement is much more common in cats than in dogs with liver disease. Dogs more often have reduced liver size because of chronic hepatitis with fibrosis. The pattern of liver enlargement may be generalized or focal, depending on the cause. Infiltrative and congestive disease processes or those that stimulate hepatocellular hypertrophy or mononuclear-phagocytic system (MPS) hyperplasia tend to result in smooth or slightly irregular, firm, diffuse hepatomegaly. Focal or asymmetrical hepatic enlargement is often seen with proliferative or expansive diseases that form solid or cystic mass lesions. Examples of such diseases are listed in Table 35-1.
 TABLE 35-1 Differential Diagnoses for Changes in Hepatic Size
TABLE 35-1 Differential Diagnoses for Changes in Hepatic Size
| DIAGNOSIS | SPECIES |
|---|---|
| Hepatomegaly | |
| Generalized | |
| Infiltration | |
| Primary or metastatic neoplasia | C, D |
| Cholangitis | C |
| Extramedullary hematopoiesis* | C, D |
| Mononuclear-phagocytic cell hyperplasia* | C, D |
| Amyloidosis (rare) | C, D |
| Passive congestion* | |
| Right-sided heart failure | C, D |
| Pericardial disease | D |
| Caudal vena cava obstruction | D |
| Caval syndrome | D |
| Budd-Chiari syndrome (rare) | C, D |
| Lipidosis | C (moderate to marked), D (mild) |
| Hypercortisolism (steroid hepatopathy) | D |
| Anticonvulsant drug therapy | D |
| Acute extrahepatic bile duct obstruction | C, D |
| Acute hepatotoxicity | C, D |
| Focal or asymmetric | |
| Primary or metastatic neoplasia | C, D |
| Nodular hyperplasia | D |
| Chronic hepatic disease with fibrosis and nodular regeneration | D |
| Abscess(es) (rare) | C, D |
| Cysts (rare) | C, D |
| Microhepatia (Generalized Only) | |
| Reduced hepatic mass† | |
| Chronic hepatic disease with progressive loss of hepatocytes | D |
| Decreased portal blood flow with hepatocellular atrophy | |
| Congenital portosystemic shunt | C, D |
| Intrahepatic portal vein hypoplasia | D |
| Chronic portal vein thrombosis | D |
| Hypovolemia | |
| Shock? | ? |
| Addison’s disease | D |
C, Primarily cats; D, primarily dogs; C, D, cats and dogs.
* Concurrent splenomegaly likely.
† Loss of portal blood flow to one lobe can cause the lobe to atrophy.
Smooth, generalized hepatosplenomegaly may be associated with nonhepatic causes, such as increased intravascular hydrostatic pressure (passive congestion) secondary to right-sided congestive heart failure or pericardial disease. In rare instances hepatic vein occlusion (Budd-Chiari syndrome) results in similar findings. Hepatosplenomegaly in icteric dogs or cats may be attributable to benign MPS hyperplasia and extramedullary hematopoiesis secondary to immune-mediated hemolytic anemia or to infiltrative processes such as lymphoma, systemic mast cell disease, or myeloid leukemia.
Another cause of hepatosplenomegaly is primary hepatic parenchymal disease with sustained intrahepatic portal hypertension. In dogs and cats with this syndrome, the liver is usually firm and irregular on palpation and often the liver itself is reduced in size as a result of fibrosis; however, the spleen can be enlarged and congested as a result of portal hypertension. For conditions that involve primarily the spleen, see Chapter 88.
ABDOMINAL EFFUSION
Abdominal effusion is much more common in dogs than in cats with liver disease. With the exception of liver disease associated with feline infectious peritonitis (FIP), cats with liver disease rarely have ascites. The pathogenesis of abdominal effusion in cats and dogs with hepatobiliary disease is determined by chemical and cytologic analysis of a fluid specimen (Fig. 35-1; see also Table 36-1). On the basis of cell and protein content, abdominal fluids are classified by standard criteria as transudates, modified transudates (moderate to low cellularity with moderate to low protein concentration), exudates (high cellularity and protein concentration), or chyle or blood (see Table 36-1). The term ascites is reserved for fluid of low to moderate protein content and low to moderate cell count (transudate or modified transudate) and is usually related to disorders of hepatic or cardiovascular origin or severe protein-losing enteropathy or nephropathy. A small amount of effusion is suspected when abdominal palpation yields a “slippery” sensation during physical examination. Moderate-to-large-volume effusion is frequently conspicuous but may distend the abdomen so much that details of abdominal organs are obscured during palpation. Whether there is small- or large-volume effusion, the general pathogeneses of third-space fluid accumulation (excessive formation by increased venous hydrostatic pressure, decreased intravascular oncotic pressure, or altered vascular permeability and insufficient resorption), singly or in combination, apply to cats and dogs with hepatobiliary diseases. In addition, an important part of the mechanisms of ascites formation in dogs with liver disease is activation of the renin-angiotensin-aldosterone system (RAAS) with sodium retention by the kidneys and increased circulating fluid volume. This RAAS activation is triggered by a decrease in systemic blood pressure caused by pooling of a significant proportion of the circulating blood volume in the splanchnic circulation. It has been observed that, in many cases, overt ascites does not develop until sodium retention by the kidneys is increased, altering the balance of fluid formation and reabsorption. Therefore aldosterone antagonists play a key role in the treatment of ascites associated with liver disease.
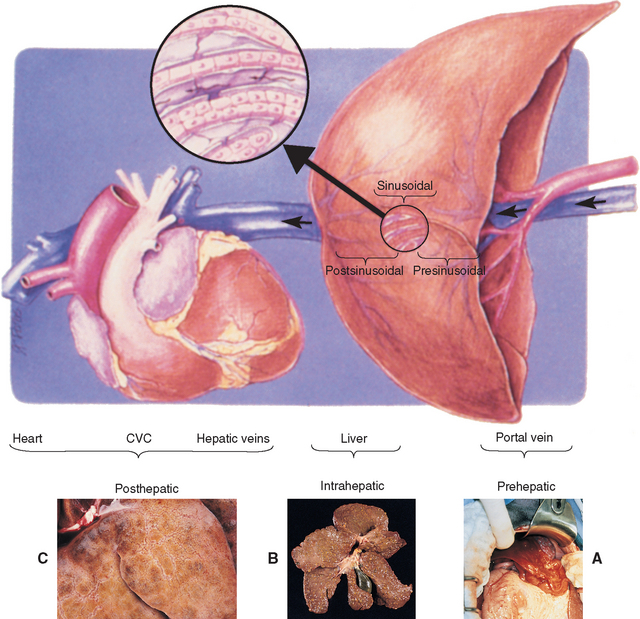
FIG 35-1 Mechanisms of abdominal fluid accumulation associated with altered portal and hepatic blood flow and clinical correlates. PREHEPATIC: arteriovenous fistula (A) or portal vein obstruction or hypoplasia; INTRAHEPATIC presinusoidal: periportal fibrosis or portal venule hypoplasia; INTRAHEPATIC sinusoidal: cellular infiltrates or collagen (B); INTRAHEPATIC postsinusoidal: central (terminal hepatic) venular fibrosis; POSTHEPATIC (passive congestion): obstruction of hepatic veins or intrathoracic caudal vena cava, right-sided heart failure (C) or pericardial disease. Arrow indicates direction of venous blood flow.
(From Johnson SE: Portal hypertension. I. Pathophysiology and clinical consequences, Compend Contin Educ 9:741, 1987.)
Intrahepatic portal venous hypertension is the most common mechanism leading to ascites in companion animals, particularly dogs, with hepatobiliary diseases. The formation of abdominal effusion depends on the site, rate, and degree of defective venous outflow. Sustained resistance to intrahepatic portal blood flow at the level of the portal triad favors exudation of fluid from more proximal (in the direction of portal blood flow; i.e., intestinal) lymphatics into the abdominal cavity. The fluid is generally of low protein content and is hypocellular. However, if the fluid is present in the abdomen for any amount of time, it becomes “modified” with an increase in protein content. The exception to this is in the animal with marked hypoalbuminemia associated with liver disease in which the ascites remains a low-protein transudate. Inflammatory or neoplastic cellular infiltrates or fibrosis in this region of the liver are the pathologic processes most often responsible for this type of effusion. Sinusoidal obstruction caused by regenerative nodules, collagen deposition, or cellular infiltrates causes effusion of a fluid composed of a mixture of hepatic and intestinal lymph that has a variable protein content and generally low cell count.
Prehepatic portal venous occlusion or the presence of a large arteriovenous fistula, leading to portal venous volume overload, and associated high intrahepatic vascular resistance triggered by increased portal flow also produces a low to moderate protein, hypocellular effusion, as would diffuse mesenteric lymphatic obstruction associated with lymphoma. The latter can also sometimes result in a chylous effusions. Examples of causes of portal venous occlusion include intraluminal obstructive masses (e.g., thrombus), extraluminal compressive masses (e.g., mesenteric lymph node, neoplasm), and portal vein hypoplasia or atresia.
Venous congestion from disease of the major hepatic veins and/or distally (i.e., thoracic caudal vena cava, heart; posthepatic venous congestion) increases formation of hepatic lymph, which exudes from superficial hepatic lymphatics. Because the endothelial cell-lined sinusoids are highly permeable, hepatic lymph is of high protein content. Abdominal effusion formed under these conditions is more likely to develop in dogs than in cats. Reactive hepatic veins that behave as postsinusoidal sphincters have been identified in dogs and are speculated to add to venous outflow impingement. Concurrent hypoalbuminemia (≤1.5 g/dl) in dogs (and rarely cats) associated with hepatic parenchymal failure may further enhance movement of fluid into the peritoneal space. Perivenular pyogranulomatous infiltrates in the visceral and parietal peritoneum of cats with the effusive form of FIP increase vascular permeability and promote exudation of straw-colored, protein-rich fluid into the peritoneal space. Typically, the fluid is of low to moderate cellularity, with a mixed cell population of neutrophils and macrophages, and with a moderate to high protein concentration. It is usually classified as an exudate but occasionally is a modified transudate.
Hepatobiliary malignancies or other intraabdominal carcinomas that have disseminated to the peritoneum can elicit an inflammatory reaction, with subsequent exudation of lymph and fibrin. The fluid may be serosanguineous, hemorrhagic, or chylous in appearance. Regardless of the gross appearance of the fluid, the protein content is variable, and the fluid may contain exfoliated malignant cells if the primary neoplasm is a carcinoma, mesothelioma, or lymphoma, although often it does not, in which case further investigations are required to diagnose the neoplasm.
Extravasation of bile from a ruptured biliary tract elicits a strong inflammatory response and stimulates transudation of lymph by serosal surfaces. In experimental animal models, the damaging component of bile has been identified as bile acids. Unlike with most other causes of abdominal effusion associated with hepatobiliary disease, there may be evidence of cranial abdominal or diffuse abdominal pain identified during physical examination in cats and dogs with bile peritonitis. The fluid appears characteristically dark orange, yellow, or green and has a high bilirubin content on analysis, and the predominant cell type is the healthy neutrophil, except when the biliary tract is infected. Because normal bile is sterile, the initial phase of bile peritonitis is nonseptic, but unless treatment is initiated rapidly, secondary infection, usually with anaerobes, may become life-threatening.
ABDOMINAL MUSCULAR HYPOTONIA
The presence of a distended abdomen in the absence of organomegaly or abdominal effusion suggests abdominal muscular hypotonia. Either the catabolic effects of severe malnutrition or (more commonly in dogs) excess endogenous or exogenous corticosteroids reduce muscular strength, giving the appearance of an enlarged abdomen. In both dogs and (much less commonly) cats with hyperadrenocorticism, the combination of generalized hepatomegaly (mild and associated with diabetes mellitus in cats), redistribution of fat stores to the abdomen, and muscular weakness causes abdominal distention.
On the basis of the physical examination findings, the problem of abdominal enlargement should be refined to the level of organomegaly, abdominal effusion, or poor muscular tone, as shown in Fig. 35-2. Additional tests are required to obtain a definitive diagnosis.
JAUNDICE, BILIRUBINURIA, AND CHANGE IN FECAL COLOR
By definition, jaundice in cats and dogs is the yellow staining of serum or tissues by an excessive amount of bile pigment or bilirubin (Fig. 35-3); the terms jaundice and icterus may be used interchangeably. Because the normal liver has the ability to take up and excrete a large amount of bilirubin, there must be either a large, persistent increase in the production of bile pigment (hyperbilirubinemia) or a major impairment in bile excretion (cholestasis with hyperbilirubinemia) before jaundice is detectable as yellow-stained tissues (serum bilirubin concentration ≥2 mg/dl) or serum (serum bilirubin concentration ≥1.5 mg/dl).
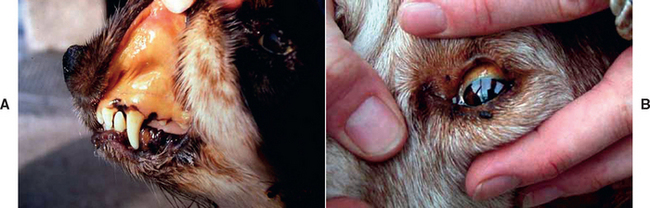
FIG 35-3 Jaundiced mucous membranes in a dog (A, gum, and B, sclera). Note that this dog had jaundice because of immune-mediated hemolytic anemia and not liver disease—hence the mucous membranes are pale and yellow (which makes them more easily photographed).
(Photographs courtesy Sara Gould.)
In normal animals bilirubin is a waste product of heme protein degradation. The primary source of heme proteins is senescent erythrocytes, with a small contribution by myoglobin and heme-containing enzyme systems in the liver. After phagocytosis by cells of the MPS, primarily in the bone marrow and spleen, heme oxygenase opens the protoporphyrin ring of hemoglobin, forming biliverdin. Biliverdin reductase converts biliverdin to fat-soluble bilirubin IXa, which is released into the circulation, where it is bound to albumin for transport to hepatic sinusoidal membranes. After uptake, transhepatocellular movement, and conjugation to various carbohydrates, conjugated bilirubin, now water soluble, is excreted into the bile canaliculi. Conjugated bilirubin is then incorporated into micelles and stored with other bile constituents in the gallbladder until it is discharged into the duodenum. However, in dogs it has been noted that only 29% to 53% of bile produced is stored in the gall bladder; the rest is secreted directly into the duodenum (Rothuizen et al., 1990). After arrival in the intestine, conjugated bilirubin undergoes bacterial deconjugation and then reduction to urobilinogen, with most urobilinogen being resorbed into the enterohepatic circulation. A small fraction of urobilinogen is then excreted in the urine, and a small portion remains in the intestinal tract to be converted to stercobilin, which imparts normal fecal color.
Inherited abnormalities of bilirubin metabolism have not been identified in cats and dogs; thus in the absence of massive increases in bile pigment production by hemolysis, jaundice is attributable to impaired excretion of bilirubin (and usually other constituents of bile) by diffuse intrahepatic hepatocellular or biliary disease or by interrupted delivery of bile to the duodenum. The inability to take up, intracellularly process, or excrete bilirubin into the bile canaliculi (the rate-limiting step) is the mechanism of cholestasis believed to be operational in many primary hepatocellular diseases. Jaundice is more likely to be a clinical feature if the liver disorder involves primarily the periportal (zone 1) hepatocytes (Fig. 35-4) than if the lesion involves centrilobular (zone 3) hepatocytes. Inflammation and swelling of larger intrahepatic biliary structures could similarly delay bile excretion.
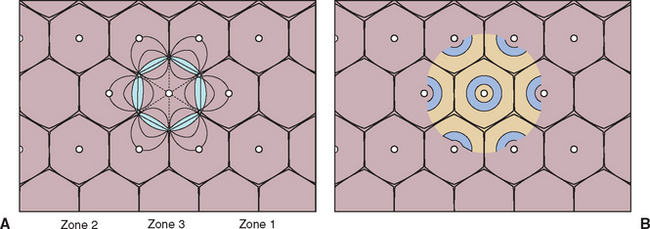
FIG 35-4 A, Rappaport scheme of the hepatic functional lobule (acinus), organized according to biochemical considerations (1958). For example, zone 1 cells are responsible for protein synthesis, urea and cholesterol production, gluconeogenesis, bile formation, and cytogenesis; zone 2 cells also produce albumin and are actively involved in glycolysis and pigment formation; and zone 3 cells are the major site of liponeogenesis, ketogenesis, and drug metabolism. Zone 3 hepatocytes, being farther from the hepatic artery and hepatic portal veins, also have the lowest oxygen supply and are therefore most susceptible to hypoxic damage. Conversely, zone 1 hepatocytes, being closest to the hepatic portal vein, are most susceptible to damage by toxins from the gut. B, Outdated theory of hepatic functional lobule, as first proposed in 1833. The apparent hexagonal boundaries have little to do with functional arrangement.
Obstruction of the bile duct near the duodenum results in increased intraluminal biliary tract pressure, interhepatocellular regurgitation of bile constituents into the circulation, and jaundice. If only one of the hepatic bile ducts exiting the liver is blocked or if only the cystic duct exiting the gallbladder is obstructed for some reason, there may be biochemical clues for localized cholestasis, such as high serum alkaline phosphatase activity; however, the liver’s overall ability to excrete is preserved, and jaundice does not ensue. Traumatic or pathologic biliary tract rupture allows leakage of bile into the peritoneal space and some absorption of bile components. Depending on the underlying cause and the time elapsed between biliary rupture and diagnosis, the degree of jaundice may be mild to moderate. If biliary rupture has occurred, the total bilirubin content of the abdominal effusion is greater than that of serum.
Reference ranges for serum total bilirubin concentrations in dogs and cats may vary from laboratory to laboratory, but most published resources agree that concentrations over 0.3 mg/dl in cats and 0.6 mg/dl in dogs are abnormal. When results of laboratory tests are assessed, species differences in the formation and renal processing of bilirubin between cats and dogs must be taken into account. Canine renal tubules have a low resorptive threshold for bilirubin. Dogs (males to a greater extent than females) have the necessary renal enzyme systems to process bilirubin to a limited extent; therefore bilirubinuria (up to 2+ to 3+ reaction by dipstick analysis) may be a normal finding in canine urine specimens of specific gravity greater than 1.025. Cats do not have this ability, and they have a ninefold higher tubular absorptive capacity for bilirubin than dogs. Bilirubinuria in cats is associated with hyperbilirubinemia and is always pathologic. Because unconjugated and most conjugated bilirubin is albumin-bound in the circulation, only the small amount of nonprotein-bound conjugated bilirubin is expected to appear in the urine in physiologic and pathologic states. In dogs with hepatobiliary disease, increasing bilirubinuria often precedes the development of hyperbilirubinemia and clinical jaundice and may be the first sign of illness detected by owners.
Several nonhepatobiliary disorders impede bilirubin excretion by poorly understood means. Jaundice with evidence of hepatocellular dysfunction but minimal histopathologic changes in the liver has been described in septic human, feline, and canine patients. Certain products released by bacteria, such as endotoxin, are known to reversibly interfere with bile flow. As yet unexplained mild hyperbilirubinemia (≤2.5 mg/dl) may also be detected in approximately 20% of hyperthyroid cats. Experimental investigations of thyrotoxicosis in laboratory animals have demonstrated increased production of bilirubin, which has been proposed to be associated with increased degradation of hepatic heme proteins. There is no histologic evidence of cholestasis at the light microscopic level in affected cats, and the hyperbilirubinemia resolves with return to euthyroidism. Guidelines for initial evaluation of the icteric cat or dog are given in Fig. 35-5. Finally, lipemia is a common cause of pseudohyperbilirubinemia in dogs as a result of interference with the laboratory test.
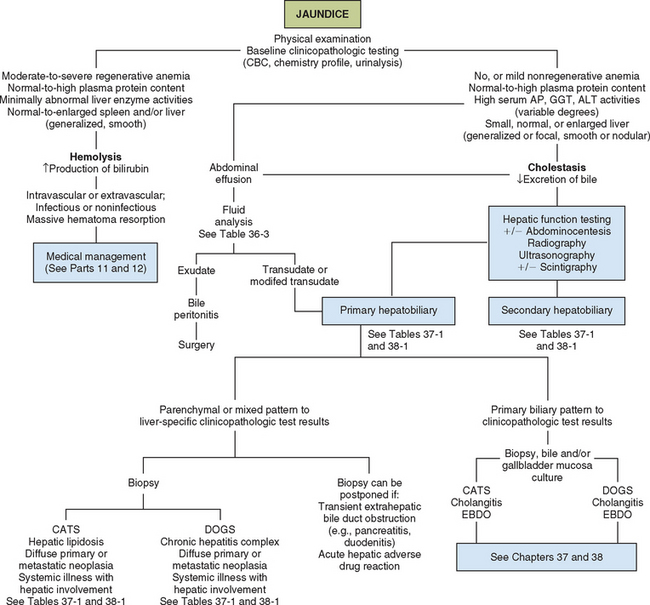
FIG 35-5 Algorithm for preliminary evaluation of the icteric cat or dog. AP, Alkaline phosphatase; GGT, γ-glutamyltransferase; ALT, alanine transaminase; EBDO, extrahepatic bile duct obstruction.
Acholic feces result from total absence of bile pigment in the intestine (Fig. 35-6). Only a small amount of bile pigment is needed to be changed to stercobilin and yield normal fecal color; therefore bile flow into the intestine must be completely discontinued in order to form acholic feces, and this is very rare in both dogs and cats. In addition to appearing pale from lack of stercobilin and other pigments, acholic feces are pale because of steatorrhea resulting from the lack of bile acids to facilitate fat absorption. Mechanical diseases of the extrahepatic biliary tract (e.g., unremitting complete extrahepatic bile duct obstruction [EBDO], traumatic bile duct avulsion from the duodenum) are the most common causes of acholic feces in cats and dogs. Total inability to take up, conjugate, and excrete bilirubin because of generalized hepatocellular failure is theoretically possible. However, because the functional organization of the liver is heterogeneous (see Fig. 35-4) and because primary hepatic diseases do not affect all hepatocytes uniformly, the overall ability of the liver to process bilirubin may be altered, although it is usually preserved. A condition has been reported rarely in cats with severe cholangitis in which bile flow ceases. Under these circumstances, “bile” consists of only clear, viscous biliary epithelial secretions, and this may result in the production of acholic feces. A similar finding, known as “white bile syndrome,” has been associated with prolonged total biliary obstruction and is thought to be the result of resorption of bile pigments. The true frequency of white bile in cats or dogs with severe cholestasis is not known.

HEPATIC ENCEPHALOPATHY
Signs of abnormal mentation and neurologic dysfunction develop in dogs and cats with serious hepatobiliary disease as a result of exposure of the cerebral cortex to absorbed intestinal toxins that have not been removed by the liver. Substances that have been implicated as important in the genesis of hepatic encephalopathy (HE), singly or in combination, are ammonia, mercaptans, short-chain fatty acids, skatoles, indoles, and aromatic amino acids. Either there is marked reduction in functional hepatic mass or portal blood flow has been diverted by the development of portosystemic venous anastomoses, thus preventing detoxification of gastrointestinal (GI) toxins, or there is a combination of these two processes. Portosystemic shunting can occur via the presence of a macroscopic vascular pattern that results from a congenital vascular miscommunication or by a complex of acquired “relief valves” that open in response to sustained portal hypertension secondary to severe primary hepatobiliary disease (Fig. 35-7). Intrahepatic, microscopic portosystemic shunting or widespread hepatocellular inability to detoxify noxious enteric substances accounts for HE when an abnormal portal vascular pattern cannot be demonstrated. Rarely, if congenital portovascular anomalies and severe primary hepatobiliary disease with acquired shunting have been ruled out, congenital urea enzyme cycle deficiencies and organic acidemias, in which ammonia cannot be degraded to urea, are considered. HE has also been reported in congenital cobalamin deficiency in dogs (Battersby et al., 2005). Animals with systemic diseases having hepatic manifestations do not undergo sufficient loss of hepatic mass or change in hepatic blood flow to develop signs of HE.
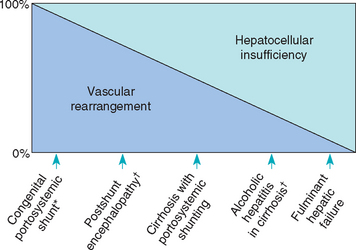
FIG 35-7 Spectrum of hepatic encephalopathy in cats and dogs ranging from pure vascular to pure hepatocellular causes. *, Clinically relevant only in dogs and cats; †, clinically relevant only in human patients.
(Modified from Schafer DF et al: Hepatic encephalopathy. In Zakim D et al, editors: Hepatology: a textbook of liver disease, Philadelphia, 1990, WB Saunders.)
The pathogenesis of this reversible abnormality in cerebral metabolism currently is incompletely understood. Increased ammonia (NH3) in the blood remains the most important cause of HE. Most of the precipitating factors and treatment recommendations for HE primarily affect blood NH3 concentrations. The effects on neurotransmitters and the cerebrospinal fluid (CSF) environment are complex. The brain is very sensitive to the toxic effects of NH3 but does not have a urea cycle, so NH3 in the CSF is detoxified to glutamine. CSF glutamine concentrations in dogs with portosystemic shunts (PSS) correlate better with clinical signs than CSF or blood NH3 levels (Fig. 35-8). Dogs with congenital PSS also have increased CSF concentrations of aromatic amino acids, particularly tryptophan and its metabolites, and this appears to be directly related to NH3 concentrations in the CSF because they share an antiport transporter. Also implicated are changes in central nervous system (CNS) serotonin activity (which is often reduced); stimulation of NMDA (N-methyl-D-aspartic acid) receptors, peripheral-type benzodiazepine receptors, and altered astrocyte receptors and handling of glutamate. Most of these changes are related to increased NH3.

FIG 35-8 Two dogs with similar fasting plasma ammonia concentrations, emphasizing the lack of correlation between plasma ammonia content and severity of encephalopathic signs. A, Female Miniature Poodle with congenital portosystemic shunt. The plasma ammonia concentration was 454 μg/dl. B, Male mixed-breed dog with chronic hepatic failure and acquired portosystemic shunting. The plasma ammonia concentration was 390 μg/dl.
The sources of increased blood ammonia in animals with liver disease are outlined in Fig. 35-9 and include the following:
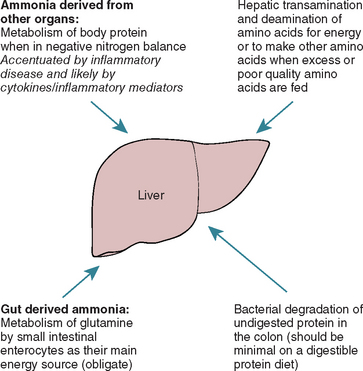
FIG 35-9 Sources of ammonia that can contribute to hepatic encephalopathy: Note that it is now believed that bacterial degradation of undigested protein in the colon is the least important of these on normal diets.
It is very important to realize that the traditional view that the toxins causing HE are predominantly of dietary origin is misleading; although the gut is an important source of NH3 in animals on high-protein diets, in many animals, particularly those with protein-calorie malnutrition, endogenous sources of NH3 may be more important and further dietary protein restriction just worsens the hyperammonemia in these cases.
Subtle, nonspecific signs of HE in cats and dogs that could be noted at any time and that represent chronic or subclinical HE include anorexia, depression, weight loss, lethargy, nausea, fever, hypersalivation (particularly in cats), intermittent vomiting, and diarrhea. Certain events might precipitate an acute episode of HE with severe neurologic signs (see Chapter 39). Nearly any CNS sign may be observed in cats and dogs with HE, although typical signs tend to be nonlo calizing, suggesting generalized brain involvement: trembling, ataxia, hysteria, dementia, marked personality change (usually toward aggressiveness), circling, head pressing, cortical blindness, or seizures (see Box 35-2). Ocassionally, animals with hyperammonemia have asymmetric, localizing neurologic signs that regress with appropriate treatment for HE.
COAGULOPATHIES
Because of the integral role of the liver in hemostasis, hemorrhagic tendencies can be a presenting sign in cats and dogs with severe hepatobiliary disease. Despite the fact that most coagulation proteins and inhibitors, except for von Willebrand’s factor (vWF) and possibly factor VIII, are synthesized in the liver (Box 35-3), the overall frequency of clinical sequelae of disturbances in hemostasis is low. Inability to synthesize vitamin K–dependent factors (II, VII, IX, and X) because of the absence of bile acid–dependent fat absorption secondary to complete EBDO or a transected bile duct from abdominal trauma can cause clinically apparent bleeding. Subclinical and clinical coagulopathies are also noted in animals with severe diseases of the hepatic parenchyma. Some animals with severe hepatic disease and relatively unremarkable results of routine coagulation tests have high serum activity of proteins induced by vitamin K antagonism (PIVKA) that could impart bleeding tendencies. In early studies of the mechanism of impaired coagulation after partial hepatectomy in dogs, after surgical removal of 70% of the hepatic mass, dogs developed significant alterations in plasma clotting factor concentrations without spontaneous hemorrhage. Having severe hepatic parenchymal disease predisposes a dog or cat not only to changes in coagulation factor activity from hepatocellular dysfunction but also to disseminated intravascular coagulation (DIC), particularly in those with acute disease (see Chapter 38). In dogs with acute hepatic necrosis, some clinicians have observed thrombocytopenia, thought to be associated with increased platelet use or sequestration.
Other than noticeable imbalances in coagulation factor activity, the only other mechanism by which bleeding might occur in a cat or dog with severe hepatic disease is portal hypertension–induced vascular congestion and fragility. In such cases, which are expected considerably more often in dogs than in cats because of the types of hepatobiliary diseases they acquire, the common site affected is the upper GI tract (stomach, duodenum); therefore hematemesis and melena are common bleeding presentations and a common cause of death in dogs with chronic liver disease. In contrast to human patients, in whom fragile esophageal varices develop and can burst, causing severe and often fatal hemorrhage, the mechanism of GI hemorrhage in companion animals is unknown but is suspected to be related to poor mucosal perfusion and reduced epithelial cell turnover associated with portal hypertension and splanchnic pooling of blood. Hypergastrinemia was observed in dogs made cirrhotic under experimental conditions and was theorized to have been provoked by excess serum bile acid concentrations. More recent studies have not borne out this theory; in fact, gastrin is often low in dogs with liver disease, and the ulcers are often duodenal and not gastric.
POLYURIA AND POLYDIPSIA
Increased thirst and volume of urination can be clinical signs of serious hepatocellular dysfunction and also of portosystemic shunts. Several factors are suspected to contribute to polydipsia (PD) and polyuria (PU), which are seen primarily in dogs and rarely in cats, with marked hepatic dysfunction. Altered sense of thirst may be a manifestation of HE. Dogs with congenital and acquired PSS have hypercortisolemia associated with reduced metabolism of cortisol in the liver and decreased cortisol binding protein concentration in the plasma. Excess secretion of adrenocorticotropic hormone stimulated by abnormal neurotransmitters leads to excess cortisol secretion and altered threshold for antidiuretic hormone release in dogs with HE. Secondary hyperaldosteronism from delayed excretion of aldosterone, which is accomplished normally by the liver, leads to sodium retention and increased water intake with compensatory PU. Changes in the function of portal vein osmoreceptors that stimulate thirst without hyperosmolality are also thought to be partly responsible for PD. Loss of the renal medullary concentrating gradient for urea because of the inability to produce urea from ammonia would first cause PU and then compensatory PD. Delayed cortisol excretion and persistent hypokalemia may also contribute to the renal concentrating defect. Investigation of polydipsia in dogs with congenital PSS has identified partial renal concentrating ability in response to water deprivation, with resolution of PD when normal portal blood flow was reestablished.
Battersby IA, et al. Hyperammonaemic encephalopathy secondary to selective cobalamin deficiency in a juvenile Border collie. J Small Anim Pract. 2005;46:339.
Maddison JE. Newest insights into hepatic encephalopathy. Eur J Compar Gastroenterol. 2000;5:17.
Moore KP, et al. Guidelines on the management of ascites in cirrhosis. Gut. 2006;55(Suppl VI):vi1.
Rothuizen J, et al. Postprandial and cholecystokinin-induced emptying of the gall bladder in dogs. Vet Rec. 1990;19:126.
Rothuizen J, et al. Chronic glucocorticoid excess and impaired osmoregulation of vasopressin release in dogs with hepatic encephalopathy. Dom Anim Endocrinol. 1995;12:13.
Shawcross D, Jalan R. Dispelling myths in the treatment of hepatic encephalopathy. Lancet. 2005;365:431.
Sterczer A, et al. Fast resolution of hypercortisolism in dogs with portosystemic encephalopathy after surgical shunt closure. Res Vet Sci. 1999;66:63.
Wright KN, et al. Peritoneal effusion in cats: 65 cases (1981–1997). J Am Vet Med Assoc. 1999;214:375.