ALTERATIONS IN COGNITIVE SYSTEMS, CEREBRAL HEMODYNAMICS, AND MOTOR FUNCTION


A person achieves functional adequacy (competence) through complex integrated processes. Three major neural systems account for this functional adequacy: cognitive systems, sensory systems, and motor systems. Alterations in any or all of these affect functional adequacy. Alterations in cognitive and sensory systems and motor function are associated with many central and peripheral nervous system injuries and pathologies. The purpose of this chapter is to present the concepts and processes of these alterations as an approach to understanding the manifestation of neurologic dysfunction. Some specific diseases are also presented (i.e., Parkinson and Huntington disease) because they fit best here. The manifestations of these concepts and processes are integrated with specific central and peripheral nervous system disorders and are presented in Chapter 17. Alterations in sensory function are presented in Chapter 15.
The neural systems essential to the cognitive sphere are (1) attentional systems that provide arousal and maintenance of attention over time; and (2) memory and language systems by which information is communicated. These core systems are fundamental to the processes of abstract thinking and reasoning. The products of abstraction and reasoning are organized and made operational through the executive system. The normal functioning of these systems manifests through the motor system in a behavioral array viewed by others as being appropriate to human activity and successful living.
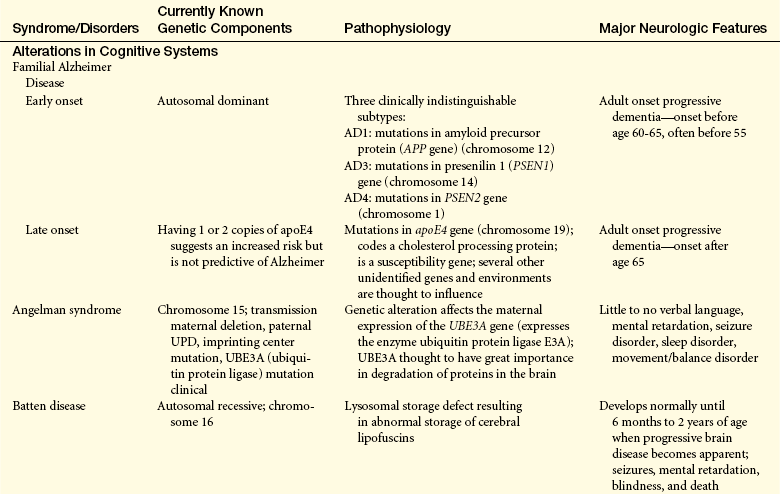
Genetics and the genetic basis of disease are becoming increasingly important in the study of pathophysiology; selected neurologic disorders that have a genetic basis are presented in Table 16-1.



ALTERATIONS IN COGNITIVE SYSTEMS
Full consciousness, in its broadest sense, is a state of awareness both of oneself and the environment and a set of responses to that environment. Full consciousness implies that the individual responds to external stimuli with a wide array of responses. Any decrease in this state of awareness and varied responses is thus a decrease in consciousness.
Consciousness often is viewed as having two distinct components: arousal and awareness. Arousal, an attentional system, is the state of awakeness that an individual exhibits. Level of arousal is mediated by the reticular activating system, which extends from the medulla to the diencephalon. The reticular activating system provides arousal to the cerebral hemispheres (see Figure 14-6). Severe alterations in arousal can occur with brain injury, both in the acute phase of injury and on a long-term basis. Approximately 30% to 40% of survivors of severe brain injury remain in prolonged states of severely reduced consciousness. Awareness encompasses all cognitive functions that embody awareness of self, environment, and affective states (i.e., moods). Content of thought is mediated by attentional systems, memory systems, language systems, and executive systems.
Coma
Possible causes of an altered level of arousal with acute onset may be separated into three major groups: structural, metabolic, and psychogenic arousal alterations. Structural causes are divided according to original location of the pathologic condition or lesion: supratentorial (above the tentorium cerebelli), infratentorial (subtentorial, below the tentorium cerebelli), subdural (below the dura mater), extracerebral (outside the brain tissue), and intracerebral (within the brain tissue). Metabolic causes may be further divided into interruption in delivery of energy substrates (hypoglycemia, ischemia, hypoxia) or alteration in neuronal excitability (drug and alcohol intoxication, anesthesia, and epilepsy).1 All the systemic diseases that eventually produce nervous system dysfunction are part of this metabolic category. Causes of altered level of arousal also are grouped according to pathologic process: infectious, vascular, neoplastic, traumatic, congenital (developmental), degenerative, polygenic, and metabolic.
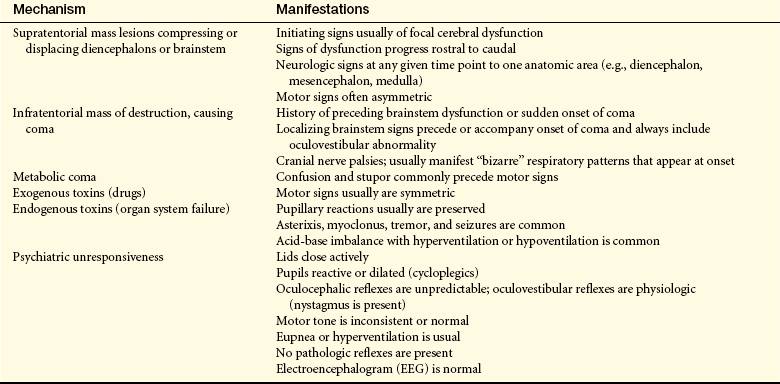
PATHOPHYSIOLOGY Coma is produced by either (1) bilateral hemisphere damage or suppression by means of metabolic derangement, such as hypoxia, hypoglycemia, uremia, or toxins, such as ammonia from liver failure; or (2) a brainstem lesion or metabolic derangement that damages or suppresses the reticular activating system (RAS/thalamocortical alerting system) or its projections.2,3 Supratentorial disorders produce a decreased level of arousal by one of three mechanisms: (1) diffuse bilateral cortical dysfunction, (2) bilateral subcortical dysfunction, or (3) localized hemispheric dysfunction. Disease processes may produce diffuse bilateral cortical dysfunction (e.g., encephalitis) and actually may occur in either the cerebral cortex or the underlying subcortical white matter. Bilateral subcortical dysfunction involves destructive disease that compromises the RAS (e.g., brainstem trauma or cerebrovascular accident) and probably surrounding structures as well. Localized hemispheric dysfunction generally is caused by masses that directly impinge on deep diencephalic structures or that secondarily compress these structures in the process of herniation. Such localized destructive processes directly impair function of the thalamic or hypothalamic activating systems.
Extracerebral disorders also can produce diffuse bilateral cortical dysfunction. Extracerebral disorders include neoplasms, closed-head trauma with subsequent bleeding, and subdural empyema (accumulation of pus). Intracerebral disorders (those within the brain substance) function primarily as masses. These disorders include bleeding, infarcts and emboli, and tumors.
Infratentorial disorders produce a reduction in arousal in one of two ways: (1) there may be direct destruction of the RAS and its pathways, or (2) the brainstem may be destroyed either by direct invasion or by indirect impairment of its blood supply. The most common cause of direct destruction is cerebrovascular disease, but demyelinating diseases, neoplasms, granulomas, abscesses, and head injury also may cause brainstem destruction. In addition, decreased level of consciousness may result from compression of the RAS by a disease process. This compression may occur because of (1) direct pressure on the pons and midbrain, producing ischemia and edema of the neurons of the RAS; (2) upward herniation of the cerebellum through the tentorial notch, thus compressing the upper midbrain and diencephalon; or (3) downward herniation of the cerebellum through the foramen magnum, compressing and displacing the medulla oblongata. Specific causes of compression of the brainstem include hematomas, hemorrhage, and aneurysm; cerebellar hemorrhage, infarcts, abscesses, and neoplasms; and demyelinating disorders.
A wide spectrum of diseases may produce a metabolically induced alteration in arousal. In encephalopathic conditions, widespread direct or indirect interference with neuronal metabolism occurs throughout much of the brain, such as occurs with liver failure (hepatic encephalopathy) and renal failure. Psychogenic unresponsiveness, although uncommon, may signal general psychiatric disorders. Despite apparent unconsciousness, the person actually is physiologically awake and the neurologic examination reflects normal response.
EVALUATION Evaluating and treating an altered level of arousal requires distinguishing between organic and functional causes. A further distinction between metabolic and structural causes is made because the treatments are different and disease progression can be rapid (Table 16-2). If the cause is structural, the pathologic condition must be localized.
Table 16-2
Clinical Manifestations of Metabolic and Structural Causes of Comas
| Manifestation | Metabolically Induced Coma | Structurally Induced Coma |
| Blink to threat (cranial nerves II, VII) | Equal | Asymmetric |
| Discs (cranial nerve II) | Flat, good pulsation | Papilledema |
| Extraocular movement (cranial nerves III, IV, VI) | Roving eye movements; normal doll’s eyes and calorics | Gaze paresis, nerve III palsy, medial longitudinal fasciculus (MLF) syndrome (internuclear ophthalmoplegia) |
| Pupils (cranial nerves II, III) | Equal and reactive, may be large (e.g., atropine), pinpoint (e.g., opiates), or midposition and fixed (e.g., glutethimide [Doriden]) | Asymmetric and/or nonreactive; may be midposition (midbrain injury), pinpoint (pons injury), large (tectal injury) |
| Corneal reflex (cranial nerve V, VII) | Symmetric response | Asymmetric response |
| Grimace to pain (cranial nerve VII) | Symmetric response | Asymmetric response |
| Motor function movement | Symmetric | Asymmetric |
| Tone | Symmetric | Paratonic, spastic, flaccid, especially if asymmetric |
| Posture | Symmetric | Decorticate, especially if symmetric; decerebrate, especially if asymmetric |
| Deep tendon reflexes | Symmetric | Asymmetric |
| Babinski sign | Absent or symmetric response | Present |
| Sensation | Symmetric | Asymmetric |
CLINICAL MANIFESTATIONS Patterns of clinical manifestations and their evolution have been identified. The patterns of clinical manifestation are important because they help in determining the extent of brain dysfunction and they serve as indexes for identifying increasing or decreasing central nervous system (CNS) function. The specific clusters of manifestations of abnormal function and their evolution suggest whether the cause of the altered arousal state is supratentorial, infratentorial, metabolic, or psychogenic (Table 16-3). Five categories of neurologic function are critical to the evaluation process: (1) level of consciousness, (2) pattern of breathing, (3) size and reactivity of pupils, (4) eye position and reflexive responses, and (5) skeletal muscle motor responses.
Level of Consciousness: Level of consciousness is the most critical clinical index of nervous system function or dysfunction. An alteration in consciousness indicates either improvement or deterioration of the individual’s condition. A person who is alert and oriented to self, others, place, and time is considered to be functioning at the highest level of consciousness, which implies full use of all the person’s cognitive capacities.
Because many different terms are used to indicate level of consciousness, definition becomes necessary. The term unconscious, for example, has no specific clinical definition and signifies different things to different people. From the normal alert state, levels of consciousness diminish in stages, each of which is clinically defined in Table 16-4.
Table 16-4
| State | Definition |
| Confusion | Loss of ability to think rapidly and clearly; impaired judgment and decision making |
| Disorientation | Beginning loss of consciousness; disorientation to time followed by disorientation to place and impaired memory; lost last is recognition of self |
| Lethargy | Limited spontaneous movement or speech; easy arousal with normal speech or touch; may not be oriented to time, place, or person |
| Obtundation | Mild to moderate reduction in arousal (awakeness) with limited response to the environment; falls asleep unless stimulated verbally or tactilely; answers questions with minimum response |
| Stupor | A condition of deep sleep or unresponsiveness from which the person may be aroused or caused to open eyes only by vigorous and repeated stimulation; response is often withdrawal or grabbing at stimulus |
| Coma | No verbal response to the external environment or to any stimuli; noxious stimuli such as deep pain or suctioning yields motor movement |
| Light coma | Associated with purposeful movement on stimulation |
| Coma | Associated with nonpurposeful movement only on stimulation |
| Deep coma | Associated with unresponsiveness or no response to any stimulus |
Pattern of Breathing: Several characteristic respiratory patterns are helpful in evaluating level of brain dysfunction and level of coma. Among these characteristics are rate, rhythm, and pattern of breathing. The breathing patterns can be categorized as hemispheric or brainstem breathing patterns (Table 16-5 and Figure 16-1).
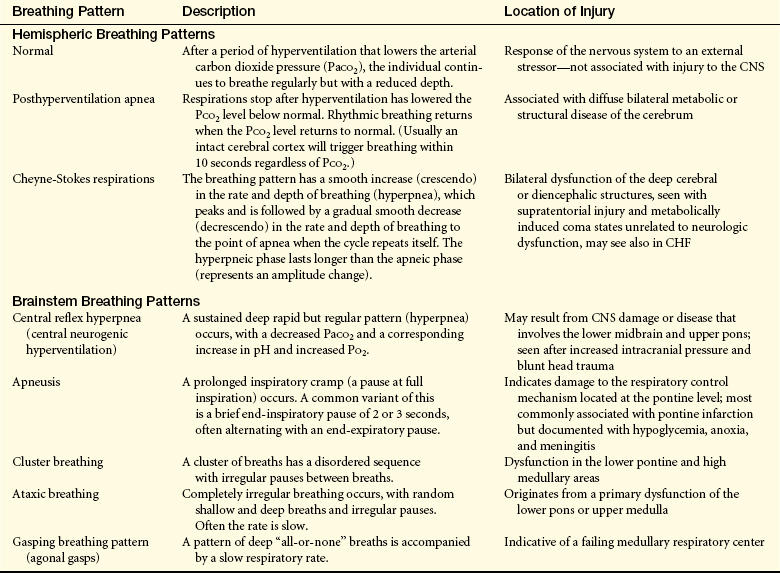
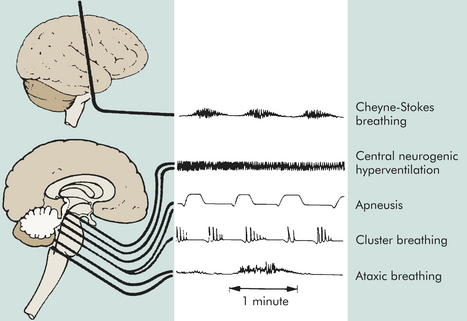
Figure 16-1 Abnormal respiratory patterns with corresponding level of central nervous system activity.
With normal breathing, a neural center believed to be located in the forebrain (cerebrum) produces a rhythmic breathing pattern despite lowered arterial carbon dioxide pressure (PaCO2). When neural control at this center is lost as consciousness decreases, the lower brainstem centers regulate the breathing pattern by responding only to changes in PaCO2 levels. The result is the irregular breathing associated with posthyperventilation apnea (PHVA).
Cheyne-Stokes respiration is an abnormal rhythm of breathing (periodic breathing) that alternates between hyperventilation and apnea. The pathophysiology of Cheyne-Stokes respiration involves a hyperventilatory response to carbon dioxide stimulation. In the damaged brain higher levels of PaCO2 (hypercapnia) are required to stimulate ventilation, and the response is hyperventilation. As a result, the PaCO2 level decreases to below normal and breathing stops (PHVA) until the carbon dioxide reaccumulates and stimulates hyperventilation. In cases of opiate or sedative drug overdose, the respiratory center is depressed and the rate of breathing gradually decreases until respiratory failure occurs.
Certain motor activities related to breathing signify the level of brain dysfunction. Yawning, vomiting, and hiccups are complex reflex-like motor responses that are integrated by neural mechanisms in the lower brainstem. These responses may be produced by compression or diseases that involve tissues in the medulla oblongata. Such disorders include infection, neoplasm, or infarct. Similar responses are produced by dysfunction in the lower brainstem through direct stimulation.
Most CNS disorders produce nausea and vomiting. Vomiting with no associated nausea indicates direct involvement of the central neural mechanism. Vomiting is associated particularly with CNS injuries that (1) involve the vestibular nuclei (located in the lower pons and medulla oblongata) or their immediate projections, particularly when double vision (diplopia) also is present; (2) impinge directly on the floor of the fourth ventricle; or (3) produce brainstem compression secondary to increased intracranial pressure.
Pupillary Changes: Anatomically, brainstem areas that control arousal are adjacent to areas that control pupils. Pupillary changes thus are a valuable guide to evaluating the presence and level of brainstem dysfunction (Figure 16-2).
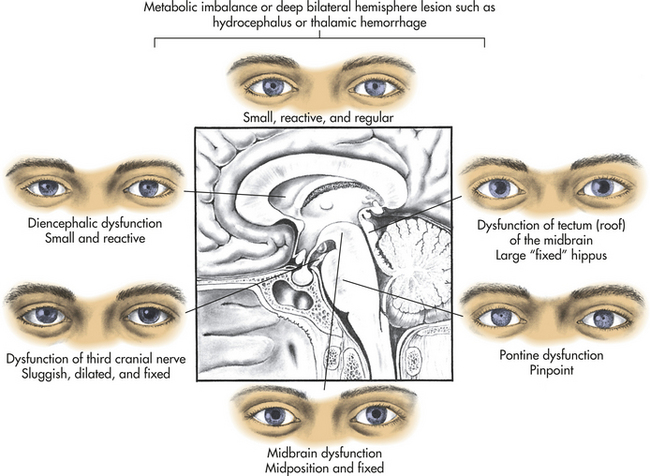
Certain drugs that affect pupils must be considered in the evaluation of pupillary response in comatose states. Atropine, scopolamine, amphetamines, mydriatics, and cycloplegics in large concentrations fully dilate and fix pupils. Glutethimide in amounts sufficient to produce a coma causes the pupils to become midposition or moderately dilated (4 to 5 mm in diameter), unequal, and frequently fixed to light. Opiates (heroin and morphine) and barbiturates, as well as extensive pontine damage, cause pinhole pupils (1 mm). Severe barbiturate intoxication may produce fixed pupils.
Severe ischemia and hypoxia produce bilaterally wide (5 mm) and fixed pupils in most instances caused by severe midbrain damage. Occasionally the pupils remain small (1 to 2.5 mm) or midposition even in the presence of profound hypoxia. Hypothermia also may cause fixed pupils.
Oculomotor Responses: Resting, spontaneous, and reflexive eye movements (oculocephalic [doll’s head, doll’s eyes] and oculovestibular [caloric] reflexes) undergo change at various levels of brain dysfunction (Table 16-6). Persons with metabolically induced coma, except in cases of barbiturate-hypnotic and phenytoin (Dilantin) poisoning, generally do retain ocular reflexes, however, even when other signs of brainstem damage, such as central neurogenic hyperventilation, are present.
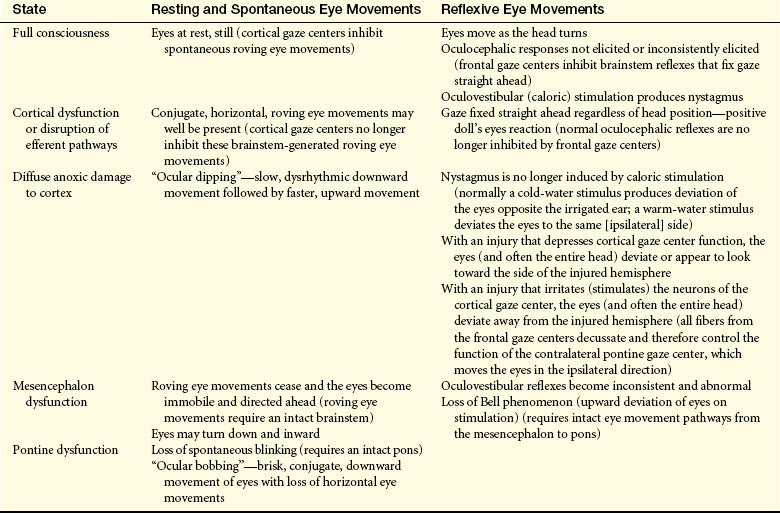
The presence of brisk oculocephalic reflexes and roving eye movements, as well as the failure to elicit nystagmus with instillation of cold or warm water into the external ear canal, indicates a decrease in consciousness (loss of cortical influence) but an intact brainstem (Figures 16-3 and 16-4).
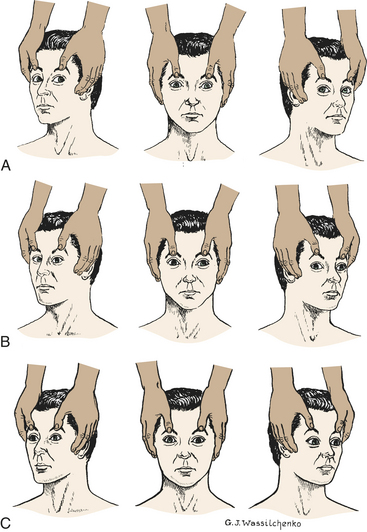
Figure 16-3 Test for oculocephalic reflex response (doll’s eyes phenomenon). A, Normal response—eyes turn together to side opposite from turn of head. B, Abnormal response—eyes do not turn in conjugate manner. C, Absent response—eyes do not turn as head position changes. (A and C from Rudy EB: Advanced neurological and neurosurgical nursing, St Louis, 1984, Mosby.)
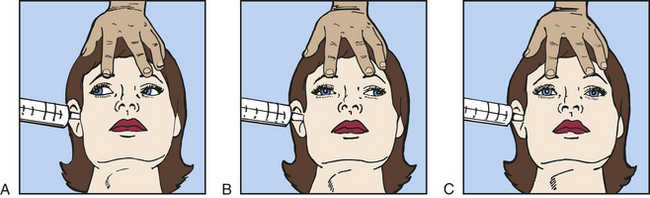
Figure 16-4 Test for oculovestibular reflex (caloric ice-water test). A, Normal response—conjugate eye movements. B, Abnormal response—dysconjugate or asymmetric eye movements. C, Absent response—no eye movements.
Destructive or compressive injury to the brainstem causes specific abnormalities of the oculocephalic and oculovestibular reflexes. For example, a skewed deviation, in which one eye diverges downward and the other looks upward, indicates brainstem dysfunction. Destructive or compressive disease processes that involve an oculomotor nucleus or nerve cause the involved eye to deviate outward, producing a resting disconjugate lateral position of the eyes (each eye diverges laterally). Unilateral abducens paralysis (paralysis of cranial nerve VI) results in an upward deviation of the ipsilateral eye. With bilateral abducens paralysis, the eyes come together (converge). Reflexive eye movements may be suppressed by drugs, most commonly phenytoin, tricyclics, and barbiturates. Occasionally alcohol, phenothiazines, and diazepam may alter reflex eye movements.
Motor Responses: Motor responses contribute to evaluating the level of brain dysfunction and determining the side of the brain that is maximally damaged. The pattern of response is described as (1) purposeful (a defensive or withdrawal movement of limbs to noxious stimuli); (2) inappropriate, or not purposeful (generalized motor movement, posturing, grimacing, or groaning); or (3) not present (unresponsive, no motor response). Purposeful movement requires an intact corticospinal system. Nonpurposeful movement is evidence of severe dysfunction of the corticospinal system.
Motor signs indicating loss of cortical inhibition that are commonly associated with decreased consciousness include contralateral or bilateral (depending on whether the process is localized or diffuse) reflex grasping, reflex sucking, snout reflex, palmomental reflex, and rigidity (paratonia) (Figure 16-5). Abnormal flexor and extensor responses in the upper and lower extremities are defined in Table 16-7 and illustrated in Figure 16-6.
Table 16-7
Abnormal Motor Responses with Decreased Responsiveness
| Motor Response | Description of Motor Responses | Location of Injury |
| Decorticate posturing/rigidity: upper extremity flexion, lower extremity extension | Slowly developing flexion of the arm, wrist, and fingers with abduction in the upper extremity and extension, internal rotation, and plantar flexion of the lower extremity | Hemispheric damage above midbrain releasing medullary and pontine reticulospinal systems |
| Decerebrate posturing/rigidity: upper and lower extremity extensor responses | Opisthotonos (hyperextension of the vertebral column) with clenching of the teeth; extension, abduction, and hyperpronation of the arms; and extension of the lower extremities | Associated with severe damage involving midbrain or upper pons |
| In acute brain injury, shivering and hyperpnea may accompany unelicited recurrent decerebrate spasms | Acute brain injury may cause limb extension regardless of location | |
| Extensor responses in the upper extremities accompanied by flexion in the lower extremities | Pons | |
| Flaccid state with little or no motor response to stimuli | Lower pons and upper medulla |
Data from Goetz CG: Textbook of clinical neurology, ed 3, Philadelphia, 2007 Saunders; Nadeau SE et al: Medical neuroscience, Philadelphia, 2004, Saunders.
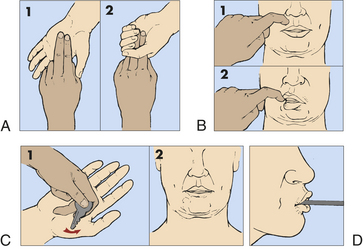
Figure 16-5 Pathologic reflexes. A, Grasp reflex. B, Snout reflex. C, Palmomental reflex. D, Suck reflex.
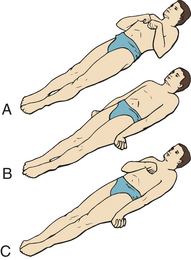
Figure 16-6 Decorticate and decerebrate responses. A, Decorticate response. Flexion of arms, wrists, and fingers with adduction in upper extremities. Extension, internal rotation, and plantar flexion in lower extremities. Both sides B, Decerebrate response. All four extremities in rigid extension, with hyperpronation of forearms and plantar extension of feet. C, Decorticate response on right side of body and decerebrate response on left side of body. (From Rudy EB: Advanced neurological and neurosurgical nursing, St Louis, 1984, Mosby.)
Outcomes: Categories of prognostic indicators related to outcome of coma include demographic variables, severity indices, neurologic signs, neuroimaging studies, neuromedical markers, psychologic ratings, and outcome scale scores.4,5 Outcome domains fall into two divisions—mortality and extent of disability (morbidity). For coma, the extent of disability division has four domains—recovery of consciousness, residual cognitive dysfunction, psychosocial (functional) domain, and vocational domain. These coma outcomes differ depending on the etiology of the injury—traumatic brain injury (TBI) or nontraumatic brain injury (NTBI). The pathophysiology underlying TBI is focal or diffuse trauma-induced injury (see Chapter 17 for further discussion). The pathophysiology of NTBIs is one of hypoxia and ischemia. This may be due, for example, to vascular insult, tumor, hydrocephalus, infection, or anorexia.
Related to mortality, two forms of neurologic death—brain death (brainstem death) and cerebral death—may result from severe TBI and NTBI. Brain death (brainstem death) occurs when irreversible brain damage is so extensive that the brain has no potential for recovery and no longer can maintain the body’s internal homeostasis. Destruction of the neuronal contents of the intracranial cavity includes the brainstem and cerebellum. On postmortem examination the brain is autolyzing (self-digesting) or already autolyzed.
Clinical criteria for brain death include the absence of discernible evidence of cerebral hemisphere function or function of the brainstem’s vital centers for an extended period. There is no detectable function above the level of the foramen magnum so there is whole brain death.6–8 In addition, the abnormality of brain function must result from structural or known metabolic disease and not be caused by a depressant drug, alcohol poisoning, neuromuscular blockage, or hypothermia. An isoelectric, or flat, electroencephalogram (EEG) (electrocerebral silence) for a period of 6 to 12 hours in a person who is not hypothermic and has not ingested depressant drugs indicates that no mental recovery is possible and usually means that the brain is already dead. A task force to determine brain death in children recommended the same criteria as for adults9 but with a longer observation period.
The following summary of medical criteria determines brain death2,3,6,10:
1. Completion of all appropriate and therapeutic procedures
2. Unresponsive coma (absence of motor and reflex movements)
3. No spontaneous respiration (apnea)—a PaCO2 that rises above 60 mmHg without breathing efforts, providing evidence of a nonfunctioning respiratory center (apnea challenge)
4. Absent cephalic reflexes (no ocular responses to head turning or caloric stimulation) with dilated, fixed pupils
5. Isoelectric (flat) EEG (electrocerebral silence)
6. Persistence of these signs for 30 minutes to 1 hour and for 6 hours after onset of coma and apnea
7. Confirming test indicating absence of cerebral circulation (optional)
Cerebral death (irreversible coma) is death of the cerebral hemispheres exclusive of the brainstem and cerebellum. Brain damage is permanent and sufficiently severe that the individual is unable to ever respond behaviorally in any significant way to the environment. The brain, however, may continue to maintain internal homeostasis (normal respiratory and cardiovascular functions, normal temperature control, and normal gastrointestinal function).
Prognosis in coma is related to the extent of disability—the recovery spectrum of neurobehavioral manifestations (in diagnostic terms, clinical states) after severe brain injury include (1) coma, (2) vegetative state, (3) akinetic mutism, (4) minimally conscious state, and (5) locked-in syndrome (Table 16-8).
Table 16-8
Comparative Clinical Features of Coma, Vegetative State, and Minimally Conscious State
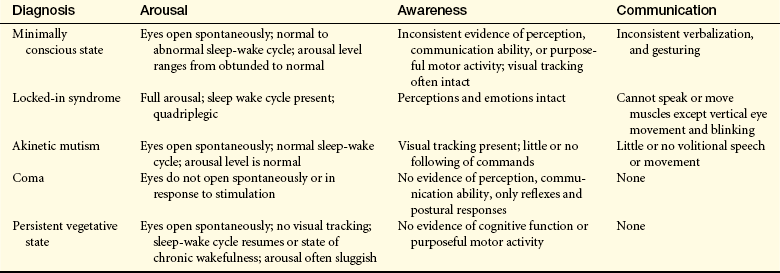
Data from Giacino JT et al: Neurology 58(3):349-353, 2002; Owen AM: Ann N Y Acad Sci 1125:225-238, 2008.
The survivor of cerebral death may remain in a coma or emerge into a vegetative state. In coma, a state of unarousable neurobehavioral unresponsiveness, the eyes are usually closed with no evidence of eye opening either spontaneously or in response to external stimuli. The person does not follow commands, does not verbalize or mouth words, and has no goal-directed or volitional behavior. There is no sustained visual pursuit movements beyond a 45-degree arc.
A vegetative state (VS) has been called a wakeful unconscious state. The Multi-Society Task Force on Persistent Vegetative States (MSTF) identified the diagnostic criteria for VS as (1) periods of eye opening (spontaneous or following stimulation); (2) the potential for subcortical responses to external stimuli, including generalized physiologic responses to pain, such as posturing, tachycardia, and diaphoresis, and subcortical motor responses, such as grasp reflex; (3) return of so-called vegetative (autonomic) functions, including sleep-wake cycles and normalization of respiratory and digestive system functions; and (4) occasional roving eye movements without concomitant visual tracking ability.11 The person’s eyes open spontaneously or following stimulation, or both. There may be random hand, extremity, or head movements. The individual maintains blood pressure and breathing without support. Brainstem reflexes (pupillary, oculocephalic, chewing, swallowing) are intact. No discrete localizing motor responses are present, and the individual does not speak any comprehensible words or follow commands. There is no awareness of self or the environment.12
Some survivors of coma progress to a minimally conscious state. The term minimally conscious state (MCS) was first used by the International Working Party on Vegetative States and supported by the Brain Injury Interdisciplinary Special Interest Group of the American Congress of Rehabilitation Medicine (ACRM). ACRM defined MCS as a condition of severely altered consciousness in which the person demonstrates minimal but defined behavioral evidence of self or environmental awareness.11,13 The clinical features include (1) following simple commands, (2) manipulation of objects, (3) gestural or verbal “yes/no” responses, (4) intelligible verbalization, and (5) stereotypic movements (e.g., blinking, smiling) that occur in a meaningful relationship to the eliciting stimulus and are not attributable to reflexive activity.
Akinetic mutism (AM) is a neurobehavioral state characterized by a severe disturbance in behavioral drive (motivation). Generally, these individuals evidence eye opening with visual tracking and have little or no spontaneous speech or following of commands. Little movement is present. This is not attributable to decreased wakefulness or motor weakness or impairment. The pathology involves damage to the frontal lobe or cingulate gyrus.14
With locked-in syndrome there is injury to the ventral pons. Both the content of thought and level of arousal are intact, but the efferent pathways are disrupted with quadriplegia and anarthria.15 Thus the individual cannot communicate either through speech or through body movement but is fully conscious, with intact cognitive function. The upper cranial nerves (I through IV) often are preserved, however, so that the person possesses vertical eye movement and blinking as a means of communication.
Prognostic Indicators for Emergence from Coma: To date, no indicators except those of brain death predict outcome of coma. Etiology of injury and time since onset of coma are currently the best prognostic indicators of recovery of consciousness or functional outcome. In NTBI, the prognosis can be established earlier than with traumatic brain injury. In traumatic coma, there is a 95% death rate in individuals whose pupillary reflexes or reflective eye movements are absent 6 hours after onset of coma and a 91% death rate if pupils are nonreactive at 24 hours.11 In nontraumatic coma, absence of any two of the following is an unfavorable sign in the first hours after admission: pupil reflexes, corneal reflexes, or oculovestibular responses. Absence of eye opening and muscle tone in 24 hours predicts death or severe disability.11
Recovery of consciousness within 2 weeks is associated with favorable outcomes. Recovery of consciousness after 6 months is correlated with severe disability on the Glasgow Outcome Scale.11 No recovery without severe disability has ever been documented after 1 year in coma.11 No emergence from a VS can be expected after 3 months in a VS from hypoxic-ischemic injury and after 1 year from TBI.
Emergence from MCS is confirmed when there is reliable and consistent demonstration of either (1) interactive communication, that is, the ability to answer basic “yes/no” or “single word answered” personal or environmental questions, and (2) functional use of objects, that is, the ability to appropriately discriminate among objects.11 Failure to emerge from MCS within 12 months predicts the likelihood of remaining in an MCS.11
Seizures
A seizure is caused by abnormal excessive hypersynchronous discharges of CNS neurons16 and is characterized by a sudden, transient alteration in brain function. Depending on the distribution of the discharges, this abnormal CNS activity can be various manifestations16 involving motor, sensory, autonomic, or psychic clinical manifestations and an alteration in level of arousal. The alteration in level of arousal is temporary. The term convulsion, sometimes applied to seizures, refers to the tonic-clonic (jerky, contract-relax) movement associated with some seizures. A seizure produces a brief disruption in brain electrical function.16
Types of Seizures
Seizures are classified in different ways—by clinical manifestations, site of origin, EEG correlates, or response to therapy.17 A simplified version of the international classification of epileptic seizures is presented in Table 16-9. Generalized seizures, 30% of seizures, involve neurons bilaterally, often do not have a local (focal) onset, and usually originate from a subcortical or deeper brain focus. Generalized seizures result from cellular, biochemical, or structural abnormalities of a widespread nature.16 With a generalized seizure, consciousness always is impaired or lost. Partial seizures (focal seizures) involve neurons only unilaterally, often have a local (focal) onset, and originate from discrete areas usually associated with structural abnormalities localized to the cortical brain tissue, thereby having a superficial focus. Consciousness may be maintained as long as the seizure activity is limited to one hemisphere in simple partial seizures, but partial seizures may become generalized to involve neurons of the other hemisphere and the deeper brain nuclei. This process is called secondary generalization. Consciousness is lost at the point of generalization. In complex partial seizures, consciousness is impaired; that is, the person is unable to respond normally to exogenous stimuli. Sixty percent of seizures are either complex partial seizures or seizures with secondary generalization.

Status epilepticus in adults is a state of continuous seizures lasting more than 5 minutes or rapidly recurring seizures before the person has fully regained consciousness from the preceding seizure or a single seizure lasting more than 30 minutes.18 The person is still in a postictal state (state that follows a seizure) when the next seizure begins. Status epilepticus most often results from abrupt discontinuation of antiseizure medications but also may occur in untreated or inadequately treated persons with seizure disorders. The situation is a medical emergency because of the resulting cerebral hypoxia. Mental retardation, dementia, other brain damage, and even death are serious threats. Aspiration also is a great risk. (Terminology associated with seizure activity is defined in Table 16-10.)
Table 16-10
Terminology Used to Describe a Seizure
| Term | Definition |
| Aura | A partial seizure experienced as a peculiar sensation preceding the onset of a generalized seizure or complex partial seizure that may take the form of gustatory, visual, or auditory experience; a feeling of dizziness or numbness; or just “a funny feeling” |
| Prodroma | Early clinical manifestations, such as malaise, headache, or a sense of depression, that may occur hours to a few days before the onset of a seizure |
| Tonic phase | A state of muscle contraction in which there is excessive muscle tone |
| Clonic phase | A state of alternating contraction and relaxation of muscles |
| Postictal state | The period immediately following the cessation of seizure activity |
Diseases and Conditions Associated with Seizure Disorders
Any condition that changes the neuronal environment may produce seizure activity, so in theory, anyone can have a seizure. Diseases or other processes that involve the nervous system may cause a seizure disorder. The onset of seizures may point to the presence of an ongoing primary neurologic disease. Etiologic factors in seizures include (1) cerebral lesions, (2) biochemical disorders, (3) cerebral trauma, and (4) epilepsy. Conditions that may produce a seizure are metabolic defects, congenital malformations, genetic predisposition, perinatal injury, postnatal trauma, myoclonic syndromes, infection, brain tumor, vascular disease, fever, and drug or alcohol abuse or both.
Seizures also may be precipitated by hypoglycemia, fatigue or lack of sleep, emotional or physical stress, febrile illness, large amount of water ingestion, constipation, use of stimulant drugs, withdrawal from depressant drugs (including alcohol), hyperventilation (respiratory alkalosis), and some environmental stimuli, such as blinking lights, a poorly adjusted television screen, loud noises, certain odors, or merely being startled. Women immediately before or during menses may have increased seizure activity.
Basic Pathologic Mechanisms
Seizures occur when there is disruption in the balance of excitation and inhibition.19 The primary abnormality may be a membrane defect leading to instability in resting potential, abnormalities of potassium conductance or calcium channels, defects of the gamma-aminobutyric acid (GABA) inhibitory system, or an abnormality in excitatory transmission enhancement, particularly of the N-methyl-D-aspartate type. In animal models, a defect in the GABA inhibitory system is the mechanism causing generalized seizures. Three groups of physiologic mechanisms are involved in seizures and epilepsies: (1) mechanisms of seizure initiation and propagation (excitation and inhibition), (2) mechanisms of epileptogenesis, and (3) genetics.19
Seizure initiation is characterized by two simultaneous events in a group of neurons: (1) high-frequency bursts of action potentials and (2) hypersynchronization. The burst activity is produced by a relatively long-lasting depolarization of the neuron caused by an influx of extracellular calcium that opens the voltage-dependent sodium channel. The influx of sodium generates repetitive action potentials.16 The firing of involved neurons becomes increasingly greater in frequency and amplitude. With sufficient neuronal activation, recruitment of surrounding neurons occurs through a variety of mechanisms. The discharge spreads or propagates to adjacent normal neurons through corticocortical synapses. If uninhibited at this point, the cortical excitation spreads through interhemispheric tracts to the contralateral cortex and through projection pathways to the subcortical areas of the basal ganglia, thalamus, and brainstem. The excitation spread to the subcortical, thalamic, and brainstem areas corresponds to the tonic phase (phase of muscle contraction with increased muscle tone) and is associated with loss of consciousness. Autonomic clinical manifestations also may emerge at this point, and apnea may be present for a few seconds. The excitation is further projected downward to the spinal cord neurons through the corticospinal and reticulospinal pathways.
The clonic phase (phase of alternating contraction and relaxation of muscles) begins as inhibitory neurons in the cortex, anterior thalamus, and basal ganglia begin to inhibit the cortical excitation. This inhibition causes an interruption in the seizure discharge, producing an intermittent contract-relax pattern of muscle contractions. The intermittent clonic bursts gradually become more and more infrequent until they finally cease. At this point the epileptogenic neurons are exhausted and the neuronal membranes probably are hyperpolarized.
The maintenance of seizure activity demands a 250% increase in adenosine triphosphate (ATP). Cerebral oxygen consumption is increased by 60%. Although cerebral blood flow also increases approximately 250% during seizure activity, available glucose and oxygen are readily depleted. With a severe seizure the brain tissue may require more ATP than can be produced by the tissues from the available oxygen and glucose. A deficiency of ATP, phosphocreatine, and glucose then occurs, and lactate accumulates in the brain tissues. Severe seizures thus may produce secondary hypoxia, acidosis, and lactate accumulation, all of which are imbalances that may result in progressive brain tissue injury and destruction. Cellular exhaustion and destruction are consequences of these events.
Epileptogenesis refers to the transformation of a normal neuronal network into one that is chronically hyperexcitable (epileptogenic focus).20 A delay of months to years often occurs between the initiating injury and the first seizure. Some forms of epileptogenesis involve structural changes in the neuronal network. Reorganization or “sprouting” of surviving neurons also has been found to affect the excitability of the network.
If a seizure focus is active for a prolonged period, a secondary focus, called a mirror focus, may develop in normal tissue. This process apparently is caused by the interhemispheric communication, inasmuch as the mirror focus is located in the contralateral cortical area. Seizure threshold in some individuals is genetically lower. Research is in progress to identify alterations in gene transcription that affect seizure threshold.21
Types of Seizure Syndromes: Seizure disorders, the second most common neurologic disorder, represent a syndrome, not a specific disease entity. The term epilepsy, meaning “to be seized by a force from without,” generally is applied to conditions in which no underlying correctable cause for the seizures is found so that the seizure activity recurs without treatment because of a primary underlying brain abnormality. Epilepsy therefore is a general term for the primary condition that causes the seizures. Epileptic syndromes are epileptic disorders characterized by specific clusters of signs and symptoms.22 The three categories are based on clinical history, EEG manifestations, and etiology: (1) localization related, (2) generalized, and (3) undetermined. Localization-related epilepsies and syndromes are typified by seizures that originate from a localized cortical region and are characterized by seizures that have a focal or partial onset. Generalized and undetermined epilepsies and epilepsy syndromes are characterized by seizures with initial activation of neurons within both cerebral hemispheres.
Epilepsy syndromes are further subdivided into idiopathic, symptomatic, or cryptogenic. Idiopathic epilepsy refers to syndromes that arise spontaneously without a known cause, presumably having a genetic basis. The genetic basis may be through a specific inherited trait in which the seizures are the principal expression of the genetic defect (e.g., childhood absence epilepsy). In two thirds of cases, the etiology of the epilepsy is not identified. Symptomatic epilepsy denotes epilepsies with an identified cause. One third of seizures can be classified as symptomatic (provoked or secondary). Some symptomatic epilepsies also have a genetic basis in which the inherited trait is expressed in a neurologic or systemic disorder that is associated with seizures (e.g., neurofibromatosis). The term cryogenic epilepsy describes syndromes that are presumed to be symptomatic but have no known etiology, and occur in persons with or without abnormalities on neurologic examination. Box 16-1 presents the international classification of epilepsies, and Table 16-11 groups the etiology of recurrent seizures by age group.
Table 16-11
Causes of Recurrent Seizures in Different Age Groups

Data from Goetz CG, editor: Textbook of clinical neurology, ed 3, Philadelphia, 2007, Saunders; Nabbout R, Dulac O: Curr Opin Neurol 21(2):16106, 2008; Waterhouse E, Towne A: Cleve Clin J Med 72(Suppl 3):S26-S37, 2005.
Epilepsy is estimated to affect 5 to 10 people per 1000 in the United States.16,23 Forty to 50 new cases per 100,000 persons develop yearly. Three percent of people are diagnosed with epilepsy.23 Incidence is highest in early childhood and declines to plateau in adulthood. However, incidence rises again in older people to early childhood levels.
CLINICAL MANIFESTATIONS The clinical manifestations associated with seizure depend on the type of seizure (Table 16-12). Two types of symptoms often signal an impending generalized tonic-clonic seizure: an aura, a partial seizure that immediately precedes the onset of a generalized tonic-clonic seizure, and a prodroma, an early manifestation that may occur hours to days before a seizure (see Table 16-10). Both manifestations may become familiar to the person experiencing recurrent generalized seizures and so may help in preventing injuries during the seizure.




EVALUATION AND TREATMENT Health history is the most critical aspect in diagnosing a seizure disorder and establishing the cause. The health history is supplemented by the physical examination and laboratory tests of blood and urine (blood glucose, serum calcium, blood urea nitrogen, urine sodium, and creatinine clearance) to identify any systemic diseases known to have seizures as a clinical manifestation. Skull x-ray films, computed tomography (CT) scan, magnetic resonance imaging (MRI), and cerebrospinal fluid (CSF) examination are useful for identifying any neurologic diseases associated with seizures. The EEG is useful in assessing the type of seizure and may help determine its focus (Figure 16-7).
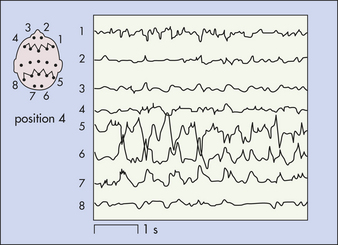
Figure 16-7 Electroencephalogram showing right posterior temporal sharp activity in individual with a microglioma. (From Perkin GD: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
Treatment for a seizure disorder is first to correct or control its cause, if possible. If this is not possible, the major means of management is the judicious administration of antiseizure medications. The therapeutic goal is complete suppression of seizure activity without intolerable side effects of the drug or drug resistance. Temporal lobectomy, amygdalohippocampectomy, or vagus nerve stimulation can improve seizure control and quality of life in people with drug-resistant temporal lobe epilepsy.24 Vagus nerve stimulation can reduce seizure frequency in persons with drug-resistant partial seizures.25 Educational programs may reduce seizure frequency and improve psychologic functioning but it is not known if behavioral and psychologic treatments are beneficial.26
Alterations in Awareness
Selective attention (orienting), or a second attentional network, refers to the ability to select from available, competing environmental and internal stimuli-specific information to be consciously processed (orienting to specific information of interest).27 Certain structures have been demonstrated to contribute to selective attention. The disengagement mechanism is mediated by the right parietal lobe. The move component is mediated by the superior colliculi for visual orienting. The engage component is mediated by the pulvinar nucleus of the thalamus (Figure 16-8). A weak orienting network results in a neglect syndrome.
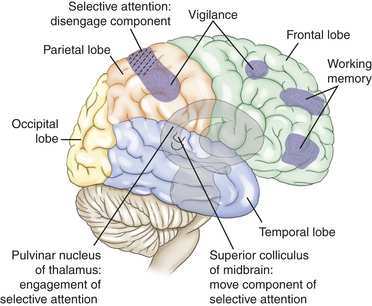
Figure 16-8 Right cortical, subcortical, and brainstem areas of the brain-mediating cognitive functions. (From Boss BJ, Wilkerson R: Communication: language and pragmatics. In Hoeman SP, editor: Rehabilitation nursing: prevention, intervention, & outcomes, ed 4, p 508, St Louis, 2008, Mosby.)
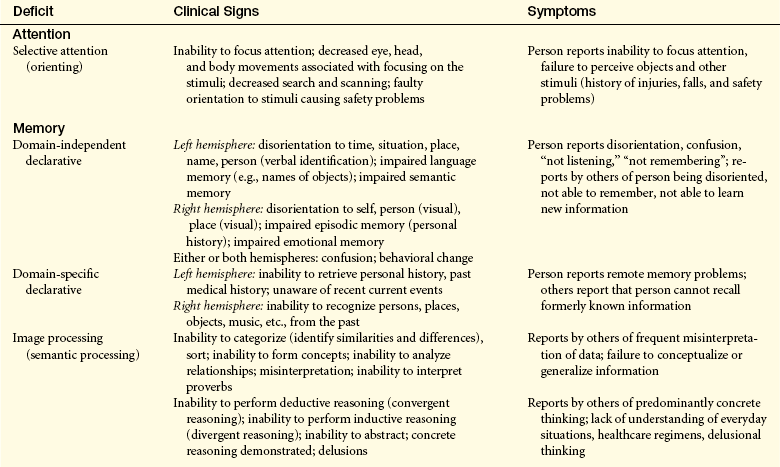
Sensory inattentiveness is a form of neglect and may be visual, auditory, or tactile. The person with sensory inattentiveness is able to recognize individual sensory input from the dysfunctional side when called on to do so but ignores (i.e., neglects, extinguishes) the sensory input from the dysfunctional side when stimulated from both sides. This phenomenon is called extinction. The entire complex of denial of dysfunction, loss of recognition of one’s own body parts, and extinction is sometimes referred to as the neglect syndrome.
An isolated (pure) selective attention deficit (orientation), which manifests as a neglect syndrome, rarely, if ever, occurs clinically because typically other deficits also are present. A neglect syndrome may appear temporarily as a result of seizure activity or a postictal state. Temporary or permanent deficits may occur with contusions or subdural hematomas, encephalitis, and ischemic stroke. Progressive neglect deficits may be found with gliomas or metastatic tumor and in Alzheimer and Pick diseases.
Memory is the recording, retention, and retrieval of knowledge. Two types of memory exist: declarative and nondeclarative. Declarative memory involves the learning and remembrance of episodic memories (personal history, events, and experiences) and semantic memories (facts and information). Declarative memory is mediated by domain-specific cortical areas of the association areas of the temporal, parietal, and occipital lobes (Figure 16-9) where long-term memories are thought to be stored and by domain-independent areas of the medial temporal lobe, the diencephalon, and the basal forebrain (Figure 16-10) where it is thought distinct domain-specific features of an experience are related or bound.28
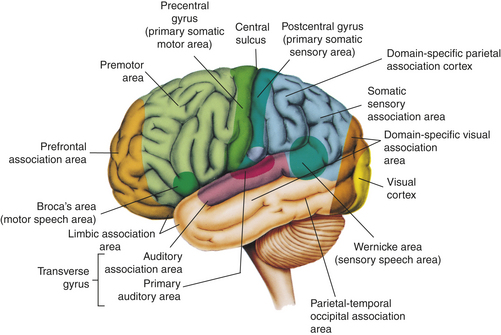
Figure 16-9 Cortical areas of the left (dominant) hemisphere. (From Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
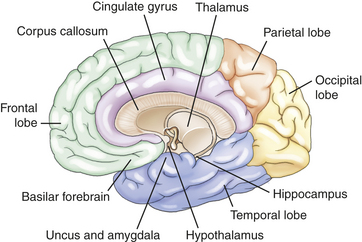
Nondeclarative memory (nonconscious), also called reflexive, procedural, or implicit memory, is the memory for actions, behaviors (habits), skills, and outcomes.29 It is not a language memory but a motor memory. Nondeclarative memory involves the laying down of the motor pattern for the motor performance so that the action, behavior, or skill becomes more and more automatic. The striatum of the basal ganglia supports this learning across trials (stimulus-response learning), as well as probabilistic classification learning, which supports outcome prediction.30 All skills and habits are stored in this memory network. Cerebellar memory was originally thought to be related to only motor learning but it is now believed to involve nonmotor functions.31 Emotional memory is mediated by the amygdala (see Figure 16-10). The amygdala attaches positive or negative dispositions to stimuli in the absence of conscious recollection of the circumstances of the emotional experience. Additionally, the amygdala modulates the event memory during and after the event (memory-enhancing effect).32
Dysmnesia is a disorder of the domain-independent declarative memory network defined as the loss of past memories (retrograde amnesia) coupled with an inability to form new memories (anterograde amnesia) despite intact attentional networks.28 Isolated (pure) domain-independent dysmnesia is caused by only a limited number of conditions, such as transient global dysmnesia (episodic global dysmnesia), amnestic stroke, and Korsakoff psychosis (amnestic or dysmnestic syndrome), as well as after temporal lobectomy. Many disorders may temporarily or permanently produce domain-independent dysmnesia that accompanies other deficits of the cognitive systems. A temporary domain-independent dysmnesia is found during complex partial seizures that persist for a time in the postictal state, in postconcussive states, and in mild posttraumatic brain injury states. A permanent domain-independent dysmnesia may be seen after subarachnoid hemorrhage or moderate or severe posttraumatic brain injury states; in carbon dioxide poisoning and other hypoxic or anoxic states; in Wernicke encephalopathy, viral encephalitis, and granulomatous meningitides; in tumors; and in Alzheimer and Pick diseases.
A pure auditory or visual domain–specific declarative memory deficit manifests as an isolated agnosia (see Table 16-14). An isolated (pure) domain-specific declarative memory deficit of tactile sensations rarely occurs clinically because selective attention would likely be affected as well. A temporary auditory, visual, or tactile pattern recognition (remote memory) deficit may appear as a result of seizure activity or a postictal state. A temporary or permanent deficit can occur with temporal, occipital, or parietal lobe contusion; with subdural hematoma or ischemic stroke; and in encephalitis. A progressive domain-specific declarative memory deficit may occur in temporal, occipital, or parietal gliomas; in metastatic tumors; and in Alzheimer and Pick diseases.
The prefrontal areas mediate several cognitive functions, called executive attention functions. The vigilance system provides the person with the ability to maintain a sustained state of alertness for searching and scanning activities and involves the right frontal areas and the locus coeruleus (LC) located in the rostral pons (see Figure 16-8). Through the neurotransmitter norepinephrine from the LC, the speed of the orienting (selective attention) network is increased and the detection function of the anterior cingulate gyrus (see Figure 16-10) is decreased.
Detection is the recognition of the object’s identity and the realization that the object fulfills a sought-after goal (i.e., target selection among competing, complex contingencies). There is conscious execution of an instruction, ensuring that the instructions are followed. The anterior cingulate cortex inhibits automatic responses so that a less routine response can be given. The basal ganglia and cingulate, as well as other frontal areas, function in color, motion, and form detection.
The anterior cingulate plus the ventrolateral and dorsolateral prefrontal cortex (see Figure 16-8) are involved in the representations of information in the absence of a stimulus, such as spatial position of visual events in memory when the event is removed from view. This is called working memory (short-term representation memory). Control of activation of these memories is also in these areas. This gives the person control over information processing. These temporary storage areas permit the brain to retrieve instructions and other information needed to guide behavior. A person holds and manipulates information in working memory. Two components are described: (1) sustained attention (concentration-over-time) and (2) tracking, the ability to maintain focus despite the presence of competing stimuli or the need to engage in alternating tasks.
Isolated (pure) vigilance, detection, and working memory deficits have been discussed in the literature, but their individual occurrence is uncommon because these deficits generally are present simultaneously. Akinetic mutism exemplifies a detection deficit. The person orients to external stimuli and can follow with his or her eyes but does not initiate other voluntary activity. There are no goals generated and no plans for carrying out the goals. The combination of vigilance, detection, and working memory deficits, accompanied by other deficits of the cognitive systems, is much more common. Whether the deficits are temporary or permanent depends on the cause and severity of injury. Deficits caused by CNS-depressant drugs, by seizure activity, and it is hoped, by neurosurgical procedures involving retraction of the frontal lobes are temporary. Deficits in postconcussive and mild traumatic brain injury states may prove to be temporary and resolve over time. Permanent deficits are more likely to be found with frontal lobe contusions, moderate or severe posttraumatic brain injury states, ischemic frontal lobe stroke, and neurosurgery that requires frontal lobe resection. Progressive deficits in vigilance, detection, and working memory functions are caused by frontal lobe gliomas, frontal lobe infarcts associated with hypertensive vascular disease, and late Alzheimer and Pick diseases. People with schizophrenia have difficulty in clearing working memory of information that is irrelevant to the task. Additionally, recently encountered visual material that is no longer in plain view cannot be preserved in working memory.
Higher-level thought involves the same neural areas used for sensory-specific computations, but when used voluntarily in thought, these areas are activated from the detection and work memory networks (top-down processing from the prefrontal cortex) rather than from bottom-up automatic processing (from the medial temporal lobe) beginning in sensory areas with a specific sensory stimulus. There is a voluntary search for a feature. By reordering component computation, a person produces novel thoughts.
PATHOPHYSIOLOGY Individuals with a disease affecting the superior colliculi have a disturbance in the move component of selective attention, which manifests as a slowness in orienting attention. People with parietal lobe disease may experience selective attention deficits related to disengagement from a stimulus. Those with parietal lobe dysfunction, especially the right parietal lobe, also may experience a unilateral neglect syndrome, the prototype of a selective attention disorder. People with a disease affecting the pulvinar of the thalamus have a disturbance in the engage component of selective attention.
A disorder in vigilance may be produced by disease in the right frontal areas. A pathologic condition in the frontal areas also may produce detection and working memory deficits. Impaired higher-level thought may result from a pathologic process in the cortical association areas of the parietal, temporal, and occipital lobes.
The exact pathophysiology of the various disorders of cognitive systems is not fully known. Researchers are studying the defects in the elementary operations (components) of each cognitive system. In the past, pathophysiology related to the memory systems was the most studied. Dysmnesia, also known as amnesia, originates from pathologic conditions in the hippocampus and related temporal lobe structures. Orienting and the executive attention network are receiving intense study.33,34
As a highly general statement, the primary pathophysiologic mechanisms that operate in cognitive systems disorders are (1) direct destruction because of direct ischemia and hypoxia or indirect destruction as a result of compression and (2) the effects of toxins and chemicals. Disinhibition resulting in overactivity, such as seen in some drug withdrawal states, is a pathologic mechanism that can produce detection deficits or a hypervigilant state. The pathophysiologic processes are summarized in Figure 16-11.
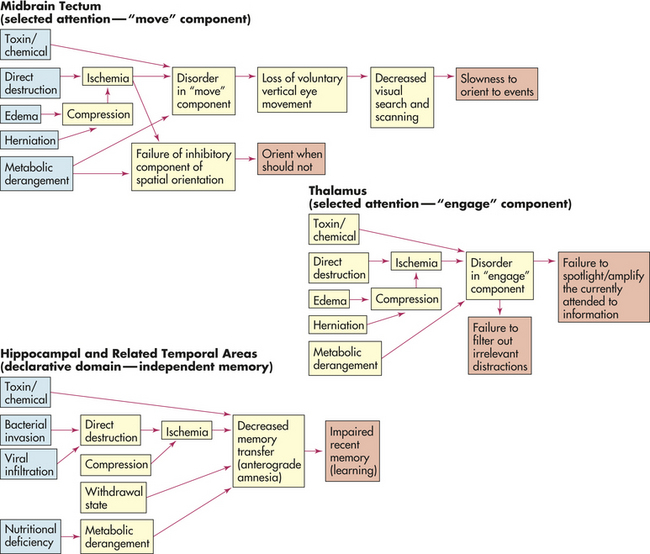
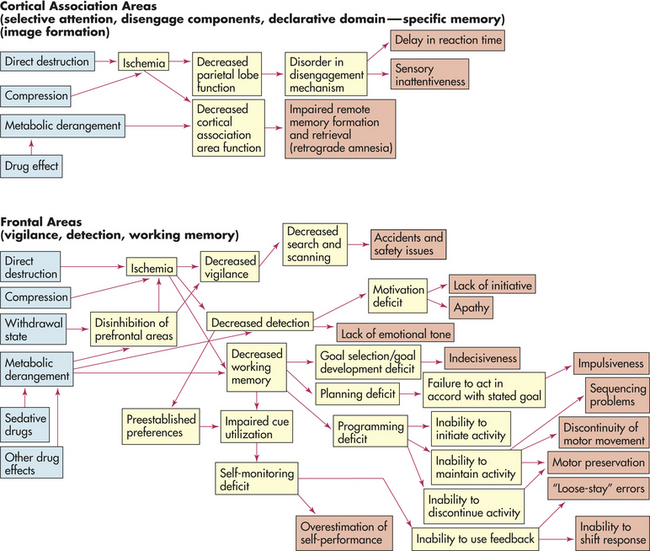
Figure 16-11 Cognitive network deficits. General pathophysiologic mechanisms underlying cognitive network deficits.
CLINICAL MANIFESTATIONS Clinical manifestations of selective attention deficits; domain-independent and domain-specific declarative deficits; and vigilance, detection, and working memory deficits are presented in Table 16-13.

EVALUATION AND TREATMENT Immediate medical management is directed at diagnosing the cause and treating reversible factors. Rehabilitative measures for cognitive system deficits generally are either compensatory or restorative in nature and have been greatly facilitated by computer technology and other electronic-assisted devices. Approaches based on behavioral techniques tend to be compensatory, whereas process-oriented approaches, it is hoped, are restorative.
Selective attention and executive attention deficits masquerade as other cognitive deficits. Differential diagnosis of other cognitive deficits is blocked, and learning potential is largely obscured, by the presence of an attention deficit. Therefore, diagnosis and treatment of attention deficits are fundamental.
Data Processing Deficits
Agnosia is a defect of pattern recognition—a failure to recognize the form and nature of objects. The disorder involves the loss of recognition through one sense, although the object or person may still be recognized by other senses. Agnosia can be tactile, visual, or auditory. For example, an individual may be unable to identify a safety pin by touching it with a hand but be able to name it when looking at it. Agnosia may be as minimal as a finger agnosia (failure to identify by name the fingers of one’s hand) or more extensive, such as a color agnosia.
Agnosia is produced by dysfunction in the primary sensory area or in the interpretive areas of the cerebral cortex (see Figure 16-9). (The types of agnosia and the associated area that is most commonly involved with each are presented in Table 16-14.) Although agnosia most commonly is associated with cerebrovascular accidents, it may arise from any pathologic process that injures these specific areas of the brain.
Table 16-14
Types of Agnosia (Concept Disorders)
| Type of Agnosia | Definition | Location of Injury |
| Tactile agnosia (astereognosis) | Inability to recognize objects by touch | Parietal lobe |
| Spatial agnosia | Incapacity to find one’s way around familiar places; disturbance of perception of space (disorders of [1] topographic [extrapersonal] orientation or [2] topographic and geographic memory [construction]) | Parietal lobe |
| Gerstmann syndrome | Loss of spatial orientation of fingers, body, sides, and numbers | Left angular gyrus (parietal lobe) |
| Finger agnosia (digital agnosia) | Inability to identify the names of one’s fingers | |
| Right-left confusion | Inability to distinguish right from left | |
| Agraphia | Inability to write | |
| Acalculia | Inability to perform mathematic calculations | |
| Visual agnosia | ||
| Object agnosia | Inability to recognize objects and pictures | Temporo-occipital area |
| Prosopagnosia | Inability to recognize faces | Temporo-occipital ventromesial region |
| Color agnosia | Inability to understand colors as qualities of objects; faulty color concepts and inability to evoke color images in the absence of color blindness; specific types: (1) “hue” problem, (2) color anomia (cannot name color) | Inferior occipital cortex in left hemisphere |
| Body image agnosias (may be spatial) | ||
| Anosognosia | Ignorance or denial of existence of the disease | Right parietal lobe |
| Autotopagnosia | Loss of ability to identify the body, in whole or in part, or to recognize relationships among various parts | Right parietal lobe |
| Word blindness (alexia/dyslexia) | Inability to recognize written symbols | Left parietotemporal region |
| Auditory agnosia (pure word deafness) | Inability to recognize speech sounds | Superior temporal area |
| Amusia (music deafness) | Loss of capacity to recognize tones and melodies | Right superior temporal area |
Dysphasia
Dysphasia is impairment of comprehension or production of language (semantic processing). With dysphasia, comprehension or use of symbols, in either written or verbal language, is disturbed or lost. Aphasia is loss of the comprehension or production of language.
Dysphasias usually are associated with cerebrovascular accidents involving the middle cerebral artery or one of its many branches. The language disorders, however, may arise from a variety of injuries and diseases—vascular, neoplastic, traumatic, degenerative, metabolic, or infectious. Dysphasia results from dysfunction in the left cerebral hemisphere, most commonly in the frontotemporal region, particularly around the insula (see Figures 14-7 and 14-9). Genes located on several chromosomes have been linked to language development and disorders.35,36 Most language disorders are caused by acute processes that either resolve or cause a chronic residual deficit. Some language disorders are caused by degenerative disorders that make the dysfunction progressive.
Dysphasias have been classified anatomically and functionally. Other classifications are linguistic and describe fluency, volume, or quantity of speech. Pure forms of any language dysfunction, however, are rare. Expressive dysphasias are characterized primarily by expressive deficits, but a verbal comprehension (auditory-receptive element) deficit may be present. Receptive dysphasias may have expressive deficits. (Table 16-15 compares types of dysphasias; Table 16-16 illustrates some of the language disturbances.)
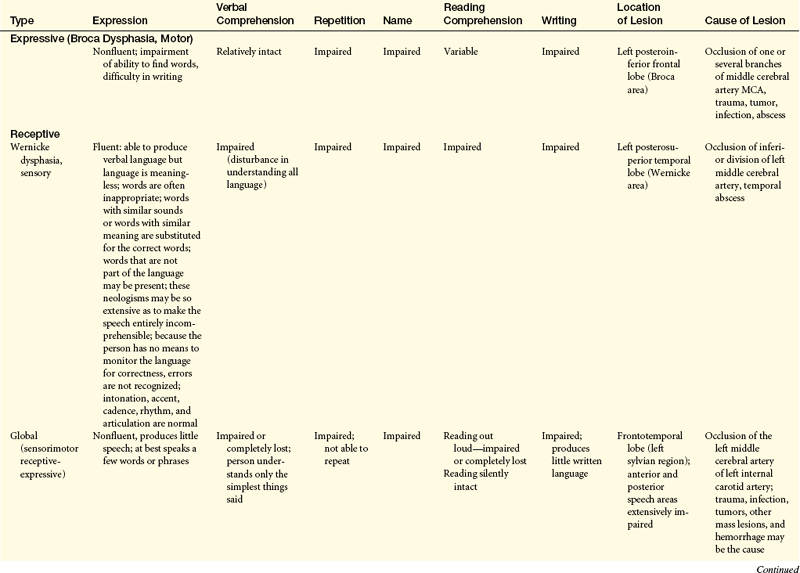
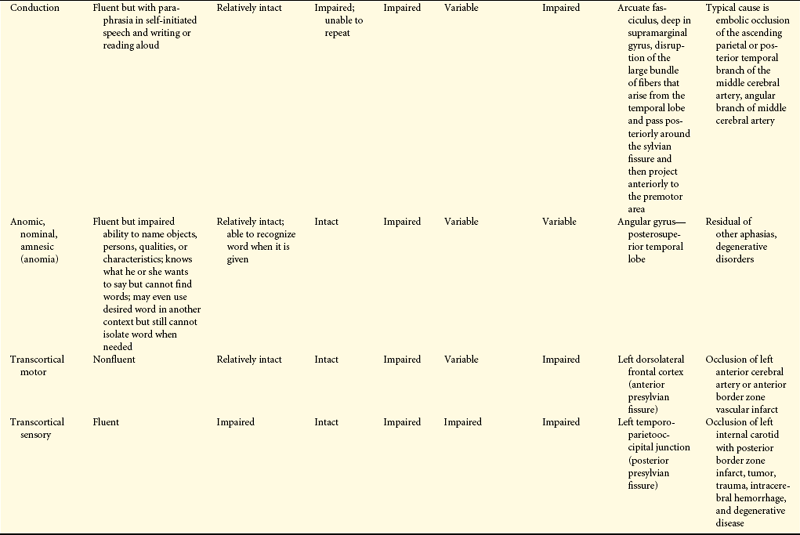
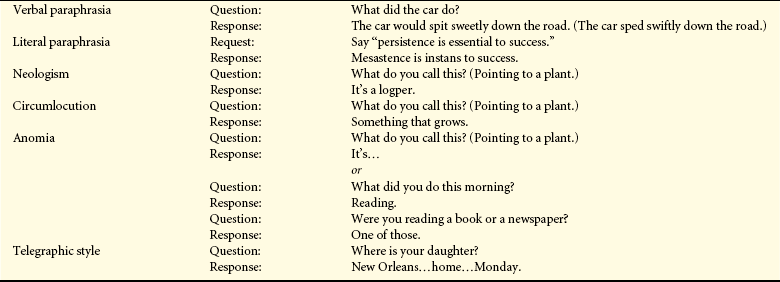
Table 16-16
Examples of Language Disturbances
| Disorder | Example | |
| Verbal paraphrasia | Question: | What did the car do? |
| Response: | The car would spit sweetly down the road. (The car sped swiftly down the road.) | |
| Literal paraphrasia | Request: | Say “persistence is essential to success.” |
| Response: | Mesastence is instans to success. | |
| Neologism | Question: | What do you call this? (Pointing to a plant.) |
| Response: | It’s a logper. | |
| Circumlocution | Question: | What do you call this? (Pointing to a plant.) |
| Response: | Something that grows. | |
| Anomia | Question: | What do you call this? (Pointing to a plant.) |
| Response: | It’s… | |
| or | ||
| Question: | What did you do this morning? | |
| Response: | Reading. | |
| Question: | Were you reading a book or a newspaper? | |
| Response: | One of those. | |
| Telegraphic style | Question: | Where is your daughter? |
| Response: | New Orleans…home…Monday. |
From Boss BJ: J Neurosurg Nurs 16(3):151, 1984.
Dysphasias, referred to as transcortical dysphasias (transcortical sensory dysphasia, mixed transcortical dysphasia, isolated speech center), involve the ability to repeat (called echolalia) and recite. Speech is fluent but with striking paraphrases. The individual cannot read and write, and comprehension is impaired.
Transcortical dysphasias are caused by hypoxia from prolonged hypotension, carbon monoxide poisoning, or other mechanisms that destroy the border zone (watershed area) of the anterior, middle, and posterior cerebral arteries (see Figure 14-19). Blood supply is marginal in this region. Hypoxia in this area occasionally may isolate the posterior speech areas or all the speech areas from the remainder of the cortex, although both areas remain intact. The sensory and motor speech areas therefore are functional, but connections with other sensory or motor areas are impaired. Information from the remaining areas of the cortex cannot be transmitted to the Wernicke area to be transformed into language.
Acute Confusional States
Acute confusional states (acute cerebral failure or acute brain failure) is an acquired mental disorder characterized by deficits in attention and coherence of thoughts and actions often associated with an altered level of arousal, global cognitive dysfunction, perceptual disturbances, sleep-wake cycle disruption, affective disturbance, and emotional liability.37 Acute confusional states result from dysfunction secondary to such causes as drug intoxication, metabolic disorders, or nervous system disease. A common cause of an acute confusional state is withdrawal from alcohol, barbiturate, or other sedative drug ingestion. Acute confusional states of toxic origin may have either sudden or gradual onset, depending on the amount of exposure to the toxin. These states often occur with febrile illnesses, with systemic diseases such as heart failure, after head injury or anesthesia, postnatally, or with certain focal cerebral lesions.38
PATHOPHYSIOLOGY Acute confusional states arise from disruption of a widely distributed neural network involving the reticular activating system of the upper brainstem and its projections to the thalamus, basal ganglion, and specific association areas of the cortex and limbic areas. Delirium (hyperkinetic confusional states) is associated with right middle temporal gyrus or left temporo-occipital junction disruption.37 These areas receive extensive input from the limbic areas and modulate motivational and affective aspects of attention. Hypokinetic confusional states are more likely associated with right-sided frontal-basal ganglion disruption.37 These areas modulate motor exploratory aspects of attention.
Most metabolic disturbances that produce a confusional state interfere with neuronal metabolism or synaptic transmission. Many drugs and toxins also interfere with neurotransmission function at the synapse. Cholinergic pathways critical for attention and arousal are often disrupted.
CLINICAL MANIFESTATIONS The predominant features of an acute confusional state are impaired or lost detection. Because of dysfunction of the anterior cingulate gyrus (see Figures 16-10 and 14-7), the ability to focus, sustain, or shift attentional focus is seriously impaired or completely lost. The person is highly distractible and unable to concentrate on incoming sensory information or on any particular mental or motor task. Besides impaired attention, the person loses coherence of thought and actions. The person may persist in thoughts or actions that are no longer appropriate (perseveration) and be unable to monitor the environment for events of importance (impaired vigilance). The person demonstrates irrelevant or inappropriate responses.
The onset of an acute confusional state usually is abrupt rather than insidious. The first clinical manifestations are difficulty in concentration, restlessness, irritability, tremulousness, insomnia, and poor appetite. Later there are top-down processing problems, including misperception, illusion, hallucination, and delirium. Obsessions, compulsive behavior, and rituals may be evident.
In hypokinetic acute confusional states, the individual exhibits decreases in mental function. Alertness is decreased, as are attention span, accurate perception, and interpretation of the environment. Forgetfulness is prominent. Reactions to the environment are slowed and indecisive. The individual dozes frequently.
Delirium, an acute hyperkinetic confusional state, typically develops over 2 to 3 days. Early clinical manifestations include difficulty in concentrating, restlessness, irritability, insomnia, tremulousness, and poor appetite. Some persons experience seizures. Unpleasant, even terrifying, dreams may occur.
In a fully developed delirium state, the individual is completely inattentive and perceptions are grossly altered. Misperception and misinterpretation are predominant. Hallucinations may be present. The person appears distressed and often very perplexed. Conversation is incoherent. Frank tremor is evident, and a great deal of restless movement is common. Violent behavior may be present. The individual cannot sleep, is flushed, and has dilated pupils, a rapid pulse (tachycardia), temperature elevation, and perfuse sweating (diaphoresis). Delirium typically abates suddenly or gradually in 2 to 3 days, although occasional delirium states persist for several weeks.
EVALUATION AND TREATMENT An acute confusional state is an acute medical problem. The initial goal is to establish that the individual is confused, and the cause must be distinguished as organic or functional (Table 16-17). Next the goal is to determine whether the confusion is delirium, an acute hypokinetic confusional state, or an underlying dementia. The precise cause of an acute confusional state is established through the complete history and physical examination. Laboratory tests include an electrocardiogram and blood, urine, CSF, and radiologic studies.
Table 16-17
Differences between Organic and Functional Confusion
| Factor | Organic Confusion | Functional Confusion |
| Memory impairment | Recent, more impaired than remote | No consistent difference between recent and remote |
| Disorientation | ||
| Time | Within own lifetime or reasonably near future | May not be related to individual’s lifetime |
| Place | Familiar place or one where person might easily be | Bizarre or unfamiliar places |
| Person | Sense of identity usually preserved | Sense of identity diminished |
| Misidentification of others as familiar | Misidentification of others based on delusion system | |
| Hallucinations | Visual, vivid | Auditory more frequent |
| Animals and insects common | Bizarre and symbolic | |
| Illusions | Common | Not prominent |
| Delusions | Concern everyday occurrences and people | Bizarre and symbolic |
| Confusion | Spotty confusion | More consistent |
| Clear intervals mixed with confused episodes | No tendency to become worse at night | |
| Worse at night |
From Morris M, Rhodes M: Am J Nurs 72(9):1632, 1972.
Once the cause is established, treatment is directed at controlling the primary disorder. In an acute confusional state, all drugs that may be contributing to or causing the condition are discontinued unless the problem is the result of drug withdrawal. Supportive measures are designed to enhance coping skills and to minimize the individual’s need for altered cortical functions. Supportive and protective management also involves maintaining the person’s intact cortical functions by promoting use of these functions. Agitated behavior is managed with neuroleptic medication.
Dementing Processes
Dementia is a syndrome that may be caused by a number of different illnesses. Dementia is the progressive failure (an acquired deterioration) of many cerebral functions that is not caused by an impaired level of consciousness.39,40 Memory is the most common cognitive ability lost41 but the dementias are all characterized by reduction in cognitive functions (intellectual function). Mental abilities are impaired, with a decrease in orienting, recent memory, remote memory, language, executive attentional functions, and alterations in behavior (Box 16-2). The greatest risk factor is age.41
Dementias can be classified according to etiologic factors (e.g., trauma, tumors, vascular disorders, infections) and to associated clinical and laboratory signs. Dementing processes have been grouped as cortical, subcortical, or both. Box 16-3 lists the most and least common causes of dementia. Alzheimer disease (AD) is the most common cause followed by vascular disease, then dementia associated with Parkinson disease.41 In people younger than 60 years, frontotemporal dementia (FTD) rivals AD in terms of frequency.41 Disruption in cerebral neural circuits is present. The culmination of a progressive dementing process is nerve cell degeneration and brain atrophy involving the cerebral cortex, diencephalon, and basal ganglia.
PATHOPHYSIOLOGY Mechanisms in dementing processes include (1) degeneration possibly caused by genetics, inflammation, or biochemical alterations; (2) atherosclerosis, multiple foci of infarction throughout the thalami, basal ganglia, cerebral projection pathways, and associated areas; (3) trauma, lesions in the cerebral convolutions, mainly frontal and temporal, corpus callosum, and mesencephalon; and (4) compression, increased intracranial pressure, and chronic hydrocephalus.
The major degenerative dementias are AD, FTD, dementia with Lewy bodies (DLB), Huntington disease (HD), and prion disorders including Creutzfeldt-Jakob disease (CJD). The molecular basis for these dementias is contrasted in Table 16-18. In some instances a familial history of dementia increases by four times the likelihood that dementia will develop. Environmental influences also may play a role in the pathogenesis of dementia. The exact nature of the influence of environmental factors, such as aluminum, is not clearly understood as yet.
Table 16-18
The Molecular Basis for Degenerative Dementia

apoE, amyloid precursor protein (APP), apolipoprotein E; PRNP, prion protein; PrPSC, prion protein; PSEN, presenilin.
From Belin AC, Westerlund M: FEBS J 275(7):1377-1383, 2008; Borroni B et al: Acta Neurol Scand 117(5):359-366, 2008; Goldman JS et al: Am J Alzheimers Dis Other Demen 22(6):507-515, 2008; Graff-Radford NR, Woodruff BK: Semin Neurol 27(1):48-57, 2007; Waring SC, Rosenberg RN: Arch Neurol 65(3):329-334, 2007.
CLINICAL MANIFESTATIONS A summary of the clinical manifestations of the degenerative dementias is presented in Table 16-19.
Table 16-19
Clinical Differentiation of the Major Degenerative Dementias
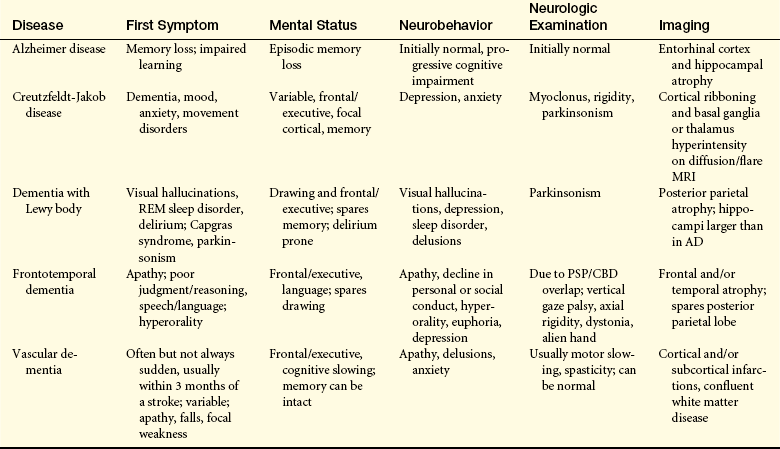
AD, Alzheimer disease; CBD, cortical basal degeneration; MRI, magnetic resonance imaging; PSP, progressive supranuclear palsy; REM, rapid eye movement.
Adapted from Bird TD, Miller BL: Dementia. In Fauci AS et al, editors: Harrison’s principles of internal medicine, ed 15, p 2538, New York, 2008, McGraw-Hill.
EVALUATION AND TREATMENT Establishing the cause for a dementing process may be complicated, but anyone evidencing the clinical manifestations of dementia should be evaluated with laboratory and neuropsychologic testing to identify underlying conditions that may be treatable.
If a specific treatable cause is identified, the appropriate treatment is initiated. For example, an infectious process requires the appropriate antibiotic, and a potentially resectable mass may require neurosurgery. Nutritional deficiencies are corrected. If the cause is metabolic, the imbalance is corrected or the metabolic disorder is treated, or both.
Unfortunately no specific treatment or cure exists for most progressive dementias. In such instances, therapy is directed at maintaining and maximizing the remaining capacities, restoring functions if possible, accommodating to lost abilities, and controlling behavioral changes. Delusions, paranoia, and hallucinations often respond to neuroleptic medications. If coexisting depression is suspected, a trial of antidepressants is appropriate. Assisting the family to understand the dementing process and to learn ways to assist the demented individual is an essential component of supportive management.
Alzheimer Disease
Alzheimer disease (dementia of Alzheimer type [DAT], senile disease complex) is a common neurologic disorder. Formerly believed to occur mostly in people younger than 65 years (familial, early onset dementia), AD has been demonstrated to be one of the most common causes of severe cognitive dysfunction in older adults. Its more prevalent forms are late-onset familial Alzheimer dementia (FAD) and nonhereditary, or sporadic, late-onset AD (70% of cases). FAD and sporadic, late-onset AD are known as senile dementia of the Alzheimer type (SDAT). AD is also associated with Down syndrome. It is estimated that 5 million Americans have AD.42
Early-onset FAD includes at least three gene defects: amyloid precursor protein (APP) gene on chromosome 21, presenilin 1 (PSEN1) on chromosome 14, and PSEN2 on chromosome 1.43,44 Late-onset FAD is linked to a defect in the apolipoprotein E (apoE4) gene on chromosome 19.45 Presence of the apoE4 allele is a marker of increased susceptibility rather than a genetic determinant.40 The 3% of those who are homozygous for apoE4 have an 85% risk, whereas the 25% who are heterozygous have a 45% to 50% risk.40 The greatest risk factors are age and familial disposition (family history).40,41,46
Other risk factors include atherosclerosis, low education level, head injury, cardiovascular diseases, elevated serum homocysteine and cholesterol levels, and female gender estrogen deficit (Figure 16-12).47 Protective factors include lifelong activity, apoE2, antioxidant substances, estrogen replacement, low caloric diet, nonsteroidal anti-inflammatory agents, and statins.47
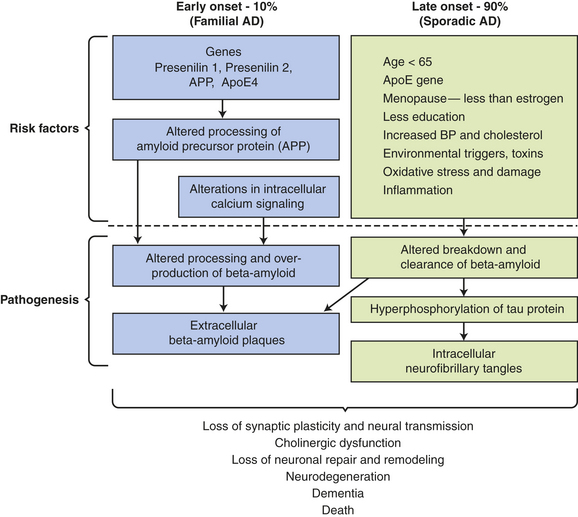
Figure 16-12 Proposed risk factors and pathogenesis of Alzheimer disease. apoE, apolipoprotein E; APP, amyloid precursor protein; BP, blood pressure. (Data from Bojarski L, Herms J, Kuznicki J: Neurochem Inst 52[4-5]:621-633, 2008; Ding Q, Dimayuga E, Keller JN: Curr Alzheimer Res 4[1]:73-79, 2007; Shah RS et al: Biomed Pharmacother 6[4]:199-207, 2008; Waring SC, Rosenberg RN: Arch Neurol 65[3]:329-334, 2008.)
PATHOPHYSIOLOGY The exact cause of AD is unknown. Several possible theories being investigated include loss of neurotransmitter stimulation by choline acetyltransferase; mutation for encoding amyloid precursor protein; alteration in apoE, which binds amyloid-beta48; and pathologic activation of N-methyl-D-aspartate (NMDA) receptors resulting in an influx of excess calcium.
The pathogenesis of AD is linked to amyloid-beta (AB) peptide. AB peptide is derived from proteolysis of APP and released as AB 30 to AB 46, with AB 40 and AB 42 the most abundant isoforms produced.47 These peptides have a strong tendency to form clusters of fibrils, especially AB 42.47 A balance between production and catabolism (involving microglia, macrophages, and bulk flow across the blood-brain-barrier) is required.47 Altered production and failure of clearance of amyloid from the brain occur in AD initiating accumulation (Figure 16-13). Fine diffuse plaques (senile plaques) are the initial accumulation of AB 42. This accumulation is followed by other AB depositions along with tau protein, activated glia, and, eventually, neurofibrillary tangles.47 The abnormal AB is neurotoxic.
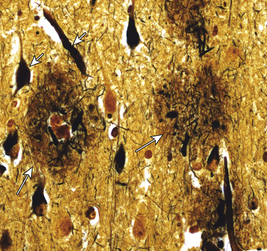
Figure 16-13 Major histopathologic changes in Alzheimer disease. Beta-amyloid protein deposits (plaques) in the neurophil (long arrow) and neurofibrillary tangles (short arrow). (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 8, p 893, Philadelphia, 2007, Saunders.)
Microscopically the tau protein that normally stabilizes the microtubular transport system in the neurons detaches from the microtubule and forms insoluble helical filaments40 called neurofibrillary tangles (see Figure 16-13). Tangles are flame shaped. Cortical nerve cell processes become twisted and dilated because of accumulation of the same filaments that form tangles. Amyloid also is deposited in cerebral arteries, causing an amyloid angiopathy. Senile plaques and neurofibrillary tangles are more concentrated in the cerebral cortex and hippocampus. The greater the number of senile plaques and neurofibrillary tangles, the more dysfunction associated with AD. Figure 16-14 shows the disturbance in blood flow in AD. In addition, aging and injury may result in changes that contribute to the development of this disease. Such changes could include decreased oxygen and glucose transport, loss of the blood-brain barrier, and mitochondrial defects that alter cell metabolism and processing of proteins, including amyloid (apoE4). Macroscopically, the brain in AD decreases in volume and weight, the sulci widen, and the gyri thin, especially in the temporal and frontal lobes. The ventricles enlarge to fill the space (Figure 16-15).
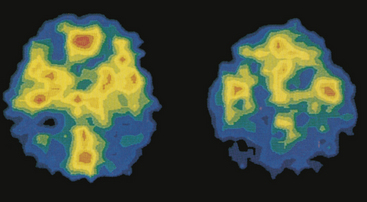
Figure 16-14 Altered cerebral blood flow in Alzheimer disease. Single photon emission computerized tomography scan showing reduction of temporoparietal blood flow (right) compared with normal blood flow (left). (From Perkin GD: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
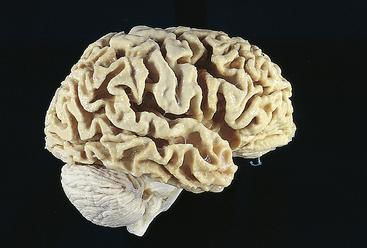
Figure 16-15 Alzheimer disease. The brain decreases in volume and weight, the sulci widen, and the gyri thin, especially in the temporal and frontal lobes. The ventricles enlarge to fill the space. (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7 p 843, Philadelphia, 2007, Saunders.)
CLINICAL MANIFESTATIONS Initial clinical manifestations are subtle and nonspecific and often attributed to forgetfulness, emotional upset, or other illness. The individual becomes progressively more forgetful over time, particularly in relation to recent events. Memory loss increases as the disorder advances, and the person becomes disoriented and confused. The ability to concentrate declines. Abstraction, problem solving, and judgment gradually deteriorate. A failure in mathematic calculation ability, language, and visuospatial orientation occurs. Dyspraxia may appear. The mental status changes induce behavioral changes, including irritability, agitation, and restlessness. Mood changes also result from the deterioration in cognition. The person may become anxious, depressed, hostile, emotionally labile, and prone to mood swings. Motor changes may occur if the posterior frontal lobes are involved. The individual exhibits rigidity (paratonia, gegenhalten), with flexion posturing, propulsion, and retropulsion. Great variability in age of onset, intensity and sequence of symptoms, and location and extent of brain abnormalities occurs among individuals with AD. Table 16-20 presents the clinical findings in each stage of AD.
Table 16-20
Progression of Alzheimer Disease

ADL, Activities of daily living; IADL, instrumental activities of daily living.
Adapted from National Conference of Gerontological Nurse Practitioners and the National Gerontological Nursing Association, Counseling Points 1(1):6, 2008.
EVALUATION AND TREATMENT The diagnosis of AD is made by ruling out other causes of a dementing process by CT and blood tests. The clinical history, cognitive testing, and the course of the illness are used for diagnosis. The course of the disorder is highly variable, usually developing over 5 years or more. Genetic tests to screen for early onset AD genes are PSEN1, PSEN2, APP, and apoE4.49,50
There are no disease-arresting therapies available for AD. Cholinesterase inhibitors (ChE-Is) are used in mild to moderate AD to enhance cholinergic transmission. Drugs for
moderate to severe AD block the activity of glutamate and work as an uncompetitive NMDA receptor antagonist.51,52
Treatment of AD also is directed at decreasing the need for the impaired cognitive function by a compensation technique, such as memory aids, maintaining those cognitive functions that are not impaired, and maintaining or improving the general state of hygiene, nutrition, and health. Environmental management, counseling, education, pharmacotherapy, and health promotion measures provide the foundation on which a comprehensive treatment program is built.51
ALTERATIONS IN CEREBRAL HEMODYNAMICS
An injured brain reacts with structural, chemical, and pathophysiologic changes. Critical variables related to cerebral oxygenation include intracranial pressure, blood flow, and oxygen delivery. The pressure and oxygen delivery are critical management issues.
Cerebral Hemodynamics
Increased intracranial pressure (ICP) was the central management issue for many years. It is now recognized that cerebral oxygenation is the critical issue. Several relevant features of cerebral hemodynamics—cerebral blood volume (CBV), cerebral blood flow (CBF), and cerebral perfusion pressure (CPP)—relate to cerebral oxygenation (Box 16-4).
To guide therapeutic management, three critical categories related to cerebral hemodynamics are possible in the injured brain: (1) cerebral oligemia (also called jugular fibrillation), (2) CPP in the normal range (60 to 100 mmHg) but with an elevated ICP, and (3) cerebral hyperemia. In the treatment algorithms, oxygen saturation measured in the internal jugular vein (SjO2) is categorized as less than 55%, greater than 55% but less than 70%, or greater than 75%. After SjO2 is categorized, the ICP must be added to the equation as less than 20 mmHg or greater than 20 mmHg. Treatment algorithms are implemented depending on the SjO2 and ICP that address not only ICP but also CPP. The therapeutic goal is to balance ICP and SjO2. Target values for relevant clinical parameters are presented in Table 16-21.
Table 16-21
Therapeutic Management Goals for Individuals with Altered Cerebral Hemodynamics
| Clinical Parameter | Target Value |
| Central perfusion pressure | >70 mmHg |
| Intracranial pressure | <20 mmHg |
| Arterial CO2 pressure (PaCO2) | 35 mmHg |
| Mean arterial pressure | 90 mmHg |
| Temperature | 34°-36° C (93.2-96.8 F) |
| Pulmonary capillary wedge pressure | 10-15 mmHg |
Increased Intracranial Pressure
Intracranial pressure normally is 5 to 15 mmHg, or 60 to 180 cm H2O. Increased intracranial pressure may result from an increase in intracranial content (as occurs with tumor growth), edema, excess CSF, or hemorrhage. A rise in intracranial pressure necessitates an equal reduction in volume of the other contents. The most readily displaced content of the cranial vault is CSF. If intracranial pressure remains high after CSF displacement out of the cranial vault, cerebral blood volume is altered, which causes stage 1 intracranial hypertension. Vasoconstriction and external compression of the venous system occur in an attempt to further decrease the intracranial pressure. Thus during the first stage of intracranial hypertension, intracranial pressure may not change because of the effective compensatory mechanisms. CSF is reduced through increased reabsorption. Blood volume is reduced by compression of intracranial veins. Small increases in volume, however, cause an increase in pressure, and the pressure may take longer to return to baseline. Clinical manifestations at this stage usually are subtle and often transient and include episodes of confusion, drowsiness, and slight pupillary and breathing changes.
If ICP is still high, a state of intracranial hypertension occurs. With continued expansion of the intracranial content, the resulting increase in ICP may exceed the brain’s compensatory capacity to adjust to the increasing pressure. In this state, the pressure begins to compromise neuronal oxygenation, and systemic arterial vasoconstriction occurs in an attempt to elevate the systemic blood pressure sufficiently to overcome the increased intracranial pressure. This is stage 2 of intracranial hypertension.
As intracranial pressure begins to approach arterial pressure, the brain tissues begin to experience hypoxia and hypercapnia and the individual’s condition rapidly deteriorates. Clinical manifestations include decreasing levels of arousal, Cheyne-Stokes respiration or central neurogenic hyperventilation, pupils that become sluggish and dilated, widened pulse pressure, and bradycardia.
Dramatic sustained rises in intracranial pressure are not seen until all the compensatory mechanisms have been exhausted. Once decompensation begins, dramatic rises in ICP occur over a very short period. Autoregulation, the compensatory alteration in the diameter of the intracranial blood vessels designed to maintain a constant blood flow during changes in cerebral perfusion pressure, is lost with progressively increased intracranial pressure. Accumulating carbon dioxide may still cause vasodilation at the local tissue level, but now, without autoregulation, this vasodilation causes the hydrostatic (blood) pressure in the vessels to drop and blood volume to increase. The brain volume is thus further enhanced, and ICP continues to rise. This is stage 3 of intracranial hypertension. Small increases in volume cause dramatic increases in ICP, and the pressure takes much longer to return to baseline. As the ICP begins to approach systemic blood pressure, cerebral perfusion pressure falls and cerebral perfusion slows dramatically. The brain tissues experience severe hypoxia and acidosis.
Increased ICP in one compartment of the cranial vault is not evenly distributed throughout the other vault compartments. In stage 4, the last stage of intracranial hypertension, brain tissue shifts (herniates) from the compartment of greater pressure to a compartment of lesser pressure (Figure 16-16). With this shift in brain tissue, the herniating brain tissue’s blood supply is compromised, causing further ischemia and hypoxia in the herniating tissues. The herniated brain tissues increase the volume of content within the lower-pressure compartment, exerting pressure on the brain tissue that normally occupies that compartment, thus impairing that tissue’s blood supply. Small hemorrhages frequently develop in the involved brain tissue. Obstructive hydrocephalus may develop. The herniation process markedly and rapidly increases ICP. Mean systolic arterial pressure soon equals ICP, and cerebral blood flow ceases at this point.
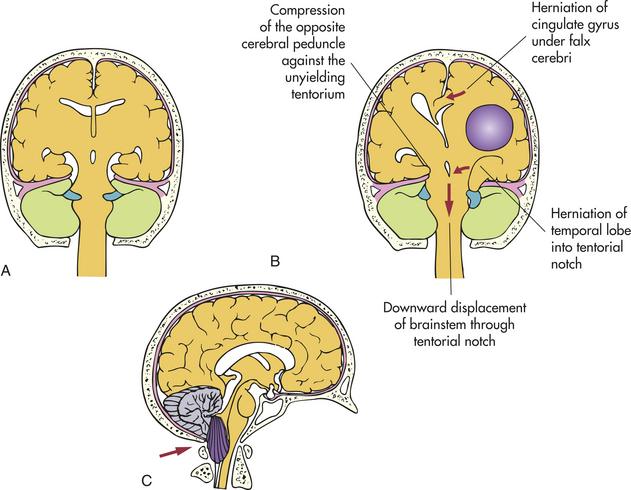
Herniation Syndromes
The three types of supratentorial herniation syndromes are (1) uncal (temporal lobe, lateral transtentorial) herniation, (2) central (transtentorial) herniation, and (3) cingulate gyrus herniation. Uncal herniation (hippocampal herniation, lateral mass herniation) occurs when the uncus or hippocampal gyrus (or both) shifts from the middle fossa through the tentorial notch into the posterior fossa, compressing the ipsilateral third cranial nerve impairing parasympathetic function carried in periphery of the nerve, then the contralateral third cranial nerve, and finally the mesencephalon-inducing coma. Uncal herniation generally is caused by an expanding mass in the lateral region of the middle fossa. The earliest signs of uncal herniation are poor concentration, drowsiness, and the bilateral corticospinal tract signs of increased tone and a positive Babinski sign caused by pressure on the opposite cerebral peduncle in some cases.2,6 The classic manifestations of uncal herniation are a decreasing level of consciousness, pupils that become sluggish before fixing and dilating (first the ipsilateral, then the contralateral pupil), Cheyne-Stokes respirations (which later shift to central neurogenic hyperventilation), the appearance of decorticate, then later decerebrate, posturing, and ipsilateral hemiplegia because of contralateral corticospinal tract compression.6
Central transtentorial herniation is the straight downward shift of the diencephalons (thalamic medial structures) through the tentorial notch. Causes of central herniation are injuries or masses located around the outer perimeter of the frontal, parietal, or occipital lobes; extracerebral injuries around the central apex (top) of the cranium; bilaterally positioned injuries or masses; and unilateral cingulate gyrus herniation. The heralding signs are miotic pupils and drowsiness.2 The individual experiencing transtentorial herniation rapidly passes from a conscious to an unconscious state; from Cheyne-Stokes respirations to apnea; from small, reactive pupils to dilated and fixed pupils; and from decortication to decerebration.
Cingulate gyrus herniation (subfalcine or transfalcial herniation) occurs when the cingulate gyrus shifts under the falx cerebri. Little is known about the clinical manifestations of this type of herniation except that there are signs of a mass causing increased intracranial pressure.6
Infratentorial (Foraminal) Herniation
Two types of infratentorial (foraminal) herniation syndromes may occur. In the most common infratentorial herniation syndrome, a cerebellar tonsil shifts through the foramen magnum because of increased pressure within the posterior fossa. The clinical manifestations of this downward infratentorial herniation are an arched, stiff neck; paresthesias in the shoulder area; decreased consciousness; respiratory abnormalities and arrest; and pulse rate variations. Occasionally the pressure force is such that an upward transtentorial herniation of a cerebellar tonsil or the lower brainstem results. No specific set of clinical manifestations is associated with this infratentorial herniation syndrome.
Cerebral Edema
Cerebral edema is an increase in the fluid content of brain tissue, a net accumulation of water within the brain (Figures 16-17 and 16-18). Cerebral edema causes an increase in extracellular or intracellular tissue volume after brain insult from trauma, infection, hemorrhage, tumor, ischemia, infarct, or hypoxia. The harmful effects of cerebral edema are caused by the distortion of blood vessels, the displacement of brain tissues, and the eventual herniation of brain tissue from one brain compartment to another.
Figure 16-17 Cerebral edema. This coronal section of cerebrum demonstrates marked compression in the lateral ventricles (long arrows) and flattening of gyri (short arrows) from extensive bilateral cerebral edema. Edema increases intracranial pressure, leading to herniation. (From Klatt EC: Robbins and Cotran atlas of pathology, p 449, Philadelphia, 2006, Saunders.)
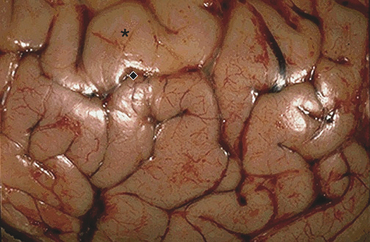
Figure 16-18 Cerebral edema, gross. The surface of the meninges of the brain with cerebral edema shows widened, flattened gyri (∗) with narrowed sulci ( ). (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders, p 449.)
). (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders, p 449.)
Three types of cerebral edema are (1) vasogenic edema, (2) cytotoxic (metabolic) edema, and (3) interstitial edema.53 Vasogenic edema is clinically the most important type. It is caused by the increased permeability of the capillary endothelium of the brain after injury to the vascular structure. The result is a disruption in the blood-brain barrier. Plasma proteins leak into the extracellular spaces, drawing water to them, and the water content of the brain parenchyma increases. Vasogenic edema starts in the area of injury and spreads with preferential accumulation in the white matter of the ipsilateral side because the parallel myelinated fibers separate more easily. Edema then promotes more edema because of ischemia from increasing pressure.
Clinical manifestations of vasogenic edema include focal neurologic deficits, disturbances of consciousness, and a severe increase in intracranial pressure. Vasogenic edema resolves by slow diffusion.
In cytotoxic (metabolic) edema, toxic factors directly affect the cellular elements of the brain parenchyma (neuronal, glial, and endothelial cells), causing failure of the active transport systems. The blood-brain barrier is not disrupted. The cells lose their potassium and gain larger amounts of sodium. Water follows by osmosis into the cell so that the cells swell. Cytotoxic edema occurs principally in the gray matter and may increase vasogenic edema.
Interstitial edema is seen most often with noncommunicating hydrocephalus (see below and Chapter 19). The edema is caused by transependymal movement of CSF from the ventricles into the extracellular spaces of the brain tissues. The brain fluid volume thus is increased predominantly around the ventricles. The hydrostatic pressure within the white matter increases, and the size of the white matter is reduced because of the rapid disappearance of myelin lipids.
Hydrocephalus
The term hydrocephalus refers to a variety of conditions characterized by an excess of fluid within the cranial vault, subarachnoid space, or both. Hydrocephalus occurs because of interference with CSF flow caused by increased fluid production, obstruction within the ventricular system, or defective reabsorption of the fluid. A papilloma (i.e., epithelial tumor) may, in rare instances, cause overproduction of CSF (Figure 16-19).
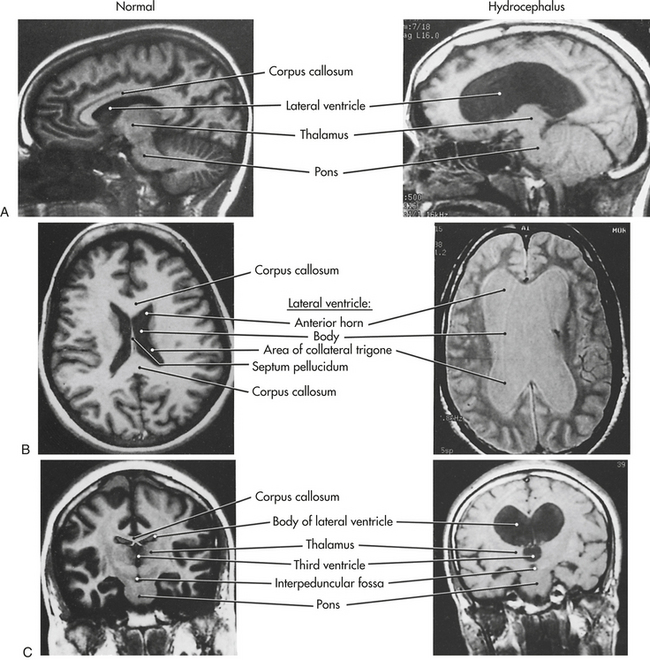
Figure 16-19 Comparison of normal and hydrocephalic brains. A, Sagittal; B, axial; and C, coronal planes as seen in magnetic resonance imaging (MRI). (From Haines DE, editor: Fundamental neuroscience, Philadelphia, 1997, Churchill Livingstone.)
Types of Hydrocephalus
Obstruction within the ventricular system, called noncommunicating hydrocephalus or internal (intraventricular) hydrocephalus, may result from congenital abnormalities in the ventricular system or mass lesions such as a tumor that compresses one of the structures of the ventricular system (see Chapter 19 for additional discussion). Impaired absorption of CSF from the subarachnoid space occurs when an obstructive process disrupts the flow of CSF through the subarachnoid space. The fluid is prevented from reaching the convex portion of the cerebrum, where the arachnoid granulations are located.
Hydrocephalus from impaired absorption may be caused by adhesions from inflammation, as with a meningitis or subarachnoid hemorrhage; compression of the subarachnoid space by a mass, such as a tumor; congenital abnormalities of the subarachnoid space; or high venous pressure within the sagittal sinus. This type of hydrocephalus is termed communicating (extraventricular) hydrocephalus. The most common causes of communicating hydrocephalus are subarachnoid hemorrhage, developmental malformation, head injury, and neoplasm.
One form of communicating hydrocephalus is hydrocephalus ex vacuo, which arises from cerebral atrophy. CSF fills the unoccupied space. The amount of CSF is increased, but the fluid is not under pressure. Another form of communicating hydrocephalus is normal-pressure hydrocephalus (low-pressure, adult, or occult hydrocephalus), which occurs mostly in late middle age. The cause is thought to be arachnoid adhesions and thickening of the arachnoid that obstructs the subarachnoid space. This form of hydrocephalus is most often seen as a complication of head injury and subarachnoid hemorrhage.
Course of the Disease
Hydrocephalus may develop from infancy through adulthood. Congenital hydrocephalus (i.e., ventricular enlargement before birth) is rare. Noncommunicating hydrocephalus is more commonly seen in children. The more common type of hydrocephalus in adults is the communicating type. (Hydrocephalus in children is discussed in Chapter 19.)
Most cases of hydrocephalus develop gradually and insidiously over time. Acute hydrocephalus, however, may develop in several hours in persons who have sustained head injuries. Acute hydrocephalus contributes significantly to increased ICP.
PATHOPHYSIOLOGY The obstruction of CSF flow associated with hydrocephalus produces dilation of the ventricles proximal to the obstruction. Obstructed CSF is under pressure, causing atrophy of the cerebral cortex and degeneration of the white matter tracts. There is selective preservation of gray matter. When excess CSF fills a defect caused by atrophy, a degenerative disorder, or a surgical excision, this fluid is not under pressure; therefore, atrophy and degenerative changes are not induced.
CLINICAL MANIFESTATIONS The presentation of acute hydrocephalus is one of rapidly developing increased intracranial pressure. The person deteriorates rapidly into a deep coma if not promptly treated. Normal-pressure hydrocephalus has a long-term presentation and develops slowly over time. The individual or family of the individual complains of declining memory and cognitive function. An unsteady, broad-based gait with a history of falling is common. Additional clinical manifestations are apathy; inattentiveness; and indifference to self, family, and the environment. Urinary incontinence is present.54
EVALUATION AND TREATMENT The diagnosis is made on the basis of physical examination, CT scan, and MRI. A radioisotopic cisternogram may be performed to aid in diagnosing normal-pressure hydrocephalus. Hydrocephalus can be treated by surgery to resect cysts, neoplasms, or hematomas or by ventricular bypass into the normal intracranial channel or into an extracranial compartment using a shunt. Excision or coagulation of the choroid plexus is needed occasionally when a papilloma is present. In normal-pressure hydrocephalus, reduction in CSF through a diuresis regimen often is used.
ALTERATIONS IN MOTOR FUNCTION
Movements are complex patterns of activity controlled by the CNS. Movements are influenced by the cerebral cortex, the pyramidal system, the extrapyramidal system, and the motor units. Dysfunction in any of these areas can cause motor dysfunction. General motor dysfunctions may produce changes in muscle tone, movement, and complex motor performance.
Alterations in Muscle Tone
Normal muscle tone involves a slight resistance to passive movement. The resistance is smooth, constant, and even throughout the range of motion. Abnormalities of muscle tone are presented in Table 16-22.

Hypotonia
In hypotonia (decreased muscle tone), passive movement of a muscle occurs with little or no resistance. Hypotonia is thought to be caused by decreased muscle spindle activity secondary to decreased excitability of neurons. Hypotonia is caused by pure pyramidal tract damage (a rare occurrence) and cerebellar damage. A pure pyramidal tract injury produces hypotonia and weakness. The hypotonia contributes to the ataxia and intention tremor in cerebellar damage and manifests with minimal weakness, with normal or slightly exaggerated reflexes. Hypotonia, often described as flaccidity (a state in which the muscle may be moved rapidly without resistance), occurs when nerve impulses necessary for muscle tone are lost, such as in spinal cord injury or cerebrovascular accident.
Individuals with hypotonia report that they tire easily (asthenia) or are weak, signs that can be observed during their activity attempts. They may have difficulty rising from a sitting position, sitting down without using arm support, and walking up and down stairs, as well as an inability to stand on their toes. Because of their weakness, accident proneness during locomotion and self-care activities is common. Inasmuch as the joints become hyperflexible in hypotonic states, people with hypotonia may be able to assume positions that require extreme joint mobility. The joints may appear loose, and the knee jerks are pendulous.
The muscle mass atrophies because of decreased input entering the motor unit. Muscle cells gradually are replaced by connective tissue and fat. The muscles are flabby on palpation and are flat in appearance. Fasciculations may be present in some cases.
Hypertonia
In hypertonia (increased muscle tone), passive movement of a muscle occurs with resistance. Four types of hypertonia are described: spasticity, gegenhalten (paratonia), dystonia, and rigidity.
Spasticity results from hyperexcitability of the stretch reflexes (overactivation of the alpha motor neurons) and is associated with damage to the motor, premotor, and supplementary motor areas, as well as lateral corticospinal tract damage (Figure 16-20). Spasticity is accompanied by increased deep tendon reflexes (hyperreflexia) and the spread of reflexes (clonus).

Figure 16-20 Left-sided hemifacial spasm. (From Perkin GD: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
Gegenhalten (paratonia) manifests as resistance to passive movement that varies in direct proportion to the force applied and is associated with frontal lobe injury. Paratonia is not truly an increase in tone but an increase in resistance by the person. Dystonia manifests as sustained, involuntary twisting movements caused by slow muscle contraction and may be caused by a failure in appropriate reciprocal inhibition of the muscles (Figures 16-21 and 16-22). Injury to the putamen or its outflow tracts also is associated with hemidystonia.
Figure 16-21 Dystonic posturing of the hand and foot. (From Perkin GD: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
Figure 16-22 Spasmodic torticollis. A characteristic head posture. (From Perkin GD: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
Rigidity produced by tonic reflex activity mediated by gamma motor neurons may be continuous or intermittent. The involved muscles are firm and tense; the increase in muscle movement is even and uniform throughout the range of passive movement. Four types of rigidity are described: plastic, or lead-pipe; cogwheel; gamma; and alpha (Table 16-23).
Table 16-23
UK Medical Research Council Classification of Muscle Power
| Grade | Definition |
| 0 | Total paralysis |
| 1 | Flicker of contraction |
| 2 | Movement with gravity eliminated |
| 3 | Movement against gravity |
| 4 | Movement against resistance but incomplete |
| 5 | Normal power |
Individuals with hypertonia may tire easily (asthenia) or be weak. Passive and active movement is equally affected, except in paratonia, in which more active than passive movement is possible. As a result of hypertonia and weakness, accident proneness during locomotion and self-care activities is common.
The muscles may atrophy because of decreased use of the muscles. However, hypertrophy occasionally may occur in some diseases. Hypertrophy results from overstimulation of muscle fibers. Overstimulation occurs when the motor unit reflex arc remains intact and functioning but is not inhibited by higher centers. The loss of inhibition and the constant state of excitation cause continual muscle contraction, resulting in enlargement of the muscle mass (Figure 16-23). The muscles are firm on palpation.
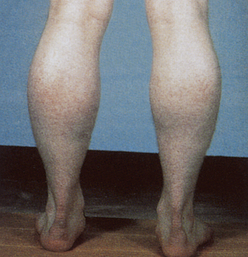
Alterations in Movement
Movement requires a change in the contractile state of muscles. Abnormal movements may occur when a variety of CNS dysfunctions alter muscular innervation. Movement disorders are not well understood. Current knowledge has come predominantly from the areas of neuropharmacology and experimental therapeutics. The neurotransmitter dopamine has an apparent role in motor function. Some movement disorders (e.g., the akinesias) result from too little dopaminergic activity, whereas others (e.g., chorea, ballism, tardive dyskinesia) result from too much dopaminergic activity. Still others are not related primarily to dopamine function. Movement disorders are not associated necessarily with mass, strength, or tone but are neurologic dysfunctions with either a decreased amount of movement or an excess of movement. Muscle strength is quantitatively evaluated on a scale of 0 to 4+ or 0 to 5, in which 4+ or 5 is normal and 0 indicates an inability to move against gravity (see Table 16-23).
Paresis and Paralysis
Paresis (weakness) is impairment of motor function, that is, partial paralysis with incomplete loss of muscle power. Paralysis is loss of motor function, that is, inability of a muscle group to overcome gravity. Two subtypes of paresis and paralysis are described: upper motor neuron paresis and paralysis and lower motor neuron paresis and paralysis (Table 16-24).
Table 16-24
Upper and Lower Motor Neuron Syndromes
| Factor | Upper Motor Neuron Syndromes∗ | Lower Motor Neuron Syndromes† |
| Distribution of affected muscles | Muscle groups are affected; when movement is possible, the proper relationship among agonists, antagonists, synergists, and fixators is preserved | Individual muscles may be affected |
| Synkinesias (residual movements) are present; attempts to move paralyzed part cause a variety of associated movements; movements of normal limb may cause imitative or mirror movements in the paralyzed limb | Individual muscles may be affected | |
| Muscle tone | Hypertonia, specifically spasticity | Hypotonia, flaccidity |
| Tendon reflexes | Hyperreflexia with extensor plantar reflex present | Hyporeflexia, no abnormal reflexes present |
| Atrophy | Slight, caused by disuse | Pronounced atrophy |
| Fasciculations | Absent | May be present |
Upper Motor Neuron Syndromes: Upper motor neuron paresis and paralysis is known also as spastic paresis and paralysis, and many different terms are used to describe a specific paresis or paralysis. Hemiparesis or hemiplegia is paresis or paralysis, respectively, of the upper and lower extremities on one side. Diplegia is the paralysis of both upper or lower extremities as a result of cerebral hemisphere injuries. Paraparesis or paraplegia refers to weakness or paralysis, respectively, of the lower extremities. Quadriparesis or quadriplegia refers to paresis or paralysis of all four extremities. Paraparesis or paraplegia and quadriparesis or quadriplegia may be caused by dysfunction of the spinal cord. Upper cord damage results in quadriparesis or quadriplegia, and lower cord damage preserves upper extremity function and causes paraparesis or paraplegia (spinal cord injury is discussed in Chapter 17).
Upper motor neuron paresis or paralysis is associated with a pyramidal motor syndrome. The pyramidal motor syndrome is a series of motor dysfunctions that result from interruption of the pyramidal system (Figures 16-24 and 16-25). The injury may be in the cerebral cortex, the subcortical white matter, the internal capsule, the brainstem, or the spinal cord. The clinical manifestations of a pure pyramidal injury without other damage are not known, but bilateral interruption of the pyramidal system in monkeys causes hypotonic paralysis, although much control of movement eventually returns. In humans, however, injury generally involves more than merely the interruption of the pyramidal system, so that an upper motor neuron paralysis occurs, which indicates involvement of several motor pathways.
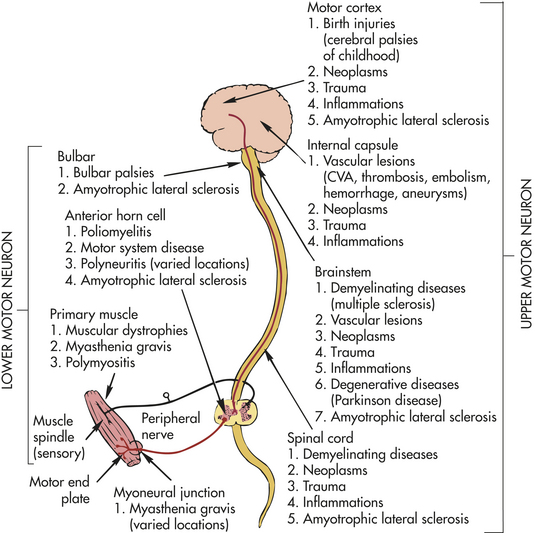
Figure 16-24 Disturbances in motor function. Disturbances in motor function are classified pathologically along upper and lower motor neuron structures. It should be noted that neoplasms occur at more than one site in an upper motor neuron (above right). A few pathologic conditions, such as amyotrophic lateral sclerosis, involve upper and lower motor neuron structures. Other lesion sites include myoneural junction and primary muscle, making it possible to classify conditions as neuromuscular and muscular, respectively. CVA, Cerebrovascular accident.
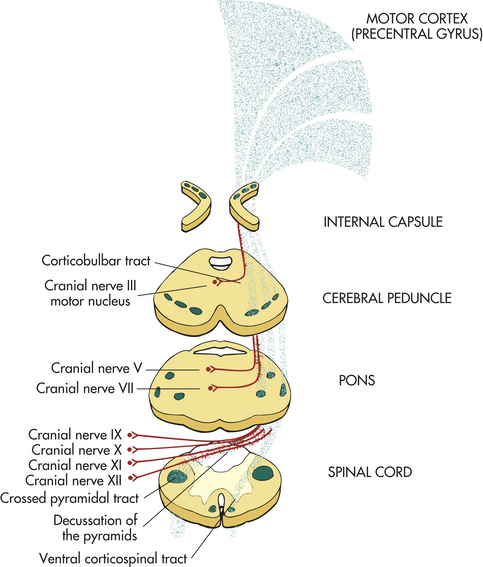
Figure 16-25 Component structure of the upper motor neuron, or pyramidal, system. Pyramidal system fibers are shown to originate primarily in the cells in the precentral gyrus of the motor cortex; to converge at the internal capsule; to descend to form the central third of the cerebral peduncle; to descend farther through the pons, where small fibers are given off to cranial nerve motor nuclei (lower motor neurons) along the way; to form pyramids at the medulla, where most of the fibers decussate; and then to continue to descend in the lateral column of the white matter of the spinal cord. A few fibers descend without crossing at the medulla level.
The distribution of clinical manifestations varies, depending on the location of the lesion, although certain features are constant. Excessive movements such as clonus and spasms occur regularly, and much variation exists, depending on the suddenness of onset and the age of the individual.
When the pyramidal system is destroyed below the level of the pons, spinal shock occurs. Spinal shock is the complete cessation of spinal cord functions below the lesion. It is characterized by complete flaccid paralysis, absence of reflexes, and marked disturbances of bowel and bladder function. The reasons for spinal shock are not fully understood, but a major factor is the sudden destruction of the efferent pathways. If destruction occurs more slowly, spinal shock may not develop (see Chapter 17).
If the pyramidal system is interrupted above the level of the pons, the hand and arm muscles are greatly affected. Paralysis rarely involves all the muscles on one side of the body, however, even when the hemiplegia results from complete damage to the internal capsule. Bilateral movements, such as those of the eye, jaw, and larynx, are affected only slightly, if at all. Predominantly the limbs are affected. Because of their bilateral control, trunk muscles are much less influenced.
Paralysis associated with a pyramidal motor syndrome rarely remains flaccid for a prolonged time. After a few days or weeks, a gradual return of spinal reflexes marks the end of spinal shock. Reflexes then become hyperactive, and muscle tone is increased significantly, particularly in antigravity muscles. Spasticity is common, although rigidity occasionally occurs. Most often, passive range of motion causes the “clasp-knife” phenomenon, probably because of the activation of the two varieties of stretch receptors: (1) the muscle spindles and (2) the Golgi tendon organ. (Muscle function is discussed in Chapter 41.) With pyramidal motor syndrome, predominantly the flexors of the arms and extensors of the legs are affected.
Lower Motor Neuron Syndromes: Lower (primary, alpha) motor neurons are the large motor neurons in the anterior (ventral) horn of the spinal cord and the motor nuclei of the brainstem. The axons from these nerve cell bodies bring nerve impulses from upper motor neurons to the skeletal muscles via the anterior spinal roots or cranial nerves. (Figure 16-26). Dysfunction in this motor system impairs voluntary and involuntary movement. The degree of paralysis or paresis is proportional to the number of lower motor neurons affected. If only a portion of the motor units that supply a muscle is affected, only partial paralysis or paresis results. If all the motor units are affected, a complete paralysis results. Other clinical manifestations also are proportional to the degree of dysfunction, but the precise manifestations depend on the location of the dysfunction in the motor unit and in the CNS.
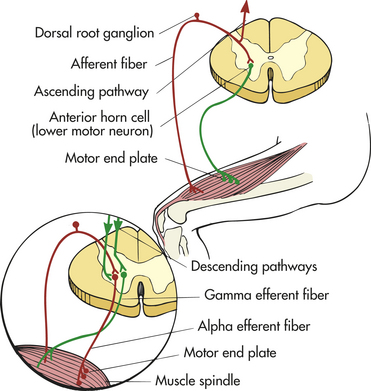
Figure 16-26 Component structure of a lower motor neuron, including motor (efferent) and sensory (afferent) elements. Top, Anterior horn cell (in anterior gray column of spinal cord and its axon), terminating in motor end plate as it innervates extrafusal muscle fibers in the quadriceps muscle. Detailed enlargement, Sensory and motor elements of the gamma loop system. The gamma efferent fiber is shown innervating the polar, or end, region of the muscle spindle (sensory receptor of skeletal muscle). Contraction of muscle spindle fibers stretches the central portion of the spindle and causes the afferent spindle fiber to transmit the impulse centrally to the cord. Muscle spindle afferent fibers in turn synapse on the anterior horn cell and are transmitted by way of gamma-efferent fibers to skeletal (extrafusal) muscle, causing it to contract. Muscle spindle discharge is interrupted by active contraction of extrafusal muscle fibers.
Small motor (gamma) neurons, which function to maintain muscle tone and protect the muscle from injury, also are necessary for normal motor movement. These neurons depend on input from the muscle spindle (arriving through an afferent limb rising to the cord). Dysfunction in this motor system impairs tone and reduces the tendon reflexes, causing hyporeflexia. The muscle is lax and soft, with a decrease in normal tone, or hypotonia, which impairs voluntary and involuntary motor movements. The muscles become susceptible to damage from hyperextensibility because the normal protective mechanisms that prevent muscle fiber injury are impaired. The degree of tone loss and the loss of tendon reflexes are proportional to the dysfunction in these reflex motor units.
Generally in a pathologic process the large and small motor neuron systems are equally affected. Therefore, the paresis and paralysis caused by a disorder of the lower motor neurons are called flaccid paresis and flaccid paralysis, respectively, because the muscle has reduced or absent tone and is accompanied by hyporeflexia or areflexia (loss of tendon reflexes).
A few gamma neuropathies (small motor neuron disorders) affect only the gamma motor system. A manifestation of these disorders is a marked reduction in the deep tendon reflexes, which are strikingly out of proportion to the degree of muscle weakness present.
Denervated muscles (i.e., muscles that have lost their nervous system input) undergo atrophy over weeks to months, mostly from disuse. Denervated muscles also demonstrate fasciculations, which are seen as muscle rippling or quivering under the skin. Occasionally denervated muscles cramp. Fibrillation (isolated contraction of a single muscle fiber) also may occur, although this manifestation is not visible clinically.
Amyotrophies: Lower motor neuron syndromes originating in the anterior horn cells or the motor nuclei of the cranial nerves are called amyotrophies. Paralytic poliomyelitis is the prototype of these disorders. It involves a severe inflammatory reaction in motor neurons, some of which do not survive, leaving a permanent lower motor neuron syndrome.
Several pathologic processes may give rise to an amyotrophy. A virally induced or postinfectious or postvaccination inflammatory process may injure or destroy anterior horn cells or cranial nerve cell bodies. Most of these inflammatory processes are mild and are followed by rapid cellular recovery (see What’s New? Bell Palsy).
In the amyotrophies, muscle strength, muscle tone, and muscle bulk are affected in the muscles innervated by the involved motor neurons. The paresis and paralysis associated with anterior horn cell injury are segmental, but because each
muscle is supplied by two or more roots, the segmental character of the weakness may be difficult to recognize. When cranial nerve motor nuclei are affected (these lack nerve roots and have only small rootlets near the point of exit from the brainstem), the distribution of the motor weakness follows that of the peripheral nerve. The weakness may involve distal muscles, proximal muscles, and the muscles of midline structures. Hypotonia and hyporeflexia or areflexia are present.
The atrophy associated with amyotrophy is segmental when the anterior horn cells of the spinal cord are involved and follows the distribution of the peripheral nerve when the motor nuclei of the cranial nerves are affected. The atrophy may be in distal, proximal, or midline muscles. Fasciculations are particularly associated with primary motor neuron injury, and muscle cramps are common. Mild fatigue is a common complaint. If the pathologic process is limited to the primary motor neuron, no sensory changes are evident.
Because degenerative disorders cause loss of nerve cells in the anterior horn or motor nuclei, the surviving cells are small, shrunken, and filled with lipofuscin. Lost neurons are replaced by astrocytes. The roots or rootlets are thin, and the muscles show denervation and atrophy.
Several brainstem syndromes involve damage to one or more of the cranial nerve nuclei. These are called nuclear palsies (Table 16-25) and may be caused by vascular occlusion, tumor, aneurysm, tuberculosis, or hemorrhage.
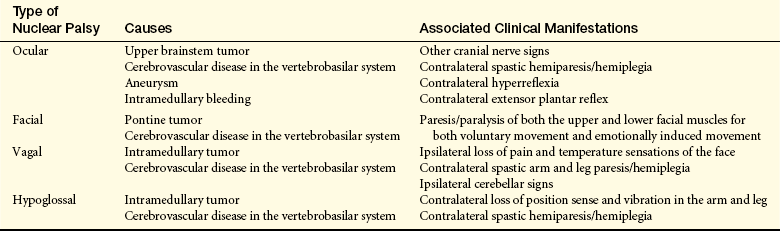
The anterior horn cells and the motor nuclei of the cranial nerves may be affected secondarily in many severe pathologic processes that primarily involve the peripheral nerves. The condition may extend proximally to affect the nerve roots or rootlets and the motor neurons themselves, a process commonly seen, for example, in Guillain-Barré syndrome. If sufficient numbers of motor neurons are destroyed, permanent loss of motor function results because regeneration of the damaged axons requires a living neuronal cell body.
A group of degenerative disorders principally cause progressive motor cell atrophy. One of these is progressive spinal muscular atrophy, in which the anterior horn cells of the spinal cord are the affected motor neurons. This disorder occurs in adults and closely resembles the familial progressive muscular atrophies that occur in infants and children and are considered inherited metabolic disorders (see Chapter 43). If the motor nuclei of the cranial nerves are affected instead of the anterior horn cells, the disorder is labeled progressive bulbar palsy, so named because the myelencephalon originally was called the bulb and a degenerative process causes a progressively more serious condition. When any lower motor neuron syndrome involves the cranial nerves that arise from the bulb (i.e., cranial nerves IX, X, and XII), the dysfunction is called a bulbar palsy.
The clinical manifestations of bulbar palsy include paresis or paralysis of the jaw, face, pharynx, and tongue musculature. Articulation is affected, especially articulation of the lingual (r, n, l), labial (b, m, p, f), dental (d, t), and palatal (k, g) consonants. Modulation is impaired, making the voice rasping or nasal. Pharyngeal reflexes are diminished or lost. Palate and vocal cord movement during phonation is impaired, and chewing and swallowing are affected. The facial muscles are weak, and the face appears to droop. The jaw jerk is decreased. Atrophy eventually becomes apparent, as do fasciculations. All these manifestations become progressively worse, leading to aspiration, malnutrition, possible dehydration, and an inability to communicate verbally.
Hyperkinesia
Hyperkinesia (excessive movement) represents the second broad category of abnormal movements. Within this category are a number of specific hyperkinesia syndromes (Table 16-26). Also included in the general category of hyperkinesias are dyskinesias, that is, abnormal involuntary movements.
Table 16-26
Types of Hyperkinesia Syndromes
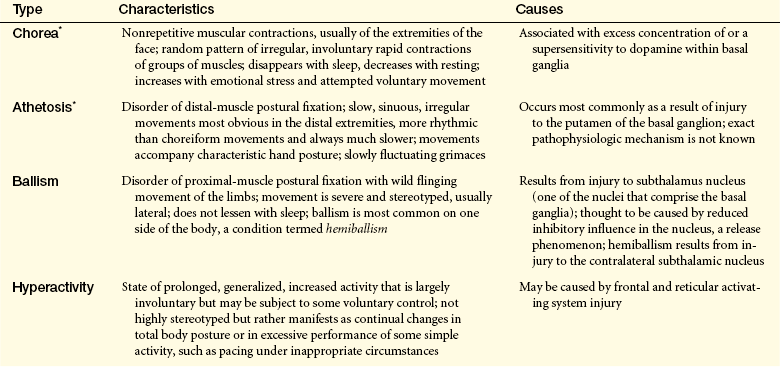


∗Choreoathetosis involves chorea and athetosis; precise pathophysiology unknown.
Paroxysmal dyskinesias are abnormal, involuntary movements that occur as spasms. The type of dyskinesia varies depending on the specific disorder.
Tardive dyskinesia is the involuntary movement of the face, trunk, and extremities. Although the condition occurs occasionally in individuals with Parkinson disease, it usually occurs as a side effect of prolonged first- or second-generation antipsychotic drugs.55 The antipsychotic drugs cause denervation hypersensitivity so that it mimics the effect of too much dopamine. The most common symptom of tardive dyskinesia is rapid, repetitive, stereotypic movements. Most characteristic is continual chewing with intermittent protrusions of the tongue, lip smacking, and facial grimacing. Stereotypic movements are believed to be a form of excessive dopaminergic activity.
Other movement disorders under this category are (1) complex repetitive movements, including automatism, stereotype, complex tics, compulsions, perseverations, and mannerisms; (2) positivism (excessive reactions to certain stimuli); and (3) paroxysmal excessive activity, including cataplexy and excessive startle reaction.
Huntington Disease
Huntington disease (HD), also known as chorea, is a relatively rare, hereditary-degenerative disorder diffusely involving the basal ganglia and cerebral cortex. The onset of HD is usually between 25 and 45 years of age, when the trait may already have been passed to the victim’s children. The disorder has a prevalence rate of approximately 2 to 8 per 100,000 persons and occurs in all races.56
PATHOPHYSIOLOGY HD is inherited as an autosomal dominant trait with high penetrance. The genetic defect is on the short arm of chromosome 4. There is an abnormally long polyglutamine tract in the huntingtin protein that is toxic to neurons caused by a cytosine-adenine-guanine (CAG) trinucleotide repeat expansion (40 to 70 repeats instead of 9 to 34).57 Age of onset of symptoms is related to the length of the repeat sequences and mechanisms of toxicity. Increased length leads to progressively earlier presentations.
The principal pathologic feature of HD is severe degeneration of the basal ganglia, particularly the caudate and putamen nuclei, and the frontal cerebral cortex (Figures 16-27 and 16-28). Tangles of protein collect in brain cells and chains of glutamine on the abnormal molecules stick to each other.58 Early in the disease, selective loss of the striatal GABA/enkephalin pathway to the lateral aspect of the pallidum occurs. The basal ganglia normally contain a preponderance of GABAergic (GABA-secreting) neurons, including the pathway between the basal ganglia and substantia nigra (pallidonigral pathway). Basal ganglia and nigral depletion of GABA, an inhibitory neurotransmitter, is the principal biochemical alteration in HD. Degeneration of the GABAergic pallidonigral pathway causes GABA depletion in the substantia nigra with decreased inhibitory GABA activity on dopaminergic neurons in the substantia nigra and a relative excess of dopaminergic activity in the basal ganglial feedback circuit within the cerebral cortex. A relative excess of dopaminergic activity in this circuit, as in HD, is manifested by hypotonia and hyperkinesia (involuntary, fragmentary movements such as chorea). Loss of excitatory glutamate may liberate the pathway from the thalamus to the premotor cortex, impairing modulation of movement later in the course of the disease. Within the neurons, producing the fuel for brain activity is difficult, with a resultant buildup of lactic acid.
Figure 16-27 Coronal MRI through frontal lobe and head of the caudate nucleus. The head of the caudate normally forms a prominent bulge into the anterior horn of the lateral ventricle (A, inversion recovery image). Profound cell loss in the neostriatum of an individual with Huntington disease greatly diminishes the size of the caudate and renders the lateral wall of the ventricle flat (B, T1-weighted image). The slightly wavy appearance of the magnetic resonance imaging (MRI) in B is the result of movement (tremor) while the scan was being done. (From Haines DE, editor: Fundamental neuroscience, Philadelphia, 1997, Churchill Livingstone.)
Figure 16-28 Caudate blood flow in Huntington disease. Single photon emission computed tomography scan showing reduced caudate blood flow (A) in Huntington disease compared with normal blood flow (B). (From Perkin DG: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
CLINICAL MANIFESTATIONS The classic manifestations of HD are abnormal movement and progressive dysfunction of intellectual processes (dementia) and thought processes. Any one of these features may mark the onset of the disease. Chorea is the most common type of abnormal movement affecting individuals with HD. Choreiform movements begin in the face and arms, eventually affecting the entire body. Symptoms of frontal lobe dysfunction include executive attention deficits, short-term memory loss (working memory); reduced capacity to plan, organize, and sequence, as well as bradyphrenia (slow thinking); and apathy. Restlessness, disinhibition, and irritability are common. Affectively, euphoria or depression or both may be present.
EVALUATION AND TREATMENT The diagnosis of HD is based on family history, clinical presentation of the disorder, and genetic testing. No known treatment is effective in halting the degeneration or progression of symptoms. The discovery in 1983 of the HD marker, called G8, on chromosome 4 paves the way for presymptomatic diagnosis of the disorder and isolation of the HD gene. Recombinant genetic techniques and neuroprotective drug strategies may someday prevent or control the disorder.59
Hypokinesia
Hypokinesia (decreased movement) is loss of voluntary movement despite preserved consciousness and normal peripheral nerve and muscle function. Types of hypokinesia include akinesia, bradykinesia, and loss of associated movement.
Akinesia: Akinesia is an absence, poverty, or lack of control of associated and voluntary muscle movements. There is a disturbance in the time it takes to perform a movement. Akinesia is related to dysfunction of the extrapyramidal system, as in parkinsonism. Pathogenesis is related to either a deficiency of dopamine or a defect of the postsynaptic dopamine receptors, which occurs in parkinsonism (see Parkinson disease, page 572).
Bradykinesia: Bradykinesia is slowness of voluntary movements. There is a disturbance in the time it takes to perform a movement. In bradykinesia all voluntary movements become slow, labored, and deliberate. Bradykinesia consists of (1) difficulty in initiating movements, (2) difficulty in continuing movements smoothly, and (3) difficulty in performing synchronous (at the same time) and consecutive tasks. Difficulty in initiating movements ranges from slight hesitancy to severe freezing (transient, helpless immobility). Each intended movement requires effort. Difficulty in continuing motions smoothly causes jerky, irregular, rapid movements, which then decrease in rate and amplitude until they stop. The individual is scarcely aware of the cessation. Difficulty in performing synchronous and consecutive tasks means that each motor act is performed separately. The individual is unable to integrate two acts or to change from one motor pattern to the next with a single smooth motion.
Loss of Associated Movement: In hypokinesia the normal, habitually associated movements that provide skill, grace, and balance to voluntary movements are lost. Decreased associated movements accompanying emotional expression cause an expressionless face, a statue-like posture, absence of speech inflection, and absence of spontaneous gestures. Decreased associated movements accompanying locomotion cause reduction in arm and shoulder movements, in hip swinging, and in rotary motion of the cervical spine.
Parkinson Disease
Parkinson disease (PD) is a commonly occurring degenerative disorder of the basal ganglia (corpus striatum, globus pallidus, subthalamic nucleus, and substantia nigra) involving the dopaminergic (dopamine-secreting) nigrostriatal pathway. Nigrostriatal disorders produce a syndrome of abnormal movement called parkinsonism (Parkinson syndrome, parkinsonian syndrome).
Etiologic classification of parkinsonism includes primary parkinsonism and secondary parkinsonism (Box 16-5). The onset of PD occurs after 40 years of age, with mean onset of 60 years of age.60 Equal incidence occurs in both sexes.23 PD is one of the most prevalent of the primary CNS disorders and a leading cause of neurologic disability in individuals older than 60 years. Approximately 1% to 2% of the U.S. population older than the age of 60 is affected—an estimated 1.5 million individuals. The familial form represents about 10% of PD; however, the majority of cases are sporadic or idiopathic. Several PD genes have been identified, the most significant of which are identified in Table 16-18.
PATHOPHYSIOLOGY The hallmark pathologic features of PD are loss of dopaminergic pigmented neurons in the substantia nigra (SN) pars compacta with dopaminergic deficiency in the putamen portion of the striatum (the striatum includes the putamen and caudate nucleus) (Figure 16-29). Dopamine loss in other brain areas including the brainstem, thalamus, and cortex also occurs.61 Degeneration of the dopaminergic nigrostriatal pathway to the basal ganglia results in underactivity of the direct motor pathway (normally facilitates movement) (Figure 16-30) and overactivity of the indirect motor loop (normally inhibits movement). This results in inhibition of the motor cortex manifested with bradykinesia and rigidity. The subthalamic nucleus (STN) overactivity also influences the limbic system60 accounting for emotional signs and symptoms. Neuronal loss within the cerebral cortex is found in one half of individuals with PD.
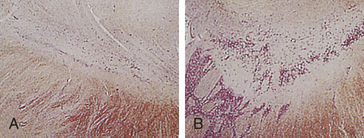
Figure 16-29 Atrophic substantia nigra (A) compared with normal control (B). (From Perkin DG: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
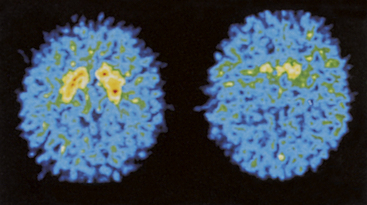
Figure 16-30 Reduced fluorodopa in Parkinson disease. Positron-emission tomography scan showing reduced fluorodopa uptake in the basal ganglia (right) compared with a normal control (left). (From Perkin DG: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
Lewy bodies, fibrillar intracellular eosinophilic inclusions, and high concentrations of alpha-synuclein, ubiquitin, tau protein, tuberculin, and other proteins, are found in the SN, LC, and other areas of the brain and are a marker for neuronal degeneration.62 Degeneration of the LC, which contains noradrenergic neurons, also occurs in PD. Norepinephrine is thought to be neuroprotective and loss of LC neurons may be associated with a worsening of disease progression and the behavioral symptoms of PD.63 Molecular events thought to be associated with the neurodegeneration of PD include mitochondrial dysfunction, oxidative stress, abnormal folding and accumulation of alpha-synuclein, abnormal phosphorylation, and dysfunction of the ubiquitin proteosome system64,65 (Figure 16-31).
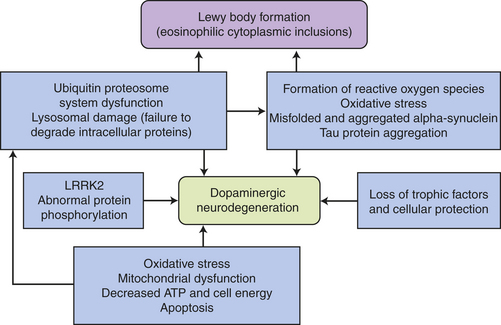
Figure 16-31 Proposed pathogenesis of dopaminergic neurodegeneration in Parkinson disease. ATP, Adenosine triphosphate; LRRK2, leucine-rich repeat kinase -2. (Data from Engelender S: Autophagy 4[3]:372-374, 2008; Lim KL, Tan JM: BMC Biochem 22:[8 Suppl 1]:S13, 2007; Mizuno Y et al: Philos Trans R Soc Lond B Biol Sci 363[1500]:2215-2227, 2008.)
CLINICAL MANIFESTATIONS Onset of symptoms is insidious and symptoms appear after a 70% to 80% loss of pigmented nigral neurons and a loss of 60% to 90% of striatal dopamine.66 The classic motor manifestations of PD are bradykinesia, tremor at rest (resting tremor), rigidity (muscle stiffness), hypoakinesia (poverty of movement), and postural abnormalities. These manifestations may develop alone or in combination, but as the disease progresses, all four are usually present to at least some degree. There is no true paralysis. A modified Hoehn and Yahr scale67 can be used to assess progression of symptoms:
1. Unilateral involvement, may have tremor of one limb
2. Bilateral involvement, balance intact
3. Bilateral involvement, slowing of body movement, mild to moderate postural instability, and gait difficulty
4. Bilateral involvement with severe postural instability, rigidity, and bradykinesia present
5. Bilateral involvement with inability to walk, confinement to wheelchair, cachexia present
In early stages of the disease, reflex, sensory, and mental status are usually normal. Nonmotor symptoms associated with PD include hyponosmia, fatigue, pain, autonomic dysfunction, sleep fragmentation, depression, and dementia with or without psychosis.68
Parkinsonian tremor, the most conspicuous and most variable symptom, is usually the first motor symptom to appear. It is an asymmetric, regular, rhythmic, low-amplitude tremor (4 to 6 cycles/second, with slowly alternating flexion-extension contraction).69 Later the tremor becomes symmetric at 7 to 12 cycles per second. It is a tremor at rest, disappearing briefly during the course of a voluntary movement and reappearing when the limb is held in a stationary position. Intensity and amplitude of the tremor vary. The arm is more affected than the leg. The head is rarely involved. Seventy percent of individuals with PD have this tremor, and 20% have a postural (kinesic) tremor or both tremor types. All tremors are increased by stress and anxiety.
Parkinsonian tremor appears to result from instability of feedback from the basal ganglia to the cerebral cortex caused by loss of the inhibitory influence of dopamine in the basal ganglia. Increased oscillation in the normal feedback cycles of the motor outflow feedback circuit when the muscles are at rest produces the tremor. When the individual performs voluntary movements, the tremor becomes temporarily blocked, presumably because other motor control signals arriving in the thalamus override the abnormal basal ganglial signals. As the disorder worsens, tremor may lessen as rigidity supervenes. The postural tremor is associated with damage to the cerebellofugal pathway to the red nucleus, a pathway that subserves communication from muscle spindles to the thalamus and motor cortex.
Parkinsonian rigidity is an increased resistance to the passive movement of a joint that impedes active and passive movement. The first symptoms of rigidity may be painful muscle cramps in the toes or hands. More commonly the limb feels stiff, heavy, tired, or aching. Plastic rigidity is constant throughout the entire range of motion and is felt as lead-pipe resistance during passive movement. Cogwheel rigidity, brief palpable jerks, is accompanied by tremor. The mechanism underlying rigidity is unclear, but there is increased resting muscle activity of antagonistic muscle groups with enhancement of the long-latency component of the stretch reflex.60
Parkinsonian bradykinesia is poverty of associated and voluntary movements. It is the most prevalent and crippling symptom and often is overlooked in the early stages. The pathophysiology underlying the bradykinesia is an overactive subthalamic nucleus (STN) that inhibits the motor thalamus and motor cortex.60 It is associated with dopamine deficiency and failure of the mechanism programming movement patterns manifested as a defect in the voluntary production of smooth motions at different speeds.70
All striated muscles—extremity, trunk, ocular, facial—are affected eventually, including muscles of mastication (chewing), deglutition (swallowing), and articulation. Micrographia is present. Extreme underactivity in the individual with PD makes the person appear stiff, even when resistance to passive movement cannot be felt. Bradykinesia is a separate phenomenon from rigidity and may be severe even in the presence of rigidity. Individuals state that they feel “wooden” (as though moving against resistance) and complain of rapid, severe fatigue.
Hypokinesia, or decreased frequency or absence of associated movements, is one of the earliest akinetic symptoms. Individuals with PD sit and lie motionless for long periods without the little shifts a normal person makes to prevent discomfort and stiffness. Bradykinesia, or slowness of voluntary movements, is characterized by difficulty initiating, continuing, or synchronizing movements. Both associated and voluntary movements are interspersed by freezing (an inability to continue movement). Freezing may be precipitated by (1) increasing the effort to move, (2) turning, and (3) initiating certain types of tactile and visual contact.
Postural Abnormalities: Three types of postural abnormalities occur in individuals with PD: (1) disorders of postural fixation, (2) disorders of equilibrium, and (3) disorders of righting. The disorder of postural fixation associated with PD is involuntary flexion of the head and neck. The individual is unable to maintain an upright position of the trunk while standing or walking. The stooped (flexed, forward leaning) posture is characteristic (Figure 16-32). Postural abnormalities of the hands and feet also occur. Postural abnormalities are caused by a loss of normal postural reflexes.
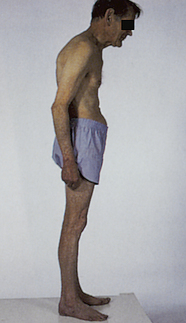
Figure 16-32 Stooped posture of Parkinson disease. (From Perkin DG: Mosby’s color atlas and text of neurology, London, 1998, Mosby-Wolfe.)
Disorders of equilibrium result from loss of postural stability. The person with PD is unable to make the appropriate postural adjustment to tilting or falling and falls like a post when starting to tilt. The festinating gait (short, accelerating steps) of the person with PD is an attempt to maintain an upright position while walking (see Figure 16-32). Individuals also are unable to right themselves when changing from a reclining or crouching position to a standing position and when rolling over from a supine to a lateral or prone position.
Autonomic and Neuroendocrine Symptoms: Autonomic and neuroendocrine dysfunctions in PD produce nonmotor symptoms that are distressing but not incapacitating. The basal ganglia influences hypothalamic function (autonomic and neuroendocrine) through pathways connecting the hypothalamus with the basal ganglia and cerebral cortex. Common autonomic symptoms in PD include inappropriate diaphoresis, gastric retention, constipation, and urinary retention. Cardiac sympathetic denervation is common and causes neurogenic orthostatic hypotension.71 A symptom attributed to neuroendocrine dysfunction is seborrhea. Hypothalamic hypersecretion of hormone-releasing factors acting on the anterior pituitary causes hypersecretion of androgenotropic hormones producing sebum hypersecretion by sebaceous glands. The resulting seborrhea is characterized by oily skin with seborrheic dermatitis along the hairline and in chin-nasal creases.
Cognitive-Affective Symptoms: Fifty percent of people with PD have a depression that is now believed to be an inherent part of the pathologic state of the disease (an endogenous depression), not a response to the situation. Thirty percent of individuals treated on an outpatient basis for PD have a dementia, and 80% of those with PD requiring institutional care have dementia as well. Dementia is more common in individuals older than 70 years. Pathologically, in those with dementia, findings include loss of cholinergic cells in the basal nucleus of Meynert; neuronal loss, senile plaques, and neurofibrillary tangles in the neocortex; and amyloid changes in small blood vessels. Lewy bodies are distributed diffusely in many neocortical neurons, making this a Lewy body dementia. The individual evidences disorientation; confusion; memory loss; distractibility; and difficulty with concept formation, abstraction, calculations, thinking, and judgment. Although the symptoms fluctuate, they progressively worsen. Anxiety disorders; impulse-control disorders; and punding, a disorder of stereotypical motor behavior in which there is intense fascination with repetitive handling and examining of mechanical objects, are recognized features of PD. A cognitive disorder unassociated with either a dementia or depression, called bradyphrenia, also is present. This disorder may appear early in the course of the disease and may progress to dementia. Bradyphrenia is caused by disruption of the caudal basal ganglion connections and outflows. The clinical manifestations are slowness of thinking, poverty of thought (diminished imagination and insight), and difficulty formulating thoughts (decreased ability to conceptualize, plan, decide, or improvise).
Nonmotor symptoms are common in PD including sensory dysfunction with anosmia, ageusia, pain, paresthesias, and disturbances in the sleep-wake cycle. Excessive daytime sleepiness is experienced in more than 50% of persons.72,73
Influence of Symptoms: Early in the disease, people often experience a sleep benefit; that is, symptoms decrease with sleep. Also symptoms fluctuate in an on-off pattern. Stress influences symptoms adversely, but the underlying mechanism is unclear. The person’s mental status may be further compromised by the side effects of the medication taken to control symptoms.
The combination of all the parkinsonian symptoms gives the individual a characteristic appearance: a wide-eyed, unblinking, staring expression with the facial muscles smoothed out and almost immobile. Saliva frequently drools from the corners of the slightly open mouth. The skin of the face is frequently greasy. The gait is pathognomonic: the individual walks with slow, short, shuffling steps; the arms are flexed, abducted, and held stiffly at the side; and the trunk is bent slightly forward. The person may break into a run spontaneously or when pushed forward or backward. Because of the disorder of postural fixation, the tendency is to fall to the side. Postural instability, sleep disorders, and difficulty concentrating are some of the most depressing symptoms for people with PD.74
EVALUATION AND TREATMENT The diagnosis of PD is made on the basis of two of the four cardinal symptoms: (1) resting tremor, (2) bradykinesia, (3) cogwheel rigidity, and (4) postural instability. One of the two symptoms must be resting tremor or bradykinesia (criteria from Core Assessment Program for Intracerebral Transplantation [CAPIT]).75 A combination of imaging techniques, clinical evaluations, biochemical markers, and genetic tests support the diagnosis of PD.76 Median time between diagnosis and death is 9 years.23
The aim of drug therapy is to restore striatal dopamine using oral drugs such as levodopa (L-dopa), a precursor of dopamine (dopamine does not cross the blood-brain barrier), dopamine agonists that directly stimulate dopamine receptors, anticholinergic drugs, antihistamines, and amantadine. L-dopa is effective in reducing symptoms in early PD but can cause motor fluctuations, “off” periods, and dyskinesia in the long term. Monoamine oxidase B inhibitors, which inhibit the breakdown of endogenous dopamine, may improve symptoms, reduce motor fluctuations, and delay the need for L-dopa but can cause adverse effects. Adding catechol-O-methyltransferase (COMT) inhibitor prolongs the half-life of dopamine (COMT metabolizes dopamine in the synapse). Dopamine agonists to L-dopa or a dopamine agonist alone may reduce “off” time or improve symptoms but can also increase disability.77 Surgery may be considered in later stages of PD. Thalamotomy and pallidotomy are being replaced with deep brain stimulation as an approach to controlling medically resistant motor symptoms and L-dopa–related peak dose dyskinesia.78,79 Implants of stem cells, fetal cells, and gene therapy hold promise for future treatment.80
Dysphagia and general immobility are special problems of the individual with PD requiring preventive, symptomatic, supportive, and rehabilitative management, such as physiotherapy and speech therapy. Nursing interventions, occupational therapy (OT), physical therapy (PT), speech, language, and swallowing therapy are considered effective and safe for improving functional status.81,82
Alterations in Complex Motor Performance
The alterations in complex motor performance include disorders of posture (stance), disorders of gait, and disorders of expression.
Disorders of Posture (Stance)
An inequality of tone in muscle groups because of a loss of normal postural reflexes results in a posturing of limbs. Many reflex systems govern tone and posture, but the most important factor in posture control is the stretch reflex, in which stretching of extensor (antigravity) muscles causes increased extensor tone and inhibited flexor tone. Four types of disorders of posture are described: (1) dystonic posture, (2) decerebrate posture, (3) basal ganglion posture, and (4) senile posture. Equilibrium and balance are disrupted when postural disorders are present.
Dystonia is the maintenance of an abnormal posture through muscular contractions. When muscular contractions are sustained for several seconds, they are called dystonic movements, such as in choreoathetoid movements associated with high levels of L-dopa; when contractions last for longer periods, they are called dystonic postures, such as in torticollis. Dystonic postures may last for weeks, causing permanent fixed contractures. Dystonia has been associated with basal ganglia abnormality, but the exact pathophysiologic mechanisms are unknown (Box 16-6). One particularly relevant dystonic posture already discussed in this chapter is decorticate (striatal posture or upper motor neuron dysfunction posture), which may be unilateral or bilateral in occurrence. Decorticate posture (also referred to as antigravity posture or hemiplegic posture) is characterized by upper extremities flexed at the elbows and held close to the body and by lower extremities that are externally rotated and extended. Decorticate posture is believed to occur when the brainstem, which facilitates the antigravity position, is not inhibited by the motor function of the cerebral cortex. Upper motor neuron posture is more commonly described as the arm flexed at the elbow, with a wristdrop; the leg inadequately bent at the knee, with the hip excessively circumabducted; and the presence of a footdrop.
Decerebrate posture refers to increased tone in extensor muscles and trunk muscles, with active tonic neck reflexes. When the head is in a neutral position, all four limbs are rigidly extended. The decerebrate posture is caused by severe injury to the brain and brainstem, resulting in overstimulation of the postural righting and vestibular reflexes.
Basal ganglion posture refers to a stooped, hyperflexed posture with a narrow-based, short-stepped gait. This posture abnormality results from the loss of normal postural reflexes and not from defects in proprioceptive, labyrinthine, or visual function. Dysfunctional equilibrium results from the loss of postural stability, and thus the individual is unable to make the appropriate postural adjustment to tilting or loss of balance and falls instead. Dysfunctional righting is the inability to right oneself when changing from a lying or crouching to a standing position or when rolling from the supine to the lateral or prone position. Dysfunctional postural fixation is the involuntary flexion of the head and neck, causing the person difficulty in maintaining an upright trunk position while standing or walking. Basal ganglion dysfunction accounts for this posture.
Senile posture is characterized by an increasingly flexed posture similar to that caused by basal ganglion dysfunction. The posture is associated with frontal lobe dysfunction, but the primary pathophysiology is not well described.
Disorders of Gait
Four predominant types of gait disorder are (1) upper motor neuron dysfunction gait, (2) cerebellar (ataxic) gait, (3) basal ganglion gait, and (4) senile (frontal lobe, pseudoparkinsonian) gait. As with posture, equilibrium and balance are affected with gait disturbances.
Several upper motor neuron gaits exist. In the presence of mild upper motor neuron dysfunction, a footdrop may appear only with fatigue. The individual may complain of hip and leg pain. A spastic gait, which is associated with unilateral injury, is manifested by a shuffling gait with the leg extended and held stiff, causing a scraping over the floor surface. An impaired leg swing around the body rather than an appropriate lifting and placing of the leg is noted. The foot may drag on the ground, and the person tends to fall to the affected side. A scissors gait is associated with bilateral injury and spasticity. The legs are abducted, causing them to touch each other. As the person walks, the legs are still swung around the body but then cross in front of each other because of adduction. Injury to the pyramidal system accounts for these gaits.
A cerebellar gait manifests as a wide-based gait with the feet apart and often turned outward or inward for greater stability. The pelvis is held stiff, and it seems to be independent of the trunk. The individual staggers when walking. Cerebellar dysfunction accounts for this particular gait.
A basal ganglion gait and a senile gait are both broad-based gaits. The person walks with small steps and a decreased arm swing. The head and body are flexed and the arms are semiflexed and abducted, whereas the legs are flexed and rigid in more advanced states. Basal ganglion and frontal lobe dysfunction, respectively, account for these two gaits.
Disorders of Expression
Disorders of expression involve the motor aspects of communication and include (1) hypermimesis, (2) hypomimesis, and (3) dyspraxias and apraxias. Hypermimesis is a disinhibition phenomenon that most commonly manifests as pathologic laughter or crying. Pathologic laughter is associated with right hemisphere injury, and pathologic crying is associated with left hemisphere injury. The exact pathophysiology is not known. Hypomimesis manifests as aprosody, or the loss of voice modulation (pitch, speed, emphasis, emotion). Receptive aprosody involves an inability to understand emotion in speech and facial expression, whereas expressive aprosody involves the inability to express emotion in speech and facial expression. Aprosody is associated with right hemisphere damage.
Dyspraxia is the partial inability and apraxia is the complete inability to perform purposeful or skilled motor acts in the absence of paralysis, sensory loss, abnormal posture and tone, abnormal involuntary movement, incoordination, or inattentiveness. These are disorders of learned skilled movements.83 Dyspraxia and apraxia are associated with vascular disorders, trauma, tumor, degenerative disorders, infections, and metabolic disorders. The medial premotor cortex, including the supplementary motor area (SMA), appears to play a role in skilled movements as does the convexity premotor areas83 (Table 16-27).
Table 16-27
| Types | Description | Location |
| Ideomotor apraxia | Impairment in selecting, sequencing, and spatial orientation of movements involved in gestures (spatial and temporal production errors) | Left parietal cortex (angular gyrus) or supramarginal gyrus |
| Posterior form | Difficulty performing in response to command and imitation; cannot discriminate well between poorly performed and well-performed acts | Left parietal cortex (angular gyrus or supramarginal gyrus) lesion |
| Anterior form | Performs poorly to command and imitation but comprehends and discriminates pantomime | Lesions anterior to the supramarginal gyrus, which disconnects visual kinesthetic motor engrams from premotor and motor areas |
| Conduction apraxia | Greater impairment in performance when imitating movements than when pantomiming to command; comprehends pantomime and gesture but cannot perform the movements | Location unknown at this time |
| Disassociation apraxia | Inability to gesture normally to command and required verbal mediation has good performance with imitation and actual tools and objects | Callosal abnormalities but not all locations known |
| Ideational apraxia | Inability to carry out an ideational plan or a series of acts in the proper sequence | Location unclear at this time |
| Conceptual apraxia | Cannot recall type of action associated with specific tools, utensils, or objects (content and tool selection errors; may be unable to recall which tool is associated with a specific object or may have impaired mechanical knowledge) | Bilateral frontal and parietal dysfunction |
True dyspraxias occur when the connecting pathways between the left and right cortical areas are interrupted causing language-motor and motor representation disconnections between the hemispheres (Figure 16-33). Dyspraxias may result from any pathologic process that disrupts the cortical areas necessary for the conceptualization and execution of a complex motor act or the communication pathways within the left hemisphere or between the hemispheres.
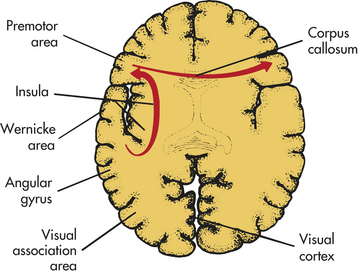
Figure 16-33 Pathways disrupted in dyspraxias. Formulation of the idea of the motor act is believed to originate in the region of the supramarginal gyrus in the inferior left parietal lobe. This area is connected via associational pathways to the left premotor cortex. The left premotor cortex is connected through the corpus callosum to the right premotor and motor areas. An injury that interrupts the pathways between the left supramarginal gyrus and the premotor region produces a dyspraxia that involves the entire body. An injury that disrupts the callosal pathways produces a dyspraxia of the left side of the body only.
Extrapyramidal Motor Syndromes
Because the extrapyramidal system encompasses all the motor pathways except the pyramidal system, two types of motor dysfunction make up the extrapyramidal motor syndromes: (1) the basal ganglia motor syndromes and (2) the cerebellar motor syndromes. Unlike pyramidal motor syndromes, both extrapyramidal motor syndromes result in movement or posture disturbance without significant paralysis, along with other distinctive symptoms (Table 16-28).
Table 16-28
Pyramidal versus Extrapyramidal Motor Syndromes
| Manifestations | Pyramidal Motor Syndrome | Extrapyramidal Motor Syndrome |
| Unilateral movement | Paralysis of voluntary movement | Little or no paralysis of voluntary movement |
| Tendon reflexes | Increased tendon reflexes | Normal or slightly increased tendon reflexes |
| Babinski sign | Present | Absent |
| Involuntary movements | Absence of involuntary movements | Presence of tremor, chorea, athetosis, or dystonia |
| Muscle tone | Spasticity in muscles (e.g., clasp-knife phenomenon) | Plastic (equal throughout movement) rigidity or intermittent (cogwheel) rigidity generalized but predominate in flexors of limbs and trunk |
| Hypertonia present in flexors of arms and extensors of legs | Hypotonia in cerebellar disease |
Basal Ganglia Motor Syndromes
Basal ganglia motor syndromes are movement disorders that involve either a paucity or an excess of movements. Stress and nervous tension typically worsen the symptoms, whereas relaxation improves motor performance. Akinesia may occur despite normal strength. Involuntary movements, such as tremor, chorea, ballism, athetosis, and dystonia, also may occur and probably are caused by the loss of the normal modulating effects of the corpus striatum and other parts of the basal ganglia.
Basal ganglia motor syndromes also are characterized by alterations in muscle tone and posture. Rigidity, together with the cogwheel phenomenon, is present in all muscle groups but is most prominent in those that maintain flexed position. Postural abnormalities result from the loss of normal postural reflexes. Dysfunctional equilibrium results from the loss of postural stability.
Cerebellar Motor Syndromes
Cerebellar motor syndromes involve the cerebellum and may result in (1) acute loss of muscle tone; (2) difficulty with coordination of voluntary movements (ataxia); (3) minor degrees of muscle weakness, tendency toward fatigue, and impairment of associated movements; and (4) disorders of equilibrium, posture, and gait. Cerebellar effects are chiefly ipsilateral (primarily affecting the same side of the body), so damage to the right cerebellum generally causes symptoms on the right side of the body. Predominant symptoms depend on the area of damage within the cerebellum. The three cerebellar syndromes are the rostral vermis, caudal vermis, and lateral syndromes84 (Table 16-29).
Table 16-29
| Anatomic Location of Dysfunction | Characteristics |
| Rostral vermis (so-called anterior lobe) | Ataxia of stance and gait with varying degrees of instability of the trunk and ataxia of legs; anteroposterior body sway; presence of Romberg sign |
| Caudal vermis (including fl occulonodular lobe) | Truncal, postural, and gait ataxia; omnidirectional body sway; Romberg negative; tendency to fall; saccadic slow pursuit, nystagmus; inability to suppress vestibulo-ocular reflex (doll's eyes) |
| Cerebellar hemisphere (neocerebellar syndrome) | Severe disturbance in ipsilateral limb movements; hypotonia in acute situation; dysmetria (extremity overshooting its target); decomposition of movement; kinetic tremor, past-pointing; deviation of gait; dysarthria |
| Pancerebellum | Ataxia of trunk and bilateral limbs; ataxia of gait and stance; dysarthria; oculomotor disturbance |
Data from Timmann D, Diener HC: Coordination and ataxia. In Goetz GC, editor, Textbook of clinical neurology, St Louis, 2007, Saunders.
Diagnosis of a cerebellar motor syndrome is based on the symptoms, but these may vary because of the individual’s attempts at compensation. Further, the nervous system often can operate well despite destruction of parts of the cerebellum, although the mechanisms responsible for this retained function are not fully understood.
Acute confusional state (acute cerebral failure, acute brain failure) 548
Acute hydrocephalus 560
Agnosia 546
Akinesia 571
Akinetic mutism (AM) 535
Alzheimer disease (dementia of Alzheimer type [DAT], senile disease complex) 553
Amyotrophy 567
Aphasia 546
Apraxia 577
Areflexia 566
Aura 542
Autoregulation 558
Basal ganglia motor syndrome 577
Basal ganglion gait 576
Basal ganglion posture 576
Bradykinesia 571
Brain death (brainstem death) 534
Bulbar palsy 568
Central (transtentorial) herniation 559
Cerebellar gait 576
Cerebellar motor syndrome 577
Cerebral death (irreversible coma) 534
Cerebral edema 559
Cheyne-Stokes respiration 531
Cingulate gyrus herniation (subfalcine herniation, transfalcial herniation) 559
Clonic phase 538
Cogwheel rigidity 573
Coma 528
Communicating (extraventricular) hydrocephalus 560
Content of thought 528
Convulsion 536
Cryogenic epilepsy 542
Cytotoxic (metabolic) edema 560
Decerebrate posture 575
Declarative memory 543
Decorticate posture (antigravity posture, hemiplegic posture) 575
Dementia 543
Diplegia 564
Dysmnesia 543
Dysphasia 546
Dyspraxia 577
Dystonia 575
Dystonic movement 575
Dystonic posture 575
Echolalia 548
Epilepsy 539
Epileptogenesis 538
Epileptogenic focus 538
Extinction 542
Extrapyramidal motor syndrome 577
Flaccid paralysis 566
Flaccid paresis 566
Freezing 571
Gamma neuropathy 566
Gegenhalten (paratonia) 563
Generalized seizure 536
Hemiparesis 564
Hemiplegia 564
Huntington disease (HD) 570
Hydrocephalus 560
Hydrocephalus ex vacuo 560
Hyperkinesia, (excessive movement) 568
Hypertonia (increased muscle tone) 562
Hypokinesia (decreased movement) 571
Hypotonia (decreased muscle tone) 562
Idiopathic epilepsy 539
Increased intracranial pressure 557
Interstitial edema 560
Intracranial pressure 557
Isolated (pure) vigilance defect 544
Locked-in syndrome 535
Memory 543
Minimally conscious state (MCS) 535
Mirror focus 539
Neglect syndrome 542
Neurofibrillary tangle 554
Noncommunicating hydrocephalus 560
Nondeclarative memory (nonconscious) 543
Normal-pressure hydrocephalus (low, adult, occult hydrocephalus) 560
Nuclear palsy 567
Paralysis 564
Paraparesis 564
Paraplegia 564
Paresis (weakness) 564
Parkinson disease (PD) 572
Parkinsonian bradykinesia 573
Parkinsonian rigidity 523
Parkinsonian tremor 573
Parkinsonism (Parkinson syndrome, parkinsonian syndrome) 572
Paroxysmal dyskinesia 569
Partial seizure (focal seizure) 536
Plastic rigidity 573
Posthyperventilation apnea (PHVA) 531
Postictal state 536
Prodroma 542
Prognosis in coma 535
Progressive bulbar palsy 568
Progressive spinal muscular atrophy 567
Pyramidal motor syndrome 564
Quadriparesis 564
Quadriplegia 564
Rigidity 563
Scissors gait 576
Secondary generalization 536
Seizure 536
Seizure initiation 538
Selective attention deficit (orientation) 543
Senile gait 576
Senile plaque 554
Senile posture 576
Sensory inattentiveness 542
Spastic gait 576
Spasticity 563
Spinal shock 565
Status epilepticus 536
Symptomatic epilepsy 539
Tardive dyskinesia 569
Tonic phase 538
Transcortical dysphasia (transcortical sensory dysphasia, mixed transcortical dysphasia, isolated speech center) 548
Uncal herniation (hippocampal herniation, lateral mass herniation) 559
Vasogenic edema 560
Vegetative state (VS) 535
Vigilance 544
Working memory deficit 544
REFERENCES
1. Stevens, R.D., Nyquist, P.A. Types of brain dysfunction in critical illness. Neurol Clin. 2008;26(2):469–486.
2. Ropper, A.H. Coma. In Fauci A.S., et al, eds.: Harrison’s principles of internal medicine, ed 15, New York: McGraw-Hill, 2008.
3. Posner, J.B., et al. Plum and Posner’s diagnosis of stupor and coma, ed 4. New York: Oxford University Press; 2007.
4. McNett, M. A review of the predictive ability of Glasgow Coma Scale scores in head-injured patients. J Neurosci Nurs. 2007;39(2):68–75.
5. Saatman, K.E., et al. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25(7):719–738.
6. Bleck, T.P. Levels of consciousness and attention. In: Gottez C.G., ed. Textbook of clinical neurology. Philadelphia: Saunders, 2003.
7. Drazkowski, J. Determining brain death: back to the basics. Semin Neurol. 2007;27(4):393–399.
8. Greer, D.M., et al. Variability of brain death determination guidelines in leading US neurologic institutions. Neurology. 2008;70(4):284–289.
9. Task Force for the Determination of Brain Death in Children: guidelines for the determination of brain death in children. Arch Neurol. 1987;44(6):587–588.
10. Young, G.B., et al. Brief review: the role of ancillary test in the neurological determination of death. Can J Anaesth. 2006;53(6):62–67.
11. Boss, B.J., Fletcher, A. Severe brain injury rehabilitation: what’s going to happen after critical care? Crit Care Nurs Clin. 2001;13(3):421–431.
12. Owen, A.M., et al. Detecting awareness in the vegetative state. Science. 2006;313(5792):1402.
13. Owen, A.M. Disorders of consciousness. Ann N Y Acad Sci. 2008;1124:225–238.
14. Widjdicks, E.F., Cranford, R.E. clinical diagnosis of prolonged states of impaired consciousness in adults. Mayo Clin Proc. 2005;80(8):1037–1046.
15. Smith, E., Delargy, M. Locked-in syndrome. BMJ. 2005;330(7488):406–409.
16. Lowenstein, D.H. Seizures and epilepsy. In Fauci A.S., et al, eds.: Harrison’s principles of internal medicine, ed 15, New York: McGraw-Hill, 2008.
17. Tuxhorn, I., Kotagal, P. Classification. Semin Neurol. 2008;28(3):277–288.
18. Feen, E.S., Bershad, E.M., Suarez, J.I. Status epilepticus. South Med J. 2008;101(4):400–406.
19. Scharfman, H.E. The neurobiology of epilepsy. Curr Neurol Neurosci Rep. 2007;7(4):348–354.
20. Pitkanen, A., et al. Epileptogenesis in experimental models. Epilepsia. 2007;48(Suppl 2):13–20.
21. Crino, P.B. Gene expression, genetics, and genomics in epilepsy: some answers, more questions. Epilepsia. 2007;48(Suppl 2):42–50.
22. Berg, A.T., Blackstone, N.W. Concepts in classification and their relevance to epilepsy. Epilepsy Res. 2006;70(Suppl 1):S11–S19.
23. BMJ clinical evidence handbook. London: BMJ Publishing Company; 2007.
24. Bate, H., et al. The seizure outcome after amygdalohippocampectomy and temporal lobectomy. Eur J Neurol. 2007;14(1):90–94.
25. Ramani, R. Vagus nerve stimulation therapy for seizures. J Neurosurg Anesthesiol. 2008;20(1):29–35.
26. Elliott, J., Shneker, B. Patient, caregiver, and health care practitioner knowledge of, beliefs about, and attitudes toward epilepsy. Epilepsy Behav. 2008;12(4):547–556.
27. Boss, B.J., Wilkerson, R. Communication: language and pragmatics. In Hoeman S.P., ed.: Rehabilitation nursing: prevention, intervention, & outcomes, ed 4, St Louis: Mosby, 2008.
28. Gabrieli, J.D.E., et al. Memory. In: Goetz C.G., ed. Textbook of clinical neurology. Philadelphia: Saunders, 2003.
29. LaVoie, D.J., Cobia, D.J. Recollecting, recognizing and other act of remembering: an overview of human memory. J Neurol Phys Ther. 2007;31(3):135–144.
30. Manns, J.R., Eichenbaum, H. Learning and memory: brain systems. In Squire L.R., et al, eds.: Fundamental neuroscience, ed 3, Burlington, MA: Academic Press, 2008.
31. Timmann, D., Daum, I. Cerebellar contributions to cognitive functions: a progress report after two decades of research. Cerebellum. 2007;6(3):159–161.
32. Buchanan, T.W. Retrieval of emotional memories. Psychol Bull. 2007;133(5):761–779.
33. Carbeza, R. Role of the parietal regions in episodic memory retrieval: the dual attentional processes hypothesis. Neurophsychologia. 2008;46(7):1813–1827.
34. Osada, T., et al. Towards understanding of the cortical network underlying associative memory. Philos Trans R Soc Lond B Biol Sci. 2008;363(1500):2187–2199.
35. Fisher, S. On genes, speech and language. N Engl J Med. 2005;353(16):1655–1657.
36. Haines, J., Camarata, S. Examination of candidate genes in language disorder: a model of genetic association for treatment studies. Ment Retard Develop Res Rev. 2004;10(3):208–217.
37. Josephson, S.A., Miller, B.L. Confusion and delirium. In Fauci A.S., et al, eds.: Harrison’s principles of internal medicine, ed 15, New York: McGraw-Hill, 2008.
38. Guntehr, M.L., Morandi, A., Ely, E.W. Pathophysiology of delirium in the intensive care unit. Crit Care Clin. 2008;24(1):45–65.
39. Drachman, D.A. Aging of the brain, entropy, and Alzheimer disease. Neurology. 2006;67(8):1340–1352.
40. Caselli, R.J., Boeve, B.F. The degenerative dementias. In: Goetz C.G., ed. Textbook of clinical neurology. Philadelphia: Saunders, 2003.
41. Bird, T.D., Miller, B.L. Dementia. In Fauci A.S., et al, eds.: Harrison’s principles of internal medicine, ed 15, New York: McGraw-Hill, 2008.
42. Alzheimer’s Association: 2006 national public policy program to conquer Alzheimer’s disease. Available at www.alz.org. Accessed November 30, 2007.
43. Bertram, L., Tanzi, R.E. Thirty years of Alzheimer’s disease genetics: the implications of systemic meta-analyses. Nat Rev Neurosci. 2008;9(10):768–778.
44. Bird, T.D. Genetic aspects of Alzheimer disease. Genet Med. 2008;10(4):231–239.
45. Ertekin-Taner, N. Genetics of Alzheimer’s disease: a centennial review. Neurol Clin. 2007;25(3):611–667.
46. Auerhalm, C. Recognition of risk factors and screening tools: optimizing care for patients with Alzheimer’s disease: emerging treatment strategies. J Acad Nurs Pract (Suppl). 2004;16(1):4–5.
47. Jellinger, K.A. Alzhiemer’s disease. In Gilman S., ed.: Neurobiology of disease, ed 1, Burlington, MA: Academic Press, 2007.
48. Mele, D. Cognitive, functional and behavioral decline in Alzheimer’s disease: optimizing care for patients with Alzheimer’s disease: emerging treatment strategies. J Acad Nurs Pract (Suppl). 2004;16(1):6–7.
49. Feldman, H.H., et al. Diagnosis and treatment of dementia: 2 Diagnosis. CMAJ. 2008;178(7):825–836.
50. Wang, X.P., Ding, H.L. Alzheimer’s disease: epidemiology, genetics and beyond. Neurosci Bull. 2008;24(2):105–109.
51. Hogan, D.B., et al. Diagnosis and treatment of dementia: Approach to management of mild to moderate dementia. CMAJ. 2008;179(8):787–793.
52. van Marum, R.J. Current and future therapy in Alzheimer’s disease. Fund Clin Pharmacol. 2008;22(3):265–274.
53. Marmarou, A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22(5):E1.
54. Factora, R., Luciano, M. When to consider normal pressure hydrocephalus in the patient with gait disturbance. Geriatrics. 2008;63(2):332–337.
55. Correll, C.U., Schenk, E.M. Tardive dyskinesia and new antipsychotics. Curr Opin Psychiatry. 2008;21(2):151–156.
56. Olanow, C.W. Hyperkinetic movement disorders. In Fauci A.S., et al, eds.: Harrison’s principles of internal medicine, ed 15, New York: McGraw-Hill, 2008.
57. Imarisio, S., et al. Huntington’s disease: from pathology and genetics to potential therapies. Biochem J. 2008;412(2):191–209.
58. Jankovic, J. Movement disorders. In: Goetz C.G., ed. Textbook of clinical neurology. Philadelphia: Saunders, 2003.
59. Hersch, S.M., Rosas, H.D. Neuroprotection for Huntington’s disease. Neurotherapeutics. 2008;5(2):226–236.
60. Galvez-Jiminez, H. Parkinson’s disease. In Gilman S., ed.: Neurobiology of disease, ed 1, Burlington, MA: Academic Press, 2007.
61. Galvan, A., Wichmann, T. Pathophysiology of parkinsonism. Clin Neurophysiol. 2008;119(7):1459–1474.
62. Wakabayashi, K., et al. The Lewy body in Parkinson’s disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology. 2007;27(5):494–506.
63. Rommelfanger, K.S., Weinshenker, D. Norepinephrine: the redheaded stepchild of Parkinson’s disease. Biochem Pharmacol. 2007;74(2):177–190.
64. Fasano, M., Lopiano, L. Alpha-synuclein and Parkinson’s disease: a proteomic view. Exp Rev Proteomics. 2008;5(2):239–248.
65. Thomas, B., Beal, M.F. Parkinson’s disease. Hum Mol Genet. 2007;16(Spec No 2):R183–R194.
66. Probst, A., Block, A., Tolnay, M. New insights into the pathology of Parkinson’s disease: does the peripheral autonomic nervous system become central? Eur J Neurol. 2008;15(Suppl 1):1–4.
67. Hoehn, M., Yahr, M. Parkinsonism: onset, progression and mortality. Neurology. 1967;17(5):427–442.
68. Wolters, E. Variablility in the clinical expression of Parkinson’s disease. J Neurol Sci. 2008;266(1-2):197–203.
69. Rivlin-Etzion, M., et al. Basal ganglia oscillations and pathophysiology of movement disorders. Curr Opin Neurobiol. 2006;16(6):629–637.
70. Jankovic, J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–376.
71. Goldstein, D.S. Cardiac denervation in patients with Parkinson disease. Cleve Clin J Med. 2007;74(Suppl 1):S91–S94.
72. Dhawan, V., et al. Sleep-related problems of Parkinson’s disease. Age Ageing. 2006;35(3):220–228.
73. Troung, D.D., Bhidayasiri, R., Wikters, E. Management of non-motor symptoms in advanced Parkinson disease. J Neurol Sci. 2008;266(1-2):216–228.
74. Baker, J.H. The symptom experience of patients with Parkinson disease. J Neurosci Nurs. 2006;38(1):51–57.
75. Defer, G.L., et al. Core assessment program for surgical interventional therapies in Parkinson’s disease (CAPSIT-PD). Mov Disord. 1999;14(4):572–584.
76. Geriach, M., et al. Early detection of Parkinson’s disease: unmet needs. Neurodegener Dis. 2008;5(3-4):137–139.
77. Jankovic, J., Stacy, M. Medical management of levodopa-associated motor complications in patients with Parkinson’s disease. CNS Drugs. 2007;21(8):677–692.
78. Guridi, J., et al. L-Dopa-induced dyskinesia and stereotactic surgery for Parkinson’s disease. Neurosurgery. 2008;62(2):311–323.
79. Yu, H., Neimat, J.S. The treatment of movement disorders by deep brain stimulation. Neurotherapeutics. 2008;5(1):26–36.
80. Goya, R.L., Kuah, W.L., Barker, R.A. The future of cell therapies in the treatment of Parkinson’s disease. Expert Opin Biol Ther. 2007;7(10):1487–1498.
81. Chan, D.K., Cordato, D.J., O’Rourke, F. Management for motor and non-motor complications in late Parkinson’s disease. Geriatrics. 2008;63(5):22–27.
82. Sapir, S., Ramig, L., Fox, C. Speech and swallowing disorders in Parkinson disease. Curr Opin Otolaryngol Head Neck Surg. 2008;16(3):205–210.
83. Heilman, K.M., Watson, R.T., Gonzalez-Rothi, L.J. Praxis. In: Goetz C.G., ed. Textbook of clinical neurology. Philadelphia: Saunders, 2003.
84. Timmann, D., Diener, H.C. Coordination and ataxia. In: Goetz G.C., ed. Textbook of clinical neurology. Philadelphia: Saunders, 2003.