38 NEUROBIOLOGY OF MENTAL ILLNESS
INTRODUCTION
Historical records show that mental illness has been recognised for millennia. This long history has been turbulent and many people with mental illness have been treated unfairly, often because of a lack of understanding about the condition. However, following the accidental discovery of effective medications in the 1950s much progress has been made in understanding the neurobiology of mental illness. Neurobiology is the study of the organisation of brain structures, the individual elements that form these structures and the processes both within and between these elements. Neurobiology can provide insights into the underlying physical mechanisms that are thought to mediate symptoms of mental illness and also help guide the development of effective treatments. Increased understanding of the actions of these medications in the brain has greatly enhanced our understanding of the mechanisms involved in the manifestation of symptoms of mental illnesses. However, despite enormous progress there is still much that remains unknown. The brain is an extraordinarily complex organ and it is virtually impossible to ascertain all the neurophysiological conditions that occur across the brain. Imaging sciences have made it possible to more accurately observe brain physiology and therefore our understanding of the pathophysiology of mental illness has increased. Thus the contemporary view of mental illness is now very different from that of earlier eras and while the pathophysiology is complex, our understanding of the neurobiology and pathogenesis of mental illness has been greatly enhanced.
New treatments are developed from an understanding of neurobiological processes and it is important for us to understand the underlying biological processes. In this chapter we cover the neurobiology of the more common mental illnesses such as anxiety disorders, mood disorders, psychotic illnesses, substance use disorders and eating disorders.
THE EPIDEMIOLOGY OF MENTAL ILLNESS IN AUSTRALIA AND NEW ZEALAND
Mental illness is one of the leading causes of non-fatal burden of disease and injury in Australasia. It is associated with increased exposure to health risk factors, poorer physical health and higher rates of death from many causes including suicide. Mental health problems are responsible for a large proportion of disability cases, incur high direct and indirect costs, result in large numbers of hospitalisations and impose a heavy burden of human suffering, including the stigmatisation of people with mental health problems and their families.
Mental health problems have an enormous impact on the health of Australia and New Zealand. When grouped, mental health problems are estimated to account for almost 5% of all general practitioner visits and it has been estimated that each year there are more than 300,000 hospitalisations due to mental-health-related diagnoses in Australia. Furthermore, mental or behavioural disorders are the underlying cause of death of more than 3000 Australians each year. In monetary terms, the direct cost of mental health problems has been estimated at over $3 billion per year.1
Evidence suggests that about 1 in 5 Australian adults will experience a mental illness over the course of their life.2 In addition, it has been estimated that yearly, 18% of Australian adults have had symptoms of a mental illness.2 Anxiety disorders were the most common problem, affecting 14% of the population. Mood disorders such as depression affected 6% of the population and bipolar disorder about 1.8%.3 Substance use disorders affected about 5% of the Australian population.3 The actions of the drug in substance abuse cases can have a direct effect on a person’s mental health. For instance, the development of psychotic symptoms from amphetamine use may occur, or substance abuse may negatively affect mental illness treatment and cause worsening of the symptoms. In contrast, co-morbid substance use in individuals with mental illnesses is quite common and in many cases this is an attempt to relieve the symptoms of the illness. Although it is probably one of the most recognised mental illnesses, schizophrenia is relatively rare in the population, with about 1.4% of the total population thought to have the disorder.1
In New Zealand the figures are similar to those for the Australian population. It has been estimated that 15% of the population experience anxiety disorders, 8% experience mood disorders and 3.5% have substance use disorders.4 Approximately 40% of the population have experienced a mental disorder at some time in their lives while 20% have experienced problems in the last 12 months. Therefore, across Australia and New Zealand, large numbers of individuals have or have experienced a mental illness.
NEUROTRANSMITTERS
Most of the current theories and a large component of the understanding of the aetiology of mental illnesses focus on problems with brain function and chemistry. As you will recall, the brain is responsible for higher functions such as reasoning, intelligence, personality and mood, and emotions. Sophisticated, coordinated neuronal activity within the brain structures enables this higher functioning, and the interplay between functionally discrete regions of the brain is likely to have an impact in mental illness. For instance, the amygdala is composed of a group of nuclei residing deep within the temporal lobe and is involved in emotional responses and the memory associated with these responses. It is a critical component of the limbic system, which also plays a major role in emotional control, and has connections with the hypothalamus and prefrontal cortex. The associations between structures such as these have been heavily implicated in the pathogenesis of mental illness. However, the communication between these regions is dependent on neurotransmission and the type and concentration of neurotransmitters is vitally important to brain functioning (see Chapter 6). To date, more than 50 neurotransmitters in the brain have been identified including dopamine, serotonin, noradrenaline, gamma (γ)-aminobutyric acid (GABA), and glutamate. However, the interaction between the neurotransmitters and receptor subtypes is complex and the aetiological implications in mental illness remain unclear.
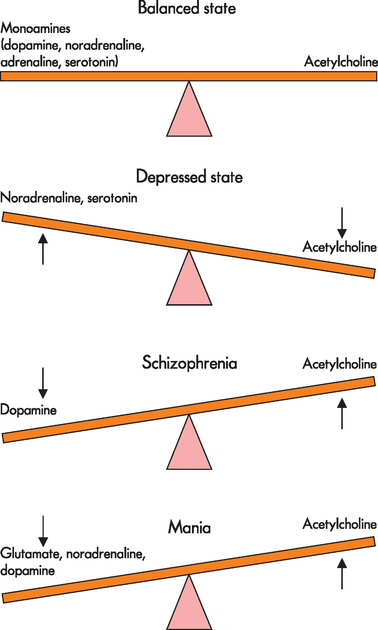
In many mental illnesses it has been found that particular regions of the brain have either an excess or a deficiency in the amount of neurotransmitter. In addition, receptor populations increase or decrease in response to the concentration of neurotransmitter or from other pathological changes. The effects of psychotropic (‘affecting the mind’) medication on individuals with mental illness caused researchers to hypothesise about neurotransmitter concentration and the pathogenesis of mental illness. For instance, the dopamine hypothesis arose from findings that demonstrated an improvement in the clinical manifestations of schizophrenia when psychotropic medication, which blocked dopaminergic D2 receptors, was taken. Furthermore, it was found that indirect dopamine agonists (drugs that increased the amount of dopamine) can result in psychosis in healthy individuals, suggesting that hyperactivity of dopamine transmission at the D2 receptor may be responsible for the pathophysiology of schizophrenia.5 However, this theory may oversimplify the pathophysiology, and upregulation of D2 receptors may also be involved. Nevertheless, despite limitations in the neurotransmitter hypotheses it is thought that neurotransmitter imbalances have a central role in mental illness pathophysiology and use of these hypotheses has greatly enhanced understanding of pharmacological therapies. A simplified version of some of the proposed neurotransmitter imbalances is shown in Figure 38-1.
CLASSIFICATION SYSTEMS
Diagnostic classification systems for mental illness have been in use since the 1850s when psychiatry first began as a specialist branch of medicine.
Two systems for the diagnosis of mental illness are currently in use. The Diagnostic and Statistical Manual of Mental Disorders, version four — text revision (DSM-IV-TR) is compiled and published by the American Psychiatric Association and is widely used in Australia and New Zealand.6 The International Classification of Diseases, 10th edition (ICD-10) is compiled and published by the World Health Organization.7 Both systems have more similarities than differences, with only minor variations in the names given to various disorders. The advantage of the ICD-10 is that it contains classifications for all diseases while DSM-IV-TR covers only psychiatric illnesses.
ANXIETY DISORDERS
One of the problems with the concept of anxiety is that it is a normal response. Anxiety is experienced when an individual feels threatened, evoking physiological responses that cause psychological and physical symptoms. The anxiety response prepares the individual for fight or flight and is thought to be a very primitive response present in most mammals as an adaptive evolutionary survival response.8 Common symptoms accompanying the anxiety response are listed in Box 38-1.
These symptoms are mediated predominantly by the autonomic nervous system. The initial physical symptoms can be very unpleasant and it has been suggested that this alerts the individual to act immediately to avoid danger.9 In many cases, the individual reacts and does not experience lasting effects from the symptoms. However, anxiety disorders arise when the symptoms impact negatively on the individual’s capacity to function normally. In 2007 approximately, 14% of Australian adults had an anxiety disorder; it is the most common mental health problem in Australia and New Zealand.3,4
The exact reasons why anxiety disorders develop in some people and not others are still the subject of conjecture, with a number of cognitive and neurobiological theories being advanced. Briefly, cognitive theories use mental (emotional and thinking) models while neurobiological models are constructed using brain structures and functions to explain symptoms. There is quite a wide range of anxiety disorders (see Table 38-1). Despite the differences between the symptoms of the various anxiety disorders they all share the common core symptoms of fear and worry.10 We shall now consider some of the more common anxiety disorders, namely obsessive-compulsive disorder, post-traumatic stress disorder and panic disorder. To provide an overview, the pathophysiology of fear and worry is discussed and related to the features of the anxiety disorders.
| TYPE OF DISORDER | DESCRIPTION |
|---|---|
| Acute stress disorder | Similar to the symptoms of post-traumatic stress disorder but occurs immediately after an extremely traumatic event |
| Agoraphobia | Anxiety generated when the individual feels that escape will be difficult from places or situations; causes avoidance of such places or situations |
| Generalised anxiety disorder | Excessive anxiety and worry that persists for 6 months or more |
| Obsessive-compulsive disorder | Obsessions that cause marked anxiety and/or compulsions which relieve the anxiety |
| Panic attack | A discrete period in which there is sudden onset of intense apprehension, fearfulness or terror. Symptoms are consistent with intense sympathetic nervous system activation and fear of losing control |
| Post-traumatic stress disorder | The re-experiencing of an extreme traumatic event with symptoms of increased arousal and avoidance of stimuli that trigger the trauma |
| Social phobia | Clinically significant anxiety brought on by exposure to social or performance situations which can lead to avoidance |
| Specific phobia | Clinically significant anxiety brought on by exposure to particular social, object or performance situations which can lead to avoidance behaviour |
Source: Modified from American Psychiatric Association (APA). Diagnostic and statistical manual of mental disorders. 4th edn, text revision, international version. Washington: APA; 2000.
THE PATHOPHYSIOLOGY OF FEAR AND WORRY
The neurobiology of the core symptoms of fear and worry is quite well understood. The factors that differentiate one anxiety disorder from another may not be due to anatomical localisation or specific neurotransmitters but to the specific nature of malfunction within the brain regions and circuits involved in fear and worry. It has been suggested that the risk of developing anxiety disorder symptoms is associated with genetic, psychological and biological variation.11
Fear is primarily mediated through the amygdala, a small almond-shaped area of brain tissue located near the hippocampus on the lower surface of the brain within the medial temporal lobe (see Figure 38-2). The amygdala is involved in fear, anxiety and aggression and forms part of the limbic system (see Chapter 6). It is connected to the medial temporal cortex, prefrontal cortex, thalamus, insula, hypothalamus and brainstem nuclei.10,12 These regions are involved in complex cognitive behaviours, termed executive functions, such as social control and personality. An increase in cerebral metabolic activity, particularly in the frontal cortex, has been shown repeatedly in patients with general anxiety disorder. This is thought to cause an excess production of neural activity generating signals that cause patients to feel anxious.13 In addition, the connections to the locus coeruleus, a nucleus within the brainstem, give rise to arousal and changes in autonomic functions (heart rate, breathing and blood pressure control). Collectively, these connections control emotions, giving rise to the subjective feeling of fear and the sympathetic activation (see Figure 38-3). In addition, there are neurohormonal responses that are mediated between the amygdala and the hypothalamus, which cause activation of the pituitary and adrenal glands. This activation is known as the hypothalamic-pituitary-adrenal axis (HPA) which is involved in the stress response, primarily through the release of cortisol and the catecholamines adrenaline and noradrenaline (see Chapter 10).
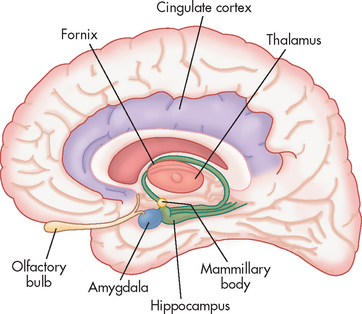
FIGURE 38-2 The amygdala and associated structures of the limbic system.
Source: McCance KL, Huether SE. Pathophysiology: the biologic basis for disease in adults and children. St Louis: Mosby; 2010.
Worry, which can include apprehension, catastrophic thinking, obsessional thinking and anxious misery, is thought to be mediated through feedback loops in the cortico-striatal-thalamic-cortical circuits (see Figure 38-4). These circuits connect different functional sections of the brain and are a major area of neurobiological research into mental health. The cortico-striatal-thalamic-cortical circuit involved in impulsivity/compulsivity (hallmarks of obsessive-compulsive disorder) originates in the orbital frontal cortex at the base of the frontal lobe.14 This connects with the base of the caudate then through the thalamus and back to the orbital frontal cortex. The caudate is involved in learning and memory and has been hypothesised to be dysfunctional when processing fear and worry signals, whereas the orbital frontal cortex participates in decision-making components and is therefore crucial to fear and worry interpretation. Signals in these circuits originate in neurons within the cortex. The outputs and actions within these circuits dictate how the response is processed. There are actually a number of different cortico-striatal-thalamic-cortical loops thought to be involved in operations such as executive function, attention and other functions. However, it is presumed that alterations to these circuits are integral to the development of an anxiety disorder. This may involve changes in neuronal activity (see Figure 38-5), neurotransmitter levels, pathological changes to the neural tissues or even autoimmune changes to the circuit.15 Therefore, pharmacological therapies target the neurotransmitters within these circuits and mitigate the perceived apprehension.
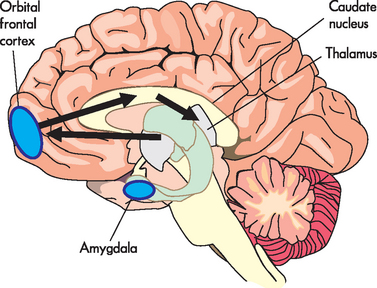
FIGURE 38-4 Cortico-striatal-thalamic-cortical loops.
These loops connect the caudate with the thalamus and the orbital frontal cortex which are associated with impulsivity and compulsivity.
Source: Modified from Stahl SM. Stahl’s essential psychopharmacology. 3rd edn. Cambridge: Cambridge University Press; 2008.
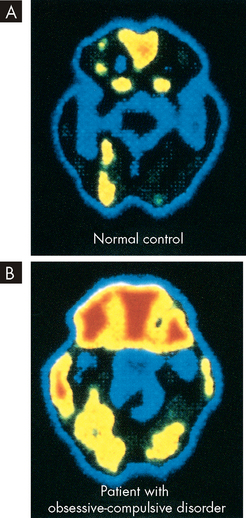
FIGURE 38-5 Hyperactivity of the orbitofrontal cortex in obsessive-compulsive disorder.
A Positron emission tomographic (PET) scan of an individual without obsessive-compulsive disorder. B Positron emission tomographic (PET) scan of an individual with obsessive-compulsive disorder with increased metabolic activity in the frontal and orbital cortex.
Source: Copstead L-EC, Banasik JL. Pathophysiology. 4th edn. St Louis: Saunders; 2009.
Obsessive-compulsive disorder
As the name suggests the main feature of obsessive-compulsive disorder is the presence of obsessions and/or compulsions that are recognised as excessive and interfere with the person’s normal routine. This is an important differentiation from, for example, the normally neat and tidy person. Some obsessionality can be healthy and useful, as seen in the behaviour of the neat and tidy individual, but it becomes obsessive-compulsive when the individual makes irrational changes to normal daily routines. Obsessions are recurrent and persistent thoughts, impulses or images that are experienced as intrusive and inappropriate and cause marked anxiety. The thoughts, impulses or images are not simply excessive worries about real-life problems which the person attempts to ignore or suppress or to neutralise with some other thought or action.16 In addition, the person recognises that the obsessional thoughts, impulses or images are products of their own mind. Examples of obsessional thoughts are illustrated in Box 38-2.
With compulsions the person feels driven to perform repetitive behaviours of mental acts in response to an obsession or according to rules that the person deems must be rigidly applied.16 The behaviours or mental acts are aimed at preventing or reducing distress or preventing some dreaded event or situation; however, these behaviours or mental acts either are not connected in a realistic way with what they are intended to neutralise or prevent or are clearly excessive. Common examples of compulsions are listed in Box 38-3.
Obsessive-compulsive disorder can be extremely disabling. Some people have enormous difficulty making even very minor decisions or can be very distressed by their thoughts while others spend extraordinary amounts of time engaging in rituals. This disorder often appears in childhood or adolescence and frequently family members are coerced into facilitating the rituals that commonly accompany this disorder.
A comprehensive mental health assessment to gather information is required to ascertain the nature of the anxiety. Cognitive behavioural therapy involving exposure and response prevention is a well-established treatment for obsessive-compulsive disorder.17 This therapy involves teaching the person relaxation techniques which will oppose the ritualistic response to their obsessive thinking. Medications, primarily selective serotonin re-uptake inhibitor antidepressants, and tricyclic antidepressants are also used for the treatment of obsessive-compulsive disorder.18
Post-traumatic stress disorder
Post-traumatic stress disorder (PTSD) is unique among the anxiety disorders in that it usually has an acute onset following exposure to some form of trauma. The person may have been functioning well prior to the traumatic event but may become quite disabled following this event. The trauma may be a single event such as being involved in or witnessing an accident, being attacked or threatened with attack (e.g. being exposed to an armed hold-up while working in a bank), or it may be a series of traumas over a short or long period of time such as the traumas experienced by soldiers in the battlefield or by paramedics or police who deal with accident victims on a daily basis. The symptoms of PTSD can last for many years, even an entire lifetime. Symptoms can include nightmares, flashbacks where the event comes vividly back to mind without any apparent stimulus, hypervigilance, insomnia, exaggerated startle reflex, persistent anxiety, difficulty concentrating and emotional numbing.
The exact neurobiology of PTSD is complex; evidence shows that the hippocampus is reduced in volume and the medial prefrontal cortex is not activated, while the amygdala neuronal activity is increased during episodes of stress.19 This is consistent with the cortico-striatal-thalamic-cortical circuit alteration.
Panic disorder
Panic disorder is literally a fear of having panic attacks. The disorder usually develops after a person has experienced a panic attack without any apparent stimulus. Panic attacks can be very frightening and distressing and people often present to hospital emergency departments. Panic attacks can affect people to the extent that they become agoraphobic.
It is suggested that the neurobiological basis of panic attacks is a result of increases in sympathetic activation and physiological instability which lead to apprehension, fearfulness and the fear of losing control.20 The symptoms of panic disorder are mostly physical and are mediated by the autonomic nervous system (see Figure 38-6). The enhanced sympathetic activation sets off a cascade of physiological events which cause the individual to sense more panic. For instance, increased ventilation causes the costal muscles of the chest to increase contractions which leads to feelings of tightness and pain. The person may also have feelings of impending doom, extreme distress, fear and worry.
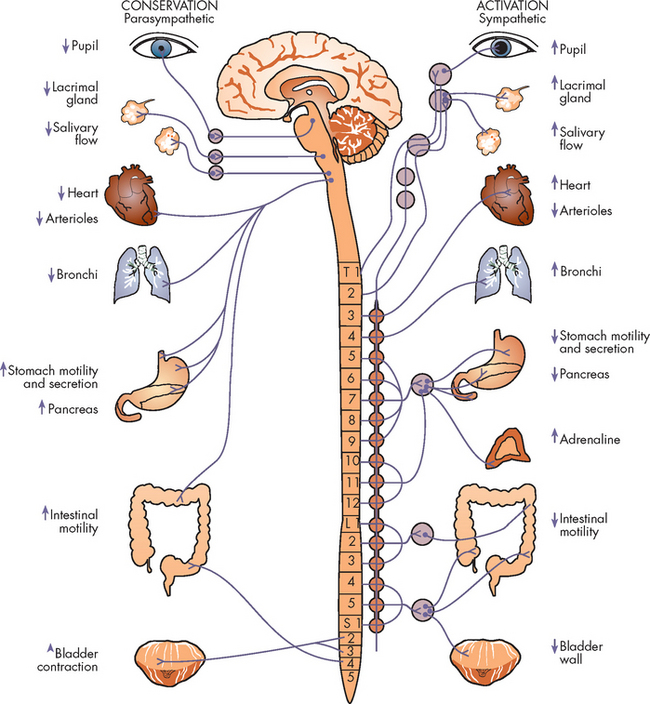
FIGURE 38-6 Heightened anxiety causes activation of the sympathetic nervous system.
Source: Stuart GW. Principles and practice of psychiatric nursing. 9th edn. St Louis: Mosby; 2008.
Treatment consists of situational in vivo exposure and cognitive behavioural therapy for panic disorder where mild agoraphobia is present. These therapies focus on cognitive strategies, exposure to interceptive sensations similar to physiological panic sensations, and breathing. Selective serotonin re-uptake inhibitor antidepressants are the first-line pharmacological treatment for panic disorder.21
MOOD DISORDERS
The term ‘mood disorders’ is used to describe a group of disorders in which the primary symptom is a disturbance of mood; it includes unipolar depression, dysthymia, bipolar depression and mania. The term bipolar affective disorder (BAD) is used where there is a history of fluctuation of mood from depression to mania. Co-morbidities are very common in people with depression, with many people experiencing anxiety and engaging in alcohol misuse.22 Less commonly (about 10% of all cases), people may have psychotic symptoms along with their depression. In these cases this is considered psychotic depression. There is also a disorder known as schizo-affective disorder, which is similar to psychotic depression but which has a preponderance of psychotic symptoms with either depressive or manic symptoms.
There are two major divisions with mood disorders: unipolar depression and bipolar affective disorder. These terms refer to the episodic properties of the mood disorder. Unipolar or major depression is the most common mood disorder presentation and is associated with a range of disorders such as coronary heart disease, diabetes mellitus and obesity.23 The term unipolar infers that the mood is displaced in one direction only, whereas bipolar affective disorder has characteristics of both depression and mania. There are two types of bipolar affective disorder: type I and type II. Type I is characterised by a manic episode and at least one depressive period which do not have to follow each other. Type II is characterised by at least one hypomanic episode and at least one major depressive period. Hence bipolar affective disorder is also referred to as manic depressive illness. Unipolar depression and bipolar depression are very similar in symptom presentation and it is impossible to differentiate the two without a longitudinal history. Bipolar depression is diagnosed when there is a history of mood fluctuations from depression to mania. In the absence of any history of manic episodes unipolar depression is the diagnosis.
PATHOPHYSIOLOGY
As with many mental illnesses there are a number of theories for the aetiology of mood disorders. There is strong evidence of a genetic factor in the development of mood disorders; however, the causative genes or robust genetic risk factors have not been identified.24,25 Therefore, many have suggested that gene–environment interaction is the trigger for the pathogenesis of mood disorders. (The complex nature of genetics and environmental risk factors is discussed in Chapter 37.) Evidence for an interaction between the serotonin transporter genotype and depression has been reported. There are two variants of the serotonin transporter gene: one with a short allele, one with a long allele. It has been shown that individuals with the short allele have reduced gene transcription and the serotonin transporter activity is diminished, thereby increasing the level of serotonin in the synapse. This has been associated with a greater rate of depression when these individuals are exposed to a stressful event.26,27 Moreover, depression and bipolar affective disorder have been shown to have a strong familial link.28 Therefore, the potential for gene–environment interplay in the development of mood disorders is high and current research is examining this relationship.
There are also a number of psychological, psychodynamic, learning and behavioural theories for the aetiology of depression. The dominant biological theory for the aetiology of depression is known as the monoamine hypothesis.29 In the past it was thought that a decrease in the neurotransmitters caused the development of mood disorders. This arose from studies which showed that some individuals who were taking drugs which depleted monoamines experienced depression. Monoamines are one of the main types of neurotransmitter in the brain, and those principally involved in depression and mania are serotonin, dopamine and noradrenaline. Therefore, it was proposed that a decrease in monoamine receptor activity caused mood disorders. However, this thinking has been modified over the years and it is now hypothesised that deficient activity in these neurotransmitters causes upregulation (an increase in the number and density) of receptors, which causes changes in gene expression within a neuron that leads to depression or mania. One candidate for altered gene expression is brain-derived neurotrophic factor which is responsible for sustaining the neuronal viability. However, under stress, the brain-derived neurotrophic factor gene may be suppressed, leading to atrophy and apoptosis of neurons, especially in the hippocampus.30 Therefore, the monoamine receptor hypothesis arose, which states that the monoamine receptors are modified in mood disorder pathogenesis and that this may be mediated by lowered neurotransmitter levels.
As with anxiety, it is hypothesised that the symptoms of mood disorders are linked to specific brain circuits. The serotonin and noradrenaline systems have been implicated (see Figures 38-7 and 38-8) and it has been hypothesised that there may be inefficient information processing in these circuits. The serotonin system originates in raphe nuclei in the brainstem and projects extensively throughout the brain. It has been shown that serotonin receptors have been down-regulated in individuals with depression and that the system is likely to be implicated in the pathophysiology of mood disorders. Similarly, the noradrenaline system contains a collection of neurons in the locus coeruleus of the brainstem with multiple projections throughout the brain. Evidence suggests that both the noradrenaline and serotonin systems are implicated in the onset of depression. However, it remains unknown if the level of neurotransmitter or regulatory changes in the number and type of noradrenaline or serotonin receptors is the causative agent in mood disorders.31
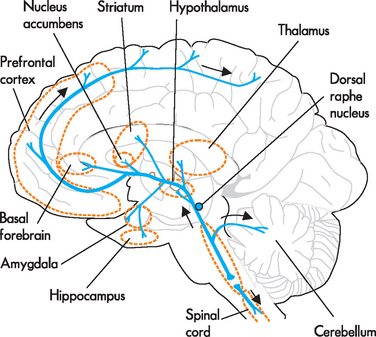
FIGURE 38-7 Serotonin pathways.
Serotonin neurons are located in the brainstem raphe nuclei. They project diffusely to all regions of the cortex, limbic regions, hypothalamus, basal nuclei, cerebellum and spinal cord.
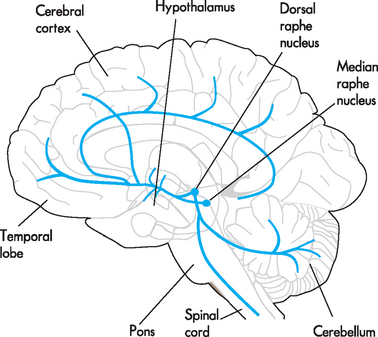
FIGURE 38-8 The noradrenaline system.
The noradrenaline cell bodies originate in the locus coeruleus and project throughout the brain. The projections include the hypothalamus, temporal lobe, entire cerebral cortex, cerebellum and spinal cord.
Source: McCance KL, Huether SE. Pathophysiology: the biologic basis for disease in adults and children. St Louis: Mosby; 2010.
Depression and dysthymia
Depression is a sustained state of low mood along with other symptoms. Dysthymia is diagnosed when there is a sustained low mood without the full range of depressive symptoms and is sometimes called sub-syndromal depression.
CLINICAL MANIFESTATIONS
The primary symptom of depression is a sustained low mood. The low mood must be present over the entire day for at least two weeks. The other primary symptom is loss of pleasure or enjoyment. Feelings of hopelessness, helplessness, worthlessness, inappropriate guilt, poor concentration and lack of motivation are also frequently present. Physical symptoms include lack of energy, insomnia, reduced libido, motor slowing or agitation and loss of appetite and associated weight loss.
Dysthymic disorder is characterised by a sustained low mood present most of the time for at least two years and at least one of the other symptoms of depression described above, but without meeting the criteria for major depressive disorder.
EVALUATION AND TREATMENT
Selective serotonin re-uptake inhibitors (SSRIs) are the newest class of antidepressants and tend to be used as first-line treatment in most new cases of depression. Their therapeutic effect is to increase serotonin levels by blocking the re-uptake of serotonin into the presynaptic neuron (see Figure 38-9).
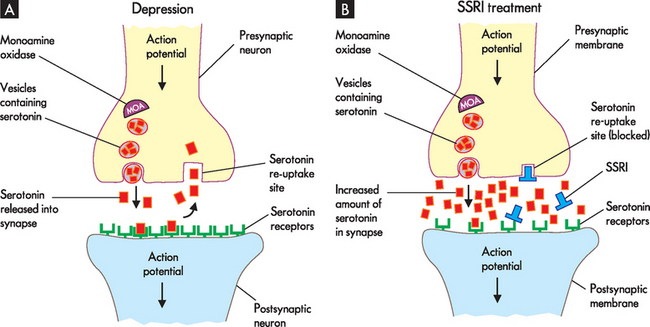
FIGURE 38-9 Action of selective serotonin re-uptake inhibitor drug.
A In individuals who have depression, the amount of neurotransmitter is reduced and the number of postsynaptic receptors increases (up-regulation) in response to lower levels of neurotransmitter. Re-uptake of serotonin occurs which contributes to the decreased amount of neurotransmitter in the synapse. B When patients are treated with selective serotonin re-uptake inhibitors (SSRIs), serotonin levels increase as the re-uptake is blocked. Treatment with SSRIs will also cause the number of postsynaptic receptors to decrease over time (down-regulation) due to an increased availability of neurotransmitter.
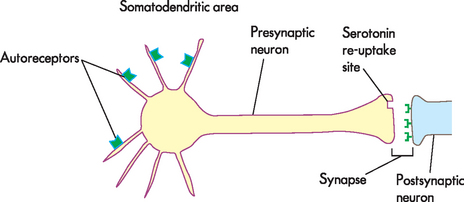
As you will recall, it is hypothesised that people with depression have up-regulated or an increased number of receptors, involving the presynaptic and postsynaptic receptors (receptors located on the presynaptic and postsynaptic neurons, respectively). In addition, autoreceptors are located on the presynaptic membrane and interact with neurotransmitters released by the same neuron. They do not induce changes in the membrane potential, but rather they function to control synthesis and neurotransmitter release. In depression, it is thought that autoreceptors detect serotonin levels in the immediate environment of the neuron and inhibit action potentials when elevated levels of serotonin are detected. Reduced neuronal activity causes a reduction in serotonin in the synapse. SSRIs increase serotonin by blocking at serotonin-re-uptake sites but also at the somatodendritic area of the neuron (see Figure 38-10). The autoreceptors at this site are desensitised or down-regulated. The effect takes days and explains why there is a delay in the onset of therapeutic action. Once the autoreceptors are down-regulated, nerve transmission through the neuron is normalised and the serotonin level at the synapse is also normalised, thereby causing an elevation in mood. As a consequence, postsynaptic receptors down-regulate, which establishes neurotransmission homeostasis (see Figure 38-9).
Psychological therapies are also effective with depression, particularly cognitive behavioural therapy and interpersonal therapy. A combination of medication and psychological therapies may be more effective than either therapy on its own.
Bipolar affective disorder
Bipolar affective disorder is characterised by significant swings in mood from depression to mania. In some people, bipolar affective disorder is characterised by manic episodes only; however, it is more common as depressive-manic swings. The depressive phase of bipolar affective disorder has the same symptoms and presentation as major depressive disorder. Bipolar affective disorder is not common and occurs in only about 1% of the adult population.32 Manic symptoms range from mild to severe and most people fall into the range known as hypomania; that is, moderately severe symptoms.
CLINICAL MANIFESTATIONS
The primary symptom of mania is an elevated, expansive, sometimes irritable mood. This is accompanied by grandiose thinking, increased energy, decreased need for sleep, inattention to basic needs and occasionally psychotic espisodes. Flight of ideas and pressure of speech are common and thought processing can be impaired.
EVALUATION AND TREATMENT
Elevated or euphoric mood is the key symptom in mania. Symptoms need to present for at least three days to differentiate between mania and drug intoxication. Stimulant drugs such as amphetamines, methamphetamine, cocaine and ecstasy can produce a mania-like state. However, a combination of bipolar affective disorder and substance-use disorders are common in individuals. In fact, of all mental illnesses, substance-use disorders have the highest prevalence in bipolar affective disorder.33
Lithium carbonate was the first effective psychiatric medication to be used. The exact mechanism of action remains unknown. However, evidence suggests that it alters sodium transport across the cell membrane by interfering with the sodium–potassium pump, thereby restricting the influx of sodium into the cell. It may also inhibit intracellular second messengers, such as adenylate cyclase or phosphatidylinositol, thereby preventing the neuron’s response to neurotransmitters, such as dopamine and noradrenaline.34 Nevertheless, lithium is often very effective in the treatment of bipolar affective disorder as it restores mood imbalances.
The primary problem with lithium is the narrow therapeutic range which requires constant monitoring of blood serum levels. Lithium toxicity can be a life-threatening condition which affects not only the central nervous system but also the renal, gastrointestinal, endocrine and cardiovascular systems.
Antipsychotic medications are often used in the acute stages of a manic episode due to the presence of psychotic symptoms. Benzodiazepines can also be used, with the added benefit of inducing sleep.
The range of symptoms presenting in bipolar affective disorder may include psychotic symptoms, depression and mania and each of these symptoms requires a different pharmacological approach.
SCHIZOPHRENIA
Schizophrenia is a low-prevalence mental illness that affects approximately 1.4% of the Australian population.3 Schizophrenia appears in all cultures across the world and has been recognised since the early 1900s. While the prevalence is low, especially compared to anxiety disorders, the effects can be devastating for the individuals and families concerned. It accounts for the largest proportion of hospitalised days and has been estimated to cost $1.85 billion per year in healthcare.2,35
While schizophrenia is commonly referred to as a single illness, it is more correct to conceptualise schizophrenia as a group of illnesses with similar symptoms but with varying causes, outcomes and responses to treatment. Approximately 25% of people who present with psychotic symptoms will recover, while another 25% will have an episodic course of illness with periods of relatively good mental health. The remaining 50% have either a continuing illness with fluctuating symptom severity and reasonably mild disability or a continuing illness with more severe disability. A small percentage of people with schizophrenia do not respond to any treatments.36
People with schizophrenia exhibit psychotic symptoms and the terms psychosis and schizophrenia tend to be used interchangeably. Schizophrenia is a time-based diagnosis and symptoms need to be present for at least six months before the diagnosis can be made.6 Therefore, in the early stages of schizophrenia the terms psychotic symptoms or psychosis are often used.
There are a number of subtypes of schizophrenia although there is little agreement about the utility of these subtypes. Nevertheless, the most common subtypes are paranoid schizophrenia, catatonic schizophrenia, disorganised schizophrenia and undifferentiated type. There are also some sub-syndromal forms of schizophrenia known as schizophreniform disorder and schizotypal personality. These refer to presentations where there are some low-intensity psychotic symptoms on a background of odd, eccentric or somewhat bizarre behaviour and thinking. Another psychotic disorder is known as delusional disorder; the primary feature is intensely held, non-bizarre delusions but with relatively intact functioning and behaviour.
PATHOPHYSIOLOGY
The aetiology of schizophrenia is complex. No single cause has been identified and it is likely, given the heterogeneity of the schizophrenia phenotype, that multiple aetiological factors are responsible. There is a strong genetic influence in schizophrenia with studies showing a strong familial relationship. Monozygotic (identical) and dizygotic (fraternal) twin studies have revealed a higher incidence rate of schizophrenia than in the general population (see Table 38-2). However, while the monozygotic twin incidence rate is high, inferring that the genetic influence on aetiology is strong, it is not absolute, and identification of an isolated gene interaction has not been found. Indeed, it is presumed that multiple, potentially interacting genes with small effects may be more likely in the development of schizophrenia.5,37
Table 38-2 GENETIC RISK FOR SCHIZOPHRENIA
| RELATIONSHIP | RISK OF DEVELOPING SCHIZOPHRENIA |
|---|---|
| Identical twin affected | 50% |
| Fraternal twin affected | 15% |
| Brother or sister affected | 10% |
| One parent affected | 15% |
| Both parents affected | 35% |
| Second-degree relative affected | 2–3% |
| No affected relative | 1% |
Source: Modified from Roberts GW, Leigh PN, Weinberger DR. Neuropsychiatric disorders. London: Mosby Europe; 1993.
Despite the strong implication of genetic factors, evidence suggests that environmental factors also have a role. It has been shown in large population-based studies that being born in urban environments and around late winter and early spring has a strong association with schizophrenia.38 In addition, a neurodevelopmental aetiology has been proposed with the possibility that in utero exposure to different factors may precipitate the development of schizophrenia. These include infectious agents such as influenza virus, immunological changes (elevated interleukin-8) or nutritional alterations.39–42 40 41 42
In addition, much research has explored neuroanatomical changes in patients with schizophrenia. Neuroimaging has shown a number of changes in the brains of people with schizophrenia but the significance of these changes is poorly understood. It has been demonstrated repeatedly in magnetic resonance imaging (MRI) scans that the ventricular volume in people with schizophrenia is enlarged (see Figure 38-11). Moreover, autopsy studies of schizophrenic patients have shown altered gene expression responsible for myelin and oligodendrocytes.37 The area of the brain in which this was evident was the dorsolateral prefrontal cortex, which has been implicated with a loss of grey matter. It is suggested that the reduction in myelin may lead to problems with electrical insulation between neurons and, therefore, abnormal neuronal activity in particular brain regions.37 Further research is required to explore this possibility.
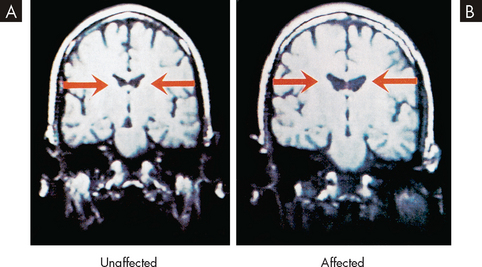
FIGURE 38-11 Enlargement of ventricular volume in patients with schizophrenia.
A MRI scan of a 44-year-old twin without schizophrenia. B Scan of the twin with schizophrenia. Note the ventricles of the person with schizophrenia are larger, suggesting structural brain changes associated with the illness.
Source: Copstead L-EC, Banasik JL. Pathophysiology. 4th edn. St Louis: Saunders; 2009.
The dominant neurotransmitter hypothesis in the neurobiology of schizophrenia is the dopamine hypothesis.43 The hypothesis arose initially when a strong correlation was found between antipsychotic drugs and the affinity for dopamine receptors, specifically type D2 receptor. In addition, it was found that dopamine agonists such as cocaine and amphetamines are able to induce psychosis in people without evidence of mental illness.5 However, since that finding, it has been suggested that up-regulation of D2 receptor expression (appearance of more D2 receptors on the cell) may explain these findings, rather than altered level of neurotransmitter.
There are different dopamine pathways in the brain and it is thought that these pathways are directly involved in positive and negative symptoms of schizophrenia. They include the mesolimbic, mesocortical and nigrostriatal pathways (see Figure 38-12). The mesolimbic pathway is thought to be involved in production of the positive symptoms of schizophrenia (see ‘Clinical manifestations’ below) through an overactive dopamine system. In contrast, the mesocortical pathway, which connects the ventral tegmental area to the nucleus accumbens, is thought to be involved with production of negative symptoms (see ‘Clinical manifestations’) through dopamine underactivity.44,45 The nigrostriatal pathway is involved with extra-pyramidal side effects, such as tremor, slurred speech and dystonia.
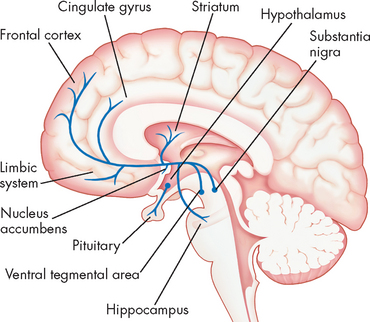
FIGURE 38-12 Dopamine pathways throughout the brain.
Dopamine cell bodies are located in the substantia nigra, where they project to the stratum (nigrostriatal pathway); and in the ventral tegmental area where they project to the frontal and cingulated cortex (mesocortical pathway). The striatum, hippocampus and limbic structures form the mesolimbic pathway. Dopamine nuclei are also located in the hypothalamus and project to the pituitary gland.
Source: McCance KL, Huether SE. Pathophysiology: the biologic basis for disease in adults and children. St Louis: Mosby; 2010.
CLINICAL MANIFESTATIONS
Symptoms of schizophrenia have been classified and grouped in different ways over time. The general consensus now is that symptoms are referred to as positive and negative. Positive symptoms are an exaggeration or distortion of normal function and include thought disorders such as delusions and illogical or incoherent thinking, perceptual disturbances such as hallucinations, and disorganised speech and behaviour. Negative symptoms are a diminution of normal function and include apathy, lack of motivation and energy, inability to initiate action, poor concentration, emotional blunting, reduced thought and speech content, lack of pleasure, inability to maintain social relationships and sleep disturbance, mainly hypersomnia.46
The onset of symptoms in schizophrenia can be quite slow or very rapid. The more usual course is for symptoms to appear gradually over time with vague, strange ideas and behaviours the first indicator of problems. A prodromal phase has been described which consists primarily of negative symptoms. However, this concept has limited utility as a predictor of patients who progress to psychosis. The severity of symptoms in schizophrenia varies enormously.
EVALUATION AND TREATMENT
The primary treatment for psychotic symptoms is antipsychotic medications. These medications were first used in 1953 when chlorpromazine (Largactil) was introduced. This was the first medication found to be effective in reducing psychotic symptoms and although its antipsychotic actions were discovered serendipitously, understanding of its actions led to the development of other antipsychotic medications.
There are two broad classes of antipsychotic: first generation (older) or typical antipsychotics such as chlorpromazine, haloperidol and trifluoperazine and second generation (newer) drugs or atypical antipsychotics such as risperidone, olanzapine and quetiapine. The latter are now the preferred type. Both classes are equally effective in reducing positive symptoms; neither class is really effective in reducing negative symptoms, although it is thought that atypical antipsychotics may not have as much negative impact on this group of symptoms.
The primary antipsychotic effect of the typical antipsychotics is due to their capacity to antagonise (or block) dopamine D2 receptors. A range of dopamine receptors has been identified but it appears that only D2 are involved in psychotic symptoms. These medications are not selective and antagonise or block all dopamine receptors throughout the brain and body. They also block many other types of receptors including histaminergic, α-adrenergic and muscarinic receptors.
Atypical antipsychotics also antagonise serotonin receptors and it is thought that this additional action is what helps to change the side effect profiles of these medications. There is a complex interaction between dopamine and serotonin neurons in the brain which can increase or dampen the actions of either type of neuron, depending on location and usual function. It is thought that serotonin receptors in the nigrostriatal pathway, responsible for extra-pyramidal side effects, are blocked with atypical antipsychotics, which allows dopamine receptors to function normally. That a similar action in the mesocortical pathways may reduce the negative effects of dopamine blockade on an already underactive system has also been proposed.
Side effect profiles are the main differentiating factor between these classes of medications. The typical medications tend to cause more movement and muscle disorders, known as extra-pyramidal side effects, which can be extremely uncomfortable and distressing. These often manifest in ways similar to Parkinson’s disease (Chapter 9). Long-term use of these medications can produce tardive dyskinesia, a permanent movement disorder mainly affecting the face, neck and trunk. These medications also have other adverse effects such as blurred vision, dry mouth and constipation.
The atypical medications can cause serious side effects. While they have less propensity to cause extra-pyramidal side effects they are known to produce increased appetite leading to weight gain, insulin dysregulation and increased prolactin levels. Weight gain and insulin dysregulation are risk factors for cardiometabolic syndrome which can lead to significant cardiac problems. Clozapine, the original atypical antipsychotic, has an additional risk for agranulocytosis, a disorder of lymphocytes and neutrophils which diminishes the immune response and can be fatal if not detected and treated quickly. This occurs in approximately 1% of people taking clozapine. For this reason, anyone taking clozapine must have regular monitoring of their lymphocyte and neutrophil levels and because of the expense involved in this, clozapine is reserved for those who are treatment resistant. Clozapine has the most complex binding properties of any antipsychotic and the exact mechanisms involved in weight gain, insulin dysregulation and other side effects are not well understood.
Smoking in schizophrenia
It has been recognised for many years that many patients with schizophrenia are heavy tobacco smokers. The reason is thought to arise from the biochemical changes induced with nicotine. Dopamine axons from the ventral tegmental area to the nucleus accumbens are involved in the reward pathway and nicotine increases dopamine release at the nucleus accumbens. In addition, nicotine affects neural signals in the mesocortical pathway thereby affecting the prefrontal cortex. It is presumed that negative symptoms are associated with reduced levels of dopamine and nicotine may negate these symptoms. Therefore, if nicotine increases dopamine in these areas and the dopamine pathways, the negative symptoms of schizophrenia may be diminished and patients are treating themselves despite the systemic deleterious health effect of smoking.
Source: Nisell M, Nomikos GG, Svensson TH. Nicotine dependence, midbrain dopamine systems and psychiatric disorders, Pharmacology and Toxicology 2009; 76:157–162; Zhang T et al. Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine. Journal of Neuroscience 2009; 29(13):4035–4043.
An important addition to medication is psychosocial interventions that enhance people’s social role and functioning. Psychosocial interventions focus on areas such as vocation, employment, education, social, leisure and life skills. Good social interventions can significantly enhance people’s quality of life and overall functioning. Cognitive behavioural therapy is also used to help people manage delusions and hallucinations more effectively.47
SUBSTANCE ABUSE
Substance abuse is a large and complex topic with many aspects that may be considered. This section focuses primarily on the pathophysiology and neurobiology of addiction rather than the myriad other issues. Substance abuse is common in Western societies and some aspects of substance use are considered to be socially acceptable, for example, alcohol consumption. Yet, alcohol abuse accounts for a significant burden on society and the healthcare system. Australia and New Zealand have two of the highest rates of alcohol consumption in the English-speaking world, with only Ireland and the United Kingdom having higher rates. Australians consume an average of 9.9 litres of alcohol annually while New Zealanders consume 9.97 and the Irish consume 14.45 litres.48
Substance abuse includes both illicit and licit substances. Legal drugs such as alcohol, caffeine and tobacco, prescribed and over-the-counter medications such as benzodiazepines, opiates, antihistamines and asthma medications are all subject to abuse. Illicit drugs can be classified according to their actions, that is, depressants, stimulants and hallucinogenics. Heroin and other opiates are examples of depressants while amphetamines, cocaine and ecstasy are examples of stimulants. LSD is a hallucinogenic. Some drugs such as marijuana can fit into all three classes. Also abused are naturally occurring drugs such as mushrooms with hallucinogenic compounds, often referred to as magic mushrooms, cacti which contain psychoactive compound (peyote) and inhalants such as petrol and glue.
Alcohol misuse can cause serious problems, eventually resulting in death if sufficient alcohol is ingested. This can be a particularly serious problem in young people who binge drink and are not aware of the risks involved.
THE PATHOPHYSIOLOGY OF REWARD AND ADDICTION
The mesolimbic dopamine pathway is hypothesised to be the final common pathway (meaning that regardless of what initiates the pathway, the end is the same) of reward and reinforcement in the brain. This pathway originates in the ventral tegmental area of the brainstem and terminates in neurons in the nucleus accumbens in the limbic system (see Figure 38-13). The mesolimbic dopamine neurons release dopamine in response to numerous stimuli. Natural highs can be triggered through accomplishments such as academic or sporting success, enjoying music or experiencing an orgasm. The brain has its own pharmacy of neurotransmitters that mediate these natural highs.
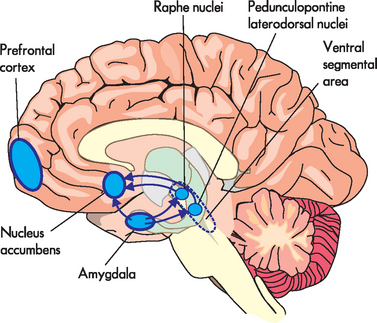
FIGURE 38-13 Common reward pathway.
The amygdala is connected to both ends of the mesocortical dopamine pathway.
Drugs of abuse bypass these natural neurotransmitters and act directly on the mesolimbic pathways. Unlike a natural high, a drug-induced high causes an increase in dopamine at the limbic dopamine receptors. The amygdala is also involved with connections at both ends of the mesolimbic dopamine pathway. The amygdala is an important site of emotional learning and learning about reward. The connections from the ventral tegmental area to the amygdala cause the amygdala to develop changes that condition it to remember the rewards of drug abuse. The connection between the amygdala and the nucleus accumbens is involved in memory, which is thought to be triggered by stimuli related to the substance abuse. Repeated exposure to drugs creates pathological learning that effectively hijacks the entire reward system and addiction develops. Therefore, people in this state are unable to make decisions that take into account the long-term consequences of their behaviour.
CLINICAL MANIFESTATIONS
Given the wide range of drugs of addiction, abuse and misuse it is not possible to list all the clinical manifestations involved. Rather, a generic description of what constitutes substance abuse is provided. The DSM-IV-TR diagnostic criteria define substance dependence as:
… a maladaptive pattern of substance use leading to clinically significant impairment or distress as manifested by one (or more) of the following occurring within a 12-month period:
One of the paradoxes of drug dependence is the persistent use of substances despite the negative consequences involved. Often these negative consequences contradict the original motive for using the drug. As explained in the pathophysiology of reward, continuing drug use sets up a vicious cycle where rational thinking may not be possible.
There is a wide range of mind-altering substances that are used for a variety of reasons. Each class of drugs has a different effect, ranging from increased energy and feelings of elation to sedation and calming effects. Table 38-3 provides more information about a range of commonly used drugs.
Table 38-3 SOCIAL OR STREET DRUGS AND THEIR EFFECTS
Source: Modified from Cotran RS, Kumar V, Collins T. Robbins pathologic basis of disease. 7th edn. Philadelphia: Elsevier/Saunders; 2005; Nahas G, Sutin K, Bennett WM. Review of marijuana and medicine. N Engl J Med 2000; 343(7):514.
EVALUATION AND TREATMENT
A full drug use history includes information about the types of drugs used, when last used, methods of ingestion or administration, the length and frequency of use, quantities used, positive and negative social and personal consequences, and effects on mental and physical health.
Pharmacological treatments for drug use disorders are available to help people withdraw from drugs by reducing unpleasant side effects. Nicotine replacement therapies help reduce the unpleasant effects of nicotine withdrawal and newer medications such as varenicline, a nicotinic partial agonist, work directly on nicotine receptors on the dopamine neurons in the ventral tegmental area of the brainstem. Benzodiazepines and antipsychotics are used in alcohol withdrawal. Other treatments, such as methadone as a maintenance treatment for heroin addiction, provide safer substitutes for the harmful drug. Codeine and naltrexone are also used to help people withdraw from opiates.
The issue of abstinence versus harm minimisation is somewhat contentious and ultimately comes down to the needs of the individual and their capacity to manage their drug use.
Abstinence programs such as Alcoholics Anonymous and Narcotics Anonymous do not suit everyone but can be very effective. Regular attendance at meetings and support from fellow addicts can help keep people abstinent from drugs and alcohol for life or for very long periods of time.
EATING DISORDERS
Many young adults and adolescents are affected by two complex and related eating disorders: anorexia nervosa and bulimia nervosa. Both conditions have a familial tendency and may be associated with other disorders such as anxiety, depression and obsessive-compulsive disorder.49
Anorexia nervosa is a psychological and physiological syndrome characterised by extremely low food intake and a low body weight. It usually develops during adolescence, but is rare, occurring in only 0.2 to 0.5% of women, and is less common in men.50 Although this disorder is not very common, it often receives media attention in Australia and New Zealand and therefore is known within the community. The incidence of anorexia nervosa appears to be increasing in Australia.51
Anorexia nervosa is characterised by the following:
Issues such as personal self-esteem, psychological control and the patient’s perception of their control over their own body contribute to the complexity of this disorder.52
Persons with anorexia nervosa often deny they have an eating problem. As the disease progresses, muscle and fat depletion give the individual a skeleton-like appearance. Postural hypotension, oedema, bradycardia, hypothermia, constipation and sleep disturbances may ensue. The loss of 25% to 30% of ideal body weight can eventually lead to death caused by starvation-induced cardiac failure.
Diagnosis of anorexia nervosa involves a thorough medical history, a physical and psychological examination and ruling out other causes of anorexia and malnutrition. Body weight is monitored through the course of this disorder; some patients will be intentionally deceptive with practices such as consuming a large amount of water prior to weight checks, so alternative measures (such as arm circumference) may more accurately reflect the patient’s progress.53
Due to the complex psychological nature of this disorder, a lenient approach to restoring weight is usually received better by patients than a more disciplinary approach.50 Treatment objectives for anorexia nervosa include reversing the compromised physical state, setting mutual goals, promoting interaction with family members, restoring developmental growth, modifying food habits and restoring weight. Correction of nutritional status can require hospitalisation. When the individual demonstrates willingness to eat food for nourishment, dietary protein, carbohydrate and fat are introduced in tolerable amounts. Psychotherapy begins as soon as the physical symptoms are stabilised and may continue for several years.54 Patients who are experiencing accompanying depression may also require antidepressant medications.50
Bulimia nervosa is characterised by bingeing — the consumption of large amounts of food at a time — followed by self-induced vomiting or purging of the intestines with laxatives. This cycle is usually kept secret. The group at risk is the same as that for anorexia nervosa, except that bulimia nervosa tends to occur in slightly older, less affluent women. Approximately 50% of individuals with anorexia nervosa are bulimic as well.55 Many young women stimulate vomiting inappropriately to control weight but are not classified as bulimic unless the pattern is obsessional or normal health or activity is interrupted. The rate of bulimia nervosa is increasing in Australia51; bulimia nervosa is more prevalent than anorexia nervosa in New Zealand.56
Diagnosis of bulimia nervosa is based on the following findings:
Although individuals with bulimia nervosa are afraid of gaining weight, their weight usually remains within normal range. They may binge and purge as often as 20 times each day. Continual vomiting of acidic chyme can cause pitted teeth, pharyngeal and oesophageal inflammation and tracheo-oesophageal fistulae (open connection between the trachea and oesophagus). Overuse of laxatives can cause rectal bleeding. Secret bingeing isolates the bulimic individual and leads to depression and anger that is turned inwards. A vicious cycle of depression, overeating to try to feel better, vomiting and purging to maintain a normal weight and returning depression perpetuates this eating disorder.
Because persons with bulimia are usually older than individuals with anorexia nervosa and have usually separated from a family core, individual or group counselling is the treatment focus. Individuals with bulimia nervosa rarely have physical problems requiring hospital care.
The epidemiology of mental illness in Australia and New Zealand
Anxiety disorders
 Fear and worry are core symptoms of anxiety and are mediated through the amygdala and associated structures, prefrontal cortex, medial temporal cortex, thalamus, insula, hypothalamus and brainstem nuclei. Gamma-aminobutyric acid (GABA) is the main neurotransmitter involved in anxiety symptoms. It has an inhibitory and modulating effect in the brain and it is thought anxiety disorders are driven by insufficient GABA. Worry is likely to be mediated through feedback loops in the cortico-striatal-thalamic-cortical circuits. Obsessive-compulsive disorder is characterised by obsessive thinking and compulsions to act to reduce the anxiety produced by the obsessive thoughts. Post-traumatic stress disorder requires exposure to acute or chronic trauma and is characterised by nightmares, flashbacks, hypervigilance, exaggerated startle reflex, persistent anxiety, poor concentration and emotional numbing.
Fear and worry are core symptoms of anxiety and are mediated through the amygdala and associated structures, prefrontal cortex, medial temporal cortex, thalamus, insula, hypothalamus and brainstem nuclei. Gamma-aminobutyric acid (GABA) is the main neurotransmitter involved in anxiety symptoms. It has an inhibitory and modulating effect in the brain and it is thought anxiety disorders are driven by insufficient GABA. Worry is likely to be mediated through feedback loops in the cortico-striatal-thalamic-cortical circuits. Obsessive-compulsive disorder is characterised by obsessive thinking and compulsions to act to reduce the anxiety produced by the obsessive thoughts. Post-traumatic stress disorder requires exposure to acute or chronic trauma and is characterised by nightmares, flashbacks, hypervigilance, exaggerated startle reflex, persistent anxiety, poor concentration and emotional numbing.Mood disorders
Genetic factors are likely in the development of mood disorders; however, causative genes or robust genetic risk factors have not been identified. The dominant neurobiological theory for mood disorders is the monoamine hypothesis. This may relate to reductions or increases in the neurotransmitters dopamine, serotonin and noradrenaline. Depression and dysthymia (a sub-syndromal form of depression) are relatively common with approximately 8% of the population experiencing depression in any 12-month period. Depression is characterised by sustained low mood, loss of pleasure and enjoyment, feelings of guilt, hopelessness and helplessness, loss of energy and changes in appetite and sleep. Dysthymia is characterised by persistent low mood and at least one of the other symptoms of depression. Mania is characterised by elevated mood, increased energy and decreased need for sleep, grandiose ideas and inflated self-importance, flight of ideas and sometimes delusions and hallucinations.Schizophrenia
Genetic factors are clearly implicated in schizophrenia with studies showing a strong familial relationship. Environmental factors, such as perinatal infection and immunological changes, have been shown to be associated with schizophrenia. Therefore, a gene–environment interaction is a strong possibility. Neuroanatomical changes in schizophrenic patients such as an enlarged ventricular volume have been documented. Alterations in the dopamine pathways have been implicated in schizophrenia. It is thought that the mesolimbic pathway causes positive symptoms while a reduction of dopamine within the mesocortical pathway causes negative symptoms. Serotonin is also thought to be involved. Schizophrenia is characterised by positive symptoms such as hallucinations, delusions and thought disorder and negative symptoms such as apathy, poor motivation, poor concentration, inability to initiate action and reduced thought and speech content.Substance abuse
A wide range of substances are abused including alcohol, tobacco, prescription medications, over-the-counter medications, heroin, marijuana, cocaine, petrol and glue. A common pathway of reward is thought to be involved in the problems of reward and addiction that characterise substance abuse.Eating disorders
Eating disorders are relatively uncommon with approximately 0.2% to 0.5% of the population affected. They are more common in females but do occur in males. Anorexia nervosa is characterised by fear of becoming obese, distorted body image, weight 15% less than normal and absence of menstrual periods in females. Starvation can cause serious physical health problems and there are risks associated with refeeding a person who has been malnourished. Disturbances in phosphate, magnesium and potassium levels need to be carefully monitored and managed during refeeding.Joan is a 78-year-old woman who lives alone in her own home following the death of her husband 2 years ago. She was diagnosed with breast cancer 15 years ago and had a left mastectomy. She also gave up smoking at this time, but had accumulated 40-pack-years of smoking (one packet of cigarettes per day for 40 years). She has annual check-ups with her oncologist and so far has had no recurrence of cancer and is due to have her next annual check-up in a week’s time. She is generally physically well but has had a history of generalised anxiety for much of her life, which has become worse following the death of her husband. She had an acute episode of labyrinthitis about 6 months ago and her symptoms have been well managed with medication.
Joan presents to the emergency department late in the afternoon with an acute episode of labyrinthitis. She was out on a bus trip with her senior citizens group and this morning had driven 20 kilometres from her home to meet the bus for the outing. She has been treated and her nausea and vertigo are under control and she is ready for discharge. It is now late in the evening. Upon discharge the nurse finds that Joan is short of breath, is complaining of a tight feeling in her chest and says her heart is pounding. She also looks apprehensive and mildly agitated.