CHAPTER 39 Treatment of Complications of Hepatic Disease and Failure
GENERAL CONSIDERATIONS
The following problems are common in dogs with hepatic failure and are usually related to sudden or chronic progressive loss of functional hepatocyte mass, intrahepatic portal hypertension resulting from primary hepatobiliary disease, acquired portosystemic shunts (PSSs), or a combination of these factors. The clinical syndrome of portal hypertension with abdominal effusion, acquired PSSs, and high risk of gastrointestinal (GI) ulceration is observed frequently in dogs with chronic liver disease but rarely in cats, whereas coagulopathies are common in cats because of the additional effects of concurrent biliary tract, pancreatic, and small intestinal disease. Hepatic encephalopathy (HE) resulting from congenital PSS is relatively common in both species. Protein-calorie malnutrition is common in both species, particularly in association with chronic disease. Effective management of these problems is vital to achieve a reasonable quality of life for the patient and to enable hepatic recovery while specific therapy is taking effect or when the underlying cause cannot be eradicated.
HEPATIC ENCEPHALOPATHY
CHRONIC HEPATIC ENCEPHALOPATHY
Treatment
The goal of treatment in cats and dogs with HE is to restore normal neurologic function by decreasing formation of gut-derived and peripherally derived encephalotoxins, eliminating precipitating factors, and correcting acid-base and electrolyte abnormalities. A variety of encephalotoxins are implicated in causing HE (see Chapter 35), but the most important from the point of view of treatment is ammonia. It was once believed that the most important source of ammonia was undigested protein in the gut, but emphasis has now shifted to interorgan metabolism of ammonia in patients with HE, whereas dietary protein itself is a less important source (Wright et al., 2007; see Chapter 35). Inflammatory mediators are also thought to be important precipitators of HE in their own right. It is known that clinically relevant episodes of HE in dogs and cats with congenital or acquired PSS are often precipitated not just by feeding but also by stress and infections, emphasizing the role of hypermetabolism, inflammation, and breakdown of body protein in the development of HE. In fact, particularly in dogs with acquired PSS and protein-calorie malnutrition, HE is often triggered by negative nitrogen balance and breaking down muscle mass (Fig. 39-1), and in these circumstances starvation and protein restriction will worsen the HE.
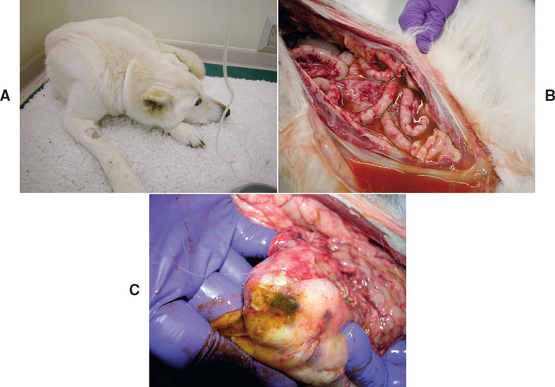
FIG 39-1 A, A 9-year-old neutered female German Shepherd Dog with previously stable noncirrhotic portal hypertension treated medically for 8 years presented very depressed with a week-long history of anorexia (same dog as Fig. 38-12 in Chapter 38). B and C, In spite of immediate institution of tube feeding on admission, the dog rapidly developed fatal septic peritonitis as a result of rupture of an ulcer at the gastroduodenal junction. It was found that the dog had developed asymptomatic pyelonephritis. The referring veterinarian had recognized the hepatic encephalopathy but tried to manage it by starvation for a week which likely increased rather than decreased ammonia production through breakdown of muscle and also increased the risk of GI ulceration because of a lack of intraluminal gut nutrition.
A combination of careful dietary manipulation, locally acting agents that discourage formation of readily absorbable ammonia and hasten evacuation of the intestinal tract, antibiotics to suppress bacterial populations that generate ammonia and other gut-derived encephalotoxins, and treatment of any precipitating cause is the standard approach for long-term management of chronic HE (Box 39-1). Dietary management and treatment of the underlying cause are the most important approaches, but advice has changed over the last few years with respect to protein restriction, and it is now clear that dogs and cats with congenital or acquired PSS have higher protein requirements; long-term feeding of a protein-restricted diet not only is not indicated but will result in protein-calorie malnutrition.
 BOX 39-1 Long-term Medical Management of Hepatic Encephalopathy
BOX 39-1 Long-term Medical Management of Hepatic Encephalopathy
Dietary Management
Lactulose
Whether it is due to congenital PSS in dogs and cats or acquired PSS (mainly in dogs), treatment of HE is much the same. The main difference is that acquired PSSs are usually the result of portal hypertension, so treatment of the other manifestations of this and the underlying liver disease will also be necessary in these cases (see the discussion of portal hypertension below). Recent studies in human medicine have questioned the actual efficacy of some of the treatment recommendations for HE, including lactulose. Controlled trials have not been conducted in animals to determine the optimal treatment for HE and for each stage (mild, moderate, severe) of HE; therefore current recommendations are based on studies in human medicine and on anecdotal reports in dogs and cats.
Diet
The ideal diet for long-term management of HE is the same as the diet recommended in chronic liver disease in dogs; dietary recommendations are outlined in Table 38-2 and Box 39-1. Protein restriction has long been recommended in patients with HE owing to the fact that undigested protein in the gut broken down by bacteria is a source of gut-derived ammonia. However, as has recently been pointed out, gut bacteria will metabolize only undigested protein that reaches the colon. This should not occur if the protein in the diet is very digestible and not in such excessive amounts that it overwhelms the digestive capacity of the small intestine. There are high amounts of ammonia in the portal circulation, particularly after a meal, but the main source of these is obligate catabolism of glutamine by small intestinal enterocytes as their main energy source, and intestinal glutaminase activity seems to increase for unknown reasons in humans with cirrhosis, increasing gut ammonia production. Studies in dogs with experimental PSS and animals and humans with acquired PSS have actually shown a higher protein requirement than in normal animals or people. Therefore the current recommendation is to feed animals with congenital or acquired PSS normal to only slightly reduced quantities of protein that is highly digestible and of high biological value in order to minimize the amounts of undigested protein reaching the colon and “wastage” of excess nonessential amino acids by transamination or deamination for energy. Some experts recommend that diets should have low amounts of aromatic amino acids, because these have been implicated in HE, but in fact there is no evidence that the ratio of dietary aromatic amino acid : branched chain amino acid has any effect on HE. Food should be fed in small amounts and often to avoid overwhelming the ability of the liver to metabolize it. Diets manufactured for dogs with liver disease are a good starting point (Hill’s canine LD; Royal-Canin canine hepatic) but are rather protein-restricted, so they should be supplemented with a high-quality protein such as cottage cheese or chicken. An alternative is to feed a veterinary diet marketed for intestinal disease; these diets contain high-quality, highly digestible protein sources (Hill’s canine or feline ID; Iam’s canine or feline intestinal formula; Royal-Canin canine or feline digestive lowfat). Most, if not all, dogs with congenital or acquired PSS can tolerate normal protein concentrations if other measures are also implemented, as outlined in the subsequent paragraphs and in Box 39-1. A few require more marked restriction in the short term, but every effort should be made to increase to a normal protein concentration over the long term.
Lactulose
Lactulose (β-galactosidofructose) is a semisynthetic disaccharide that is not digestible by mammals and therefore passes into the colon, where it is degraded by bacteria into short chain fatty acids (SCFA), particularly lactic and acetic acid. These SCFAs help control signs of HE by acidification of the intestinal contents, which traps ammonium ions in the colon, and by promoting osmotic diarrhea. In addition, SCFAs are used as an energy source by colonic bacteria, allowing them to grow and thus incorporate colonic ammonia into their own bacterial protein, which is subsequently lost with the bacteria in the feces (a type of bacterial “ammonia trap”).
The dose is adjusted until there are two to three soft stools per day (see Box 39-1); overdosing results in watery diarrhea. There are no known complications of chronic lactulose use in animals (other than diarrhea). However, the efficacy of lactulose has never been critically evaluated in dogs and cats with HE, and recent studies in humans suggest that it may not be as helpful as previously thought. Lactulose can also be given by enema in animals with acute HE (Box 39-2). Many cats and dogs object strongly to the sweet taste of lactulose; an attractive alternative is lactitol (β-galactosidosorbitol), which is a relative of lactulose and can be used as a powder (500 mg/kg/day in three to four doses, adjusted to produce two to three soft stools daily). Currently, lactitol is available in the United States as a food sweetener, but it has not been studied in dogs and cats with HE
BOX 39-2 Treatment of Acute Encephalopathic Crisis
Antibiotic Treatment
If dietary therapy alone or in combination with lactulose is insufficient to control signs of HE, other medications may be added. Antibacterial drugs that are effective for anaerobic organisms (metronidazole, 7.5 mg/kg administered PO q8-12h; amoxicillin, 22 mg/kg administered PO q12h) are preferable. Antibiotics effective for gram-negative, urea-splitting organisms (neomycin sulfate, 20 mg/kg administered PO q12h) may also be used, although neomycin is more useful in acute HE rather than in long-term use because intestinal bacteria tend to become resistant to neomycin. In addition, it is not systemically absorbed and remains within the gastrointestinal tract; it is preferable to use a systemically absorbed antibiotic over the long term to protect against bacteremia. The low dose of metronidazole is given to avert neurotoxicity as a potential adverse effect of delayed hepatic excretion. Other therapeutic strategies investigated in humans with chronic HE include ornithine aspartate supplementation (see Box 39-1) and probiotics to increase numbers of beneficial bacteria. These may show benefit in dogs in the future, but there are currently no published studies documenting their use in small animals.
Controlling Precipitating Factors
Certain conditions are known to accentuate or precipitate HE and should be avoided or treated aggressively when detected (Box 39-3). In fact, in many cases it is the precipitating factors (rather than the diet) that are most important in triggering HE. It is particularly important to identify and treat any concurrent inflammatory disease because even infections as apparently mild as cystitis or middle ear disease can trigger HE episodes in susceptible individuals. Recent work in humans and experimental animals has highlighted the importance of inflammation and inflammatory cytokines in triggering HE (Wright et al., 2007).
 BOX 39-3 Precipitating Factors for Hepatic Encephalopathy in a Susceptible Individual
BOX 39-3 Precipitating Factors for Hepatic Encephalopathy in a Susceptible Individual
Increased Generation of Ammonia in the Intestine
Increased Generation of Ammonia Systemically
ACUTE HEPATIC ENCEPHALOPATHY
Treatment
Acute HE is a true medical emergency. Fortunately, it is much less common than chronic, waxing and waning HE. Animals may present in seizure or comatose, and although HE initially causes no permanent brain damage, prolonged seizures, status epilepticus, or coma will; prolonged severe HE by itself may lead to serious cerebral edema as a result of accumulation of the osmolyte glutamine (from ammonia detoxification) in astrocytes. In addition, the effects of acute HE, particularly hypoglycemia, can be fatal if not recognized and treated. The treatment of acute encephalopathic crises is outlined in Box 39-2. Intensive management is required. However, treatment is worthwhile because some animals can go on to complete recovery and successful long-term medical management, particularly if the acute crisis was triggered by a definable event (e.g., acute gastrointestinal bleeding in a dog with chronic liver disease and portal hypertension). Nothing by mouth (NPO), administration of enemas, and intravenous fluid therapy constitute the basic therapeutic approach. Warm water cleansing enemas may be useful simply by removing colonic contents and preventing absorption of intestinal encephalotoxins. Lactulose or dilute vinegar may be added to acidify the colon and decrease absorption of ammonia. The most effective enema contains three parts lactulose to seven parts water at a total dose of 20 ml/kg. The solution is left in place, with the aid of a Foley catheter, as a retention enema for 15 to 20 minutes. For lactulose to be beneficial, the pH of the evacuated colon contents must be 6 or lower. These enemas can be given every 4 to 6 hours. Because lactulose is osmotically active, dehydration can occur if enemas are used too aggressively without careful attention to fluid intake. Fluids chosen for replacement of losses, volume expansion, and maintenance should not contain lactate, which is converted to bicarbonate, because alkalinizing solutions may precipitate or worsen HE by promoting formation of the more readily diffusible form of ammonia. Half-strength (0.45%) saline solution in 2.5% dextrose is a good empirical choice, with potassium added according to its serum concentration (see Table 55-1). Serum electrolyte concentrations in dogs with HE are extremely variable; until the results become available, 20 mEq KCl/L in administered fluids is a safe amount to add. Seizuring dogs can be stabilized with low-dose propofol infusions (Fig. 39-2) or phenobarbital. The dose of propofol is calculated by giving an initial bolus to effect (usually about 1mg/kg), timing how long it takes for the animal to show mild signs of seizuring, such as mild limb paddling again, and then dividing the dose by the time to calculate an infusion rate. For example, if after a bolus of 1 mg/kg of propofol the dog began to show signs of seizure activity again after 10 minutes, the infusion rate to give would be 1/10 = 0.1 mg/kg/min. In practice, the dose of propofol to give by constant rate infusion is usually about 0.1 to 0.2 mg/kg/min. Dogs sometimes need to remain on the infusion for hours or days, but the rate can be gradually reduced to control seizures while still allowing the dog to regain consciousness—in some cases, even enough to start eating.
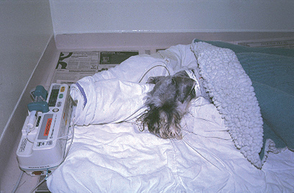
FIG 39-2 A Miniature Schnauzer with a congenital portosystemic shunt that had postligation seizures is stabilized with a propofol infusion.
In spite of some early promising reports, there is still no convincing evidence in support of other pharmacological treatments for HE, apart from antibiotics and lactulose, and therefore other drugs cannot currently be recommended for use in dogs. Trials of the benzodiazepine receptor antagonist flumazenil in human patients with refractory acute HE have had mixed results, and although flumazenil has been studied in animals for its ability to reverse the action of benzodiazepine tranquilizers, there have been no clinical studies on its use in acute HE in animals.
PORTAL HYPERTENSION
Pathogenesis
Portal hypertension is the sustained increase in blood pressure in the portal system and is seen most frequently in dogs with chronic liver disease, although it may also occasionally occur in dogs with acute liver disease. Portal hypertension is extremely uncommon in cats. It is caused by the increased resistance to blood flow through the sinusoids of the liver or (less commonly) by more direct obstructions to the portal vein such as thromboemboli. Early in chronic liver disease, portal hypertension can be the result of multiplication and phenotypic transformation of hepatic Ito (stellate) cells, which become contractile myofibroblasts that surround the sinusoids and cause constriction. In the longer term, fibrous tissue laid down by these transformed stellate cells results in more irreversible sinusoidal obstruction. The most common cause of portal hypertension is therefore chronic hepatitis progressing to cirrhosis in dogs (Fig. 39-3). It can also occur in association with hepatic neoplasia or diffuse hepatic swelling.
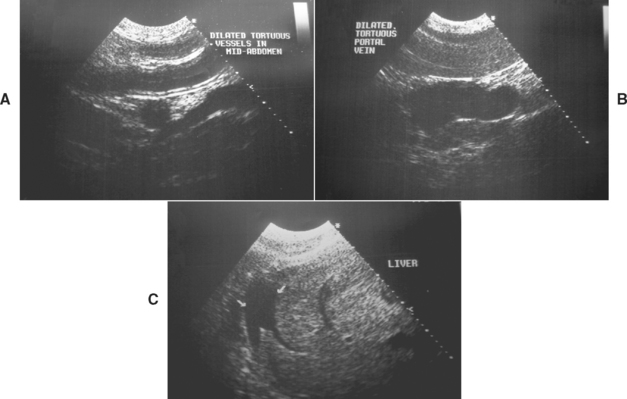
FIG 39-3 Ultrasonographic images demonstrating the progressive development of ascites with portal hypertension in a dog with cirrhosis: Ultrasonography on the first visit showed no evidence of free abdominal fluid, but dilated vessels in the midabdomen (including splenic congestion, A) and also a dilated portal vein (B). When the dog returned for a liver biopsy 2 weeks later, ultrasonography now revealed the development of mild early ascites (C).
(Courtesy Diagnostic Imaging Dept, Queen’s Veterinary School Hospital, University of Cambridge.)
The changes in hemodynamics associated with “back pressure” in the portal circulation result in one or more of the typical triad of intestinal wall edema/ulceration, ascites, and acquired PSSs. Acquired PSSs occur as “escape valves” when the portal vein pressure is consistently higher than the pressure in the caudal vena cava (see Fig. 38-2). They are always multiple and occur as a result of the opening up of previously nonfunctional veloomental vessels. They are an important compensatory mechanism because they dissipate some of the increased portal pressure, limiting the increase in splanchnic pressure and thus reducing the risk of gastrointestinal ulceration. In humans with chronic portal hypertension, acquired PSSs have been demonstrated to prolong life expectancy by reducing the chance of serious gastrointestinal or esophageal bleeding—to the point that if they are not already present, they are often created surgically. Similar survival data are not available for dogs, but it is clear that ligation of acquired PSS is contraindicated and will result in fatal splanchnic congestion. Acquired PSSs result in HE in a similar way to congenital PSSs; treatment is outlined in the preceding section.
SPLANCHNIC CONGESTION AND GASTROINTESTINAL ULCERATION
Pathogenesis
Splanchnic congestion is a common and early complication of portal hypertension, the result of the pooling of blood in the splanchnic circulation and reduced flow into the portal system (see Fig. 39-3). This can cause visible congestion and edema of the gut wall that can be detected either ultrasonographically (where there may be thickening and loss of layering of the gut) or during surgery. It occurs before the onset of ascites and persists after ascites resolves (see Fig. 39-3). The congested gut wall is at increased risk of GI ulceration. Catastrophic gastrointestinal or esophageal ulceration is the most common cause of death in humans with portal hypertension who do not undergo a liver transplant, and it appears also to be the most common cause of death in dogs with stable chronic liver disease (see Fig. 39-1). Ulceration associated with portal hypertension in humans often takes the form of bleeding esophageal varices, whereas in dogs the ulceration is most commonly in the proximal duodenum, presumably reflecting a difference in the anatomy of the portal system in the two species. Preventing gastrointestinal ulceration is therefore vital, and for this reason it is very important to refrain from using ulcerogenic drugs (e.g., steroids) in dogs with portal hypertension whenever possible. Corticosteroids have been shown to shorten the life expectancy of humans with chronic hepatitis and concurrent portal hypertension and should not be used in dogs with portal hypertension unless there is a very good reason for it. If they are deemed necessary, the owners should be fully informed of their potentially serious adverse effects. Other triggers for GI ulceration in dogs with portal hypertension are sepsis and protein-calorie malnutrition (discussed in more detail later), particularly if combined with a period of anorexia (see Fig. 39-1). The small intestine requires luminal glutamine and other nutrients to permit effective healing, and prolonged anorexia results in an increased risk of gastrointestinal ulceration as a result of glutamine depletion.
The clinician must be aware that GI ulceration may occur acutely in dogs with splanchic congestion and serious clinical deterioration may occur before melena is apparent because it takes several hours for the blood to pass from the small to the large intestine. Before this occurs, it is possible for the animal to show sudden onset and marked signs of HE because blood is a “high-protein meal” in the small intestine (see preceding section) or even for the ulcer to perforate and cause peritonitis (see Fig. 39-1).
Treatment
Treatment of gastrointestinal ulceration largely revolves around its prevention (i.e., avoiding triggers as much as pos sible, such as the use of steroids or nonsteroidal antiinflammatory drugs, and avoiding hypotension during any surgery). It is particularly important that any dog with portal hypertension that undergoes a prolonged period of anorexia is fed because these individuals will be at high risk of gastrointestinal ulceration if they do not receive nourishment (see Fig. 39-1). Parenteral nutrition is not an effective alternative in these dogs because it does not supply luminal nutrients for enterocyte healing (in fact, upper gastrointestinal ulceration is a common adverse effect of total parenteral nutrition in humans, even in those without portal hypertension), and some form of enteral support should be instituted as soon as possible. The use of gastric acid secretory inhibitors (H2 blockers or proton pump inhibitors) is of questionable benefit in patients with portal hypertension because it is usually the duodenum that is ulcerated (rather than the stomach); also, there have been reports that the gastric pH in dogs with liver disease may already be higher than normal as a result of changes in gastrin metabolism. However, in the face of active ulceration and melena, they are often used in the hope that they will help. In these circumstances, cimetidine is contraindicated because of its effect on hepatic P450 enzymes; therefore ranitidine (2 mg/kg administered orally or via slow IV administration q12h) or famotidine (0.5 to 1 mg/kg administered PO q12-24h) are recommended. Likewise, sucralfate (Carafate™) is of questionable efficacy; it is most effective against gastric ulceration (i.e., in association with a low pH), but it is often used (at a dosage of 500 mg to 1 g per dog PO q8h). Hemostasis profiles should also be evaluated, and any coagulopathy treated with vitamin K (see the section on coagulopathy) or plasma transfusions.
ASCITES
Pathogenesis
The development of ascites (defined as the accumulation of a transudate or modified transudate in the peritoneal cavity) is another consequence of portal hypertension (see Fig. 39-3), but the pathogenesis is complex and has really been studied only in humans; it is assumed that the mechanisms of ascites are similar in dogs. One way in which dogs differ from humans is that dogs do not develop the “spontaneous” infection of ascites of liver origin by extension of gut bacteria into the fluid that results in peritonitis, which is commonly reported in people. The presence of ascites is a poor prognostic indicator in humans with chronic hepatitis, and the same appears to be true in dogs. Hypoalbuminemia contributes to the development of ascites but by itself is rarely sufficient to cause fluid accumulation; portal hypertension is a critical contributing factor. The development of ascites in patients with liver disease also seems to lead to sodium retention by the kidneys. In many cases there is systemic hypotension and increased renal sodium retention, partly as a result of reduced glomerular filtration rate and decreased sodium delivery to the tubules and partly as a result of increased release of renin-angiotensin-aldosterone (RAAS) that results in increased sodium retention in the distal tubules. This leads to an increase in circulating fluid volume, precipitating the formation of ascites, which in turn reduces venous return because of increased pressure on the caudal vena cava and initiates a vicious cycle of renal sodium retention and ascites. Therefore aldosterone antagonists are usually most effective in dogs with ascites secondary to portal hypertension, whereas loop diuretics, such as furosemide used alone, can be ineffective or even, in some cases, actually increase the volume of effusion by causing a further decrease in systemic blood pressure as a result of hemoconcentration and secondary increases in RAAS activation.
Treatment
Treatment of ascites associated with liver failure revolves around the use of diuretics: first aldosterone antagonists (spironolactone, 1 to 2 mg/kg administered PO q12h), but then with the addition of furosemide (2-4 mg/kg administered PO q12h) if necessary in refractory cases. Spironolactone usually takes 2 or 3 days to reach full effect, and the resolution of ascites can be monitored by weighing the patient daily (any acute changes in weight will be due to fluid shifts). Dietary sodium restriction has also been recommended, although it is unclear how effective or important this is. However, it is certainly wise to refrain from feeding the patient high-salt snacks and treats.
It is very important to monitor serum electrolyte concentrations (mainly sodium and potassium) daily during the first few days of treatment and every few weeks to months thereafter, depending on how stable the dog and drug doses are. Hypokalemia should be avoided because it can precipitate HE (see preceding section), but it is less likely in a dog on both aldosterone antagonists and loop diuretics than in a dog on furosemide alone. Hyponatremia can also occur; if it is marked, the diuretics should be stopped and the patient given careful intravenous replacement until the sodium is normalized.
Therapeutic paracentesis is indicated only in patients with ascites that is severe enough to compromise breathing. This is actually unusual and is manifested by severe, drumlike ascites; the dog is unable to settle and lie down. Paracentesis should be accompanied by concurrent intravenous administration of a colloid plasma expander, plasma, or albumin; removal of a large volume of fluid containing albumin can result in a precipitous hypoalbuminemia and decrease in oncotic pressure, leading to pulmonary edema. This is a real problem in dogs with chronic liver disease in which the liver’s capacity to manufacture albumin is reduced. Clear recommendations for dogs have not been published, but the recommendations for humans, adapted for dogs, are outlined in Box 39-4.
BOX 39-4 Guidelines for Therapeutic Paracentesis in Dogs with Ascites Resulting from Liver Disease
Adapted from Moore et al: Guidelines on the management of ascites in cirrhosis, Gut 55 (suppl 6):vi1, 2006.
Reserve for use ONLY in cases with severe, refractory ascites:
COAGULOPATHY
Pathogenesis
The liver plays a central role in both the coagulation and fibrinolytic systems. The liver synthesizes all the coagulation factors with the exception of factor VIII and also makes the inhibitors of coagulation and fibrinolysis. Factors II, VII, IX, and X also require hepatic activation by a vitamin K–dependent carboxylation reaction. Hemostatic abnormalities are quite common in both dogs and cats with liver disease; in one study 50% and 75% of dogs with liver disease had prolongation of the one-stage prothrombin time (OSPT) and activated partial thromboplastin time (APTT), respectively (Badylak et al., 1983). In another study 82% of cats with liver disease had hemostatic abnormalities (Lisciandro et al., 1998). Cats appear to be particularly susceptible to prolongation of clotting times; this is at least partly due to reduced vitamin K absorption. Dogs and cats with vitamin K–responsive coagulopathies have prolongation of both the OSPT and APTT (and the OSPT may actually be longer than the APTT). Vitamin K is a fat-soluble vitamin, and its absorption is decreased in association with biliary tract disease (which is common in cats); bile acid secretion into the small intestine is also reduced. Moreover, the inflammatory bowel disease commonly seen concurrently in cats with chronic biliary tract disease results in reduced fat absorption. Finally, some cats with chronic biliary tract disease have concurrent chronic pancreatitis, and as this progresses to exocrine pancreatic insufficiency, fat absorption (and thus vitamin K absorption) will decline further.
In contrast, dogs with chronic liver disease rarely have clinically relevant prolongation of clotting times. However, in both species severe diffuse liver disease, particularly acute infiltration such as lipidosis (cats) and lymphoma (cats and dogs), will cause a decrease in the activity of clotting factors in many cases as a result of hepatocyte damage and reduced synthesis in the liver. In patients with lymphoma or lipidosis this decreased activity of clotting factors is rapidly reversible if the underlying disease can be successfully treated, thus allowing recovery of hepatocyte function. In one study of cats coagulopathies were seen most commonly in cats with hepatic lipidosis and cats with inflammatory bowel disease and concurrent cholangitis (Center et al., 2000).
Coagulopathies can also occur in dogs and cats with liver disease as a result of disseminated intravascular coagulation (DIC) with resultant prolongation of clotting times and thrombocytopenia. DIC is particularly a complication of acute, fulminating hepatitis and also some hepatic tumors; it carries a very poor prognosis.
Clinical Features and Diagnosis
Despite the presence of hemostatic abnormalities, spontaneous bleeding is uncommon in patients with chronic liver disease but relatively common in those with acute disease. Because dogs with portal hypertension and gastrointestinal hemorrhage (see previous section) may also have a coagulopathy predisposing to their bleeding, they should be thoroughly evaluated. However, the risk of hemorrhage increases after a challenge to hemostasis, such as liver biopsy; therefore it is very important to evaluate hemostasis before performing liver biopsy. One study (Bigge et al., 2001) suggested that thrombocytopenia was a more significant predictor of bleeding complications after ultrasonography-guided biopsies in dogs and cats than prolongation of the OSPT and APTT. Therefore clinicians must perform a platelet count in dogs and cats before performing a liver biopsy. A platelet estimate can be can be done manually on the blood smear (Chapter 87) The platelet count (per μL) can be estimated by counting the number of platelets in 10 oil immersion fields and multiplying the average number per field by 15,000 to 20,000. Prolongation of coagulation times may also increase the risk of bleeding; in the same study, prolongation of the OSPT in dogs and the APTT in cats was significantly associated with bleeding complications after biopsy. Ideally, therefore, both OSPT and APTT should be evaluated in cats and dogs before hepatic biopsy; however, a practical alternative could be assessment of at least an activated clotting time (ACT) in a glass tube containing diatomaceous earth as a contact activator, although theoretically this is more useful in cats than dogs because it assesses the intrinsic pathway (=APTT) and final common pathway only.
Because factor depletion must be greater than 70% to result in prolongation of the OSPT or APTT, many more dogs and cats may have subtle abnormalities in the concentration of individual coagulation factors. These can be detected by more sensitive tests, such as measuring the concentration of individual clotting factors or the PIVKA (proteins induced by vitamin K absence) test, although its clinical efficacy in large numbers of dogs and cats is untested. If available, thromboelastography may allow for rapid quantification of hemostasis (see Chapter 87).
In dogs and cats with severe acute liver disease, spontaneous bleeding may result from depletion of clotting factors; in addition, there is a potential for developing DIC (see Chapter 87). In patients with DIC, APTT and OSPT may be prolonged, but it is impossible to distinguish this from reduced hepatic production of clotting factors. However, measurement of increased D-dimers and/or fibrin degradation products, combined with decreases in platelet count, increases the index of suspicion for DIC. D-dimer concen trations are often mildly to moderately increased in dogs with liver disease because of reduced clearance in the liver, and this does not necessarily mean that the dog has a thrombus or DIC. More marked elevations are suggestive of DIC.
Treatment
Dogs and cats with prolonged clotting times associated with chronic liver disease often respond to parenteral vitamin K supplementation alone. It is recommended that all patients receive vitamin K1 (phytomenadione), at a dosage of 0.5 to 2.0 mg/kg administered IM or SQ 12 hours before biopsy and repeated q12h for 3 days as necessary.
It is important to monitor clotting during long-term therapy (OSPT + APTT or PIVKA) and stop when they normalize because it is possible to overdose on vitamin K, which can result in Heinz body hemolysis. If the coagulopathy fails to respond to vitamin K treatment alone or if there are clinical signs of hemorrhage associated with the disease (which is more common with acute disease), administration of fresh frozen plasma or stored plasma is indicated to replenish depleted clotting factors. A starting dose of 10 ml/kg given slowly is recommended; the dose of plasma is titrated on the basis of the results of the OSPT and APTT. Again, liver biopsy, surgery or the placement of central venous catheters should not be contemplated until coagulation times have been normalized.
The treatment of DIC is difficult and frequently unsuccessful. The most effective treatment is to remove the inciting cause, which in acute liver failure in humans means rapid liver transplant. Without this option in dogs and cats, the mortality in DIC of acute fulminant hepatitis is likely to be 100%. Recommended therapies include plasma transfusion to replace depleted clotting factors and careful heparin therapy during the hypercoagulable phase. However, the efficacy of heparin therapy in DIC has recently been called into question in humans, and there are no clinical data supporting its use in dogs and cats.
PROTEIN-CALORIE MALNUTRITION
Pathogenesis
Protein-calorie malnutrition is very common in dogs with chronic hepatitis as a result of reduced intake caused by anorexia, vomiting, and diarrhea and increased loss/wastage of calories caused by hypermetabolism and poor liver function. Protein-calorie malnutrition is likely to have a serious impact on both longevity and quality of life in affected dogs. There are no studies specifically addressing the effect of malnutrition on survival and infections of dogs with liver disease, but in other canine diseases it is known to increase the risk of septic complications. This is true in humans with portal hypertension and also likely in dogs. In humans with portal hypertension malnutrition also predisposes to gut ulceration. In addition, negative nitrogen balance and reduced muscle mass predispose to HE. Breakdown of body protein results in more ammonia production, and also in a normal individual up to 50% of arterial ammonia is metabolized in skeletal muscle by conversion of glutamate to glutamine, so loss of muscle mass will reduce the ability to detoxify ammonia. What gives the most cause for concern regarding protein-calorie malnutrition in the veterinary patient is that it is often partly caused by well-meaning but unhelpful manipulations by the clinician or even by a lack of recognition and attention (discussed in greater detail later). For this reason, it is very important that clinicians treating dogs with chronic liver disease remain alert to the possibility of protein-calorie malnutrition.
Malnutrition can also be seen in dogs and cats with congenital PSS, both as a result of reduced liver synthetic capability or as a result of inappropriately severe protein restriction by the attending clinician. Cats with chronic liver disease may have negative energy balance, often as a result of the effects of concurrent intestinal and pancreatic disease reducing digestion and absorption of food. In addition, cats in negative nitrogen balance are at a particular risk of developing acute hepatic lipidosis (see Chapter 37) so protein-calorie malnutrition in this species requires particularly aggressive management.
Clinical Signs and Diagnosis
When suffering from severe malnutrition, dogs and cats appear cachectic, with reduced muscle mass. However, loss of muscle mass occurs relatively late in the process, and in the earlier stages of protein-calorie malnutrition the animal’s body condition score may be normal and yet many potentially deleterious effects on the immune system and gut wall will already be under way. There is no simple blood test that allows diagnosis of malnutrition. The most effective means to do this is by taking a careful history as well as performing a clinical examination. Any animal with liver disease should be considered as being at risk of protein-calorie malnutrition. A history of partial or complete anorexia for more than 3 days or recent weight loss of >10% not associated with fluid shifts should trigger rapid and aggressive nutritional management.
Treatment
The treatment is to feed the patient an appropriate diet. Protein restriction should be avoided as much as possible—and in some cases of chronic liver disease associated with obvious cachexia, supplementation of a maintenance diet with extra high-quality protein (such as dairy protein) is even indicated. If the patient will not eat voluntarily, some form of assisted tube feeding should be instituted short term. This is particularly important in cats with hepatic lipidosis, which almost invariably refuse to eat independently and require gastrostomy or esophagostomy tube feeding (see Chapter 37). A search should then be made for any underlying cause of anorexia, such as concurrent infections (see Fig. 39-1).
It is very important to avoid iatrogenic malnutrition while the patient is hospitalized. Withholding food for several days to allow multiple tests (e.g., liver biopsy or endoscopy) is a common problem; tests should be spread out over a longer period if necessary to allow feeding between them. It is also possible for malnutrition to develop unnoticed in the hospital as a result of inadequate record keeping and frequent staff turnover. Finally, feeding an excessively protein-restricted diet to a dog or cat with liver disease can also result in negative nitrogen balance.
Aronson LR, et al. Endogenous benzodiazepine activity in the peripheral and portal blood of dogs with congenital portosystemic shunts. Vet Surg. 1997;26:189.
Badylak SF, et al. Alterations of prothrombin time and activated partial thromboplastin time in dogs with hepatic disease. Am J Vet Res. 1981;42:2053.
Badylak SF, et al. Plasma coagulation factor abnormalities in dogs with naturally occurring hepatic disease. Am J Vet Res. 1983;44:2336.
Bigge LA, et al. Correlation between coagulation profile findings and bleeding complications after ultrasound-guided biopsies: 434 cases (1993–1996). J Am Anim Hosp Assoc. 2001;37:228.
Center SA, et al. Proteins invoked by vitamin K absence and clotting times in clinically ill cats. J Vet Intern Med. 2000;14:292.
Griffen A, et al. Evaluation of a canine D-dimer point-of-care test kit for use in samples obtained from dogs with disseminated intravascular coagulation, thromboembolic disease, and hemorrhage. Am J Vet Res. 2003;64:1562.
Kummeling A, et al. Coagulation profiles in dogs with congenital portosystemic shunts before and after surgical attenuation. J Vet Intern Med. 2006;20:1319.
Laflamme DP, et al. Apparent dietary protein requirement of dogs with portosystemic shunt. Am J Vet Res. 1993;54:719.
Lisciandro SC, et al. Coagulation abnormalities in 22 cats with naturally occurring liver disease. J Vet Intern Med. 1998;12:71.
Mount ME, et al. Use of a test for proteins induced by vitamin K absence or antagonism in diagnosis of anticoagulant poisoning in dogs: 325 cases (1987–1997). J Am Vet Med Assoc. 2003;222:194.
Moore, et al. Guidelines on the management of ascites in cirrhosis. Gut. 2006;55(suppl 6):vi1.
Niles JD, et al. Hemostatic profiles in 39 dogs with congenital portosystemic shunts. Vet Surg. 2001;30:97.
Shawcross D, et al. Dispelling myths in the treatment of hepatic encephalopathy. Lancet. 2005;365:431.
Wright G, et al. Management of hepatic encephalopathy in patients with cirrhosis. Best Pract Res Clin Gastroenterol. 2007;21:95.