Ipid Metabolism Ii: Lipoprotein Lipid Metabolism Ii: Lipoprotenins, Cholesterol And Prostaglandins
Compared to glucose and amino acids, which are freely transported in the aqueous plasma, transport of water insoluble lipids involves the participation of a range of complex molecules. Unesterified (“free”) fatty acids are transported in non-covalent binding to serum albumin, but triacylglycerols and cholesterol esters are too hydrophobic for such transport. They are, therefore, packaged together with proteins (apoproteins) and amphipathic lipids by hepatocytes and enterocytes into particles called lipoproteins. Lipoproteins are important for they provide means for fat transport between different organs and tissues. In this chapter, a detailed account of normal functions of lipoproteins and their relationship to abnormal states, particularly atherosclerosis, is given. The chapter also deals with derived lipids such as cholesterol and eicosanoids. Cholesterol is a major component of cell membranes and serves as a precursor of bile salts and steroid hormones. Eicosanoids, which include prostaglandins, thromboxanes and leukotrienes act as regulators of cellular functions. After going through this chapter, the student should be able to understand:
Composition, separation, properties, metabolism and significance of different types of lipoproteins; and function of each type of lipoprotein and apolipoprotein.
The de novo pathway of cholesterol biosynthesis and its regulation, with special emphasis on role of hormones.
Classification, origin and structure of prostaglandins, thromboxanes, and leukotrienes; and important physiological effects and therapeutic uses of these compounds.
I Lipoproteins
A Overview
Lipoproteins are molecular complexes consisting of lipids and proteins that provide means for lipid transport between different organs and tissues. The lipids, by virtue of their immiscibility with aqueous solutions, depend on protein carriers for transport within bloodstream and extracellular fluids. A lipoprotein particle is a spherical structure having a hydrophobic core wrapped in an amphipathic coating.
• The core contains cholesterol esters and triacylglycerols, which, because of their non-polar nature, always avoid contact with water (Fig. 12.1 ).
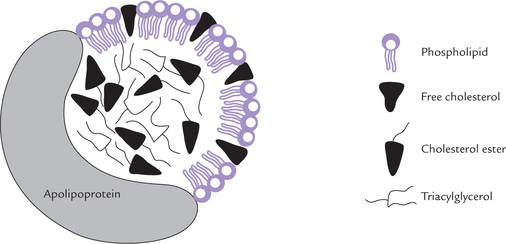
Fig. 12.1 Structure of a lipoprotein: a globular particle consisting of a hydrophobic core of triacylglycerol and cholesterol ester, surrounded by an amphipathic coat of proteins, phospholipids and cholesterol.
• The amphipathic coat contains phospholipids, free cholesterol, and proteins. It interacts with water in plasma, thereby promoting solubility of lipoproteins.
It is noteworthy that the interactions between lipids and proteins in the lipoprotein particle are of a purely non-covalent nature. Protein part of lipoprotein is called apoprotein or apolipoprotein. The apolipoproteins on surface of lipoproteins help to solubilize the lipids and target the lipoproteins to the correct tissues.
The most characteristic structural feature of apolipoproteins is the amphipathic helix,an α-helix in which one side is formed by hydrophobic- and the other by hydrophilic-amino acid side chains. The latter faces the aqueous exterior (being found at the water-lipid interface), so that lipoproteins are soluble in water.
 Most plasma lipids are components of lipoproteins, the non-covalent aggregates of lipids and proteins, that serve as transport vehicles for triacylglycerols, phospholipids and cholesterol around the body.
Most plasma lipids are components of lipoproteins, the non-covalent aggregates of lipids and proteins, that serve as transport vehicles for triacylglycerols, phospholipids and cholesterol around the body.
The amount and types of lipids can change rapidly in the circulation according to dietary habits because most lipids are exchanged readily between different lipoprotein particles. The normal range for the lipid levels are shown in Table 12.1 .
Table 12.1
Plasma lipid levels in adults (360–600 mg/dL) of which approximately 1/3rd is phospholipids and less than 1/3rd is triacylglycerols
| Lipids* | Concentration (mg/dL) |
| Triacylglycerol (TAG) | 40–140 |
| Cholesterol (C) | 150–200 |
| Phospholipids (PL) | 145–250 |
| Free fatty acids (FFA) | 4–20 |
| Total | 360–600 |
*TAG, C and PL are carried by lipoproteins but FFAs are transported after forming complexes with serum albumin.
B Nomenclature
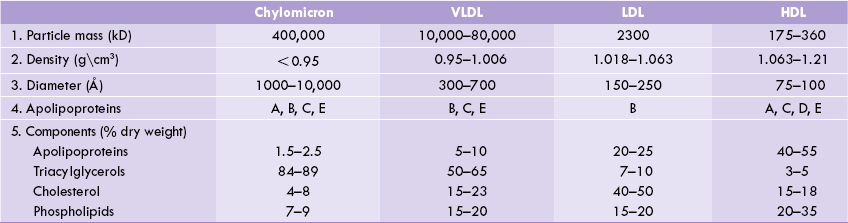
Several different classes of lipoproteins exist, each having characteristic lipid and protein compositions. Their densities are inversely related to their lipid content: the higher the lipid content, the lower the density (Table 12.2 ). Apart from the largest species, the chylomicron, lipoproteins are named according to their density (because they are most commonly isolated by ultracentrifugation). The various classes are: (a) chylomicrons (CM), (b) very low density lipoproteins (VLDL), (c) low density lipoproteins (LDL), and (d) high density lipoproteins (HDL).
C Functions
The lipoprotein particles have three important functions in human body. First, they transport the dietary fat from the intestinal mucosa, where it is absorbed, to liver by exogenous lipid transport (this role is performed by chylomicrons). The second role is to transfer triacylglycerols and cholesterol from liver to other tissues by endogenous lipid transport (VLDL and LDL carry out this function). The third function is to transport cholesterol from extrahepatic tissues to the liver by reverse cholesterol transport (HDL performs this role).
D Constituents
Lipids
Typical analysis of the lipid composition of plasma lipoproteins are shown in Table 12.2. The following observations are noteworthy:
1. All lipoproteins contain various types of lipids: triacylglycerols, phospholipids, and cholesterol-free and esterified,although the quantities and relative proportions of these vary widely.
2. The bulk of plasma cholesterol is transported associated with LDL, and the bulk of plasma triacylglycerol is transported associated with chylomicrons or VLDL. Therefore, elevated cholesterol level most often reflects an increase in the LDL fraction,whereas elevation of the plasma triacylglycerol level is usually caused by increased VLDL or chylomicrons.
3. Density is inversely proportional to lipid content and directly proportional to the protein content.
Proteins
Five main classes of apolipoproteins (A-E) have been identified in lipoproteins. They are mainly synthesized in liver, although smaller quantities are produced in almost all organs. The group-A are found in highest amounts in HDL, while the group-B are associated with VLDL, LDL and chylomicrons. The latter are tightly integrated into the phospholipid bilayer, whereas members of C and E group are less tightly associated and exchange among lipoproteins with HDL acting as the distributor.
Functions of Apolipoproteins
The apolipoproteins participate in lipoprotein metabolism by performing following functions:
1. Transport: The primary role is to impart a hydrophilic character to the lipoprotein particles so that they can be transported in the aqueous plasma.
2. Enzyme activation: They activate various enzymes of lipoprotein metabolism. For example, apoC-II activates the enzyme lipoprotein lipase.The activated enzyme then catalyzes degradation of the triacylglycerol component of VLDL and chylomicrons.
3. Ligand: The apoproteins recognize the cell surface receptors, thus serving as ligands. For instance, the apoB-100 serves as a ligand for the LDL receptor.
4. They facilitate the transfer of lipids between different lipoprotein classes or between lipoproteins and cells. ApoD, for example, acts to facilitate the transfer of esters of cholesterol between lipoproteins.
Further details about roles of apolipoproteins are given in Table 12.3 .
Table 12.3
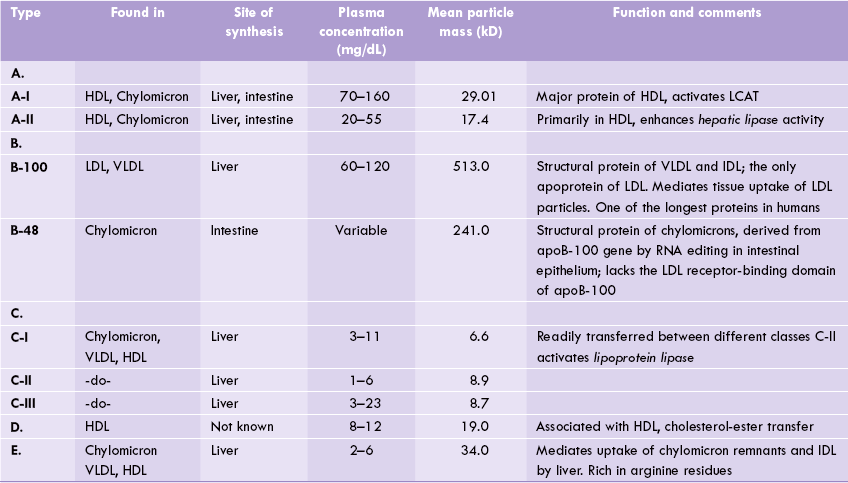
HDL = high density liporotein, IDL = intermediate density lipoproteins, LCAT = lecithin cholesterol acyl transferase, LDL = low density lipoprotein, VLDL = very low density lipoproteins.
In some cases there is more than one gene for a particular class of apolipoprotein and these are specified by post-scripts (e.g. apoA-I, apoC-II). In addition, different alleles at a particular locus have been recognized within a given population, i.e. there is polymorphism. Presence of a particular allele is sometimes found to be associated with increased susceptibility to certain diseases. For example, there are four alleles of apoE designated as ε1 to ε4 and the individuals expressing the ε4 allele are at greatly increased risk of developing Alzheimer’s disease and senile dementia.
E Separation of Lipoproteins
Plasma lipoproteins can be separated by either their electrophoretic mobility or their density. The techniques most commonly used are electrophoresis on agarose gel, and ultracentrifugation.
Electrophoresis
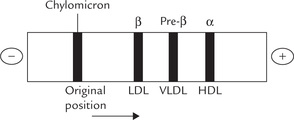
This method separates lipoproteins on the basis of their charge and mass to yield four major bands. These bands are shown in Figure 12.2, which also shows the alternative names used to specify lipoproteins. In mildly alkaline pH (8.3), with the fasting serum or plasma, two most prominent lipoprotein bands are found in the α and the β fractions. They are designated accordingly as α-lipoprotein and β-lipoprotein, which consist respectively of HDL and LDL. A less prominent band the pre-β-lipoprotein ( = VLDL) moves slightly ahead of the β-lipoprotein. After a fatty meal, a fourth band, which does not show any mobility in electrophoresis, consists of chylomicrons.
Ultracentrifugation
This method allows separation according to density and also yields four major fractions. Since lipids are less dense (density approximately 0.9 g/cm3) than proteins, the lipoproteins with high lipid content have a low density (from 0.9 g/cm3 to 1.006 g/cm3) and so float on centrifugation. With increasing protein content, the densities of lipoprotein particles increase to well above 1.0 g/cm3, and so they sediment easily.
Currently, the terms used to designate density of the lipoproteins are also used in naming them. In the order of increasing density, these are very low density lipoproteins, VLDL; low density lipoproteins, LDL and high density lipoproteins, HDL (density of chylomicrons are comparable to that of VLDL).
F Lipoprotein Metabolism
Chylomicrons
The dietary triacylglycerols are transported by chylomicrons, the largest of lipoproteins. They are synthesized in the intestine and transport dietary triacylglycerols to skeletal muscles and adipose tissue, and dietary cholesterol to the liver. As the chylomicrons pass through the capillaries of various tissues, the triacylglycerols are hydrolyzed by the action of lipoprotein lipase(LPL), an enzyme located on the endothelial cells. The fatty acids and monoacylglycerols released by the hydrolysis diffuse directly into the tissue either for metabolism or for storage.
Synthesis
Following absorption in intestine, the dietary lipids are incorporated in chylomicrons. Since chylomicrons carry lipids (mainly triacylglycerols) of dietary origin, they are synthesized and appear in circulation only after a meal rich in fats.
The relative content of triacylglycerols, cholesterol, phospholipids and fat-soluble vitamins present in chylomicrons reflect the lipid composition of the preceding meal. These lipid components are assembled in the SER and the Golgi apparatus of the mucosal cells. Then the apolipoproteins (B-48 and A) synthesized in RER are also incorporated. The particles so formed, called nascent chylomicrons, are exocytosed into the laceteals of the intestinal villi. From these lymph vessels (i.e. laceteals) the nascent chylomicrons reach the blood circulation via the thoracic duct.
Metabolism
Initially, as the chylomicrons are synthesized by intestinal cells, they contain only apoB-48 and apoA. But upon entering the circulation, the nascent particles acquire apoC and apoE from plasma HDL to form mature chylomicrons (Fig. 12.3 ). The acquisition of these apolipoproteins makes the mature chylomicrons functionally competent. In particular, the apo C-II in mature particles activates the enzyme lipoprotein lipase(LPL).
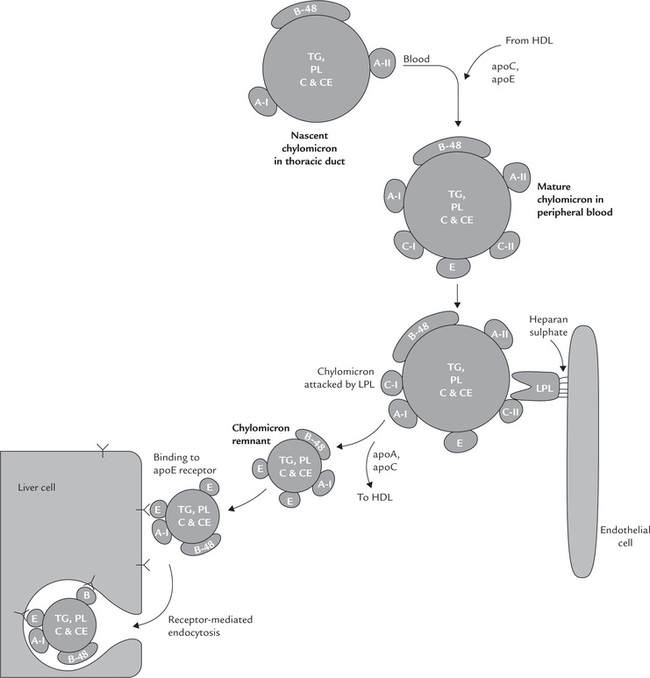
Fig. 12.3 Metabolism of chylomicrons showing their role in transporting dietery cholesterol to liver. (TAG = triacylglycerol, PL = phospholipid, C = free cholesterol, CE = cholesterol ester, LPL = lipoprotein lipase).
In the peripheral tissues, such as muscles and adipose tissue, the activated LPL causes hydrolysis of about 80–90% of the chylomicron triacylglycerols. This is accompanied by the transfer of most of the A- and C-apolipoproteins to HDL. These changes convert the chylomicron into a smaller particle, known as a chylomicron remnant. The fatty acids released from the hydrolyzed triacylglycerols enter muscle and adipose tissue cells, and the glycerol part enters the liver, where it is used for synthesis of TAG. Decreased degradation of chylomicron triacylglycerols due to impaired LPL activity results in elevated levels of chylomicron even after overnight fast (Case 12.1).
Fate of Chylomicron Remnant
The chylomicron remnants formed by off loading of triacylglycerols are finally removed from blood circulation by liver. They bind to lipoprotein receptors on the surface of hepatocytes, including the LDL receptor and the LDL-receptor-related protein (LRP). This binding requires apoE. After binding to one of these receptors, the whole remnant particle is taken up by hepatocytes (i.e. endocy-tosis). Intracellularly, the endocytosed vesicles are carried to the lysosomes where they are degraded to release the constituents. Hence, much of the cholesterol taken in the diet is delivered to liver, together with fat-soluble vitamins. The lifetime of a chylomicron, from its secretion by the intestinal cell to the uptake of remnant by the liver is less than 1 hour.
Very Low Density Lipoproteins (VLDL)
Synthesis and Secretion
Synthesis of nascent VLDL particles occurs in the hepatocytes by a similar set of reactions as described for the synthesis of nascent chylomicrons. It involves assembly of triacylglycerols, phospholipids, cholesterol and apoB-100. The biosynthetic process requires participation of several organelles:
• RER is involved in the production of apoB-100 and SER in its assembly with the lipid components.
• Glycosylation occurs in the Golgi to produce nascent VLDL.
For additional information on synthesis of apoB-100, refer to Box 12.1.
Secretion of the nascent VLDL particles occurs in the same way as in case of the chylomicrons. However, the nascent VLDL particles are released directly into blood circulation, where they obtain more of apoE and C-apolipoproteins from HDL to form mature VLDL particles (Fig. 12.4 ).
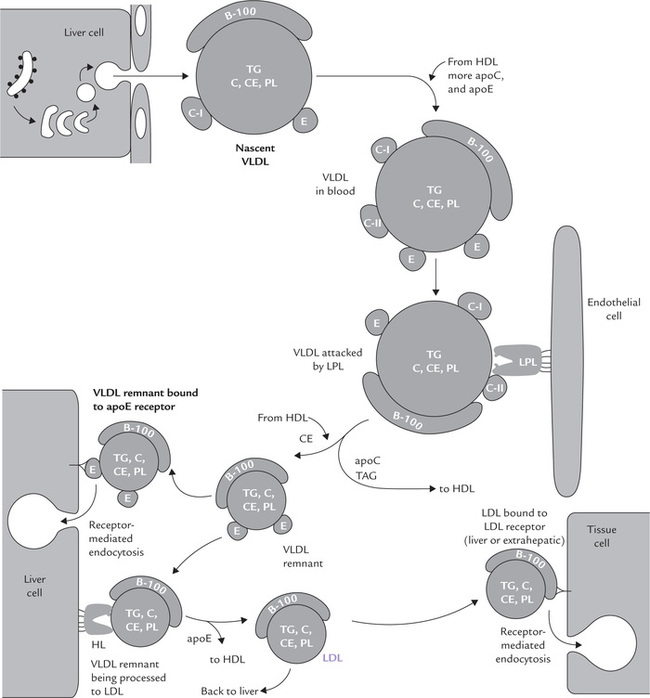
Fig. 12.4 Metabolic fate of VLDL and LDL (TAG = triacylglycerol, PL = phospholipid; C = free cholesterol, CE = cholesterol esters, HL = hepatic lipase).
The TAG-rich mature VLDL exchanges TAGs for cholesterol esters with HDL, as discussed latter.
Metabolism of VLDL
Like chylomicrons, the mature VLDL is acted upon by lipoprotein lipase in capillaries of peripheral tissues. The enzyme is activated by apoC-II of mature VLDL. (Refer to Box 12.2 for details on action of LPL.)
VLDLs are synthesized in the liver and transport triacylglycerols, cholesterol and phospholipids to other tissues, where lipoprotein lipase hydrolyzes the TAGs and releases the fatty acids for uptake.
The VLDL triacylglycerols are hydrolyzed more slowly compared to those in chylomicrons with a residence time in blood of 15–60 minutes (5–10 minutes for chylomicron triacylglycerols). Half-life of VLDL in serum is 1–3 hours.
Progressive stripping of triacylglycerols is accompanied by the transfer of apoC to HDL. Like the chylomi-cron remnants, the VLDL remnants possess apoE, which would mediate their uptake into the liver. The fatty acids released from the degraded triacylglycerols are taken up by mainly by peripheral tissues.
Processing of VLDL remnants to IDL and LDL: Initial hydrolysis of the VLDL triacylglycerols by lipoprotein lipase in peripheral tissues produces smaller particles called VLDL remnants (Fig. 12.4). The VLDL remnant is smaller and contains relatively more cholesterol and apoE than VLDL. At this stage, apoE becomes responsible for further metabolism of the remnant particles. A proportion of these particles are internalized in the liver through mediation of receptors (that recognize the apoE) and the rest are further degraded by hepatic lipase (a liver specific enzyme present in space of Disse) to yield intermediate density lipoproteins (IDL), and then LDL. The transformation (IDL to LDL) is effected by the enzyme hepatic lipase (HL) by further loss of triacylglycerols as well as transfer of excess apolipoproteins to HDL. The HL, which has structural homology with LPL, is found on cell surfaces of hepatocytes, anchored to heparan sulphate proteoglycans. It hydrolyzes lipids of IDL (and HDL, as discussed later). Finally, by the time IDL is converted to LDL, it has lost all apolipoproteins except apoB-100. The LDL now acquires a conformation that enables it to recognize the LDL receptors on cell surfaces of peripheral tissues.
The VLDL remnants are transformed first to IDL and then to LDL as all of their apoproteins other than B100 are removed, and their triacylglycerols hydrolyzed.
Mode of action of hepatic lipase: Hepatic lipase is also released by heparin, like LPL. But unlike LPL the latter it is not activated by apoC and does not attack chylomi-crons and VLDL. Its physiological substrates are triacylglycerols (and phosphoglycerides) in VLDL remnant, IDL and HDL. In addition to degrading TAGs in these lipoproteins, HL also promotes uptake of remnant particles into hepatocytes, as discussed later.
Low Density Lipoproteins (LDL)
Most of the LDL is derived from VLDL and IDL but a smaller amount is released directly from intestine. During its formation from VLDL, apoB-100 is the only apolipoprotein that is retained in LDL. Most of the triacylglycerols are also lost during this transition, so that LDL particles contain relatively higher concentration of cholesterol, and cholesterol esters. ApoB-100 serves as ligand for the LDL receptors.
LDL Receptors
These receptors are present in all cells but are most abundant on hepatocytes and adrenal cortex (Fig. 12.4).
Helped by apoB-100, these receptors mediate uptake of cholesterol rich LDL molecules either by liver (approximately 70%) or by peripheral tissues (approximately 30%), especially fibroblasts, vascular smooth muscles and lymphocytes. It is a highly regulated process of receptor-mediated endocytosis occurring via clathrin-coated pits (Chapter 7). The LDL receptor is sometimes referred to as the apoB-100/apoE receptor because it has affinity for apoE also (but only at high concentration of apoE), and can mediate uptake of IDL particles as well. This step of receptor-mediated endocytosis of LDL is an important facet of whole-body cholesterol homoeostasis. The subsequent degradation of LDL (and IDL) in the lysosomal compartment of cells results in the release of cholesterol. The free receptor can then return to the cell surface for further uptake of LDL molecules. Goldstein and Brown were awarded Nobel prize in 1985 for their work on LDL receptor.
LDL is removed by receptor-mediated endocytosis both by the liver (two-third) and by extrahepatic tissues (one-third). The removal is initiated by binding of apoB-100 to the LDL receptor.
A summary of metabolic fate of chylomicrons, VLDL and LDL is shown in Figure 12.5 . Defective LDL receptors lead to decreased clearance of circulating LDL particles by peripheral tissues. The resulting condition is called familial hypercholesterolaemia (Case 12.2).
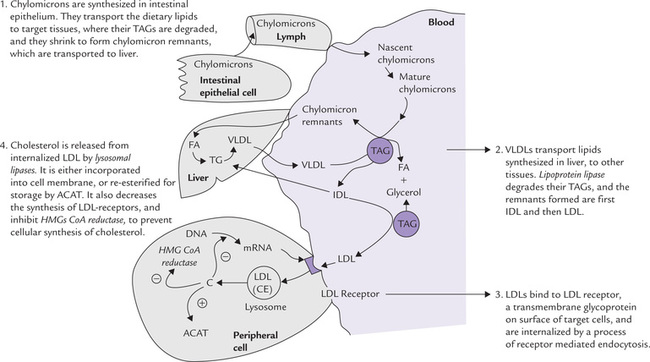
Fig. 12.5 Summary of metabolic fate of chylomicrons and very low density lipoproteins (VLDL), and regulation of low density lipoproteins (LDL) receptors (ACAT = acyl cholesterol acyl transferase, C = cholesterol, CE = cholesterol ester, FA = fatty acids, TAG = triacylglycerol, IDL = intermediate density lipoproteins).
Regulation of LDL Receptors
The number of LDL receptors present on the cell surface is regulated by the cellular needs. For example, when the intracellular cholesterol level falls, it induces increased expression of receptors on the cell membrane. This process, known as up regulation, enables the cells to internalize more circulating LDL particles and utilize their cholesterol content. Conversely, when cell has adequate supply of cholesterol, number of receptors decrease to prevent any further uptake of LDL. This process is called down regulation, and is seen in the following conditions:
1. Raised intracellular cholesterol concentration, which occurs following cellular uptake of the circulating LDL particles.
2. Hypercholesterolaemia, when the blood circulation is flooded with cholesterol and excessive uptake is to be avoided.
Increased intracellular cholesterol concentration depresses transcription of the genes for LDL receptors (Fig. 12.5) thus reducing the number of receptors on cell surface.
Fate of the Internalized LDL
The internalized LDL is acted upon by lysosomal enzymes, and cholesterol esters are hydrolyzed to cholesterol and fatty acids (Fig. 12.5). The free cholesterol can then be used directly as a component of membranes (the fatty acids can have any of the catabolic or anabolic fates discussed earlier).
Cholesterol, not needed for membrane synthesis can have following effects:
1. It decreases cholesterol biosynthesis by inhibiting activity of the key enzyme, HMG CoA reductase. Decreased de novo synthesis of cholesterol prevents any further elevation of the cellular cholesterol content. This point has important implications: the dietary cholesterol suppresses the synthesis of cholesterol by the body, especially in tissues other than liver.
2. Free cholesterol in cell inhibits synthesis of LDL receptors. As a result, cellular uptake of LDL is inhibited, and the circulating cholesterol in bloodstream increases.
3. The intracellular cholesterol activates the intracellular enzyme acyl CoA cholesterol acyl transferase (ACAT). This enzyme catalyzes transfer of an acyl group from a fatty acid derivative to cholesterol, resulting in the formation of esterified cholesterol. By this mechanism, the surplus cholesterol that is not immediately required for utilization by the cells is converted to cholesterol ester. The cholesterol ester is stored in the cell for subsequent use.
Some cholesterol is expelled from the cell and transferred to liver by HDL.
LDL Receptors on Macrophages are not Down-Regulated
The circulating LDL infiltrates through arterial walls and taken up by macrophages through another type of receptors which are different from the LDL receptors. These receptors on macrophages readily take up the LDLs, especially when they are chemically modified, either by acetylation or oxidation of apoB-100. They are not down regulated by intracellular cholesterol content of the macrophages. Therefore, uptake of cholesterol by macrophages continues even after intracellular cholesterol oncentration is substantially built up. Furthermore, the intracellular cholesterol content does not depress activity of HMG CoA reductase within the macrophages. Thus, macrophages are capable of unchecked and excessive uptake of circulating cholesterol. When level of circulating cholesterol rises (i.e. hypercho-lesterolaemia) the intracellular cholesterol concentration (of macrophages) builds up substantially. This causes transformation of the macrophages into “foam cells” which play an important role in development of the atherosclerotic plaques.
Macrophages play a role in atherogenesis because of unchecked and excessive uptake of circulating cholesterol.
LDL is bad cholesterol
The cholesterol rich LDL particles (about 75% of the serum cholesterol is contained in LDL) serve to transport cholesterol from liver to peripheral tissues. Therefore, elevated serum concentration of LDL is deleterious for having a positive correlation with cardiovascular disease. Hence, the LDL cholesterol is lethally dangerous, and commonly referred to as the “bad cholesterol”.
High Density Lipoprotein (HDL)
The HDL particles are referred to as scavengers because their primary role is to remove free (unesterified) cholesterol from the extrahepatic tissues, which is then excreted through bile. Evidently, this a crucial mechanism that prevents the inappropriate accumulation of cholesterol in peripheral tissues. Because accumulation of cholesterol in tissues is strongly associated with the development of atherosclerosis, the level of HDL in serum is inversely related to the incidence of myocardial infarction. Thus, HDL is cardioprotective or anti-atherogenic in nature, and is referred to as the “good cholesterol”.
A key role in reverse transport of cholesterol from extrahepatic tissues to the liver is played by HDL, which also serves as circulating reservoir of apolipoproteins.
Metabolism
The intestinal cells and hepatocytes synthesize various components of HDL and release them into bloodstream. These newly assembled particles are called nascent HDL. They are found to be discoid in shape (Fig. 12.6 ). The disc consists of a phospholipid bilayer, associated with which are a number of apolipoproteins (A-I, A-II, E and C). ApoA-I is the principal apolipoprotein initially, others are acquired later. Once in circulation, the nascent HDL attracts free, unesterified cholesterol from cells. It (the nascent HDL) binds these cells via apoA-I or apoE receptors, and acquires cholesterol from them.
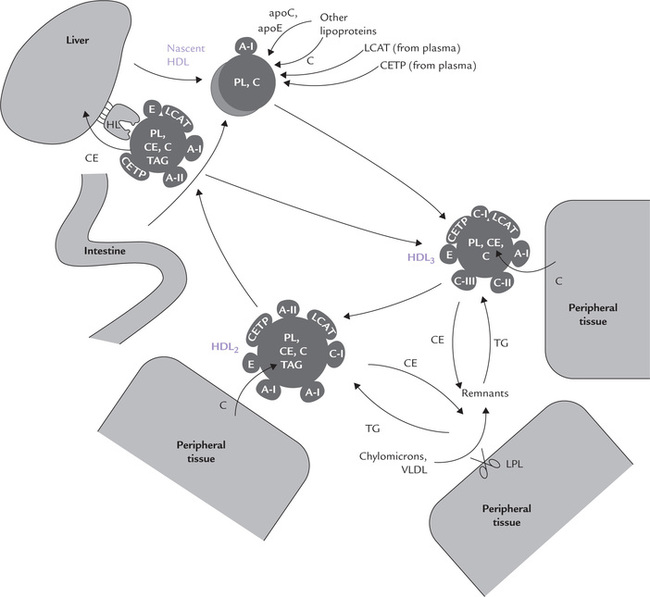
Fig. 12.6 Metabolism of HDL (TAG = triglyceride, PL = phospholipids, C = cholesterol esters, CETP = cholesterol ester transfer protein, LPL = lipoprotein, HL = hepatic lipase. All HDL apolipoproteins can be exchanged with other lipoprotein classes).
Further fate of this cell-derived cholesterol depends on a soluble, plasma enzyme, lecithin cholesterol acyl trans-ferase (LCAT). This enzyme binds to the disc (i.e. HDL) and gets activated by the apoA-I. The activated enzyme catalyzes transfer of fatty acyl residues from lecithin (phosphatidylcholine) to cholesterol to form lysolecithin and cholesterol ester:
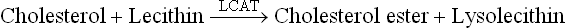
The lysolecithin is transferred to albumin while the hydrophobic cholesterol ester sinks between the two leaflets of the bilayer of the discoid, thereby forcing them apart. The originally flat HDL now bulges into a spherical, mature HDL particles called HDL3 with a hydrophobic core of cholesterol esters. The latter may then be transferred to other lipoproteins, such as chylomicrons, VLDL or remnants, either alone or in exchange for triacylglycerols (Fig. 12.7 ). The exchange is effected by cholesterol ester transfer protein (CETP; or apoD). The triacylglycerols thus acquired increase the particle size and the smaller HDL3 thus changes to larger HDL2.
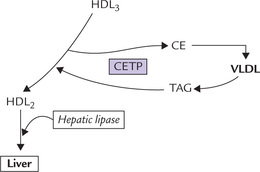
Fig. 12.7 Transformation of HDL3 to HDL2 by transfer of cholesterol esters to VLDL in exchange for triacylglycerols (CE = cholesterol esters, CETP = cholesterol ester transfer protein, HDL = high density lipoprotein, TAG = triacylglycerol, VLDL = very low density lipoprotein).
At the liver surface, HDL2 particles bind to a receptor called SR-B1. They (HDL2) are substrate for hepatic lipase (HL) which removes triacylglycerols in the liver. Cholesterol and cholesterol esters are also off-loaded from the degrading particles and transferred into liver cells. The lipid depleted, apoA rich HDL particles now return to plasma and continue to extract cholesterol from all membranes. A minority of these HDL particles acquire multiple copies of apoE from other lipoproteins and are taken up by liver cells by apoE-mediated receptor mechanism for degradation.
Functions
1. HDL extracts cholesterol from cell membrances for their subsequent excretion. This process termed reverse cholesterol transport (Box 12.3), ultimately lowers the level of intracellular cholesterol (since the cholesterol esters stored in the cell will be mobilized to replace the cholesterol removed from the cell membrane).
2. HDL serves as the circulating reservoir of apolipoproteins, transferring apoC and apoE to the nascent VLDL and chylomicrons (Figs 12.3and 12.4).
G Lipoprotein Receptors
A number of receptors present on hepatocytes or peripheral cells play an important role in the metabolism of lipoproteins.
1. The LDL receptor protein is 839 amino acid long and spans the cell membrane. Its gene is located on chromosome 19. It binds with two apolipoproteins (apoE and apoB-100) and therefore participates in endocytosis of apoB-100-containing LDL particles, and apoE-containing IDL particles. ApoE binds to the LDL receptor with higher affinity than apoB-100. ApoB-48 does not bind to the LDL receptor, so chylomicrons uptake is not mediated by LDL receptor.
2. Chylomicrons use a different type of hepatic receptor which recognizes apoE and apoB-48 and belongs to LDL-receptor gene family.
3. A third lipoprotein receptor, VLDL receptor has a high degree of homology with the LDL receptor. It has been shown to bind apoE containing VLDL, though its exact function in humans is uncertain.
4. The fourth type of receptor binds apoE and mediates uptake of HDL in liver cells.
H Lipoprotein(a)
Lipoprotein(a) has nearly identical structure as LDL. However, it contains an additional apolipoprotein molecule, called apolipoprotein(a), which is covalently linked to apoB-100.
Presence of lipoprotein(a) in blood in excessive amount increases risk of coronary heart disease. This could possibly be due to structural resemblance of this apolipoprotein with plasminogen. These two proteins have about 80% sequence homology. Plasminogen is the precursor of a blood protease, plasmin that degrades fibrin, the major component of blood clots. Because of close structural resemblance, apolipoprotein(a) competes with plasminogen, thereby decreasing its conversion to plasmin.
Reduced plasmin activity results in decreased degradation of clots. This, in turn, increases susceptibility of an individual to atherosclerosis.
Recent studies, however, do not substantiate this hypothesis. Clinical trials are underway to investigate this point.
I Disorders of Plasma Lipoproteins (Dyslipoproteinaemia)
The disorders of lipoprotein metabolism are divided in two categories: hyperlipoproteinaemias and hypolipoproteinaemias.
Hyperlipoproteinaemia
There is elevation of circulating lipoprotein levels, which may be primary or secondary to some chronic disease (e.g. diabetes-mellitus, alcoholism, or hypothyroidism). In most cases, however, the hyperlipoproteinaemia is multifactorial, resulting from the interaction between a sedentary lifestyle and the genetic constitution. Most widely accepted Fredrickson’s classification (later modified by WHO in 1970) catalogues hyperlipoproteinaemias into five types (Table 12.4 ), based on plasma levels of chylomicrons, VLDL, LDL, HDL, cholesterol and triacylglycerols after a 14-hour fast. These types are not diseases but rather phenotypes that occur in a variety of contexts, e.g. defect in apoproteins, receptors or enzymes. All of these except the type I are associated with atheromatous vascular disease. The deposition of lipids in subcutaneous tissue leads to xanthomas. Cholesterol deposition in cornea leads to corneal arcus; it indicates hypercholesterolaemia.
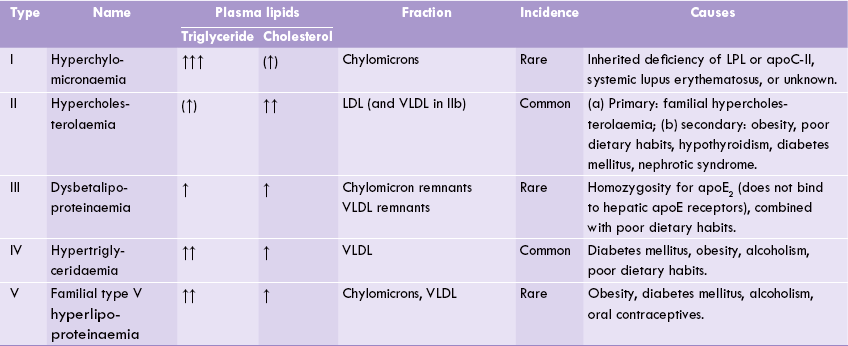
Type I (Hyperchylomicronaemia)
It is a rare form (prevalence 1 in 10,000) in which hydrolysis of chylomicrons is impaired due either to
In other cases, no specific cause can be identified.
Chylomicron band is prominent in fasting plasma and plasma triacylglycerols are markedly elevated (close to 1000 mg/dL). The patients complain of abdominal pain, have eruptive cutaneous xanthomas, and at risk of developing dangerously lethal pancreatitis in case the severe hypertriglyceridaemia (exceeding 2000 mg/dL) persists.
Type IIa (Familial-hypercholesterolaemia)
Cellular uptake of LDL is impaired resulting in high LDL and hypercholesterolaemia. The underlying causes is deficiency of functional LDL receptors in liver and extrahepatic tissues. This pattern is a major risk factor for atherosclerosis and coronary heart disease (CHD). The inheritance is autosomal dominant. The receptor deficiency may be 50% of normal in affected heterozygotes, and in homozygotes very few receptors may be there or even absent altogether. Xanthomas, especially in the form of tendon xanthomas, develop in most patients, but more serious is coronary atherosclerosis, which results in premature death by CHD (see Case 12.2). Fortunately, homozygosity for familial-hypercholesterolaemia (FH) is rare: 1 in 106.
Recently, four classes of mutations have been defined in FH.
The class 1 defect (most common) results in loss of receptor synthesis (due to gene deletion—partial or complete).
The class 2 defect result in synthesis of receptor proteins that are poorly translocated to the plasma membrane.
The class 3 defect results in receptors that bind poorly to apoB-100.
The class 4 defects result in receptors that are not internalized properly after binding LDL.
Type IIb (Hypercholesterolaemia)
In contrast to major inherited abnormality of type IIa, this type includes multifactorial and secondary forms that are far more common, affecting 2% to 8% of the total population. There is combined elevation of LDL and VLDL, therefore, both cholesterol and triacylglycerols are elevated in plasma. Obesity, diabetes mellitus, hypothyroidism, nephrotic syndrome and high cholesterol diet and saturated fats are important causes of this pattern.
Type III (Dysbetalipoproteinaemia)
A genetic variant of apoE, termed apoE2 is expressed, which cannot bind to the hepatic apoE receptors. This results in accumulation of remnant particles, e.g. IDL, chylomicron remnants, and IDL-like VLDL remnants in plasma, resulting in hypercholesterolaemia and hypertriglyceridaemia. Palmar xanthomas and tubo-eruptive xanthomas on knees, elbows and buttock are seen and there is high incidence of atheromatous vascular diseases. The atherosclerotic lesions are seen in peripheral arteries and coronary arteries, and so risk of CHD is also increased.
Type IV (Familial endogenous hypertriglyceridaemia)
It is a very common type caused by overproduction of VLDL associated with glucose intolerance and hyperinsulinaemia. Serum triglyceride levels are markedly elevated but milder elevation of cholesterol is also seen, so that incidence of atherosclerosis is increased. These manifestations generally set in after the fourth decade of life. Weight gain and obesity, diabetes mellitus (type 2), impaired glucose tolerance, chronic alcoholism, oral contraceptives, and excess dietary carbohydrates (especially sugars) are all frequently associated with this condition.
Type V
There is combined increase of chylomicrons and VLDL (cause unknown), so triglyceride levels are increased. Mostly this pattern has a familial background, but may also be secondary to other causes, such as diabetes mellitus, obesity, alcoholism and renal disease.
Other Causes
Some other causes of hyperlipoproteinemia, apart from those explained above, are as below:
1. Familial combined hyperlipoproteinemia: A dominantly inherited gene that increases synthesis of apoB-100 is present in this condition. However, hyperlipoproteinaemia is multifactorial because, depending on a variety of genetic and environmental factors, either the type II or the type IV pattern is expressed in these patients. These two most common patterns may occur in different members of the same family.
2. Familial LCAT deficiency: Cholesterol cannot be esterified, so free-cholesterol : cholesterol-ester ratio is increased enormously in all classes of lipoproteins. The reverse cholesterol transport and consequently cholesterol excretion is impaired and a variable hypercholesterolaemia and hypertriglyceridaemia develops.
3. CETP deficiency: Reverse cholesterol transport is decreased in this benign condition. Affected homozygotes have approximately a fourfold elevation of HDL cholesterol, but LDL cholesterol is low or normal (40–150 mg/dL). The circulating HDL particles are oversized, with lot of cholesterol esters, but little triacylglycerols.
4. Deficiency of hepatic lipase or cholesterol ester hydrolase in lysosomes (Wolman’s disease): It causes elevated VLDL levels in blood.
5. Excess of Lp(a): It often causes premature coronary heart disease (CHD).
Hypolipoproteinaemias
In this group of disorders concentration of one or more lipoproteins in plasma is decreased. The commonest of these disorders are abetalipoproteinaemia, characterized by absence of betalipoprotein (LDL) because of defect in synthesis or secretion of apoB-100 containing lipoprotein; hypobetalipoproteinaemia in which inability to synthesize apoB-100 and apoB-48 is the underlying defect; and hypoalphalipoproteinaemia in which the underlying defect has not been clearly defined (Case 12.3).
Abetalipoproteinaemia
It is a rare autosomal recessive disorder in which the liver and the intestinal cells cannot incorporate lipids in the apoB containing lipoproteins, i.e. chylomicrons, VLDL, and LDL. These lipoproteins are either absent or greatly decreased in plasma. Extremely low plasma levels of cholesterol and triacylglycerols occur. Severe fat malabsorption, and accumulation of triacylglycerols in the intestinal mucosa and the liver occur. Absorption of fat-soluble vitamins is severely impaired resulting in degenerative changes in retinal (atypical form of retinitis pigmentosum), myopathy, and spiny projections on erythrocytes (acanthocytes). Many patients die in childhood.
Hypobetalipoproteinaemia
This is an autosomal dominant disorder. There is decreased synthesis of apoB due to apoB gene mutations (approximately 20 different mutations identified) and so the apoB-containing lipoproteins are synthesized at a lower rate. LDL concentrations are 10–20% of the normal, VLDL is slightly lower and HDL is normal. Plasma cholesterol and triacylglycerols are decreased. Clinical abnormalities are mild and the condition is compatible with life.
Hypoalphalipoproteinaemia
It is an autosomal recessive disorder in which HDL level is reduced to less than 5% of normal. It is also known as Tangier disease as it was first reported on the Tangier island. Although the primary genetic defect is not yet known, there is marked deficiency of the major apolipopro-teins of HDL (apoA-I and apoA-II), probably because of accelerated catabolism. This results in decreased level of HDL in plasma and impaired reverse-transport of cholesterol. Cholesterol esters are accumulated in liver, spleen, tonsils, lymph nodes, Schwann cells, and the patients develop neuropathy, hepatospleenomegaly and lymphadenopathy. An orange discolouration of the tonsils is the most obvious clinical sign.
Despite HDL deficiency and consequent impaired reverse cholesterol transport, there is only a mild tendency for early atherosclerosis because the decrease of LDL cholesterol reduce the need for reverse cholesterol transport (LDL levels are reduced to approximately one third of the normal).
II Fatty Liver Syndrome
A Overview
The total lipid content of a hepatocyte is around 50% of the total cell mass and triacylglycerols account for a third of it. When excessive amounts of triacylglycerols are accumulated in the liver to such an extent that it may be observed by naked eye, the condition is referred to as fatty liver. As the fat accumulation becomes chronic, fibrotic changes that later progress to liver cirrhosis are induced in the hepatic tissue.
Excessive accumulation of triacylglycerols may occur due to:
Synthesis
In hepatocytes, the TAG molecules are synthesized from the fatty acids that are derived from two sources:
1. Hydrolysis of the TAG stores of adipose tissue by hormone sensitive lipase.
2. Hydrolysis of chylomicrons and VLDL triacylglycerols by lipoprotein lipase.
The synthesis of triacylglycerols in liver cells occurs within the cisternae of the endoplasmic reticulum.
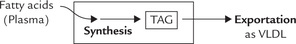
Exportation
Subsequently, these triacylglycerol molecules are complexed with apolipoprotein B100 and other biomolecules to form very low density lipoprotein (i.e. VLDL) which then leave the liver cells to enter the blood circulation (Fig. 12.8 ).
B Causes of Fatty Liver
The TAG accumulation initially produces morphological changes in liver cells, called fatty change. Metabolic derangements and a host of agents, including chemicals as well as the bacterial or viral infections induce fatty change in liver cells.
As shown in Figure 12.8, the TAG accumulation may result from interference at some points in the metabolism in the liver cells: either TAGs are synthesized excessively, or less TAGs are exported from hepatocytes.
Excessive Synthesis of Triacylglycerols
It occurs when plasma fatty acid levels rise, and therefore, excessive amount of fatty acids enter the liver cells for TAG synthesis. For example, in starvation and diabetes mellitus, depot fats are mobilized and increased amount of fatty acids are brought to the liver where they are synthesized into triacylglycerols. Intake of alcohol causes elevation of plasma triacylglycerols and induces esterification of fatty acids; however, other mechanisms may also be involved.
Impaired Exportation of Triacylglycerols as VLDL
It may occur as a consequence of (a) diminished VLDL synthesis or (b) impaired release of VLDL into the circulation.
Major causes of impairment of the lipoprotein production are as here:
Inhibition of Protein Synthesis
Inhibition of protein synthesis in rough endoplasmic reticulum is caused by substances like puromycin, carbon tetrachloride, phosphorus, lead and arsenic. By inhibiting protein synthesis, these substances decrease availability of apolipoprotein B-100, which is the major apolipoprotein of VLDL. This results in decreased production of VLDL.
Impaired Formation of the Lipid Moiety
It occurs when there is a dietary deficiency of choline or its precursor methionine or betaine. These substances, known as the lipotropic factors, are required for the formation of phospholipids.
• Choline: It is an essential component of the phosphatidylcholine (lecithin), the major phospholipid of VLDL. Deficiency of choline, therefore, leads to diminished synthesis of VLDL.
• Methionine: Activated methionine, termed S-adenosyl methionine (SAM), participates in synthesis of lecithin by transmethylation reaction. It involves transfer of methyl groups from active methionine (S-adenosyl-methionine; SAM) to cephalin.
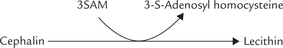
Therefore, methionine is actively involved in synthesis of lecithin and thus acts as a lipotropic substance.
Betaine which contains three methyl groups, also serves as a lipotropic factor.
Lipotropic factors are required for synthesis of phospholipid, and hence VLDL. Their deficiency leads to decreased VLDL synthesis—an important cause of fatty liver.
Deficiency of lipotropic factors leads to fatty liver by additional mechanism such as by inducing deficiency of phosphocholine and carnitine.
(a) Phosphocholine deficiency: Phosphocholine is required for stimulating glycosylation of apoB-100. Since this step is important for the production of nascent VLDL particles, its impairment leads to a decreased production of the latter.
(b) Carnitine deficiency: This compound contains three methyl groups and is required in β-oxidation. Its deficiency results in decreased utilization and hence increased cellular content of fatty acids, which generate excessive triacylglycerol deposits.
Fatty liver is an excessive accumulation of triacylglycerols in liver, which results because of imbalance in the rate of triacylglycerol synthesis (↑) and export (↓) from liver. Impaired export is due to decreased VLDL synthesis, which is mostly associated with the deficiency of lipotropic factors: choline, methionine, betaine.
C Role of Chemicals and Other Agents
A number of chemicals and other agents are known to cause fatty liver. Substance like carbon tetrachloride, phosphorus, lead and arsenic interfere with the function of the ER, where assemblage of various components ofVLDL occurs. They bring about disruption of the ER membrane through generation of free radicals. Protection against such damage is provided by antioxidants including vitamin E. When adequate vitamin E is not supplemented in the diet, it may lead to enhanced oxidative damage and thus fatty liver.
Carbon tetrachloride has an additional damaging effect: it causes an impaired secretion of VLDL from hepatocytes.
Ethionine acts by a competitive mechanism because of its structural similarity with methionine. It replaces methionine in S-adenosyl methionine (SAM). This traps the available adenine and thus prevents synthesis of ATP. Administration of ATP or adenine, therefore, reverses the inhibition caused by ethionine.
Orotic acid causes fatty liver by interfering with glycosylation of apoB-100. This inhibits the formation and release of nascent VLDL.
Clostridium diphtheriae exotoxin is also known to induce fatty liver. It acts by interfering with metabolism of carnitine.
Deficiency of essential fatty acids (EFA) can also lead to fatty liver because EFA are needed for the synthesis of phospholipids. The second carbon position of the glycerol backbone is usually esterified with EFA. Deficiency of EFA therefore, inhibits formation of the phospholipids, which in turn inhibit synthesis of VLDL. Excess cholesterol also induces deficiency of EFAs. In this condition most EFAs are used for esterification of cholesterol and less are left for phospholipid synthesis.
Chronic alcoholism also causes fatty liver, which may later lead to cirrhosis. The triacylglycerols are accumulated due to increased endogenous synthesis of triacylglycerols following alcohol metabolism (Chapter 15).
Finally, the significance and interpretation of fatty change depends on the underlying pathogenic mechanisms. A milder form of fatty change may have no effect on the cellular function while it may be fatal if severe. In most cases the fatty change is reversible. However, if some vital intracellular process is irreversibly impaired, the consequences may be serious.
III Metabolism of Cholesterol
A Overview
Cholesterol (in Greek, chole means bile, steros means solid, ol means alcohol) is the most important sterol in animal cells. It is the only important membrane steroid in animals and humans. In a 70 kg man, approximately 140 g cholesterol is present, the majority of which is “free” (unesterified) cholesterol in the cellular membranes. Cholesterol is most abundant in those tissues that contain large amounts of membrane lipids, namely the nervous system; brain contains approximately 30 g cholesterol. The condensed ring structure of cholesterol makes it rigid, and this is believed to alter the stiffness of membrane and its permeability.
Cholesterol molecule (27 carbons) comprises a non-aromatic ring system known as cyclopentano-perhydrophenanthrene ring, which is decorated with a hydroxyl group, two methyl groups and a branched hydrocarbon chain (Chapter 3). The hydroxyl group, present at C-3 of the ring, is esterified with a long-chain fatty acid, to form cholesterol ester (CE), which is used as intracellular storage form of cholesterol. CE is abundant in some steroid hormone producing tissues, particularly the adrenal cortex where the cholesterol ester stores may be so extensive that the cells resemble adipocytes in possessing visible lipid droplets. CEs are also prominent in the plasma lipoproteins, wherein approximately 70% of the cholesterol is esterified. Cholesterol esters make up about two third of the total circulating cholesterol.
Cholesterol is a product of animal metabolism. It is present in various foodstuffs of animal origin such as egg yolk, meat, and liver; a vegetarian diet is essentially cholesterol free. Plants contain other sterols, which are collectively called phytosterols. Ergosterol (mostly in fungi) and beta-sitosterol (in higher plants) are examples of phytosterols. They are poorly absorbed from dietary sources and therefore are present only in small amounts in the human body.
B Biosynthesis of Cholesterol
All nucleated cells of the body can synthesize cholesterol, including liver, adrenal cortex, intestine, ovaries, testes, skin, arterial walls, etc., but liver is the most important site, accounting for at least 50% of the total. Some sterol hormone producing endocrine tissues, such as the adrenal cortex and the corpus luteum, have very high rates of cholesterol synthesis. Endogenous cholesterol synthesis amounts to 0.5–1 g per day, depending on the dietary supply. Low cholesterol diet enhances the synthesis, whereas high-cholesterol diet suppresses it.
Acetyl CoA is the source of all the 27 carbon atoms of cholesterol, as reported by Konrad Bloch in 1940. Formation of a 27-carbon molecule from a two-carbon precursor (i.e. acetyl CoA) involves a series of condensation reactions. These reactions require input of a large amount of energy. The energy is provided by the high energy thioester bonds, as also by the ATP molecules. These reactions also require reducing equivalents, which are provided by NADPH.
The enzymes of the biosynthetic pathway, which number close to 30, are found in the cytosol and the endoplasmic reticulum (ER). An outline of various stages of the pathway is as below:
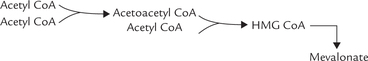
Stage I: Formation of 6-C Compound, Mevalonate
By a sequential action of three enzymes, mevalonate is produced from acetyl CoA molecules. Two acetyl CoA molecules condense first to form a four-carbon compound the acetoacetyl CoA. The reaction is catalyzed by the enzyme thiolase. One more acetyl CoA molecule then condenses with the acetoacetyl CoA to form a six carbon compound called hydroxymethyl glutaryl CoA (HMG CoA). This reaction is catalyzed by HMG CoA synthase (Fig. 12.9 ). Liver parenchyma cells contain two types of HMG CoA synthase: cytosolic and mitochondrial. The cytosolic form participates in cholesterol synthesis, whereas the mitochondrial enzyme is involved in the synthesis of the ketone bodies.
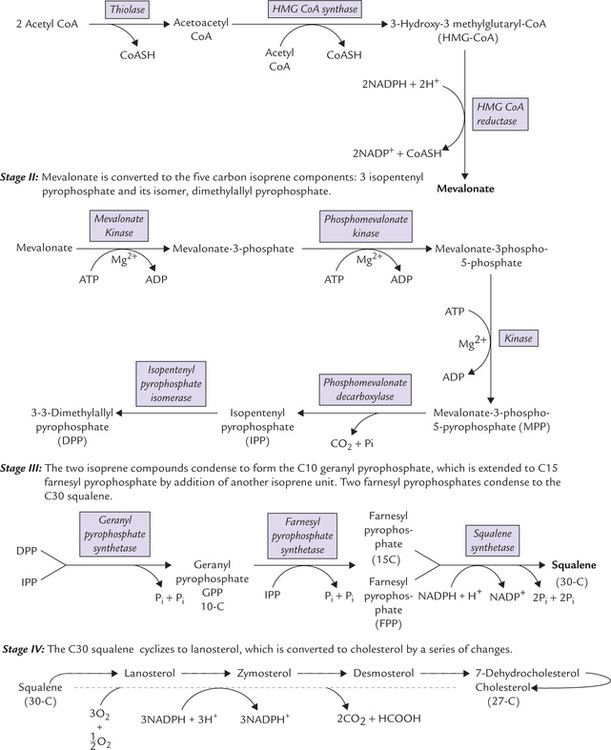
In the next step, HMG CoA is reduced by the enzyme HMG CoA reductase to form mevalonate; two molecules of NADPH are used to provide the reducing equivalents. This step is rate-limiting for the pathway. Activity of HMG CoA reductase is regulated by a number of modulators, as discussed later in this chapter.
Like other biosynthetic reactions, these three reactions are also endergonic, which require input of energy. For this purpose, the precursors for these reactions are energized: acetyl CoA represents the energized form of acetate.
Stage II: Formation of Energized 5-C Units: Isopentenyl Pyrophosphate and Dimethylallyl Pyrophosphate
Mevalonate is further activated to form the energized “5 carbon units” which are capable of participating in the more complex endergonic reactions that follow. Formation of these 5-C units occurs in two phases:
• First, mevalonate is activated by three consecutive phosphorylation reactions to form mevalonate-3-phospho-5-pyrophosphate. These reactions are driven by the ATP energy.
• Decarboxylation of mevalonate-3-phospho-5-pyrophosphate follows to form the 5-carbon isopentenyl pyrophosphate (IPP). The reaction is again driven forward by energy provided by ATP. IPP is then isomerized to 3–3 dimethylallyl pyrophosphate (DPP).
Both IPP and DPP are energized compounds and are therefore, highly reactive. They are referred to as the iso-prene units.
Stage III: Squalene Formation by Condensation of Six Isoprene Units
Formation of 30-C squalene from the 5-C (isoprene) units is evidently a complex process (Fig. 12.9). It involves successive condensation reaction as below:
1. IPP and DPP condense to form a ten carbon compound, geranyl pyrophosphate (GPP). The reaction is driven forward by pyrophosphate hydrolysis.
2. A second molecule of IPP condenses with GPP to form farnesyl pyrophosphate (FPP), a 15-carbon compound. This compound stands at branch point of various biosynthetic pathways; it can enter the pathways leading to biosynthesis of cholesterol, dolichol or ubiquinone.
3. Two molecules of FPP (15-C each) further condense to yield a 30-carbon compound, called squalene. Squalene synthase is the enzyme catalyzing this reaction. Two molecules of pyrophosphate are released in this step (one from each of the two molecules of farnesyl pyrophosphate), which ensures irreversibility of this complex reaction. Two NADPH molecules are used for providing the reducing equivalents.
Stage IV: Formation of 27-C Cholesterol from Squalene
A series of modifications in the ER produce cholesterol from squalene. In order to understand these modifications, it is essential to know the structural differences between these two compounds. These are:
1. Squalene contains 30 carbon atoms, whereas cholesterol is a 27-carbon compound.
2. Squalene is a linear molecule, whereas cholesterol has a cyclic ring structure.
3. Squalene contains six double bonds, whereas cholesterol contains only one.
4. Squalene does not contain a hydroxyl group, whereas cholesterol has one at the third carbon position.
Therefore, the following changes are introduced in the squalene molecule during its conversion to cholesterol:
• Loss of 3 carbon atoms by decarboxylation reactions
Squalene to cholesterol conversion is a multi-step process which has been proposed to include 19 different enzymatic reactions. All the enzymes catalyzing these reactions are located in the endoplasmic reticulum. This is in contrast to enzymes of the stage I, II and III reactions which are located in cytosol.
Some of the important intermediates of the pathway are lanosterol, zymosterol and desmosterol (Fig. 12.9).
The committed step in cholesterogenesis is formation of mevalonate from HMG CoA (enzyme: HMG CoA reductase). Mevalonate is processed to the branched-chain 5-C compounds (isoprene units), six of which condense to form the C-30 compound, squalene, which cyclizes to yield lanosterol—the steroid precursor of cholesterol.
C Regulation of Cholesterol Biosynthesis
The rate limiting enzyme of cholesterol biosynthesis, HMG CoA reductase, is an intrinsic membrane protein in the endoplasmic reticulum. Its active site extends into the cytosol. Activity of this enzyme is regulated by a number of factors:
Feedback Inhibition
Activity of HMG CoA reductase is inhibited by cholesterol. Thus, amount of cholesterol in diet significantly influences the endogenous cholesterol synthesis; average diet contains 300–400 mg of cholesterol, and the synthesis increases when cholesterol in diet is low.
Hormonal Regulation
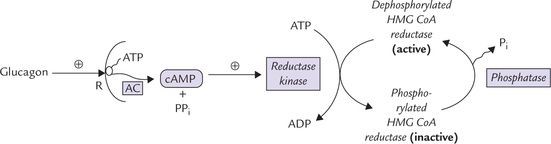
The cAMP-directed cascade, which is involved in regulation of glycogen metabolism controls the activity of HMG CoA reductase on short-term basis (Fig. 12.10 ). For example, glucagon increases intracellular cAMP level, which activates the enzyme reductase kinase. The latter in turn favours phosphorylation of the HMG CoA reductase. Since phosphorylated form of this enzyme is inactive, rate of cholesterol biosynthesis decreases by this sequence of reactions. In contrast, insulin favours dephosphorylation and hence enhances formation of the active (dephosphorylated) enzyme. This results in an increased rate of cholesterol biosynthesis.
Sterol-mediated Regulation of Transcription
Rise in intracellular cholesterol concentration inhibits cholesterol biosynthesis by decreasing the transcription of the HMG CoA reductase gene. The effect may also be mediated at post-transcriptional level. Thus, intracellular levels of cholesterol, when elevated, inhibit both the activity as well as the synthesis of HMG CoA reductase. Since HMG CoA has a lifespan of approximately 4 hours, a change in its rate of synthesis can effect cholesterol synthesis rather rapidly.
Inhibition by Drugs
Some drugs inhibit cholesterol synthesis by acting as reversible competitive inhibitors of HMG CoA reductase. Examples include mevastatin and lovastatin. They are used as cholesterol lowering agents in patients having elevated serum cholesterol concentration. Action of various hypolipidaemic drugs is described later in this chapter.
D Cholesterol Homeostasis in General
Despite popular belief, cholesterol is not a health-hazard, but an essential component of cell membranes and the basis of several steroid hormones. It creates a problem only if it is in excess and in this respect the maintenance of cholesterol homeostasis is of vital significance. Normally, the body has to deal with about one gram of cholesterol a day (Fig. 12.11 ), derived from both exogenous sources (i.e. dietary intake) and endogenous sources (i.e. de novo synthesis).
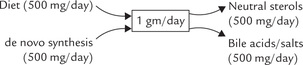
Fig. 12.11 Cholesterol can either be obtained in diet or synthesized in liver, to a total quantity = 1gram/day. Bile acids (salts) and neutral sterols are the major excretory forms of cholesterol.
Normally, the de novo synthesis and dietary sources make roughly equal contributions of about 500 mg each. Total quantity (1 g a day) remains fairly constant because if dietary intake changes, compensatory changes occur in the biosynthesis of the sterol in the tissues.
Homeostasis is maintained by the loss of cholesterol metabolites in faeces: as bile acids or neutral sterols.
1. Cholesterol from diet and de novo synthesis: Dietarycholesterol is absorbed in form of chylomicrons, which are delivered to blood via lymphatic systemand then presented to the liver. Plant sterols decreaseintestinal absorption of cholesterol. The absorption depends on the presence of bile salts and is influenced by the amount of dietary cholesterol: it may beabove 60% on a low cholesterol diet, but an absorption of 30% is more typical for a high-cholesterol diet.
As mentioned earlier, there is a tight control of biosynthesis so that it is responsive to changes in dietary supplies.
2. Excretion of cholesterol: Daily excretion of cholesterolamounts to about one gram, which corresponds todaily cholesterol load from diet plus endogenoussynthesis. In humans the sterol ring of cholesterolcannot be cleaved. Therefore, the cholesterol has tobe disposed off after some modifications in its sidechain. These modifications result in formation of neutral sterols and bile acids.
Neutral sterols: Cholesterol present in bladder bile or that present in epithelial cells desquamated into intestinal flora serve as precursor for the neutral sterols. Coprostenol is the principal neutral sterol, formed from cholesterol in the lower intestine by the bacterial flora therein, and is eliminated in faeces. The fecal sterols account for elimination of 500 mg cholesterol each day.
Bile acids (salts): These are formed from cholesterol in hepatocytes and secreted actively into biliary canaliculi. They aid the digestion of dietary lipids. A majority of the secreted bile acids are recovered via the enterohepatic circulation, but daily faecal losses equivalent to 500 mg cholesterol occurs (Fig. 12.12 ).
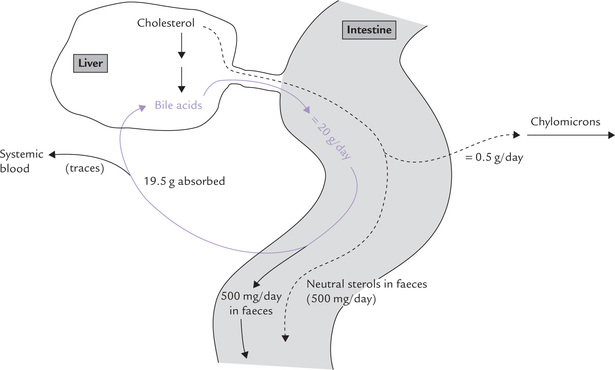
Fig. 12.12 Disposition of cholesterol in enterohepatic system of about 20 grams bile acids daily secreted into intestine, about 19.5 grams are recovered by enterohepatic circulation. Fecal loss equivalent to 500 mg cholesterol occurs as bile acids, and another 500 mg as neutral sterols.
Metabolism of Bile Acids and Salts
The 24-carbon bile acids are synthesized in hepatocytes and stored in the gall bladder, and secreted via biliary canaliculi into intestine where they play a vital role in lipid digestion. They are more appropriately called bile salts because they are present in the deprotonated form at pH of 7.0.
Synthesis
About half of the cholesterol in the body is ultimately metabolized to bile acids. The primary bile acids, which are synthesized in the liver include cholic acid and cheno-deoxycholic acid, cholic acid being more abundant. They are synthesized from cholesterol by:
(a) Hydroxylation - addition of hydroxyl groups to the ring, (b) Chain cleavage - removal of a 3-carbon fragment from the hydrocarbon chain attached at C-17 of the steroid nucleus, (c) Reduction of the B-ring of the steroid nucleus, and (d) Oxidation of the terminal carbon of the hydrocarbon chain to carboxyl group.
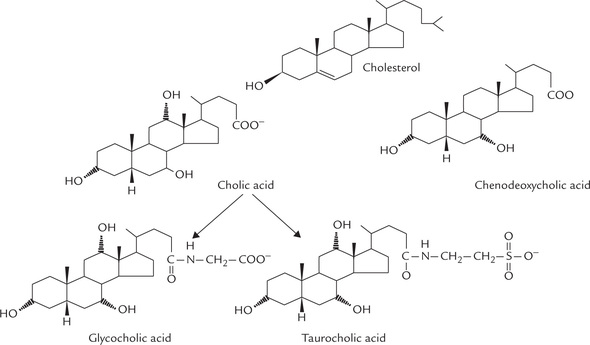
Liver releases the bile acids not in the free form but as conjugation products with glycine or taurine (Fig. 12.13 ).
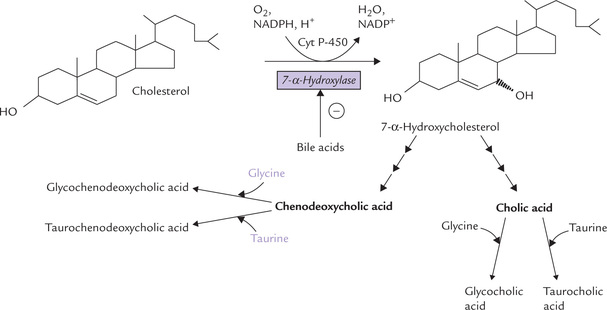
Fig. 12.13 Synthesis of the primary bile acids by ER-associated enzymes in hepatocytes. Cholesterol is converted into cholic acid, which combines with either glycine (to form glycocholic acid) or taurine (to form taurocholic acid). The other primary bile acid, chenodeoxycholic acid, also forms similar conjugated products, which have stronger detergent effect.
The primary bile acids, cholic acid and chenodeoxycholic acids are conjugated with glycine taurine, and are excreted through the bile, where they exist as sodium or potassium salts of bile acids, called bile salts.
Structure of primary bile acids are shown in Figure 12.14 .
Regulation of Biosythesis
• Synthesis of 7-α-hydroxylase, the rate limiting enzyme, is inhibited by bile acids (via reducing transcription of its gene).
• Dietary cholesterol induces the transcription of this enzyme.
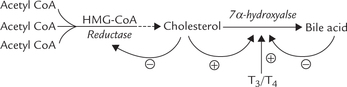
Because cholesterol also exerts a stimulatory effect on 7-α-hydroxylase, it not only inhibits further synthesis of cholesterol, but also channels the existing cholesterol for bile acid synthesis. These regulatory mechanisms maintain an adequate pool of free cholesterol in the liver.
Thyroid hormones (T3, T4) induce transcription of 7-α-hydroxylase; thus in patients with hypothyroidism plasma cholesterol tends to rise.
Secondary bile acids
The primary bile acids are modified in the lower parts of small intestine by bacterial enzymes to form secondary bile acids, e.g. deoxycholic acid and lithocholic acid. Only a proportion of primary bile acids are converted into secondary bile acids, a process which requires:
In these reactions, cholic acid is converted to deoxycholic acid and chenodeoxycholic acid to lithocholic acid (Fig. 12.15 ). They are mostly returned to the liver (98%) through the portal circulation where they are conjugated and secreted in the bile. Because of this enterohepatic circulation, discussed below, both primary and secondary bile acids are present in the bile.
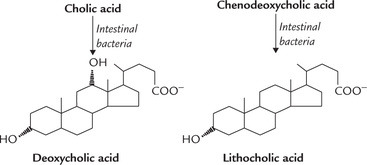
Functions of bile acids
Bile acids act as detergents (because they contain polar and non-polar regions) assisting the emulsification of ingested lipids into very small globules; this aids enzymatic digestion of dietary fats. They form molecular aggregates called micelle, which respectively bring about the enzymatic digestion and absorption of dietary fat (Chapter 26).
Enterohepatic circulation
Approximately 20 g bile acids pass from the bile duct into the intestine each day but only 2% of this (500 mg) is lost through faeces (Fig. 12.12). The rest (19.5 g) is deconjugated and reabsorbed. Passive reabsorption of bile acids occurs in the jejunum and colon but the majority is reabsorbed in the ileum by a process of active transport. The reabsorbed bile acid molecules are non-covalently bound to albumin and transported in blood via the portal vein to liver, and finally re-secreted through bile.
This enterohepatic circulation of bile acids allows large amounts of bile acids (20–30 g) to be delivered to the intestine daily from a relatively small total bile acid pool (3–5 g). Only 500 mg is synthesized per day but an average bile acid molecule is recycled five to eight times every day, and it remains in the system for approximately a week before it is finally excreted. The daily loss and daily synthesis (500 mg each) balance each other.
E Specialized Products Derived from Cholesterol
Cholesterol is not only an essential component of cell membrane, but also a precursor of the following biologically active compounds.
1. Vitamin D: Formation of vitamin D3 occurs by ultraviolet irradiation of the skin. It converts 7-dehydrocholesterol into choecalciferol by spontaneous cleaving of the B-ring of the cholesterol.
2. Adrenocortical hormone: The steroid hormones of the adrenal cortex are produced from cholesterol by a series of changes (Chapter 30).
3. Sex hormones: The male sex hormone, testosterone (19-C), and the female sex hormone, oestrogen (18-C) are also derived from cholesterol.
4. Bile acids and bile salts: These have already been described.
IV Atherosclerosis
Atherosclerosis is a condition in which arteries are blocked to a greater or lesser extent by the deposition of cholesterol plaques, leading most commonly to coronary heart disease by blocking of coronary arteries. Involvement of cerebral arteries causes cerebral thrombosis, and of arteries supplying blood to lower limbs to peripheral vascular diseases.
A Role of Hyperlipidaemia in Atherosclerotic Diseases
Several studies throughout the world have provided substantial evidence that raised plasma concentration of lipid parameters are linked to atheromatous vascular diseases (although a vast number of individuals manifesting hyperlipidaemia do not suffer from one of these monogenic diseases). This provides a major stimulus for estimation of plasma lipids in clinical practice. As a preliminary to the discussion on the role of certain forms of hyperlipidaemias in causing atherosclerosis, it is useful to consider normal lipid profi le (hereafter called atherogenic profi le) and its interpretation.
B Atherogenic Profile
The lipid parameters commonly estimated are: total cholesterol (TC), LDL-cholesterol, HDL-cholesterol, triacylglycerols and phospholipids. The sample should be collected only after 12–14 hours of fasting. Total plasma lipids are estimated to be 400–600 mg/dL, of which, roughly speaking, one-third is cholesterol, one-third is triacylglycerols and one-third is phospholipids (Table 12.1). Several methods are available for the estimation of total cholesterol, such as Liebermann-Burchard reaction, Carr and Dructor method and, more recently cholesterol oxidase method. HDL-cholesterol is estimated after adding polyethylene glycol (PEG), which precipitates LDL and VLDL. VLDL is equivalent to one-fifth of the plasma triacylglycerols in the fasted state. LDL-cholesterol can be calculated by the following formula:
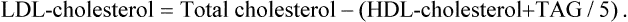
Cholesterol is present in plasma mainly in LDL (70%; the “bad cholesterol”) and in smaller amounts in HDL (“the good cholesterol”). Normal values of total cholesterol and LDL- and HDL-cholesterol, and the associated risk of atherosclerosis in adults are shown in Table 12.5 .
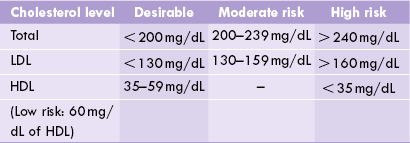
The values mentioned in Table 12.5 are recommended by the American Heart Foundation, but cannot be applied to Indians for Indians have higher incidence of other risk factors, such as central obesity with high waist: hip ratio and Lp(a). Lower values of total cholesterol (< 180 mg/dl) are therefore desirable.
In addition to the usual lipid parameters described above, measurement of apolipoproteins are carried out in more advanced set-ups for additional information. It has been reported that plasma levels of apoproteins A-I and B are more informative than HDL and LDL. A rise in A-I indicates a decreased risk of CHD whereas a rise in apoB indicates an increased risk. A ratio of A-I : A-III exceeding 3.0 is interpreted as highly cardioprotective.
Age Related Changes in Atherogenic Profile
Age related changes are observed in the above parameters; for example, after the fourth decade of life plasma cholesterol levels tend to slowly rise in men and post-menopausal women. Serum level of LDL for adult males of 30–40 years = 80–175, and for 40–60 years = 90–200 mg/dL have been reported.
Pathological Variations
Serum cholesterol is subject to variations in a number of conditions. Increased concentration (hypercholesterolaemia) is characteristically seen in the following conditions:
1. Hypothyroidism: This is due to decreased synthesis of 7-α-hydroxylase that causes conversion of cholesterol to bile acids (thyroid hormones induce synthesis of this enzyme). Moreover, there is decreased expression of HDL receptors on hepatocytes in hypothyroidism.
2. Diabetes mellitus: This is because of increased formation of acetyl CoA (by β-oxidation), the precursor of cholesterol synthesis.
3. Obstructive jaundice: Impaired excretion of cholesterol through bile leads to plasma accumulation.
4. Nephrotic syndrome: Urinary loss of albumin leads to compensatory increase in synthesis of globulins. When lipoproteins are thus increased, there is a corresponding elevation in cholesterol.
C The Assessment of Cardiovascular Risk
Several epidemiologic studies and clinical trials have demonstrated that there is a relationship between the levels of lipids and lipoproteins in plasma and the incidence of atherosclerosis and coronary heart disease. Though total plasma cholesterol level is believed to be the prime risk factor, other lipid parameters also play a role. Furthermore, several studies have shown that a sustained reduction in plasma cholesterol levels leads to reduced mortality and morbidity from coronary heart disease.
1. Role of cholesterol: The risk of CHD is related to plasma levels of total cholesterol and LDL-cholesterol. Framingham Heart Study with 30 years of follow up reported a linear increase in coronary “risk” when the total cholesterol level crosses 200 mg/dL. Conversely, a 1% decrease in total plasma cholesterol predicts a 2% reduction in CHD risk. The Multiple Risk Factor Intervention Trial in the USA also suggests a link between prevalence of CHD and serum cholesterol levels.
2. Role of LDL and HDL: LDL carries about 70% the oftotal cholesterol, transporting them to tissues, henceenhancing the “risk” of CHD, and HDL has a beneficial,cardioprotective role. Recent studies have shown that atany given level of LDL the probability of CHD increasesas the level of HDL decreases. The prevalence of CHD at35 mg/dL plasma HDL-cholesterol is eight times the CHD rate of persons with HDL levels of 65 mg/dL orgreater. In addition to removing cholesterol via the hepatobiliary route, the other antiatherogenic effects of HDLcholesterol include:
• Inhibition of conversion of LDL to oxidized LDL, which is preferentially taken up by the tissue macro-phages to form fatty streaks.
• Prevention of adhesion of monocytes to the endo-thelium.
• Prolongation of the half-life of prostacyclin produced by endothelial cells; this promotes vasodilatory effects.
Finally, decreased HDL levels are associated with male sex, obesity, physical inactivity, cigarette smoking and hypertriglyceridaemia—the other risk factors for CAD, described later.
3. Role of triacylglycerols: Some of clinical studies suggestthat an increased triacylglycerides level is associated withan increased risk for CHD.
4. Other contributing risk factors: Lipids are not the onlyfactors that determine the development of atherosclerotic disease. A number of other risk factors are alsoinvolved, as summarized in Table 12.6 . These factorshave additive effect on the risk of CHD, meaning thatpresence of these risk factors increases the risk associatedwith dyslipidaemia. Therefore, the desirable cholesterollevels are lower in individuals with other risk factors,such as diabetes or hypertension.
Table 12.6
Risk factors for atherosclerotic diseases
| Risk factor | Comment | Remarks |
| Male sex | Female hormonal background provides a measure of protection, so the difference in “risk” equalizes in post-menopausal women. | – |
| Sedentary life | Greatly increases risk. Beneficial effects of exercise are related to: lipid-lowering and physiological processes, e.g. dilatation of the arteries of heart, increased blood flow. | Lifestyle changes |
| Age | Increased risk with age may relate to age-related decline in physical activity, and other factors. | – |
| Smoking | Nicotine causes transient vasoconstriction of coronary and carotid arteries, and also enhances adipose lipolysis to increase free fatty acids and serum cholesterol. | Smoking cessation |
| Stress | Catecholamines mobilize free fatty acids from adipose tissue. Increase synthesis of VLDL. | Lifestyle changes |
| Diet | Dietary fat, particularly cholesterol and saturated fats are especially hazardous. | Diet low in saturated fat, and where appropriate, cholesterol-lowering drugs |
| Apo(a) | Independent risk factor for CHD. | – |
| Homocysteine | Increased level in blood is a risk factor for CHD (normal < 200 mg/dL). | – |
| Hypertension | Systolic > 160 mmHg is a major risk factor for stroke, and a risk for CHD. Roughly an increase by 10 mmHg reduces life expectancy by 10 years. | Control blood pressure, diet, drugs |
| Obesity | Greatly enhances risk of CHD. | Attain ideal body weight |
| Diabetes | TAG stores of adipose mobilized; dyslipidaemia common; glycosylation of LDL prolongs half-life, and oxidized LDL more readily taken up, thus enhancing risk of atherosclerosis. Cardiovascular disease is the main cause of death in diabetes. |
CHD = coronary heart diseases, LDL = low density lipoproteins, TAG = triacylglycerols, VLDL = very low density lipoproteins.
Atherosclerosis is a multifactorial disease characterized by accumulation of atheromatous plaques, consisting of a core of cholesterol esters surrounded by an area of fibrosis. Lipoproteins are important risk factors: elevated VLDL, IDL or LDL are associated with atherosclerosis, whereas high levels of HDL have a protective effect.
D Pathogenesis and Consequences of Atherosclerosis
Atherosclerosis is a set of pathological changes in intima of large arteries that can lead to serious and often fatal complications. The characteristic lesion in the intima is atheromatous plaque having a core of soft lipid (athere), surrounded by an area of fibrous (sclerosis) component. The lipid core mainly consists of cholesterol esters which are derived from LDL particles. The latter enter intima by transcytosis where they are endocytosed by macrophages through the scavenger receptors. Since the scavenger receptor is not regulated, the macrophages become overloaded with cholesterol and become foam cells. The conglomerates of foam cells form fatty streaks, yellow patches visible in the arterial wall. Fatty streaks are an early reversible lesion: most of the fatty streaks regress spontaneously but some may develop into atheromatous plaques by fibroproliferative response.
The fibroproliferative response starts when surrounding smooth muscle cells and the endothelial cells start secreting a range of small peptides such as cytokines and growth factors that regulate cell growth. Examples include platelet-derived growth factor, interleukin-1, and tumour necrosis factor. These peptides stimulate smooth muscle cells to proliferate and to migrate toward the luminal side of the arterial wall. The proliferating muscle cells start synthesizing extracellular matrix, particularly collagen. These changes (migration of smooth-muscle cells and accumulation of collagen rich extracellular matrix) result in the formation of a cap that surrounds the lipid rich core. This is mature atherosclerotic plaque. The plaque protrudes into the arterial lumen, which impedes the blood flow and makes it turbulent. The turbulent flow increases the risk of clot formation. In due course of time the atheromatous plaques may get dislodged because macrophages and T-cells residing at the edges of the plaque secrete enzymes (called metalloproteinases) that degrade extracellular matrix. The degradation exposes collagen to the blood stream. This leads to adherence and aggregation of platelets to the exposed collagen and initiates the formation of blood clot. The resulting occlusion of a major blood vessel leads to ischaemic death of the affected tissue.
Consequences are serious; for example, block in coronary artery manifests as acute myocardial infraction, blockage in arteries supplying blood to brain causes stroke and block in arteries supplying to lower limbs results in peripheral vascular diseases.
E Management of Hyperlipidaemia: Biochemical Basis
The management guidelines for hyperlipidaemia rely heavily on biochemical results. The aim is to reduce cholesterol and triacylglycerols to acceptable limits: total cholesterol < 200mg/dL, LDL < 130mg/dL, HDL > 40mg/dL and TAG < 140 mg/dL.
F Dietary Management
Some of the management guidelines are:
1. Reduce calorie intake (coupled with exercises) to achieve ideal body weight. In addition to beneficial effect on lipid profile, this frequently improves glucose tolerance and lowers blood pressure.
2. Reduce total fat intake to provide only about 30% of total calories.
3. Reduce saturated fat intake to provide only about 30% of total fat intake, and instead take polyunsaturated fatty acids since they are required in esterification and final excretion of cholesterol, thereby reducing serum cholesterol level.
4. In essence, it is recommended that consumption of red meat, dairy products and fried food be reduced while that of dietary fibres (fruits and vegetables), pulses be increased. Fibres increase intestinal mobility and reduce reabsorption of bile salts. Increased excretion of bile acids leads to increased conversion of cholesterol to bile acids and an up-regulation of hepatic LDL receptors, resulting in reduction in plasma LDL levels. Vegetables also contain plant sterols (sitosterol) which decrease the absorption of cholesterol. Moderate alcohol may raise HDL level.
Drug Therapy
Drug therapy may be considered only if the patient does not adequately respond to dietary management and other lifestyle changes. Currently, a range of lipid-lowering drugs are available. More commonly (though erroneously) referred to as hypocholesterolaemic drugs, they are divided in three classes based on their action.
1. Bile acid sequestrant resins (colestipol and cholestyramine): They are insoluble, non-absorbable anion exchangers which bind bile acids in lumen of intestine. This blocks their reabsorption and interrupts their enterohepatic circulation, so that instead of returning to the liver, the bile salts are excreted in stools. Feedback inhibition of 7-α-hydroxylase by bile salts is also released, so that conversion of cholesterol to bile salts is increased. The resulting depletion of the cellular cholesterol pool leads to an up-regulation of hepatic LDL receptors and a reduction of the LDL level in the plasma.
2. Inhibitors of HMG CoA reductase: These are the commonest drugs for the treatment of primary hypercholester-olaemia with least side effects, and are also called statins. By causing enzyme inhibition, they decrease endogenous cholesterol synthesis, so that the cells become dependent on the supply of cholesterol from plasma lipoproteins. Decreased endogenous synthesis also causes increased synthesis of LDL receptors. By this mechanism, the HMG CoA inhibitors reduce LDL cholesterol. Lovastatin and mevastatin are the oldest members of this class; the newer ones are pravastatin, simvastatin, fluvastatin, etc.
3. Vibrates: These are the commonest drugs for the treatment of primary hypertriglyceridaemia or combined hyperlipidaemia. Commonest drugs of this group are clofibrate, fenfibrate and gemfibrozil. They activate lipo-protein lipase to lower triglyceride, total and LDL cholesterol, and in some cases increase HDL cholesterol.
Nicotinic acid in large doses decreases the serum cholesterol level, though the mechanism is not clear.
V.Prostaglandins
A Overview
Prostaglandins (PG) are a group of 20-carbon compounds derived from arachidonic acid, which act as local hormones. They elicit a vast range of biochemical effects, and are widely used as therapeutic agents.
Prostaglandins and related compounds—prostacyclins, thromboxanes and leukotrienes are collectively known as eicosanoids because they are all C-20 compounds (Greek: eikosi means twenty). Prostaglandins contain a fivemembered cyclopentane ring. Prostacyclins (also called prostaglandin I) contain a five-membered oxygen containing ring, and the thromboxanes have a six-membered oxygen-containing ring instead of the cyclopentane ring. Eicosanoids act at very low concentrations and are involved in the production of pain and fever, and in the regulation of blood pressure, blood coagulation and reproduction.
They tend to act locally, close to the cells that produce them, and are metabolized rapidly (within seconds or minutes), which limits their effects on nearby tissues.
Prostaglandins (and other eicosanoids) have been often referred to as local hormones. However, they differ from the true hormones in some important aspects:
1. Site of production: Prostaglandins are produced by almost all tissues (except erythrocytes) in contrast to hormones which are produced in specialized endocrine organs.
2. Site of action: Prostaglandins generally act locally unlike the hormones which are transported through blood circulation to the specific target tissues where their action is elicited.
3. Metabolism: Prostaglandins are not stored, and are metabolized to inactive produces near the site of their synthesis only; this is in contrast to the hormones which are metabolized at sites distant from the site of origin.
B Historical Perspective
Prostaglandins were discovered in 1933 by Ulf von Euler, a Swedish scientist. It had been observed that muscle contraction was induced in uterus by human semen, and von Euler speculated that this action was elicited by a group of compounds originally synthesized in prostate. Hence, he named them prostaglandins. Though it was discovered later that prostaglandins were synthesized in almost all tissues, the name prostaglandin was by then widely accepted, and hence continued to be used.
Biosynthetic pathway for prostaglandins was elucidated by Bengt Samuelsson (Nobel Prize, 1982). Ellas Corey was awarded Nobel Prize in 1990 for chemical synthesis of prostaglandins.
C Chemical Nature
Chemically, the prostaglandins are derivatives of the hypothetical parent compound, prostanoic acid which resembles a 20-carbon saturated fatty acid, but has a cyclic five membered ring (Fig. 12.16 ). This cyclopentane ring is formed by carbon atoms 8 to 12.
Numerous types of prostaglandins are known, which are designated by alphabetical letters (A, B, C, etc.) according to the nature of the substituent on the cyclopentane ring (Table 12.7 ). They are characterized further by subscript indicating the number of double bonds outside the cyclopentane ring (Fig. 12.17 ).
Table 12.7
Different prostaglandins named according to substituent groups on their cyclopentane ring
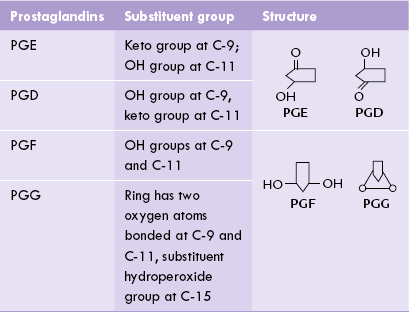
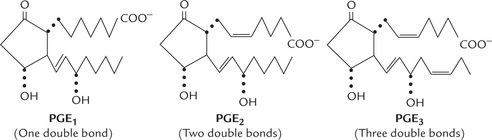
Fig. 12.17 Prostaglandins are characterized by subscript indicating number of double bonds outside the cyclopentane ring. Number of double bonds affects the potency of the product.
D Metabolism
Synthesis
In humans, the most important precursor for prostaglandins is arachidonic acid, a polyunsaturated fatty acid with four double bonds (eicosatetraenoicacid). It is stored in cell membranes as the C2 ester of phosphatidylinositol and other phospholipids. The biosynthetic pathway, called cyclooxygenase pathway, occurs in the following steps:
1. Release of the arachidonate from the membrane bound phospholipids: It occurs by action of phospholipase A2. This reaction is rate-limiting for the pathway and is promoted by epinephrine and bradykinin.
2. Oxidation and cyclization of arachidonic acid: It produces PGG2, which is then converted to PGH2 (Fig. 12.18 ). The reactions are brought about by the micro-somal prostaglandin synthase complex, which consists of the cyclooxygenase and peroxide components. The overall reaction consumes two moles of oxygen and two moles of reduced glutathione. The enzyme is short lived and gets rapidly inactivated, which prevents excessive production of prostaglandins.
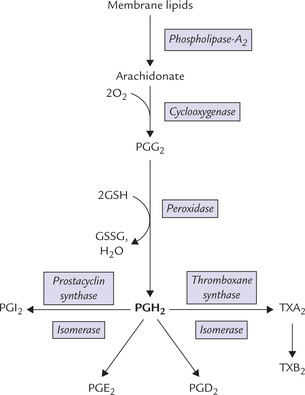
Fig. 12.18 Synthesis of prostaglandins (PGG2, PGH2, PGD2, PGE2, PGI2) and thromboxanes (TXA2, TXB2) by cyclooxygenase pathway. The key enzyme is the microsomal prostaglandin synthase complex, which has cyclooxygenase and peroxidase components (PG = prostaglandin, TX = thromboxane, GSH = reduced glutathione, GSSG = oxidized glutathione).
PGH2 is the first prostaglandin to arise in the cyclooxygenase pathway, hence called primary prostaglandin.
3. Conversion to primary prostaglandin to other eicosanoids: PGH2 serves as the immediate precursor for anumber of other prostaglandins, as also of prostacyclinsand thromboxanes. The enzymes for the conversions ofH-prostaglandin to the other prostanoids are cell specific.Each cell type produces only one or a few major prostanoids. Thus, vascular endothelium produces PGE andPGI, and platelets produce thromboxanes.
Figure 12.18 shows various enzymes responsible for various eicosanoids from PGH2.
E Anti-inflammatory Drugs Inhibit Prostaglandin Synthesis
Two important classes of anti-inflammatory drugs are in current use: the steroids (glucocorticoids) and the non-steroidal anti-inflammatory drugs (NSAIDs). Both of these drug classes act on the synthesis of prostaglandins.
• The steroids inhibit the action of phospholipase A2, which normally is rate limiting in eicosanoid synthesis, and
• NSAIDs are inhibitors of cyclooxygenase (Fig. 12.19 ).
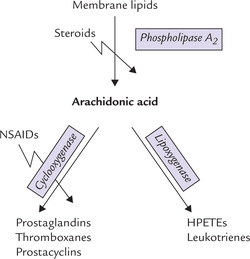
The steroids reduce the synthesis of all eicosanoids, but the NSAIDs inhibit synthesis of only prostaglandins. Synthesis of leukotrienes (formed from arachidonate by the lipoxygenase pathway) is not affected and may even be increased because more arachidonic acid is available for the lipoxygenase pathway.
Aspirin Irreversibly Inhibits Cyclooxyganase Activity
Prostaglandins are potent mediators of inflammation, pain and fever; and aspirin (acetyl salicylic acid) is an inhibitor of prostaglandin synthesis. Therefore, aspirin is widely used as an anti-inflammatory, analgesic (pain relieving), and antipyretic (fever reducing) agent. Its use was prevalent much before its exact mechanism of action was elucidated in 1971 by John Vane. Aspirin acts by acetylating a specific serine residue of the PG synthase, which prevents the arachidonate from reaching the active site of this enzyme. This causes irreversible inhibition of the cyclooxygenase activity of the enzyme, thereby inhibiting prostaglandin synthesis.
Another useful effect of aspirin is decreased production of thromboxane A2, which results in decreased vasoconstriction and platelet aggregation. This explains the utility of aspirin in prevention of heart attack.
However, since aspirin may enhance the lipoxygenase pathway, it is contraindicated in asthma, a disease in which bronchoconstriction is induced by leukotrienes rather than prostaglandins.
F Biological Action and Uses
Prostaglandins participate in several physiological processes. They are involved in production of pain and fever, in regulation of blood pressure, blood coagulation, and reproduction. Almost every cell in the human body responds to one or several types of prostaglandins; often opposing actions are induced in the same tissue by different PGs. Only some of the more important functions of prostaglandins are discussed here.
1. Control of blood pressure: PGE and PGI produced by vascular endothelium are potent vasodilators. This lowers peripheral resistance to decrease the blood pressure. However, these PGs cannot be used as antihypertensives because of their rapid inactivation and numerous extravascular effects.
PGE1 is used in infants with pulmonary stenosis to keep patency of the ductus arteriosus till surgical correction can be performed.
2. Mediators of inflammation: PGE2 and PGE1 are synthesized in macrophages and other cells as local mediators of inflammation (they produce inflammation mainly by increasing capillary permeability). Together with cytokines, histamine and bradykinin, they mediate the cardinal signs of inflammation.
3. Platelet aggregation and thrombosis: PGI (prostacyclins) inhibit platelet aggregation, and PGE2 promotes it. Thromboxanes also promote platelet aggregation and blood clotting, which may lead to thrombosis, as described in the next section.
4. Reproduction: PGE2 and PGF2, which are produced in the endometrium, induce uterine contraction. Their levels increase massively at parturition, and together with oxytocin they participate in the normal induction of labour. They may be used for this purpose and for the medical termination of pregnancy via intrauterine, intravenous or intra-amniotic administration. Unlike oxytocin, the prostaglandins contract the uterus not only at term of pregnancy but also at all times, so they are suitable for the inducing abortion.
5. Regulation of gastric secretion: Synthetic prostaglandins of PGE2 series inhibit gastric secretion, which makes them therapeutically useful in treatment of acid peptic disease. There is evidence that healing of the gastric ulcers is also accelerated by the prostaglandins.
6. Relief of asthma: Prostaglandins are bronchodilators and, hence may be of pharmacological use in bronchial asthma.
7. Influence on renal functions: Prostaglandins increase the tubular reabsorption of water and sodium.
8. Effects on metabolism: Prostaglandins inhibit lipolysis in adipose tissue.
9. Control of fever response: Fever is mediated by interleu-kin-1 (IL-1), which is released from stimulated mono-cytes and macrophages. IL-1 binds to vascular receptors in the preoptic area of the hypothalamus, where it induces the formation of PGE2. The latter causes fever by a direct action on the thermoregulatory centre.
G Thromboxanes, Prostacyclins and Leukotrienes
1. Thromboxanes A2 are structurally similar to prostaglandins, but have an oxygen atom in the cyclic ring (Fig.12.20 ). Synthesis of TX2, the major thromboxane, occurs inplatelets from PGH2 by the enzyme thromboxane synthase(Fig. 12.18). It has a half-life of about 30 seconds in blood;it is rapidly transformed into the inactive thromboxane B2.
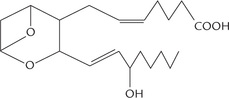
Its major effects are vasoconstriction and platelet aggregation. The effect on platelet aggregation is mediated via cAMP. Dipyrimadole, an inhibitor of thromboxane synthase, is a widely used therapeutic agent.
2. Prostacyclins are formed in vascular endothelium inaorta and probably in other artery walls. Action of theprostacyclins is mediated via cAMP. Their action is antagonistic to thromboxane and they are inhibitors of plateletaggregation. They are highly potent; as little as 1 mg/mLof PGI2 is capable of preventing platelet aggregation. Thishas a protective effect on vessel wall against depositionof platelets, hence minimizing the risk of thrombosis.But any injury to vessel wall would inhibit the prostacyclin synthesis so that platelet aggregation occurs to promote thrombus formation at the site of injury.
In addition, prostacyclins cause relaxation of vascular smooth muscles. Because of these actions, the prostacyclins decrease the risk of myocardial infarction.
2. Leukotrienes (LT) are 20-carbon polyenoic fatty acids having a number of substituents. Depending on the substituents, they are divided into LTA, LTB, LTC, LTD and LTE. Each type is subdivided depending on the number of double bonds, which vary between one and five. The most important leukotrienes are LTB4, LTC4, LTD4, and LTE4 (each has four double bonds).
Thromboxanes and prostacyclins are antagonistic. Whereas thromboxane TXA2 (as also prostaglandin E1) promotes platelet aggregation, the prostacyclin PGI2 inhibits platelet aggregation.
Leukotrienes are produced from arachidonic acid by an alternate pathway called lipoxygenase pathway (Fig. 12.19). The lipoxygenases are dioxygenases that produce hydroperoxy derivatives known as hydroxyperoxy eicosa-tetraenoic acids (HPETEs). Formation of various leukot-rienes then occurs from these intermediates in leukocytes, platelets, mast cells, and the vascular tissue of heart and lungs. Synthesis of various leukotrienes from the intermediate 5-HPETE is shown in Figure 12.21 .
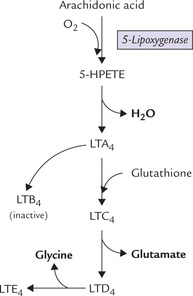
Leukotrienes are powerful constrictors of bronchial and intestinal smooth muscles. They are mainly involved in hypersensitivity reactions and inflammation. Unlike the other eicosanoids, leukotrienes are relatively stable and persist for a few hours in the tissues. The slow reacting substance of anaphylaxis (SRS-A) has three leukotrienes as its active component (e.g. LTC4, LTD4, and LDE4). It is far more potent than histamine (or prostaglandins) in stimulating allergic reactions. In addition, LTB4 attracts leucocytes at the site of inflammation.
Leukotrienes, produced from arachidonic acid by lipoxygenase pathway, are involved in hypersensitivity reactions and inflammation.
Fish foods are cardioprotective. Certain fish oils are rich in an unsaturated fatty acid, namely eicosapentaenoic acid (EPA; 5 double bonds at carbons 5, 8, 11, 14, 17). The prostacyclin derived from EPA (PGI3) is a highly potent anti aggregator of platelets the thromboxane (TX3) derived from it is a weak aggregator. As a result the anti-aggregating effect gains predominance, which accounts for lower incidence of myocardial infarction among Eskimos.
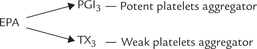
Moreover, both PGI3 and TX3 inhibit the release of arachidonate from phospholipids, thereby decreasing the formation of the aggregators, e.g. PG2 and TX2.
Exercises
Essay type questions
1. Describe synthesis of cholesterol and its regulation. Add a note on the significance of plasma cholesterol estimation.
2. Why do serum cholesterol levels depend on LDL receptor activity? Discuss the role of cholesterol in atherosclerosis and describe the mechanism of action of hypocholesterolaemic drugs.
3. Describe eicosanoids and their functions. Which drugs act by inhibiting eicosanoid synthesis?
4. Describe the synthesis and catabolism of various lipoprotein fractions. Explain role of various lipoproteins in atherogenesis.