Complex Lipids
Learning objectives
After reading this chapter you should be able to:
 Describe how the various glycerol-based phospholipids are synthesized and how they are interconverted.
Describe how the various glycerol-based phospholipids are synthesized and how they are interconverted.
Describe the multiple roles of cytidine nucleotides in activation of intermediates in phospholipid synthesis.
Describe the various types of sphingolipids and glycolipids that occur in mammalian cells and their functions.
Explain the etiology of lysosomal storage diseases, their pathology, and the rationale for enzyme replacement therapy for treatment of these diseases.
Introduction
Complex lipids encompass the glycerophospholipids, introduced in Chapter 3, and the sphingolipids. These molecules are found mostly in two locations, either embedded in biological membranes or in circulating lipoproteins. The sphingolipids are almost exclusively in cell membranes, primarily the plasma membrane. They carry a wide range of carbohydrate structures which face into the exterior environment and, like glycoproteins, have a range of recognition functions. A major difference between these two classes of lipids is that glycerophospholipids are saponifiable (except plasmalogens), while sphingolipids contain no alkali-labile ester bonds. Thus, it was convenient to isolate sphingolipids from tissues by saponification, and then extract the remaining lipids into organic solvent. Once isolated, the characterization of the glycan structure of the sphingolipids was technically challenging. Therefore, the structures were, for a long time, unknown and mysterious, leading to their name: sphinx-like or sphingolipids.
This chapter discusses the structure, biosynthesis, and function of the two major classes of polar lipids: glycerophospholipids and sphingolipids. In preparation for this chapter, it might help to review the structure of phospholipids in Chapter 3.
Synthesis and turnover of glycerophospholipids
Synthesis of glycerophospholipids
There are many species of glycerophospholipids with a distinct composition of polar head groups and hydrophobic acyl groups (see Chapter 3). With regard to the acyl groups, saturated fatty acids are usually esterified at the sn-1 position, whereas unsaturated fatty acids are esterified at the sn-2 position. Biosynthesis of glycerophospholipids first proceeds by the de novo pathway, and subsequently the originally attached fatty acids in the de novo pathway are replaced with new ones in the remodeling pathway. Through this remodeling pathway, diversity and asymmetry of the acyl groups are generated.
De novo pathway
Phospholipids are in a constant state of synthesis, turnover, and remodeling
The de novo pathway begins with sequential reactions, in which glycerol-3-P is acylated by transfer of two fatty acids from acyl-CoA to produce phosphatidic acid (PA) via the intermediate, lysophosphatidic acid (see Fig. 28.1). Then PA is dephosphorylated to diacylglycerol (DAG) by a specific cytosolic phosphatase. Alternatively, PA reacts with cytidine triphosphate (CTP) to yield the activated phosphatidic acid, cytidine diphosphate (CDP)-DAG. Phosphatidic acid and DAG are common intermediates in the synthesis of both triglycerides (triacylglycerols) and phospholipids. All animal cells, except for erythrocytes, are able to synthesize phospholipids de novo, whereas triglyceride synthesis occurs mainly in liver, adipose tissue, and intestinal cells. The starting material, glycerol-3-phosphate, is formed in most tissues by reduction of the glycolytic intermediate dihydroxyacetone phosphate (DHAP). In liver, kidney and intestine, glycerol-3-P can also be formed directly via phosphorylation of glycerol by a specific kinase. DHAP may also be acylated by addition of a fatty acid to the 1-hydroxyl group; this intermediate is then reduced and acylated to PA.
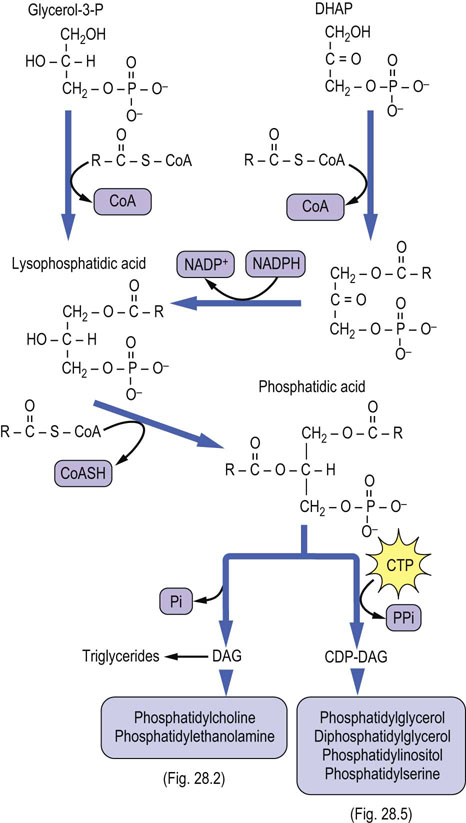
Fig. 28.1 De novo pathway to synthesis of glycerophospholipids.
CDP, cytidine diphosphate; CDP-DAG, CDP-diacylglycerol; CTP, cytidine triphosphate; CoAS, coenzyme A; DHAP, dihydroxyacetone phosphate; Pi, inorganic phosphate; PPi, inorganic pyrophosphate.
The biosynthesis of the major phospholipid phosphatidylcholine (PC; also known as lecithin) from DAG requires activation of choline to CDP-choline. In this series of reactions, shown in Figure 28.2, the choline ‘head group’ is converted to phosphocholine and then activated to CDP-choline by a pyrophosphorylase reaction. The pyrophosphate bond is cleaved and phosphocholine (choline phosphate) is transferred to DAG to form PC. This reaction is analogous to the transfer of GlcNAc-6-P to dolichol or the high-mannose core of lysosomal enzymes – both the sugar and a phosphate are transferred from the nucleotide derivative. Phosphatidylethanolamine (PE) is formed by a similar pathway using CTP and phosphoethanolamine, to form CDP-ethanolamine. Both PC and PE can react with free serine by an exchange reaction to form phosphatidylserine (PS) and the free base, choline or ethanolamine (Fig. 28.3).
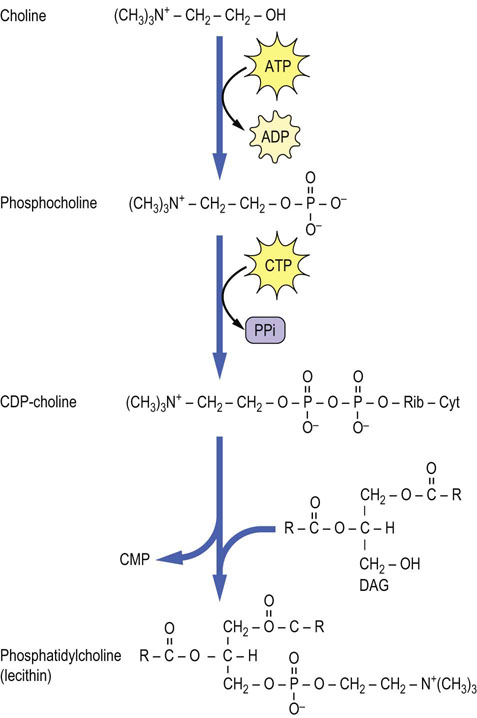
Fig. 28.2 Formation of phosphatidylcholine by the CDP-choline pathway.
This pathway is an extension of the bottom left side of Figure 28.1. Cyt, cytosine; CDP, cytidine diphosphate; CMP, cytidine monophosphate; DAG, diacylglycerol; Rib, ribose; CTP, cytidine triphosphate.
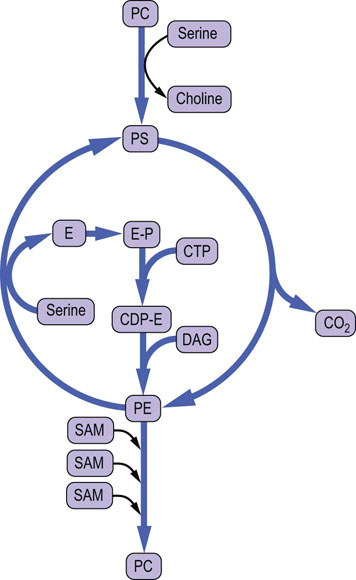
Fig. 28.3 Pathways of interconversion of phospholipids by exchange of head groups, by methylation or by decarboxylation.
CDP, cytidine diphosphate; CTP, cytidine triphosphate; DAG, diacylglycerol; E, ethanolamine; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PC, phosphatidylserine; SAM, S-adenosylmethionine.
Large amounts of PC are needed in liver for biosynthesis of lipoproteins and bile in liver. In a secondary pathway, which is a necessary supplement to the above CDP-choline pathway in liver in times of starvation, PC can also be formed by methylation of PE with the methyl donor S-adenosylmethionine (SAM) (Figs 28.3 and 28.4). This methylation pathway involves the sequential transfer of three activated methyl groups from three molecules of SAM. PE used in this pathway is supplied from PS by a specific mitochondrial decarboxylase (Fig. 28.3).
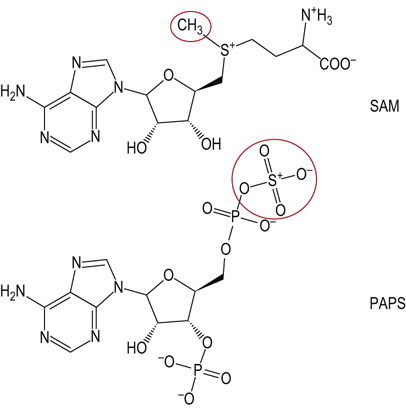
Fig. 28.4 Structures of the methyl and sulfate donors involved in the synthesis of membrane lipids.
SAM, S-adenosylmethionine; PAPS, 3'-phosphoadenosine-5'-phosphosulfate (active sulfate).
PS and other phospholipids with an alcohol head group, e.g. phosphatidylglycerol (PG) and phosphatidylinositol (PI), are synthesized by an alternative pathway. In this case, PA is activated by CTP, yielding CDP-DAG (see Fig. 28.5); the PA group is then transferred to free serine, glycerol or inositol, to form PS, PG or PI, respectively. A second PA may also be added to phosphatidylglycerol to form 1,3-diphosphatidylglycerol (DPG). This lipid, known commonly as cardiolipin, is found almost exclusively in the inner mitochondrial membrane; it represents about 20% of phospholipids in heart mitochondria and is required for efficient activity of electron transport complexes III and IV and the ATP:ADP translocase.
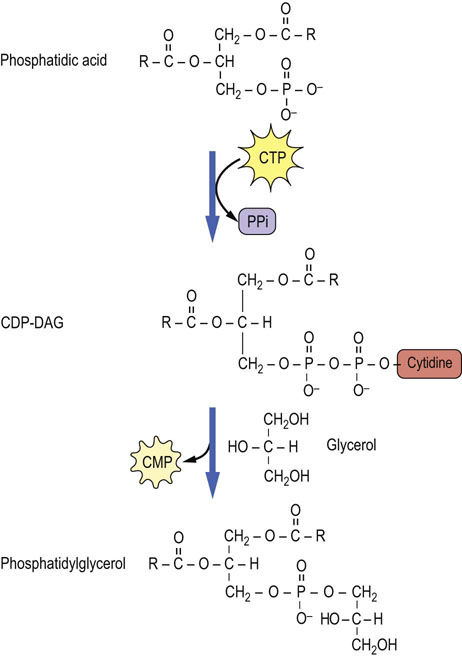
Fig. 28.5 Formation of phosphatidylglycerol by activation of phosphatidic acid to form CDP-DAG, and transfer of DAG to glycerol.
This pathway is an extension of the lower right side of Figure 28.1. CMP, cytidine monophosphate; CTP, cytidine triphosphate.
Plasmalogens are a second major class of mitochondrial lipids, and are enriched in nerve and muscle tissue; in the heart they account for nearly 50% of total phospholipids. The biosynthesis of plasmalogens proceeds from DHAP: it is first acylated at C-1; then the acyl group exchanges with a lipid alcohol to form an ether lipid. The ether lipid is desaturated, leading eventually to a 1-alkenylether-2-acyl-phospholipid. The function of plasmalogens versus diacylphospholipids is not clear, but there is some evidence that they are more resistant to oxidative damage, which may provide protection against oxidative stress in tissues with active aerobic metabolism (see Chapter 37).
 Clinical box Surfactant function of phospholipids: the acute respiratory distress syndrome
Clinical box Surfactant function of phospholipids: the acute respiratory distress syndrome
Acute respiratory distress syndrome (ARDS) accounts for 15–20% of neonatal mortality in Western countries. The disease affects premature infants and its incidence is directly related to the degree of prematurity.
Immature lungs do not have enough type II epithelial cells to synthesize sufficient amounts of the phospholipid dipalmitoylphosphatidylcholine (DPPC). This phospholipid makes up more than 80% of the total phospholipids of the extracellular lipid layer that lines the alveoli of normal lungs. DPPC decreases the surface tension of the aqueous surface layer of the lungs, facilitating opening of the alveoli during inspiration. Lack of surfactant causes the lungs to collapse during the expiration phase of breathing, leading to ARDS. The maturity of the fetal lung can be assessed by measuring the lecithin:sphingomyelin ratio in amniotic fluid. If there is a potential problem, a mother can be treated with a glucocorticoid to accelerate maturation of the fetal lung. ARDS is also seen in adults in whom the type II epithelial cells have been destroyed as a result of the use of immunosuppressive drugs or certain chemotherapeutic agents.
Remodeling pathway
The acyl groups of glycerophospholipids are highly diverse and distributed in an asymmetric manner between the sn-1 and sn-2 position of glycerol; polyunsaturated fatty acids, such as arachidonate, are found predominately at the sn-2 position. The composition of fatty acyl groups in phospholipids also varies among tissues and membranes, and with the nature of the head group: choline, ethanolamine, serine, inositol or glycerol. The diversity and asymmetry of phospholipids are not explained by the de novo pathway since phosphatidic acid and DAG are common precursors of both triglycerides and phospholipids. Instead, the redistribution of fatty acids in phospholipids is accomplished by remodeling pathways through the concerted action of phospholipase A2 (PLA2) and lysophospholipid acyltransferases (LPLAT), which remove, replace and, in the process, redistribute fatty acids in phospholipids. Not until the last decade have the LPLAT enzymes been identified; they play an essential role in (re)incorporation of polyunsaturated fatty acids into phospholipids.
Turnover of phospholipids
Phospholipids are in a continuous state of turnover in most membranes. This occurs as a result of oxidative damage, during inflammation, and through activation of phospholipases, particularly in response to hormonal stimuli. As shown in Figure 28.6, there are a number of phospholipases that act on specific bonds in the phospholipid structure. Phospholipase A2 (PLA2) and phospholipase C (PLC) are particularly active during the inflammatory response and in signal transduction. Phospholipase B (not shown) is a lysophospholipase that removes the second acyl group after action of PLA1 or PLA2. The lysophospholipids may be degraded or recycled (reacylated).
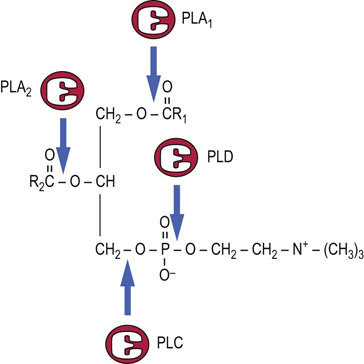
Fig. 28.6 Sites of action of phospholipases on phosphatidylcholine.
PLA1, PLA2, PLC, PLD are phospholipases A1, A2, C, and D, respectively.
 Advanced concept box Glycosylphosphatidylinositol membrane anchors
Advanced concept box Glycosylphosphatidylinositol membrane anchors
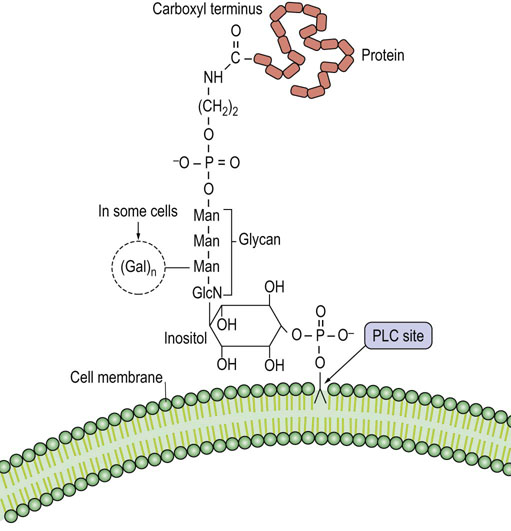
Phosphatidylinositol is an integral component of the glycosylphosphatidylinositol (GPI) structure that anchors various proteins to the plasma membrane (Fig. 28.7). In contrast to other membrane phospholipids, including most of the membrane phosphatidylinositol, GPI has a glycan chain containing glucosamine and mannose attached to the inositol. Ethanolamine connects the GPI-glycan to the carboxyl terminus of the protein. Many membrane proteins in eukaryotic cells are anchored by a GPI structure, including alkaline phosphatase and acetylcholinesterase, which have roles in bone mineralization and nerve transmission, respectively. In contrast to integral or peripheral membrane proteins, GPI-anchored proteins may be released from the cell surface by phospholipase C in response to regulatory processes.
Sphingolipids
Structure and biosynthesis of sphingosine
Sphingolipids are a complex group of amphipathic, polar lipids. They are built on a core structure of the long-chain amino alcohol sphingosine, which is formed by oxidative decarboxylation and condensation of palmitate with serine. In all sphingolipids, the long-chain fatty acid is attached to the amino group of the sphingosine in an amide linkage (Fig. 28.8). Because of the alkaline stability of amides, compared to esters, sphingolipids are nonsaponifiable, which facilitates their separation from alkali-labile glycerolipids.
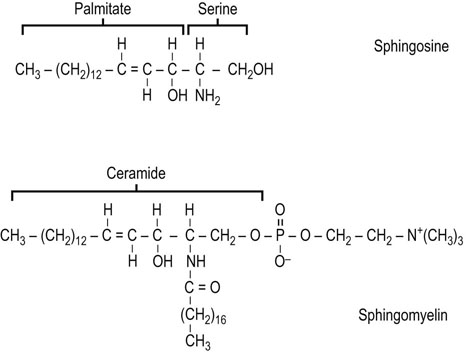
The synthesis of the sphingosine base of sphingolipids involves condensation of palmitoyl-CoA with serine, in which the carbon-1 of serine is lost as carbon dioxide. The product of this reaction is converted in several steps to sphingosine, which is then N-acylated to form ceramide (N-acylsphingosine). Ceramide (see Fig. 28.9) is the precursor and backbone structure of both sphingomyelin and glycosphingolipids.
Advanced concept box Variable surface antigens of trypanosomes
The parasitic trypanosome that causes sleeping sickness, Trypanosoma brucei, has a protein called the variable surface antigen bound to its cell surface by a GPI anchor. This variable surface antigen elicits the formation of specific antibodies in the host, and these antibodies can attack and kill the parasite. However, some of the parasites evade immune surveillance by shedding this antigen, as if they were shedding a coat.
Trypanosomes and some other pathogens are able to shed their surface antigens because they have an enzyme, phospholipase C, that cleaves the GPI anchor at the phosphate–diacylglycerol bond, releasing the protein-glycan component into the external fluid. Surviving cells rapidly make a new coat with a different antigenic structure that will not be recognized by the antibody. Of course, this new coat will elicit the formation of new specific antibodies but the parasite can again shed this coat, and so on, in a random sequence to evade the host immune system.
Clinical box Defects in GPI anchoring in hematopoietic cells: paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria (PNH) is a complex hematologic disorder characterized by hemolytic anemia, venous thrombosis in unusual sites, and deficient hematopoiesis. The diagnosis of this disease is based on the unusual sensitivity of the red blood cells to the hemolytic action of complement (Chapter 38), because red cells from patients with PNH lack several proteins that are involved in regulating the activation of complement at the cell surface.
One of these cell surface proteins is decay accelerating factor, a GPI-anchored protein that inactivates a hemolytic complex formed during complement activation; in its absence, there is increased hemolysis. PNH is an acquired genetic disease due to a hematopoietic stem cell mutation defect. One of these mutations involves a defect in the GlcNAc transferase that adds N-acetylglucosamine to the inositol moiety of phosphatidylinositol, the first step in GPI anchor formation (see Fig. 28.7).
Sphingomyelin
Sphingomyelin is the only sphingolipid that contains phosphate and is the major phospholipid in the myelin sheath of nerves
Sphingomyelin (see Fig. 28.8) is found in plasma membranes, subcellular organelles, endoplasmic reticulum, and mitochondria. It comprises 5–20% of the total phospholipids in most cell types, and is mostly localized in the plasma membrane. The phosphocholine group in sphingomyelin is transferred to the terminal hydroxyl group of sphingosine by a transesterification reaction with phosphatidylcholine. The fatty acid composition varies, but long-chain fatty acids are common, including lignoceric (24 : 0), cerebronic (2-hydroxylignoceric) and nervonic (24 : 1) acids.
Glycolipids
Sphingolipids containing covalently bound sugars are known as glycosphingolipids or glycolipids. As with glycoconjugates, in general, the structure of the oligosaccharide chains is highly variable. In addition, the glycosyltransferase distribution and glycosphingolipid content of cells varies during development and in response to regulatory processes.
Glycolipids can be classified into three main groups: neutral glycolipids, sulfatides and gangliosides. In all of these compounds, the polar head-group, comprising the sugars, is attached to ceramide by a glycosidic bond at the terminal hydroxyl group of sphingosine. Figure 28.9 illustrates the structure and biosynthesis of some of the simpler glycolipids. Neutral glycolipids contain only neutral and amino sugars. Glucosylceramide (GlcCer) and galactosylceramide (GalCer) are the smallest members of this class of compounds and serve as the nucleus for elaboration of more complex structures. Sulfatides are formed by addition of sulfate from the sulfate donor, 3'-phosphoadenosine-5'-phosphosulfate (PAPS) (Fig. 28.4), yielding, for example, GalCer 3-sulfate. Finally, glycolipids containing sialic acids (N-acetylneuraminic acid, NeuAc) are termed gangliosides.

Structure and nomenclature of gangliosides
Gangliosides are glycosphingolipids containing sialic (N-acetylneuraminic) acid
The term ‘ganglioside’ refers to glycolipids that were originally identified in high concentrations in ganglionic cells of the central nervous system. In general, more than 50% of the sialic acid in these cells is present in gangliosides. Gangliosides are also found in the surface membranes of cells of most extraneural tissues, but in these tissues they account for less than 10% of the total sialic acid.
The nomenclature used to identify the various gangliosides is based on the number of sialic acid residues contained in the molecule, and on the sequence of the carbohydrates (Fig. 28.10). GM refers to a ganglioside with a single (mono) sialic acid, whereas GD, GT and GQ would indicate two, three and four sialic acid residues in the molecule, respectively. The number after the GM, e.g. GM1, refers to the structure of the oligosaccharide. These numbers were derived from the relative mobility of the glycolipids on thin layer chromatograms; the larger, GM1, gangliosides migrate the most slowly.
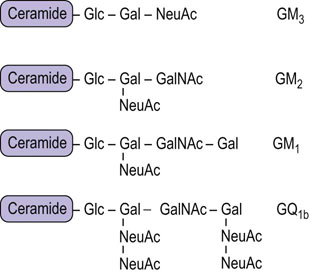
Fig. 28.10 Generalized structures of gangliosides.
Glc, glucose; Gal, galactose; NeuAc, N-acetylneuraminic acid; GalNAc, N-acetylgalactosamine.
Clinical box Sphingolipidoses and gangliosidoses
Tay–Sachs disease is a gangliosidosis in which ganglioside GM2 accumulates as a result of an absence of hexosaminidase A (see Fig. 28.11). Individuals with this disease usually have mental retardation and blindness and die between 2 and 3 years of age. Fabry's disease is a sphingolipidosis resulting from deficiency of α-galactosidase and accumulation of globotriaosylceramide (Gb3) (see Table 28.1). The symptoms of Fabry's disease are skin rash, kidney failure, and pain in the lower extremities. Patients with this condition benefit from kidney transplants and usually live into early to mid-adulthood. Most of these lysosomal storage diseases appear in several forms (variants), resulting from different mutations in the genome. Some lysosomal storage diseases and some variants are more severe and debilitating than others. Although lysosomal storage diseases are relatively rare, they have had a major impact on our understanding of the function and importance of lysosomes.
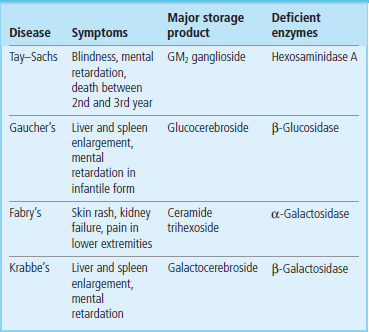
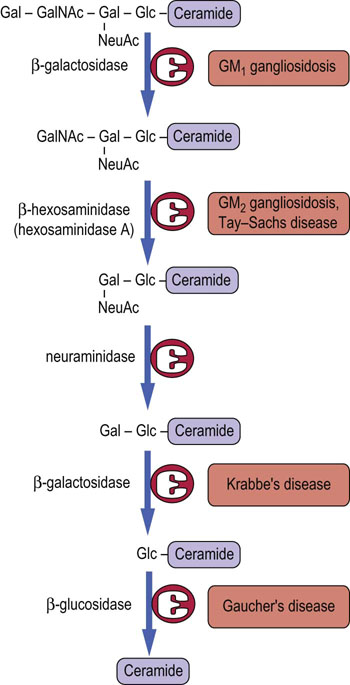
Fig. 28.11 Lysosomal pathway for turnover (degradation) of ganglioside GM1 in human cells.
Various enzymes may be missing in specific lipid storage diseases, as indicated in Table 28.1. Gal, galactose; GalNAc, N-acetylgalactosamine; Glc, glucose; NeuAc, N-acetylneuraminic acid.
When cells die, biomolecules, including glycosphingolipids and glycoproteins, are degraded to their individual components. Figure 28.11 presents the pathway for the degradation of ganglioside GM1 in the lysosomes. A number of lysosomal diseases result from the absence of an essential glycosidase (see Table 28.1). The sphingolipidoses are characterized by lysosomal accumulation of the substrate of the missing enzyme, which interferes with normal lysosomal function in turnover of biomolecules.
Clinical box Gaucher's disease: a model for enzyme replacement therapy
Gaucher's disease is a lysosomal storage disease in which afflicted individuals are missing the enzyme β-glucosidase (also known as glucocerebrosidase). This enzyme removes the final sugar from the ceramide, allowing the lipid portion to be further degraded in the lysosomes. This disease is characterized by hepatomegaly and neuro-degeneration, but there are milder variants that are amenable to treatment by enzyme replacement therapy.
For treatment of Gaucher's disease, exogenous β-glucosidase was successfully targeted to the lysosomes of macrophages. To do this, it was necessary to produce the recombinant replacement enzyme with N-glycan chains containing terminal mannose residues. This was done by cleaving the glycans of the enzyme produced in mammalian cells with a combination of sialidase (neuraminidase), β-galactosidase and β-hexosaminidase to trim the complex chains down to the mannose core. An alternative recombinant glucosidase has been produced in a baculovirus-infected insect cell system. Both enzymes have a high mannose oligosaccharide; although they do not contain Man-6-P residues, they are recognized by a macrophage cell surface receptor for high-mannose oligosaccharides; the enzymes are endocytosed and end up in the lysosomal compartment where they hydrolyze glucosyl-ceramide. The recombinant enzymes are administered intravenously. The success in using recombinant glucocerebrosidase for treatment of Gaucher's disease has stimulated the development of other lysosomal hydrolases for treatment of lysosomal storage diseases.
Lysosomal storage diseases resulting from defects in glycolipid degradation
The complex oligosaccharides on glycolipids are built up, one sugar residue at a time, in the Golgi apparatus and are degraded in a similar stepwise fashion but in the opposite direction by a series of exoglycosidases in lysosomes (Fig. 28.11). Defects in sequential degradation of glycolipids lead to a number of lysosomal storage diseases, known as sphingolipidoses and gangliosidoses (Table 28.1). These diseases are autosomal recessive in inheritance. Heterozygotes are asymptomatic, indicating that a single copy of the gene for a functional enzyme is sufficient for apparently normal turnover of glycolipids. Like I-cell disease (Chapter 27), the sphingolipidoses are characterized by accumulation of undigested lipids in inclusion bodies in the cells.
Clinical box Fabry's disease (incidence 1 in 100,000)
A 30-year-old man was found to have proteinuria at an insurance medical examination. He had been seen over a number of years from around age 10 with headaches, vertigo and shooting pains in his arms and legs. No diagnosis was made and he had grown accustomed to these problems. The physician carefully examined his perineum and scrotum, identifying small, raised, red angiokeratoma.
This man was diagnosed with Fabry's disease, which often takes years before a diagnosis is confirmed by measuring α-galactosidase A activity. The principal endothelial depositions of a ceramide trihexoside (Gal-α1-4-Gal-β1-4-Glc-β-ceramide; Gb3) occur in the kidney (leading to proteinuria and renal failure), the heart and brain (leading to myocardial infarction and stroke), and around blood vessels supplying nerves (leading to painful paresthesiae). Historically, most patients experienced end-stage renal disease, requiring transplantation. However, recombinant enzyme replacement therapy appears to clear the deposited Gb3 and initial studies suggest that renal function is maintained.
ABO blood group antigens
Blood transfusion replenishes the oxygen-carrying capacity of blood in persons who suffer from blood loss or anemia (see Chapter 5). The term ‘blood transfusion’ is something of a misnomer, because it involves only the infusion of washed and preserved red cells. The membranes of red blood cells contain a number of blood group antigens, of which the ABO blood group system is the best understood and most widely studied.
The ABO blood group antigens are complex carbohydrates present as components of glycoproteins or glycosphingolipids of red cell membranes (Fig. 28.12). The H locus codes for a fucosyltransferase, which adds fucose to a galactose residue in a glycan chain. Individuals with type A blood have, in addition to the H substance, an A gene that codes for a specific GalNAc transferase that adds GalNAc α1,3 to the galactose residue of H substance, to form the A antigen. Individuals with type B blood have a B gene that codes for a galactosyl transferase that adds galactose α1,3 to the galactose residue of H substance, to form the B antigen. Individuals with type AB blood have both the GalNAc and the Gal transferases, and their red blood cells contain a mixture of A and B substances. Those with type O blood have only H substance on their red cell membranes; they do not make either enzyme. Enzymes such as coffee bean α-galactosidase can remove the galactose from type B red cells, an approach that is being tested for increasing the supply of type O (universal donor) red cells.
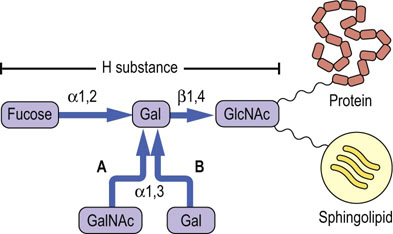
Fig. 28.12 Relationship between the H, A, and B blood group substances.
The terminal oligosaccharide is linked through other sugars to proteins and lipids of the red cell membrane. GlcNAc, N-acetylglucosamine; GalNAc, N-acetylgalactosamine; Gal, galactose.
An individual may have type A, type B, type AB or type O blood. Individuals with type A cells develop natural antibodies in their plasma that are directed against and will agglutinate type B and type AB red blood cells; those with type B red cells develop antibodies against A substance and will agglutinate type A and type AB blood. Persons with type AB blood have neither A nor B antibodies and are called ‘universal recipients’, as they can be transfused with cells of either blood type. Individuals with type O blood have only H substance, not A or B substance, on their red blood cells and are ‘universal donors’, as their red blood cells are not agglutinated by either A or B antibodies; however, they may accept blood only from a type O donor.
The ABO antigens are present on most cells in the body but they are referred to as blood group antigens because of their association with transfusion reactions. The transfusion reaction is the result of reaction of host antibodies with transfused red cells, resulting in complement-mediated hemolysis (see Chapter 38). While the transfusion reaction demonstrates the role of carbohydrates in recognition of the foreign red cells, the physiologic function of blood group substances is unclear. Persons with an O genotype are generally as healthy as those with A or B genotype. However, there is some evidence that specific phenotypes may confer differential resistance to disease; for example, people with type A and type O blood appear to be more susceptible to smallpox and cholera, respectively.
Other blood group substances
Advanced concept box Glycolipids are binding sites for bacteria and bacterial toxins
Bacteria have evolved proteins called adhesins that recognize and interact with specific carbohydrate structures on glycolipids, glycoproteins and even proteoglycans. Many of the bacterial adhesins are protein subunits of pili, hair-like structures on the surfaces of the bacteria. The carbohydrate recognition domains are usually located at the tip of the pili. Most bacteria also have several different kinds of adhesins on their surfaces, each having different carbohydrate recognition sites, and these adhesins define the range of susceptible tissues that the bacteria can bind to and perhaps invade. Each individual adhesin binding is of low affinity and the binding is weak, but there are many copies of a given adhesin on the bacterial surface and they cluster together, so the total interaction is polyvalent rather than monovalent and binding becomes quite strong. The interaction of the adhesin with its receptor can activate signal transduction pathways and lead to events that are critical for colonization and perhaps infection.
A number of bacterial adhesins target Galβ1-4Glc-containing oligosaccharides. This is the disaccharide structure that is found in the glycolipid lactosylceramide, and this structure may be present as such or it may be capped with other sugars, as in the ABO blood group antigens. But some bacteria secrete enzymes (glycosidases) that can remove these terminal sugars to expose the lactose structure for binding to their adhesin. The epithelial cells of the large intestine express lactosylceramide, whereas the cells lining the small intestine do not express this glycolipid. As a result, Bacteroides, Clostridium, Escherichia coli and Lactobacillus only colonize the large intestine under normal conditions.
In addition to the bacterial binding, a number of toxins that are secreted from bacterial cells also bind to specific glycolipids. The best studied of these toxins is cholera toxin, the toxin produced by Vibrio cholerae, which binds to GM1. The toxin from Shigella dysenteriae also binds to cells of the large intestine, but it recognizes a different glycolipid, in this case Gb3. These two examples show quite clearly how subtle changes in the structures of carbohydrate molecules can be recognized by different proteins, and why nature has selected carbohydrates molecules as providers of chemical recognition information. There are other toxins, such as the tetanus toxin produced by Clostridium tetani or botulinum toxin by Clostridium botulinum, which also bind to glycolipids on nerve cell membranes. These toxins recognize much more complicated glycolipids. For example, tetanus toxin binds to the ganglioside GT1b.
Clinical box Ganglioside receptor for cholera toxin
The galactose-containing glycolipids in the plasma membranes of intestinal epithelial cells are binding sites for bacteria. The glycolipids appear to assist in retention of normal intestinal flora (symbionts) in the intestine but, conversely, binding of pathogenic bacteria to these and other glycolipids is believed to facilitate infection of the epithelial cells. The difference between symbiotic and parasitic bacteria depends, in part, on their ability to secrete toxins or to penetrate the host cell after the binding reaction.
Intestinal mucosal cells contain ganglioside GM1 (see Fig. 28.10). This ganglioside serves as the receptor to which cholera toxin binds as the first step in its penetration of intestinal cells. Cholera toxin is a hexameric protein secreted by the bacterium Vibrio cholerae. The protein is composed of one A subunit and five B subunits. The protein binds to gangliosides by multiple interactions through the B subunits, which enables the A subunit to enter the cell and activate adenylate cyclase on the inner surface of the membrane. The cyclic AMP that is formed then stimulates intestinal cells to export chloride ions, leading to osmotic diarrhea, electrolyte imbalances, and malnutrition. Cholera remains the number one killer of children in the world today.
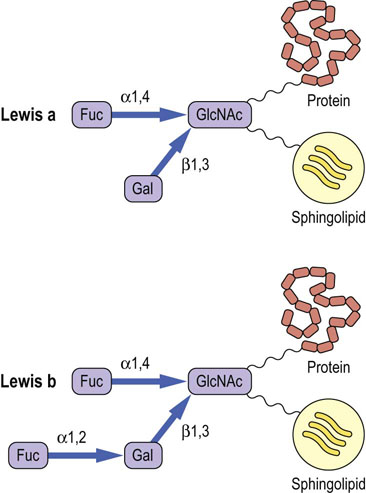
The Lewis blood group antigens correspond to a set of fucosylated glycan structures. The Lewis-A antigen (Lewisa) is synthesized by a fucosyltransferase that transfers a fucose residue to a GlcNAc residue in a glycan chain (Fig. 28.13), while the Lewisb antigen is synthesized by the action of a second fucosyltransferase which transfers fucose to the terminal galactose residue in the same glycan chain. Note the similarity of these structures to the sialyl Lewis-X antigen in Figure 27.9 and the ABO antigens in Figure 28.12. There are 13 fucosyltransferase genes in the human genome. Changes in fucosylation of glycans are associated with differentiation, development, carcinogenesis and metastasis.
The P blood group antigens expressed on the globo-series glycosphingolipids are distributed in red cells and other tissues. Again, the glycans in this blood group are synthesized by the sequential action of distinct glycosyltransferases. The physiologic function of these blood groups is unknown, but P antigens are associated with urinary tract infections and parvovirus infections. Uropathogenic strains of E. coli express lectins that bind to the Galα1,4Gal moiety of the Pk and P1 antigens. More work is needed to understand the genetics and biochemistry of these and other blood group antigens as well as their roles in physiology and disease.
Summary
Complex polar lipids are essential components of all cell membranes.
Phospholipids are the major structural lipids of all membranes, but they also have important functional properties as surfactants, as cofactors for membrane enzymes, and as components of signal transduction systems.
The primary route for de novo biosynthesis of phospholipids involves the activation of one of the components (either DAG or the head group) with CTP to form a high-energy intermediate, such as CDP-diacylglycerol or CDP-choline.
Phospholipids undergo maturation in the remodeling pathway, in which acyl groups at the sn-2 position are replaced with new ones, yielding diversity and asymmetry of the hydrophobic moiety of phospholipids.
Glycosphigolipids function as receptors for cell–cell recognition and interactions, and as binding sites for symbiotic and pathogenic bacteria and for viruses. Carbohydrate structures on glycosphingolipids of red cell membranes are also antigenic determinants responsible for the ABO and other blood types.
Glycosphingolipids are degraded in lysosomes by a sequence of reactions that involve a stepwise removal of sugars from the nonreducing end of the molecule, with each step involving a specific lysosomal exoglycosidase. A number of inherited lysosomal storage diseases result from defects in degradation of sphingolipids.
Beck, M. Therapy for lysosomal storage diseases. IUBMB Life. 2010; 62:33–40.
Brown, JR, Crawford, BE, Esko, JD. Glycan antagonists and inhibitors: a fount for drug discovery. Crit Rev Biochem Mol Biol. 2007; 42:481–515.
Cipolla, L, Araujo, AC, Bini, D, et al. Discovery and design of carbohydrate-based therapeutics. Expert Opin Drug Discov. 2010; 5:721–737.
Desnick, RJ, Schuchman, EH. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Hum Genet. 2012; 13:307–335.
Kulkarni, AA, Weiss, AA, Iyer, SS. Glycan-based high affinity ligands for toxins and pathogen receptors. Med Res Review. 2010; 30:327–393.
Mignani, R, Feriozzi, S, Schaefer, RM, et al. Dialysis and transplantation in Fabry disease: indications for enzyme replacement therapy. Clin J Am Soc Nephrol. 2010; 5:379–385.
Sindou, H, Shimizu, T. Acyl-CoA:lysophospholipid acyltransferases. J Biol Chem. 2009; 284:1–5.
Van Meer, G, Voelker, DR, Feigenson, GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008; 9:112–124.
Yamamoto, F, Cid, E, Yamamoto, M, Blancher, A. ABO research in the modern era of genomics. Transfus Med Rev. 2012; 26:103–118.
Xu, YH, Barnes, S, Sun, Y, Grabowski, GA. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J Lipid Res. 2010; 51:1643–1675.
Blood groups. www.bloodbook.com/type-sys.html.
Gaucher's disease. www.ninds.nih.gov/disorders/gauchers/gauchers.htm.
Paroxysmal nocturnal hemoglobinuria. http://emedicine.medscape.com/article/207468-overview.
Tay–Sachs disease. www.ninds.nih.gov/disorders/taysachs/taysachs.htm.