9 ALTERATIONS OF NEUROLOGICAL FUNCTION ACROSS THE LIFE SPAN
INTRODUCTION
Alterations in central nervous system function are caused by traumatic injury, vascular disorders, tumour growth, infectious and inflammatory processes, metabolic derangements (including those arising from nutritional deficiencies and drugs or chemicals) and degenerative processes. Alterations in peripheral nervous system function and neuromuscular function occur also. The disruptions to homeostasis of the nervous system described in this chapter are often disabling and debilitating, which reflects the absolute importance of this system.
In this chapter, we begin by considering the most significant alteration of the neurological system in our community — cerebrovascular disorders, with the main one being stroke (or cerebrovascular accident). Stroke is among the leading causes of death and disability in our society and therefore its impact is substantial. For those who survive stroke, the disability is often severe and they may require substantial support. Stroke is often nowadays referred to as a ‘brain attack’, which indicates that it deserves the same awareness and urgency of medical care associated with a heart attack.
Throughout the remainder of the chapter, we discuss a range of other conditions that affect the brain and spinal cord and hence can cause symptoms as diverse as forgetfulness, abnormal motor function and sensory deficits. We have included some diseases that are relatively rare in the population considering their incidence and prevalence. However, these conditions are included because they are actually quite significant in the healthcare setting as the chronic nature of many of these diseases is such that they will progressively worsen to the point where more medical attention is necessary.
CEREBROVASCULAR DISORDERS
Cerebrovascular disease is the most frequently occurring neurological disorder and is responsible for more than 8% of all Australian deaths.1 Any abnormality of the brain caused by a pathological process in the blood vessels is referred to as a cerebrovascular disease. The brain abnormalities induced by cerebrovascular disease are either (1) ischaemia, with or without infarction (death of brain tissue) or (2) haemorrhage. The common clinical manifestation of cerebrovascular disease is a stroke (cerebrovascular accident), which is a sudden, non-convulsive focal neurological deficit.
Stroke
Stroke (or cerebrovascular accident, CVA) is a leading cause of disability in Australia and New Zealand and is the second-highest cause of death behind coronary heart disease (discussed in Chapter 23; see Table 9-1).1 Stroke occurs mainly among those older than 60 years of age, but can affect younger people as well, with 20% of strokes occurring in individuals younger than 60.2 The average age of people suffering from their first stroke in Australia is 74 years old.3 Stroke is more common in females, perhaps due to the fact that it occurs mainly in old age and females tend to live approximately five years longer than males in Australia and New Zealand (see Chapter 33 for more on life expectancy). In addition, people with both hypertension and type 2 diabetes mellitus are four times more likely to have a stroke and eight times more likely to die from stroke.4 In its mildest form, a cerebrovascular accident is so minimal as to be almost unnoticed; but in its most severe state, paralysis, coma and death result.
Table 9-1 LEADING CAUSES OF DEATH IN AUSTRALIA
| CAUSE OF DEATH | 2007 | |
|---|---|---|
| Number | Rank | |
| Coronary heart disease | 22,729 | 1 |
| Stroke | 11,491 | 2 |
| Trachea and lung cancer | 7,626 | 3 |
| Dementia and Alzheimer’s disease | 7,320 | 4 |
| Chronic lower respiratory diseases | 5,762 | 5 |
| Colon and rectal cancer | 4,107 | 6 |
| Diabetes | 3,810 | 7 |
| Blood and lymph cancer (including leukaemia) | 3,603 | 8 |
| Heart failure | 3,444 | 9 |
| Diseases of the kidney and urinary system | 3,230 | 10 |
| Prostate cancer | 2,938 | 11 |
| Breast cancer | 2,706 | 12 |
| Influenza and pneumonia | 2,623 | 13 |
| Pancreatic cancer | 2,248 | 14 |
| Suicide | 1,880 | 15 |
| Skin cancer | 1,727 | 16 |
| Hypertensive diseases | 1,627 | 17 |
| Cirrhosis and other diseases of the liver | 1,437 | 18 |
| Cardiac arrhythmias | 1,397 | 19 |
| Land transport accidents | 1,273 | 20 |
Source: Australian Bureau of Statistics. Causes of death, 2007. 3303.0. Canberra: Commonwealth of Australia; 2009.
Each year, approximately 45,000 Australians are affected by stroke, with more than 10,000 of these cases being fatal.1,5 Overall, the rates of stroke have declined in recent years (see Figure 9-1), which may be due to factors such as increased public awareness of the symptoms of stroke and improvements in the early diagnosis of stroke. The percentage of people who require hospitalisation for stroke is much higher for Indigenous than for non-Indigenous Australians.6
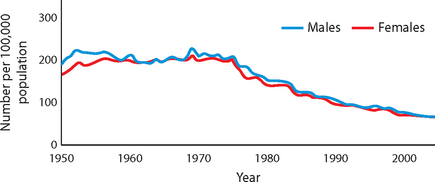
FIGURE 9-1 Death rates from stroke, Australia, 1950–2002 (per 100,000 population).
Source: Based on Australian Institute of Health and Welfare. Heart, stroke and vascular diseases: Australian facts, 2004. AIHW cat. no. CVD 27. Canberra: Australian Institute of Health and Welfare and National Heart Foundation of Australia; 2004.
A stroke occurs when a blood vessel (artery) supplying the brain is altered, usually by either a lack of flow or bleeding. Neurons can survive without oxygen for only a very limited amount of time, so if the blood flow is insufficient for more than a few minutes, death of the neurons occurs. Neurons within the brain do not regenerate (refer to Chapter 6) so the damage is permanent if blood flow is not corrected immediately. Death of neurons is called a cerebral infarct; hence a cerebral infarction is another name for a stroke. The type and severity of the symptoms of stroke depend on which blood vessel is affected; however, impairment of blood supply to the vital brain structures, such as the brainstem, is often fatal.
Strokes are classified according to pathophysiology (see Figure 9-2):
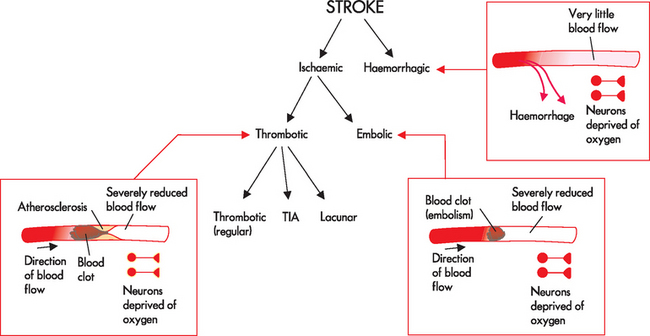
The main types of stroke are ischaemic and haemorrhagic. See the text for details of the different types of stroke.
Ischaemic strokes
The vast majority of strokes are ischaemic (80%).5 This is because more people have trouble with unwanted clotting than bleeding into the brain. Ischaemic strokes occur due to a blockage in the blood vessels supplying the brain. The two subtypes of ischaemic strokes are thrombotic and embolic. In addition, a transient ischaemic attack is a short-term (less than 24 hours) obstruction of blood supply; if this lasts for longer than 24 hours, then it is classified as a thrombotic stroke.
Thrombotic stroke
A thrombotic stroke (cerebral thrombosis) occurs when there is a blockage inside a blood vessel that supplies the brain tissue (see Figure 9-2). This may occur in arteries entering the brain, commonly in the internal carotid artery (carotid stenosis),7,8 or from the smaller vessels within the brain (the intracranial vessels). The deposits are usually formed by fatty plaques known as atherosclerosis, which contain substances including cholesterol (details in Chapter 23). People with high blood cholesterol have a much higher risk of ischaemic stroke than those with normal cholesterol levels.9 The atherosclerotic plaque creates an inviting location for blood to accumulate into a clot, forming a thrombus. This thrombus occupies so much of the vessel lumen that it forms a blockage so that blood flow cannot proceed past it; hence neurons that are downstream of the thrombus are deprived of oxygen.
A thrombotic stroke results in alterations in neuronal function that persist for more than 24 hours. Because neurons cannot survive without a constant supply of oxygen, death of neurons occurs within a few minutes. Larger areas of tissue disintegration appear by 48 to 72 hours after infarction. Permanent disruptions to brain function occur and strokes are often fatal. The longer the blockage remains, the more extensive the neuronal damage.
Transient ischaemic attacks (TIAs) are temporary decreases in brain blood flow resulting in brief changes in brain function, such as changes in vision, speech, motor function or symptoms of dizziness or loss of consciousness. TIAs may be due to blood clots or a vessel undergoing a spasm and narrowing, causing a transient (temporary) blockage of circulation. All neurological deficits are completely clear within 24 hours, leaving no dysfunction or permanent brain injury; in most cases, the TIA is actually resolved within the first hour.10,11 TIAs are a warning that cerebrovascular disease is developing and that another TIA or more severe stroke is likely to occur soon afterwards: the recurrence of a neurological event, such as another TIA, may be as soon as 24–72 hours after the first TIA.12 After a TIA, the patient is educated about modifying the risk of subsequent stroke. The TIA on its own can actually be a significant event; in one study, more than 10% of patients suffered disability or death two weeks after the initial TIA, despite receiving the recommended treatment.7
Lacunar infarcts are particularly small thrombotic strokes less than 1 cm in diameter and involve the small arteries, predominantly in deep brain regions such as the basal nuclei and pons. They are associated with smoking, hypertension and diabetes mellitus.13 Because of their location and small area of infarction, these strokes may have motor and sensory deficits only, rather than the wide range of symptoms seen with larger areas of infarction. Furthermore, evidence demonstrates that mortality and morbidity is reduced in patients following lacunar infarcts compared with other types of strokes.14
Embolic stroke
An embolic stroke involves fragments that break from a thrombus formed outside the brain — the most common is when a fragment in the heart breaks away during abnormal heart function (during atrial fibrillation; see Chapter 23) and travels to the brain. If an embolic stroke is suspected, investigations of the heart are necessary to establish the cause. The embolus becomes wedged in small brain vessels causing obstruction and ischaemia to the brain tissue distal to the occlusion. In people who experience an embolic stroke, usually a second stroke follows because the source of emboli continues to exist. The characteristic feature to distinguish between a thrombotic stroke and an embolic stroke is that the clot originates in the brain vessels for thrombotic strokes and in a vessel outside of the brain for embolic strokes. Fat emboli are less common and sometimes develop with fractures of long bones, as the fatty material from within the bone marrow can enter the bloodstream.
Haemorrhagic stroke
In contrast to ischaemic stroke where the neuronal damage is due to inadequate blood flow, haemorrhagic stroke occurs in response to bleeding in the brain. There are two main types of haemorrhagic stroke:
 Intracerebral haemorrhage accounts for about 10% of all strokes and is related to hypertension (high blood pressure) and ruptured aneurysms (see the next section). Other causes include bleeding into a tumour, haemorrhage associated with bleeding disorders, anticoagulation medications (drugs that delay clotting), head trauma and illicit drug use (particularly sniffing cocaine). Subarachnoid haemorrhage accounts for about 5% of all strokes and results in bleeding into the subarachnoid space that contains CSF. This type of haemorrhage (loss of blood from the vessel) often causes significant haematomas (clotted masses of blood within tissues), which cause an increase in intracranial pressure. A haemorrhage often leads to a haematoma within the cranium as the blood cannot escape from the area.
Intracerebral haemorrhage accounts for about 10% of all strokes and is related to hypertension (high blood pressure) and ruptured aneurysms (see the next section). Other causes include bleeding into a tumour, haemorrhage associated with bleeding disorders, anticoagulation medications (drugs that delay clotting), head trauma and illicit drug use (particularly sniffing cocaine). Subarachnoid haemorrhage accounts for about 5% of all strokes and results in bleeding into the subarachnoid space that contains CSF. This type of haemorrhage (loss of blood from the vessel) often causes significant haematomas (clotted masses of blood within tissues), which cause an increase in intracranial pressure. A haemorrhage often leads to a haematoma within the cranium as the blood cannot escape from the area.With a subarachnoid haemorrhage, blood escapes from a defective or injured vessel into the subarachnoid space (see Figure 9-3). The bleed increases intracranial volume15 and impairs the circulation of the CSF; together, these lead to an immediate increase in intracranial pressure, which returns to near baseline in about 10 minutes. Cerebral blood flow also decreases, which lowers the blood supply to the brain. The expanding mass of clotted blood, now known as a haematoma, compresses and displaces brain tissue (see Chapter 8). The blood is also extremely irritating to the neural tissues and produces an inflammatory reaction, as macrophages and astrocytes appear to clear away the blood. The cerebral haemorrhage resolves through reabsorption and a cavity forms, surrounded by a dense gliosis after removal of the blood. Mortality in subarachnoid haemorrhage is 50% at one month.
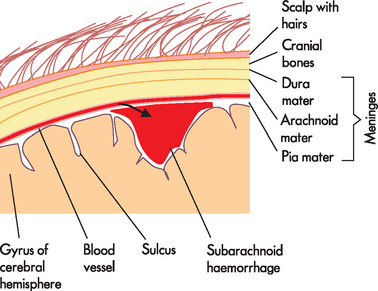
FIGURE 9-3 Subarachnoid haemorrhage.
The growing blood volume in the area underneath the arachnoid mater — the subarachnoid space — compresses the nearby brain tissue.
Haemorrhages are described as massive, small, slit or petechial. Massive haemorrhages are several centimetres in diameter; small haemorrhages are 1–2 cm in diameter; slit haemorrhages lie in the sub-cortical area; and petechial haemorrhages are the size of a pinhead bleed. The most common sites for hypertensive haemorrhages are in the basal nuclei (55%), the thalamus (10%), the cortex and sub-cortex (15%), the pons (10%) and the cerebellar hemispheres (10%).
The most common risk factors for stroke are:
Because these risk factors are highly preventable with adequate patient education, stroke has been described as highly preventable.16
CLINICAL MANIFESTATIONS
The noticeable signs and symptoms of stroke are shown in Table 9-2 and these are used to educate the public and those at risk about stroke.
| If you have the following: call an ambulance immediately. |
Source: Stroke Foundation. www.strokefoundation.com.au/are-you-are-having-a-stroke, accessed July 2010.
Ischaemic stroke
Following ischaemic stroke, fluid accumulates between neurons, which results in cellular oedema. Cerebral oedema reaches its maximum in about 72 hours and takes about 2 weeks to subside. Most people survive an initial hemispheric ischaemic stroke unless there is massive cerebral oedema, which is nearly always fatal.
Clinical manifestations of thrombotic stroke vary, depending on the artery obstructed. Different sites of obstruction create different occlusion syndromes, and if the area of damage is restricted to one side of the brain, then symptoms will only be seen on the opposite side of the body (see Figure 9-4). For example, if the Broca’s speech area in the frontal lobe (which is responsible for the motor aspects of speech) is affected, then the classical sign of stroke of difficulty in speaking will occur and the patient may say only a couple of short words rather than full sentences. If the blood flow to brain areas involved in speech remains normal, then speech will not be affected. Another classical manifestation of stroke regarding speech involves Wernicke’s area in the temporal lobe, which interprets speech. In this case, the person will be able to articulate, but may use words and sentences that do not make sense. Since the patient cannot interpret what they are saying, they will be unaware of their errors. These are examples of aphasia (refer to Chapter 8).
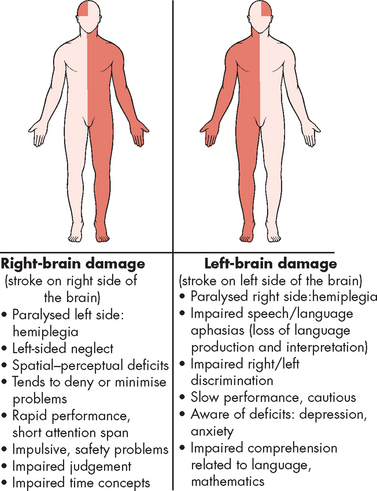
Haemorrhagic stroke
Individuals experiencing intracranial haemorrhage from a ruptured or leaking aneurysm (discussed in the next section) have one of three sets of symptoms: (1) onset of an excruciating generalised headache with an almost immediate lapse into an unresponsive state; (2) headache but with consciousness maintained; or (3) sudden lapse into unconsciousness. Once a deep unresponsive state occurs, the person rarely survives. The immediate prognosis is grave, but if the person survives, recovery of function is often possible, although the individual may never recover full function like that before the stroke.
If the haemorrhage is confined to the subarachnoid space, there may be no local signs. If bleeding spreads into the brain tissue, paralysis on one side of the body (hemiparesis), difficulty using or understanding language (dysphasia) or blindness in half of the visual field may be present. Warning signs of an impending aneurysm rupture may be present and include headache and temporary changes of unilateral weakness, numbness and tingling, and speech disturbance. Rapid worsening of headache is much more common in haemorrhagic stroke than ischaemic stroke.10
A ruptured vessel causes a sudden ‘explosive’ headache, accompanied by nausea and vomiting, visual disturbances, motor deficits and loss of consciousness related to a dramatic rise in intracranial pressure. Meningeal irritation and inflammation often occur, causing neck stiffness (nuchal rigidity), photophobia, blurred vision, irritability, restlessness and low-grade fever. A positive Kernig’s sign (straightening the knee with the hip and knee in a flexed position produces pain in the back and neck regions) and Brudzinski’s sign (passive flexion of the neck produces neck pain and increased rigidity) may appear. No localising signs are present if the bleed is confined completely to the subarachnoid space.
EVALUATION AND TREATMENT
It is essential to distinguish between ischaemic and haemorrhagic causes of stroke to guide treatment options. A range of investigations can assist with this diagnosis and all major hospitals have a dedicated stroke care unit to provided more efficient care; however, these units are less common in rural and remote settings.17 The patient’s medical history is important, particularly since hypertension is the main risk factor for haemorrhagic stroke. Knowing the time that the symptoms commenced is important: if the patient was experiencing stroke on waking, the time that symptoms commenced is assumed to be the time when they were last awake and asymptomatic. The lack of pain with ischaemic strokes means these strokes do not usually wake the patient, but haemorrhagic strokes do.10
The Australian guidelines for predicting those at early risk of stroke after a TIA have recently been modified to the new ABCD2 score, which assigns points based on a patient’s symptoms of age, blood pressure, clinical features (unilateral weakness, speech impairment), duration of symptoms and diabetes (see Table 9-3).11,18 This allows patients to be classified as low or high risk of subsequent stroke — those at high risk should have a CT scan of the brain within 24 hours.11
Table 9-3 ABCD2 SCORE FOR PREDICTING EARLY RISK OF STROKE
| The newly adopted ABCD2 scoring for predicting those with a TIA at a high risk of early stroke. A score above 4 indicates those at high risk, who require an urgent CT scan. | ||
| Symptoms | Points | |
| A | Age: 60 or older | 1 |
| B | Blood pressure: 140/90 mmHg or higher | 1 |
| C | Clinical features: | |
| D | Duration: | |
| D | Diabetes | 1 |
Total: maximum of 7
Source: Johnston S et al. Validation and refinement of scores to predict very early stroke risk after transient ischaemic attack. Lancet 2007; 369: 283–292.
A brain scan is essential to confirm the diagnosis of stroke or TIA and to eliminate other causes of the symptoms (such as a tumour). The CT scan should be performed as soon as possible but at least within 24 hours of symptoms commencing if the patient is at high risk of stroke (see Figure 9-5). While an MRI scan is preferable for distinguishing between ischaemic and haemorrhagic stroke, the cost and time involved with this test means that many facilities will rely on the CT scan.11,19 A cerebral angiograph can be used to visualise the blood vessels to identify areas of blockage or aneurysm. This entails inserting a catheter into an artery (femoral or brachial) and guiding it into the internal carotid artery. Dye is then injected into the intracranial arteries and X-rays are used to determine the vascular anatomy and any abnormalities, such as aneurysms.
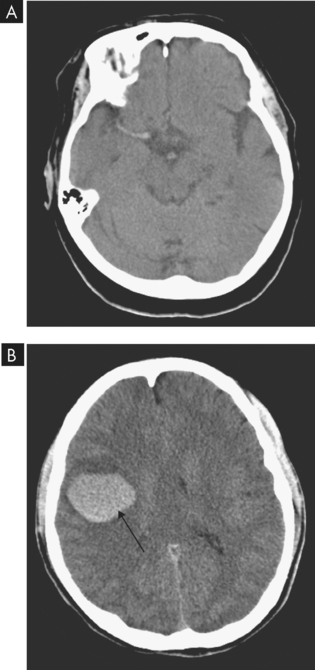
FIGURE 9-5 A CT scan is essential to confirm the diagnosis of stroke.
A CT image of an ischaemic stroke due to blockage in the middle cerebral artery. B CT image of a haemorrhagic stroke (arrow) in a patient due to use of the drug ecstasy.
Source: A Haaga JR. CT and MRI of the whole body. 5th edn. St Louis: Mosby; 2008. B Soto JA, Lucey BC. Emergency radiology: the requisites. Philadelphia: Mosby; 2009.
In thrombotic stroke, thrombolytic therapy using a drug that dissolves clots (known as tissue plasminogen activator, commonly abbreviated to tPA) by breaking down fibrin is given as soon as possible. It is recommended that the tPA (alteplase) be administered at least within six hours of the stroke,20 but preferably under three hours. This requires coordination of services for the patient to ensure quick diagnosis and treatment.21 Brain scans are essential prior to commencement of thrombolytic therapy to confirm an ischaemic stroke: if the stroke is haemorrhagic, it is imperative that thrombolytic therapy not be used, as it will worsen the condition. Treatment is directed at restoration of adequate blood flow, prevention of ischaemic injury and supportive management to control cerebral oedema and increased intracranial pressure. In those patients who have atherosclerosis in the carotid artery, a carotid endarterectomy (removal of atheromas from the artery) may be performed to surgically remove this build-up (see Figure 9-6).16 Arresting the disease process by control of risk factors is critical, and anticoagulant therapy using aspirin is usually instituted to minimise the risk of subsequent stroke.22
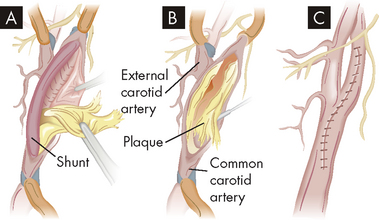
FIGURE 9-6 Carotid endarterectomy is performed to prevent stroke.
The procedure can be performed by clamping the artery to temporarily prevent blood flow, with the other carotid artery supplying enough blood to the brain, or redirecting blood flow using a vascular shunt. A A shunt (tube) is inserted above and below the blockage to allow blood flow to the brain. B Atherosclerotic plaque in the common carotid artery is removed. C Once the artery is stitched closed, the shunt is removed and blood flow through the artery is restored.
Source: Brown D, Edwards H. Lewis’s medical–surgical nursing. 2nd edn. Sydney: Elsevier; 2008.
The diagnosis of a subarachnoid haemorrhage is based on the clinical presentation, a CT scan and a lumbar puncture (insertion of a needle into the subarachnoid space to sample the CSF) — this is to see whether blood is present in the CSF. Treatment is directed at controlling intracranial pressure, preventing ischaemia and hypoxia of the neural tissues, and preventing rebleeding episodes. Treatment of an intracranial bleed, regardless of the cause, focuses on stopping or reducing the bleeding, controlling the increased intracranial pressure, preventing rebleeding and preventing vasospasm (a condition where arteries spasm, leading to vasoconstriction and potentially ischaemia). Intravenous administration of a drug that promotes blood clotting (recombinant activated factor VII) within 4 hours of onset of a cerebral bleed is currently under study.23 Some surgeons prefer to drain the blood, but the benefit is not documented in studies.
It has been proposed that patients with TIA or stroke should be treated with the same degree of urgency and attention as those with acute coronary disorders, particularly in the light of the risk of disability in these high-risk patients.24,25 The risk of recurrence is high — without diagnosis and treatment, 80–90% have a recurrence of symptoms by 1 year.26,27 Even with adequate treatment, almost 10% suffer a recurring event.3 Smoking is an important risk factor for stroke; however, of those people who had been smoking when they had a stroke, five years later less than 40% had quit.28 Preventing stroke recurrence is a balance between education, patient modification of risk factors and improved research.
The longer-term consequences of stroke
Patients need to be evaluated for dysphagia (difficulty swallowing). Nasogastric feeding (semi-liquid diet infused directly into the stomach via a tube through the nose) may be necessary for the first month if swallowing is not functional.11 Urinary incontinence (inability to control emptying of the bladder) may require a combination of exercise and medication. Although faecal incontinence (inability to control bowel emptying) may occur in one-third of stroke patients, this improves to only 11% remaining incontinent after several months.29 Stroke patients need to be assessed for depression, anxiety and mood alterations, and appropriate medications may be used to treat these. Other healthcare professionals may be involved in the rehabilitation of patients after stroke, including physiotherapists and speech pathologists.
Two new warning signs found for impending stroke
Prevention of stroke is far more preferable than treatment once a stroke has occurred. Researchers have been searching for ways to identify what signs and individuals may be associated with higher stroke risk. It has been shown that individuals with elevated biochemistry markers of lipoprotein-associated phospholipase A2 (a proinflammatory enzyme secreted by macrophages that binds to low-density lipoprotein) and highly sensitive C-reactive protein (CRP, a protein produced by the liver during periods of inflammation) have a significantly increased risk for ischaemic stroke. In addition, those with obstructive sleep apnoea also have a greater risk for stroke independent of other risk factors. More research is needed to find the exact reasons for the increased risk.
Source: Elkind MS et al. High-sensitivity C-reactive protein, lipoprotein-associated phospholipase A2 and outcome after ischaemic stroke. Arch Intern Med 2006; 166(19):2073–2080; Munoz R et al. Severe sleep apnea and risk of ischaemic stroke in the elderly. Stroke 2006; 37(9):2317–2321; Yaggi HK et al. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005; 353(19):2034–2041.
Stroke survivors may need to be assessed by Aged Care Assessment Teams (ACATs) or an equivalent service, which can determine those who are eligible for specific assistance programs. A wide range of techniques and equipment are available to assist those with motor difficulties in the longer term. Many patients will require palliative care and almost 20% of stroke survivors require nursing home or other support.17
Intracranial aneurysm
An aneurysm is a region of the blood vessel that is overfilled with blood, giving it a swollen, balloon-like appearance. Intracranial aneurysms may result from atherosclerosis (blockages in arteries), congenital abnormality, trauma, inflammation and cocaine use. The size may vary from 2 mm to 3 cm. Most aneurysms are located at bifurcations (blood vessel branches) in or near the circle of Willis, in the vertebrobasilar arteries or within the carotid vessels. Aneurysms may be single, but in 10% of cases, more than one is present.30 Because the vessel is swollen, it is stretched and overfilled with blood and prone to rupture. Peak incidence of rupture occurs in persons aged 50 to 60 and the incidence is slightly higher in women than in men. If an aneurysm ruptures, a massive haemorrhage may occur within the cranium, which is one of the main causes of a haemorrhagic stroke.
PATHOPHYSIOLOGY
Aneurysms are classified on the basis of their shape and form. Saccular aneurysms (berry aneurysms) occur frequently (in approximately 2% of the population30) and probably result from congenital abnormalities in the media of the arterial wall as well as degenerative changes.13 The sac gradually grows over time. A saccular aneurysm may be: (1) round, with a narrow stalk connecting it to the parent artery; (2) broad-based without a stalk; or (3) cylindrical (see Figure 9-7).
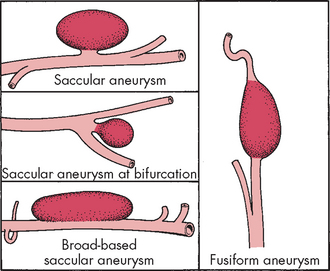
Fusiform aneurysms (giant aneurysms) occur as a result of diffuse atherosclerotic changes and are found most commonly in the basilar arteries or internal carotid arteries. They occupy space within the cranium (refer to Chapter 8).
Aneurysms rupture through thin areas, causing haemorrhage into the subarachnoid space that spreads rapidly, producing localised changes in the cerebral cortex and focal irritation of nerves. Bleeding ceases when a fibrin-platelet plug forms at the point of rupture and as a result of compression. The bleed undergoes reabsorption through arachnoid villi within 3 weeks.
CLINICAL MANIFESTATIONS
Aneurysms often are asymptomatic. Of all those undergoing routine autopsy, 5% are found to have one or more intracranial aneurysms. Clinical manifestations may arise from cranial nerve compression, but the signs vary, depending on the location and size of the aneurysm. Cranial nerves III, IV, V and VI are affected most often. Unfortunately, the most common first indication of the presence of an aneurysm is an acute subarachnoid haemorrhage or intracerebral haemorrhage, which may cause a haemorrhagic stroke (see previous section).
EVALUATION AND TREATMENT
The Hunt and Hess subarachnoid haemorrhage grading system is based on a description of the clinical manifestations (see Table 9-4).31 Rebleeding is a significant risk, with a high mortality (up to 70%). The period of greatest risk is within the first month, with the peak incidence of rebleeding occurring during the first 2 weeks after the initial bleed. Rebleeding is manifested by a sudden increase in blood pressure and intracranial pressure, along with a deteriorating neurological status.
Table 9-4 SUBARACHNOID HAEMORRHAGE CLASSIFICATION SCALE
| CATEGORY | DESCRIPTION |
|---|---|
| Grade I | Neurological status intact; mild headache, slight nuchal rigidity (neck stiffness) |
| Grade II | Neurological deficit evidenced by cranial nerve involvement; moderate to severe headache with more pronounced meningeal signs (e.g. photophobia, nuchal rigidity) |
| Grade III | Drowsiness and confusion with or without focal neurological deficits; pronounced meningeal signs |
| Grade IV | Stuporous with pronounced neurological deficits (e.g. hemiparesis, dysphasia); nuchal rigidity |
| Grade V | Deep coma state with decerebrate posturing and other brainstem functioning — poor survival rate |
Source: Cook HS. Aneurysmal subarachnoid hemorrhage: neuroscience frontiers and nursing challenges. In: Winkelman C (ed.). AACN clinical issues in critical nursing. Philadelphia: Lippincott; 1991.
Diagnosis before a bleeding episode is made through intracranial angiography. After a subarachnoid or intracerebral haemorrhage, a tentative diagnosis of an aneurysm is based on clinical manifestations, a history, CT scanning and MRI. The treatment of choice for an aneurysm is surgical management. Small coils may be packed into the aneurysm, which encourages blood clotting and tissue growth over the area, reducing the risk of it rupturing (see Figure 9-8). This technique is currently under study and appears to be a viable option.32,33 The location and size of the aneurysm and the person’s clinical status determine whether invasive therapy is feasible.
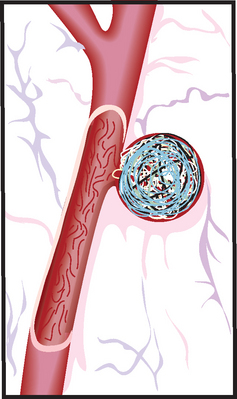
FIGURE 9-8 Coil inserted in aneurysm.
Platinum coils attached to a thin wire are inserted into the catheter and then placed in the aneurysm until the aneurysm is filled with coils. Packing the aneurysm with coils prevents the blood from circulating through the aneurysm, reducing the risk of rupture.
Source: Brown D, Edwards H. Lewis’s medical–surgical nursing. 2nd edn. Sydney: Elsevier; 2008.
Vascular malformation
An arteriovenous malformation (AVM) is a tangled mass of dilated blood vessels creating abnormal channels between the arterial and venous systems. AVMs may occur in any part of the brain and vary in size from a few millimetres to large malformations extending from the cortex to the ventricle. AVMs occur equally in males and females and occasionally occur in families. Although AVMs are usually present at birth, symptoms exhibit a delayed age of onset and commonly occur before 30 years of age.
PATHOPHYSIOLOGY
AVMs do not have a normal blood vessel structure and are abnormally thin. One or several arteries may feed the AVM and become tortuous and dilated over time. With moderate to large AVMs, sufficient blood is shunted into the malformation to deprive surrounding tissue of adequate blood perfusion.
CLINICAL MANIFESTATIONS
About 20% of people with an AVM have a characteristic chronic, nondescript headache, although some experience migraine. On average, 50% of people experience seizures caused by compression, and the other 50% experience an intracerebral, subarachnoid or subdural haemorrhage. Bleeding from an AVM into the subarachnoid space causes symptoms identical to those associated with a ruptured aneurysm. If bleeding occurs into the brain tissue, focal signs that develop resemble a stroke-in-evolution; 10% of people experience hemiparesis or other focal signs. At times, hydrocephalus develops with a large AVM that extends into the ventricular lining.
EVALUATION AND TREATMENT
It is difficult to know that a person has an AVM in their cerebral vascular anatomy. A bruit (an abnormal sound heard with a stethoscope) over the carotid artery in the neck, the mastoid process or the eyeball in a young person is almost diagnostic of an AVM. Confirming diagnosis is made by CT scan and MRI followed by MRA (magnetic resonance angiography). Treatment options are direct surgical intervention, embolisation or radiotherapy.
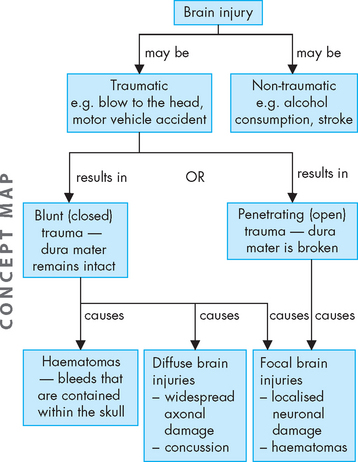
TRAUMA TO THE CENTRAL NERVOUS SYSTEM
Brain trauma
Traumatic brain injury occurs from a blow to the head. In Australia and New Zealand, such injuries are usually caused by a motor vehicle or sporting accident (see Table 9-5). Non-traumatic brain injury commonly arises from alcohol consumption, as well as from stroke (discussed previously). As a result of the brain injury, the individual suffers functional impairments, such as physical, emotional or cognitive deterioration.34 Males are twice as likely to be hospitalised for traumatic brain injury than females, with most people who are hospitalised being in their late teens and early 20s, and also in the over-60 age group.35 Furthermore, Indigenous Australians have a hospitalisation rate for traumatic brain injury more than double that for non-Indigenous Australians.34
Table 9-5 CAUSES OF BRAIN INJURY
| TYPE OF INJURY | MECHANISM |
|---|---|
| Coup | Injury is directly below site of forceful impact |
| Contrecoup | Injury is on opposite side of brain from site of forceful impact |
| Extradural haematoma | Vehicular accidents, falls, sporting accidents |
| Subdural haematoma | Vehicular accidents or falls, especially in the elderly or those with chronic alcohol abuse |
| Intracerebral haemorrhage | Contusions caused by forceful impact, usually vehicular accidents or falls from a distance |
| Compound fracture | Objects strike head with great force or head strikes object forcefully; temporal blows, occipital blows, upward impact of cervical vertebrae (basilar skull fracture) |
| Penetrating injury | Missiles (bullets) or sharp projectiles (knives, ice picks, axes, screwdrivers) that enter the cranial vault |
| Diffuse axonal injury | Moving head strikes hard, unyielding surface or moving object strikes stationary head; vehicular accidents (occupant or pedestrian); torsional head motion |
There are two types of injuries to the brain:
Focal brain injuries account for most head injury deaths, while the more severely disabled survivors, including those in an unresponsive state or reduced level of consciousness, have mainly diffuse axonal injuries.
Head injuries are broadly categorised into blunt (closed) trauma and penetrating (open) trauma (see Figure 9-9). Blunt trauma is more common and involves either the head striking a hard surface or a rapidly moving object striking the head. The dura mater of the meninges (brain covering within the skull) remains intact, so the brain is not exposed to the environment. Blunt trauma may result in both localised focal brain injuries and widespread diffuse axonal injuries. Penetration of the dura results in exposure of the cranial contents to the environment, which is caused mainly by open trauma and results in focal brain injuries.
In recent years, there has been a focus on reducing the severity of injury (such as motor vehicle air bags) and improved medical management both at the scene of the injury and in the healthcare facility. As a result, more people are surviving and those with severe traumatic brain injuries are being admitted to rehabilitation programs.
PATHOPHYSIOLOGY
Focal brain injury
The force of impact from a focal brain injury typically produces contusions — injury to brain tissue without breaking the inner pia mater. Damage results from compression of the skull at the point of impact and rebound effect, and may be coup or countercoup (see Figure 9-10). Contusion and bleeding occur from small tears in blood vessels caused by these forces. Brain oedema forms around and in damaged neural tissues, contributing to the increasing intracranial pressure. The maximum effects of these injuries peak 18–36 hours after severe head injury. Contusions are found most commonly in the frontal and temporal lobes of the cerebral cortex. They result in changes in attention, memory, emotion and behaviour. Focal cerebral contusions are superficial, involving just the cerebral cortex.
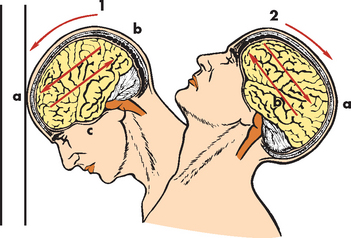
FIGURE 9-10 Coup and contrecoup head injury after blunt trauma.
1 Coup injury: impact against object; a, site of impact and direct trauma to brain; b, shearing of subdural veins; c, trauma to base of brain. 2 Contrecoup injury: impact within skull; a, site of impact from brain hitting opposite side of skull; b, shearing forces through brain. These injuries occur in one continuous motion — the head strikes the wall (coup) and then rebounds (contrecoup).
Source: Modified from Rudy EB. Advanced neurological and neurosurgical nursing. St Louis: Mosby; 1984.
One of the important consequences of brain injury is the development of a haematoma — a clotted mass of blood that is contained within tissues (whereas a haemorrhage refers to the actual loss of blood from the vessel — as blood from a haemorrhage cannot easily exit the cranium, development of haematoma is likely). A serious complication of intracranial haematoma is that the accumulation of blood can increase the intracranial pressure and compress the brain. This leads to severe consequences such as brain herniation, whereby the brain is forced downwards towards the brainstem and spinal cord, which is usually fatal (review using Chapter 8). Haematomas may be epidural, subdural or intracranial (see Figure 9-11).
Epidural haematomas are due to bleeds from arteries or veins and are usually emergencies. The bleed occurs just exterior to the outer layer of the meninges (the dura mater), between the meninges and the skull. As the volume of blood occupies space within the skull, it causes pressure on the brain tissue, causing it to herniate (move) into the ventricles or down towards the spinal cord. Subdural haematomas may be acute, subacute or chronic. Acute subdural haematomas develop rapidly (within 48 hours) and are usually at the top of the skull. Subacute subdural haematomas develop more slowly over 2 weeks. Chronic subdural haematomas are common in the elderly and those who abuse alcohol and have brain atrophy (shrinkage); these develop over weeks or months and the increased intracranial pressure eventually compresses the bleeding vessels. Intracerebral haematomas are common in the frontal and temporal lobes and in the cerebral hemisphere deep white matter. Penetrating forces associated with the speed of head movement (or the colliding object) traumatise small blood vessels. The increasing intracranial pressure causes oedema (fluid accumulation in the brain tissue). Delayed intracerebral haematomas may appear 3–10 days after the head injury.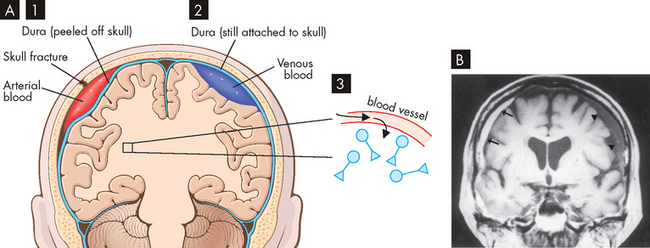
FIGURE 9-11 Types of Haematomas.
A 1 Epidural, 2 subdural and 3 intracerebral. B Acute subdural haematoma on the right side (arrows) from a 76-year-old man who fell 10 days prior to imaging (MRI). Chronic subdural haematoma (arrowheads) on the left side is also present.
Source: A Based on Kumar et al. Robbins & Cotran pathological basis of disease. 8th edn. Philadelphia: Saunders; 2010. B Based on Bradley WG et al. Neurology in clinical practice. 5th edn. Philadelphia: Butterworth-Heinemann; 2008.
Open trauma produces discrete (focal) injuries. A compound fracture (break in the skull and associated opening of the skin) exposes the brain to the environment and should be investigated whenever there are head lacerations (cuts or wounds). Debris from skull injury can cause damage by penetrating the brain, as well as stretching nearby blood vessels and nerves.
Diffuse brain injury
Immediately following concussion (altered consciousness and neuronal function), all cells in the brain send action potentials at once (massive electrical discharges), causing release of the neurotransmitter glutamate; this opens potassium channels and potassium exits the cells. In response, calcium enters the cells and its mitochondria, causing them to dysfunction. As mitochondria are involved in cellular energy production, mitochondrial dysfunction creates a cellular energy crisis. This prohibits the cells from restoring their electrolyte balance that is necessary for neuronal action potentials and therefore neurons are functionally impaired (see Chapter 6).
Diffuse brain injury to axons (known as diffuse axonal injury or DAI) results from a shaking effect associated with high levels of acceleration and deceleration, seen in road traffic crashes. Rotational acceleration or twisting movements produce strains and distortions within the brain. The most severe DAIs cause extensive cognitive impairments, as seen in survivors of traumatic brain injury resulting from motor vehicle crashes. This damage reduces the speed of information processing and responding and disrupts attention; often, the patient requires lifelong assistance with activities of daily living.
CLINICAL MANIFESTATIONS
Focal brain injury
A contusion may be evidenced by some short-term consequences (immediate to a few minutes): loss of consciousness, loss of reflexes (the individual falls to the ground, as muscle reflexes involved in posture are impaired), absence of breathing, slow heart rate (bradycardia) and decreased blood pressure. Increased CSF pressure occurs, as do changes in the electrical activity of the heart and brain (observed by ECG and EEG, respectively). Vital signs may stabilise to normal in a few seconds, reflexes return and the person regains consciousness over minutes to days. Residual deficits may persist and some people never regain a full level of consciousness.
Epidural haematomas. Individuals lose consciousness on injury and then many become lucid (normal cognitive function) for a few minutes to a few days. As the haematoma accumulates, a headache of increasing severity, vomiting, drowsiness, confusion, seizure and hemiparesis (weakness on one side of the body) may develop. As temporal lobe herniation occurs, level of consciousness is rapidly lost, with ipsilateral pupillary dilation (on the side of haematoma) and contralateral hemiparesis. The prognosis is good if intervention is initiated before bilateral dilation of the pupils. Epidural haematomas are almost always medical emergencies. Subdural haematomas. In acute, rapidly developing subdural haematomas, the expanding clots compress the brain and can actually close the broken vessel to stop the bleeding. However, cerebral compression and displacement of brain tissue can cause temporal lobe herniation. An acute subdural haematoma classically begins with headache, drowsiness, restlessness or agitation, slowed cognition and confusion. These symptoms worsen over time and progress to loss of consciousness, respiratory pattern changes and pupillary dilation (i.e. the symptoms of temporal lobe herniation). Defective vision in either the right or the left visual field and disconjugate gaze (where the eyes do not move equally) may also occur. Most people affected by chronic subdural haematomas have chronic headaches and tenderness over the haematoma on palpation. Most people appear to have a progressive dementia with generalised rigidity (paratonia). Intracerebral haematomas. Intracerebral haematomas cause decreasing consciousness. Coma or a confusional state from other injuries, however, can make the cause of this increasing unresponsiveness difficult to detect. Contralateral hemiplegia also may occur and as intracranial pressure rises, temporal lobe herniation may appear. In delayed intracerebral haematoma, the presentation is similar to that of a hypertensive brain haemorrhage: sudden, rapidly progressive decreased level of consciousness with pupillary dilation, breathing pattern changes, paralysis on one side of the body (hemiplegia) and a positive Babinski reflex. The Babinski reflex is present in children up to the age of 2 years. It gradually disappears as the child’s neurological system becomes more mature. The reflex is elicited by stimulating the lateral side of the sole from the heel to the toes and observing for the fanning out of the toes. If present in adults, it strongly indicates damage to the corticospinal tract, which is a series of axons between the cerebral cortex and spinal cord.Diffuse brain injury
Diffuse axonal injury results in the following:
physical consequences: spastic paralysis (whereby the muscles are in a constant state of spasm so that they cannot contribute to function), peripheral nerve injury, swallowing disorders, dysarthria (difficulty articulating words), visual and hearing impairments, taste and smell deficits cognitive deficits: disorientation and confusion, short attention span, memory deficits, learning difficulties, dysphasia, poor judgement, perceptual deficits behavioural manifestations: agitation, impulsiveness, blunted affect, social withdrawal, depression.The most common result of traumatic brain injury is concussion — altered consciousness and altered neuronal activity; this may be mild or classical cerebral concussion (see Table 9-6). Mild concussion is characterised by immediate but temporary clinical manifestations. CSF pressure rises and ECG and EEG changes occur without loss of consciousness. The initial confusional state lasts for one to several minutes, possibly with amnesia (memory loss) for events prior to the trauma (retrograde amnesia). Anterograde amnesia for events after the trauma may also occur. Some people experience headache and complain of nervousness and ‘not being themselves’ for up to a few days.
Table 9-6 CATEGORIES OF DIFFUSE BRAIN INJURY
| TYPE OF INJURY | MECHANISM |
|---|---|
| Mild concussion | Temporary axonal disturbance affecting attentional and memory systems; consciousness not lost |
| Grade I | Confusion and disorientation with amnesia (momentary) |
| Grade II | Momentary confusion and retrograde amnesia after 5–10 minutes |
| Grade III | Confusion and retrograde amnesia from impact; also anterograde amnesia |
| Classic cerebral concussion (Grade IV) | Diffuse cerebral disconnection from brainstem reticular activating system; physiological/neurological dysfunction without substantial anatomical disruption; immediate loss of consciousness lasting less than 6 hours; retrograde and anterograde amnesia (posttraumatic) |
| Diffuse axonal injury | Prolonged traumatic coma (longer than 6 hours) |
| Mild | Posttraumatic coma lasts 6–24 hours; death uncommon; persistent residual cognitive, psychological and sensorimotor deficits; rare — only 8% of severe head injuries |
| Moderate | Widespread physiological impairment throughout the cerebral cortex and diencephalon; actual tearing of axons in both hemispheres; prolonged coma (longer than 24 hours); incomplete recovery among survivors; common — 20% of severe head injuries |
| Severe | Formerly called primary brainstem injury or brainstem contusion; severe mechanical disruption of axons in both hemispheres, diencephalon and brainstem; 16% of severe head injuries |
In classic cerebral concussion, consciousness is lost for up to 6 hours and reflexes fail, causing falls. Breathing stops, the heart rate and blood pressure fall temporarily and vital signs quickly stabilise to within normal limits. Amnesia before and after the incident occurs, along with a confusional state lasting for hours to days. Head pain, nausea, fatigue, attentional and memory system impairments (inability to concentrate and forgetfulness) and mood changes (anxiety, depression, irritability, fatigability, insomnia) occur. A postconcussive syndrome, including headache, nervousness or anxiety, irritability, insomnia, depression, inability to concentrate, forgetfulness and fatigability, may exist. Treatment entails reassurance and symptomatic relief in addition to 24 hours of close observation.
EVALUATION AND TREATMENT
Evaluation includes a complete history and physical examination. Brain imaging including X-rays, CT and MRI scans is used to distinguish between focal and diffuse injuries, as well as to locate any haematoma. Large contusions and lacerations with haemorrhage may be surgically excised (cut out). Otherwise, treatment is directed at controlling intracranial pressure and managing symptoms.
The degree of brain injury can be clinically assessed using the Glasgow coma scale, as a series of responses to verbal and painful stimuli (refer to Chapter 8). The patient may have a score of 3 (if there are no detectable responses) through to 15 (if no abnormalities are detected). The patient may improve from a low score to a higher score within one day if the injury is only mild. The time that it takes for the patient to regain memories is also considered.34
Treatment of haematoma depends on its location. Epidural haematomas are treated using surgical ligation (tying off), while chronic subdural haematomas require a craniotomy (opening the skull) to remove the gelatinous blood. Evacuation of a singular intracerebral haematoma has only occasionally been helpful. Otherwise, treatment is directed at reducing intracranial pressure and allowing the haematoma to reabsorb slowly (as described in the section on haemorrhagic stroke earlier in the chapter).
Open-head injuries often require debridement to remove the dead and traumatised tissues to prevent infection and to remove blood clots, thereby reducing intracranial pressure. Intracranial pressure is managed with corticosteroids and diuretics (drugs that cause fluid excretion). Broad-spectrum antibiotics are administered to prevent infection.
Early and late seizures must be prevented and controlled — phenytoin (an antiepileptic agent) is effective in preventing early seizures (within 7 days), but prevention of later seizures is more difficult.36 Acquired brain injuries result in disability in many cases, the most common being physical, intellectual and psychiatric, with others including vision, hearing and speech disability.34
Spinal cord trauma
The incidence of spinal cord trauma is quite low in Australia, with fewer than 300 new cases each year: 83% of these cases are males, with the highest incidence being in the 15–24 age group, followed by the elderly (over 65). Over half of all cases are transport-related, and another third are due to falls.37
PATHOPHYSIOLOGY
Spinal cord injuries most commonly occur because of injuries to the vertebral column (spine), which contains the spinal cord (refer to Chapter 20). Vertebral injuries in adults occur most often at the cervical vertebrae and between the last few thoracic and first few lumbar vertebrae, the most mobile portions of the vertebral column. Injuries to the cord are summarised in Table 9-7. Quadriplegia occurs if all limbs are paralysed (with injury to the cervical region), while paraplegia involves paralysis of the torso, which occurs if the injury is lower than T1.
Table 9-7 SPINAL CORD INJURIES
| INJURY | DESCRIPTION |
|---|---|
| Cord concussion | Results in a temporary disruption of cord-mediated functions |
| Cord contusion | Bruising of the neural tissue causing swelling and temporary loss of cord-mediated functions |
| Cord compression | Pressure on the cord causing ischaemia to tissues; must be relieved (decompressed) to prevent permanent damage to the spinal cord |
| Laceration | Tearing of the neural tissues of the spinal cord; may be reversible if only slight damage is sustained by the neural tissues; may result in permanent loss of cord-mediated functions if the spinal tracts are disrupted |
| Transection | Severing of the spinal cord, causing permanent loss of function |
| Complete | All tracts in the spinal cord are completely disrupted; all cord-mediated functions below the transection are completely and permanently lost |
| Incomplete | Some tracts in the spinal cord remain intact, together with functions mediated by these tracts; has the potential for recovery, although function is temporarily lost |
| Preserved sensation only | Some demonstrable sensation below the level of injury |
| Preserved motor non-functional | Preserved motor function without useful purpose; sensory function may or may not be preserved |
| Preserved motor functional | Preserved voluntary motor function that is functionally useful |
| Haemorrhage | Bleeding into the neural tissue as a result of blood vessel damage; usually no major loss of function |
| Damage or obstruction of spinal blood supply | Causes local ischaemia |
With injury, microscopic haemorrhages appear in the central grey matter and the pia mater as well; these increase in size causing necrosis (cell death). Oedema in the white matter occurs, impairing the microcirculation of the cord. The haemorrhaging and oedema obstruct blood flow and lead to development of ischaemic areas. Temporary cord swelling causes impairment, which is difficult to distinguish from permanent dysfunction. In the cervical region, cord swelling may be life-threatening if it impairs the diaphragm function and functions mediated by the medulla oblongata.
CLINICAL MANIFESTATIONS
Normal activity of the spinal cord cells at and below the level of injury ceases after cord injury, thus causing spinal shock. Reflex function is completely lost in all segments below the lesion, including all skeletal muscles; bladder, bowel and sexual function; and autonomic control. Severe impairment below the level of the lesion is obvious and it includes paralysis of the muscles and absence of sensation. The condition also results in disturbed thermal control by the hypothalamus, because the sympathetic nervous system is damaged and blood vessels cannot be constricted to maintain body heat; therefore, the individual assumes the temperature of the air (poikilothermia). Spinal shock generally lasts days to months. It terminates with the reappearance of reflex activity, such as reflex emptying of the bladder.
Autonomic hyperreflexia may occur after spinal shock resolves. This is a massive cardiovascular response to stimulation of the sympathetic nervous system (see Figure 9-12). The condition is life-threatening and requires immediate treatment. Individuals most likely to be affected have lesions at the T6 level or above. Characteristics include hypertension (up to 300 mmHg systolic), pounding headache, blurred vision, sweating above the level of the lesion with flushing of the skin, nasal congestion, nausea, piloerection caused by pilomotor spasm and very slow heart rate (bradycardia, 30–40 beats/minute).
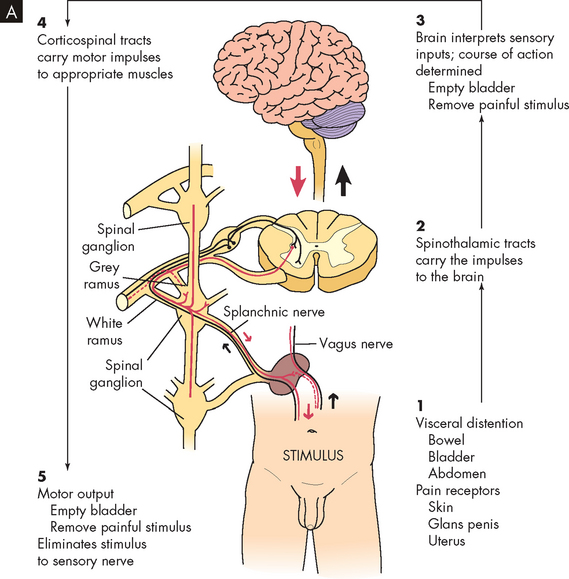
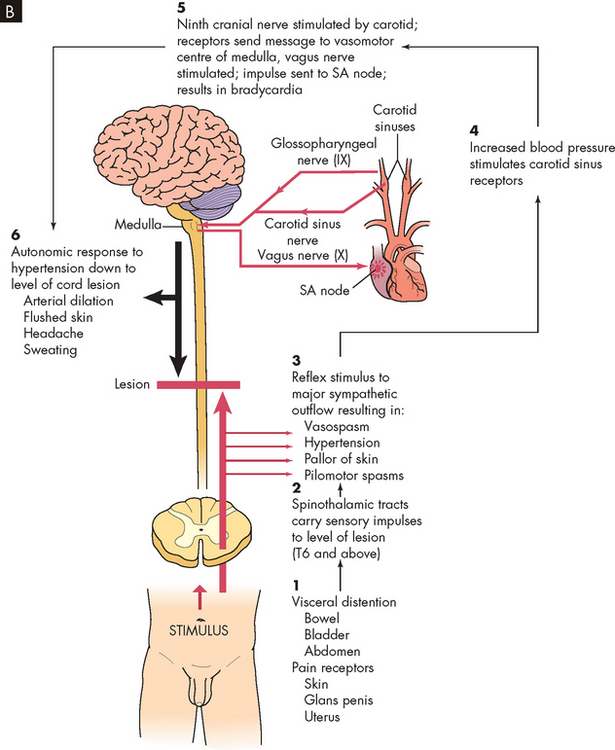
FIGURE 9-12 Autonomic hyperreflexia.
B Autonomic hyperreflexia pathway. SA, sinoatrial.
Source: Modified from Rudy EB. Advanced neurological and neurosurgical nursing. St Louis: Mosby; 1984.
In autonomic hyperreflexia, sensory receptors below the level of the cord lesion are stimulated. The intact autonomic nervous system reflexively responds with an arteriolar spasm that increases blood pressure. Baroreceptors in the cerebral vessels, the carotid sinus and the aorta sense the hypertension and stimulate the parasympathetic system. The heart rate decreases, but the visceral and peripheral vessels do not dilate because efferent impulses cannot pass through the cord.
The most common cause of autonomic hyperreflexia is a distended bladder or rectum, but any sensory stimulation can elicit autonomic hyperreflexia. Stimulation of the skin or pain receptors may cause autonomic hyperreflexia. Bladder or bowel emptying usually relieves the syndrome and drugs such as phenoxybenzamine (to block α-adrenergic receptors) may facilitate this result.
EVALUATION AND TREATMENT
Diagnosis of spinal cord injury is based on physical examination, CT scan, MRI and myelography (X-ray of the spinal cord using dye). For a suspected or confirmed vertebral injury, immediate immobilisation of the spine is essential to prevent further trauma. Decompression and surgical fixation may be necessary. Corticosteroids decrease additional cord injury from inflammation. Nutrition, lung function, skin integrity and bladder and bowel management must be addressed. Plans for rehabilitation need early consideration. In cases of autonomic hyperreflexia, intervention must be prompt because cerebrovascular accident is possible. Antihypertensive medications may be used if blood pressure remains elevated.
DEGENERATIVE DISORDERS OF THE CENTRAL NERVOUS SYSTEM
Up until this point, we have explored conditions that cause acute damage to the central nervous system. Patients with these conditions may experience significant neurological impairment, but many have the capacity to recover some, if not all, neurological function. However, there are many disorders that can significantly incapacitate individuals, although this may occur over several years. The most prevalent of these in Australia and New Zealand, especially in the older population, are the degenerative disorders — those that cause a progressive, irreversible decline in neurological function. The most common of the degenerative disorders are dementias, with Alzheimer’s disease being the most prevalent.
Alzheimer’s disease
Alzheimer’s disease has been demonstrated to be one of the most common causes of severe cognitive dysfunction in older people. In Australia, Alzheimer’s disease accounts for approximately 50–70% of all cases of dementia. To put this in perspective, dementia is the most prevalent condition for people aged 65 years and older: almost 1 in 15 people aged over 65 are diagnosed with dementia, and this rises to 1 in 4 for those aged over 85.38 Furthermore, the number of people with dementia is projected to increase, with estimations that by 2050 almost three-quarters of a million Australians will have dementia.
PATHOPHYSIOLOGY
The exact cause of Alzheimer’s disease is unknown, but it is characterised by a decreased brain size — there is evidence that the cerebral cortex (grey matter) actually shrinks in Alzheimer’s disease (see Figure 9-13). In addition to loss of neurons, there is also a loss of neuronal synapses (or connections). Synapses are the key to the formation of memories, so loss of synapses causes loss of the memories that were stored in that area.39 There are also differences in neurotransmitters: an increased level of glutamate (excitatory neurotransmitter) and a decreased level of acetylcholine.
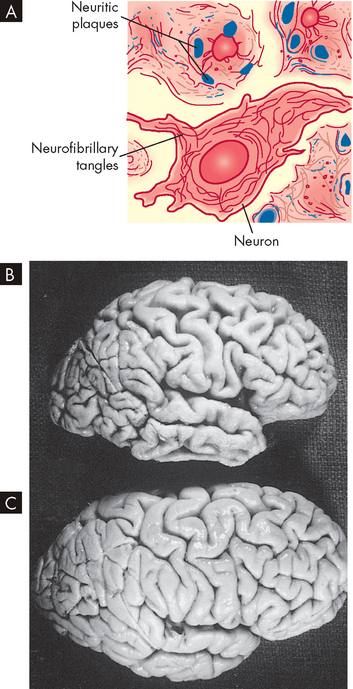
FIGURE 9-13 Pathological changes in Alzheimer’s disease.
A Schematic representation of neuritic plaque and neurofibrillary tangle. B The gross brain of an Alzheimer’s patient: note the reduced size, narrow gyri and wide sulci, mainly in the frontal and temporal lobes. C An age- and sex-matched brain of an individual without Alzheimer’s disease.
Source: A Brown D, Edwards H. Lewis’s medical–surgical nursing. 2nd edn. Sydney: Elsevier; 2008. B & C Damjanov I, Linder J (eds). Anderson’s pathology. 10th edn. St Louis: Mosby; 1996.
At the cellular level, there are two key changes in the neurons that can be observed microscopically (see Figure 9-13):
Senile plaques and neurofibrillary tangles are more concentrated in the cerebral cortex — the area of most apparent brain shrinkage — and hippocampus. The decline in cognitive processes is directly related to the number of these abnormalities seen in postmortem examination, with the plaques being strongly associated with early symptoms.40,41
CLINICAL MANIFESTATIONS
Initial clinical manifestations are insidious and often attributed to forgetfulness, emotional upset or other illness — these vague manifestations mean that Alzheimer’s disease cannot be conclusively diagnosed in the early stages. The individual becomes progressively more forgetful over time, particularly in relation to recent events. Memory loss increases as the disorder advances and the person becomes disoriented and confused and loses the ability to concentrate. Abstraction, problem solving and judgement gradually deteriorate. Behavioural changes include irritability, agitation and restlessness. Mood changes result and the person may become anxious, depressed, hostile and prone to mood swings. Interestingly, the mood changes may also seem more positive, so someone who previously was difficult to get along with may become more compliant. Motor changes may occur if the posterior frontal lobes are involved, causing rigidity (paratonia) with flexion posturing (leaning forward). Great variability in age of onset, intensity and sequence of symptoms, and location and extent of brain abnormalities is common. For example, those who develop Alzheimer’s at a younger age often deteriorate more quickly than those who develop the condition at a much older age.
EVALUATION AND TREATMENT
The diagnosis of Alzheimer’s disease is made by ruling out other causes. The history and course of the illness, which may span 5 years or more, is used in diagnosis. The patient undergoes a mental status examination, which has a number of questions such as the date, calculations and recall. Brain imaging using CT, MRI or PET is used to assist in excluding other causes (see Figure 9-14). Because it takes a long time to confirm the diagnosis, the disease is usually advanced by this stage.42 In addition, genetic testing for substances with key roles in the senile plaques is possible (test for amyloid precursor protein and apolipoprotein E43). However, these genetic factors cannot be used as a predictor of who will get the disease, as not all people with the genetic abnormalities have Alzheimer’s disease.
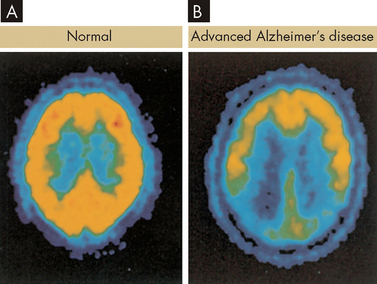
FIGURE 9-14 Positron emission tomography (PET) used in the diagnosis of Alzheimer’s disease.
Radioactive fluorine is applied to glucose, which is taken up by metabolically active cells (yellow areas). A Normal brain. B Advanced Alzheimer’s disease is recognised by hypometabolism in many areas of the brain.
Source: Brown D, Edwards H. Lewis’s medical–surgical nursing. 2nd edn. Sydney: Elsevier; 2008.
Treatment is directed at using devices to compensate for the impaired cognitive function, such as memory aids; maintaining unimpaired cognitive function; and maintaining or improving the general state of hygiene, nutrition and health. There are a few drugs available that increase the concentration of acetylcholine (mainly by limiting its breakdown) and these have had a modest effect on cognitive function in the early stage of Alzheimer’s disease (donepezil, galantamine, rivastigmine). Memantine is a drug that interacts with the other main neurotransmitter activity — it blocks glutamate activity and is used to slow progression of disease in moderate to severe Alzheimer’s disease.44–46 4 6 There is currently no drug available that is able to correct the damage to the neurons.
Parkinson’s disease
Parkinson’s disease is a commonly occurring degenerative disorder of the basal nuclei (previously known as basal ganglia) (corpus striatum) involving failure of the neurons that secrete dopamine.
Parkinson’s disease begins after the age of 40 years, with onset for most people around the age of 60.47 It is slightly more common in males. This disease is one of the most prevalent of the primary CNS disorders and is a leading cause of neurological disability in individuals older than 60 years. It is estimated that at least 54,000 individuals were diagnosed with Parkinson’s disease in Australia in 2005. However, there is difficulty with the diagnosis and the exact figure remains unclear. Nevertheless, the prevalence increases substantially with age and in Australia it is estimated that 290 per 100,000 people aged 55–64 years have Parkinson’s, and this increases to 2940 per 100,000 for people aged over 85 years.48
PATHOPHYSIOLOGY
The cause of Parkinson’s disease is unknown. Loss of neurons in the substantia nigra is key; these neurons release dopamine into the basal nuclei, hence Parkinson’s is characterised by loss of neurons with depletion of dopamine (an inhibitory neurotransmitter); (see Figure 9-15A). Dopamine depletion in the basal nuclei results in a relative excess of cholinergic activity in the feedback circuit. This is manifested by hypertonia (tremor and rigidity) and akinesia (‘a’ refers to without, thus meaning without movement).
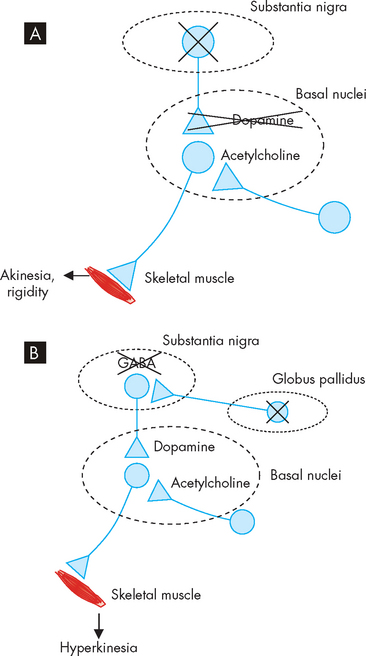
FIGURE 9-15 Neurotransmitter imbalances in Parkinson’s disease and Huntington’s disease.
A In Parkinson’s disease, loss of neurons from the substantia nigra results in less dopamine transmission; as a result, the amount of acetylcholine is increased and the muscle movement becomes lessened, resulting in rigidity. B In Huntington’s disease, loss of neurons from the globus pallidus results in loss of the inhibitory neurotransmitter GABA; as a result, the amount of dopamine is effectively increased, resulting in excessive movements.
CLINICAL MANIFESTATIONS
The classic manifestations of Parkinson’s disease are tremor at rest, rigidity (muscle stiffness) and bradykinesia (slow movements). Others are postural disturbance, difficulty speaking (dysarthria) and difficulty swallowing (dysphagia). As the disease progresses, all are usually present. Because the onset is insidious, the beginning of symptoms is difficult to document. Early in the disease, reflex status, sensory status and mental status usually are normal. Postural abnormalities (flexed, forward leaning; see Figure 9-16), difficulty walking and weakness develop. Depression is common. Other difficulties due to autonomic nervous system imbalance include inappropriate diaphoresis (sweating), orthostatic hypotension, drooling, gastric retention, constipation and urinary retention.
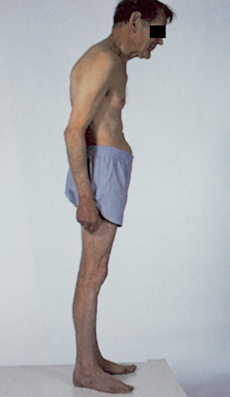
FIGURE 9-16 The stooped posture of Parkinson’s disease.
Source: Perkin DG. Mosby’s color atlas and text of neurology. 2nd edn. London: Mosby; 2002.
Disorders of equilibrium result from postural abnormalities. The person with Parkinson’s disease cannot make the appropriate postural adjustment to tilting or falling and falls like a post when starting to tilt. The festinating gait (short, accelerating steps) of the individual with Parkinson’s disease is an attempt to maintain an upright position while walking. Individuals are also unable to right themselves when changing from a reclining or crouching position to a standing position and when rolling over from a supine to a lateral or prone position. Excessive daytime sleepiness is experienced in more than 50% of cases.49 Postural instability, sleep disturbance and difficulty concentrating are some of the most depressing symptoms for persons with the disease.50
Progressive dementia may be associated with the disease and is more common in those older than 70 years. The person’s mental status may be further compromised by the side effects of the medication taken to control symptoms.
Surgery for Parkinson’s disease
Surgery to reduce parkinsonian motor symptoms has been an effective treatment procedure for more than 40 years. Precise lesions are made in the subthalamic nucleus, globus pallidus and thalamus, resulting in rapid reduction of symptoms in a high number of individuals with the disease. The use of concise imaging techniques allows precision in the placement of therapeutic lesions. Advances in new technologies, including gamma knife radiosurgery and deep brain stimulation from implanted electrodes, provide relief for individuals with advanced disease. Gene therapy to promote dopaminergic neurons and implantation of genetically engineered stem cells is also being explored.
Source: Jankovic J. Update on the treatment of Parkinson’s disease. Mt Sinai J Med 2006; 73(4):682–689; Pereira EA, Aziz TZ. Surgical insights into Parkinson’s disease. J R Soc Med 2006; 99(5):238–244.
EVALUATION AND TREATMENT
The diagnosis of Parkinson’s disease is based on the history and physical examination; PET scans are also useful. Treatment of Parkinson’s disease is symptomatic, involving drug therapy to increase dopamine levels such as levodopa (which the body uses to produce dopamine) or a dopamine agonist (such as carbidopa, with several others now available). Because of troublesome side effects and loss of effectiveness, drug therapy may not be started until the symptoms become incapacitating. However, advanced Parkinson’s disease is often unresponsive to these dopamine-related agents. Surgical interventions include pallidotomy to surgically inactivate the globus pallidus (see the box ‘Health alert: surgery for Parkinson’s disease’).51,52 Parkinson’s disease progresses slowly for 15–20 years before producing severe immobility and dependence.
Huntington’s disease
Huntington’s disease, also known as chorea (from the Greek word for ‘dancing’, because the movements can appear dance-like) is a relatively rare, hereditary disorder. In Australia, Huntington’s disease affects approximately 6–7 people in every 100,000.53 The disease affects several brain areas, mainly the basal nuclei. There is a gradual onset from the age of 30–50 years with uncontrollable movements (chorea), which progresses to a general lack of coordination. Other symptoms include difficulty in decision making, irritability and mood fluctuations. As the disease continues, further debilitating symptoms arise, such as a decrease in intellectual capacity, until death, usually 20 years after onset of the disease. Offspring of an individual with Huntington’s disease have a 50% chance of inheriting the disease. Those at high risk of the disease may choose to undergo prenatal genetic testing to ascertain the likely health of their offspring; although the overall usage of this testing is low, some people use alternative reproductive options to avoid passing on the genetic abnormality.54 Regardless of reproductive decisions, an individual who is asymptomatic but has family members affected may opt for genetic testing to ascertain their own status of carrying the altered gene.
PATHOPHYSIOLOGY
Huntington’s disease is inherited as an autosomal dominant trait, which means that offspring only need to inherit one copy of this abnormal gene in order to exhibit symptoms of the disease (see Chapter 5 on genes). The principal pathological feature of Huntington’s disease is severe degeneration within the basal nuclei of the neurons that send inhibitory signals (using the neurotransmitter gamma-aminobutyric acid, GABA) in control of motor function; therefore, degeneration in this area allows excessive motor output and more abnormal movements. Neuronal loss removes the inhibitory pathway, thereby causing decreased inhibitory GABA activity on dopaminergic neurons in the substantia nigra. As a consequence, there is a relative excess of dopaminergic activity in the basal nuclei feedback circuit with the cerebral cortex. In some ways, Huntington’s disease is the opposite of Parkinson’s in terms of neurotransmitters — whereas Parkinson’s is characterised by a deficiency of dopamine, Huntington’s has excess dopamine (see Figure 9-15B).
CLINICAL MANIFESTATIONS
The classic manifestations of Huntington’s disease are abnormal movement and progressive dysfunction of intellectual and thought processes (dementia). Any one of these features may mark the onset of the disease. Chorea, the most common type of abnormal movement affecting these individuals, begins in the face and arms, eventually affecting the entire body. A range of neurological deficits include loss of working memory and reduced capacity to plan, organise and sequence. Thinking is slow and patients may experience apathy, irritability or depression.
EVALUATION AND TREATMENT
The diagnosis of Huntington’s disease is based on family history and clinical presentation of the disorder. No known treatment is effective in halting the degeneration or progression of symptoms. Depression or psychosis is treated with drug therapy. The patient requires adequate support and palliative care; the dependency on others providing adequate care increases as the disease progresses, leading to a long-term requirement for palliative care.55,56 One important drug treatment is tetrabenazine, which depletes the stores of dopamine in the brain.57 However, the dosage must be slowly increased to produce the desired effect on movements; a dose too high can result in a Parkinson’s-like syndrome. Another adverse effect is depression,58 so caution must be taken to avoid worsening the patient’s existing depression.
Multiple sclerosis
Multiple sclerosis (MS) is a relatively common disorder involving destruction of previously normal myelin of axons within the CNS. The onset of multiple sclerosis is usually between 20 and 50 years of age. It affects three times more females than males59 and is a leading cause of neurological disability in early adulthood. Although the disorder does not exhibit a defined inheritance pattern, 15% of those with MS have an affected relative. The prevalence in Australia is approximately 2.4 per 100,000 people.60
PATHOPHYSIOLOGY
Multiple sclerosis involves an autoimmune process that develops when a previous viral insult to the nervous system has occurred in a genetically susceptible individual. This gene is activated in astrocytes of persons with multiple sclerosis, resulting in some inflammatory processes that lead to the destruction of oligodendrites, the myelin-forming cells.61 Pathological features of this process are: (1) interaction between the immune system and the CNS; and (2) demyelination of the white matter.
The acute (early) stage of plaque formation is characterised by demyelination with inflammatory oedema. Symptoms usually remit, partially or completely, weeks after the onset of an early episode. The chronic stage of demyelination and plaque formation is characterised by gliosis (glial scarring with late degeneration of axons). Progressive loss of function leads to permanent disability, usually after 20 years or more.
CLINICAL MANIFESTATIONS
Various events occur immediately before the onset or exacerbation of symptoms and are regarded as precipitating factors. Infection, trauma and pregnancy are the least debated. Most of the pregnancy-related exacerbations occur 3 months postpartum, suggesting a relation to the stresses of labour and the increased fatigue during the postpartum period rather than to the pregnancy itself.
The major manifestations of MS are the initial syndromes (see Table 9-8), followed by remissions (apparent absence of disease) and established syndromes with no remissions. Usually people with late MS predominantly have one of the following established syndromes: mixed (generalised), spinal or cerebellar. The syndrome depends on the portion of the CNS most involved. Early cognitive changes are now being found on testing of asymptomatic persons. After several years, 50% of individuals appear to have established syndromes of mixed involvement.
Table 9-8 SYMPTOMS OF MULTIPLE SCLEROSIS
Source: Brain Foundation. Multiple sclerosis. 2009. Available at www.brainaustralia.org.au/AZ_of_Brain_Disorders2/multiple_sclerosis.
Short-lived attacks of neurological deficits are the temporary appearance or worsening of symptoms. The mechanism of these attacks is complete, reversible conduction block in partially demyelinated axons. Conditions that cause short-lived attacks include: (1) minor increases in body temperature or serum calcium concentration; and (2) functional demands exceeding the conduction capacity of the neurons. An increase in body temperature or serum calcium level increases current leakage through demyelinated neurons, so action potentials become transmitted less effectively.
Paroxysmal attacks (sudden recurrence) are sensory or motor symptoms of abrupt onset and short duration (a few seconds or minutes) and include dysarthria (difficulty speaking) and ataxia (lack of coordination) and tonic head turning. The mechanism of these attacks is due to nerve impulses being directly transmitted between adjacent demyelinated axons. A classical paroxysmal symptom, called Lhermitte’s sign, is the momentary paraesthesia (shock-like or tingling sensation) that shoots down the trunk or limbs during flexion of the neck. Paroxysmal attacks tend to persist for weeks or months and may be followed by progressive symptoms of multiple sclerosis.
EVALUATION AND TREATMENT
The diagnosis of MS is based on the history and physical examination, supported by findings from CSF examination, visual evoked potentials (testing the conduction efficiency of the optic nerves) and MRI.59 Persistently elevated CSF immunoglobulin G (IgG; discussed in Chapter 12) is found in most people with MS due to the inflammatory response. MRI is the most sensitive method available of detecting the disease. Signs of two separate attacks or flares with demyelination in the CNS support the diagnosis.
Neural stem cells protect and restore brain functions
Transplant of adult neural stem cells can protect and restore functions of the brain in animal models of ischaemic and inflammatory brain injury. Some subsets of stem cells replace damaged tissue and also have modulated immune responses with potential benefit in diseases like multiple sclerosis. Progress towards the use of both embryonic and adult neural stem cells for neuroprotective and regenerative interventions holds much promise for the future.
Source: Leker RR. Manipulation of endogenous neural stem cells following ischaemic brain injury. Pathophysiol Haemost Thromb 2006; 35(1–2):58–62; Sailor KA, Ming GL, Song H. Neurogenesis as a potential therapeutic strategy for neurodegenerative diseases. Expert Opin Biol Ther 2006; 6(9):879–890; Uccelli A et al. Stem cells in inflammatory demyelinating disorders: a dual role for immunosuppression and neuroprotection. Expert Opin Biol Ther 2006; 6(1):17–22.
Multiple sclerosis is treated for three purposes: (1) acute managing of relapses to prevent disability; (2) reducing the frequency of relapses and disease progression; and (3) managing symptoms.62 Drugs with anti-inflammatory properties (corticosteroids) are used to treat acute episodes. Other drugs may be useful in slowing progression of the disease by suppressing the T lymphocyte responses (which may be involved in myelin destruction) — these include interferons and glatiramer.57,59 Symptom management is also part of treatment plans. Special problems requiring preventive and symptomatic management are: fatigue; weakness; spasticity; bladder, bowel and sexual dysfunction; pain; tremor and ataxia; depression; vertigo; sensory sensations; and heat intolerance. Supportive and rehabilitative management is directed towards relieving specific symptoms and preventing the complications of immobility — especially pressure sores and infections of the pulmonary and genitourinary systems.
Motor neuron disease
Motor neuron disease (also known as amyotrophic lateral sclerosis or Lou Gehrig’s disease) is a degenerative disorder involving lower and upper motor neurons and resulting in progressive muscle weakness. The term amyotrophic (progressive muscle wasting) refers to the predominant lower motor neuron component of the syndrome. Lateral sclerosis, scarring of the corticospinal tract (refer to Figure 6-19A) in the lateral column of the spinal cord, refers to the upper motor neuron component of the syndrome. It usually begins in the early 50s, with males affected slightly more than females. Stephen Hawking (the British physicist) is one of the most famous people living with motor neuron disease. The disease affects approximately 1200 people in Australia, with 400 new cases being diagnosed each year.63
PATHOPHYSIOLOGY
The cause of motor neuron death is unknown. Current data suggest that a genetic factor is involved: 20% of people with familial motor neuron disease have a genetic mutation in the glial cells surrounding the motor neuron.64,65 The principal pathological feature of motor neuron disease is the degeneration of motor neurons in the brainstem and spinal cord. Death of the motor neuron results in axonal degeneration and secondary demyelination with glial proliferation and sclerosis (scarring). Lower motor neurons adjacent to those that have degenerated attempt to compensate by distal intramuscular sprouting, re-innervation and enlargement of motor units.
CLINICAL MANIFESTATIONS
Weakness and wasting may begin in any or all muscles of the body (see Figure 9-17). Paralysis occurs with progressive muscle atrophy. No associated mental, sensory or autonomic symptoms are present. Sensory functions are sustained until death.
FIGURE 9-17 Motor neuron disease.
A This patient has progressive muscular atrophy and presented with wasting of the muscles between the thumb and index finger on the dorsal (arrow) and palmar surfaces. B Another patient with motor neuron disease showing tongue atrophy.
Source: A Goldman L. Cecil medicine. 23rd edn. Philadelphia: Saunders; 2008. B Bertorini TE. Neuromuscular case studies. Philadelphia: Butterworth-Heinemann; 2008.
EVALUATION AND TREATMENT
Diagnosis of the syndrome is based predominantly on the history and physical examination. Electromyography and muscle biopsy verify lower motor neuron degeneration and denervation. Little treatment is available to alter the overall course of motor neuron disease. The drug riluzole has delayed progression to that requiring ventilatory assistance.66 Supportive management and rehabilitative management are directed towards preventing complications of immobility. The average duration of life is approximately 2–3 years from the appearance of symptoms, but the course of the disease may run from a few months to 15 years.
PERIPHERAL NERVOUS SYSTEM AND NEUROMUSCULAR JUNCTION DISORDERS
Disease processes may injure the axons travelling to and from the neuronal cell bodies of the CNS. The injury may affect a distinct anatomical area on the axon, or the spinal nerves may be injured at the roots, at the plexus before peripheral nerve formation or at the nerves themselves. The cranial nerves do not have roots or plexuses and are affected only within themselves. Autonomic nerve fibres may be injured as they travel in certain cranial nerves and emerge through the ventral root and plexuses to travel in the peripheral nerves of the body.
Guillain-Barré syndrome
Guillain-Barré syndrome is an autoimmune disease whereby the myelin surrounding the neuronal axons in the peripheral nervous system is destroyed by the person’s immune system. Although the myelin destruction is the main concern, the damage may even include the actual axons as well. An autoimmune disease is caused by the immune system actually targeting and destroying a normal component of the healthy body, in the same way that it might destroy something foreign, such as bacteria that infect the body (see Chapter 14). The reasons why autoimmune diseases occur are complex and more research is needed to understand these causes of disease. The incidence in Australia is approximately 1–2 per 100,000 people.67
PATHOPHYSIOLOGY
Guillain-Barré is often associated with bacterial and viral infection, so the antibodies which are produced during that infection begin to destroy the myelin as well. Interestingly, a vaccine for swine influenza that was used in the 1970s was related to the development of Guillain-Barré syndrome and the recent epidemic of swine flu in 2009 caused this issue to resurface. Importantly, the vaccine that is being used for the 2009 outbreak is different from that used in the 1970s and there is no expected risk of Guillain-Barré with the current swine flu vaccination.67
CLINICAL MANIFESTATIONS
The lack of myelination around the nerves means that fewer action potentials reach the skeletal muscles; without receiving the signal from the neurons, the muscles are unable to contract, which leads to muscle weakness and paralysis. Weakness and paralysis of the respiratory muscles can become life-threatening. Other symptoms include abnormal sensation due to demyelination of the sensory receptors as well. Guillain-Barré is described as an ‘ascending paralysis’, because the loss of muscle function and sensory deficit usually begins in the feet and then progresses up the body to affect more areas.
TREATMENT
Treatment using plasmapheresis is effective for autoimmune disorders. This procedure takes a quantity of the patient’s blood and the plasma goes through a process whereby substances such as antibodies are removed from the plasma; the ‘washed’ plasma, which no longer contains the autoimmune antibodies, is returned to the patient. Most people actually recover within weeks or years,68 but most of this time will be spent hospitalised.
Myasthenia gravis
Myasthenia gravis is a chronic autoimmune disease that affects the neuromuscular junction and is characterised by muscle weakness and fatiguability. Myasthenia gravis affects approximately 3 in every 100,000 people in Australia, with about twice as many women affected as men.69
Generalised autoimmune myasthenia involves the proximal musculature throughout the body and has several courses: (1) a course with periodic remissions; (2) a slowly progressive course; (3) a rapidly progressive course; and (4) a fulminating course. In neonatal myasthenia, some signs of myasthenia gravis are present in 10–15% of infants born to mothers with myasthenia gravis, as the antibodies from the mother’s immune system can cross the placenta. Usually, the newborn recovers in the first weeks after birth.
PATHOPHYSIOLOGY
Myasthenia gravis results from a defect in nerve impulse transmission at the neuromuscular junction. The postsynaptic acetylcholine receptors on the muscle’s cell membrane are no longer recognised as normal components of the person’s own body; because they are recognised as foreign, the immune response causes production of antibodies, which results in destruction of the receptors for acetylcholine receptors. This causes diminished transmission of the nerve impulse across the neuromuscular junction and lack of muscle depolarisation (see Figure 9-18).
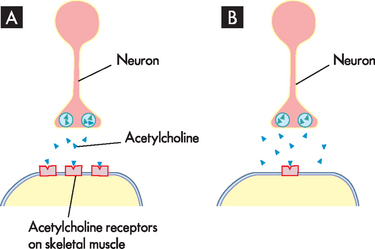
FIGURE 9-18 Pathophysiology of myasthenia gravis.
A Normally, acetylcholine is released from the neuron, which travels across the synapse and binds with acetylcholine receptors on the skeletal muscle. In response, the skeletal muscle contracts. B With myasthenia gravis, there is destruction of the acetylcholine receptors, so that there is very little acetylcholine binding on the skeletal muscle. As a result, the muscle does not receive a significant signal to undergo contraction.
CLINICAL MANIFESTATIONS
Myasthenia gravis typically has an insidious onset. Clinical manifestations may first appear during pregnancy, during the postpartum period or in conjunction with the administration of certain anaesthetic agents. The foremost complaints are muscle fatigue and progressive weakness. The first muscles to be affected are usually the eyes, as well as the face, mouth, throat and face. The person often complains of fatigue after exercise and has a recent history of recurring upper respiratory tract infections. The extraocular (eye) muscles and levator muscles are most affected. Manifestations include diplopia (double vision, due to eye muscles not working equally) and ocular palsies (paralysis). Individuals are unable to undergo strong, sustained movements of the eyes (see Figure 9-19).

FIGURE 9-19 ‘Peek’ sign in myasthenia gravis.
During sustained forced eyelid closure this patient with myasthenia gravis is unable to bury his eyelashes (left), and after 30 seconds he is unable to keep the lids fully closed (right).
Source: Reproduced with permission from Sanders DB, Massey JM. Clinical features of myasthenia gravis. In: Engel AG (ed.). Neuromuscular junction disorders. New York: Elsevier; 2008.
The muscles of facial expression, mastication (chewing), swallowing and speech are the next most involved. The results are facial droop and an expressionless face; difficulty chewing and swallowing associated with dietary changes and weight loss; drooling and episodes of choking and aspiration; and a nasal, low-volume but high-pitched monotonous speech pattern.
The muscles of the neck, shoulder girdle and hip flexors are less frequently affected. When these muscles do become involved, however, the person experiences fatigue requiring periods of rest, weakness of the arms and legs that improves with rest, and difficulty in maintaining head position. The respiratory muscles of the diaphragm and chest wall become weak and ventilation is impaired. Deep breathing and coughing difficulties result. In the advanced stage of the disease, all muscles are weak.
Myasthenic crisis occurs when severe muscle weakness causes extreme quadriplegia, respiratory insufficiency with shortness of breath and extreme difficulty in swallowing. The individual in myasthenic crisis is in danger of respiratory arrest.
Cholinergic crisis may arise from anticholinesterase drug toxicity. The clinical picture is like that of myasthenic crisis, but other symptoms are present also. Intestinal motility increases, with episodes of diarrhoea and complaints of cramping, fasciculation (muscle twitches just below the skin), bradycardia (slow heart rate), pupillary constriction, increased salivation and increased sweating. These are caused by the smooth muscle hyperactivity secondary to excessive accumulation of acetylcholine at the neuromuscular junctions and excessive parasympathetic-like activity (refer to Chapter 6 for the parasympathetic nervous system neurotransmitters). As in myasthenic crisis, the individual is in danger of respiratory arrest.
EVALUATION AND TREATMENT
The diagnosis of myasthenia gravis is made on the basis of different tests including an electromyogram to assess the amount of muscle activity. The purpose of the electromyogram is to electrically stimulate the neurons and measure the corresponding size of the response in the muscle. Testing for the response to edrophonium (Tensilon) may be used, although access to the drug is limited in Australia. This drug is an anticholinesterase drug (it limits the breakdown of neurotransmitter by acetycholinesterase), which means that it allows more acetylcholine to be available at synapses; if there is more acetylcholine at the skeletal muscle, then its effect at the remaining receptors can be maximised. With intravenous administration of the drug, immediate demonstrable improvement in muscle strength usually persists for several minutes. The progression of myasthenia gravis varies, appearing first as a mild case that spontaneously remits, with a series of relapses and symptom-free intervals ranging from weeks to months. Over time the disease can progress, leading to death.
In addition, corticosteroids and immunosuppressant drugs are used to treat myasthenia gravis and myasthenic crisis. Plasmapheresis is effective in removing the antibodies from the plasma. For individuals with cholinergic crisis, anticholinergic drugs are withheld until blood levels fall out of the toxic range, while ventilatory support is provided and respiratory complications are prevented.
INFECTION AND INFLAMMATION OF THE CENTRAL NERVOUS SYSTEM
Meningitis
Meningitis is an infection of the meninges, including the pia mater and arachnoid, subarachnoid space, ventricular system and CSF. Meningitis may be caused by a number of different microorganisms, with the most common types in Australia being bacterial and viral causes. A systemic or bloodstream infection or a direct extension from an infected area is the access route to the subarachnoid space.
Most cases of bacterial meningitis in Australia are due to meningococcus (Neisseria meningitidis), with approximately 1–3.5 people affected per 100,000 per year.70,71 Fatality rates are approximately 7% of affected people.71 This is also the most common organism to cause bacterial meningitis in children, accounting for 60% of all paediatric cases of meningitis.72 Approximately 2–5% of healthy children are carriers of N. meningitidis. The risk of developing meningitis from day-care centre contact with children with meningococcal disease is 1 per 1000.73 Meningococcal meningitis occurs predominantly in males, with epidemics predominantly affecting children and adolescents. Young people are at high risk due to their high amount of social activities and close personal contact.74
Other causes of bacterial meningitis are less common in Australia now that the childhood immunisation program includes a vaccination for N. meningitidis type C. However, type B is still prevalent — of the 281 Australians confirmed with N. meningitidis in one year, 85% were type B.75 Vaccinations for other bacterial meningitis are also included in the childhood immunisation program for Haemophilus influenzae and Streptococcus pneumoniae. In New Zealand, a national immunisation program for N. meningitidis type B was implemented after an epidemic several years ago, which reached its peak in 2001. Most of the New Zealand population under the age of 20 has received this vaccination, although it is no longer on the childhood immunisation schedule since nearing the end of the epidemic.76
Viral causes of meningitis include herpes simplex virus type 2, as well as varicella zoster virus (which causes chickenpox) and the viruses that cause measles and mumps. In the case of herpes infection, there is usually genital herpes associated with the meningitis.77 Viral meningitis is not a reportable disease in Australia, so exact population statistics are not available. However, although it is more common than the bacterial form, it usually causes a less-severe meningitis.78 It may resolve within approximately one week.
PATHOPHYSIOLOGY
The bacteria that usually cause meningitis are common inhabitants of the nasopharynx (the back of the nasal cavity; see Chapter 24), but a predisposing factor such as a prior upper respiratory infection must be present before the bacteria become blood-borne. The bacteria function as irritants and induce an inflammatory reaction by the meninges, CSF and ventricles. Blood cells (neutrophils) migrate into the subarachnoid space, producing an exudate that thickens the CSF and interferes with normal CSF flow around the brain and spinal cord — this is key in diagnosing the type of meningitis. This can obstruct arachnoid villi and produce hydrocephalus. Further inflammation occurs as the purulent exudate (pus containing bacterial and immune cells) increases rapidly, especially around the base of the brain, and extends into the sheaths of the cranial and spinal nerves and into the perivascular spaces of the cortex. Meningeal cells become oedematous and the combined exudate and oedematous cells increase intracranial pressure. Small and medium-sized subarachnoid arteries, veins and choroid plexuses become engorged, disrupting blood flow and potentially producing thrombosis. Secondary infection of the brain may occur.
For viral meningitis, the infection usually begins in the lining of the respiratory or gastrointestinal tract. From there, the virus enters the lymphatic system and travels to the blood, finally crossing the blood–brain barrier to reach the meninges.74
CLINICAL MANIFESTATIONS
Meningitis is characterised by severe headache and fever. The clinical manifestations of bacterial meningitis can be grouped into meningeal signs, infectious signs and neurological signs. The clinical manifestations of the systemic infection include rapid onset of fever, tachycardia, chills and a petechial rash. Clinical manifestations that are more specific to meningeal irritation are a generalised throbbing headache that becomes very severe, photophobia (inability to tolerate light) that becomes severe and nuchal rigidity (neck stiffness, which results in inability to flex the head forward). The neurological signs include a decrease in consciousness, cranial nerve palsies, seizures and focal neurological deficits that include weakness (hemiparesis) or paralysis (hemiplegia) on one side of the body and ataxia (uncoordination of movements). Often the vomiting centre is irritated, causing projectile vomiting. With meningococcal meningitis, a petechial or purpuric rash covers the skin and mucous membranes (see Figure 9-20). As intracranial pressure increases, papilloedema develops and delirium may progress to unconsciousness.
FIGURE 9-20 Skin lesions with meningitis.
Characteristic purpura with petechiae and ecchymoses in a patient who has severe sepsis and meningitis due to Neisseria meningitidis.
Source: Courtesy of Professor W Zimmerli, University of Basel, Switzerland. From Cohen J, Powderly WG. Infectious diseases. 2nd edn. St Louis: Mosby; 2004.
The clinical manifestations of viral meningitis are similar although mild compared with those associated with bacterial meningitis. Mild, generalised throbbing headache, mild photophobia, mild neck pain, stiffness, fever and malaise accompany viral meningitis.
EVALUATION AND TREATMENT
Diagnosis of meningitis is based on physical examination, nasopharyngeal smear and antigen tests. CSF cultures may be needed to confirm the cause. One of the distinguishing features between bacterial and viral causes of meningitis is determined by laboratory analysis of the main leucocytes (white blood cells) in the CSF — in bacterial meningitis, the neutrophils predominate, whereas the lymphocytes predominate in viral meningitis.77
Antibiotic therapy (refer to Chapter 12 on the immune system) for bacterial causes includes rifampicin or ciprofloxacin and other supportive measures — treatment for those exposed to meningococcal meningitis involves using these for prevention (prophylaxis) and vaccination.79 Corticosteroids may be useful in minimising inflammation.80 There are no specific treatments for viral causes, other than supportive care and rest.
Encephalitis
Encephalitis is an acute febrile illness, usually of viral origin, which causes an inflammation of the nervous tissue. If inadequately treated, it can be fatal in most cases, and for the survivors, there are long-term health alterations.77 It differs from meningitis, which is inflammation of the meninges rather than neural tissue. The most common forms are caused by mosquito-borne (arthropod) viruses, such as Murray Valley encephalitis and herpes simplex type 1 (which is associated with lesions around the mouth, such as cold sores). Encephalitis may occur as a complication of systemic viral diseases such as poliomyelitis or mononucleosis, or it may arise after recovery from viral infections such as rubella. Encephalitis may also follow vaccination with a live attenuated virus vaccine if the vaccine has an encephalitis component — for example, measles, mumps and rubella.
PATHOPHYSIOLOGY
Meningeal involvement is present in all types of viral encephalitis. Altered consciousness is more common with encephalitis than meningitis.81 There may be widespread nerve cell degeneration. Oedema, necrosis with or without haemorrhage and increased intracranial pressure develop. Infectious encephalitis may result from a postinfectious autoimmune response to the virus or from direct invasion of the CNS.
CLINICAL MANIFESTATIONS
Encephalitis ranges from a mild infectious disease to a life-threatening disorder. Dramatic clinical manifestations include fever, delirium or confusion progressing to unconsciousness, seizure activity, cranial nerve palsies, paresis and paralysis, involuntary movement and abnormal reflexes. Signs of marked intracranial pressure may be present.
EVALUATION AND TREATMENT
Diagnosis is made by the history and clinical presentation aided by CSF examination and culture, serology, white blood cell count, CT scan or MRI. Encephalitis due to herpes simplex virus or varicella zoster virus is now being treated with antiviral agents, such as aciclovir.77 Patients require intensive care, and measures to control intracranial pressure and ventilation are paramount.
Abscesses
Brain abscesses are localised collections of pus within the tissue of the brain and spinal cord. Men experience brain abscesses twice as often as women. The median age for abscess formation is 30–40 years. Abscesses can occur: (1) after open trauma and during neurosurgery; (2) in association with infection near the central nervous system, such as the middle ear, nasal cavity and nasal sinuses; and (3) through spread from a distant site, such as the heart, lungs, pelvic organs, skin, tonsils, abscessed teeth, osteomyelitis (bone infection) and dirty needles (especially in compromised hosts). Streptococci, staphylococci and Bacteroides are the most common bacteria that cause abscesses; however, yeast and fungi have been found also.
Brain abscesses are classified as extradural or intracerebral. Extradural brain abscesses are associated with osteomyelitis in a cranial bone. Unlike intracerebral brain abscesses, they rarely arise from a vascular source. Spinal cord abscesses are classified as epidural or intramedullary. Individuals with diabetes mellitus show an increased incidence of spinal epidural abscesses (which form in the epidural space), whereas debilitated individuals with sepsis more often develop intramedullary spinal cord abscesses (those within the spinal cord). Epidural spinal abscesses usually originate as osteomyelitis in a vertebra; the infection then spreads into the epidural space.
PATHOPHYSIOLOGY
Brain abscesses evolve through four stages:
Existing abscesses also tend to spread and form multiple abscesses in the nearby area.
CLINICAL MANIFESTATIONS
Clinical manifestations of brain abscesses are associated with an intracranial infection or expanding intracranial mass. Early manifestations include headache (the most common symptom), low-grade fever, neck pain and stiffness with mild nuchal rigidity, confusion, drowsiness, sensory deficits and communication deficits. Later clinical manifestations include inattentiveness (distractibility), memory deficits, decreased visual acuity and narrowed visual fields, ocular palsy, ataxia and dementia. The development of symptoms may be insidious, often making an abscess difficult to diagnose.
Extradural brain abscesses are associated with localised pain, purulent drainage from the nasal passages or auditory canal, fever, localised tenderness and neck stiffness. Occasionally the individual experiences a focal seizure.
Clinical manifestations of spinal cord abscesses have four stages: (1) spinal aching; (2) severe root pain, accompanied by spasms of the back muscles and limited vertebral movement; (3) weakness caused by progressive cord compression; and (4) paralysis.
EVALUATION AND TREATMENT
The diagnosis is suggested by clinical features and confirmed by CT scan or MRI. Aspiration (obtaining a biopsy or removing the abscess) through a burr hole (a very small hole in the cranium) is used, although excision through an open craniotomy may be unavoidable. Antibiotic therapy is used, preferably after obtaining the biopsy to guide the choice of antibiotic drug. In addition, intracranial pressure may have to be managed, and the corticosteroid dexamethasone is often used.82
As decompression is necessary, spinal cord abscesses are treated with surgical excision or aspiration. Antibiotic therapy and support therapy also are instituted.
TUMOURS OF THE NERVOUS SYSTEM
Tumours are abnormal growths that may be benign or malignant — malignant tumours are usually referred to as cancers. A complete discussion of the genetic basis of cancer, cancer growth and spread, and cancer treatments can be found in Chapter 36. Here we examine some specific information relating mainly to central nervous system cancers.
Cranial tumours
Tumours within the cranium can be either primary or secondary (metastatic), as follows:
Primary. Intracerebral tumours originate from brain structures such as neuroglia, neurons, cells of blood vessels and connective tissue. Extracerebral tumours originate outside substances of the brain and include meningiomas, acoustic nerve tumours and tumours of the pituitary and pineal glands. Primary tumours may undergo metastasis, or spread, within the brain. Although many brain tumours remain relatively benign, they can be life-threatening due to direct compression of brain tissue. Secondary or metastatic. The cancer originates outside the brain, but has spread to the brain. The concept of cancer spread by metastasis is discussed in Chapter 36.Brain tumours are fatal for more than 1200 Australians per year.83 The incidence of cranial tumours increases to age 70 years and then decreases. Interestingly, CNS tumours are the second most common group of tumours occurring in children, affecting more than 100 children per year.83 These cancers have a low survival rate, meaning that a relatively small percentage of those diagnosed will be alive in the years following diagnosis. The percentage of those surviving 5 years after diagnosis of brain cancer in Australia is only 19%.84
Local effects of cranial tumours are caused by the destructive action of the tumour itself on a particular site in the brain and by compression causing decreased cerebral blood flow. Effects include seizures, visual disturbances, unstable gait (walking) and cranial nerve dysfunction. Generalised effects result from increased intracranial pressure caused by obstruction of the ventricular system, haemorrhages in and around the tumour, or cerebral oedema (see Figure 9-21). Intracranial brain tumours do not metastasise as readily as tumours in other organs because there are no lymphatic channels within the brain tissue.
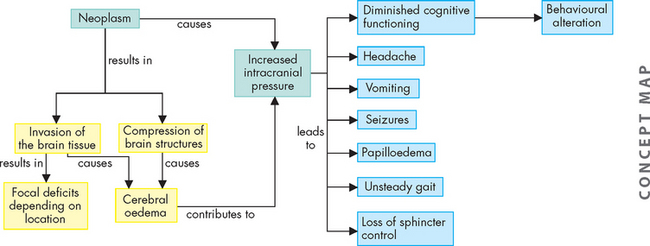
Medulloblastomas, ependymomas, astrocytomas, brainstem gliomas, craniopharyngiomas and optic nerve gliomas make up approximately 75–80% of all paediatric brain tumours. The types and characteristics of childhood brain tumours are summarised in Table 9-9.
Table 9-9 BRAIN TUMOURS IN CHILDREN
| TYPE | CHARACTERISTICS |
|---|---|
| Astrocytoma | |
| Optic nerve glioma | |
| Medulloblastoma (infiltrating glioma) | |
| Brainstem glioma | |
| Ependymoma | |
| Craniopharyngioma |
Primary intracerebral tumours
Primary intracerebral tumours may be benign and encapsulated or non-encapsulated and invasive tumours. Gliomas are tumours of the glial cells (which support the neurons) and represent 42% of all primary brain tumours and 77% of malignant brain tumours.85 Typically, these tumours invade and destroy adjacent normal CNS tissue, and more distal neural and vascular tissues are displaced and compressed, causing ischaemia (inadequate oxygen delivery to the cells), oedema and increased intracranial pressure.
Surgery to fully excise the growth, surgical decompression (surgery to decrease intracranial pressure), chemotherapy and radiotherapy are used for these tumours. Supportive treatment is directed at reducing oedema.
Astrocytomas
Astrocytomas are the most common glioma (about 40% of all tumours of the brain and spinal cord85) and are graded I–IV (see Table 9-10). Developed from astrocytes, astrocytomas expand and infiltrate into the normal surrounding brain tissues.
Table 9-10 CLASSIFICATION SYSTEMS FOR ASTROCYTOMAS
| GRADE | DESCRIPTION | |
|---|---|---|
| ASTROCYTOMAS | ||
| Grade I | Well-differentiated astrocytoma | Least malignant, grow slowly, near normal appearance |
| Grade II | More cellular and anaplastic astrocytoma | Abnormal appearance under a microscope, infiltrated and may recur at a higher grade |
| GLIOBLASTOMAS | ||
| Grade III | Poorly differentiated astrocytoma | Malignant, many cells undergoing mitosis, infiltrated and may recur at a higher grade |
| Grade IV | Poorly differentiated astrocytoma (glioblastoma multiforme) | Increased number of cells undergoing cell division, bizarre appearance under a microscope, widely infiltrated, neovascularisation, central necrosis |
One-third of astrocytomas are classified at diagnosis as grade I or grade II, which are slow-growing. These tumours are generally located in the cerebrum, hypothalamus or pons. Headache and seizures may be early signs. Onset of a focal seizure disorder between the second and sixth decades of life suggests an astrocytoma. Other general or focal neurological manifestations develop gradually, with increased intracranial pressure occurring late in the tumour’s course.
Grade I astrocytomas are treated with surgery and follow-up CT scans. Grade II astrocytomas are treated surgically if accessible or by conventional external radiation, local radiation or radiosurgery. Some 25% of people survive 5 years following surgery alone, while 55% survive 5 years when surgery is followed by radiation therapy.86 Grade I and II astrocytomas commonly progress to a higher grade tumour.
Grades III and IV astrocytomas are found predominantly in the lobes and cerebral hemispheres. Men are twice as likely to have them as women and those 45–55 years old have the highest incidence. Grade IV astrocytomas, glioblastoma multiforme, are highly vascular and extensively infiltrative. They may become large enough to extend from the meningeal surface through the ventricular wall and half of them occupy more than one lobe at the time of death.
The typical clinical presentation for grade IV astrocytomas is that of diffuse, nonspecific clinical signs, such as headache, irritability and ‘personality changes’ that progress to more clear-cut manifestations of increased intracranial pressure such as headache on position change, visual changes, seizures or vomiting. Symptoms may progress to include definite focal signs, such as hemiparesis, dysphasia, cranial nerve palsies and visual field deficits.
Diagnosis of high-grade astrocytoma most commonly takes several months from the onset of the first clinical manifestations because the person does not recognise the need to consult a healthcare provider. Grade III astrocytomas are treated surgically if they are accessible and with radiotherapy and chemotherapy. With treatment, 55–60% of people survive 1 year, 30–35% survive 2 years and 10% survive longer than 5 years. Grade IV astrocytomas are also treated with surgery, radiotherapy and chemotherapy, but average survival time is only about 1 year.87
If a young child complains of repeated and worsening headache, a thorough investigation should take place because headache is uncommon in young children. Headache caused by increased intracranial pressure usually is worse in the morning and gradually improves during the day when the child is upright and venous drainage is enhanced. The frequency of headache and other symptoms worsens as the tumour grows. Irritability and increased somnolence (drowsiness) may also result. Like headache, vomiting occurs more commonly in the morning. Often it is not preceded by nausea and may become projectile, differing from a gastrointestinal disturbance in that the child may be ready to eat immediately after vomiting.
Oligodendrogliomas
Oligodendrogliomas constitute about 4% of all intracranial gliomas85 and are typically slow-growing. The majority are found in the frontal and temporal lobes, often in the deep white matter, but they are found also in other parts of the brain. Many are found in young adults with a history of epilepsy. Approximately half of tumours classified as oligodendrogliomas are actually a mixed type of oligodendroglioma and astrocytoma. Malignant degeneration occurs in approximately one-third of those with oligodendrogliomas and the tumours are then referred to as oligodendroblastomas. If there is extension to the pia mater or ependymal wall, oligodendrogliomas may metastasise to distant CNS sites through the ventricular and arachnoid spaces.
More than 50% of individuals experience a focal or generalised seizure as the first clinical manifestation. Half of those with an oligodendroglioma have increased intracranial pressure at the time of diagnosis and surgery, and one-third develop focal manifestations. The time from the first clinical manifestation to surgical intervention ranges from 2 to 6 years. Survival time is approximately 5–10 years.86
Ependymomas
Ependymomas are gliomas that arise from ependymal cells in the walls of the ventricles and grow either into the ventricle or into adjacent brain tissue; they are not encapsulated (see Figure 9-22). They constitute about 3% of all primary brain tumours in adults and 10% in children and adolescents. Most are in the fourth ventricle, with others found in the third ventricle, lateral ventricles and spinal cord.
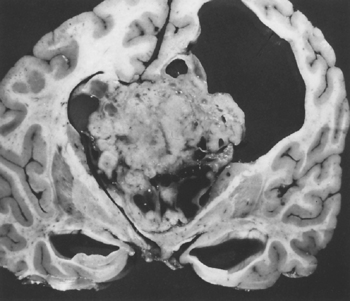
FIGURE 9-22 Large septal ependymoma.
This tumour represents the severity of tissue compression that occurs with a large brain tumour. The area in the upper-right part of the picture represents a secondary hydrocephalus that has further compressed brain tissue.
Source: Courtesy Dr JE Olivera-Rabiela, Mexico City, Mexico. From Rosai J. Ackerman’s surgical pathology. 7th edn. St Louis: Mosby; 1989.
Fourth ventricle ependymomas present with difficulty in balance, unsteady gait, uncoordinated muscle movement and difficulty with fine motor movement. The clinical manifestations of a lateral and third ventricle ependymoma that involves the cerebral hemispheres are seizures, visual changes and contralateral weakness of a body part on one side of the body. Blockage of the CSF pathway by the tumour clinically results in the presence of headache, nausea and vomiting related to the hydrocephalus produced (refer to Chapter 8).
The course of ependymomas may be short or long. The interval between the first manifestations and surgery may be as short as 4 weeks or as long as 7 or 8 years. Ependymomas are treated with radiotherapy, radiosurgery and chemotherapy: 20–50% of people survive 5 years. Some people benefit from a shunting procedure to treat hydrocephalus by draining the CSF.
Primary extracerebral tumours
Meningiomas
Meningiomas constitute about 30% of all intracranial tumours. They are considered benign because they are encapsulated and usually do not invade the surrounding brain; interestingly, although benign, it is possible to have several meningiomas. These tumours usually originate from the dura mater or arachnoid membranes. Small meningiomas (less than 2 cm in diameter) are often found on postmortem examination in middle-aged and elderly individuals who experienced no clinical manifestations and died of totally unrelated causes. The meningioma may extend to the dural surface or exhibit malignant qualities. Focal seizures are often the first manifestation. Because of the extremely slow-growing nature of these tumours, increased intracranial pressure is uncommon. There is a 20% recurrence rate, even with complete surgical excision. Radiation therapies are also used to slow growth.
Nerve sheath tumours
Nerve sheath tumours are either neurofibromas or schwannomas (neuromas, neurolemmas). Some 5% of all neuromas are attributable to neurofibromatosis (an inherited disorder);88,89 the remainder are benign tumours that arise from the myelin sheath of the Schwann cells surrounding the axons of the cranial nerves. These tumours most commonly affect people in their 20s and 30s. The vestibular division of cranial nerve VIII is most commonly affected, with cranial nerves V, VIII and IX also affected. The tumour generally originates just distal to the junction between the nerve root and brainstem. As it grows, it compresses adjacent nerves and eventually the brainstem is displaced and CSF flow is obstructed.
Initial clinical manifestations include headache, tinnitus (ringing in the ears), hearing loss, impaired balance, unsteady gait, facial pain and loss of facial sensations. Later, vertigo with nausea and vomiting, a sense of pressure in the ear and moderate to severe unsteadiness with rapid position changes may appear. CT scan or MRI can establish the diagnosis. Treatment is by surgical excision and radiotherapy of the neuroma.
Secondary (metastatic) brain carcinomas
Metastatic brain tumours that originate from systemic cancers are the most common type of brain tumour and an estimated 10–15% of people with cancer develop metastasis to the brain.86 Some 50% of metastatic brain tumours arise from the lung, 13% from melanomas, 6% from the breast and 4% from the kidneys.90 Carcinomas are disseminated to the brain through the circulation. In more than three-quarters of cases, the metastases are multiple and found scattered throughout the cerebrum and cerebellum. Metastatic tumours are often located in the meninges and near the brain surface in the grey matter and sub-cortical white matter. Metastatic brain tumours produce signs resembling those of glioblastomas. Metastatic brain tumours carry a poor prognosis. If a solitary tumour is found, surgery or radiation therapy is used, but if multiple tumours exist, symptomatic relief only is pursued.
 PAEDIATRICS
PAEDIATRICSDEVELOPMENTAL DISORDERS
Central nervous system malformations are responsible for 75% of fetal deaths and 40% of deaths during the first year of life. During the perinatal period, CNS malformations account for one-third of all apparent congenital malformations, and 90% of CNS malformations are defects of neural tube closure. Other injury to the developing brain, such as occurs with cerebral palsy, appears to occur before birth, but the causes of this are poorly understood.
Defects of neural tube closure
Neural tube defects occur in approximately 4.6 out of 10,000 live births in Australia each year, with around 25% of these babies dying within the first month after birth.91 Fetal death often occurs prior to birth as a result of the neural defects, thereby reducing the actual prevalence of neural defects at birth.92 These defects include anencephaly (an = without; enkephalos = brain), whereby a major part of the brain is missing; encephalocele, where part of the brain or meninges protrudes through an abnormal opening with the cranial bones; and spina bifida (see the next section).
The cause of neural tube defects is believed to be multifactoral — a combination of genes and environment. No single gene has been found to cause neural tube defects.93,94 Folic acid (folate) appears to be important, particularly in the very early stages of pregnancy. Folic acid is essential for healthy DNA replication, particularly for rapidly dividing cells. The early stages of nervous system growth and development for the fetus occur shortly after conception, and folic acid deficiency during the early stages of pregnancy increases the risk for neural tube defects.92,93 Preconceptional supplementation assures adequate folate levels, and in Australia it is now mandatory for folate to be added to bread flour to further lessen the risk of neural tube defects.91 New Zealand is planning on introducing mandatory fortification of bread with folate in mid-2012.95 Other risk factors include heredity, maternal blood glucose concentrations, use of anticonvulsant drugs (particularly valproic acid) and maternal hyperthermia.92,93
Spina bifida
When defects of neural tube closure occur, an accompanying vertebral defect allows the protrusion of the neural tube contents. This is called spina bifida and it is present in 3.4 per 10,000 births in Australia.91 Although the cause of spina bifida is unknown, periconceptual maternal folate deficiency and genetic alterations are commonly associated with the defect.96 It also may occur without any visible exposure of the meninges or neural tissue, for which the term spina bifida occulta (occult = unseen) is used. In spina bifida occulta, the posterior vertebral laminae have failed to fuse. This is a more mild form of the condition, as the extent of the defect is less — extremely common, the defect occurs to some degree in 10–25% of infants. Approximately 80% of these vertebral defects are located in the lumbosacral regions, most commonly in the fifth lumbar vertebra and the first sacral vertebra; they may be detected prenatally with ultrasonic scanning and amniotic fluid alpha-fetoprotein (AFP) testing. About 3% of normal adults have spina bifida occulta of the atlas (cervical vertebra 1).
Certain cutaneous or subcutaneous abnormalities suggest underlying spina bifida, including the following (see Figure 9-23):
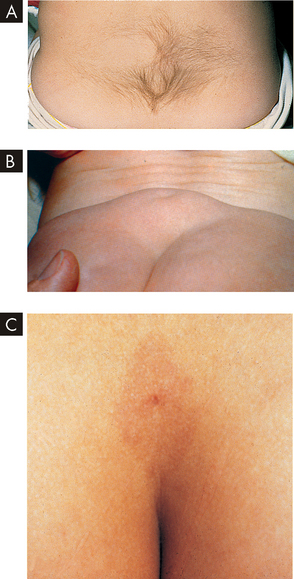
FIGURE 9-23 Cutaneous and subcutaneous markers of occult spina bifida.
A Note the hairy patch over the lumbar region. B The soft subcutaneous mass seen overlying the sacrum of this infant was determined to be a lipoma. C Sacral sinus tract associated with intraspinal dermoid tumour.
Source: Courtesy Michael J Painter, MD, Children’s Hospital of Pittsburgh. From Zitelli BJ, Davis HW. Atlas of pediatric physical diagnosis. 5th edn. Elsevier; 2007.
Spina bifida occulta usually causes no serious neurological dysfunctions. When dysfunctions occur, the common lumbosacral defects cause gait abnormalities, positional deformities of the feet as a result of muscle weakness, or sphincter disturbances of the bladder and bowel. These dysfunctions become evident during periods of rapid growth. Surgical closure is usually completed in the neonatal period and techniques are being developed for intrauterine closure.97
Cerebral palsy
Cerebral palsy is the term given to a diverse group of non-progressive syndromes that affect the brain and cause motor dysfunction beginning in early infancy. The cause was thought to include prenatal cerebral hypoxia or trauma; however, there is also evidence that hypoxia and other injuries during childbirth are not associated.98 There is evidence that genetic factors, specific inflammatory processes and maternal infection with viruses such as herpes simplex virus, varicella zoster virus and Epstein-Barr virus that can cross the placenta may be all associated with the development of cerebral palsy.99,100
Cerebral palsy can be classified on the basis of neurological signs and symptoms and the types of muscle movements that result. The different types of cerebral palsy are due to different brain regions being affected. A range of neurological and cognitive processes are altered, as well as difficulty moving due to the damage to motor pathways. Cerebral palsy affects 1 in 400 births101 and is one of the most common crippling disorders of childhood.
One of the distinguishing features of cerebral palsy is the inability to walk with the foot flat on the floor due to stiffness of muscles in the lower leg (gastrocnemius) (see Figure 9-24). Spastic cerebral palsy is associated with increased muscle tone, causing stiffness and difficulty with movements (including reflexes). This accounts for approximately 70–80% of cerebral palsy cases. Dyskinetic cerebral palsy is associated with extreme difficulty in fine motor coordination and purposeful movements. Movements are jerky and uncontrolled. This form of cerebral palsy accounts for approximately 10–20% of cases. Ataxic cerebral palsy manifests with gait disturbances and instability. The infant with this form of cerebral palsy may have hypotonia (low muscle tone) at birth, but stiffness of the trunk muscles develops by late infancy. Persistence of this increased tone in truncal muscles affects the child’s gait and ability to maintain equilibrium. This form of cerebral palsy accounts for approximately 5–10% of cases. A child may have symptoms of each of these cerebral palsy types, which leads to a mixed disorder accounting for approximately 13% of cases.102
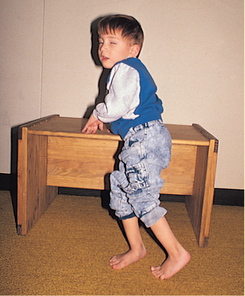
A 4-year-old child with cerebral palsy cruises on furniture. Notice that the child is crouched because of hamstring tightness and is toe-walking because of gastrocnemius tightness.
Source: From Zitelli BJ, Davis HW. Atlas of pediatric physical diagnosis. 5th edn. Elsevier; 2007.
Children with cerebral palsy often have associated neurological disorders, such as seizures (about 50%) and intellectual impairment ranging from mild to severe (about 67%). Other complications include visual impairment, communication disorders, respiratory problems, bowel and bladder problems, and orthopaedic disabilities.102
Although the brain injury is static (unchanging), the chemical picture of cerebral palsy may change with growth and development. Therefore, a fundamental component of an effective treatment regimen includes ongoing assessment, evaluation and revision of the child’s overall management plan. The use of baclofen (a muscle relaxant drug) and botulinum toxin has shown some improvement in some children with cerebral palsy (see Box 9-1). Family-focused interdisciplinary team management provides the best treatment outcomes.103,104
Box 9-1 BOTOX AND CEREBRAL PALSY
In recent years, there has been a surge in the awareness and usage of Botox for cosmetic procedures. ‘Botox’ is an abbreviation of botulinum toxin, which is produced by the bacteria Clostridium botulinum. These bacteria cause severe illness and may be fatal in the case of food poisoning. The toxin blocks the release of acetylcholine at the neuromuscular junction, so it should be no surprise that it may be detrimental if ingested in food, as in high levels it can produce muscle paralysis, including those involved in respiration.
When Botox is used cosmetically, a controlled dosing system is followed, so that it decreases muscle activity in a dose-dependent manner — for example, a small dose decreases muscle activity by a small amount, while a larger dose results in a more substantial decrease in muscle contractions. Botox is injected into facial muscles to lessen the wrinkles associated with ageing (brow furrow or glabellar lines, and crow’s feet). It paralyses the muscles that contract and pull on the skin and cause wrinkles — relaxing these muscles makes the wrinkles less obvious.
Perhaps less well-known is the value of Botox in the treatment of cerebral palsy, by injecting the toxin into the spastic muscles to allow them to become partially relaxed. Muscle tightness can render the muscles ineffective, but partially relaxing them using Botox provides the way for more controlled usage of muscle contractions. After the toxin is injected, splinting is used at night to maximise the benefit of the drug in maintaining muscle elongation.
Botox is also used in combination with other therapies, such as occupational therapy, and other drugs. This important drug is increasing muscle function not only with cerebral palsy, but also with other conditions associated with muscle contraction.
Source: Hoare BJ et al. Botulinum toxin A as an adjunct to treatment in the management of the upper limb in children with spastic cerebral palsy (update). Cochrane Database System Rev 2010; 20(1):CD003469.
Neuroblastomas
Neuroblastoma is a common childhood tumour originating in tissues that normally give rise to the sympathetic ganglia and the adrenal medulla (part of the adrenal gland, involved with the sympathetic nervous system). It is most commonly diagnosed during the first 2 years of life and 75% of neuroblastomas are found before the child is 5 years old. Occasionally, these tumours are diagnosed at birth with metastasis actually apparent in the placenta. Although it accounts for only 8–10% of paediatric malignancies, neuroblastoma causes 15% of cancer deaths in children.
The most common location of neuroblastoma is in the adrenal medulla. The tumour is evident as an abdominal mass and may cause anorexia, bowel and bladder alteration and sometimes spinal cord compression. Neuroblastoma is also found in the mediastinum (the area separating the lungs). There the tumour may cause dyspnoea or infection related to airway obstruction.
A number of systemic signs and symptoms are characteristic of neuroblastoma, including weight loss, irritability, fatigue and fever. Intractable diarrhoea occurs in 7–9% of children. More than 90% of children with neuroblastoma have increased amounts of adrenaline and noradrenaline and associated metabolites in their urine; higher levels of these are associated with a poorer prognosis.
Cerebrovascular disorders
Cerebrovascular disease is the most frequently occurring neurological disorder. Any abnormality of the blood vessels of the brain is referred to as a cerebrovascular disease. The main types of cerebrovascular disease are stroke, aneurysm and vascular malformation. While strokes occur mainly in the elderly, one-fifth of sufferers are actually younger than 60 years of age. Cerebrovascular accidents or strokes are classified according to pathophysiology and include ischaemic (thrombotic or embolic) and haemorrhagic (intracranial haemorrhage). Thrombotic stroke is caused by a blockage that develops inside cranial blood vessels, which obstructs the blood supply to areas of brain tissue. Alterations in neuronal function occur and last for more than 24 hours. Permanent damage to the affected area of the brain occurs and the stroke may be fatal. Less severe types of ischaemic stroke are transient ischaemic attacks (TIAs), which are temporary decreases in brain blood flow, and lacunar infarcts, in which the thrombosis is particularly small and localised. An embolic stroke occurs when a fragment from elsewhere in the body becomes lodged within a cerebral vessel. A subsequent stroke is likely after embolic stroke. Symptoms of ischaemic stroke depend on the blood vessels involved. Classical symptoms of difficulty speaking occur if the Broca’s speech area or Wernicke’s area for speech interpretation is affected. Haemorrhagic stroke is caused by bleeding of a cerebral blood vessel. An intracerebral haemorrhage is strongly related to hypertension. A subarachnoid haemorrhage can cause a substantial haematoma that increases cerebral pressure. Symptoms of haemorrhagic stroke include severe headache (which may be rapidly worsening) and unconsciousness. Paralysis of one side of the body may occur. Distinguishing the type of stroke is essential to guide treatments. A CT scan is most commonly used, and MRI, cerebral angiograph or lumbar puncture may also be used. In the case of thrombotic stroke, tissue-plasminogen activator (or tPA) is administered in the form of alteplase within 6 hours of the onset of symptoms. For subarachnoid haemorrhage, limiting the bleeding is essential.Trauma to the central nervous system
Motor vehicle accidents or sporting injuries are the major cause of traumatic CNS injury. Traumatic injuries to the head are classified as closed-head trauma (blunt) or open-head trauma (penetrating). Closed-head trauma is the more common type of trauma. Different types of focal brain injury include contusion (injury or bruising of the brain without breaking the pia mater), laceration (tearing of brain tissue), extradural haematoma (accumulation of blood above the dura mater), subdural haematoma (blood between the dura mater and arachnoid membrane), intracerebral haematoma (bleeding into the brain) and open-head trauma (skull fracture with exposure of the cranial vault to the environment). Diffuse brain injury (diffuse axonal injury or DAI) results from the effects of head rotation. The brain experiences shearing stresses resulting in axonal damage ranging from concussion to a severe DAI state. Spinal cord trauma is mainly transport-related or due to falls. Those with this type of trauma are most commonly young males and the elderly. Spinal cord injury involves damage to vertebral or neural tissues by compressing tissue, pulling or exerting tension on tissue or shearing tissues so that they slide into one another. Spinal cord injury may cause spinal shock with cessation of all motor, sensory, reflex and autonomic functions below any transected area. Loss of motor and sensory function depends on the level of injury.Degenerative disorders of the central nervous system
Alzheimer’s disease is characterised by decreased size of the brain, loss of the number of neurons and a decreased number of neuronal synapses. Disturbances of memory are common. Mood alterations and motor changes may also occur. Diagnosis of Alzheimer’s disease includes the history and course of the illness, mental status examination and brain imaging. There is some evidence of genetic involvement. Management involves support and therapeutic agents, but neuronal damage cannot be corrected. Parkinson’s disease is prevalent in our community and is associated with loss of brain neurons that secrete dopamine. This lack of dopamine leads to alterations in muscle function, such as hypertonia and akinesia. Tremor at rest, rigidity and postural disturbances are also common. Pharmacological agents that increase the level of dopamine are useful. Huntington’s disease is characterised by uncontrolled movements, with an average age of onset at 30–50 years. The disease progresses to dysfunction of cognition. There is degeneration of neurons within the brain that control motor function, due to a relative excess of dopamine. Multiple sclerosis (MS) is a relatively common degenerative disorder of myelin of the central nervous system. Although the pathogenesis is unknown, the demyelination is thought to result from an immunogenetic-viral cause — a previous viral insult to the nervous system in a genetically susceptible individual yields a subsequent abnormal immune response in the central nervous system.Peripheral nervous system and neuromuscular junction disorders
Guillain-Barré syndrome is a demyelinating disorder caused by an immunological reaction directed at the peripheral nerves. The clinical manifestations may vary from paresis of the legs to complete quadriplegia, respiratory insufficiency and autonomic nervous system instability. Plasmapheresis is used during the acute phase. Myasthenia gravis is a disorder of the neuromuscular junction characterised by muscle weakness and fatiguability. It is an autoimmune disease that destroys acetylcholine receptors, causing decreased transmission of the nerve impulse across the neuromuscular junction. This leads to defects in nerve impulse transmission at the neuromuscular junction.Infection and inflammation of the central nervous system
Meningitis (infection of the meninges) may be due to a number of different microorganisms, with the most common cause being bacterial. Bacterial meningitis is primarily an infection of the pia mater and arachnoid and of the fluid of the subarachnoid space. An inflammatory reaction occurs with bacterial meningitis and exudate is formed and increases rapidly. CSF flow is impaired. These changes lead to an increase in intracranial pressure. Symptoms of meningitis include decreased consciousness, seizure, muscle weakness or paralysis, vomiting and a distinct skin rash. Encephalitis is an acute, febrile illness of viral origin with nervous system involvement. The most common encephalitides are caused by arthropod-borne (mosquito-borne) viruses and herpes simplex. Meningeal involvement appears in all encephalitides. Altered consciousness is a distinct sign of encephalitis. Clinical manifestations include fever, delirium, confusion, seizures, abnormal and involuntary movement and increased intracranial pressure. Herpes encephalitis is treated with antiviral agents. No definitive treatment exists for the other causes of encephalitis. Brain abscesses often originate from infections outside the CNS. Organisms gain access to the CNS from adjacent sites or spread along the wall of a vein. A localised inflammatory process develops with formation of exudate, thrombosis of vessels and degenerating leucocytes. After a few days, the infection becomes delimited with a centre of pus and a wall of granular tissue.Tumours of the central nervous system
Two main types of tumours occur within the cranium: primary and metastatic. Primary tumours are classified as intracerebral tumours (astrocytomas, oligodendrogliomas and ependymomas) or extracerebral tumours (meningiomas or nerve sheath tumours). Metastatic tumours can be found inside or outside the brain substance.Developmental disorders
Neural tube defects are related to insufficient maternal folate; hence fortification of bread flour can be preventative for the population.Mrs Smith is 82 years old and lives in her own home. She receives assistance with physical chores such as shopping and cleaning, but is still able to do light tasks such as tidying the house and cooking her own meals. She is not overweight, but does not undertake any exercise activities. She has a history of hypertension and her blood pressure measured today is 145/90. She has not been diagnosed with diabetes or coronary heart disease.
Mrs Smith has presented today after experiencing an episode of blurred vision, difficulty speaking and dizziness. She did not lose consciousness and did not experience muscle weakness. The episode commenced about an hour ago and lasted for approximately 20 minutes. She was not asleep when the symptoms commenced. After arriving at hospital, a CT scan was performed and no tumour was observed. An MRI scan is about to be performed.