Metabolism of Carbohydrates I: Mainline Metabolic Pathways
Carbohydrates are important for both the generation of metabolic energy and biosynthetic purposes. They are the structural basis of cellulose, bones, cartilage, lubricants such as mucus, cell recognition, DNA, RNA, and some membrane lipids (sphingosine). They are source of carbon skeleton of certain amino acids and are basis of some intracellular messenger systems. As an energy source they are broken down, and when there is a surplus, they are stored as starch and glycogen.
Glucose occupies a key position in carbohydrate metabolism, being a building block of all major dietary carbohydrates and the principal transported carbohydrate in humans. Fructose and galactose, obtained from dietary sucrose and lactose respectively, can be converted to glucose in liver. Therefore, the largest portion of dietary carbohydrates reaches the consuming tissues in the form of glucose. Evidently, an understanding of glucose metabolism forms the core of study of carbohydrate metabolism.
A detailed account of metabolic pathways is given in this chapter. After going through this chapter, the student should be able to understand:
• Transport of glucose into cells: facilitated transport and secondary active transport across cell membrane.
• Pathway of glycolysis: reaction sequence, generation of ATP and regulation.
• Feeder pathways: reaction sequences for monosaccharides, disaccharides and polysaccharides, and the associated metabolic diseases.
• Tricarboxylic acid cycle: reactions, energy yield, synthetic function and regulation.
• Pathway of gluconeogenesis; bypass reactions and reversible steps; energetics and regulation.
• Glycogen metabolism reactions of glycogen synthesis (glycogenesis) and degradation (glycogenolysis), their reciprocal regulation through cAMP activated cascade; role of hormones in the regulation; and biochemical basis and clinical presentations of glycogen storage diseases.
I Transport of Glucose Into Cells
Glucose cannot passively move across the cell membrane to enter the cells. The polar nature of glucose hinders its movement across the predominantly non-polar lipid bilayer. Two kinds of transport mechanisms are present in the cell membrane, which permit glucose to cross the membranous barrier and enter the cell. These are (a) the facilitated transport, and (b) the secondary active transport.
Both are carrier-mediated processes, requiring transport protein(s).
Facilitated transport occurs along a concentration gradient and is mediated via a family of at least five transport proteins located in the cell membrane. These proteins are designated as GLUT-1, GLUT-2, GLUT-3 GLUT-4 and GLUT-5. Glucose moves from higher extracellular glucose concentration to a lower concentration inside the cell. A given transport protein exists in two conformational states, which alternate during the transport process (Fig. 7.11).
The glucose transport proteins are tissue specific. GLUT-1 is present in RBCs, whereas GLUT-4 is abundant in skeletal muscles and adipose tissue. Insulin increases number and activity of GLUT-4, thereby promoting entry of glucose in these tissues (Chapter 7).
Note
Insulin is not required for glucose uptake by some tissues, such as liver, brain and red blood cells.
Secondary active transport is the mechanism of sodium-glucose cotransport across the membranous barrier, against a concentration gradient of glucose (i.e. from lower glucose concentration to a relatively higher concentration). The "uphill movement" of glucose is powered by the movement of sodium along its concentration gradient (Chapter 7). This process is operative during the movement of glucose from lumen of intestine into the intestinal mucosal cells (Fig. 7.16). Another type of such Na+/glucose co-transporter (called SLUT-1) is known to operate in renal tubules.
Abnormalities in some of the transport proteins may lead to diseases; for example, renal glycosuria results in case of a defect in the renal transport mechanism for glucose.
 Specific integral membrane proteins facilitate movement of glucose along concentration gradient, without expenditure of any energy in facilitated transport; whereas in secondary active transport a specific transport protein moves glucose against concentration gradient through expenditure of energy; the energy comes by co-transport of sodium ions along its concentration gradient.
Specific integral membrane proteins facilitate movement of glucose along concentration gradient, without expenditure of any energy in facilitated transport; whereas in secondary active transport a specific transport protein moves glucose against concentration gradient through expenditure of energy; the energy comes by co-transport of sodium ions along its concentration gradient.
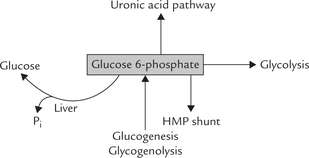
Glucose 6-phosphate: a branch-point compound: After entering the cell glucose is rapidly converted to glucose 6-phosphate. The latter serves as a common link between various metabolic pathways, therefore called a branch point compound. Figure 9.1 shows some important interconnections. Glucose 6-phosphate can enter a number of pathways, e.g. glycolysis, uronic acid pathway, HMP shunt, glycogenesis, and can again form glucose by hydrolytic removal of the phosphate group. It can be generated by glycogenolysis and gluconeogenesis also. Further, glucose 6-phosphate cannot simply diffuse out of the cell by crossing the cell membrane and hence is committed to intracel-luar metabolism. Detailed description of various pathways mentioned above has been given in Chapters 9 and 10.
II Glycolysis
Of all the metabolic transformations that glucose 6-phosphate can undergo, the sequence of reactions leading to the formation of pyruvate is quantitatively the most important. This pathway is known as glycolysis or the Embden-Meyerhof pathway. It is an anaerobic process that involves the breaking down of a molecule of glucose into two molecules of pyruvate or lactate.
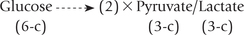
Glycolysis is common to most organisms, and in humans it occurs virtually in all tissues. Although the gly-colytic sequence usually begins with glucose, other sugars may also enter it via appropriate intermediates. Glycolysis serves a dual role:
Glycolysis occurs in 10 sequential reactions, that occur in the cytosol. The fate of pyruvate, the end product of these reactions, is different in anaerobic and aerobic conditions (Fig. 9.2 ).
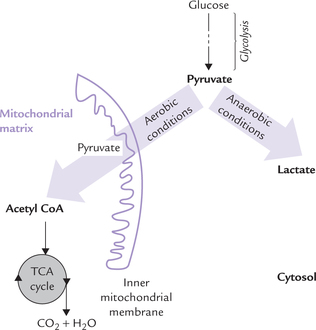
Fig. 9.2 Pyruvate metabolism under aerobic and anaerobic conditions. Under anaerobic conditions, pyruvate is reduced to lactate, whereas in aerobic conditions pyruvate is oxidized to acetyl CoA, which is further oxidized in TCA cycle.
Under aerobic conditions: Pyruvate enters the mitochondrial matrix and is oxidized to acetyl CoA, which is the major metabolic fuel for the citric acid cycle. Further oxidation of acetyl CoA in the citric acid cycle releases free energy which is used to generate ATP molecules. This process occurs in all tissues except those which lack mitochondria (e.g. erythrocytes, leucocytes) and the exercising muscles.
Under anaerobic conditions: Pyruvate is reduced in cytosol by NADH to form lactate. This process, called anaerobic glycolysis is discussed later. (Note that the term anaerobic literally means without air, but in practice it means without oxygen).
Glycolysis is a cytoplasmic reaction sequence of 10 enzyme-catalyzed reactions which produces two molecules of the three-carbon compound, pyruvate (or lac-tate), from the six-carbon substrate, glucose.
A Reactions
Glycolysis can be divided into two main stages. The stage I, which comprises the first five reactions, is an endergonic (i.e. energy-requiring) stage in which glucose molecules are activated through the expenditure of ATP molecules. Thus, this stage involves energy investment. Stage II leads to energy generation and hence referred to as the "payoff stage".
Reactions of glycolysis are shown in Figure 9.3 .
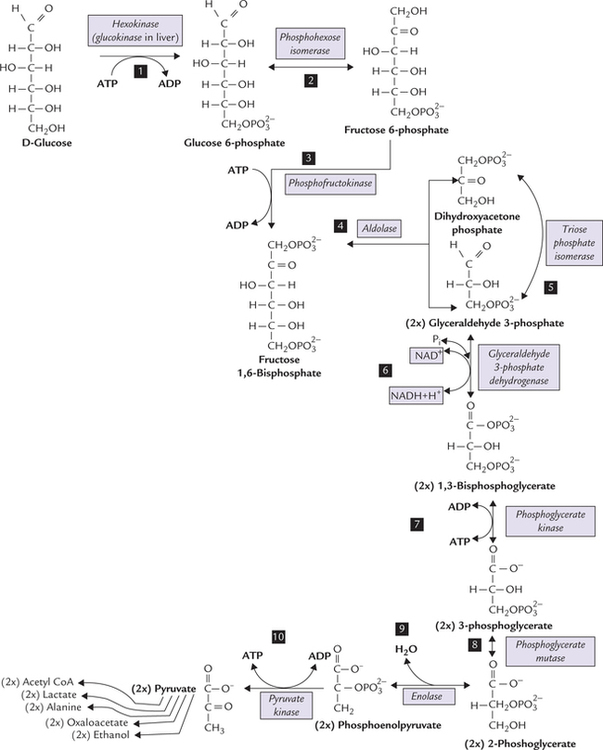
Fig. 9.3 The pathway of glycolysis and fate of pyruvate. Stage I: Reactions 1-5—energy-consuming reactions and Stage II: 6-10—energy-yielding reactions. Irreversible steps: 1st, 3rd and 10th).
Stage I: The Investment Phase
Reaction 1: Phosphorylation of Glucose
Glucose is phosphorylated at the sixth carbon to form glucose 6-phosphate. This places a large negative charge on the glucose molecule so that it cannot simply diffuse out of the cell. The phosphate group is obtained from ATP which is converted to ADP in the process, which means that energy is lost in this step. However, this is better than losing glucose out of the cell.
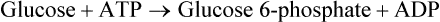
The enzyme involved in this reaction is kinase, (an enzyme that catalyzes a phosphorylation and uses ATP as its source of phosphate is called kinase). There are two possible enzymes for this reaction, depending on the tissue. One is glucokinase (found in liver) and the other is hexokinase (muscle and fat), which will catalyze phosphorylation of most hexoses, including glucose. Kinetic properties of the two enzymes are different (Table 9.1 ).
Table 9.1
Comparative properties of hexokinase and glucokinase
| Hexokinase | Glucokinase | |
| Specificity | Broad (hexoses) | Narrow (glucose only) |
| Feedback inhibition | Yes | No |
| Affinity for glucose | High | Low |
| Km value | 10-2 mmol/L | 20 mmol/L |
| Vmax value | Low | High |
Hexokinase has low Km, i.e. high affinity for glucose, low Vmax, and is subject to feedback inhibition by the reaction product, glucose 6-phosphate. The hexokinase reaction is not specific for glycolysis; it only commits glucose to intracellular metabolism because glucose 6-phosphate is not transported across the plasma membrane.
Glucokinase, on the other hand, is specific for glucose. It has high V and, unlike hexokinase, is not inhibited by the reaction product, glucose 6-phosphate. Moreover, it has high Km (20 mmol/L), i.e. low affinity for glucose and therefore, comes into play only when intracellular glucose concentration rises steeply, such as following a carbohydrate rich diet. These properties make the glucokinase suitable for rapidly phosphorylating the glucose that is presented to liver following a meal. Within the hepato-cytes, the glucose molecules are rapidly phosphorylated by glucokinase (due to high Vmax and high Km), without any hindrance to its action (due to lack of feedback inhibition). The above transformation is further enhanced because of induction of glucokinase synthesis after a meal, especially following a carbohydrate rich diet. The enzyme induction is probably brought about by insulin, that is released at such time. Large quantities of glucose 6-phosphate are thereby produced, which are channeled into the pathway of glycogen synthesis (i.e. glycogenesis).
Reaction 2: Isomerization of Glucose 6-Phosphate
Glucose 6-phosphate, an aldose sugar, is now prepared for subsequent cleavage into two 3-carbon molecules. To split anything is easier if it is bilaterally symmetrical. Glucose 6-phosphate is, however, not very symmetrical, and so converted to fructose 6-phosphate, a little more symmetrical molecule. It is a freely reversible reaction catalyzed by phosphohexose isomerase.
Reaction 3: Phosphorylation of Fructose 6-Phosphate
Fructose 6-phosphate, formed in the previous step, is made more symmetrical by esterifying a phosphate to C-1. This yields fructose 1,6-bisphosphate, a molecule that is almost symmetrical. The phosphate group comes from ATP and the enzyme is phosphofructokinase (PFK}).
Note
A diphosphate differs from bisphosphate (and a tri-phosphate from trisphosphate) in that, for a diphosphate (e.g. ADP) the phosphates are joined to each other, whereas in a bisphosphate (e.g. fructose 1, 6-bisphosphate), the phosphate groups are attached at different places on the sugar.
This reaction is irreversible and is the "rate-limiting step" for glycolysis. It is also the committed step, meaning that once fructose 1, 6-bisphosphate is formed it must go for the glycolytic pathway only. Activity of PFK1 is controlled by a variety of positive and negative regulatory modulators, which make this step the most important regulatory step of the pathway. Regulation of glycolysis is discussed later in this chapter.
Reaction 4: Cleavage of Fructose 1,6-Bisphosphate
Fructose 1,6-bisphosphate, a 6-carbon compound, is cleaved by the enzyme aldolase into two trioses: glyceral-dehyde 3-phosphate and dihydroxyacetone phosphate. This enzyme, which splits an aldose, is not specific for fructose 1,6-bisphosphate, hence its general name.
Reaction 5: Isomerization of Dihydroxyacetone Phosphate
Of the two trioses, only glyceraldehyde 3-phosphate can be further metabolized through the glycolytic sequence. Dihydroxyacetone phosphate, therefore, must be converted to glyceraldehyde 3-phosphate for further metabolism. This aldose-ketose isomerization is catalyzed by the enzyme, triose phosphate isomerase.
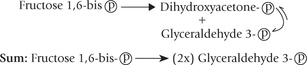
It is necessary to understand that two glyceraldehyde 3-phosphate molecules are generated from the original glucose molecule and enter this part of the pathway, and therefore, all of the following steps occur twice, once for each of the two glyceraldehyde 3-phosphate molecules.
Stage II: Energy Pay-off Phase
The next few steps are remarkable for they bring about oxidation of each of the three carbons of glyceralde-hydes. There is addition of oxygen at C-1, loss of hydrogen at C-2, and loss of phosphate at C-3. In these steps a large amount of energy is liberated, enough to replace the energy lost so far (2ATPs) plus some more.
Reaction 6: Dehydrogenation of Glyceraldehyde 3-Phosphate
Dehydrogenation of glyceraldehyde 3-phosphate is catalyzed by the enzyme glyceraldehyde 3-phosphate dehydrogenase, resulting in the formation of 1,3-bisphosphoglycerate.
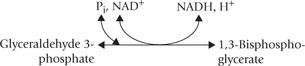
There are two interesting aspects of this reaction: firstly, the pair of hydrogen atoms removed is replaced by a phosphate group. This hydrogen is transferred to NAD+ to form NADH, which is a valuable product (its oxidation in the respiratory chain is a source of ATP). Second, the phosphate group added to the glyceralde-hyde 3-phosphate molecules is inorganic phosphate. This means that energy is not lost, which would have been the case if it had come from ATP.
Reaction 7: ATP Production from 1,3-Bisphosphoglycerate
The 1,3-bisphosphoglycerate (1,3-BPG) contains a carbox-ylic anhydride with phosphate at C-1. This type of anhydride, called acyl-phosphate, has a very high standard free energy of hydrolysis; the ΔG0′ of 1,3-bisphosphoglycerate is -11.8 kcal/mole (Chapter 8). The cell uses this energy by transferring the phosphate to ADP to form ATP (i.e. substrate level phosphorylation).
This reaction is catalyzed by the enzyme phosphoglycer-ate kinase. Since two molecules of 1,3-bisphosphoglycer-ate are produced from a single molecule of glucose, two ATPs are actually generated in this step from a single glucose molecule.
The 1,3-BPG is converted to an alternate metabolite, 2,3-bisphosphoglycerate (2,3-BPG) in erythrocytes which enhances unloading of oxygen (Box 9.1).
Reaction 8: Inter-Molecular Shift of Phosphate Group
The phosphate group is transferred from the C-3 of the 3-phosphoglycerate to the C-2 by the enzyme phosphoglycerate mutase, resulting in the formation of 2-phosphoglycerate.
Reaction 9: Dehydration of 2-Phosphoglycerate
Removal of a water molecule from 2-phosphoglycerate is catalyzed by the enzyme enolase, resulting in formation of phosphoenolpyruvate (PEP). In this reaction, redistribution of energy occurs within the same molecule, so that the phosphate ester (PEP) becomes unusually energy rich (ΔG0′ = —14.8 kcal/mole), which is sufficient for generation of ATP (i.e. substrate level phosphorylation).
Inhibition of enolase catalyzing this step has important role in clinical biochemistry (Box 9.2).
Reaction 10: ATP Production from Phosphoenol Pyruvate
The last step of the glycolytic sequence involves transfer of high energy phosphate group from PEP to ADP to yield a molecule of ATP (Fig. 9.4 ).
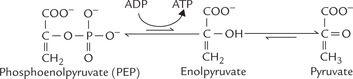
Fig. 9.4 The pyruvate kinase reaction. Pyruvate shows keto-enol tautomerism, the keto form being energetically far more stable than the enol form.
This is the third irreversible step of glycolysis (reaction 1 and 3 are the other two). Pyruvate kinase, the enzyme catalyzing this step, is a regulatory enzyme.
Deficiency of pyruvate kinase causes decreased production of ATP from glycolysis. Red blood cells are predominantly affected in this disorder. They are unable to generate sufficient ATP for their sodium pumps. Consequently, the ionic gradient across the erythrocyte membrane cannot be maintained, resulting in lysis of the cell. This results in haemolysis, and hence haemolytic anaemia.
B Fate of Pyruvate
There are several pathways into which pyruvate can enter (Fig. 9.3). The pathway chosen in a given tissue depends on its state of oxygenation and the prevailing metabolic conditions, as described below:
1. Oxidative decarboxylation to acetyl CoA: In tissues that are adequately perfused with oxygen (i.e. under aerobic conditions), pyruvate undergoes oxidative decar-boxylation to form acetyl CoA, which is further oxidized via citric acid cycle.
2. Reduction to lactate: Lactate is produced by glycolysis under anaerobic conditions and the process is called anaerobic glycolysis. The pyruvate to lactate reduction is brought about by the cytosolic enzyme lactate dehydrogenase and requires NADH.
The NADH is produced in the glyceraldehyde 3-phosphate dehydrogenase reaction (Reaction 6) of glycolysis (Fig. 9.5 ). The pyruvate to lactate conversion regenerates NAD+ for the above reaction so that glycolysis can proceed normally. It is noteworthy, that without regeneration of NAD+ by lactate dehydrogenase reaction, the Reaction 6 would stop and the glycolytic pathway would also soon be halted for lack of NAD+. Thus anaerobic glycolysis provides elegantly simple solution to ensure that glycoly-sis proceeds uninterruptedly.
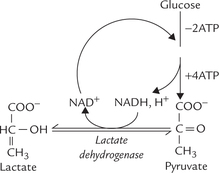
Fig. 9.5 Outline of reactions of anaerobic glycolysis. It regenerates NAD+ (for the glyceraldehyde 3-phosphate dehydrogenase reaction) so that glycolytic sequence can proceed smoothly with generation of 2ATP molecules.
More about anaerobic glycolysis is discussed at end of this section.
Under aerobic conditions, pyruvate enters the mitochondrion, where it is oxidatively decarboxylated to acetyl CoA, and under anaerobic conditions, pyruvate is reduced to regenerate NAD+ for glycolysis.
3. Carboxylation to oxaloacetate: Pyruvate is converted to oxaloacetate by biotin-dependent carboxylation catalyzed by the enzyme pyruvate carboxylase. The oxaloac-etate can form glucose via gluconeogenesis.
4. Conversion to malate: Malic enzyme converts pyruvate to malate, which can also enter gluconeogenic sequence.
5. Conversion to alanine: Pyruvate may form the amino acid, alanine by transamination.
6. Reduction to ethanol: Conversion of pyruvate to ethanol occurs in yeasts and some bacteria, including those of the intestinal flora.
Pyruvate can be funneled into various metabolic pathways. It can also be generated from a number of compounds, as shown in Figure 9.6 . Evidently, pyruvate is said to be at the metabolic cross-roads.
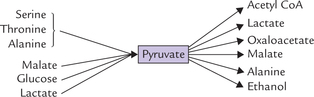
Fig. 9.6 Pyruvate can be generated from several compounds, and can be channeled into various pathways.
Anaerobic Glycolysis
Anaerobic glycolysis occurs in tissues that lack mitochondria, such as erythrocytes, lens and cornea of the eye, renal medulla, and leukocytes. In oxygen deficient tissues such as severely exercising muscles, this pathway is chosen. Oxygen deficiency occurs because the blood is squeezed out of the contracting muscles. Moreover, production of pyruvate in exercising muscle is very fast due to rapid rate of glycolysis, but its oxidation is much slower due to oxygen deficiency. The surplus pyruvate is diverted for lactate formation (Fig. 9.5).
Finally, during hypoxia resulting from vascular obstruction or other causes, for example, in acute myocardial infarction - anaerobic glycolysis occurs to help the cells survive the brief episodes of oxygen depletion.
Significance
Anaerobic glycolysis provides less energy: net gain of ATPs is only two molecules (Fig. 9.5). However, it is an important process because of two reasons:
1. NAD+ is generated during their process, which is required for step 6 of glycolysis (Fig. 9.5). As discussed in the previous section, NAD+ regeneration must occur for, glycolysis to continue.
2. Carbohydrates are the only metabolic substrates that can produce ATP under anaerobic conditions.
C Generation of ATP by Glycolysis
Energy yield in the anaerobic and the aerobic states are different, and therefore, discussed separately.
Aerobic State
In course of conversion of one molecule of glucose through glycolytic sequence to two molecules of pyru-vate, there occurs generation of four molecules of ATP and, utilization of two molecules of ATP.
Initially, two ATPs are utilized to phosphorylate glucose to glucose 6-phosphate and fructose 6-phosphate to fructose 1,6-bisphosphate (see reactions 1 and 3; Fig. 9.3). Subsequently, cleavage of fructose 1,6-bisphosphate, followed by isomerization of dihydroxyacetone phosphate, produces two molecules of glyceraldehyde 3-phosphate. Therefore, from each molecule of glucose, two molecules of triose phosphate are produced. Each molecule of tri-ose phosphate then generates two molecules of ATP: one ATP is produced by phosphoglycerate kinase (Reaction 7) and the other by pyruvate kinase (Reaction 10). Therefore, two molecules of triose-phosphate generates four ATPs. With two molecules of ATP invested and four generated, net gain of ATPs is two molecules (Table 9.2 ).
Table 9.2
Energy yield during the conversion of one molecule of glucose to two molecules of pyruvate in aerobic glycolysis*
| Reaction | Enzyme | Product |
| 1st | Hexokinase | -1 ATP |
| 3rd | Phosphofructokinase | -1 ATP |
| 6th | Glyceraldehyde 3-phosphate dehydrogenase | + 2 NADH |
| 7th | Phosphoglycerate kinase | + 2 ATP |
| 10th | Pyruvate kinase | + 2 ATP |
| 2 ATP + 2 NADH |
*Note that all reactions beyond the aldolase reaction occur twice for each glucose molecule.
In addition to these, generation of more ATPs occurs by oxidation of the NADH, (generated earlier in glyceralde-hyde 3-P dehydrogenase reaction). Each of these NADH molecules yields three ATPs by oxidative phosphorylation (Chapter 14). Thus, a total of six ATPs are produced from this source. When two ATPs generated earlier are added, the net production of ATPs in aerobic glycolysis becomes eight.
Anaerobic State
Generation of two molecules of ATP and two molecules of NADH occurs initially in this type also. The latter are oxidized to NAD+ during reduction of pyruvate. Thus, there is no net production of NADH (Fig. 9.5) in anaerobic glycolysis and, therefore, net generation of only two ATP occurs.
Lactate, the end product of anaerobic glycolysis, still contains a large amount of inherent free energy, but is not degraded further. Thus anaerobic glycolysis releases only a small fraction of the energy of the glucose molecules. Still it is a valuable source of energy in tissues lacking mitochondria. Moreover, in case of oxygen depletion, the affected tissues depend more on anaerobic glycolysis for getting the required energy. Production of lactate is thereby increased. However, excessive production of lac-tate may have serious consequences (Case 9.1).
Cori Cycle or Lactic Acid Cycle
In an actively exercising muscle, less than 10% of pyru-vate is utilized by the citric acid cycle, and the rest is reduced to lactate (i.e. anaerobic glycolysis). Accumulation of lactate and consequent fall in pH is potentially hazardous and should be prevented. Mechanisms exist in body which prevent such excessive accumulation of lactate.
The lactate first diffuses out into the blood circulation, the plasma membrane of muscle cells being freely permeable to lactate. It is carried to liver, and is oxidized to pyruvate in hepatocytes by the lactate dehydrogenase reaction. Pyruvate, so produced, enters the gluconeogenic sequence and converted to glucose, which is then transported to skeletal muscles (Fig. 9.7 ).
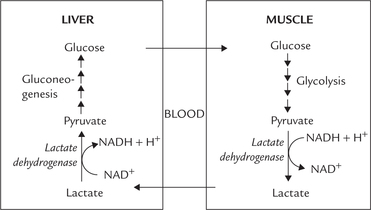
Fig. 9.7 Cori cycle. The exercising muscles generate lactate via anaerobic glycolysis, which is transported to liver for resynthesis of glucose.
Thus, a cyclic process is set up between liver and muscle, which ensures efficient reutilization of lactate by the body. It is referred to as the Cori cycle, named after Carl Cori and Gerty Cori who were awarded Nobel prize in 1947 for this discovery.
D Regulation
Glycolysis is central to metabolism and is integrated with a number of other metabolic pathways. Therefore, the regulatory enzymes of glycolysis respond to appropriate signals from several other pathways. This makes the regulation of glycolysis a complex event. There are three glycolytic reactions catalyzed by hexokinase (or glucoki-nase), phosphofructokinase (PFK1) and pyruvate kinase, which are metabolically irreversible and subject to regulation. The main control step is that catalyzed by PFK1 but hexokinase and pyruvate kinase are additional control sites.
Regulation of Hexokinase/Glucokinase
Hexokinase activity is subject to feedback inhibition by glucose 6-phosphate. This prevents intracellular accumulation of glucose 6-phosphate.
Glucokinase, that specifically effects phosphorylation of glucose in liver, is an inducible enzyme. Availability of substrate glucose induces the enzyme synthesis, probably through insulin. It is free from feedback inhibition by glucose 6-phosphate.
Regulation of Phosphofructokinase
Most important control point for glycolysis is through regulation of phosphofructokinase (PFK1), a complex allosteric enzyme. Its activity is influenced by cellular energy charge (high energy charge inhibits PFK1 and low energy charge stimulates it; Box 9.3), and by a number of positive and negative allosteric modulators (Table 9.3 ):
Table 9.3
Allosteric modulators of phosphofructokinase (PFK1)
| Activators | Inhibitors |
| AMP | ATP |
| Fructose 2,6-bisphosphate | Citrate |
| ADP | Ca2+ |
| K+ | Mg2+ |
| Phosphate | Low pH |
Box 9.3 Rate of Glycolysis is Regulated by Cellular Energy Charge
An overview of regulation of glycolysis shows that its rate is adjusted according to the energy content of the cell. In general, when the cellular energy content is low, the catabolic pathways are accelerated so as to provide the required energy; and conversely, when the cell has sufficient energy, inhibition of these pathways occurs. The anabolic pathways are regulated in a reciprocal manner, i.e. stimulated by high energy content and inhibited by low energy content of the cell. Evidently, cellular energy content is an important determinant of the rate of various metabolic pathways, including glycolysis. It is reflected by the following equation:
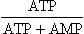
It is commonly referred to as the energy charge of the cell. Low energy charge signals that the cell needs energy, and accordingly the catabolic pathways, including glycolysis, are stimulated. When the energy charge is high, inhibition of glycolysis occurs.
1. ATP: ATP is an important inhibitory modulator of the enzyme activity.
• When the intracellular ATP level rises (indicating high cellular energy charge), the enzyme activity is inhibited. This is because of binding of ATP to the allo-steric site of the PFK1, which results in diminished affinity of the enzyme for fructose 6-phosphate.
• Conversely, low intracellular ATP level results in removal of ATP from the allosteric site, which results in enhanced activity of the enzyme.
2. Citrate: The inhibitory effect of ATP on PFK1 is enhanced by citrate, an intermediate of TCA cycle. Regulation of glycolysis by an intermediate of citric acid cycle ensures that rates of these two pathways, keep pace with each other. Whenever TCA cycle is impeded, accumulation of citrate results, which in turn slows down glycolysis as well.
3. AMP: Effect of AMP on PFK1 is stimulatory since it opposes the inhibitory effect of ATP on this enzyme. Reciprocal influences of ATP and AMP have additive effect on PFK1. This is because when intracellular AMP level is high, ATP level is correspondingly low and both (low ATP and high AMP) stimulate PFK1, thereby speeding up the rate of glycolysis.
4. Fructose 2,6-bisphosphate (F-2,6-BP): This compound is the most important allosteric modulator (activator) of PFK1 and, therefore, of glycolysis in liver. It is synthesized from fructose 6-phosphate by phosphofructokinase 2 (PFK2), a different enzyme from PFK1, and is hydro-lyzed back to fructose 6-phosphate by fructose 2,6-bisphosphatase 2 (FBPase2). Interestingly, both PFK2 and FBPase2 are activities catalyzed by the same poly-peptide; hence this is a bifunctional enzyme (Fig. 9.8 ).
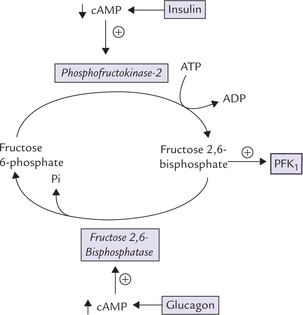
Covalent Modulation of PFK2
Activity of PFK2 is regulated by phosphorylation-dephos-phorylation mechanism.
The following hdormones regulate interconversion of the phosphorylate/dephosphorylated forms: Glucagon: It acts through the second messenger cAMP (and cAMP activated protein kinase A) and phosphory-lates the enzyme. This results in activation of the phospha-tase activity of PFK2, causing hydrolysis of fructose 2,6-bisphosphate. This decreases activity of PFK1 and, therefore, the rate of glycolysis falls. Thus, the overall effect is:
Glucagon → cAMP induced phosphorylation → stimulation of phosphatase activity of PFK2 → ↓ concentration of fructose 2,6-bisphosphate → ↓ activity of PFK1, and decreased rate of glycolysis.
Insulin
opposes glucagon by lowering the cellular cAMP concentration that promotes dephosphorylation of the enzyme and hence stimulates its kinase activity.
Thus, by adjusting the cellular concentration of fructose 2,6-bisphosphate, insulin and glucagon can influence the glycolytic activity (and gluconeogenic activity, discussed later) within minutes.
Fructose 6-phosphate is a potent modulator of PFK2-activity. It not only stimulates the synthesis of F-2,6-BP, but also inhibits its hydrolysis. F-2,6-BP in turn strongly activates PFK, and hence stimulates glycolysis. The overall effect is that when fructose 6-phosphate levels are high, such as after meals, the PFK, (and hence) gly-colysis is stimulated.
Because of various aforementioned regulatory influences the activity of PFK1 can increase up to 700-fold from the basal resting level.
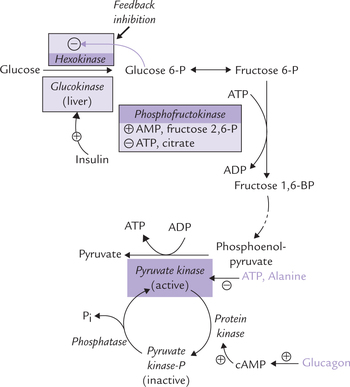
Regulation of Pyruvate Kinase
Pyruvate kinase is next in importance to PFK1 in the regulation of glycolysis. It has three isozyme forms, all catalyzing the final reaction (Reaction 10) of the glycolytic sequence. Its activity is regulated allosterically and by covalent modulation.
Allosteric regulation
Pyruvate kinase is inhibited allosterically by ATP and alanine and activated by fructose 1,6-bisphos-phate. The concentration of fructose 1,6-bisphosphate is increased whenever PFK1 activity increases, and so its effect on pyruvate kinase is an example of feed-forward stimulation.
Covalent modulation
Pyruvate kinase is inhibited by the cAMP-induced phosphorylation of a serine side chain in the enzyme protein (Fig. 9.9 ). Thus glucagon, which acts via cAMP-dependent protein kinase, inhibits the enzyme activity (thereby inhibiting hepatic glycolysis); and conversely insulin stimulates it.
E Diseases Associated with Glycolysis
Excessive accumulation of lactic acid occurs in a number of conditions, described in Chapter 1, to cause lactic acidosis. It causes severe metabolic acidosis, and is a potentially lethal condition (Case 9.1).
Deficiencies of glycolytic enzymes, most commonly of pyruvate kinase and hexokinase, have been reported. In these rare conditions, the predominant clinical manifestation is haemolytic anaemia because energy depleted erythrocytes are easily destroyed. A rare inherited disease causing pyruvate dehydrogenase (PDH) depletion has been reported (incidence 1 in 250,000 births). Metabolism of pyruvate being blocked, lactic acidosis frequently develops. Blockage of the PDH reaction can also occur in beriberi (thiamine pyrophosphate being a cofactor in PDH) and among alcoholics (Case 18.1).
III Feeder Pathways
Glycolysis is not exclusively for the catabolism of glucose; many other carbohydrates enter the glycolytic sequence in course of their metabolism. The metabolic pathways by which various monosaccharides, disaccharides and polysac-charides enter the glycolytic sequence are called feeder pathways. Separate feeder pathways exist for various monosaccha-rides, disaccharides and polysaccharides.
A Feeder Pathways for Monosaccharides
Metabolism of Fructose
D-Fructose is present in free form in many fruits, and honey. The major dietary source is sucrose (cane sugar), a disaccharide consisting of glucose and fructose. In the body, entry of fructose into the cells is not dependent on insulin. This is in contrast to glucose, which requires insulin for this purpose.
The Pathway
Some of the fructose in the cells is phosphorylated to the glycolytic intermediate fructose 6-phosphate by hexoki-nase in muscle and adipose tissue. Affinity of hexokinase for fructose, however, is extremely low (Km approximately 20 times higher than that for glucose), so this pathway is normally insignificant, and is important only when the fructose concentration is very high. Most of the fructose is phosphorylated by the enzyme fructokinase in the liver, kidney, and intestine. The liver accounts for nearly half of the total fructose metabolism. Fructokinase catalyzes phosphorylation of fructose at C-1 to form fructose 1-phosphate (Fig. 9.10 ). The latter is cleaved into dihydroxyacetone phosphate (DHAP) and glyceral-dehyde by the enzyme aldolase B. Defective action of this enzyme leads to a disorder called hereditary fructose intolerance (Case 9.2).
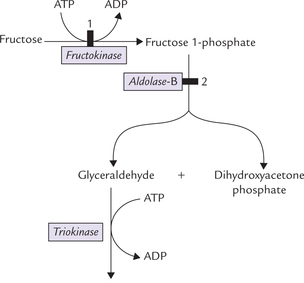
Fig. 9.10 Conversion of fructose to intermediates of glycolysis in liver. ( = metabolic blocks: 1. in essential fructokinase, 2. in hereditary fructose intolerance).
= metabolic blocks: 1. in essential fructokinase, 2. in hereditary fructose intolerance).
Aldolase B is an isoenzyme of the aldolase (see Reaction 4-glycolysis), and it can cleave both fructose 1,6-bisphosphate and fructose 1-phosphate. Glyceraldehyde is phosphorylated by the enzyme triokinase to glyceral-dehyde 3-phosphate, which along with DHAP, is metabolized further by glycolysis or gluconeogenesis.
Fructose is absorbed from the intestine less rapidly than glucose, but once in the blood it is metabolized nearly twice as rapidly as glucose. It bypasses the phospho-fructokinase step in liver, and therefore, its metabolism is simpler than that of glucose (Box 9.4).
Disorders of Fructose Metabolism
Essential fructosuria
It is a rare medical problem associated with fructose metabolism. The enzyme fructokinase is deficient in this disorder and therefore, fructose cannot be metabolized as rapidly as in normal subjects. Plasma fructose levels rise, and fructose may appear in urine. The condition is asymptomatic, sometimes detected incidentally during urinalysis when “reducing sugar” (fructose) is present. Restriction of dietary fructose is effective to treat this.
Hereditary fructose intolerance
These are disorders caused by the deficiency of one of the fructose metabolizing enzymes. The enzymes involved are:
1. Aldolase B: Deficiency of aldolase B results in intracel-lular accumulation of fructose 1-phosphate. There is vomiting, jaundice, and hypoglycaemia caused by intra-cellular accumulation of fructose 1-phosphate. This compound allosterically inhibits liver phosphorylase to block glycogenolysis, thus leading to hypoglycaemia. Besides tying up phosphate, and thereby impairing ATP synthesis, fructose 1-phosphate inhibits aldolase and phosphohexose isomerase; these changes result in liver damage, and jaundice is commonly seen. More about hereditary fructose intolerance is given in Case 9.2.
2. Fructose 1,6-bisphosphatase: Deficiency of this gluco-neogenic enzyme causes fructose intolerance similar to aldolase-B deficiency, but these patients also have fasting hypoglycaemia. They can form glucose from stored glycogen, but gluconeogenesis is blocked. Glycogen degradation can maintain a reasonably normal blood glucose level for many hours, but the defect in gluconeogenesis results in dangerous hypoglycaemia when the period of fasting exceeds 14-18 hours.
Excess Fructose is Hazardous for Body
Based on the reasoning that insulin-dependent PFK1 reaction is bypassed by fructose, diabetic diets were formulated in the past with fructose as the predominant carbohydrate. It was soon found, however, that excess fructose is toxic because it can cause damaged liver and cause increased lactate formation, hypertriglyceridaemia and hyperuricaemia, not only in individuals with diabetes but in normal subjects as well.
Liver damage: The activity of fructokinase far exceeds that of aldolase B, so fructose 1-phosphate tends to accumulate (concentration in liver can reach up to 10 jumol/g). This compound allosterically affects several enzymes of carbohydrate metabolism and also ties up substantial portion of the phosphate in the cell. This can impair oxidative phosphorylation, and lower the synthesis of ATP from ADP, with consequent damage to liver cells.
Hyperlactataemia: This can be traced to a rapid metabolism of fructose to pyruvate, which is in equilibrium with lactate.
Hypertriglyceridaemia: Fructose is rapidly metabolized to yield acetyl CoA, which is channeled into lipogenesis (fatty acids and triglycerides synthesis) in the liver.
Hyperuricaemia: This is due to excessive accumulation of ADP and AMP (due to lack of Pi) followed by their degradation to uric acid.
Metabolism of Galactose
D-Galactose is present in milk as one of the monomeric constituents of lactose; the other one is glucose. Galactose is liberated in the intestine by action of the brush border enzyme, lactase.
The Pathway
Entry of galactose into peripheral cells is independent of insulin. Galactose is phosphorylated at C-1 by the enzyme galactokinase to form galactose 1-phosphate. Galactose 1-phosphate is converted to glucose 1-phosphate by the action of galactose 1-phosphate uridyl transferase. In this reaction, uridine diphosphate (UDP) acts as a carrier of hexose molecule (Fig. 9.11 ). The net reaction of the pathway is the ATP-dependent conversion of galactose to glucose 1-phosphate. The latter enters glycolytic sequence via glucose 6-phosphate.
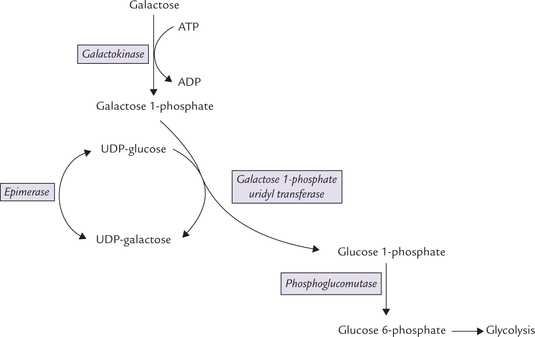
Fig. 9.11 Galactose entering glycolysis via the galactose-glucose interconversion pathway, a four-step reaction sequence.
UDP-galactose is an important intermediate formed in the second reaction (from galactose 1-phosphate and UDP-glucose). It can be epimerized to UDP-glucose, as shown in the Figure 9.11. The reversibility of epimerization makes it possible to obtain UDP-galactose from UDP-glucose, so that galactose is not an essential nutrient in diet.
UDP-Galactose has synthetic roles, being involved in the following reactions:
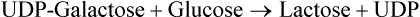
Galactosaemia
Inborn errors due to deficiency of one of the enzymes of galactose metabolism cause galactosaemia. The deficient enzyme may be galactokinase, galactose 1-phosphate uridyl transferase, or rarely epimerase. Deficiency of the uridyl transferase, known as classical galactosaemia, is best known.
Classical galactosaemia
This is an autosomal recessive disease with an estimated population incidence of 1 in 40,000. Deficiency of the uridyltransferase leads to accumulation of galactose and galactose 1-phosphate after ingestion of milk or food containing galactose . There is elevated concentration of galactose in blood (galactosaemia) and urinary elimination (galactosuria). Other salient features are as follows:
1. Excess galactose is reduced to galactitol (dulcitol) by aldose reductase, the same enzyme that forms sorbitol from glucose in the polyol pathway. Galactitol, like sorbitol, accumulates in lens and causes cataract.
2. Accumulation of galactose 1-phosphate and concomitant depletion of inorganic phosphate causes liver dysfunction. It is evident within weeks after birth: feeding difficulties and vomiting result in poor weight gain, and jaundice is present that often is misdiagnosed as physiological jaundice of newborn. Excess galactose 1-phosphate inhibits glycogen phosphorylase and phos-phoglucomutase; both impede glycogenolysis and glyco-gen accumulates in liver.
3. The central nervous system is affected as well, as evidenced by an abnormal lethargy or irritability. Hypo-glycaemia is commonly observed because galactosaemia provides persistent stimulus for insulin secretion from the pancreas. Galactose in erythrocytes inhibits glucose 6-phosphate dehydrogenase and hence HMP-shunt, which reduces the supply of NADPH. This results in haemolysis, as discussed in Chapter 10.
4. In severe, untreated cases of galactosaemia, mental deficiency, liver cirrhosis, cataract, aminoaciduria and albuminuria develop.
Classical galactosaemia, caused by deficiency of enzyme galactose 1-phosphate uridyl transferase, results in accumulation of galactose and galactose 1-phosphate. Diversion of galactose in the galactitol is implicated in pathogenesis of cataract.
Diagnosis: Presence of reducing material (galactose) in urine with a negative glucose oxidase suggests diagnosis. In addition, galactose 1-phosphate uridyl transferase activity can be measured in RBCs: the patients completely lack the enzyme, and the unaffected heterozygotes have a reduced enzyme activity.
Treatment: The patient is placed on a milk-free diet. Synthetic diets like soya milk, which are free of galactose, are recommended. The galactosaemic child, even when sustained on galactose-free diet, thrives well because galac-tose needed for the synthesis of biomolecules, can be obtained from UDP-glucose by epimerization reaction.
B Feeder Pathways for Disaccharides and Polysaccharides
The disaccharides are cleaved into the constituent mono-saccharides by disaccharidases such as lactase, maltase, and sucrase. Lactose is cleaved by lactase into glucose and galactose, sucrose by sucrase into glucose and fructose, and maltase cleaves maltose into two glucose molecules.
The disaccharidases are present in outer surface of the epithelial cell lining of the small intestine. The monosaccha-rides, generated by their action are carried to liver by portal blood. In liver, the monosaccharides are further metabolized through glycolytic sequence, as described earlier.
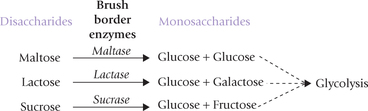
Similarly, the polysaccharides are first hydrolyzed into the constituent monosaccharides, which are then funneled into the central glycolytic sequence.
Monosaccharides, e.g. fructose, galactose, and man-nose, as also the common disaccharides and polysac-charides, are enzymatically channeled into the pathway of glucose metabolism.
Lactose intolerance is a commonly encountered clinical condition, due to deficiency of the intestinal lactase. Lactose cannot be digested and is oxidized by bacteria in gut, producing gas, bloating and watery diarrhoea. Deficiency disorders of some other disaccharidases are also known to exist.
Metabolism of Amino Sugars
The amino sugars are required for the synthesis of glyco-lipids, glycoproteins, and proteoglycans. They are synthesized from fructose 6-phosphate. The amino group is derived from amide group of glutamine, the reaction being catalyzed by amido transferase.
Glucosamine 6-phosphate forms its acetylated derivative (N-acetyl glucosamine 6-phosphate) by condensing with acetyl CoA. A mutase enzyme then converts it to N-acetyl glucosamine 1-phosphate, which reacts with UTP to produce the nucleotide activated form, UDP-N-acetyl glucos-amine. The latter is epimerized to form UDP-N-acetyl galactosamine (Fig. 9.12 ).
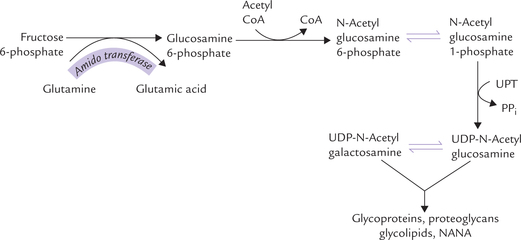
Both the nucleotide activated forms (UDP-N-acetyl glucosamine and UDP-N-acetyl galactosamine) are then used for synthesis of complex molecules, e.g. glycopro-teins, proteoglycans and glycolipids. UDP-N-acetyl glu-cosamine can enter an alternate pathway to form NANA.
IV Tricarboxylic Acid Cycle
Tricarboxylic acid (TCA) cycle is also called Krebs cycle or the citric acid cycle. It is a cyclic pathway occurring in mitochondria, also referred to as the “central catabolic pathway”. It occupies a central place in catabolism because catabolic pathways involving various biomolecules of diverse origin and chemical nature ultimately terminate in TCA cycle. The end product of these pathways is one of the intermediates of TCA cycle. For example, the glycogenic amino acids yield pyruvate, oxaloacetate, or α-ketogluta-rate; the ketogenic amino acids yield acetyl CoA (which is the main precursor of TCA cycle), and the end product of degradation of various fatty acids and carbohydrates is also acetyl CoA (Fig. 9.13 ). Thus, TCA cycle is spoken of as the meeting point where catabolic pathways converge.
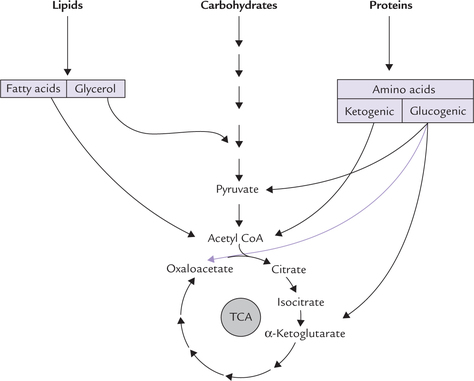
TCA cycle is the final common pathway of the oxidation of the acetyl CoA, formed from carbohydrates, fatty acids and amino acids.
TCA cycle serves two main functions in metabolism:
(a) to provide an important pool of metabolic intermediates for synthesis of many useful compounds, and
(b) to break down some compounds for generation of energy. The energy obtained is captured by reducing NAD+ and FAD to NADH and FADH2, respectively. To obtain full usage of the energy generated in this cycle, NADH and FADH2, are processed by another pathway (oxidative phosphorylation) where their energy is converted to ATP.
A Pyruvate Dehydrogenase Complex: A Bridge Between Glycolysis and TCA Cycle
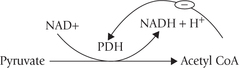
Acetyl CoA, one of the two major reactants in the first reaction of TCA cycle (the other being oxaloacetate), is mainly generated from pyruvate. This conversion involves a series of complex reactions, catalyzed by a multienzyme complex, called pyruvate dehydrogenase complex (PDH). Strictly speaking, this reaction sequence is not a part of the TCA cycle. It is, however, discussed here along with TCA cycle, because it serves as a bridge between glycolysis and TCA cycle.
The pyruvate dehydrogenase complex is located in the mitochondrial matrix. It is a multimolecular aggregate (MW 9 × 106) consisting of three enzymes and five coenzymes.
Enzymes
Pyruvate decarboxylase, dihydrolipoyl transacety-lase and dihydrolipoyl dehydrogenase are the three enzymes present in the complex. There are about 60 molecules of dihydrolipoyltransacetylase and about 20-30 molecules each of the other two enzymes in each complex.
Coenzymes
Lipoic acid, FAD, NAD+, coenzyme A and thiamine pyrophosphate (TPP) are the coenzymes in this complex. They serve as transient carriers of functional groups or participate in the oxidation-reduction reactions.
Aggregation of various enzymes in a complex greatly increase the efficiency of the transformation. This is because the intermediate products are not released to the medium but are channeled to the succeeding enzymes of the pathway. Thus a metabolic intermediate is available for the next reaction immediately after its production. Outline of the reaction sequence and role of various enzymes and coenzymes of this complex are summarized in Figure 9.14 .
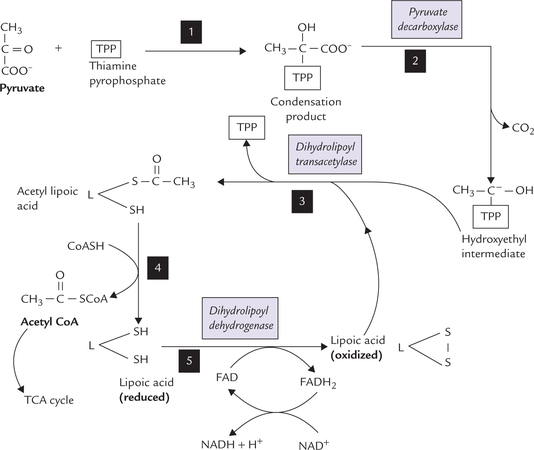
Fig. 9.14 Mechanism of conversion of pyruvate to acetyl CoA by pyruvate dehydrogenase complex. Step 1: Pyruvate and thia-mine pyrophosphate (TPP) combine to form a condensation product. Step 2: Pyruvate decarboxylase catalyzes release of carbon dioxide from the condensation product to form a hydroxyethyl intermediate. The latter is attached to the reactive carbon of the TPP. Step 3: Transfer of acetyl group from the hydroxyethyl intermediate to the lipoic acid (oxidized) occurs to form acetyl lipoic acid. The reaction is catalyzed by dihydrolipoyl transacetylase. Step 4: Transfer of an acetyl group from the acetyl lipoic acid to coenzyme A (CoASH) occurs next, converting the latter to acetyl CoA. The acetyl CoA then enters the TCA cycle. The other product of this reaction is reduced lipoic acid. Step 5: This step regenerates the oxidized lipoic acid (from the reduced lipoic acid), which then participates in the next cycle of reactions. This conversion is catalyzed by the dihydrolipoyl dehydrogenase component of the enzyme complex, which catalyzes transfer of the reducing equivalents first to FAD and then to NAD+. Overall: Pyruvate + NAD+ + CoASH → Acetyl CoA + CO2 + NADH + H+.
B TCA Cycle as a Cyclic Pathway
TCA cycle consists of eight sequential reactions. It is begins with condensation of a four carbon oxaloacetate (OAA) molecule with an acetyl CoA molecule (2-carbon) to form a six carbon citrate molecule (Reaction 1; Fig. 9.15 ). In subsequent reactions of TCA cycle, two carbon atoms are lost from the citrate in the form of CO2 (Reactions 3 and 4, Fig. 9.14). A series of modifications occur, in the remaining four carbon atoms, in successive steps, to ultimately form oxaloacetate (OAA). Thus, the last intermediate of one cycle (i.e. OAA) is ready for use as a substrate in the next cycle. In this way, there is no net generation of OAA, or of any of the cycle intermediates.
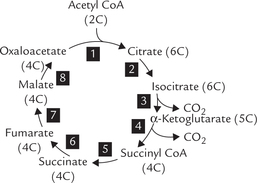
Fig. 9.15 TCA cycle: Oxaloacetate (OAA), the last intermediate is also a reactant in the first step. The eight reactions of the pathway are numbered 1 to 8.
The reactions of TCA cycle take place in the mitochondrial matrix. This is in close proximity to the reactions of oxidative phosphorylation, which occur in the inner mitochondrial membrane (Chapter 14). This arrangement ensures that the free energy liberated during reactions of the TCA cycle is promptly trapped in the form of ATP.
C Reactions of TCA Cycle
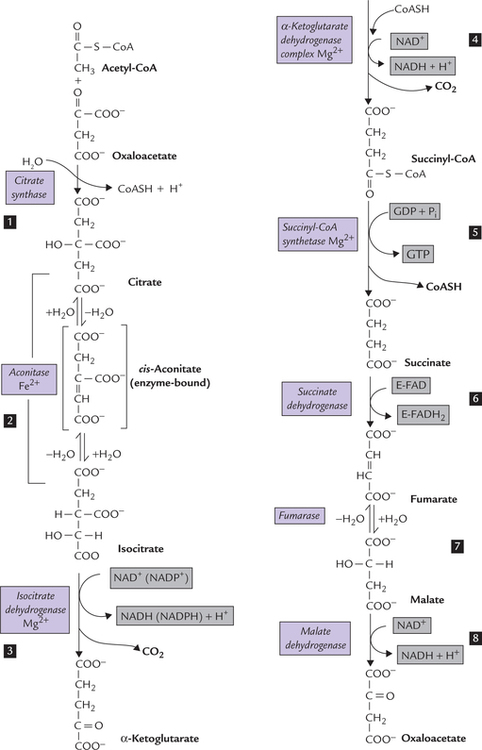
The reactions brought about by different enzymes of the TCA cycle are depicted in Figure 9.16 . All the enzymes are present in mitochondrial matrix except succinate dehydrogenase which is located in the inner mitochon-drial membrane.
Reaction 1: Synthesis of Citrate from Acetyl CoA and Oxaloacetate
Citrate is produced by condensation of acetyl CoA with oxaloacetate in an irreverssible reaction catalyzed by the enzyme, citrate synthase. The reaction equilibrium lies far towards the right (i.e. towards the formation of citrate).
TCA cycle derives its other name (citric acid cycle) from this first intermediate, citrate. However, citrate may leave the citric acid cycle to participate in other metabolic pathways as well (Fig. 9.17 ). Excessive citrate crosses the inner mitochondrial membrane through specific tricar-boxylate carriers and reaches the cytosol, where it provides acetyl CoA:
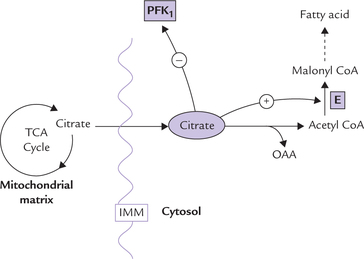
Fig. 9.17 Citrate, a TCA intermediate, plays an important regulatory role in lipogenesis and glycolysis by modulation of activities of the enzymes, phosphofructokinase (PFK1) and acetyl CoA carboxylase (E) (IMM = inner mitochondrial membrane).
In cytosol, acetyl CoA serves as a precursor for fatty acid synthesis (lipogenesis). Moreover, citrate stimulates acetyl CoA carboxylase, the rate-limiting enzyme of fatty acid synthesis. Thus citrate enhances fatty acid synthesis by:
• Providing the substrate (i.e. acetyl CoA).
• Stimulating the key lipogenic enzyme (i.e. acetyl CoA carboxylase).
In the cytosol, citrate has another important regulatory role to play. It adjusts the rate of glycolysis to that of TCA cycle. It does so by inhibiting phosphofructokinase (PFK1) the rate-limiting enzyme of glycolysis (Table 9.3). Inhibition of this enzyme decreases the rate of glycolysis, and therefore, the production of acetyl CoA falls. This results in decreased rate of TCA cycle since acetyl CoA is needed in the first reaction of TCA cycle. In this way, rates of glycolysis and TCA keep pace with each other.
Thus, when citrate concentration is high, implying that the cell is adequately supplied with fuel molecules and, therefore, further production of energy is not required, the energy-yielding catabolic pathways (e.g. glycolysis and TCA cycle) are inhibited. The biosynthetic pathway (i.e. fatty acid synthesis) is favoured at the same time. This illustrates a fundamental principle of biochemistry, i.e. the catabolic and the anabolic pathways are regulated reciprocally. If one pathway is favoured, the other is inhibited.
Reaction 2: Isomerization of Citrate
Isomerization of citrate by the enzyme aconitase yields isocitrate. During this reaction, a transient enzyme-bound intermediate, cis-aconitate is formed.
Reaction 3: Oxidative Decarboxylation of Isocitrate
The enzyme isocitrate dehydrogenase (IDH) catalyzes removal of two hydrogen atoms from isocitrate (i.e. oxidation) with a concomitant release of a CO2 molecule (i.e. decarboxylation). α-Ketoglutarate is the reaction product.
NAD+ serves as a coenzyme in this step, and is converted to NADH by accepting a pair of hydrogen atoms. NADP+ can also serve as a coenzyme in this reaction. Isocitrate dehydrogenase is one of the few enzymes that are capable of using both NAD+ and NADP+ as coenzymes.
Reaction 4: Oxidative Decarboxylation of α-Ketoglutarate
Like the previous step, this one also involves oxidation and decarboxylation. The reaction is catalyzed by the enzyme α-ketoglutarate dehydrogenase, to produce succi-nyl CoA.
The mechanism of this conversion is similar to that of the conversion of pyruvate to acetyl CoA (Fig. 9.14). The same set of coenzymes, i.e. thiamine pyrophosphate, lipoic acid, FAD, NAD+, and CoASH, is used. Each of these performs a function analogous to that performed in the pyruvate dehydrogenase complex.
The reaction releases the second CO2 molecule of the cycle and produces the second NADH. The equilibrium of the reaction lies towards the right, i.e. towards succinyl CoA formation.
Reaction 5: Cleavage of Succinyl CoA
The enzyme succinyl CoA synthetase (also called succinate thiokinase) cleaves the high-energy thioester bond in suc-cinyl CoA (ΔG0′ is -8.0 kcal/mole) to release large amount of free energy, which is used to produce a GTP molecule.
This reaction provides an example of substrate level phosphorylation, since the production of a high-energy phosphate (e.g. GTP) is coupled with enzymatic transformation of a substrate molecule. GTP can produce ATP by action of the enzyme nucleoside diphosphokinase.
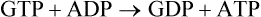
Reaction 6: Oxidation of Succinate
A pair of reducing equivalents is removed from succinate by the enzyme succinate dehydrogenase to form fumarate. FAD serves as a coenzyme in this reaction. Unlike the other enzymes of the TCA cycle, which are located in the mitochondrial matrix, succinate dehydrogenase is anchored in the inner mitochondrial membrane. It catalyzes removal of a hydrogen pair from succinate and transfers it to FAD which becomes FADH2. This in turn transfers its electrons to ubiquinone, which becomes ubiquinol, and is then transferred to complex III for oxidation (Chapter 14).
Reaction 7: Hydration of Fumarate
Addition of a water molecule to fumarate forms malate. It is a reversible reaction, catalyzed by the enzyme fumarase.
Reaction 8: Oxidation of Malate
Removal of a pair of reducing equivalents from malate by the enzyme malate dehydrogenase produces oxaloace-tate. The reaction generates the third NADH molecule of the cycle. Malate to oxaloacetate conversion is the last reaction of the cycle. Oxaloacetate, generated in this step, condenses with another molecule of acetyl CoA to initiate another cycle.
The following equation summarizes all the reactions of the TCA cycle.
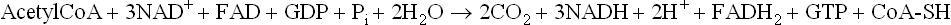
Several chemicals can inhibit reactions of the TCA cycle, as outlined in Box 9.5.
A four-carbon compound oxaloacetate reacts with acetyl CoA to form six-carbon citrate, which is then converted back to oxaloacetate in the remaining reactions of the TCA cycle.
The following points about TCA cycle are noteworthy:
1. Two carbon atoms enter the cycle as acetyl CoA (and condense with oxaloacetate) and two carbons leave in the form of two molecules of CO2. Thus, TCA basically involves oxidation of acetyl CoA to carbon dioxide and, as such there is no net consumption or regeneration of oxaloacetate or any of the other cycle intermediates.
2. Some of the reactions of TCA cycle are reversible and yet they always proceed unidirectionally. This is because equilibrium of some of the reactions of TCA cycle (e.g. Reactions 1, 3 and 4, Fig. 9.12) lies far towards the right. These reactions are irreversible and have strong tendency to proceed unidirectionally, towards the right. This generates a strong "thermodynamic pull" so that rest of the (reversible) reactions are also pulled in that direction. As mentioned earlier, fumarate to malate conversion which is freely reversible otherwise, always proceeds in direction of malate formation because of the thermodynamic pull.
D Energy Yield from TCA Cycle
There are four dehydrogenation reactions in TCA cycle (Reactions 3, 4, 6 and 8), which generate three NADH and one FADH2 molecules. These reduced coenzymes donate electrons to the electron transport chain and generate ATPs by oxidative phosphorylation. Oxidation of each NADH in this manner generates three ATPs, whereas two ATPs are produced from one FADH2 molecule. In addition, one GTP is produced during enzymatic transformation of succinyl CoA (Reaction 5), which generates an ATP through action of nucleoside diphosphokinase. Hence, each cycle of TCA cycle, produces 12 ATP molecules.
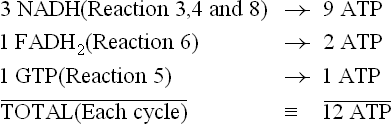
However, two acetyl CoA molecules are generated from each glucose, therefore, this cycle occurs twice, generating 6NADH, 2FADH2 and 2GTPs (Fig. 9.18 ) Hence, total 24 ATPs are generated. Thus, complete oxidation of glucose via glycolysis, pyruvate dehydrogenase, the Krebs cycle and the oxidative phosphorylation pathway yields 38 ATPs. Since two ATPs were initially used during stage I reactions of gly-colysis, the net yield is 36 ATPs. Compared with anaerobic pathway, the oxidative pathway thus yields 18 times more energy in the form of ATP. However, this is still far less than the energy obtained by burning of a molecule of glucose in calorimeter (2780 kJ), which is sufficient to generate about 91 ATPs (one ATP has energy bond equivalent to 30.5 kJ). Evidently, only about 40% of the energy locked in chemical bonds of glucose molecule is captured for ATP synthesis in our body.
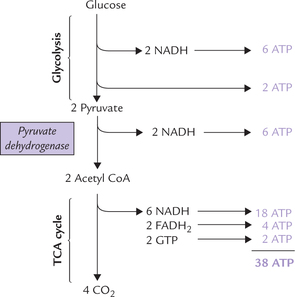
Fig. 9.18 Energetics of glucose oxidation. 38 ATPs are generated by complete oxidation of glucose via glycolysis, PDH reaction, and TCA cycle.
Pasteur Effect
It has been observed that under anaerobic condition a tissue or microorganism utilizes much more glucose than it does under aerobic conditions. This is the Pasteur effect, and it reflects inhibition of glycolysis by oxygen. It is also observed that the levels of glycolytic intermediates from fructose 1,6-bisphosphate onwards decrease while the earlier intermediates increase. Evidently, the Pasteur effect is because of inhibition of the enzyme phosphofructokinase. The inhibitory effect is caused by citrate and ATP, the compounds produced in presence of oxygen.
Crabtree Effect
When oxygen supply is kept constant and glucose concentration is increased, the oxygen consumption by cells falls. This is called Crabtree effect, which is basically opposite of Pasteur effect. It is seen in the cells that have a high rate of aerobic glycolysis. In such cells the glycolytic sequence consumes much of the available Pi and NAD+, which limits their availability for oxidative phosphorylation. As a result, rate of oxidative phosphorylation decreases, and oxygen consumption also shows a corresponding fall.
E Synthetic Functions of the TCA Cycle
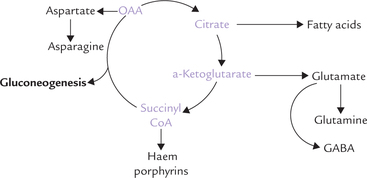
TCA is not only the final oxidative pathway for various biomolecules, its intermediates are also substrates for various biosynthetic pathways (Fig. 9.19 ). Thus, TCA is both catabolic and anabolic in nature, and hence regarded as amphibolic.
Some important synthetic pathways connected with TCA are as follows:
Synthesis of Glucose
Intermediates of TCA cycle can serve as substrates for gluconeogenesis, the pathway that produces glucose from non-carbohydrate sources. As glucose is synthesized in this manner some intermediates of TCA cycle are removed from the cycle. These intermediates may be subsequently replenished by anaplerotic reactions.
Synthesis of Amino Acids
Oxaloacetate and α-ketoglutarate can form aspartate and glutamate, respectively by transamination reactions, which in turn are required for the synthesis of other nonessential amino acids (Chapter 13), as also of purines and pyrimidines.
Others
Succinyl CoA can be diverted from TCA cycle for biosynthesis of porphyrins and haem (for further information refer to Box 9.6). The α-ketoglutarate can form gluta-mate, from which 7-aminobutyric acid (GABA), an inhibitory neuro-transmitter is produced.
F Regulation of TCA Cycle
A number of modulators regulate activities of various enzymes of TCA cycle. Two types of regulatory mechanisms—allosteric modulation and covalent modulation of the enzyme activities operate. Regulation of the pyru-vate dehydrogenase complex, the bridge between glycolysis and TCA cycle, and the regulation of the cyclic pathway are discussed below.
Regulation of the Pyruvate Dehydrogenase Complex
Activity of the pyruvate dehydrogenase complex (PDH) is switched-on or switched-off based on cellular energy needs. When the cellular energy charge is high and surplus fuel molecules are present intracellularly, inhibition of activity of the enzyme complex occurs. Two mechanisms of regulation have been recognized.
Allosteric regulation
The enzyme complex is subject to allosteric inhibition by the reaction products: acetyl CoA and NADH. It is stimulated by AMP.
Elevated intracellular concentrations of both acetyl CoA and NADH indicate surplus intracellular fuel molecules and excessive cellular energy charge. Inhibition of the PDH, under these circumstances, stops further production of energy and fuel molecules.
Covalent modulation
Thus, covalent attachment of a phosphate group renders the PDH complex inactive, whereas removal of this group results in its activation. The following enzymes are involved in interconversion of the phosphorylated-dephosphorylated forms.
1. Pyruvate dehydrogenase kinase: This enzyme phosphorylates the enzyme complex, thereby inactivating it. Increased activity of the kinase is, therefore, associated with decreased activity of PDH. Conversely, inhibition of the kinase activates the enzyme complex (Fig. 9.20 ).
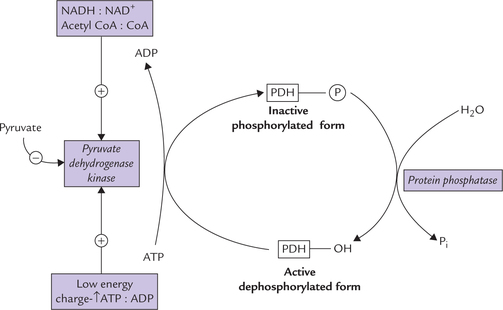
Pyruvate dehydrogenase kinase is allosterically activated by high energy charge, high [NADH] : [NAD+] ratio, and high [acetyl CoA] : [CoA] ratio and is inhibited by pyru-vate. Conversely the enzyme activity is inhibited by low energy charge, reflected by elevated ratio of ADP : ATP, which in turn results in activation of the PDH. The elevated ADP : ATP ratio signals increased demand for energy production, which is met through activation of the PDH.
2. Protein phosphatase: This enzyme removes a phosphate group from the phosphorylated form of the PDH complex to produce the active dephosphorylated form (Fig. 9.20). Thus, it activates the PDH.
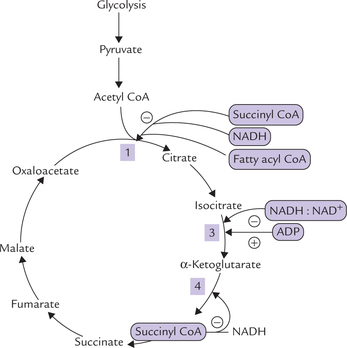
Regulation of the Cyclic Pathway (Fig. 9.21 )
Regulation of Citrate Synthesis
This is the most important regulatory step of the pathway. As in case of most metabolic pathways, where an initial step is the rate setting step for the pathway as a whole, the first step of TCA cycle is also most important in regulation. Rate of this reaction is determined by the following factors:
1. Concentration of the substrate molecules (i.e. oxaloace-tate and acetyl CoA): Concentration of oxaloacetate is the most critical factor in determining the reaction rate. Concentration of acetyl CoA is also an important determinant for pushing the reaction forward.
2. Allosteric modulation of the enzyme: Activity of citrate synthase, the enzyme catalyzing this step, is mainly regulated by succinyl CoA, which inhibits the enzyme activity by decreasing its affinity for acetyl CoA. Fatty acyl CoA and NADH also act as negative allosteric modulators of the enzyme. Raised levels of these metabolites indicate that the cell has adequate supply of fuel and energy. Under these circumstances, TCA cycle, which is an energy-yielding pathway, is inhibited so that there is no further production of energy.
Regulation of Oxidative Decarboxylation of Isocitrate
The enzyme catalyzing this step, isocitrate dehydrogenase, is activated by ADP and inhibited by high energy charge and high [NADH] : [NAD+] ratio. Elevated levels of mitochondrial ADP indicate low energy state of the cell and signals a need for generation of more high-energy phosphate molecules (i.e. ATP). Stimulation by ADP accelerates the cycle, generating the required ATP.
Regulation of Oxidative Decarboxylation of α-ketoglutarate
The enzyme catalyzing this step, α-ketoglutarate dehydro-genase, is allosterically inhibited by its own products (succinyl CoA and NADH) and by high energy charge.
It is evident that low energy charge stimulates TCA so as to generate more energy; conversely, high energy charge inhibits it. The other energy-yielding pathways discussed so far, i.e. glycolysis and oxidative decarboxyl-ation of pyruvate are also regulated similarly.
The TCA cycle is regulated at the steps catalyzed by citrate synthase, isocitrate dehydrogenase and α-ketoglutarate dehydrogenase. Regulation is accomplished mainly by substrate availability, allosteric modulation, and feedback inhibition.
Under normal conditions, the rates of glycolysis and TCA cycle are integrated with each other so that only that much glucose is metabolized as required to supply the initial substrate, i.e. the acetyl CoA for the TCA cycle. Rates of these two pathways are matched because of inhibition of phosphofructokinase (PFK1), the key regulatory enzyme of glycolysis, by a TCA cycle intermediate, citrate. ATP and NADH, produced during TCA cycle also inhibit glycoly-sis by acting as negative modulators of this enzyme.
G Glyoxylate Cycle
Synthesis of carbohydrate from fat is not possible in the mammalian cell due to irreversibility of the pyruvate dehy-drogenase reaction and lack of alternate pathways. However, plant cells and some microorganisms can accomplish this conversion because of a cyclic pathway, termed gly-oxylate cycle (Fig. 9.22 ). It shares several reactions with TCA cycle and is considered an anabolic variant of the latter. It is useful in germinating seeds where the stored fat is converted to glucose for meeting the energy needs of the cell. It operates in glyoxysomes, the specialized cellular organelles where fatty acid oxidation also occurs.
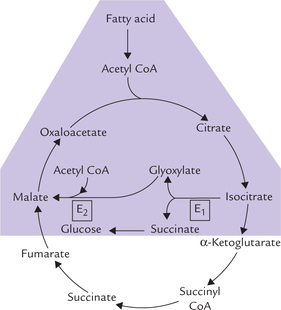
Fig. 9.22 Reactions of glyoxylate cycle (shaded area) (E1 = isocitrate lyase, E2 = malate synthase).
Reactions
The initial few reactions up to isocitrate formation, are common with TCA cycle, but the isocitrate bypasses the TCA cycle and is cleaved by the enzyme isocitrate lyase to succinate and glyoxylate. The latter compound forms malate (by condensing with another molecule of acetyl CoA), which is an intermediate of the TCA cycle. Net result of these reactions is conversion of two 2-carbon fragments of acetyl CoA to malate, which is a substrate for gluconeo-genesis, and can also enter TCA cycle.
In addition to glyoxylate, the other product of cleavage of isocitrate is succinate, which is converted to glucose via reactions of gluconeogenesis, thereby ensuring net synthesis of carbohydrate from fat.
V Gluconeogenesis
Synthesis of glucose from compounds other than carbohydrates is called gluconeogenesis (i.e. formation of new sugar). A variety of biomolecules such as lactate, pyruvate, gly-cerol (derived from triacylglycerols) and α-keto acids (derived from amino acid catabolism) are substrates for gluconeogenesis. The pathway is important because some organs, like the brain and the erythrocytes, depend exclusively on glucose for their energy needs. The brain is said to be a voracious eater of glucose, consuming about 120 grams per day (out of about 160 grams needed by the entire body) and requires a blood glucose concentration of 70-100 mg/dL. Though degradation of stored glycogen can also provide glucose, gluconeogenesis is the only source of glucose during prolonged fasting and starvation.
Gluconeogenesis occurs mainly in the liver, and to a lesser extent in renal cortex. On weight basis, the gluco-neogenic capacity of the renal cortex equals that of the liver, but because of the size difference between these tissues, the gluconeogenic activity in the kidney amounts to only 10% of that in the liver. However, in certain abnormal metabolic states, such as prolonged starvation, kidneys become the major glucose-producing organ.
The gluconeogenic pathway is located mainly in cytosol, although some precursors are produced in the mitochondria.
A Gluconeogenesis: Not a Reversal of Glycolysis
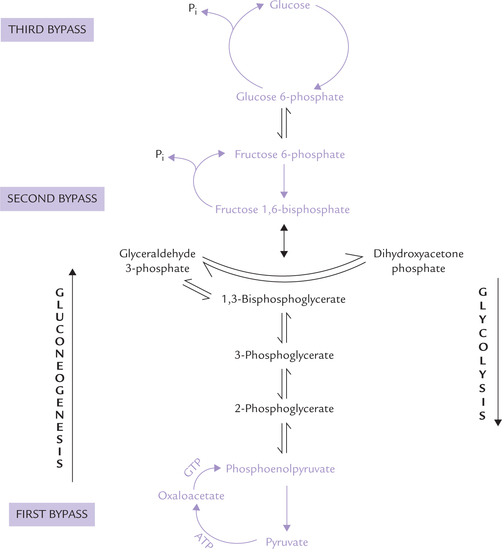
Though most of the reactions of the gluconeogenic sequence (7 out of 10) are reversal of those of glycolysis the following three reactions are unique to gluconeogen-esis, and are catalyzed by a different set of enzymes. They are called the bypass steps:
First bypass Conversion of pyruvate to phosphoenolpy-ruvate (PEP).
Second bypass Conversion of fructose 1, 6-bisphosphate to fructose 6-phosphate.
Third bypass Conversion of glucose 6-phosphate to glucose.
The corresponding steps in glycolysis are formation of pyruvate from phosphoenol pyruvate (for first bypass); fructose 1,6-bisphosphate from fructose 6-phosphate (for second bypass) and glucose 6-phosphate from glucose (for third bypass). Recall that only these three reactions in glycolysis are irreversible. Therefore, an alternate set of reactions, unique to gluconeogenesis, is needed to circumvent these three irreversible glycolytic reactions (Fig. 9.23 ). Apart from these three steps, rest of the seven steps of the glycolytic sequence are reversible and are operative in the gluconeogenesis as well.
B The Bypass Reactions of Gluconeogenesis
First Bypass: Conversion of Pyruvate to Phosphoenolpyruvate (PEP)
During conversion of phosphoenolpyruvate to pyruvate in glycolytic sequence, release of a large amount of free energy (ΔG0′-14.8 kcal/mole) occurs under standard conditions. For the reverse reaction (i.e. conversion of pyruvate to PEP during gluconeogenesis), input of an equivalent amount of free energy is required. This is a thermody-namically unfavourable situation, which is circumvented by making this conversion occur in two distinct reactions:
Sum: Pyruvate to PEP: 1 ATP + 1 GTP are hydrolyzed.
The conversion involves the following four steps, which occurs partly in cytosol and partly in mitochondrial matrix (Fig. 9.24 ):
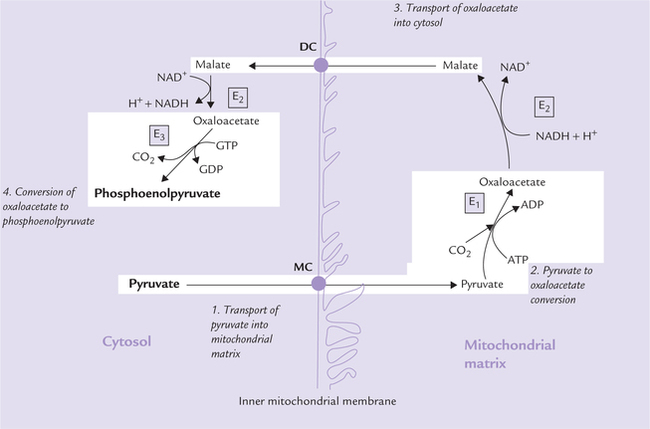
Fig. 9.24 Synthesis of phosphoenolpyruvate from pyruvate. The conversion occurs partly in cytosol, partly in mitochondria and in four steps (MC = monocarboxylate carrier, DC = dicarboxylate carrier, E1 = pyruvate carboxylase, E2 = malate dehydrogenase, E3 = phosphoenolpyruvate carboxy kinase).
1. Transport of pyruvate into mitochondrial matrix: Pyruvate moves across the inner mitochondrial membrane (IMM) to enter the mitochondrial matrix through mediation of specific transport proteins, called the monocarboxylate carriers monocarboxylate carrier monocarboxylate carriers (MC).
2. Pyruvate to oxaloacetate conversion: Pyruvate, a 3-carbon molecule is converted to 4-carbon oxaloacetate by car-boxylation reaction catalyzed by pyruvate carboxylase (E1), a biotin-dependent mitochondrial enzyme.
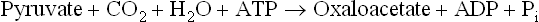
3. Transport of oxaloacetate into cytosol: Rest of the enzymes of gluconeogenesis are located in the cytosol. Therefore, the mitochondrial oxaloacetate, produced in the step 2, must be transported out of the mitochondria into cytosol. However, oxaloacetate cannot directly cross the IMM because the latter is impermeable to most molecules except for the ones for which specific carrier proteins exist (such as monocarboxylate carrier for pyruvate, dicarboxylate carrier for malate, and tricar-boxylate carrier for citrate). Therefore, the oxaloacetate is converted to malate, a molecule which is capable of crossing the IMM. The enzyme malate dehydrogenase (E2) catalyzes this reaction.
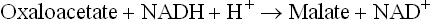
Malate is carried out of the mitochondria into cytosol by the dicarboxylate carrier (DC). In cytosol, oxaloacetate is recovered from malate by reversal of the above reaction.
4. Conversion of oxaloacetate to phosphoenolpyruvate: In cyto-sol, the oxaloacetate undergoes decarboxylation and phosphorylation to yield phosphoenolpyruvate (PEP).
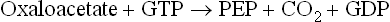
This reaction is catalyzed by the enzyme phosphoenol-pyruvate carboxykinase (PEPCK), and is driven forward by hydrolysis of GTP.
Thus, a concerted action of several enzymes and carrier proteins permits conversion of PEP to pyruvate; a conversion which is thermodynamically unfavourable otherwise. Energy of two high-energy phosphate bonds (equivalent to 14.6 kcal/mole), one each from ATP and GTP, are required to drive the conversion across the energy “road-block”. The overall equation for this set of reactions is as below:
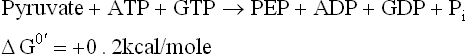
A series of reversible reactions follow next, which are shared by gluconeogenesis and glycolysis. These reactions result in conversion of PEP to fructose 1,6-bisphosphate (Fig. 9.23).
Second Bypass: Dephosphorylation of Fructose 1,6-bisphosphate
Hydrolysis of fructose 1,6-bisphosphate by fructose 1,6-bisphosphatase is the second reaction that is unique to gluconeogenesis. It bypasses the irreversible phosphofructo-kinase reaction. It also provides an energetically favourable pathway for the formation of fructose 6-phosphate (ΔG0′ = -3.9 kcal/mole). This reaction is an important control point for gluconeogenesis, discussed later in this chapter.
Third Bypass: Hydrolysis of Glucose 6-phosphate
Glucose 6-phosphatase catalyzes hydrolytic cleavage of the phosphate ester to liberate free glucose. Unlike the other gluconeogenic enzymes, which are cytoplasmic (except pyruvate carboxylase), this enzyme resides on the luminal surface of the endoplasmic reticulum (ER) membrane. Like the fructose 1,6-bisphosphatase reaction, this step is also irreversible, and so provides an energetically favourable pathway for the formation of free glucose (ΔG0′ = -2.9 kcal/mole).
C Reversible Steps of Gluconeogenesis
In addition to the three bypass steps, there are seven reversible reactions common to both these pathways (Fig. 9.23). During gluconeogenesis, their equilibrium is pushed in favour of glucose synthesis.
A summary of all reactions of gluconeogenesis, including the reversible ones is as below:
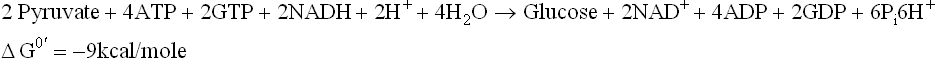
D Gluconeogenesis is an Expensive Process
Several reactions of gluconeogenesis are energetically unfavourable. It requires input of considerable free energy to drive these “uphill” reactions: Thus six phosphoanhy-dride bonds are required for the synthesis of one glucose molecule from two pyruvate molecules (Table 9.4 ).
Table 9.4
Energy expenditure for synthesis of one glucose molecule from two molecules of pyruvate
| Enzyme | Reaction | Energy consumption |
| Pyruvate carboxylase | 2 × Pyruvate → 2 × Oxaloacetate | 2 ATPs |
| PEP-carboxy-kinase | 2 × Oxaloacetate → 2 × PEP | 2 GTPs |
| Phosphoglycerate kinase | 2 × 3 Phosphoglycerate → 2 × 1,3 Bisphoglycerate | 2 ATP |
Sum: 6 High energy phosphate bonds
Pyruvate carboxylase reactions consume two ATPs, PEP-carboxykinase consume two GTPs, and phosphoglycerate kinase consume two ATPs in the reversal of the substrate level phosphorylation (see reaction 7; Fig. 9.3).
In addition, two NADH molecules are used up in the gly ceraldehyde 3-phosphate dehydrogenase reaction, for each molecule of glucose synthesized. Since each NADH can generate 3ATPs by oxidative phosphorylation, this is equivalent to the input of another six ATPs per glucose synthesized.
In spite of being expensive, gluconeogenesis is fairly efficient—the liver can make a kilogram of glucose per day by gluconeogenesis.
E Substrates for Gluconeogenesis
Pyruvate and TCA cycle intermediates are important substrates. In addition, a variety of other molecules can also yield glucose via the gluconeogenic sequence, as discussed here.
Lactate
Lactate produced in the skeletal muscles is the major substrate for gluconeogenesis (Cori’s cycle). It is converted to pyruvate (in liver) by the lactate dehydrogenase reaction, which then enters gluconeogenic sequence.
Amino Acids
Glycogenic amino acids (all except lysine and leucine) are catabolized to pyruvate or some intermediate of citric acid cycle, which can generate glucose (Chapter 13). During fasting and starvation, the glycogenic amino acids become most important precursors for gluconeo-genesis.
Glycerol
Glycerol is obtained during degradation of adipose triac-ylglycerols. It enters the gluconeogenic sequence in the liver. First, it reacts with ATP to form glycerol 3-phosphate, which is oxidized to dihydroxyacetone phosphate. The latter being a glycolytic intermediate can be converted to glucose.
Propionate
Catabolism of some amino acids (methionine, isoleu-cine) and odd chain fatty acids yields propionyl CoA, which enters a reaction sequence to ultimately yield suc-cinyl CoA (Chapter 11). Being a TCA cycle intermediate, succinyl CoA is convertible to glucose. Fats cannot generate glucose: It is important to note that fatty acids are NOT substrates for gluconeogenesis, so it is generally said that glucose cannot be synthesized from fat. This is because fatty acids are oxidized to acetyl CoA via β-oxidation (Chapter 11). Acetyl CoA cannot be converted to pyruvate, because the pyruvate dehydrogenase reaction is irreversible and there are no alternative reactions to channel acetyl CoA into gluconeogenesis. However, odd chain fatty acids are exceptions because their terminal three carbons may serve as gluconeogenic substrate.
The substances that are degraded to acetyl CoA, including ketone bodies, ethanol and even chain fatty acids, are also not substrates of gluconeogenesis.
F Regulation
Certain bypass steps of gluconeogenesis serve as important control points.
Regulation of Pyruvate to Oxaloacetate Conversion
Pyruvate carboxylase, the enzyme catalyzing this bypass step is a regulatory enzyme; it is allosterically inhibited by ADP and stimulated by acetyl CoA. So potent is the effect of acetyl CoA that the enzyme is virtually inactive in its absence. Stimulation by acetyl CoA ensures that sufficient oxaloacetate is generated (by pyruvate carboxylase) for condensing with it (acetyl CoA) to form citrate (Fig. 9.25a ).
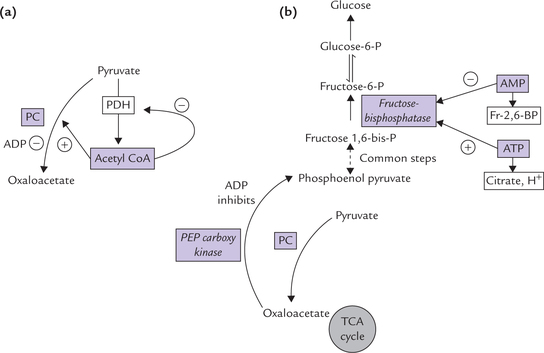
Fig. 9.25 Regulation of gluconeogenesis. (a) Acetyl CoA as the positive allosteric modulator of pyruvate carboxylase (PC) and inhibitor of pyruvate dehydrogenase (PDH) complex, (b) Role of various other allosteric modulators ( = inhibits reaction
= inhibits reaction  = accelerates reaction, Fr-2,6-BP = fructose 2,6-bisphosphate).
= accelerates reaction, Fr-2,6-BP = fructose 2,6-bisphosphate).
Regulation of Fructose 1,6-bisphosphate to Fructose 6-phosphate Conversion
ATP is a positive modulator, while AMP affects fructose 1,6-bisphosphatase negatively (Fig. 9.25b ). Therefore, rate of gluconeogenesis is determined by cellular energy charge, reflected by the ratio of ATP : (ADP + AMP)
1. When the energy charge is low, as indicated by increased intracellular AMP concentration, rate of gluconeo-genesis is impeded through inhibition of fructose 1,6-bisphosphatase by AMP.
2. Conversely, when the energy charge is high, rate of gluconeogenesis is enhanced due to stimulation of this enzyme by ATP. Since the corresponding glycolytic enzyme, PFK1, is stimulated by AMP and ADP, and inhibited by ATP, these two pathways are regulated in a reciprocal manner. For example, when energy charge of the cell is low, glycolysis is favoured but gluconeo-genesis is inhibited.
Hormonal Regulation
The above mechanisms bring about short-term regulation of gluconeogenesis. The day-to-day regulation is effected by the action of hormones on the amount of the key enzymes. Insulin represses the gluconeogenic enzymes (and induces most of the glycolytic enzymes); and these effects are counter-balanced by glucagon and other insulin antagonists.
Regulation by Glucagon
Glucagon, a peptide hormone produced by the α-cells in the endocrine pancreas stimulates gluconeogenesis by following mechanisms:
1. Glucagon raises the intracellular concentration of the second messenger cyclic AMP (cAMP), which causes conversion of the active form of pyruvate kinase to the inactive form (Fig. 9.9). This reduces the PEP to pyru-vate conversion and the former is diverted into the gluconeogenic sequence.
2. Glucagon reduces the intracellular concentration of fructose 2,6-bisphosphate (Fig. 9.8). This compound is a negative allosteric modulator of fructose 1,6-bisphosphatase and, as explained earlier, a positive modulator of phosphofructokinase (PFK1); both these factors ultimately favour gluconeogenesis.
3. Glucagon increases fatty acid mobilization and their oxidation to yield acetyl CoA, NADH and ATP; these compounds inhibit the PDH complex and acetyl CoA stimulates pyruvate carboxylase (Fig. 9.25a ). Thus, the available pyruvate is not converted to acetyl CoA but to oxaloacetate for gluconeogenesis.
4. Glucagon causes induction of the key enzymes of gluconeogenesis, i.e. phosphoenolpyruvate carboxykinase and glucose 6-phosphatase (and possibly pyruvate carboxyl-ase also).
Regulation by Insulin
Insulin causes repression of severed gluconeogenic enzymes, a detailed account of which is given in Chapter 15.
Reciprocal Regulation of Gluconeogenesis and Glycolysis
It is evident from the above discussion that glucagon not only stimulates gluconeogenesis but concomitantly inhibits glycolysis. Allosteric modulations also ensure that the opposing enzymes of these two processes are not active at the same time, which in turn prevents futile cycles (refer to Box 9.8 for details):
1. As shown in Figure 9.26, compounds reflecting high energy state of cell, e.g. ATP, citrate and acetyl CoA, allo-sterically stimulate the key gluconeogenic enzymes but inhibit the corresponding enzymes of glycolysis.
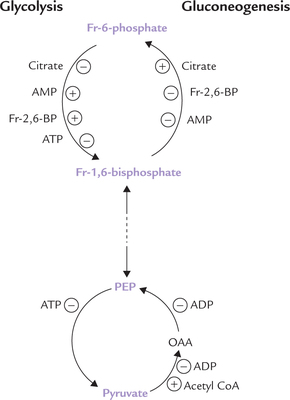
2. Fructose 2,6-bisphosphate also has opposite effects on phosphofructokinase (stimulatory) and fructose 1,6-bisphosphatase (inhibitory). Levels of fructose 2,6-bisphosphate are high in fed state (and low in starvation) so that glycolysis is accelerated and glyconeogenesis is inhibited in fed state. During starvation, glyco-neogenesis predominates because levels of fructose 2,6-bisphosphate are low (refer to Box 9.7 for details).
GN Ratio or DN Ratio
It refers to the ratio between the dextrose (glucose) to nitrogen excreted in urine. In the experimental animals treated with alloxan (causes destruction of pancreatic (β-cells) or phlorizin (causes renal tubular cell necrosis) glucose is lost through urine and body attempts to generate glucose through gluconeogenesis. The body proteins are degraded to provide amino acid substrates for gluconeogenesis, and their catabolic end-product, urea, is excreted in urine. The ratio of glucose to urea nitrogen in such animals is found to be 3.65, meaning that one gram of nitrogen (from proteins) forms 3.65 grams of glucose.
GN ratio is increased when catabolism is enhanced, such as in starvation, hyperthyroidism and cancer.
Because 3.65 grams glucose is synthesized from one gram of nitrogen of protein, and proteins contain 16% nitrogen, it is calculated that 58% of protein is glycogenic.
VI Glycogen Metabolism
Glycogen is stored mainly in liver and skeletal muscles as an energy reserve, just as starch in plants. The tissue concentration of glycogen in liver (6-8%) is higher than that in muscle (1-2%), but because of the relative masses of muscle and liver, the majority of glycogen in the body is stored in muscles. Though total hepatic glycogen stores (50-100 g) are significantly less than the muscle stores (200-300 g), they are involved in a vital activity, that is to serve as our first line of defense against declining blood glucose level, particularly between meals. Muscle glycogen is essential for muscle energy metabolism during bursts of physical activity even though muscle relies primarily on fats as a source for energy.
Glycogen is the storage form of carbohydrates in liver and muscles. Hepatic glycogen generates glucose (due to presence of glucose 6-phosphatase), whereas muscle glycogen serves as a source of metabolic fuel for use in muscle.
Glycogen is an extensively branched homopolysac-charide, consisting of chains of a(1→4) linked glucosyl residues with branches that are joined by a(1 → 6) linkages (Chapter 2). The latter are spaced about every 4-6 residues along the a-1,4 chain. The gross structure of glycogen is dendritic in appearance, expanding from a core sequence bound to a tyrosine residue in the protein, glycogenin.
A Glycogen Synthesis (Glycogenesis)
Glycogen is synthesized from glucose. Like most other anabolic processes, glycogenesis occurs in the cytosolic fraction of the cell and requires input of energy. The energy for gly-cogenesis comes from high energy nucleotides, ATP and UTP. These nucleotides provide energy for the conversion of the precursor glucose molecule to its energized form, UDP-glucose. The UDP-glucose (UDPG) serves as activated donor of glucose residues that are added to the growing glyco-gen molecule. Glycogen synthase is the major enzyme of this complex process, which occurs in following four stages:
Synthesis of UDP-glucose
Activation of glucose to form UDP-glucose (UDPG) occurs in three sequential reactions, outlined in Figure 9.27 .
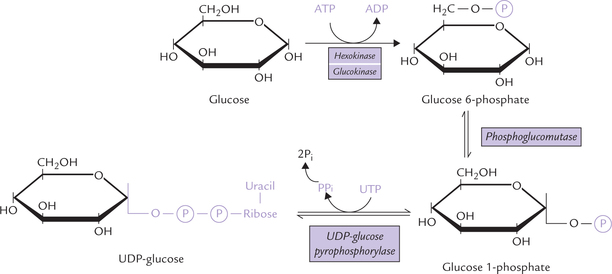
Reaction 1
Glucose is phosphorylated at C-6 by glucokinase in liver ( hexokinase in muscle) to form glucose 6-phosphate.
Glucokinase has high Km (i.e. low affinity for glucose) and high Vmax which permits it to rapidly phosphorylate large quantities of glucose after meals, when the ambient glucose concentration is high.
Synthesis of Primer to Initiate Glycogen Synthesis
UDPG donates glucose residues to an existing a(1 → 4) glucosyl chain called primer, which will accept the incoming glucose residues.
Normally a fragment of glycogen serves as a primer. Such fragments are obtained from glycogen molecules that had been partially degraded in liver during fasting or in muscle during exercise. However, when the glycogen stores are depleted, a specific protein, known as glyco-genin, provides the site at which the primer is built. Glycogenine contains a specific tyrosyl residue, which accepts the first glucose donated by UDPG.
The above reaction is catalyzed by glycogenin (autoca-talysis) or by the enzyme glycogen synthase.
More of glucosyl residues coming from UDP glucose, are attached sequentially in the same manner. Thus a short a(1 → 4) glucosyl chain (i.e. the primer) is built which is attached to the protein.
Elongation of Chains by Glycogen Synthase
The primer is elongated by sequential addition of glucose residues. The enzyme glycogen synthase catalyzes this reaction. It transfers the glucose residues from UDPG to the non-reducing end of glycogen in α (1 → 4) linkage (Fig. 9.28 ).
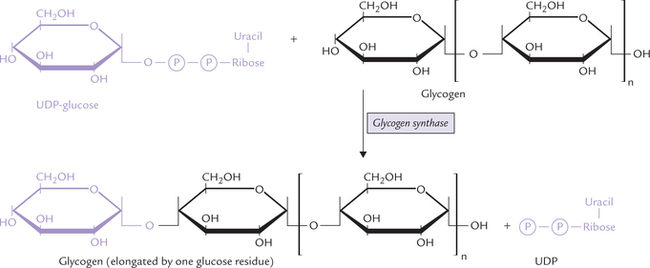
This reaction has been elaborately shown in Figure 9.28.
UDP produced in the above reaction is reconverted to UTP by the enzyme nucleoside diphosphate kinase.
Formation of Branches in Glycogen
The reactions discussed so far result in formation of linear, unbranched chains that are linked by α (1→4) linkage. However, glycogen is a highly branched structure; the branch points are created by the action of the “branching enzyme”. This enzyme comes into action after glycogen synthase has added at least ten glucosyl units (Fig. 9.29 ).
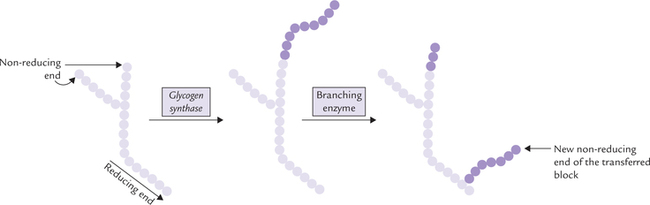
Fig. 9.29 The action of branching enzyme. It forms branches by transferring a block of glycosyl residues, typically seven in length, from the end of an unbranched chain to a more interior location (on the same or other chain). The transferred block is attached via α 1-6 linkage. → indicates non-reducing ends [each circle represents a glucose residue].
The branching enzyme removes a block of five to eight glucose residues from the non-reducing end of the chain and transfers it to a more internal location on the same or other chain. The transferred block is attached via α(1 → 6) linkage to a glucose residue at the new location. The branching enzyme is also referred to as a glucosyl 4 : 6 transferase because it forms a new α (1 → 6) bond instead of an α(1 → 4) bond.
The transfer of a block results in creation of a new non-reducing end. More of glucose residues can be added to this end. In this way, branching increases the number of non-reducing ends to which new glucose residues can be added, thereby greatly accelerating the rate at which glycogen synthesis occurs.
B Glycogen Degradation (Glycogenolysis)
The principal enzyme of glycogenolysis is glycogen phos-phorylase. It acts in association with a debranching enzyme (Fig. 9.30 ).
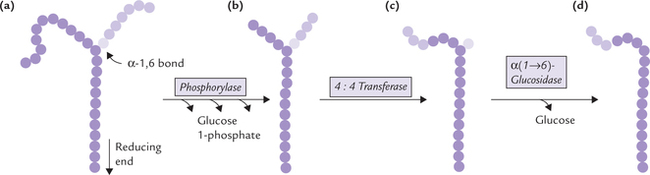
Fig. 9.30 Glycogen degradation. Designation of glucose residues as in Figure 9.28.
Action of Glycogen Phosphorylase
Glycogen phosphorylase removes glucose residues, one at a time, from the non-reducing ends of glycogen (Fig. 9.30a). It utilizes inorganic phosphate (P1) to cleave the α(1 → 4) bonds. This results in release of the terminal glucosyl residue as glucose 1-phosphate. The reaction is termed as phosphorolysis, that is, breakage of a covalent bond by addition of a phosphate group.
Pyridoxal phosphate is an essential cofactor in this reaction; it is covalently bound to the enzyme protein.
Phosphorylase is specific for α (1 → 4) linkages only, it cannot cleave α (1 → 6) linkages. Further, this enzyme cannot approach the branching glucose residues efficiently. Thus, as shown in Figure 9.30b, phosphorylase cleaves the external glucose residues until the branches are about four residues long. Then the action of glycogen phosphory-lase stops and debranching enzyme comes into play.
Removal of Branches
The key enzyme for removing branch points of glycogen is the debranching enzyme, which possesses dual activity, namely glucosyl 4 : 4 transferase activity and α (1 → 4) glucosidase activity.
4: 4 transferase activity: Three of the four external glucosyl residues that remain are removed as a trisaccharide and transferred to the non-reducing end of a nearby chain (Fig. 9.30c). This action involves cleaving of an α (1 → 4) bond at one site and formation of a new α(1 → 4) bond elsewhere.
α(1 → 6) glucosidase activity: The single glucosyl residue that remains at the branch point is removed by the α(1 → 6) glucosidase, to liberate free glucose (Fig. 9.30d).
Removal of branch point in this manner exposes another set of α (1 → 4) linkages, till the next branch point. When another branch point is reached, it is again removed by the debranching enzyme, and the cycle thus continues.
Thus, the phosphorolysis/debranching processes, acting alternately, can cleave a large glycogen molecule having thousands of glycosyl residues.
Fate of Glucosyl Units Released from Glycogen
The glucosyl units are released from glycogen in two forms: glucose 1-phosphate and free glucose. About 90% of the glucose is released as glucose 1-phosphate, and only the a-1,6 branching residue, is released as free glucose. Glucose 1-phosphate is then converted to glucose 6-phosphate by the enzyme phosphoglucomutase.
Further, fate of glucose 6-phosphate varies depending on the tissue. Liver possesses the enzyme glucose 6-phosphatase, which forms free glucose from glucose 6-phosphate. This glucose is released into blood for use by needy tissues.
Muscles lack glucose 6-phosphatase, hence cannot contribute to the blood glucose. Rather muscle glycogen is used for the generation of metabolic energy in the exercising muscles themselves.
Role of liver glycogen and muscle glycogen are different: Liver releases free glucose in blood circulation, and thereby plays an important role in glucose homeostasis. However, hepatic glycogen stores are barely sufficient for maintenance of blood glucose concentration during a 12-hour fast.
Muscle glycogen stores are used for liberating fuel molecules (i.e. glucose 6-phosphate), which in turn are used in the exercising muscles for generating metabolic energy.
C Regulation
Synthesis and breakdown of glycogen are important intracellular activities, having implications beyond the cell. Balance between the two processes is important for:
Normal blood glucose levels are especially important for the tissues that use glucose as their primary substrate, such as, brain erythrocytes, renal medulla, lens and cornea of the eye. Therefore, tight regulation of these two processes (i.e. glycogenesis and glycogenolysis) is essential.
Regulation of Activity of Glycogen Phosphorylase
The regulatory enzyme for glycogenolysis is glycogen phosphorylase. Activity of this enzyme is regulated by (a) covalent modulation through phosphorylation-dephosphorylation, (b) allosteric regulation modulation, and (c) calcium ions.
Covalent Modulation
Studies concerning regulation of glycogen phosphorylase have been mostly conducted in skeletal muscles. The muscle enzyme is a dimeric protein consisting of two identical subunits. Each subunit contains an essential serine residue, where phosphate group can covalently attach. Thus, the glycogen phosphorylase exists in two forms:
• Phosphorylated form called phosphorylase a which is catalytically active.
• Dephosphorylated form called phosphorylase b which is much less active.
These two forms of the enzyme are interconvertible. Conversion of inactive phosphorylase b to active phosphorylase a is a catalyzed by the enzyme phosphorylase kinase, which phosphorylates, and thereby activates glycogen phosphory-lase. Another enzyme, protein phosphatase-1, dephos-phorylates, and thereby inactivates glycogen phosphorylase a (Fig. 9.31 ).
Through action of these enzymes, ratio of the active and the inactive phosphorylase can be varied, which ultimately controls the rate of glycogenolysis. The same mechanism is operative in liver as well.
Allosteric Regulation
Superimposed upon the covalent modulation are the actions of allosteric effectors:
1. AMP: Glycogen phosphorylase in muscles and other extrahepatic tissues is stimulated powerfully by AMP (Fig. 9.31). This allosteric effect ensures that glycogen is degraded rapidly in severely contracting muscles, to provide substrate for an anaerobic glycolysis. Concentration of AMP quickly builds up in contracting muscles due to its rapid production by pyrophosphate cleavage of ATP
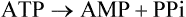
Note: Unlike other forms of stored energy, only gly-cogen can be used for ATP synthesis under anaerobic conditions.
2. Glucose: Glycogen phosphorylase in liver is inhibited by glucose. Because the intracellular glucose concentration in the liver approximates the blood glucose level, glycogen degradation in liver is regulated directly by the blood glucose level.
3. Glucose 6-phosphate: It inhibits muscle glycogenolysis.
4. ATP: Both liver and muscle glycogenolysis are inhibited by ATP.
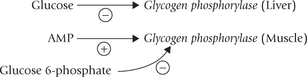
To sum up, the various mechanisms described ensure that glycogenolysis is stimulated when glucose concentration and energy levels are low, and inhibited when these are high.
Regulation by Calcium Ions
During muscle contraction, calcium is released from sar-coplasmic reticulum and activates phosphorylase kinase (via a calmodulin-calcium modulating protein; Chapter 29). This enzyme causes phosphorylation of glycogen phos-phorylase and hence enhances glycogenolysis (without involving cAMP).
Hepatic and muscle glycogenolysis serve different roles and respond to different regulatory signals. Major differences between the two are given in Table 9.5 .
Table 9.5
Difference between glycogenolysis in liver and muscles
| Muscle | Liver |
| 1. Little glucose formed, since glucose 6-phosphatase is absent | Free glucose forms by glucose 6-phosphatase |
| 2. Stimulated by adrenaline, glucagon has no effect | Glucagon is major stimulator of glycogenolysis |
| 3. Stimulated by AMP but inhibited by glucose 6-phosphate | No effect of AMP, but hepatic glycogen phosphorylase is inhibited by glucose |
Regulation of Activity of Glycogen Synthase
The regulatory enzyme of glycogenesis is glycogen synthase. Regulation of its activity is effected by the following mechanisms: (a) allosteric regulation, and (b) covalent modulation.
Covalent Modulation
Activity of glycogen synthase is also regulated by phosphorylation-dephosphorylation, as in the case of glycogen phosphorylase. However, there is a major difference: glycogen synthase is more active in the dephosphory-lated form and less active in the phosphorylated form (Fig. 9.32 ). Thus, it is activated by protein kinase and rendered less active by protein phosphatase-1 .
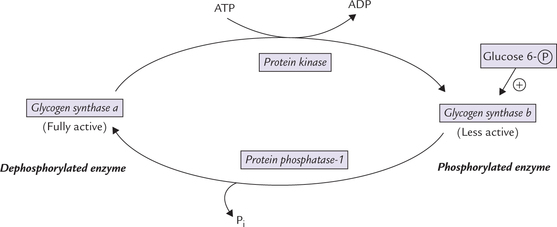
It is important to note that simultaneous phosphorylation of both glycogen-phosphorylase and synthase will switch the cell from glycogen synthesis to glycogen degradation. This prevents futile cycles (Box 9.8).
Allosteric Regulation
Glucose 6-phosphate is a potent activator of glycogene-sis. It does so by stimulating the protein phosphatase-1, so that glycogen synthase is converted to the active (dephos-phorylated) form. It also causes direct allosteric stimulation of the glycogen synthase activity. In muscle tissue, glucose 6-phosphate not only stimulates glycogen syn-thase, but also inhibits activity of glycogen phosphorylase.
Hormonal Regulation of Glycogen Metabolism
Glucagon and epinephrine, acting through cAMP, are primarily involved in regulation of the glycogen metabolism. Glucagon acts on liver cells and epinephrine acts on both liver and muscle cells. These hormones stimulate glycoge-nolysis and inhibit glycogenesis. Insulin, on the other hand, promotes glycogenesis and inhibits glycogenolysis.
Role of Glucagon and Epinephrine
Both these hormones activate the membrane enzyme adenylate cyclase, which catalyzes formation of cAMP from ATP (Fig. 9.33 ). The cAMP binds to protein kinase A (PKA, also called cAMP-dependent protein kinase), a tetrameric protein. The cAMP binding activates this enzyme by letting its catalytic subunits free (Chapter 29). Activation of PKA has the following consequences: (i) Inhibition of glycogenesis, and (ii) Stimulation of glycogenolysis.
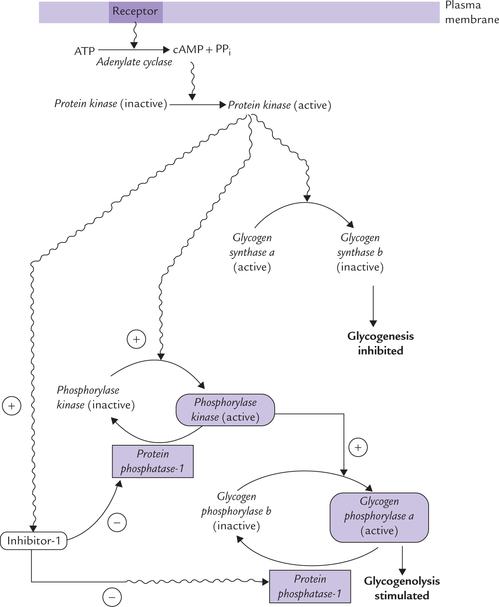
Fig. 9.33 Hormonal regulation of glycogen synthesis and degradation. The cAMP generated by hormones activates protein kinase A, which cause phosphorylation of glycogen synthase (inactivation), phosphorylase kinase (activation) and the inhibitor-1 (activation). The last inhibits protein phosphatase. Activated phosphorylase kinase causes phosphorylation of glycogen phosphorylase b, activating it to glycogen phosphorylase ( = inhibitor, = stimulator).
• Inhibition of glycogenesis: The active PKA cause phosphorylation of glycogen synthase. Since the phosphor-ylated glycogen synthase is less active, glycogenesis is inhibited.
• Stimulation of glycogenolysis: The active PKA stimulates glycogenolysis by a dual mechanism, as discussed below:
(a) It catalyzes phosphorylation of phosphorylase kinase, which exists as an inactive dephosphory-lated form and an active phosphorylated form. Thus, phosphorylase kinase is now activated, and in turn phosphorylates the glycogen phosphorylase b, converting it to phosphorylase a, that now carries out a rapid dehydration of glycogen (Fig. 9.33).
(b) Active PKA phosphorylates a protein called inhibitor-1, which thereby gets stimulated. This protein inhibits protein phosphatase-1, the enzyme that normally causes inactivation of glycogen phosphor-ylase a. The overall effect is that the glycogen phos-phorylase a dose not get inactivated, and is "locked" in activated (phosphorylated) form providing a persistent stimulus for glycogenolysis (Fig. 9.34 ).
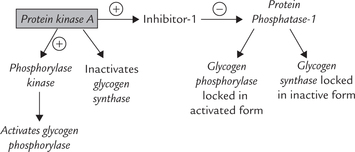
Note
Inhibition of protein phosphatase-1 locks glycogen synthase in its inactive (phosphorylated) form, hence suppressing glycogenesis.
Cascade with amplification: The cAMP activated process is a cascade in which the initial hormonal signal is amplified several folds. Single hormone molecule of glucagon or adrenaline activates the enzyme adenylate cyclase for long enough to produce hundreds of cAMPs which activate protein kinase molecules. Although four cAMP molecules are required to activate a protein kinase (Chapter 29), each active PKR molecule can convert hundreds or thousands of molecules of phosphorylase kinase to active form. Similarly, each active molecule of phosphorylase kinase, can convert thousands of molecules of glycogen phosphorylase b to phosphorylase a, and so on. The net result is that a single hormone molecule can generate thousands of molecules of glucose 1-phosphate by glycogenolysis.
Role of Insulin
Insulin is the key hormone that stimulates glycogen synthesis. It is released in the fed state and activates an intracellular phosphatase. The latter causes dephosphorylation of gly-cogen synthase. Dephosphorylation causes this enzyme to be activated, resulting in increased glycogen synthesis.
Moreover, insulin stimulates the uptake of glucose by muscle, providing substrate molecules for glycogen synthesis. Dephosphorylation of glycogen phosphorylase causes a concomitant fall in the rate of glycogenolysis. Finally, insulin promotes activity of phosphodiesterase (it degrades cAMP) in liver, which decreases cAMP levels. This enhances glycogenesis and suppresses glycogenolysis.
D Glycogen Storage Diseases
Any deficiency of a glycogen degrading enzyme causes abnormal accumulation of glycogen in the affected tissue. The resulting diseases, summarized in Table 9.6, are collectively known as the glycogen storage diseases. These diseases are quite rare (overall incidence of 1 in 40 000) and are mostly inherited as autosomal recessive trait. Normally a defect in an enzyme of glycogenolysis (or glycogenesis) leads to abnormal accumulation of glyco-gen. However, defective action of a glycolytic enzyme, phosphofructokinase, may also result in abnormal glyco-gen accumulation.
Table 9.6
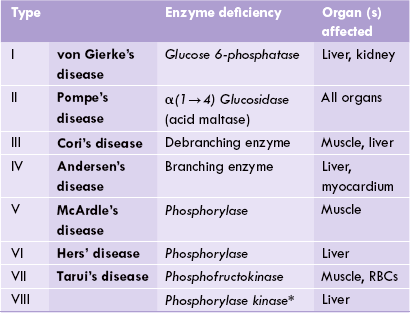
*There is also on X-linked form of phosphorylase kinase deficiency. This is sole exception as all other glycogen storage diseases are inherited as autosomal recessive trait.
In some forms, the main abnormality lies in liver, whereas in others muscle tissue is primarily affected. Accordingly, glycogen storage diseases can be classified in two major types: the hepatic forms and the myopathic forms. Certain forms (e.g. Type II) cannot be put into either of these categories. Some rare forms, e.g. IX, × and XI have also been identified which are due to defects in enzymes that activate or deactivate liver phosphorylase.
Hepatic Forms
Type I, III, IV, VI and VII are classified as the hepatic forms of glycogen storage disease. These are characterized by excessive deposition of glycogen in liver, and hence liver enlargement (hepatomegaly) is the prominent feature. The biochemical manifestations are hypoglycaemia, lactic aci-dosis, hyperuricaemia and hyperlipidaemia, which are most prominent in the type I form (von Gierke’s disease), but may occur in varying severities in the other forms. Enzymatic defects in various forms are shown in Table 9.6.
von Gierke’s disease (Type I)
It is a classical hepatic glycogen storage disease, caused by deficiency of glucose 6-phosphatase. It is a rare disorder with an incidence of 1 per 200,000 persons. It is characterized by severe hepatomegaly and dangerous hypoglycaemia, that may develop within hours of last meal (not surprising, because in between meals glucose 6-phosphatase is required for formation of glucose by both glycogenolysis and gluconeo-genesis). The patient may be kept alive only by feeding carbohydrates at regular intervals, day and night. The hypogly-caemia is unresponsive to glucagon or adrenaline because of the metabolic block in formation of free glucose from liver glycogen stores. Other manifestations of von Gierke’s disease are as follows:
1. Lactic acidosis: It may develop because the glucose 6-phosphate cannot be degraded to glucose, and is therefore, diverted into the glycolytic sequence to form pyruvate and then lactate. Lactate level in blood increases and pH is lowered (acidosis).
2. Hyperuricaemia: It may develop because accumulation of glucose 6-phosphate enhances the reactions of HMP shunt (2nd stage). This stage involves interconver-sion of hexose phosphates (e.g. glucose 6-phosphate) and pentose phosphates (e.g. ribose 5-phosphate). Therefore, accumulation of glucose 6-phosphate leads to increased formation of ribose 5-phosphate. This leads to increased uric acid production since ribose phosphate enhances formation of purine nucleotides by “salvage pathway” (Chapter 20), which are then degraded to uric acid.
Moreover, lactic acidosis inhibits renal excretion of uric acid, which aggravates hyperuricaemia.
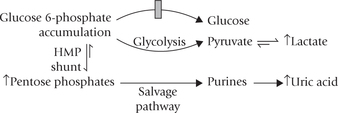
3. Hyperlipidaemia: It is the result of decreased glucose availability, leading to mobilization of the stored fats.
The above features may occur, in varying degrees, in the other hepatic forms of glycogen storage diseases (Table 9.6).
Cori’s disease (Type III)
Branched chain glycogen accumulates because of absence of the debranching enzyme. Clinical course runs like the type I, but is much milder.
Anderson’s disease (Type IV)
A rare disease in which gly-cogen having few branches accumulates because of absence of the branching enzyme. Death from liver cirrhosis usually occurs before the age of two years.
Myopathic Forms
These include types V (McArdle’s disease) and VII (Tauri’s disease). Muscle glycogen stores are very high but are not available for use. Because glycogen is the primary substrate in the exercising muscles, the latter are most severely affected. Clinical course of both type V and type VII runs similarly: the affected patients suffer from muscle cramps and pain upon exertion and are easily fatigued. Apart from this they lead a normal life. This shows that the utilization of muscle glycogen is not essential for life. Interestingly, after muscular activity, these patients do not show the expected increase in blood lactate. This demonstrates that the most important source of lactic acid during muscular activity is not the free glucose from blood but stored muscle glycogen.
Pompe’s disease
(Type II), which involves all organs, needs a special mention because of its peculiar features. In this disease, glycogen accumulation occurs in lysosomes since ability to hydrolyze it is impaired. This is because of impairment (or absence) of the lysosomal enzyme α(1 → 4) glucosidase. Consequently the lysosomes get distended with glycogen, especially in the myocardium, skeletal muscle, liver and motor nuclei of spinal cord.
The infantile form of this disease is very severe, and the child dies in first few months of life due to cardiac enlargement, that leads to cardiac failure. The juvenile form of this disease is also fatal, but the adult form is relatively milder, being marked by slowly developing myopathy.
Exercises
Essay type questions
1. Enumerate the rate regulating enzymes of glycolysis and glyconeogenesis. How are these enzymes regulated in a reciprocal manner?
2. Describe glycolysis and its regulation. Explain energetics of aerobic and anaerobic glycolysis.
3. List distinctive features of glucokinase and hexokinase. Explain why the high Km of glycokinase is important for the role of the liver in buffering blood glucose.
4. Describe the conditions under which the Cori’s cycle and the glucose-alanine cycle operate.
5. Describe the reactions of the pyruvate dehydrogenase multienzyme complex. How is entry of acetyl CoA into the TCA cycle regulated at this complex?
6. Mention the reactions of glycogenesis and glycogenolysis in flow diagram. Describe how these pathways are regulated. Describe the distinctive features of gly-cogen synthesis and degradation in liver and skeletal muscle.
7. With at least one example each related to glucose metabolism, discuss how enzyme activity is regulated by (a) allosteric mechanism, and (b) covalent modification.
8. Highlight the role played by protein kinases in glycogen metabolism. Explain the differences in glycogenolysis between liver and muscle.