Infection and Inflammation of the Central Nervous System
The CNS may be affected directly by bacteria, viruses, fungi, parasites, and mycobacteria. Viruses causing acute and chronic CNS infections are listed in Table 17-10. The infecting microorganisms gain entry to the nervous system by (1) hematogenous spread through arterial blood or (2) direct extension from another site of infection.66 Neurologic infections produce disease by several mechanisms: direct neuronal or glial infection; mass lesion formation; inflammation with subsequent edema; interruption of cerebrospinal fluid pathways; neuronal damage, or vasculopathy; and secretion of neurotoxins (Figures 17-32, 17-33, and 17-34). Syndromes are acute and subacute bacterial meningitis, epidural and brain abscess, encephalitis, peripheral neuropathy, or neurosyphilis depending on the infecting microorganism. Signs and symptoms are produced because of (1) interference with the function of the nervous system tissue being invaded or compressed or (2) the inflammatory response produced by the body in response to infection. The cardinal signs of CNS infection are fever, head or spine pain, and generalized or focal neurologic dysfunction.67
Table 17-10
Viruses Causing Acute and Chronic CNS Diseases
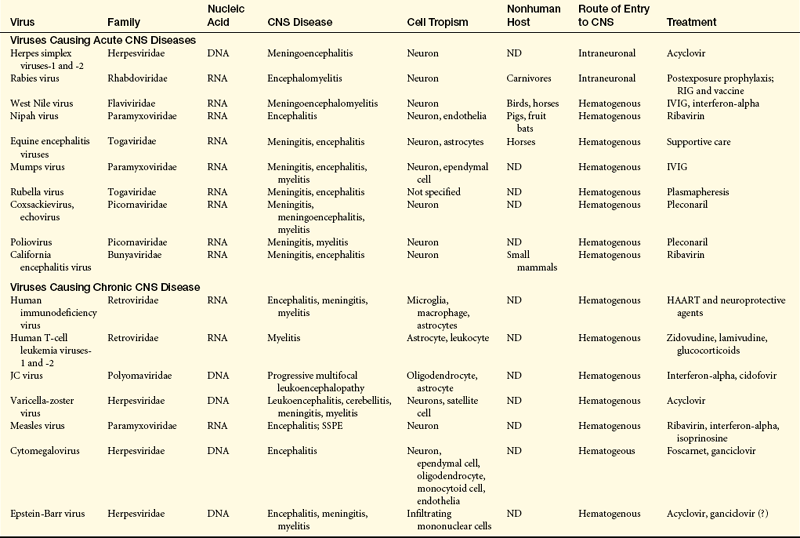
CNS, Central nervous system; DNA, deoxyribonucleic acid; HAART, highly active retroviral therapy; IVIG, intravenous immunoglobulin; JC, John Cunningham; ND, not determined; RIG, rabies immune globulin; RNA, ribonucleic acid; SSPE, subacute sclerosing panencephalitis.
From Lindquist L, Vapalahti O: Lancet 371(9627):1861-1871, 2008; Power C, Noorbakhsh F: Central nervous system viral infections: clinical aspects and pathogenic mechanism. In Gilman S, editor, Neurobiology of disease, pp 487-488, Burlington, MA, 2007, Elsevier.
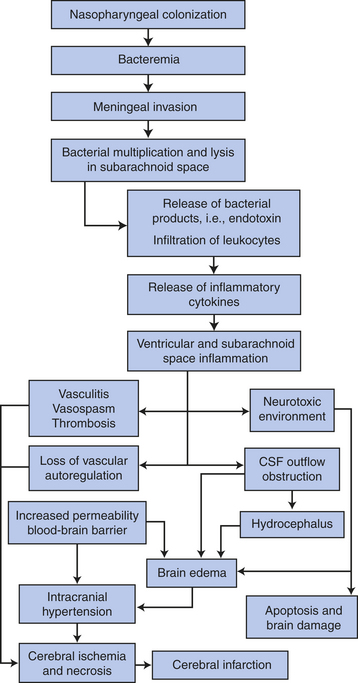
Figure 17-32 Pathogenesis of meningitis. (Adapted from Cohen J, Powderly WG: Infectious diseases, ed 2, Mosby, 2004, Edinburgh.)
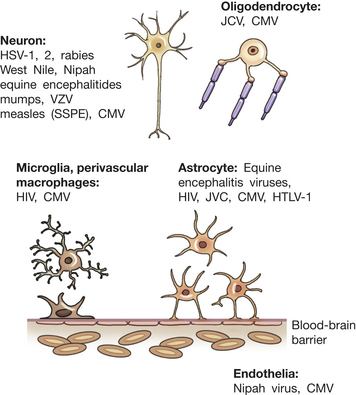
Figure 17-33 Viral infection in the central nervous system (CNS). Viruses infect specific cell types within the CNS depending on the specific properties of the virus together with individual cell membrane proteins expressed on permissive cell types. Normally the brain is protected from circulating pathogens and toxins by the blood-brain barrier. HIV, Human immunodeficiency virus; CMV, cytomegalovirus; HSV, herpes simplex virus; HTLV-1, human T-cell lymphotropic virus (causes T-cell leukemia); JCV, John Cunningham virus (a polyomavirus causing progressive multifocal leukoencephalopathy); SSPE, subacute sclerosing panencephalitis; VZV, varicella zoster virus. (Adapted from Power C, Noorbakhsh G: Central nervous system viral infections: clinical aspects and pathogenic mechanisms. In Gilman S, editor, Neurobiology of disease, p 488, Burlington, MA, 2007, Elsevier.)
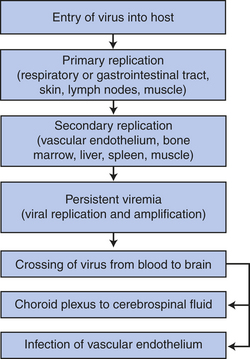
Figure 17-34 Hematogenous spread of viral pathogens to the central nervous system. (Adapted from Cohen J, Powderly WG: Infectious diseases, Mosby, 2007, Edinburgh.)
Meningitis
Meningitis (infection of the meninges) may be caused by bacteria, viruses, fungi, parasites, or other toxins. (The pathophysiology of infection is discussed in Chapter 9.) The infections are classified as acute, subacute, or chronic processes, and the pathophysiology, clinical manifestations, and treatment differ for each type of microorganism.
Bacterial meningitis is primarily an infection of the pia mater and arachnoid, the subarachnoid space, the ventricular system, and the CSF. A systemic or bloodstream infection or a direct extension from an infected area is the access route to the subarachnoid space. The bacterial infection originates in another part of the body. The incidence of bacterial meningitis is 2.5 to 3.5 cases per 100,000. The incidence is 20 per 100,000 annually for neonates, and 2 to 9 per 100,000 annually for those older than 60 years. The mortality is 25% in adults. Meningococcus (Neisseria meningitidis) and pneumococcus (Streptococcus pneumoniae) are the common causes of bacterial meningitis after the neonatal period. Pneumococcus and gram-negative enteric bacilli are the most common neonatal agents.
Meningococcus has been identified worldwide. Meningococcal meningitis occurs predominantly in men and boys and during the fall, winter, and spring. Epidemics of meningococcal meningitis occur in approximately 10-year cycles. Children and adolescents are affected predominantly; with pneumococcal meningitis, young persons and those older than 40 years are mostly affected.
Aseptic meningitis (viral meningitis, nonpurulent meningitis, lymphocytic meningitis) is an inflammation believed to be limited to the meninges. The most at-risk populations and the time of year when occurrences are seen depend on the virus. Aseptic meningitis produces a variety of symptoms and is caused by a variety of infectious agents, most of which are viruses. They include enteroviral viruses (the most common) (echovirus, coxsackievirus, and nonparalytic poliomyelitis), mumps, herpes simplex types 1 and 2, St. Louis encephalitis virus, West Nile virus, California encephalitis virus, Venezuelan equine encephalitis, Colorado tick fever, lymphocytic choriomeningitis virus, Epstein-Barr virus, and influenzavirus types A and B.68 Bacterial infections not adequately treated are another cause of aseptic meningitis.
Fungal meningitis is a chronic, much less common condition than bacterial or viral meningitis. The most common fungal infections of the nervous system are histoplasmosis, cryptococcosis, coccidioidomycosis, mucormycosis, candidiasis, and aspergillosis. Fungal meningitis most frequently occurs in persons with impaired immune responses or alterations in normal body flora. Fungal meningitis develops insidiously, usually over days or weeks. Syphilis, tuberculosis, and Lyme disease also are associated with chronic meningitis.
Tubercular meningitis, the most common and serious form of CNS tuberculosis, is again on the rise in the United States, especially in persons with acquired immunodeficiency syndrome (AIDS). Miliary tubercles form in the brain and meninges. At some point the tuberculomas erode the pia mater, and the mycobacteria enter the CSF, producing a hypersensitivity reaction that results in a purulent exudate involving the basal meninges, cerebrum, and spinal nerves. Cerebral ischemia and infarction occur from vasculitis. Symptoms include headache, low-grade fever, nausea and vomiting, irritability, difficulty sleeping, and fatigue. These signs and symptoms increase to confusion, stiff neck, significant behavioral changes, and seizures. Hydrocephalus and cranial nerve palsies or cerebral infarcts may occur. Recovery rate is 90% with early diagnosis and treatment with appropriate antituberculosis therapy.
PATHOPHYSIOLOGY The bacteria that commonly cause bacterial meningitis are common inhabitants of the nasopharynx, but a predisposing factor such as a prior upper respiratory infection must be present before the bacteria become blood-borne. Bacterial meningitis also may develop as a consequence of ear, dental, or paraspinal infections; impairment in the anatomic barrier from trauma or neurosurgery; and, rarely, when a brain abscess ruptures into the ventricular system or subarachnoid space.67 The method of CNS entry is through the choroid plexuses or areas of altered blood-brain barrier or by hematogenous spread. Bacteria multiply in the subarachnoid space. The bacteria or their toxins function as irritants and induce an inflammatory reaction by the meninges (pia mater and arachnoid), the CSF, and the ventricles. The meningeal vessels undergo change, becoming hyperemic and increasingly permeable. Blood cells (neutrophils) migrate into the subarachnoid space, producing an exudate that thickens the CSF and interferes with normal CSF flow around the brain and spinal cord (Figure 17-35). The exudate has the potential to obstruct arachnoid villi and produce hydrocephalus and interstitial edema. The amount of purulent exudate increases rapidly (especially around the base of the brain), causing further inflammation. The exudate extends into the sheaths of the cranial and spinal nerves and into the perivascular spaces of the cortex. Meningeal cells become edematous. The exudate and vasogenic edema increase ICP. The small and medium-sized subarachnoid arteries, veins, and choroid plexuses undergo inflammatory changes and become engorged, disrupting blood flow and potentially producing thrombosis. Secondary infection of the brain may occur. The cortical neurons also show some changes, including an increase in the number of microglia and astrocytes.
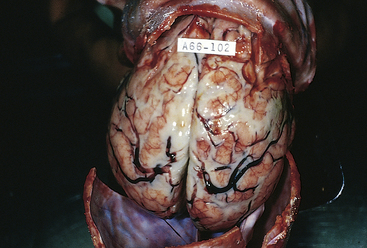
Figure 17-35 Acute leptomeningitis. The leptomeninges contain abundant creamy, purulent exudate, most prominently over the superior surface of the cerebrum. The underlying brain is swollen, and the vessels are congested. (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, Philadelphia, 2003, Saunders.)
Fungi in the nervous system usually produce a granulomatous reaction with formations of granulomas or gelatinous masses. These usually develop in the meninges at the base of the brain. Fungi also may extend along the perivascular sites in the subarachnoid space and into the brain tissue, producing arteritis with thrombosis, infarction, and communicating hydrocephalus. Meningeal fibrosis develops later in the inflammatory process. Cranial nerve dysfunction, caused by compression, often results from the granulomas and fibrosis.
CLINICAL MANIFESTATIONS The clinical manifestations of a bacterial meningitis can be grouped as (1) inflammation and irritation—generalized meningeal signs, throbbing headache that becomes more severe, photophobia that becomes more severe, nuchal rigidity, Kernig sign, and Brudzinski sign; (2) local tissue dysfunction—cranial nerve palsies, focal neurologic deficits (such as hemiparesis/hemiplegia, ataxia), and seizures; (3) mass effect—decreased level of consciousness, nausea, vomiting, and increased intracranial pressure; and (4) vascular compromise. The irritation and damage to the cranial nerves produced by the inflamed sheaths manifest as follows:
Cranial nerve II: papilledema, blindness
Cranial nerves III, IV, and VI: ptosis, visual field deficits, diplopia
Neck stiffness and pain, and possibly head retraction, reflect the irritability of spinal accessory and cervical spinal nerves. Often the vomiting center is irritated, causing projectile vomiting. Confusion and decreasing responsiveness are evidence of cortical involvement. In meningococcal meningitis, petechial or purpuric rash involving the skin and mucous membranes occurs. As ICP increases, papilledema may develop and delirium may progress to the point that the individual becomes unconscious.
The signs and symptoms of bacterial meningitis in neonates are subtle and nonspecific. Low-grade fever and mild behavioral changes with few meningeal signs may be the first signs. High fever, lethargy, irritability, hypothermia, seizures, bulging fontanels, poor feeding, vomiting, and respiratory distress may be present. Children may present with either a subacute infection that worsens over several days following an ear infection or upper respiratory infection, or as an acute fulminant illness that has developed rapidly over a few hours. Older adults often develop low-grade fever with confusion or other mild behavioral changes. Stupor or coma may appear later.
The clinical manifestations of aseptic meningitis are mild compared with those associated with bacterial meningitis. Mild generalized throbbing headache, mild photophobia, mild neck pain, stiffness, fever, and malaise are manifestations of aseptic meningitis.
Fungal meningitis develops slowly and insidiously. The first manifestations are often those of dementia or communicating hydrocephalus (see Chapter 16). The individual is characteristically afebrile.
EVALUATION AND TREATMENT Diagnosis of bacterial meningitis is based on physical examination, including skin rash, nasopharyngeal smear, and antigen tests. CSF is the gold standard for diagnosis. Bacterial meningitis and fungal meningitis are treated with appropriate antibiotic therapy, but resistant strains are an increasing problem. Other supportive measures may be needed. Aseptic meningitis is managed pharmacologically with antiviral drugs and steroids. Conjugate vaccines exist for meningococcal, pneumococcal, and hemophilic meningitis.69 Chemoprophylaxis for exposure to meningococcal meningitis is rifampin, ciprofloxacin, or ceftriaxone.70
Suppurative Cerebral Masses
Localized pus-filled masses can develop with the CNS. The mass may be an abscess within the brain or spinal cord, a subdural empyema with the infection between the dura and the subarachnoid space, or an epidural abscess with the infection between the dura and skull or vertebrae. The latter two occur less commonly but have similar predisposing conditions, infectious agents, and clinical manifestations. Treatments are also similar. Subdural empyemas and epidural abscess generally spread locally from infections in an adjoining structure.
Abscess: Abscesses are localized collections of pus within the parenchyma of the brain and spinal cord. The incidence of abscesses is about 1 per 100,000 hospital admissions. Men experience abscesses more frequently than women, with a 2:1 ratio. The median age for abscess formation is 30 to 40 years of age. Abscesses occur (1) after open trauma and during neurosurgery; (2) in association with a contiguous focus of infection, such as the middle ear, mastoid cells, nasal cavity, and nasal sinuses; (3) through metastatic or hematogenous spread from distant foci, such as the heart, lungs, pelvic organs, skin, tonsils, abscessed teeth, osteomyelitis in other than cranial bones, and dirty needles (especially in compromised hosts); and (4) cryptogenically, arising without other associated areas of infections. Streptococci, staphylococci, and bacteroids, often in combination with anaerobes, are the most common bacteria that cause abscesses; however, yeast and fungi also have been found in CNS abscesses. Toxoplasma gondii is producing an ever-increasing number of CNS abscesses in individuals with AIDS: 80% are located in the cerebrum and 20% are cerebellar. The frontal and temporal lobes are the most common sites (Figure 17-36). The abscesses are in more than one site in 5% to 20% of cases. The immunosuppressed are particularly at risk for abscesses.
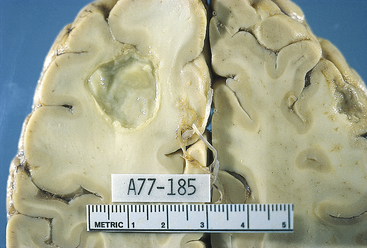
Figure 17-36 Brain abscess. The abcess is sharply demarcated, indicating that it has been present for some time. Purulent exudate is visible in the center of the abscess. Because antibiotics penetrate very poorly into abscesses, surgical drainage is often necessary to treat such lesions. (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, Philadelphia, 2003, Saunders.)
Spinal cord abscesses are classified as epidural or intramedullary. Debilitated individuals with sepsis more frequently develop intramedullary spinal cord abscesses (those within the spinal cord).
PATHOPHYSIOLOGY Microorganisms gain entrance to the CNS from adjacent sites by direct extension from osteomyelitis or spread along the wall of a vein. Infective emboli carry the microorganisms from distant sites. Brain abscess evolves through four stages regardless of infecting microorganism except in the immunosuppressed host, where the process may be incomplete. The stages are as follows:
1. Early cerebritis (days 1 to 3): localized inflammatory process in which perivascular infiltration or inflammatory cells, composed of neutrophils, plasma cells, and mononuclear cells, surround a central core of coagulative necrosis; marked cerebral edema surrounds the area
2. Late cerebritis (days 4 to 9): necrotic center is surrounded by inflammatory infiltrate of macrophages and fibroblasts; rapid new blood vessel formation occurs around the abscess; a thin capsule of fibroblasts and reticular fibers gradually develops; the area is still surrounded by cerebral edema
3. Early capsule formation (days 10 to 13): necrotic center decreases in size; inflammatory infiltrate changes in character and contains an increasing number of fibroblasts and macrophages; mature collagen evolves forming a capsule
4. Late capsule formation (days 14 and longer): well-formed necrotic center surrounded by a dense collagenous capsule67
A free (nonencapsulated) abscess is associated with a higher mortality. A mature abscess has three layers: (1) a center of polymorphonuclear leukocytes, (2) a collagenous capsule, and (3) peripheral gliosis. Existing abscesses also tend to spread and form daughter abscesses.
Abscesses arising from the ear frequently are located in the middle or inferior temporal lobe or in the anterolateral cerebellar hemispheres. Abscesses originating from the oral and nasal area most commonly are located in the frontal and temporal lobes. Abscesses from distant foci often occur in multiple numbers in the distal portion of the middle cerebral arteries. In extradural abscesses, pus and granulation tissue accumulate in the extradural space.
CLINICAL MANIFESTATIONS Clinical manifestations of brain abscesses are associated with (1) intracranial infection, such as fever and increased sedimentation rate; or (2) an expanding intracranial mass, such as headache, nausea, vomiting, decreasing cognitive abilities, paresis, and seizures. Early clinical manifestations of brain abscesses are low-grade fever, headache, neck pain and stiffness with mild nuchal rigidity, confusion, drowsiness, sensory deficits, and communication deficits. Headache is the most common early symptom. Later clinical manifestations may include inattentiveness (distractibility), memory deficits, decreased visual acuity and narrowed visual fields, papilledema, ocular palsy, ataxia, and dementia. Symptoms depend on the location of the abscess. The development of symptoms may be very insidious, often making an abscess difficult to diagnose. Extradural brain abscesses are associated with localized pain, purulent drainage from the nasal passages or auditory canal, fever, localized tenderness, and neck stiffness; occasionally the individual experiences a focal seizure. Clinical manifestations of spinal cord abscesses have four stages: (1) spinal aching; (2) root pain, which is usually severe, accompanied by spasms of the back muscles and limited vertebral movement because of pain and spasm; (3) weakness caused by progressive cord compression; and (4) paralysis.
EVALUATION AND TREATMENT The diagnosis is suggested on the basis of clinical features and confirmed by CT. MRI is helpful when the CT scan does not show an abscess even though it is suggested by clinical features. Surgery is indicated if the diagnosis is in doubt or there are space-occupying problems. Aspiration through a burr hole or excision through craniotomy with antibiotic therapy may be used. Multiple or surgically inaccessible abscesses are treated with antibiotics, often in conjunction with steroid therapy to treat the cerebral edema. In addition, ICP may have to be managed. Because decompression is necessary, spinal cord abscesses are treated with surgical excision or aspiration. Antibiotic and support therapy also is instituted.
Encephalitis
Encephalitis is an acute febrile illness, usually of viral origin, with nervous system involvement. The most common encephalitides are caused by arthropod-borne (mosquito-borne) viruses and herpes simplex, almost exclusively herpes simplex type 1 in adults (Figure 17-37). Etiologic agents for viral encephalitis are presented in Table 17-10. Referred to as infectious viral encephalitides, encephalitis also may occur as a complication of systemic viral diseases such as poliomyelitis, rabies, or mononucleosis, or it may arise after recovery from some viral infection such as rubella or rubeola. Encephalitis also may follow vaccination with a live attenuated virus vaccine if the vaccine has an encephalitis component. Such vaccines include measles, mumps, and rubella. Typhus, trichinosis, malaria, and schistosomiasis also are associated with encephalitis. Toxoplasmosis may acutely reactivate in immunosuppressed hosts when the once-dormant parasite in cyst form disseminates in brain tissues (see p. 629).
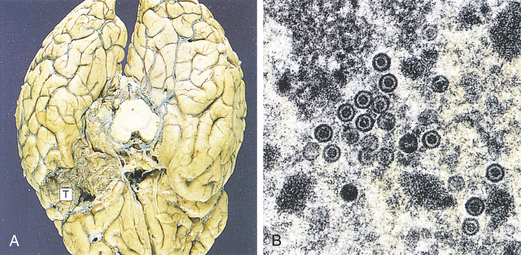
Figure 17-37 Herpes simplex encephalitis. In herpes simplex encephalitis (A) necrosis of the temporal lobes (T) is a typical development. Brain biopsy is useful in diagnosis when the virus can be seen by electron microscopy (B) as rounded particles with a dense core. Virus also can be identified by immunostaining or culture. In early cases, polymerase chain reaction (PCR) can be used to identify viral deoxyribonucleic acid (DNA) in cerebrospinal fluid samples. (From Stevens A, Lowe J: Pathology, ed 2, London, 2000, Mosby.)
With the exception of the California viral encephalitis, which is endemic, the arthropod-borne encephalitides occur in epidemics, varying in geographic and seasonal incidence (Table 17-11). Eastern equine encephalitis is the most serious but least common of the encephalitides.67 West Nile virus is presented in Box 17-5.
PATHOPHYSIOLOGY Viruses gain access to the CNS through the bloodstream or through an intraneuronal route from peripheral nerves.71 Evidence of meningeal involvement appears in all encephalitides. The arthropod-borne viral encephalitides cause widespread nerve cell degeneration. Edema and areas of necrosis with or without hemorrhage develop. Increased ICP develops and may progress to herniation. Large degenerative injuries are found in eastern equine encephalitis, whereas the other arthropod-borne viral encephalitides have microscopic areas of injury and degeneration.
Infectious encephalitis may result from a postinfectious autoimmune response to the virus or from direct invasion of the CNS by the virus. Herpes simplex type 1 has a tendency to infect the inferomedial surfaces of the temporal and frontal lobes and causes hemorrhagic necrosis.
CLINICAL MANIFESTATIONS Encephalitis may range from a mild infectious disease to a life-threatening disorder. The dramatic clinical manifestations of encephalitis are fever, delirium or confusion progressing to unconsciousness, seizure activity, cranial nerve palsies, paresis and paralysis, involuntary movement, and abnormal reflexes. Signs of marked ICP may be present.
EVALUATION AND TREATMENT Diagnosis is based on medical history and clinical presentation aided by CSF examination and culture, serologic examination, white blood cell (WBC) count, CT scan, or MRI. (Treatment available for the viral encephalitides is listed in Table 17-10.) Supportive therapy is initiated, and measures to control ICP are paramount.
Neurologic Complications of Acquired Immunodeficiency Syndrome
Approximately 40% to 60% of all persons with AIDS have neurologic complications (see Chapter 8 for the pathophysiology of AIDS). On postmortem examination, 75% have nervous system pathologic findings. The CNS pathologic findings result from (1) the primary human immunodeficiency virus (HIV) infection; (2) the immune dysregulation of early HIV infection and progressive immunosuppression in late HIV infections resulting in opportunistic infections, neoplasms, and systemic illness; and (3) complications of therapy.72
A variety of CNS complications of HIV exist (Box 17-6). Multiple CNS pathologic conditions may be experienced by one person. The most common neurologic disorder is HIV-associated dementia; others are peripheral neuropathies, vacuolar (spongy softening) myelopathy, opportunistic infections of the CNS, and neoplasms.
HIV-infected macrophages/monocytes in blood are attracted to the brain by up-regulation of proinflammatory mediators such as monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor-alpha (TNF-α), and adhesion molecules on endothelial cells. Up-regulation enables transendothelial migration of activated macrophages/monocytes.
Human Immunodeficiency Virus–Associated Dementia: HIV-associated dementia (HAD) (HIV-associated cognitive dysfunction, HIV encephalopathy, subacute encephalitis, HIV-associated dementia complex, HIV cognitive motor complex, AIDS encephalopathy, AIDS dementia complex, or AIDS-related dementia) may affect adults and children and is characterized by progressive cognitive dysfunction in conjunction with motor and behavioral alterations72 (Table 17-12). The syndrome typically develops later in the disease but may be an early or a singular manifestation. The syndrome is more prevalent in drug users with HIV.73
Table 17-12
Classification of HIV-Associated Cognitive Dysfunction
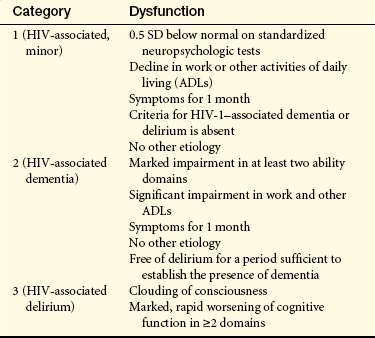
HIV, Human immunodeficiency virus; SD, standard deviation.
From Belman AL, Mirjana-Savatic M: Human immunodeficiency virus and acquired immunodeficiency syndrome. In Goetz CG, editor: Textbook of clinical neurology, ed 2, Philadelphia, 2003, Saunders.
At the time of primary HIV infection, circulating HIV-infected leukocytes (activated leukocytes), including macrophages and lymphocytes, adhere to the endothelial lining of blood vessels within the nervous system. These cells subsequently enter the brain parenchyma. HIV infects the perivascular macrophages, the microglial cells in the area, and, to a lesser degree, the astrocytes in the area.74 HIV preferentially affects the basal ganglia and deep white matter.74 Affected macrophages, macrophage-derived multinucleated cells, and microglia cause an immune-mediated demyelination process in white matter. Some viral replication occurs in some of the glial cells and occasionally within neurons. Multiple small nodules containing inflammatory cells are scattered throughout the white matter and in subcortical gray matter, such as the basal ganglia and thalami. Perivascular inflammation is present. Focal and diffuse demyelination of white matter and spongy changes of the spinal cord are present. Factors other than direct cell damage are involved, such as released toxins, lymphokines, or other substances. Elevation in CSF levels of quinolinic acid, a neurotoxic metabolite of tryptophan, is correlated with the degree of dementia. A decreased concentration of adenosine triphosphate (ATP) and inorganic phosphates in the CNS produces a decrease in metabolism.
HIV-associated dementia is insidious in onset and unpredictable in its course. Most individuals experience a steady progression of mental slowing characterized by abrupt accelerations of signs over several months to more than 1 year, although some experience an abrupt onset or an accelerated course. The triad of clinical manifestations are neurocognitive impairment, behavioral disturbance, and motor abnormalities.
Early clinical manifestations of HIV-associated dementia may be vague. Impaired concentration and short-term memory and retrieval deficits commonly occur. Apathy and lack of motivation, social withdrawal, irritability, and emotional lability may appear. Later, difficulties with language, spatial or temporal disorientation, and visual construction appear. Some individuals manifest an organic psychosis with agitation, inappropriate behavior, and hallucinosis.
Generalized cognitive system deficits occur later in the course of HIV-associated dementia, often accompanied by psychomotor slowing and decreased speech spontaneity and fluency. Progressive loss of balance, ataxia, spastic paraparesis or paralysis, and generalized hyperreflexia are common motor signs. Decreased writing ability, tremor, myoclonus, and seizure are less commonly seen.
Diagnosis is difficult, especially in early stages. The individuals medical history along with physical examination findings and supporting CSF, CT, and MRI data help establish the diagnosis. Specific screening tools have been developed to assist in diagnosis. Antiretroviral, protease inhibitors, reverse transcriptase inhibitors, and adjunctive agents may be effective along with supportive treatment. However, highly active antiretroviral therapy (HAART) protocols have shown little efficacy in reversing HAD because it does not target the cause.75
HIV Myelopathy: HIV myelopathy involving diffuse degeneration of the spinal cord may occur with HIV. Vacuolar myelopathy is believed to be a direct consequence of HIV. The lateral and posterior columns of the lumbar spinal cord are affected. A progressive spastic paraparesis with ataxia is the predominant clinical manifestation. Leg weakness, upper motor neuron signs, incontinence, and posterior column sensory loss may be present. Diagnosis is made on the basis of history, physical findings, and supporting data from diagnostic procedures. Vacuolar myelopathy is treated supportively and does not respond to antiretrovirals.
HIV Neuropathy: Some HIV-related peripheral neuropathies occur early, may coincide with seroconversion, are immune-mediated, and respond to standard immunotherapies. Other peripheral neuropathies primarily develop with advanced HIV infection and immunocompromised states and are facilitated by HIV replication, neurotoxicity from antiretroviral therapies, and coinfection with opportunistic pathogens.76
HIV neuropathy may have one or a combination of several presentations: a predominantly sensory neuropathy, an autonomic neuropathy, a mononeuritis multiplex, a Guillain-Barré–like syndrome, and a myopathy. The peripheral nervous system may sustain injury in HIV, manifesting as a peripheral neuropathy or radiculopathy. A progressive radiculopathy of predominantly the dorsal roots of the lumbar and sacral nerves may occur, involving severe myelin and axonal loss. HIV-associated distal symmetric polyneuropathy, a sensory neuropathy occurring late in the disease, is the most commonly occurring neuropathy with slowly progressive numbness and paresthesias and burning sensations in the feet.77
HIV has been isolated from peripheral nerves, so it is believed that the virus may directly infect nerves. Individuals experience painful, burning dysesthesias and paresthesias, typically in the extremities. Weakness and decreased or absent distal reflexes may be present. Diagnosis is established through history, physical findings, laboratory data, nerve conduction studies, EMG, and possibly biopsy. The most common myopathy is polymyositis; it may be present initially or develop later. The muscle fiber is infiltrated, initiating inflammation that leads to cellular degeneration and necrosis. The individual experiences muscle weakness of extremities with myalgia and fatigue. Steroids are used therapeutically in polymyositis.
Aseptic Viral Meningitis: Some people develop an acute aseptic meningitis at approximately the time of seroconversion. This may well represent the initial infection of the nervous system by the HIV. Symptoms include headache, fever, and meningismus. Cranial nerve involvement, especially of nerves V and VII, may appear, but the disease is self-limiting and requires only symptomatic treatment. Aseptic meningitis might occur at any point in the disease.
Opportunistic Infections: Opportunistic infections may be bacterial, fungal, protozoal, or viral in origin and produce nervous system disease. Typically bacterial infections are caused by unusual microorganisms. Cryptococcal infection is the most common fungal disorder and the third leading cause of neurologic disease with HIV. In Cryptococcus neoformans, small granulomas and cysts are found in the cerebral cortex and later may be present in deep cerebral tissues. The symptoms are vague, such as fever, headache, malaise, and meningismus. Herpes encephalitis and herpes varicella-zoster radiculitis may develop. Papovavirus (especially JC virus) in the immunocompromised person with HIV may produce a demyelinating disorder called progressive multifocal leukoencephalopathy (PML). This virus is found in 90% of healthy persons but is dormant. The virus reactivates to cause PML in 15% of HIV victims. Sensory and motor deficits, aphasia, and apraxia are common clinical manifestations. The condition is progressive.
Cytomegalovirus Infection: Cytomegalovirus encephalitis is common with AIDS but often not diagnosed while a person is alive. The encephalitis may be present as an acute illness with encephalitis features accompanied by nystagmus and cranial nerve signs. Retinitis is found in 50% of those affected.
Parasitic Infection: Toxoplasmosis is the most common opportunistic infection and occurs in one third of AIDS cases. CNS toxoplasmosis typically manifests as focal encephalitis. Toxoplasma gondii, a protozoan, is thought to reactivate from latent lesions to produce a well-demarcated necrotizing process. Inflammatory infiltrates, thrombotic lesions, and fibrinoid vascular walls are present at the necrotic edge. Marked edema is present adjacent to necrotic areas. Lesions may be multiple and exist throughout the cerebral hemispheres.
Clinical manifestations of CNS toxoplasmosis are focal but highly variable and include clumsiness to hemiplegia, aphasia, seizures, ataxia, cognitive changes, and constitutional symptoms. Fever and headache are common. Toxoplasmosis is difficult to diagnose but is treated effectively with pyrimethamine and sulfadiazine. Allergic response to sulfadiazine can be a problem, and other drugs can be substituted. Individuals with HIV may develop meningitis, encephalitis, or fungal, mycobacterial, and bacterial brain abscesses.
Central Nervous System Neoplasms: CNS neoplasms associated with HIV include primary CNS lymphoma, systemic non-Hodgkin lymphoma, and metastatic Kaposi sarcoma. The precise mechanism of lymphoproliferation is not known. Primary CNS lymphoma is a large-cell lymphoma that presents as rapidly developing and expanding multicentric intracranial mass lesions. The meninges are invaded and, possibly, the cranial nerves and spinal cord are invaded as well in systemic non-Hodgkin lymphoma. Metastasis of a Kaposi sarcoma to the CNS is uncommon.
Other Central Nervous System Complications: Individuals with HIV may develop multifocal ischemic infarctions, hemorrhagic infarctions, hemorrhage into tumors, subdural hematomas, and epidural hemorrhage. The precise mechanism of these cardiovascular complications is not yet known. Reported neurologic symptoms produced by HIV therapeutics include extrapyramidal movements, myoclonus, dysphasia, delirium, and acute myelopathy.
Lyme Disease
Lyme disease, a tick-borne spirochette bacterial infection, is a common arthropod-borne infection in the United States. It affects all age groups and involves the peripheral and central nervous systems. Lyme disease is caused by Borrelia burgdorferi introduced by tick bite, requiring about 36 hours of attachment.76 Transmission is most likely in the late summer or early fall. Infected ticks are endemic in the Midwest, western wooded and coastal areas, and the mid- to northeast Atlantic. The microorganism incubates for 3 to 32 days and then migrates to the skin, lymph nodes, and other body systems. The pathologic process progresses through three stages:
Stage I (acute localized): Within 1 month after the bite, the disease is characterized by a bull’s-eye–like (5 cm in diameter) burning centrifugally expanding erythema migrans rash followed by acute disseminated disease with general malaise, flulike symptoms (fever, muscle pain), stiff neck, and headache.
Stage II: With acute widespread dissemination of antibodies and immune complexes, cardiac and neurologic involvement predominates. About 10% of cases show cardiac signs and symptoms (palpitations, dizziness, shortness of breath, dysrhythmias, and first-degree heart block). Neurologic signs occur in 10% to 15% and include headache, chronic aseptic (lymphocytic) meningitis, Bell palsy, encephalitis, and radiculitis. Pathologically there is meningeal inflammation, perivascular inflammatory cell formation, and focal demyelination.
Stage III (chronic stage): The third stage may occur up to 2 years after the bite and involves arthritis and involvement of brain parenchyma with encephalitis, chronic neuropathy, and encephalopathy.
Treatment of choice is antibiotic therapy. Minor recurring symptoms are common in 50% of individuals.
Demyelinating Disorders
Demyelinating disorders are the result of damage to the myelin nerve sheath. They can occur in either the central (i.e., multiple sclerosis) or peripheral (i.e., Guillain-Barré syndrome) nervous system. Causes of the disorders include genetics, infections, autoimmune reactions, environmental toxins, and unknown factors.
Multiple Sclerosis
Multiple sclerosis (MS) is an autoimmune disorder diffusely involving degeneration of CNS myelin and loss of axons. The peripheral nervous system is not involved. MS and its variants are primary demyelinating disorders. In secondary disorders, CNS demyelination is caused by disorders other than multiple sclerosis.
MS is a diffuse and progressive CNS disease that affects white and gray matter.78 About 0.1% of the population is affected (400,000 persons in the United States and 2.5 million worldwide).79 Prevalence rates vary with geographic location (higher in temperate regions far above the equator) and racial groups (highest in whites, although it occurs in all races).
The onset of MS is usually between 20 and 40 years of age with a peak of age 30. Male:female ratio is about 1:2. MS is the most prevalent CNS demyelinating disorder and a leading cause of neurologic disability in early adulthood. Life expectancy is not greatly altered by MS and the disease course often extends over 30 years.80 Genetic and environmental factors and interactions are implicated in disease onset.81 Although the disorder does not exhibit a defined inheritance pattern, 15% of those with MS have an affected relative. Multiple genes affect risk of development of MS. A genetic link exists in the human leukocyte antigen (HLA) complex, a large cluster of genes responsible for many immune functions. Two genes, the interleukin [IL]-2 receptor gene (IL2RA) and IL7RA, have been identified.82 The first demyelinating event or “clinically isolated syndrome” (CIS), is a single episode of neurologic dysfunction lasting greater than 24 hours that can be a prelude to MS. Characteristic episodes include optic neuritis, solitary brainstem lesions, and transverse myelitis. MRI is used to assist diagnosis and assess prognosis although there are limitations to detecting the full extent of the lesions.83
PATHOPHYSIOLOGY MS is described as occurring when a previous viral insult to the nervous system has occurred in a genetically susceptible individual with a subsequent abnormal immune response in the CNS. The innate and adaptive immune systems are activated in the pathology of MS.84 Various mechanisms cause the irreversible tissue damage (inflammation, oligodendrocyte injury, demyelination, and axonal degeneration) that characterizes MS. These degenerative processes begin early in the course of the disease and continue to progress throughout a person’s life.80 Early inflammation and demyelination lead to irreversible axonal degeneration and scarring or sclerosis. Demyelinated axons are more fragile and susceptible to further damage, and when degeneration exceeds self-repair ability (remyelination), permanent disability results.85
Myelin destruction and axonal damage begin prior to symptom onset (early inflammatory demyelination). As the disease progresses, inflammatory changes in the CNS increase, and loss of brain volume progresses more rapidly.86 Box 17-7 describes the clinical courses of MS in relation to disease progression.
The immunopathology of MS involves:
1. CD8+ T-cell activation (autoreactive), cells cross the blood-brain barrier and enter CNS and attack myelin
2. IL-12 and IL-23 (proinflammatory cytokines)
3. Lack of IL-10 (an anti-inflammatory cytokine)
4. IL-17 (proinflammatory cytokine) production by T cells
5. Integrins expressed to facilitate adherence and passage of immune cells into the CNS
6. Chemokines promote the migration of immune cells and are up-regulated in MS
7. B lymphocytes and plasma cells contribute to the inflammatory response and directly damage myelin and axons; B cells are more active in chronic or progressive forms of MS (B cells produce autoantibodies, secrete inflammatory cytokines, and activate T cells by presenting antigen)87
8. Complement activation (promotes inflammation) during the acute phase; may be neuroprotective during relapse88
In addition, eosinophils, neutrophils, and macrophages are present. The demyelination disrupts sodium, calcium, and potassium ion channels and calcium influx is proinflammatory and neurotoxic in itself. Activated micro-glia and macrophages release nitric oxide and oxygen-free radicals. Activated immune cells also produce glutamate, a neurotoxin.89
MS is characterized not only by focal inflammatory changes but also diffuse injury throughout the CNS (MS lesions) (Figures 17-38 and 17-39). Four MS lesion pathologic patterns have been described (Box 17-8). MS lesions may occur anywhere in white or gray matter. In addition, other neurodegenerative processes that involve the entire CNS are taking place, including (1) changes in gray matter in the cortex, basal ganglia, brainstem, and spinal cord with substantial loss over time; (2) brain atrophy that begins early in the disease and is highly correlated with disability and progressive MS; and (3) direct dysfunction of or damage to oligodendrocytes that manufacture myelin.80,90 Normal-appearing white matter also is highly abnormal microscopically with the presence of wallerian degeneration, diffuse inflammation, extensive microglial activation, neurotoxic substances, and gene activation that disrupt cellular processes.80 In established disease the multifocal, multistaged feature of MS lesions gives rise to the aphorism that the lesions are “scattered in space and time.” Symptoms therefore are multiple and variable.
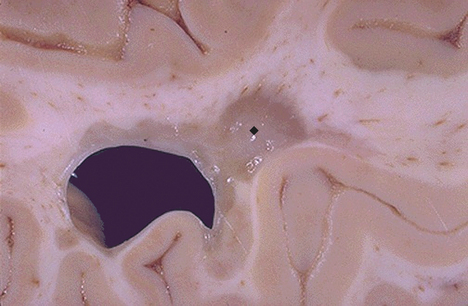
Figure 17-38 Multiple sclerosis, gross. Seen here in periventricular white matter is a large “plaque” ( ) of demyelination that has a sharp border with adjacent normal white matter. Such plaques have a gray-tan appearance and are typically associated with the clinical appearance of transient or progressive loss of neurologic function in multiple sclerosis (MS). Because MS is often multifocal, and the lesions appear in various white matter locations in the central nervous sytem over time, the clinical course and findings can be quite varied. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
) of demyelination that has a sharp border with adjacent normal white matter. Such plaques have a gray-tan appearance and are typically associated with the clinical appearance of transient or progressive loss of neurologic function in multiple sclerosis (MS). Because MS is often multifocal, and the lesions appear in various white matter locations in the central nervous sytem over time, the clinical course and findings can be quite varied. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
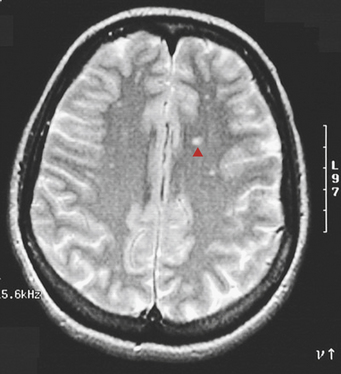
Figure 17-39 Multiple sclerosis (MS), MRI. This T2-weighted magnetic resonance image in axial view shows multiple bilateral small bright foci ( ) that represents areas of demyelinating plaque formation in an individual with an exacerbation of MS. White matter anywhere within the brain and spinal cord can be involved. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
) that represents areas of demyelinating plaque formation in an individual with an exacerbation of MS. White matter anywhere within the brain and spinal cord can be involved. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
CLINICAL MANIFESTATIONS A variety of events (e.g., infection, trauma, or pregnancy) occurring immediately before the onset or exacerbation of symptoms are regarded as precipitating factors related to MS. Most of the pregnancy-related exacerbations occur 3 months postpartum, suggesting a relation to the stresses of labor and the increased fatigue during the postpartum period rather than to the pregnancy itself.
The major classifications of MS are relapsing-remitting, primary progressive, secondary progressive, and progressive-relapsing (see Box 17-7). Initially 90% of persons present with a relapsing, remitting course; 10% present with a primary progressive course; and 90% develop a progressive course in 10 to 20 years after onset of the disease. However, once walking problems develop, disease progression occurs quickly regardless of disease type. Usually persons with late MS have one of the established syndromes—mixed, spinal, or cerebellar (Box 17-9). The initial syndrome depends on the portion of the CNS that is most involved. After years, 50% of individuals appear to have established syndromes of mixed involvement.
Mixed (General) Type: Twenty-five percent of persons initially experience retrobulbar or optic neuritis, the manifestations of optic nerve axonal loss.91 The condition usually evolves rapidly over hours to days and is highly suggestive of MS. Involvement may be unilateral or bilateral. Subjective symptoms are impaired central vision (blurring, fogginess, haziness) and impaired color perception. Signs are decreased central visual acuity; central or paracentral scotoma (area of diminished vision); acquired color vision deficit, especially to red and green; and defective pupillary reaction to light. A variety of field defects may occur. In the acute phase these symptoms may reflect optic papillitis (inflammation and swelling of the optic disc) or retrobulbar neuritis with a normal disc. One third of persons recover completely, and most others improve significantly. Later, pallor of the temporal half of the disc occurs from demyelination of a portion of the optic nerve. (Normal visual function is discussed in Chapter 15.)
The brainstem lesions involve cranial nerves III through XII at the root, nuclear, or corticobulbar (upper motor neuron) level. Internuclear ophthalmoplegia, nystagmus, and dysarthria are the most common brainstem symptoms, followed by deafness, vertigo and vomiting, tinnitus, facial weakness, and facial sensory deficit. Internuclear ophthalmoplegia is lateral gaze paralysis caused by involvement of the medial longitudinal fasciculus, the brainstem pathway that coordinates eye movement. Diplopia and eyeball pain are common complaints. Bilateral internuclear ophthalmoplegia in a young adult is virtually diagnostic of MS.
Cognitive dysfunction has been demonstrated to occur early in the disease course. The person experiences decreased short-term memory, recent memory impairment, decreased concentration, word-finding problems, and planning difficulties. Mood alterations are common in MS. Depression is far more common than euphoria.
Spinal Type: The spinal type of MS is the second most common type, chiefly involving the spinal tracts and dorsal column. Weakness, numbness, or both in one or more limbs are initial symptoms in 50% of cases of MS. Subjective corticospinal (upper motor neuron) symptoms (stiffness, slowness, weakness) are often unilateral and are a component of fatigability. Spinal signs are usually bilateral (symmetric), with lower limbs more often and more severely affected than upper limbs; spastic paraparesis is probably the most common single neurologic finding in MS.
Bladder and bowel symptoms occur with major spinal cord involvement. Urgency and hesitancy generally precede incontinence. Bladder dysfunction most often involves a small, spastic bladder, although occasionally a large, flaccid bladder may result with retention problems. Neurogenic impotence often occurs when sphincter symptoms are present. Bowel incontinence is rare, but constipation is common with severe disease. Subjective dorsal column symptoms are symmetric paresthesias (tingling and numbness) in an unpredictable pattern but with a predilection for lower extremities over upper extremities. Dorsal column signs are vibration, position, and two-point discrimination deficits. Sensory complaints often are not substantiated by objective physical findings but by further diagnostic tests.
Cerebellar Type: A nystagmus and ataxia presentation initially is not uncommon and reflects cerebellar and corticospinal involvement. Cerebellar deficits are usually symmetric, with all four limbs involved. With combined corticospinal and cerebellar involvement, the individual has a spastic ataxic gait and ataxia of the arms. Pure cerebellar symptoms are those of motor ataxia, hypotonia, and asthenia (weakness). Manifestations of motor ataxia are decomposition of movement, inability to perform rapid alternating movements (dysdiadochokinesia), and dysmetria. Charcot triad describes a combination of dysarthria, intention tremor, and nystagmus. Hypotonia is manifested by decreased resistance to passive movement, hypoactive deep tendon reflexes, and pendular knee jerk.
Short-lived attacks of neurologic deficits are the temporary appearance or worsening of symptoms. The mechanism of these attacks is complete reversible conduction block in partially demyelinated axons. Conditions that cause short-lived attacks include (1) minor increases in body temperature or serum Ca++ concentration and (2) functional demands exceeding conduction capacity. An increase in body temperature or serum Ca++ level increases current leakage through demyelinated neurons. Individuals with MS may become dramatically worse when body temperature is raised. Hypercalcemia induced by decreased serum pH may aggravate symptoms of MS. Physical and emotional stress imposes functional demands that may exceed conduction capacity of affected neurons.
Paroxysmal attacks are sensory or motor symptoms of abrupt onset and short duration (a few seconds or minutes). These symptoms include paresthesias, dysarthria and ataxia, and tonic head turning. The mechanism of paroxysmal attacks is nonsynaptic transmission in which nerve impulses are directly transmitted between adjacent demyelinated axons. These impulses arise focally and spuriously in the cervical portion of the spinal cord or in the brainstem. A common paroxysmal symptom, called Lhermitte sign, is the momentary paresthesia (shocklike or tingling sensation) that shoots down the trunk or limbs during active or passive flexion of the neck. Bending the neck evokes nonsynaptic impulses in demyelinated axons of the dorsal column in the spinal cord. A person with MS may have many paroxysmal attacks each day. Inciting events include sensory stimulation, voluntary movement, hyperventilation, and emotional stress. Paroxysmal attacks tend to persist for weeks or months and may be followed by progressive symptoms of MS.
EVALUATION AND TREATMENT The diagnostic criteria for MS were revised in 2001 and are known as the McDonald criteria. Clinical examination in combination with MRI, to demonstrate MS lesions in time and space, and CSF findings are used to make the diagnosis earlier (Table 17-13) so that treatment may begin sooner. Persistently elevated CSF immunoglobulin G (IgG) index is found in about two thirds of individuals with MS, and oligoclonal bands of IgG on electrophoresis are found in more than 90% of persons. Evoked response (ER) studies aid diagnosis by detecting decreased conduction velocity in visual, auditory, and somatosensory pathways.
Table 17-13
Diagnostic Tests for Multiple Sclerosis
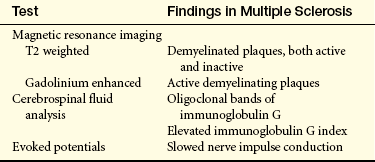
From Chisholm-Burns MA, et al: Pharmacotherapy principles and practices, p 434, New York, 2008, McGraw-Hill.
The treatment goal in MS is prevention of permanent neurologic damage. Acute relapses are treated with corticosteroids to speed recovery.92 Evidence supports that corticosteroids may improve symptoms in acute relapse situations.93 Disease-modifying drugs are used to decrease the number of relapses, prevent permanent CNS damage, and prevent disability.94 Evidence supports that in relapsing-remitting multiple sclerosis (RRMS) glatiramer acetate, intravenous (IV) immunoglobulins, and azathioprine or other drug combinations may reduce relapses.95 Interferon-beta and mitoxantrone may reduce relapses and disease progression but have serious adverse effects.2 Treatments are in development to prevent demyelination, promote remyelination and repair, and suppress selective B-cell and T-cell function.96,97
Symptom management for fatigue, weakness, vertigo, ataxia, tremor, heat intolerance, spasticity, bladder dysfunction, bowel dysfunction, sexual dysfunction, sensory sensations, pain, cognitive difficulties, depression, and psychosocial issues is essential but many treatments are only partially effective, particularly for fatigue and spasticity.2 Supportive and rehabilitative management is directed toward preventing the complications of immobility, especially pressure sores and infections of the pulmonary and genitourinary systems. Interdisciplinary inpatient rehabilitation may improve function in the short term.98
Neurodegenerative Disorders
Amyotrophic lateral sclerosis (ALS) (sporadic motor system disease, sporadic motor neuron disease, motor neuron disease) is a worldwide degenerative disorder diffusely involving lower and upper motor neurons resulting in progressive muscle weakness leading to respiratory failure and death, usually 2 to 5 years from symptom onset.99,100 There are no racial, ethnic, or socioeconomic boundaries. The prevalence rate is about 1 to 3 cases per 100,000,101 with 2 deaths per 100,000, and 5000 newly diagnosed cases per year in the United States. The term amyotrophic (without muscle nutrition or progressive muscle wasting) refers to the predominant lower motor neuron component of the syndrome. Lateral sclerosis, or scarring of the corticospinal tract in the lateral column of the spinal cord, refers to the upper motor neuron component of the syndrome. ALS differs from other motor neuron disorders in that upper and lower motor neurons are involved.
Classic ALS (Lou Gehrig disease) may begin at any time from the fourth decade of life; its peak occurrence is in the early 50s. Male/female ratio is 1:4 to 2:5,100 equalizing after menopause. In familial ALS, mutations have been found in the superoxide dismutase (SOD1) gene on chromosome 21.102 The defective SOD1 gene leads to an autosomal dominant pattern with age-dependent penetrance and accounts for 15% to 20% of the cases of familial ALS. The ALS2 gene is mapped to chromosome 2q33 and leads to a rare, recessively inherited ALS associated with deficiency of the gene product alsin and a juvenile form of ALS. The ALS4 gene (senataxin) is linked to locus 9q34 and produces a rare juvenile form of familial ALS inherited in an autosomal dominant pattern. Several genes also alter the risk of developing sporadic ALS.103
ALS presentations include crural ALS, proximal or shoulder girdle ALS, and hemiplegic (Mills) ALS. Of persons with ALS, 20% have a benign form of the disease.
PATHOPHYSIOLOGY The pathogenesis of ALS is not clear. The molecular dysfunction caused by the SOD1 is under study. Apoptotic factors, abnormal synthesis of filament units, defects in axonal transport, excitotoxicity and glutamate transports, oxidative stress, growth factors, mitochondrial dysfunction, and neuroinflammation are likewise under study for a possible contribution to the pathogenesis.
The principal pathologic feature of ALS is lower and upper motor neuron degeneration. The number of large motor neurons in the spinal cord, brainstem, and cerebral cortex (premotor and motor areas) is reduced, with ongoing degeneration in the remaining motor neurons. The nuclei of cranial nerves III, IV, and VI are not involved. Death of the motor neuron results in axonal degeneration and secondary demyelination with glial proliferation and sclerosis (scarring) along the corticospinal tract. Inclusion bodies containing the protein ubiquitin are found in surviving neurons. However, there also is widespread neural degeneration of nonmotor neurons in the spinal cord and motor cortices, as well as in the premotor, sensory, and temporal cortices.100 Altered astrocytes and microglial functions are suspected to exist.
Lower motor neuron degeneration denervates motor units. Adjacent, still-viable lower motor neurons attempt to compensate by a process of distal intramuscular sprouting, reinnervation, and enlargement of motor units. The initial symptoms of the disease may be related to lower or upper motor neuron dysfunction or both. Fifty percent of persons with ALS present with hand weakness or incoordination, dysarthria, or leg weakness or incoordination.104
CLINICAL MANIFESTATIONS Weakness may begin in any or all muscles of the body. Muscle weakness in ALS exhibits the following characteristics:
1. Paresis usually begins in a single muscle group.
2. Corresponding muscle groups are asymmetrically affected in a mottled distribution.
3. Gradual involvement occurs in all striated muscles except extraocular muscles and heart and progresses to paralysis with no remissions.
4. Flaccid and spastic paresis may coexist in a single muscle group; flaccid paresis may mask spasticity, which is usually mild.
The lower motor neuron syndrome of flaccid paresis consists of weakness of individual muscles, progressing to paralysis, associated with hypotonia and primary muscle atrophy (i.e., atrophy caused by denervation). Hypotonia is manifested by (1) decreased resistance to passive movement, (2) hypoactive or absent deep tendon reflexes, (3) absent abdominal and cremasteric reflexes, and (4) absent Babinski sign. Primary atrophy is manifested by (1) severe, irreversible muscular wasting; (2) fasciculations; (3) metabolically related changes in the skin and appendages; and (4) specific EMG findings. Fasciculations, along with fibrillations, are prominent features of ALS. Metabolic changes include (1) thinning of the skin, (2) thickening of the nails, (3) loss of body hair, and (4) decreased perspiration.
The upper motor neuron syndrome of spastic paresis consists of weakness of movement patterns, progressing to paralysis, associated with spasticity and, in some cases, atrophy secondary to disuse. Spasticity is manifested by (1) clasp-knife phenomenon, evident with passive movement; (2) hyperactive deep tendon reflexes and clonus with severe spasticity; (3) absent abdominal and cremasteric reflexes; and (4) presence of Babinski sign. The coexistence of a dementia has been demonstrated to be higher than previously thought.
EVALUATION AND TREATMENT The diagnosis of the syndrome is based predominantly on medical history and physical examination.105 EMG and muscle biopsy verify lower motor neuron degeneration and denervation. Muscle biopsy usually is not needed to confirm the diagnosis. Riluzole (Rilutek), an antiglutamate, is the standard treatment for ALS, decreasing the risk of death by 35%.100 Treatment is also directed at symptom relief, prevention of complications, maintenance of maximal function, and maintenance of optimal quality of life.104 Special problems requiring preventive and symptomatic management are communication difficulty caused by dysmasesis and dysphonia, salivation problems with either thick saliva or excessively thin saliva (sialorrhea), and dyspnea caused by diaphragmatic and intercostal weakness. Ventilatory issues become prominent. Supportive and rehabilitation management is directed toward preventing complications of immobility. Psychologic support of the affected individual and the family is extremely important in this disorder.