Chapter 64 NURSING MANAGEMENT: arthritis and connective tissue diseases
1. Compare and contrast the sequence of events leading to joint destruction in osteoarthritis and rheumatoid arthritis.
2. Detail the clinical manifestations, and multidisciplinary care and nursing management of osteoarthritis and rheumatoid arthritis.
3. Summarise the pathophysiology, clinical manifestations, and multidisciplinary care and nursing management of ankylosing spondylitis, psoriatic arthritis and reactive arthritis.
4. Describe the pathophysiology, clinical manifestations and multidisciplinary care of septic arthritis, tick-borne infection and gout.
5. Evaluate the differences in pathophysiology, clinical manifestations, and multidisciplinary care and nursing management of systemic lupus erythematosus, polymyositis, dermatomyositis and Sjögren’s syndrome.
6. Explain the drug therapy and related nursing management associated with arthritis and connective tissue diseases.
7. Compare and contrast the possible aetiologies, clinical manifestations, and collaborative and nursing management of myofascial pain syndrome, fibromyalgia syndrome and chronic fatigue syndrome.
ARTHRITIS
Arthritis refers to the inflammation of a joint, whereas rheumatic disease involves the bones and muscles, as well as the joints. It is estimated that more than 3.1 million Australians are affected by arthritis (a prevalence rate of 15.2%). Because the rate of arthritis increases as people age, the prevalence rate will increase as the population ages. Within Australia, for those aged over 75 the greater proportion of sufferers are women, but for those aged between 22 and 44 years, arthritis is slightly more common in men. Factors such as socioeconomic status have been found to affect the prevalence of the disease. Arthritis occurs less frequently among those living in high socioeconomic areas compared to those living in relatively low socioeconomic areas.1 Indigenous Australians have been found to have a higher prevalence than non-Indigenous Australians.
Within New Zealand, one in seven adults (14.8%) have been told by a doctor that they have arthritis. This equates to 460,500 adults. The prevalence of arthritis is higher in women (13.2%) than in men (10.9%), and among the different ethnic groups the prevalence is 16.1% in Europeans, 11.1% in Māori, 7.9% in Pacific Islanders and 6.2% in Asians.2
There are more than 100 types of arthritis, with the most prevalent types in the developed world being osteoarthritis, rheumatoid arthritis and gout. Arthritis and other musculoskeletal conditions account for around $4 billion in health expenditure in Australia per year.1
Osteoarthritis
Osteoarthritis (OA), the most common form of joint (articular) disease in Australia and New Zealand, is a slowly progressive non-inflammatory disorder of the diarthrodial (synovial) joints. Osteoarthritis involves the formation of new joint tissue in response to cartilage destruction.3
AETIOLOGY AND PATHOPHYSIOLOGY
OA is no longer considered to be a normal part of the ageing process, but ageing is one risk factor for disease development.4 Cartilage destruction may actually begin between 20 and 30 years of age and the majority of adults are affected by the age of 40. Few patients experience symptoms until they are 50 or 60, but more than half of those over 64 have X-ray evidence of the disease in at least one joint. Women are more often affected than men and they may have more severe OA.5
OA may occur as an idiopathic (formerly primary) or secondary disorder. The cause of idiopathic OA is unknown, whereas secondary OA is caused by a known event or condition that directly damages cartilage or causes joint instability (see Table 64-1).
TABLE 64-1 Causes of secondary osteoarthritis
| Cause | Effects on joint cartilage |
|---|---|
| Trauma | Dislocations or fractures may lead to avascular necrosis or uneven stress on cartilage |
| Mechanical stress | Repetitive physical activities (e.g. sports activities) cause cartilage deterioration |
| Inflammation | Release of enzymes in response to local inflammation can affect cartilage integrity |
| Joint instability | Damage to supporting structures causes instability, placing uneven stress on articular cartilage |
| Neurological disorders | Pain and loss of reflexes from neurological disorders, such as diabetic neuropathy and Charcot’s joint, causes abnormal movements that contribute to cartilage deterioration |
| Skeletal deformities | Congenital or acquired conditions such as Legg-Calvé-Perthes disease or dislocated hip contribute to cartilage deterioration |
| Haematological/endocrine disorders | Chronic haemarthrosis (e.g. haemophilia) can contribute to cartilage deterioration |
| Drugs | Drugs such as indomethacin, colchicine and corticosteroids can stimulate collagen-digesting enzymes in joint synovium |
Researchers have been unable to identify a single cause for OA, but a number of factors have been linked to disease development. The increased incidence of OA in ageing women is believed to be due to oestrogen reduction at menopause. Genetic factors also appear to play a significant role in the occurrence of OA. Modifiable risk factors have been identified, including obesity, which contributes to hip and knee OA. Regular moderate exercise, which also helps with weight control, has been shown to decrease the likelihood of disease development and progression. Anterior cruciate ligament injury, which is associated with quick stops and pivoting as in all types of football, netball and soccer, has been linked to an increased risk of knee OA.6 Occupations that require frequent kneeling and stooping are also linked to a higher risk of knee OA.
OA results from cartilage damage that triggers a metabolic response at the level of the chondrocytes (see Fig 64-1). Progression of OA causes the normally smooth, white, translucent articular cartilage to become dull, yellow and granular. Affected cartilage gradually becomes softer, less elastic and less able to resist wear with heavy use. The body’s attempts at cartilage repair cannot keep up with the destruction that is occurring. Continued changes in the collagen structure of the cartilage lead to fissuring and erosion of the articular surfaces. As the central cartilage becomes thinner, cartilage and bony growth (osteophytes) increase at the joint margins. The resulting incongruity in joint surfaces creates an uneven distribution of stress across the joint and contributes to a reduction in motion.
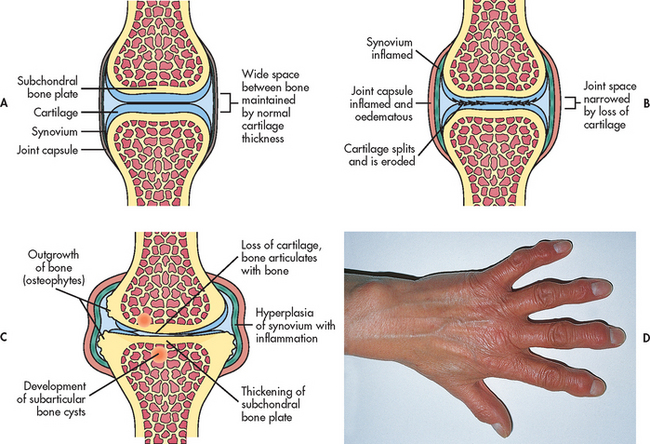
Figure 64-1 Pathological changes in osteoarthritis. A, Normal synovial joint. B, An early change in osteoarthritis is destruction of articular cartilage and narrowing of the joint space. There is inflammation and thickening of the joint capsule and synovium. C, With time, there is thickening of subarticular bone caused by constant friction of the two bone surfaces. Osteophytes form around the periphery of the joint by irregular overgrowths of bone. D, In osteoarthritis of the hands, osteophytes on the distal interphalangeal joints of the fingers are termed Heberden’s nodes and appear as small nodules.
Although inflammation is not characteristic of OA, a secondary synovitis may result when phagocytic cells try to rid the joint of small pieces of cartilage torn from the joint surface. These inflammatory changes contribute to the early pain and stiffness of OA. The pain of later disease results from contact between exposed bony joint surfaces after the articular cartilage has deteriorated completely.
CLINICAL MANIFESTATIONS
Systemic
Systemic manifestations, such as fatigue, fever and organ involvement, are not present in OA. This is an important distinction between OA and inflammatory joint disorders such as rheumatoid arthritis.
Joints
Manifestations of OA range from mild discomfort to significant disability. Joint pain is the predominant symptom and the typical reason that the patient seeks medical attention. Pain generally worsens with joint use. In the early stages of OA, joint pain is relieved by rest. In advanced disease, however, the patient may complain of pain with rest or experience sleep disruptions caused by increasing joint discomfort. Pain may also become worse as the barometric pressure falls before inclement weather. As OA progresses, increasing pain can contribute significantly to disability and loss of function. The pain of OA may be referred to the groin, buttock or medial side of the thigh or knee. Sitting down becomes difficult, as does rising from a chair when the hips are lower than the knees. As OA develops in the intervertebral (apophyseal) joints of the spine, localised pain and stiffness are common.
Unlike pain, which is typically provoked by activity, joint stiffness occurs after periods of rest or static position. Early morning stiffness is common but generally resolves within 30 minutes, a factor distinguishing OA from inflammatory arthritic disorders. Overactivity can cause a mild joint effusion that temporarily increases stiffness. Crepitation, a grating sensation caused by loose particles of cartilage in the joint cavity, can also contribute to stiffness. Crepitation indicates the loss of cartilage integrity and is present in more than 90% of patients with knee OA.
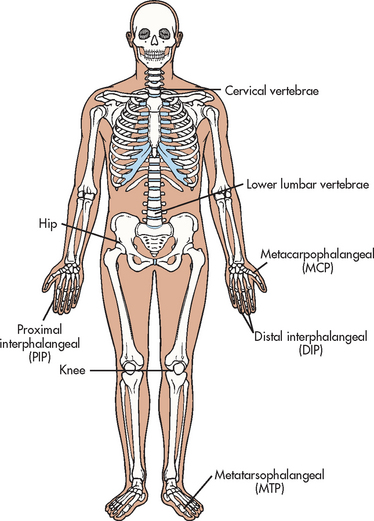
OA usually affect joints asymmetrically. The most commonly involved joints are the distal interphalangeal (DIP) and proximal interphalangeal (PIP) joints of the fingers, the metacarpophalangeal (MCP) joint of the thumb, weight-bearing joints (hips, knees), the metatarsophalangeal (MTP) joint of the foot, and the cervical and lower lumbar vertebrae (see Fig 64-2).
Deformity
Deformity or instability associated with OA is specific to the involved joint. For example, Heberden’s nodes occur on the DIP joints as an indication of osteophyte formation and loss of joint space (see Fig 64-1, D). They can appear in the OA patient as early as age 40 and tend to be seen in family members. Bouchard’s nodes on the PIP joints indicate similar disease involvement. Heberden’s and Bouchard’s nodes are often red, swollen and tender. Although these bony enlargements usually do not cause significant loss of function, the patient may be distressed by the visible disfigurement.
Knee OA often leads to joint malalignment as a result of cartilage loss in the medial compartment. The patient has a characteristic bowlegged appearance and may develop an altered gait in response to the obvious deformity. In advanced hip OA, one of the patient’s legs may become shorter from a loss of joint space.
DIAGNOSTIC STUDIES
A bone scan, computed tomography (CT) scan or magnetic resonance imaging (MRI) may be useful to diagnose OA because of the sensitivity of these tests to detect early joint changes. X-rays are helpful in confirming disease and staging the progression of joint damage. As OA progresses, X-rays typically show joint space narrowing, bony sclerosis and osteophyte formation. However, these changes do not always correlate with the degree of pain experienced by the patient. Despite significant radiological indications of disease, the patient may be relatively free of symptoms. Conversely, another patient may have severe pain with only minimal X-ray changes.
No laboratory abnormalities or biomarkers have been identified that are specific diagnostic indicators of OA. Blood tests to confirm OA remain an active area of investigation.7 The erythrocyte sedimentation rate (ESR) is normal except in instances of acute synovitis, when minimal elevations may be noted. Other routine blood tests (e.g. full blood count [FBC], renal and liver function tests) are useful only in screening for related conditions or for establishing baseline values before the initiation of therapy. Synovial fluid analysis allows differentiation between OA and other forms of inflammatory arthritis. In the presence of OA, the fluid remains clear yellow with little or no sign of inflammation.
MULTIDISCIPLINARY CARE
Because there is no cure for OA, multidisciplinary care focuses on managing pain and inflammation, preventing disability, and maintaining and improving joint function (see Box 64-1). Non-pharmacological interventions are the foundation for OA management and should be maintained throughout the patient’s treatment period. Drug therapy serves as an adjunct to non-pharmacological treatments. Symptoms of disease are often managed conservatively for many years, but the patient’s loss of joint function, unrelieved pain and diminished ability to independently perform self-care may prompt a recommendation for surgery. Reconstructive surgical procedures are discussed in Chapter 62. Arthroscopic surgery for knee OA remains widely practised although recent randomised controlled trials showed no benefit in reducing patient pain and improving function.8
MULTIDISCIPLINARY CARE
Collaborative therapy
Nutritional and weight management counselling
Rest and joint protection, use of assistive devices
Complementary and alternative therapies
Drug therapy*
*See Table 64-2.
Rest and joint protection
The OA patient must understand the importance of maintaining a balance between rest and activity. The affected joint should be rested during any periods of acute inflammation and maintained in a functional position with splints or braces if necessary. However, immobilisation should not exceed 1 week because of the risk of joint stiffness with inactivity. The patient may need to modify their usual activities to decrease stress on affected joints. For example, the patient with knee OA should avoid prolonged periods of standing, kneeling or squatting. Using an assistive device such as a cane, walker or crutches can also help decrease stress on arthritic joints.
Heat and cold applications
Applications of heat and cold may help reduce complaints of pain and stiffness. Although ice is not used as often as heat in the treatment of OA, it can be appropriate if the patient experiences acute inflammation. Heat therapy is especially helpful for stiffness, including hot packs, whirlpool or spa baths, ultrasound and paraffin wax baths.
Nutritional therapy and exercise
If the patient is overweight, a weight-reduction program is a critical part of the total treatment plan. The nurse needs to help the patient evaluate their current diet to make appropriate changes. (Ch 40 discusses ways to assist the patient in attaining and maintaining a healthy body weight.) Because the load on the joints and the degree of joint mobilisation are essential to the preservation of articular cartilage integrity, the National Health and Medical Research Council in conjunction with the Royal Australian College of General Practitioners have identified exercise as a fundamental part of OA management.9 Aerobic conditioning, range-of-motion (ROM) exercises and specific programs for strengthening the quadriceps have been beneficial for many patients with knee OA.
Complementary and alternative therapies
Complementary and alternative therapies for symptom management of arthritis have become increasingly popular with patients who have failed to find relief through traditional medical care. Acupuncture, for example, has been found to be a safe treatment resulting in decreased arthritis pain for some patients with OA.10 Other therapies include yoga, massage, guided imagery and therapeutic touch (see Ch 7). Nutritional supplements such as glucosamine and chondroitin sulfate may be helpful in some patients for relieving moderate-to-severe arthritis pain in the knees and improving joint mobility (see the Complementary & alternative therapies box).
COMPLEMENTARY & ALTERNATIVE THERAPIES
Scientific evidence
Strong evidence for use in the treatment of mild-to-moderate osteoarthritis of the knee; good evidence for use in treatment of osteoarthritis in general.*
*Based on a systematic review of scientific literature. Available at www.naturalstandard.com.
Drug therapy
Drug therapy is based on the severity of the patient’s symptoms (see Table 64-2). The patient with mild to moderate joint pain may receive relief from paracetamol. The patient may receive up to 1000 mg every 6 hours, with the daily dose not to exceed 4 g.
TABLE 64-2 Arthritis and connective tissue disorders
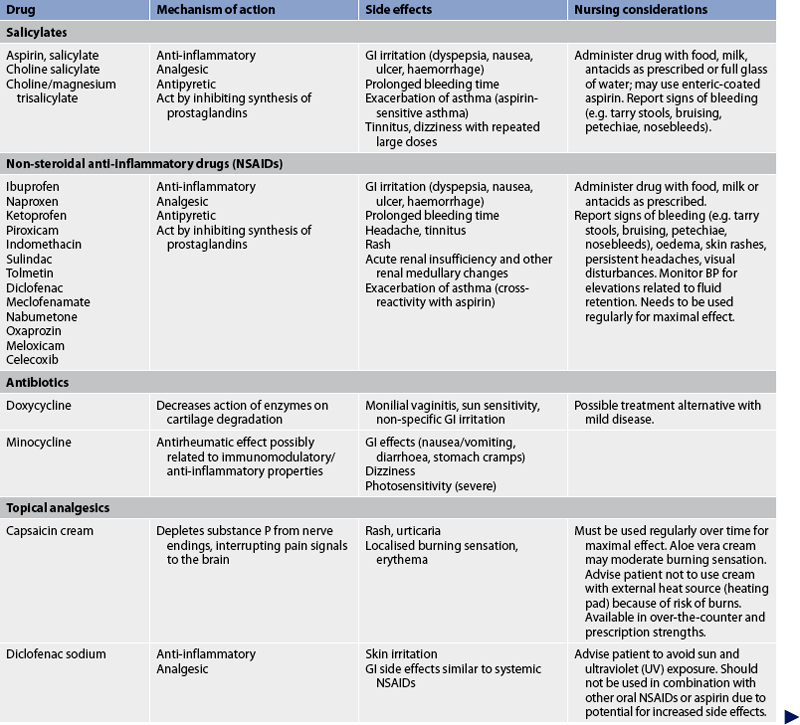
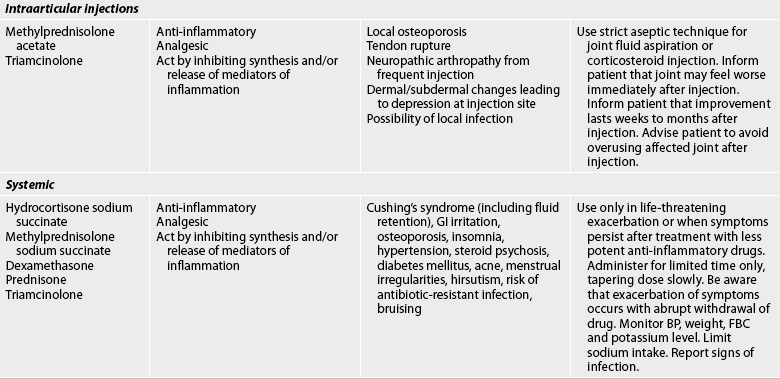
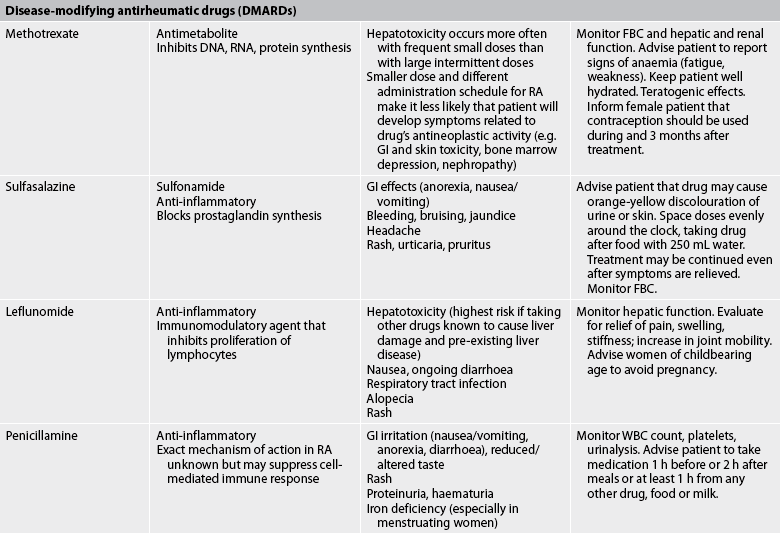
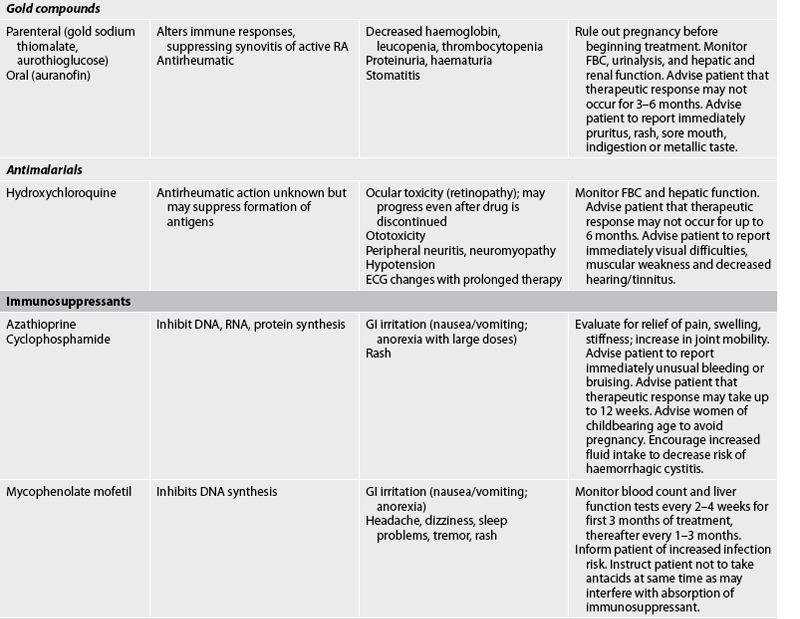
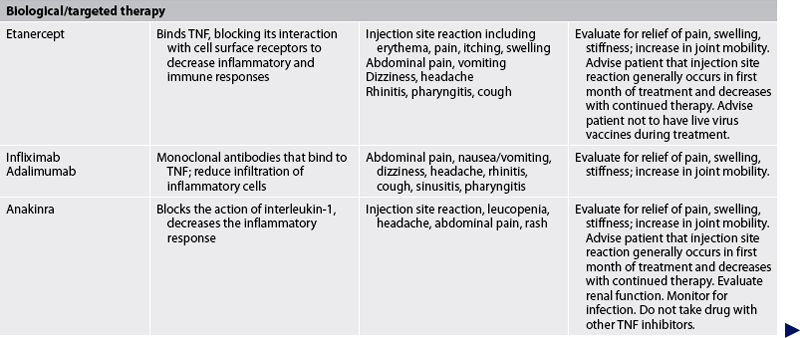
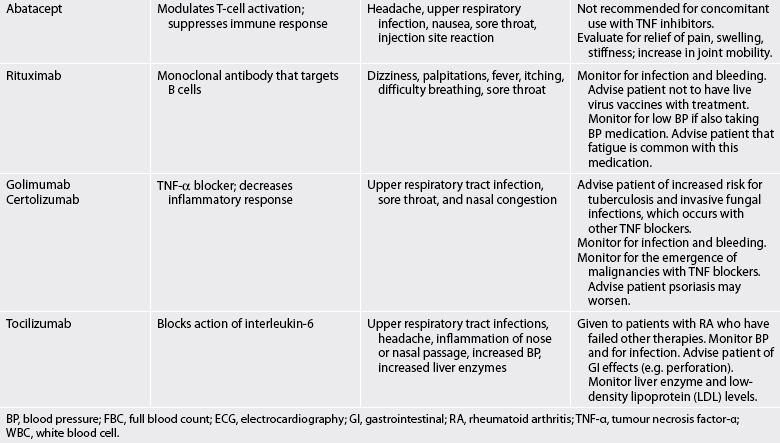
Bp, blood pressure; FBC, full blood count; ECG, electrocardiography; gi, gastrointestinal; RA, rheumatoid arthritis; TNF-α, tumour necrosis factor-α; WBC, white blood cell.
A topical agent such as capsaicin cream (a constituent of many over-the-counter creams) may also be beneficial, either alone or in conjunction with paracetamol. It blocks pain by locally interfering with substance P, which is responsible for the transmission of pain impulses. A concentrated product is available by prescription, but creams of 0.025% to 0.075% capsaicin are sold over-the-counter. Other over-the-counter products that contain camphor, eucalyptus oil and menthol may also provide temporary pain relief. Topical salicylates that can be absorbed into the blood are another alternative for patients who are able to take aspirin-containing medication. Because the effects of topical agents are not sustained, several applications may be needed daily.
For the patient who fails to obtain adequate pain management with paracetamol or for the patient with moderate-to-severe OA pain, a non-steroidal anti-inflammatory drug (NSAID) may be more effective for pain treatment. NSAID therapy is typically initiated in low-dose over-the-counter strengths (ibuprofen 200 mg up to four times daily), with the dose increased as patient symptoms indicate. If the patient is at risk for or experiences gastrointestinal (GI) side effects with a conventional NSAID, supplemental treatment with a protective agent such as misoprostol may be indicated. Arthrotec 50, a combination of misoprostol and the NSAID diclofenac, is also available. Diclofenac gel may be applied for knee and hand joint pain.
Because traditional NSAIDs block the production of prostaglandins from arachidonic acid by inhibiting the production of cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) (see Fig 8-9), the risk for GI erosion and bleeding is increased. Traditional NSAIDs affect platelet aggregation, leading to a prolonged bleeding time. Patients on both warfarin and an NSAID are at high risk for bleeding. Concerns have also been raised regarding the possible negative effects of long-term NSAID treatment on cartilage metabolism, particularly in older patients who may already have diminished cartilage integrity. As an alternative to traditional NSAIDs, treatment with the COX-2 inhibitor celecoxib may be considered in selected patients.11 However, the New Zealand Ministry of Health has restricted the use of COX-2 agents: they are not recommended for routine use in patients with rheumatoid arthritis or osteoarthritis except where the patient is at high risk of developing a serious GI adverse effect from other standard NSAIDs. COX-2 agents should not be prescribed to patients who are at high risk of heart attack or stroke and those taking aspirin. Within Australia, manufacturers of COX-2 inhibitors are required to place explicit warnings in product information about the increased risk of cardiovascular adverse events associated with this group of drugs.
When given in equivalent anti-inflammatory dosages, all NSAIDs are considered comparable in efficacy but vary widely in cost. Individual responses to NSAIDs are also variable. Aspirin is still preferred by some patients, but it is no longer a common treatment, and it should not be used in combination with NSAIDs because both inhibit platelet function and prolong bleeding time. Intraarticular injections of corticosteroids may be appropriate for the elderly patient with local inflammation and effusion. Four or more injections without relief suggest the need for additional intervention. Systemic use of corticosteroids is not indicated and it may actually accelerate the disease process.
Another treatment for mild-to-moderate knee OA is hyaluronic acid (HA), a type of viscosupplementation. HA is found in normal joint fluid and articular cartilage. It contributes to both the viscosity and the elasticity of synovial fluid, and its degradation can result in joint damage. Synthetic and naturally occurring HA derivatives are administered in three weekly injections directly into the joint space. Hylan G-F 20, a newer single-injection HA drug, may offer pain relief for up to 6 months. HA may be added to oral supplements of glucosamine and chondroitin. Few side effects have been reported with HA.
Medications thought to slow the progression of OA or support joint healing are known as disease-modifying osteoarthritis drugs (DMOADs). A number of drugs are under investigation with mixed results as to whether they are effective and safe. Among them is the antibiotic doxycycline, which may decrease the loss of cartilage in some patients with knee OA.12
 NURSING MANAGEMENT: OSTEOARTHRITIS
NURSING MANAGEMENT: OSTEOARTHRITIS
Nursing assessment
The nurse should carefully assess and document the type, location, severity, frequency and duration of the patient’s joint pain and stiffness. The patient should also be questioned about the extent to which these symptoms affect their ability to perform activities of daily living. The nurse should note pain management practices and question the patient about the duration and success of each treatment. Physical examination of the affected joint or joints includes assessment of tenderness, swelling, limitation of movement and crepitation. An involved joint should be compared with the contralateral joint if it is not affected.
Nursing diagnoses
Nursing diagnoses for the patient with OA may include, but are not limited to, the following:
• acute and chronic pain related to physical activity and lack of knowledge of pain self-management techniques
• impaired physical mobility related to weakness, stiffness or pain on ambulation
• self-care deficits related to joint deformity and pain with activity
• imbalanced nutrition: more than body requirements related to intake in excess of energy output
• chronic low self-esteem related to changing physical appearance and social and work roles.
Planning
The overall goals are that the patient with OA will: (1) maintain or improve joint function through a balance of rest and activity; (2) use joint protection measures (see Box 64-2) to improve activity tolerance; (3) achieve independence in self-care and maintain optimal role function; and (4) use pharmacological and non-pharmacological strategies to manage pain satisfactorily.
BOX 64-2 Joint protection and energy conservation
PATIENT & FAMILY TEACHING GUIDE
Include the following instructions when teaching patients with arthritis to protect joints and conserve energy.
• Maintain appropriate weight.
• Use assistive devices, if indicated.
• Avoid forceful repetitive movements.
• Avoid positions of joint deviation and stress.
• Use good posture and proper body mechanics.
• Seek assistance with necessary tasks that may cause pain.
• Develop organising and pacing techniques for routine tasks.
• Modify home and work environment to create less stressful ways to perform tasks.
Nursing implementation
Health promotion
Prevention of primary OA is not possible. However, community education should focus on the alteration of modifiable risk factors through weight loss and the reduction of occupational and recreational hazards. Sports instruction and physical fitness programs should include safety measures that protect and reduce trauma to the joint structures. Congenital conditions, such as Legg-Calvé-Perthes disease, that are known to predispose a patient to the development of OA should be treated promptly.
Acute intervention
The person with OA most often complains of pain, stiffness, limitation of function and the frustration of coping with these physical difficulties on a daily basis. The older adult may believe that OA is an inevitable part of ageing and that nothing can be done to ease the discomfort and related disability.
The patient with OA is usually treated on an outpatient basis, often by an interdisciplinary team of healthcare providers that includes a rheumatologist, a nurse, an occupational therapist and a physiotherapist. Health assessment questionnaires are often used to pinpoint areas of difficulty for the patient with arthritis. Questionnaires are updated at regular intervals to document disease and treatment progression. Treatment goals can be developed based on data from the questionnaires and the physical examination, with specific interventions to target identified problems. The patient is usually hospitalised only if joint surgery is planned (see Ch 62).
Drugs are administered for the treatment of pain and inflammation. Non-pharmacological pain management strategies may include massage, the application of heat (thermal packs) or cold (ice packs), relaxation and guided imagery. Splints may be prescribed to rest and stabilise painful or inflamed joints. Once an acute exacerbation has subsided, a physiotherapist can provide valuable assistance in planning an exercise program. Therapists may recommend Tai Chi as a low-impact form of exercise. Tai Chi can be performed by patients of all ages and may be done in a wheelchair. Nurses need to emphasise the importance of warming up beforehand to prevent stretch injuries.
Patient and family teaching related to OA is an important nursing responsibility in any care setting and is the foundation of successful disease management. The nurse should provide information about the nature and treatment of the disease, pain management, correct posture and body mechanics, correct use of assistive devices such as a cane or walker, principles of joint protection and energy conservation (see Box 64-2), nutritional choices, weight and stress management, and a therapeutic exercise program. The nurse should assure the patient that OA is a localised disease and that severe deforming arthritis is not the usual course. The patient can also gain support and understanding of the disease process through community resources available in each state in Australia and through Arthritis New Zealand. A number of organisations have self-management programs that can be suggested to people who have OA (see the Resources on p 1855).
Ambulatory and home care
Chronic pain and a loss of function of the affected joints continue to be primary concerns. Home management goals should be individualised to meet the patient’s needs, and the carer, family members and significant others should be included in goal setting and teaching. Home and work environment modification is essential for patient safety, accessibility and self-care.13 Measures include removing scatter rugs, providing rails for stairs and the bath, using night-lights and wearing well-fitting supportive shoes. Assistive devices such as canes, walkers, elevated toilet seats and grab bars also reduce the load on an affected joint and promote safety. The nurse should urge the patient to continue all prescribed therapies at home and to be open to the discussion of new approaches to symptom management.
Sexual counselling may help the patient and significant other to enjoy physical closeness by introducing the idea of alternative positions and timing for intercourse. Discussion also increases awareness of each partner’s needs. The patient can take analgesics or a warm bath to decrease joint stiffness before sexual activity.
Evaluation
The expected outcomes are that the patient with OA will: (1) maintain or improve joint function through a balance of rest and activity; (2) use joint protection measures to improve activity tolerance; (3) achieve independence in self-care and maintain optimal role function; (4) use pharmacological and non-pharmacological strategies to manage pain satisfactorily; and (5) demonstrate that they understand what OA is and develop self-management strategies in collaboration with their healthcare team.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune disease characterised by inflammation of connective tissue in the diarthrodial (synovial) joints, typically with periods of remission and exacerbation. RA is frequently accompanied by extraarticular manifestations.
RA occurs globally, affecting all ethnic groups. It can occur at any time of life, but the incidence increases with age, peaking between 30 and 50 years old. It is the second most common type of arthritis and the most common autoimmune disease. Between 1 and 2% of the populations of New Zealand and Australia are affected by RA.14 Women are affected more commonly than men.
AETIOLOGY AND PATHOPHYSIOLOGY
The cause of RA is unknown. Despite past theories, no infectious agent has been cultured from blood and synovial tissue or fluid with enough reproducibility to suggest an infectious cause for the disease. An autoimmune aetiology is currently the most widely accepted.
Autoimmunity
The autoimmune theory suggests that changes associated with RA begin when a susceptible host experiences an initial immune response to an antigen. The antigen, which is probably not the same in all patients, triggers the formation of an abnormal immunoglobulin G (IgG). RA is characterised by the presence of autoantibodies against this abnormal IgG. The autoantibodies are known as rheumatoid factor (RF) and they combine with IgG to form immune complexes that initially deposit on synovial membranes or superficial articular cartilage in the joints. Immune complex formation leads to the activation of complement, and an inflammatory response results. (Complement activation is discussed in Ch 12 and immune complex formation is discussed in Ch 13.) Neutrophils are attracted to the site of inflammation, where they release proteolytic enzymes that can damage articular cartilage and cause the synovial lining to thicken (see Fig 64-3). Other inflammatory cells include T helper (CD4) cells, which are the primary orchestrators of cell-mediated immune responses. Activated CD4 cells stimulate monocytes, macrophages and synovial fibroblasts to secrete the proinflammatory cytokines interleukin-1 (IL-1), interleukin-6 (IL-6) and tumour necrosis factor (TNF). These cytokines are the primary factors that drive the inflammatory response in RA.
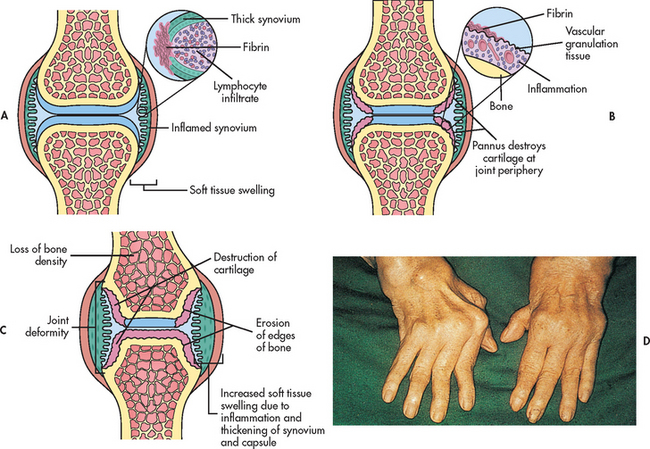
Figure 64-3 Rheumatoid arthritis. A, An early pathological change in rheumatoid arthritis is rheumatoid synovitis. The synovium is inflamed. There is a great increase in lymphocytes and plasma cells. B, With time, there is articular cartilage destruction; vascular granulation tissue grows across the surface of the cartilage (pannus) from the edges of the joint, and the articular surface shows loss of cartilage beneath the extending pannus, most marked at the joint margins. C, Inflammatory pannus causes focal destruction of bone. At the edges of the joint there is osteolytic destruction of bone, responsible for erosions seen on X-rays. This phase is associated with joint deformity. D, Characteristic deformity and soft-tissue swelling associated with severe advanced rheumatoid disease of the hands.
Joint changes from chronic inflammation begin when the hypertrophied synovial membrane invades the surrounding cartilage, ligaments, tendons and joint capsule. Pannus (highly vascular granulation tissue) forms within the joint. It eventually covers and erodes the entire surface of the articular cartilage. The production of inflammatory cytokines at the pannus–cartilage junction further contributes to cartilage destruction. The pannus also scars and shortens supporting structures such as tendons and ligaments, ultimately causing joint laxity, subluxation and contracture.
Genetic factors
Genetic predisposition appears to be important in the development of RA. For example, a higher occurrence of the disease has been noted in identical twins compared to fraternal twins. The strongest evidence for a familial influence is the increased occurrence of a human leucocyte antigen (HLA) known as HLA-DR4 in Caucasian RA patients. Other HLA variants have also been identified in patients from other ethnic groups. (HLA is discussed in Ch 13.) Smoking significantly increases the risk of RA for both men and women who are genetically predisposed to the disease.15
The pathogenesis of RA is more clearly understood than its aetiology. If unarrested, the disease progresses through four stages, which are identified in Box 64-3. The symptoms of RA and systemic lupus erythematosus can be very similar in the early stages, so it is important that the symptoms are explored properly and treated.
BOX 64-3 Anatomical stages of rheumatoid arthritis
Stage II: Moderate
X-ray evidence of osteoporosis, with or without slight bone or cartilage destruction; no joint deformities (although possibly limited joint mobility); adjacent muscle atrophy; possible presence of extraarticular soft-tissue lesions (e.g. nodules, tenosynovitis)
Stage III: Severe
X-ray evidence of cartilage and bone destruction in addition to osteoporosis; joint deformity, such as subluxation, ulnar deviation or hyperextension, without fibrous or bony ankylosis; extensive muscle atrophy; possible presence of extraarticular soft-tissue lesions (e.g. nodules, tenosynovitis)
Source: American College of Rheumatology. Classification criteria for determining progression of rheumatoid arthritis. Available at www.hopkins-arthritis.org/physician-corner/education/acr/acr.html. Used in Australia and New Zealand to classify progression.
CLINICAL MANIFESTATIONS
Joints
The onset of RA is typically insidious. Non-specific manifestations such as fatigue, anorexia, weight loss and generalised stiffness may precede the onset of arthritic complaints. The stiffness becomes more localised in the following weeks to months. Some patients report a history of a precipitating stressful event such as infection, work stress, physical exertion, childbirth, surgery or emotional upset. However, research has been unable to correlate directly such events with the onset of RA.15
Specific articular involvement is manifested clinically by pain, stiffness, limitation of motion and signs of inflammation (e.g. heat, swelling, tenderness).3 Joint symptoms occur symmetrically and frequently affect the small joints of the hands (PIP and MCP) and feet (MTP). Larger peripheral joints such as the wrists, elbows, shoulders, knees, hips, ankles and jaw may also be involved. The cervical spine may be affected, but the axial spine is generally spared. Table 64-3 compares RA and OA.
TABLE 64-3 Comparison of rheumatoid arthritis and osteoarthritis
| Parameter | Rheumatoid arthritis | Osteoarthritis |
|---|---|---|
| Age at onset | Young to middle age | Usually age >40 |
| Gender | Female/male ratio is 2:1 or 3:1; less marked gender difference after age 60 | Before age 50, more men than women; after age 50, more women than men |
| Weight | Lost or maintained weight | Often overweight |
| Disease | Systemic disease with exacerbations and remissions | Localised disease with variable, progressive course |
| Affected joints | Small joints first (PIPS, MCPs, MTPs), wrists, elbows, shoulders, knees; usually bilateral, symmetrical | Weight-bearing joints of knees and hips, small joints (MCPs, DIPs, PIPs), cervical and lumbar spine; often asymmetrical |
| Pain characteristics | Stiffness lasts 1 h to all day and may decrease with use, pain is variable, may disrupt sleep | Stiffness occurs on arising but usually subsides after 30 min, pain gradually worsens with joint use and disease progression, relieved with rest |
| Effusions | Common | Uncommon |
| Nodules | Present, especially on extensor surfaces | Heberden’s (DIPs) and Bouchard’s (PIPs) nodes |
| Synovial fluid | WBC count >20,000/μL with mostly neutrophils | WBC count <2000/μL (mild leucocytosis) |
| X-rays | Joint space narrowing and erosion with bony overgrowths, subluxation with advanced disease; osteoporosis related to corticosteroid use | Joint space narrowing, osteophytes, subchondral cysts, sclerosis |
| Laboratory findings | RF positive in 80% of patients | RF negative |
| Elevated ESR, CRP indicative of active inflammation | Transient elevation in ESR related to synovitis |
CRP, C-reactive protein; dips, distal interphalangeals; ESR, erythrocyte sedimentation rate; MCPs, metacarpophalangeals; MTPs, metatarsophalangeals; PIPs, proximal interphalangeals; RF, rheumatoid factor; WBC, white blood cell.
The patient characteristically experiences joint stiffness after periods of inactivity. Morning stiffness may last from 60 minutes to several hours or more, depending on disease activity. MCP and PIP joints are typically swollen. In early disease, the fingers may become spindle shaped from synovial hypertrophy and thickening of the joint capsule. Joints become tender, painful and warm to the touch. Joint pain increases with motion, varies in intensity and may not be proportional to the degree of inflammation. Tenosynovitis frequently affects the extensor and flexor tendons around the wrists, producing manifestations of carpal tunnel syndrome and making it difficult for the patient to grasp objects.
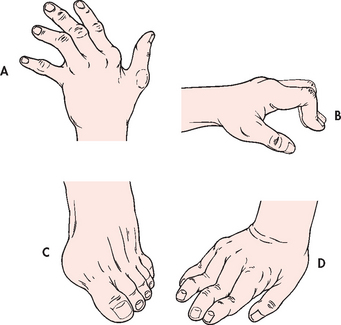
As disease activity progresses, inflammation and fibrosis of the joint capsule and supporting structures may lead to deformity and disability. Atrophy of muscles and destruction of tendons around the joint cause one articular surface to slip past the other (subluxation). Typical distortions of the hand include ulnar drift (‘zig-zag deformity’), swan neck and Boutonnière deformities (see Fig 64-4 and also Fig 64-3, D). Metatarsal head subluxation and hallux valgus (bunion) in the feet may cause pain and walking disability.
Extraarticular manifestations
RA can affect nearly every system in the body. Extraarticular manifestations of RA are depicted in Figure 64-5. The three most common are rheumatoid nodules, Sjögren’s syndrome and Felty’s syndrome.
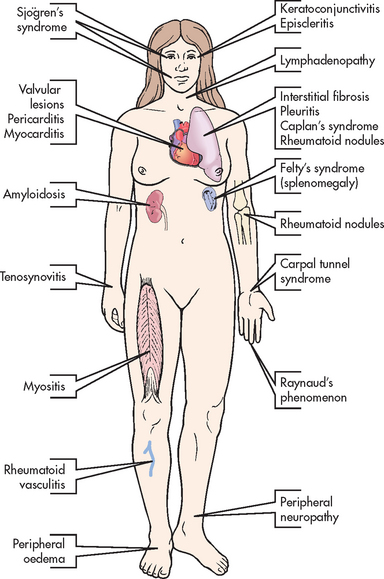
Rheumatoid nodules develop in up to 25% of all patients with RA. Those affected with nodules usually have high titres of RF. Rheumatoid nodules appear subcutaneously as firm, non-tender, granuloma-type masses and are usually located over the extensor surfaces of joints such as fingers and elbows. Nodules at the base of the spine and back of the head are common in older adults. Nodules develop insidiously and can persist or regress spontaneously. They are usually not removed because of the high probability of recurrence, but they can easily break down or become infected. Nodules that appear on the sclera or lungs indicate active disease and a poorer prognosis.
Sjögren’s syndrome is seen in 10–15% of patients with RA. It can occur as a disease by itself or in conjunction with other arthritic disorders such as RA and systemic lupus erythematosus. Affected patients have diminished lacrimal and salivary gland secretion, leading to complaints of dry mouth as well as burning, gritty, itchy eyes with decreased tearing, and photosensitivity. (Sjögren’s syndrome is discussed later in this chapter.)
Felty’s syndrome occurs most commonly in patients with severe, nodule-forming RA. It is characterised by inflammatory eye disorders, splenomegaly, lymphadenopathy, pulmonary disease and blood dyscrasias (anaemia, thrombocytopenia, granulocytopenia).
COMPLICATIONS
Without treatment, joint destruction begins as early as the first year of the disease. Flexion contractures and hand deformities cause diminished grasp strength and affect the patient’s ability to perform self-care tasks. Nodular myositis and muscle fibre degeneration can lead to pain similar to that of vascular insufficiency. Cataract development and loss of vision can result from scleral nodules. Complications can also result from rheumatoid nodules. On the skin, these nodules can ulcerate, similar to pressure ulcers. Nodules on the vocal cords lead to progressive hoarseness, and nodules in the vertebral bodies can cause bone destruction. In later disease, cardiopulmonary effects may occur. These may include pleurisy, pleural effusion, pericarditis, pericardial effusion and cardiomyopathy. Carpal tunnel syndrome of the wrist can result from swelling of the synovial membrane, which causes pressure on the median nerve and results in pain.
DIAGNOSTIC STUDIES
An accurate diagnosis is essential to the initiation of appropriate treatment and the prevention of unnecessary disability. A diagnosis is often made based on the history and physical findings, but some laboratory tests are useful for confirmation and to monitor disease progression (see Box 64-4). Positive RF occurs in approximately 80% of patients, and titres rise during active disease. ESR and C-reactive protein (CRP) are general indicators of active inflammation. Antinuclear antibody (ANA) titres are also seen in some RA patients. Antibodies to cyclic citrulline protein (anti-CCP) are showing important potential as a marker for the early detection of RA. CCP antibodies have been found in the patient’s blood for up to 10 years before symptoms appear.7
MULTIDISCIPLINARY CARE
Collaborative therapy
Nutritional and weight management counselling
Rest and joint protection, use of assistive devices
Complementary and alternative therapies
Drug therapy*
*See Table 64-2.
Synovial fluid analysis in early disease often shows a straw-coloured fluid with many fibrin flecks. The enzyme MMP-3 is increased in the fluid of the patient with RA and it may be a marker of progressive joint damage. The white blood cell (WBC) count of synovial fluid is elevated (up to 25,000/μL). Inflammatory changes in the synovium can be confirmed by tissue biopsy.
X-rays are not specifically diagnostic of RA. They may be inconclusive during early stages of the disease, revealing only soft-tissue swelling and possible bone demineralisation. In later disease, narrowing of the joint space, destruction of articular cartilage, erosion, subluxation and deformity are seen. Malalignment and ankylosis are often evident in advanced disease. Baseline films may be useful in monitoring disease progression and treatment effectiveness. Bone scans are more useful in detecting early joint changes and confirming a diagnosis so that RA treatment can be initiated.
While still used by many clinicians, the 1987 American College of Rheumatology (ACR; formerly the American Rheumatism Association) classification criteria for RA have been criticised for their lack of sensitivity in early disease. The 2009 clinical guidelines developed by the National Health and Medical Research Council and the Royal Australian College of General Practitioners are used in New Zealand and Australia and focus on identifying early RA. This has been done in order to emphasise the importance of earlier diagnosis and the implementation of effective disease-suppressing therapy to prevent or minimise the occurrence of the undesirable sequelae of RA. Early treatment and diagnostic tests are summarised in Figure 64-6.
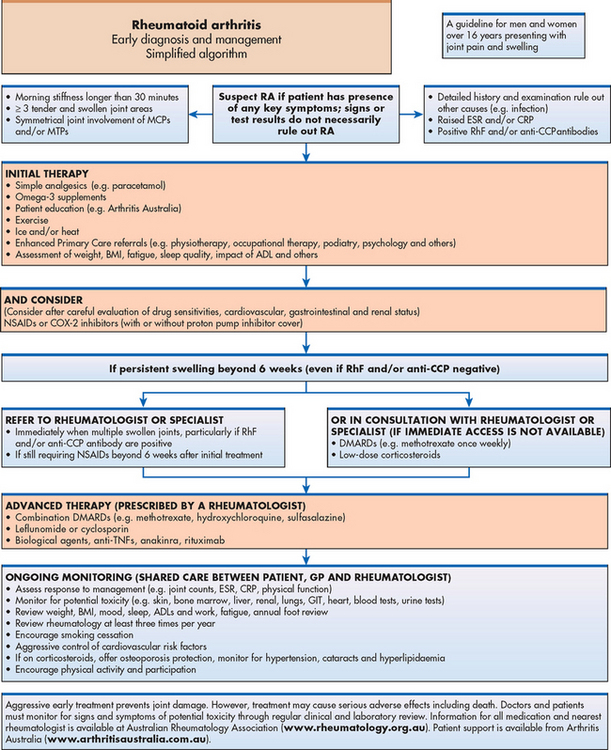
Figure 64-6 RA simplified algorithm: early diagnosis and management. ADLs, activities of daily living; BMI, body mass index; CCP, cyclic citrullinated peptide; CRP, C-reactive protein; DMARDs, disease-modifying anti-rheumatic drugs; ESR, erythrocyte sedimentation rate; GIT, gastrointestinal tract; MCP, metacarpal phalanges; MTP, metatarsal phalanges; NSAIDs, non-steroidal anti-inflammatory drugs; TNF, tumour necrosis factor.
Source: Clinical guideline for the diagnosis and management of early rheumatoid arthritis. Melbourne: Royal Australian College of General Practitioners; 2009.
MULTIDISCIPLINARY CARE
Care of the patient with RA begins with a comprehensive program of education and drug therapy. Teaching regarding drug therapy includes correct administration, reporting of side effects, and frequent medical and laboratory follow-up visits. The nurse should educate the patient and family about the disease process and home management strategies. NSAIDs are prescribed to promote physical comfort. Physiotherapy helps the patient to maintain joint motion and muscle strength. Occupational therapy develops upper extremity function and encourages joint protection with splints or other assistive devices and strategies for activity pacing.
An individualised treatment plan considers the nature of the disease activity, joint function, age, gender, family and social roles, and response to previous treatment (see Box 64-4). A caring, long-term relationship with an arthritis healthcare team can promote the patient’s self-esteem and positive coping.
Drug therapy
Drugs remain the cornerstone of RA treatment (see Table 64-2). Instead of maintaining the patient on high doses of aspirin or NSAIDs until X-rays show clear evidence of the disease, healthcare providers now aggressively prescribe disease-modifying antirheumatic drugs (DMARDs) with the knowledge that irreversible joint changes can occur as early as the first year of RA.16 These drugs have the potential to lessen the permanent effects of RA, such as joint erosion and deformity. The choice of drug is based on disease activity, the patient’s level of function and lifestyle considerations, such as the desire to bear children.
Treatment of early RA often involves therapy with methotrexate. The rapid anti-inflammatory effect of methotrexate reduces clinical symptoms in days to weeks. It is also inexpensive and has a lower toxicity compared to other drugs. Side effects include bone marrow suppression and hepatotoxicity. Methotrexate therapy requires frequent laboratory monitoring, including FBC and chemistry panel. Sulfasalazine and the antimalarial drug hydroxychloroquine may be effective DMARDs for mild-to-moderate disease. They are rapidly absorbed, relatively safe and well-tolerated medications. The synthetic DMARD leflunomide blocks immune cell overproduction. Its efficacy is similar to methotrexate and sulfasalazine with side effects of severe liver injury, diarrhoea and teratogenesis. In women of childbearing age, pregnancy must be excluded before therapy is initiated.
 DRUG ALERT—hydroxychloroquine
DRUG ALERT—hydroxychloroquineBiological/targeted therapies are also used to slow disease progression in RA. These drugs include etanercept, infliximab, adalimumab, anakinra and abatacept. They can be used to treat patients with moderate-to-severe disease who have not responded to DMARDs, or in combination therapy with an established DMARD such as methotrexate.
Etanercept is a biologically engineered copy (using recombinant deoxyribonucleic acid [DNA] technology) of the TNF cell receptor. This soluble TNF receptor binds to TNF in circulation before TNF can bind to the cell surface receptor. By inhibiting binding of TNF, etanercept inhibits the inflammatory response. This drug is given in 25-mg doses twice per week (3–4 days apart) or as a 50-mg dose once weekly in a subcutaneous injection.
Infliximab and adalimumab are monoclonal antibodies against TNF. They bind to TNF, thus preventing it from binding to TNF receptors on cells. Infliximab is given intravenously (IV) as an initial dose, followed by additional dosing at 2 and 6 weeks, and then every 8 weeks thereafter. It should be given in combination with methotrexate. Adalimumab is given subcutaneously every other week. If the response is inadequate, dosing can be increased to weekly.
Anakinra is a recombinant version of IL-1 receptor antagonist (IL-1Ra). It blocks the biological activity of IL-1 by competitively inhibiting IL-1 binding to the IL-1 receptor. Anakinra is not available for use in New Zealand and its use is restricted in Australia. It is given as a daily subcutaneous injection. Anakinra is used to reduce the pain and swelling associated with moderate to severe RA. It can be used in combination with DMARDs but not with TNF inhibitors. Concurrent use of these agents can cause serious infection and neutropenia.
Abatacept is recommended for patients who have an inadequate response to DMARDs or TNF inhibitors. Abatacept blocks T cell activation and is given by IV infusion. Similar to anakinra, it should not be used concomitantly with TNF inhibitors.
Rituximab may be used in combination with methotrexate for patients with moderate-to-severe RA not responding to TNF antagonist therapies (e.g. etanercept, infliximab). This drug is a monoclonal antibody that targets B cells (see Ch 15). It is initially given in two IV infusions separated by 2 weeks. Some patients may be treated with a second course of the drug after 4–6 months.
Two newer TNF inhibitors, golimumab and certolizumab, improve symptoms in patients with moderate-to-severe RA. Both drugs are given in combination with methotrexate. When compared to other biological therapies, certolizumab stays in the system longer and may also show a more rapid (1–2 weeks) and significant reduction in RA symptoms.17 This drug is also used in treating Crohn’s disease.
Tocilizumab is a relatively new drug used to treat patients with moderate-to-severe RA who have not adequately responded to or cannot tolerate other approved drug classes for RA. This drug works by blocking the action of interleukin-6 (IL-6), a proinflammatory cytokine.
Additional medications used infrequently for treating RA include antibiotics (minocycline), immunosuppressants (azathioprine, penicillamine) and gold compounds (auranofin, gold sodium thiomalate).
Corticosteroid therapy can be used to aid in symptom control. Intraarticular injections may temporarily reduce the pain and inflammation associated with disease flare-ups. Long-term use of oral corticosteroids should not be a mainstay of RA treatment because of the risk of complications, including osteoporosis and avascular necrosis. However, low-dose prednisone may be used for a limited time in select patients to decrease disease activity until a DMARD effect is seen.
Various NSAIDs and salicylates continue to be included in the drug regimen to treat arthritis pain and inflammation. Enteric-coated aspirin may be used in high dosages of 4–6 g per day (10–18 tablets). The ability to obtain serum salicylate levels is helpful in developing and evaluating individualised treatment plans.
NSAIDs have anti-inflammatory, analgesic and antipyretic properties. Although many NSAIDs are potent inhibitors of inflammation, they do not appear to alter the natural history of RA. Some relief may be noted within days of the start of treatment with NSAIDs, but full effectiveness may take 2–3 weeks. NSAIDs may be used when the patient cannot tolerate high doses of aspirin. Anti-inflammatory drugs that are taken only once or twice a day may improve the patient’s ability to follow the treatment regimen (see Table 64-2). The newer-generation NSAIDs, the COX-2 inhibitors, are effective in RA as well as OA. Celecoxib is currently the only available COX-2 inhibitor, though the rheumatologist will need to discuss the risks associated with this medication with the patient.
Nutritional therapy
Although there is no special diet for RA, balanced nutrition is important. Fatigue, pain, depression, limited endurance and mobility deficits often accompany RA and may cause a loss of appetite or interfere with the patient’s ability to shop for and prepare food. Weight loss may result. An occupational therapist may help the patient to modify their home environment and to use assistive devices to make food preparation easier.
Corticosteroid therapy or immobility secondary to pain may result in unwanted weight gain. A sensible weight loss program consisting of balanced nutrition and exercise reduces stress on affected joints. Corticosteroids also increase the appetite, resulting in a higher kilojoule intake. In addition, the patient may become distressed as signs and symptoms of Cushing’s syndrome, including moon face and the redistribution of fatty tissue to the trunk, change the physical appearance. The patient should be encouraged to continue a balanced diet and not to alter the corticosteroid dose or stop therapy abruptly. Weight slowly adjusts to normal several months after cessation of therapy.
NURSING MANAGEMENT: RHEUMATOID ARTHRITIS
Nursing assessment
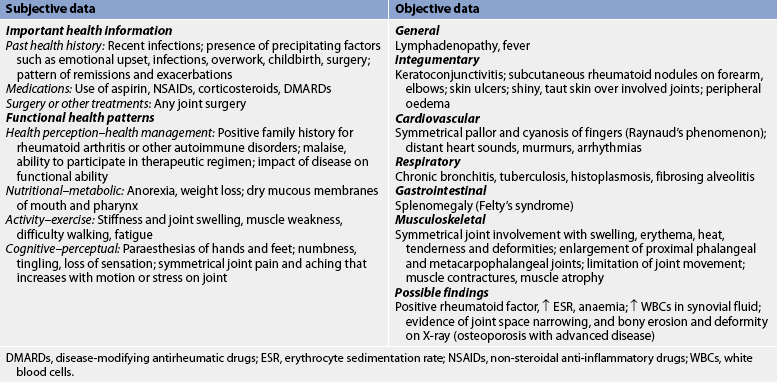
Subjective and objective data that should be obtained from the patient with RA are presented in Table 64-4.
Nursing diagnoses
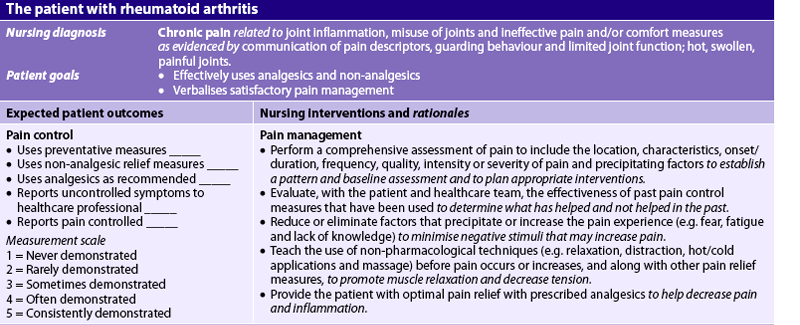
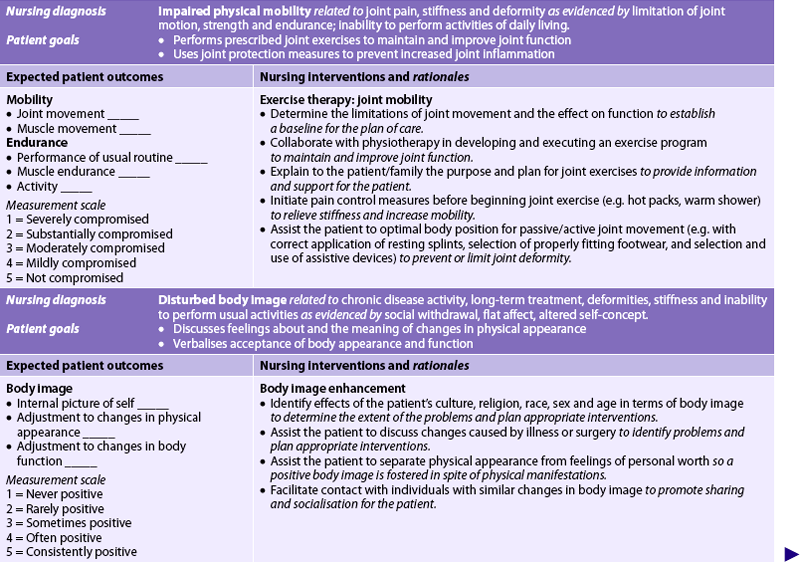
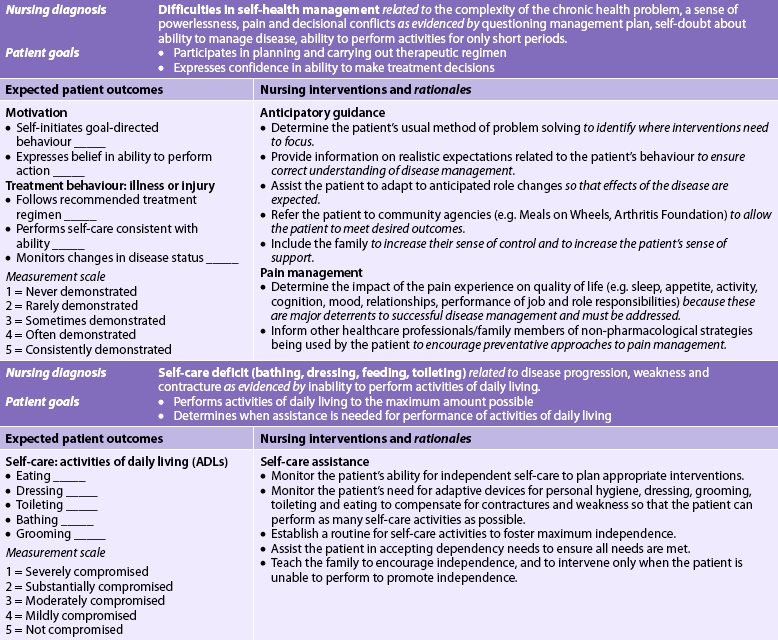
Nursing diagnoses for the patient with RA may include, but are not limited to, those presented in NCP 64-1.
Planning
The overall goals are that the patient with RA will: (1) have satisfactory pain management; (2) have minimal loss of functional ability of the affected joints; (3) participate in planning and carrying out the therapeutic regimen; (4) maintain a positive self-image; and (5) perform self-care to the maximum amount possible.
Nursing implementation
Health promotion
Prevention of RA is not yet possible. However, community education programs should focus on symptom recognition to promote early diagnosis and treatment of RA. Arthritis New Zealand and the various Arthritis Foundations in Australia offer many publications, classes and support activities to assist people (see the Resources on p 1855).
Acute intervention
The primary goals in the management of RA are the reduction of inflammation, management of pain, maintenance of joint function and prevention or minimisation of joint deformity. Goals may be met through a comprehensive program of drug therapy, rest, joint protection, heat and cold applications, exercise, and patient and family teaching. The nurse works closely with the healthcare provider, physiotherapist, occupational therapist and social worker to help the patient to restore function and make appropriate lifestyle adjustments to chronic illness.
The newly diagnosed RA patient is usually treated on an outpatient basis, although hospitalisation may be necessary for patients with extraarticular complications or advancing disease requiring reconstructive surgery for disabling deformities. Intervention begins with a careful physical assessment (e.g. joint pain, swelling, ROM and general health status) and includes an evaluation of psychosocial needs (e.g. family support, sexual satisfaction, emotional stress, financial constraints, career limitations) and environmental concerns (e.g. transportation, home or work modifications). After any problems have been identified, the nurse can coordinate a carefully planned program for rehabilitation and education with the interdisciplinary healthcare team.
Suppression of inflammation may be achieved effectively through the administration of NSAIDs, DMARDs and biological/targeted therapies. Careful attention to timing is critical to sustain a therapeutic drug level and reduce early morning stiffness. The nurse should discuss the action and side effects of each prescribed drug and the importance of necessary laboratory monitoring. Many patients with RA will take several different drugs, and the nurse needs to make the drug regimen as understandable as possible. Patients should be encouraged to develop a method for remembering to take their medications (e.g. pill containers or other system).
Non-drug management may include the use of therapeutic heat and cold, rest, relaxation techniques, joint protection (see Boxes 64-2 and 64-5), biofeedback, transcutaneous electrical nerve stimulation (see Ch 8) and hypnosis. The patient and family should choose therapies that promote optimal comfort within the parameters of their lifestyle.
BOX 64-5 Protection of small joints
PATIENT & FAMILY TEACHING GUIDE
Include the following instructions when teaching the patient with arthritis to protect small joints.
Lightweight splints may be prescribed to rest an inflamed joint and to prevent deformity from muscle spasms and contractures. The splints should be removed at regular intervals to give skin care and perform ROM exercises. After assessment has been completed and supportive care has been given, the splints are reapplied as prescribed. The occupational therapist may help to identify additional self-help devices that can assist in activities of daily living.
Nursing care and procedures should be planned around the patient’s morning stiffness. Sitting or standing in a warm shower, sitting in a bath with warm towels around the shoulders or simply soaking the hands in a basin of warm water may help relieve joint stiffness and allow the patient to perform activities of daily living more comfortably.
Ambulatory and home care
Rest
Alternating scheduled rest periods with activity throughout the day helps relieve fatigue and pain. The amount of rest needed varies according to the severity of the disease and the patient’s limitations. The patient should rest before becoming exhausted. Total bed rest is rarely necessary and should be avoided to prevent stiffness and immobility. However, even a patient with mild disease may require daytime rest in addition to 8–10 hours of sleep at night. The nurse can help the patient to identify ways to modify daily activities to avoid overexertion that can lead to fatigue and exacerbation of disease activity. For example, the patient may tolerate meal preparation more easily while sitting on a high stool in front of the sink. The nurse should assist the patient to pace activities and set priorities based on realistic goals.
Good body alignment while resting can be maintained through use of a firm mattress or bed board. Positions of extension should be encouraged, and positions of flexion should be avoided. Splints and casts may be helpful in maintaining proper alignment and promoting rest, especially when joint inflammation is present. Lying prone for half an hour twice daily is also recommended. To decrease the risk of joint contracture, pillows should never be placed under the knees. A small, flat pillow may be used under the head and shoulders.
Joint protection
Protecting joints from stress is important. The nurse can help the patient to identify ways to modify tasks to put less stress on joints during routine activities (see Box 64-5). Energy conservation requires careful planning. The emphasis is on work simplification techniques. Work should be done in short periods with scheduled rest breaks to avoid fatigue (pacing). Work should also be spread throughout the week rather than attempted at one time (e.g. all cleaning should not be done on the weekend). Activities should be organised to avoid going up and down stairs repeatedly. Carts can be used to carry supplies, or materials that are used often can be stored in a convenient, easily reached area. Time-saving joint protective devices (e.g. an electric can opener) should be used whenever possible. Tasks can also be delegated to other family members.
Patient independence may be increased by occupational therapy training with assistive devices that help simplify tasks, such as built-up utensils, buttonhooks, modified drawer handles, lightweight plastic dishes and raised toilet seats. Wearing shoes with Velcro fasteners and clothing with buttons or a zipper down the front instead of the back makes dressing easier. A cane or walker offers support and relief of pain when walking. A platform wheeled walker further minimises strain on the small joints of the hands and wrists.
Can folic acid decrease gastrointestinal side effects of methotrexate therapy?
EVIDENCE-BASED PRACTICE
Clinical question
For patients with rheumatoid arthritis (RA) on long-term methotrexate therapy (P), does folic acid supplementation (I) versus no folic acid supplementation (C) decrease gastrointestinal (GI) side effects (O)?
Critical appraisal and synthesis of evidence
Implications for nursing practice
• Consult with treating doctor regarding folic acid supplements for patients on long-term methotrexate therapy.
• Counsel the patient that folic acid supplements will probably be needed as methotrexate therapy continues.
• Use of folic acid supplements does not decrease the need to monitor laboratory values for early hepatic toxicity.
P, patient population of interest; I, intervention or area of interest; C, comparison of interest or comparison group; O, outcome(s) of interest.
Visser K, Katchamart W, Loza E, et al. Multinational evidence-based recommendations for the use of methotrexate in rheumatic disorders with a focus on rheumatoid arthritis: integrating systematic literature research and expert opinion of a broad international panel of rheumatologists in the 3E Initiative. Ann Rheum Dis. 2009;68:1086.
Heat and cold therapy and exercise
Heat and cold applications can help relieve stiffness, pain and muscle spasm. Application of ice is especially beneficial during periods of disease exacerbation, whereas moist heat appears to offer better relief of chronic stiffness. The treatment modality should be selected according to disease severity, ease of application and cost. Superficial heat sources such as heating pads, moist hot packs, paraffin baths, whirlpool or spa baths and warm baths or showers can relieve stiffness to allow participation in therapeutic exercises. Plastic bags of frozen vegetables (e.g. peas or corn), which can easily mould around the shoulder, wrists or knees, are an easy home treatment. The patient can also use ice cubes or small paper cups of frozen water to massage proximal or distal to a painful joint. Heat and cold can be used as often as desired; however, the heat application should not exceed 20 minutes at one time, and the cold application should not exceed 10–15 minutes at one time. The nurse should alert the patient to the possibility of an ice burn and to avoid the use of a heat-producing cream (e.g. capsaicin) with another external heat device.
Individualised exercise is an integral part of the treatment plan. A therapeutic exercise program is usually developed by a physiotherapist and includes exercises that improve the flexibility and strength of the affected joints, and the patient’s overall endurance. Reinforce program participation and ensure that the exercises are being done correctly. Inadequate joint movement can result in progressive joint immobility and muscle weakness, and overaggressive exercise can result in increased pain, inflammation and joint damage. Emphasise that participating in a recreational exercise program (e.g. walking, swimming) or performing usual daily activities does not eliminate the patient’s need for therapeutic exercise to maintain adequate joint motion.
Gentle ROM exercises are usually done daily to keep the joints functional. The patient should have the opportunity to practise the exercises with supervision. Careful adherence to the prescribed exercise program should be a prime goal of the teaching program. Aquatic exercises in warm water (25–30°C) allow easier joint movement because of the buoyancy of the water. At the same time, although movement seems easier, water provides two-way resistance that makes muscles work harder than they would on land. Aerobic conditioning programs have been shown to improve the physical fitness levels of patients with RA. During acute inflammation, exercise should be limited to one or two repetitions.
Psychological support
Self-management and adherence to an individualised home treatment program can be accomplished only if the patient has a thorough understanding of RA, the nature and course of the disease, and the goals of therapy. In addition, the patient’s value system and perception of the disease must be considered. The patient is challenged constantly by problems of limited function and fatigue, loss of self-esteem, altered body image and fear of disability and deformity. Alterations in sexuality should be discussed. Chronic pain or loss of function may make the patient vulnerable to unproven or even dangerous remedies through the claims of false advertising. The nurse can help the patient to recognise fears and concerns that are faced by all people who live with chronic illness.
Evaluation of the family support system is important. Financial planning may be necessary. Community resources such as a home care nurse, homemaker services and vocational rehabilitation may be considered. Self-help groups may be beneficial for some patients.
SPONDYLOARTHROPATHIES
The spondyloarthropathies are a group of interrelated multisystem inflammatory disorders that affect the spine, peripheral joints and periarticular structures. These disorders are all negative for RF. Thus they are often referred to as seronegative arthropathies. Inheritance of HLA-B27 is strongly associated with occurrence of these diseases. Both genetic and environmental factors play a role in the development of this group of diseases, which includes ankylosing spondylitis, psoriatic arthritis and reactive arthritis. (HLAs and their relationship to autoimmune diseases are discussed in Ch 13.) The spondyloarthropathies share clinical and laboratory characteristics that make it difficult to distinguish among them in early disease. A diagnosis is made when inflammatory spinal pain or asymmetrical synovitis is accompanied by one or more of the following: (1) episodes of alternating buttock pain; (2) radiographic evidence of sacroiliitis; (3) heel enthesopathy (e.g. plantar fasciitis, Achilles tendinitis); (4) positive family history of spondyloarthropathy in a first-degree relative; (5) current or documented history of psoriasis; (6) current or documented history of inflammatory bowel disease; and (7) urethritis, cervicitis or acute diarrhoea that occurred within the month preceding onset of arthritic symptoms.18
Ankylosing spondylitis
Ankylosing spondylitis (AS) is a chronic inflammatory disease that primarily affects the axial skeleton, including the sacroiliac joints, intervertebral disc spaces and costovertebral articulations. The HLA-B27 antigen is found in approximately 80–90% of people with AS. Individuals with the antigen have a 20 times greater risk of developing a spondyloarthropathy than those who are HLA-B27 negative.19 Although the usual age of onset is 15–35 years, the highest incidence of the disease is in people 25–34 years of age. Men are three to five times more likely to develop AS than women. The disease may go undetected in women because of a milder course and because the disease is not often considered in a differential diagnosis for women.
Gerontological considerations: arthritis
The prevalence of arthritis in older adults is high, and the disease is accompanied by problems unique to this age group. The most problematic areas related to connective tissue disease in older adults include the following:
1. The high incidence of OA expected in older adults may prevent their healthcare provider from considering the presence of other types of arthritis.
2. Age alone causes changes in serological profiles, making interpretation of laboratory values such as RF and ESR more difficult.
3. Polypharmacy in the older adult can result in iatrogenic arthritis.
4. Non-organic musculoskeletal pain syndromes and weakness may be related to depression and physical inactivity.
5. Diseases such as SLE, which commonly occurs in younger adults, can develop in a milder form in older adults.
Ageing brings many physical and metabolic changes that may increase the older patient’s sensitivity to both the therapeutic and the toxic effects of some drugs. The use of NSAIDs with a shorter half-life may require more frequent dosing but may also produce fewer side effects in the older patient with altered drug metabolism. The older adult who takes NSAIDs has an increased risk for side effects, particularly GI bleeding and renal toxicity. The common occurrence of polypharmacy makes the use of additional drugs in RA treatment particularly challenging in the older adult because of the increased likelihood of untoward drug interactions. The drug regimen should be simplified as much as possible to increase adherence (e.g. limited number of drugs with decreased frequency of administration), particularly for the patient without regular assistance.
A major concern of treatment in the older patient relates to the use of corticosteroid therapy. Corticosteroid-induced osteopenia can add to the problem of decreased bone density related to age and inactivity. It can also increase the occurrence of pathological fractures, especially compression fractures of vertebrae. Corticosteroid-induced myopathy can be minimised or prevented by an age-appropriate exercise program. Although important for all age groups, an adequate support system for the older adult is a critical factor in the ability to follow a treatment regimen that includes nutritional planning, exercise, general health maintenance and appropriate pharmacotherapy.
HEALTH DISPARITIES
Incidence
• HLA-B27 is present in 8% of the general population, including those without ankylosing spondylitis (AS)
• About 80–90% of people with AS have the HLA-B27 antigen
• Only 2% of people with HLA-B27 have clinically detectable disease
• AS is three to five times more common in men than in women
• It occurs more often in those of Western European descent than in other ethnic groups
Clinical implications
• Diagnosis of AS usually occurs between the ages of 18 and 40
• It is a systemic disease often affecting the eyes and heart
• Genetic and environmental factors play a role in the pathogenesis of the disease
• AS is present in about 3–10% of patients with inflammatory bowel disease (IBD)
• About 50–70% of patients with both AS and IBD are HLA-B27 positive
Source: Better Health Victoria. Ankylosing spondylitis. Available at www.betterhealth.vic.gov.au/bhcv2/bhcarticles.nsf/pages/ankylosing_spondylitis.
AETIOLOGY AND PATHOPHYSIOLOGY
The cause of AS is unknown. Genetic predisposition appears to play an important role in the disease pathogenesis, but the precise mechanisms are unknown. Aseptic synovial inflammation in the joints and adjacent tissue causes the formation of granulation tissue (pannus) and the development of dense fibrous scars that lead to fusion of articular tissues. Extraarticular inflammation can affect the eyes, lungs, heart, kidneys and peripheral nervous system.
CLINICAL MANIFESTATIONS AND COMPLICATIONS
AS is characterised by symmetrical sacroiliitis and progressive inflammatory arthritis of the axial skeleton. Symptoms of inflammatory spine pain are the first clues to a diagnosis of AS. The patient typically complains of low back pain, stiffness and limitation of motion that is worse during the night and in the morning but improves with mild activity. In women, early symptoms of disease may manifest as pain and stiffness in the neck rather than the lower back. General symptoms such as fever, fatigue, anorexia and weight loss are rarely present. Uveitis (intraocular inflammation) is the most common non-skeletal symptom. It can appear as an initial presentation of the disease years before arthritic symptoms develop. AS patients may also experience chest pain and sternal/costal cartilage tenderness that can be distressing.
Severe postural abnormalities and deformity can lead to significant disability for the patient with AS (see Fig 64-7). Impaired spinal ROM and fixed kyphosis contribute to altered visual function, raising concerns about safe ambulation. Aortic insufficiency and pulmonary fibrosis are frequent complications. Cauda equina syndrome can also result, contributing to lower extremity weakness and bladder dysfunction. In addition, the patient is at risk for spinal fracture because of osteoporosis.
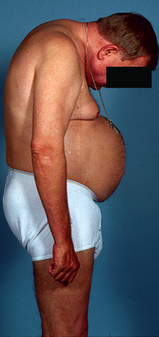
DIAGNOSTIC STUDIES
X-rays are essential for the diagnosis of AS. However, spinal X-rays are seldom useful in initial diagnosis. Instead, pelvic X-rays demonstrate characteristic changes of sacroiliitis that range from subtle erosion to completely fused joints in which the joint spaces have been obliterated. Changes on later spinal films include the appearance of ‘bamboo spine’, which is the result of calcifications (syndesmophytes) that bridge from one vertebra to another. Laboratory testing is not specific, but an elevated ESR and mild anaemia may be seen. When the suspicion of AS is high, the presence of the HLA-B27 antigen improves the likelihood of this diagnosis.
MULTIDISCIPLINARY CARE
Prevention of AS is not possible. However, families with other diagnosed HLA-B27–positive rheumatic diseases (e.g. acute anterior uveitis, juvenile spondyloarthritis) should be alert to signs of low back pain for early identification and treatment of AS.
Care of the patient with AS is aimed at maintaining maximal skeletal mobility while decreasing pain and inflammation. Heat applications can help in the relief of local symptoms. NSAIDs and salicylates are commonly prescribed. DMARDs such as sulfasalazine or methotrexate have little effect on spinal disease but may be helpful with peripheral joint disease. Local corticosteroid injections may be beneficial in relieving symptoms. Etanercept, a biological/targeted therapy, binds TNF and inhibits its action. TNF, which promotes inflammation, is found in elevated levels in the blood and certain tissues of patients with AS. Etanercept has been shown to reduce active inflammation and improve spinal mobility.20 Additional anti-TNF biological agents (infliximab, adalimumab, golimumab) may also be effective.
Once pain and stiffness are managed, exercise is essential. Postural control is important to minimise spinal deformity. The exercise regimen should include back, neck and chest stretches. Hydrotherapy has also been shown to decrease pain and facilitate spinal extension. Surgery may be indicated for severe deformity and mobility impairment. Spinal osteotomy and total joint replacement are the most commonly performed procedures (see Ch 62).
NURSING MANAGEMENT: ANKYLOSING SPONDYLITIS
A key responsibility is to educate the patient with AS about the disease and the principles of therapy. The home management program should include regular exercise and attention to posture, local moist heat applications and knowledgeable use of drugs.
Baseline ROM assessment should include chest expansion (using breathing exercises). If the patient smokes, they should be encouraged to give up smoking to decrease the risk for lung complications in those with reduced chest expansion. Ongoing physiotherapy should include gentle, graded stretching and strengthening exercises to preserve ROM and improve thoracolumbar flexion and extension. Excessive physical exertion should be discouraged during periods of active flare-up of the disease. Proper positioning at rest is essential. The mattress should be firm and the patient should sleep on their back with a flat pillow, avoiding positions that encourage flexion deformity. Postural training emphasises avoiding spinal flexion (e.g. leaning over a desk), heavy lifting and prolonged walking, standing or sitting. Sports that facilitate natural stretching, such as swimming and racquet games, should be encouraged. Family counselling and vocational rehabilitation are important.
Psoriatic arthritis
Psoriasis is a common benign, inflammatory skin disorder that appears to have a genetic predisposition (see Ch 23). Approximately 10% of the 3 million people with psoriasis develop psoriatic arthritis (PsA). PsA is now recognised as a progressive inflammatory disease that can cause significant disability. The exact cause of PsA is unknown, but a combination of immune, genetic and environmental factors is suspected. PsA can occur in five forms:
• arthritis involving primarily the small joints of the hands and feet
• asymmetrical arthritis involving joints of the extremities
• symmetrical polyarthritis resembling RA
• arthritis of the sacroiliac joints and spine (psoriatic spondylitis)
• arthritis mutilans, a rare but very deforming and destructive disease.21
On X-ray, the cartilage loss and erosion resemble that of RA. Advanced cases of PsA often reveal widened joint spaces, and a ‘pencil in cup’ deformity is common at the DIP joints as a result of osteolysis. Elevated ESR, mild anaemia and elevated blood uric acid levels can be seen in some patients. Therefore, the diagnosis of gout must be excluded. Treatment includes splinting, joint protection and physiotherapy. Intramuscular gold therapy was used to treat PsA in the past with some success, but has been replaced by newer DMARDs such as methotrexate, which are effective for both articular and cutaneous manifestations. Sulfasalazine and cyclosporin have been used with some success, and biological/targeted therapy drugs, including etanercept, golimumab, adalimumab and infliximab, are also used.
Reactive arthritis
Reactive arthritis (Reiter’s syndrome) occurs more commonly in young men than in young women. It is associated with a symptom complex that includes urethritis, conjunctivitis and mucocutaneous lesions. In women, symptoms also include cervicitis. Although the exact aetiology is unknown, reactive arthritis appears to occur after certain types of genitourinary or GI tract infections.22 Chlamydia trachomatis is most often implicated in sexually transmitted reactive arthritis. Men and women appear to have equal risk for developing dysenteric reactive arthritis, which typically occurs within days or weeks after infection with Shigella, Salmonella, Campylobacter or Yersinia. Individuals with inherited HLA-B27 are at increased risk of developing reactive arthritis after sexual contact or exposure to certain enteric pathogens, supporting the likelihood of a genetic predisposition.
Urethritis develops within 1–2 weeks after sexual contact or dysentery. Low-grade fever, conjunctivitis and arthritis may occur over the next several weeks. This type of arthritis tends to be asymmetrical, frequently involving the large joints of the lower extremities and the toes. Lower back pain may occur with severe disease. Mucocutaneous lesions commonly occur as small, painless, superficial ulcerations on the tongue, oral mucosa and glans penis. Soft-tissue manifestations commonly include enthesopathies such as Achilles tendinitis or plantar fasciitis. Few laboratory abnormalities occur, although the ESR may be elevated.
Prognosis is favourable, with most patients recovering after 2–16 weeks. Because reactive arthritis is often associated with C. trachomatis infection, treatment of patients and their sexual partners with doxycycline 100 mg twice daily for up to 3 months is widely recommended. Conjunctivitis and lesions require no treatment, but topical ophthalmic corticosteroids are typically prescribed for treatment of uveitis. Physiotherapy may be helpful during disease recovery.
Joints may heal completely, and many patients have complete remission with full joint function. Up to 50% may develop chronic or recurring disease, which can result in major disability. X-ray changes in chronic disease closely resemble those of AS. Treatment of chronic reactive arthritis is symptomatic.
Septic arthritis
Septic arthritis (infectious or bacterial arthritis) is caused by an invasion of the joint cavity with microorganisms. Bacteria can travel through the bloodstream from another site of active infection, resulting in haematogenous seeding of the joint. Organisms can also be introduced directly through trauma or surgical incision. Any bacteria can cause the infection. In the immunocompromised patient, even non-pathogenic bacteria can be responsible for the development of septic arthritis. Staphylococcus aureus is the most common causative organism. Streptococcus haemolyticus is also seen. Neisseria gonorrhoeae is the most common cause in sexually active young adults. Factors that increase the risk of infection include: (1) diseases in which there is decreased host resistance (e.g. leukaemia, diabetes mellitus); (2) treatment with corticosteroids or immunosuppressive drugs; and (3) debilitating chronic illness.
In septic arthritis, large joints such as the knee and hip are most frequently involved. Inflammation of the joint cavity causes severe pain, erythema and swelling. Because infection has often spread from a primary site elsewhere in the body, fever or shaking chills may accompany articular manifestations. A diagnosis may be made by aspiration of the joint (arthrocentesis) and culture of the synovial fluid.23 Blood cultures should also be obtained for aerobic and anaerobic organisms.
Septic arthritis requires prompt treatment to prevent joint destruction. Broad-spectrum antibiotics against Gram-negative organisms, pneumococci and staphylococci are often started before cultures identify the causative organism. Once the organism is determined, specific treatment can be determined. Infections may respond to treatment within 2 weeks or may take as long as 4–8 weeks, depending on the causative organism. Local aspiration and surgical drainage may be required. If diagnosis and treatment are delayed, destruction of articular cartilage can occur, followed by loss of joint function. Chronic infection may also develop. Septic arthritis of the hip can contribute to the development of avascular necrosis.24
The nurse should assess and monitor joint inflammation, pain and fever. To control pain with immobilisation of affected joints, resting splints or traction should be used. Local hot compresses can also help relieve pain. Gentle ROM exercises should be initiated as soon as tolerated to prevent muscle atrophy and joint contractures. The nurse should explain the need for antibiotics and the importance of their continued use until the infection is resolved. Support should be offered to the patient who requires arthrocentesis or operative drainage. It is important to use strict aseptic technique when assisting with joint aspiration procedures.
Tick-borne infection
The burden of disease attributable to tick-related illness in Australia and New Zealand appears to be small; however, as tick-related illnesses are unreportable events their true incidence is unknown. There have been a number of media reports about people who have suffered adverse effects from tick bites.25 Both the New Zealand and Australian health departments have information about the effects and prevention of tick bites (see the Resources on p 1855). A species of paralysis tick called Ixodes holocyclus can be found along Australia’s east coast and may cause tick paralysis, tick typhus and allergic reactions.
The best-described tick-borne infection and one that other tick infections mimic is Lyme disease, which is a spirochetal infection caused by Borrelia burgdorferi and transmitted by the bite of an infected deer tick. It was first identified in 1975 in Lyme, Connecticut, after an unusual clustering of arthritis in children. It is now the most common vector-borne disease in the US.26 The tick typically feeds on mice, dogs, cats, cows, horses, deer and humans. Wild animals do not exhibit the illness, but clinical Lyme disease does occur in domestic animals. Person-to-person transmission does not occur. The peak season for human infection is during the summer months.
While there is little evidence that Australian ticks cause Lyme disease, other infections carried by Australian ticks may cause an infection similar to Lyme disease. However, these infections remain poorly characterised.25 Although Lyme disease is common in parts of the US, it also exists throughout much of the world. In New Zealand, some cases have been reported, but only in people who have recently travelled from an endemic area.
Lyme disease symptoms can mimic other diseases such as multiple sclerosis, mononucleosis and meningitis. The most characteristic clinical symptom of early localised disease is erythema migrans (EM), a skin lesion that frequently occurs at the site of the tick bite within 2–30 days after exposure. The lesion begins as a red macule or papule that slowly expands to form a large round lesion with a bright-red border and central clearing. The EM lesion is often accompanied by acute viral-like symptoms, such as low-grade fever, chills, headache, stiff neck, fatigue, swollen lymph nodes, and migratory joint and muscle pain. Symptoms usually occur in a week but may be delayed for up to 30 days.
If not treated, the spirochete can disseminate within several weeks or months to the heart, joints and central nervous system (CNS). Carditis may occur with chronic arthritic pain and swelling in the large joints. Nervous system problems may include severe headaches, temporary facial paralysis (e.g. Bell’s palsy) or poor motor coordination. One neurological condition known as tertiary neuroborreliosis results in confusion and forgetfulness. Diagnosis is often based on clinical manifestations, in particular the EM lesion, and a history of exposure in an endemic area. FBC and ESR results are usually normal. Active lesions can be treated with oral antibiotics. Doxycycline, cefuroxime and amoxicillin are often effective in early-stage infection and in prevention of later stages of the disease. Doxycycline is effective in preventing Lyme disease when given within 3 days after the bite of a deer tick. Short-term therapy of 2–3 weeks is usually effective for solitary EM, but long-standing infection may require extended parenteral antibiotic therapy. Intravenous ceftriaxone is given for severe cardiac or neurological problems. In most cases Lyme disease is treated successfully with antibiotics.26
Reducing exposure to ticks is the best way to prevent tick bites and associated problems. Patient and family teaching for people living in endemic areas is outlined in Box 64-6. No vaccine is available for diseases caused by tick bites.27
BOX 64-6 Prevention and treatment of tick bites
PATIENT & FAMILY TEACHING GUIDE
How to prevent tick bites
• Ticks tend to live in coastal areas. Wear appropriate clothing when outdoors in tick areas, including long-sleeved shirt, long pants tucked into socks and a wide-brimmed hat. Ticks are more easily detected on light-coloured clothing.
• Spray clothes and hats with insect repellent and wear a repellent that contains DEET.
• When returning from an area known to have ticks, remove clothing and search for ticks, especially behind the ears, on the back of the head, in the groin, in the armpits and at the back of the knees. Be careful where clothes are placed as they may introduce ticks inside the house. Do not forget to check children and pets.
• Ticks in the nymph stage are tiny (as big as a poppy seed) but can still transmit infections so it is important to have a very close look at skin in good light to see whether any small ticks are feeding. People are sometimes unaware that they have been bitten by a tick.
• In tick-infested areas, mow grass in the backyard and keep mulch and leaf litter away from the main entrance to the house. Trim shrubs overhanging paths and play areas.
• When walking in tick-infested areas try to keep to the centre of cleared paths as much as possible and try to avoid brushing up against plants and grasses.
• The risk of tick bites is greatest in late spring, summer and early autumn when the nymph ticks are more abundant and when people visit habitats where ticks live, such as forests and other densely vegetated areas, especially areas with high grass and a lot of leaf litter.
What to do if bitten by a tick
• Remove a tick as soon as possible after locating it.
• Use fine-pointed tweezers and grasp the tick as close to the skin as possible. Gently pull the tick straight out with steady pressure. If having difficulty, seek medical attention.
• Do not try to kill the tick with methylated spirits or any other chemicals as this will cause the tick to inject more toxins. For a severe infestation of larval-stage ticks (often referred to as grass ticks) add 1 cup of bicarbonate of soda to bathwater and bathe for 30 minutes.
Source: New South Wales Department of Health. Infectious disease factsheet: Lyme disease. Updated 23 July 2010. Available at www.health.nsw.gov.au/factsheets/infectious/lyme_disease.html.
Gout
AETIOLOGY AND PATHOPHYSIOLOGY
Gout is caused by an increase in uric acid production or underexcretion of uric acid by the kidneys. Uric acid is the major end product of purine catabolism and is primarily excreted by the kidneys. Characteristic deposits of sodium urate crystals occur in articular, periarticular and subcutaneous tissues. Joint involvement includes recurrent attacks of acute arthritis. Hyperuricaemia may be the result of increased purine synthesis, decreased renal excretion, or both. A high dietary intake of purine alone has relatively little effect on uric acid levels. Hyperuricaemia may result from prolonged fasting or excessive alcohol consumption because of the increased production of ketoacids, which then inhibit uric acid excretion.
Gout may be classified as primary or secondary.28 In primary gout, a hereditary error of purine metabolism leads to the overproduction or retention of uric acid. Secondary gout may be related to another acquired disorder (see Box 64-7) or may be the result of drugs known to inhibit uric acid excretion. Secondary gout may also be caused by drugs that increase the rate of cell death, such as the chemotherapy agents used in treating leukaemia. Primary gout, which accounts for 90% of cases, occurs predominantly in middle-aged men, with almost no incidence in premenopausal women. Hyperuricaemia may also develop in patients taking thiazide diuretics, in postmenopausal women and in organ transplant recipients who are receiving immunosuppressive agents.
Obesity in men increases the risk of gout. Hypertension, diuretic use and excessive alcohol consumption are additional risk factors. A diet high in purine-rich foods (e.g. shellfish such as crab and prawns; vegetables such as lentils, asparagus and spinach; meats such as beef, chicken and pork) will not cause gout but can trigger an acute attack if a person is susceptible to gout.
CLINICAL MANIFESTATIONS AND COMPLICATIONS
In the acute phase, gouty arthritis may occur in one or more joints but usually fewer than four. Affected joints may appear dusky or cyanotic and are extremely tender. Inflammation of the great toe (podagra) is the most common initial problem. Other affected joints may include the midtarsal area of the foot, the ankle, knee and wrist. Olecranon bursae may also be involved. Acute gouty arthritis is usually precipitated by trigger events such as trauma, surgery, alcohol ingestion or systemic infection. The onset of symptoms typically occurs at night with sudden swelling and excruciating pain peaking within several hours, often accompanied by low-grade fever. Individual attacks usually subside, treated or untreated, in 2–10 days. The affected joint returns to normal and patients are often free of symptoms between attacks.
Chronic gout is characterised by multiple joint involvement and visible deposits of sodium urate crystals called tophi. These are typically noted in the synovium, subchondral bone, olecranon bursae and vertebrae; along tendons; and in the skin and cartilage (see Fig 64-8). Tophi are rarely present at the time of the initial attack and are generally noted only many years after the onset of disease.
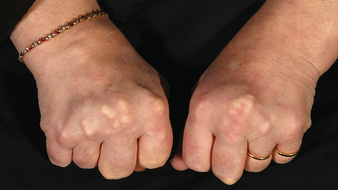
Figure 64-8 Tophi over the dorsum of the hands accentuated by finger flexion and stretching of overlying skin.
The severity of gouty arthritis is variable. The clinical course may consist of infrequent mild attacks or multiple severe episodes (up to 12 per year) associated with a slowly progressive disability. In general, the higher the serum uric acid level, the earlier the appearance of tophi and the greater the tendency towards more frequent, severe episodes of acute gout. Chronic inflammation may result in joint deformity, and cartilage destruction may predispose the joint to secondary OA. Large and unsightly tophaceous deposits may perforate overlying skin, producing draining sinuses that often become infected. Excessive uric acid excretion may lead to the formation of kidney or urinary tract stones. Pyelonephritis associated with intrarenal sodium urate deposits and obstruction may contribute to renal disease.
DIAGNOSTIC STUDIES
Serum uric acid levels are usually elevated above 400 mmol/L. However, hyperuricaemia is not specifically diagnostic of gout because increased levels may be related to a variety of drugs or may exist as a totally asymptomatic abnormality in the general population. Specimens for 24-hour urine uric acid levels may be obtained to determine whether the disease is caused by decreased renal excretion or overproduction of uric acid.
Synovial fluid aspiration is a controversial part of patient evaluation because an accurate diagnosis of gout is possible in 80% of patients based on clinical symptoms alone. However, aspiration may have therapeutic value by decompressing a swollen joint capsule. Joint aspiration is also the only reliable method to distinguish gout from septic arthritis and pseudogout (calcium phosphate crystals are formed). Affected fluid characteristically contains needle-like crystals of sodium urate.29 X-rays appear normal in the early stages of gout, with tophi, an indicator of chronic disease, appearing as eroded areas in the bone.
MULTIDISCIPLINARY CARE
Goals for the care of the patient with gout (see Box 64-8) include termination of an acute attack through use of an anti-inflammatory agent such as colchicine, with NSAIDs prescribed adjunctively for pain management. Future attacks are prevented by a maintenance dose of allopurinol in combination with weight reduction (as needed) and possible avoidance of alcohol and foods high in purine (red and organ meats). Treatment is also aimed at preventing the formation of uric acid kidney stones and the development of associated conditions such as hypertriglyceridaemia and hypertension.
MULTIDISCIPLINARY CARE
Drug therapy
Acute gouty arthritis is treated with colchicine and NSAIDs. Because colchicine has anti-inflammatory effects but no analgesic properties, an NSAID may be added to the treatment regimen for pain management. Oral administration of colchicine generally produces dramatic pain relief within 24–48 hours. Colchicine also has diagnostic merit in that a good response to this drug gives further evidence for the diagnosis of gout. Colchicine has a low threshold for toxicity and must be used with extreme care.
Recurrent gout may be prevented by combining colchicine with a xanthine oxidase inhibitor such as allopurinol or a uricosuric drug such as probenecid. Febuxostat, a selective inhibitor of xanthine oxidase, is given for long-term management of hyperuricaemia in people with chronic gout.30 Corticosteroids either orally or by intraarticular injection can be helpful in treating acute attacks. Systemic corticosteroids may be used only if routine therapies are contraindicated or ineffective. Adrenocorticotropic hormone (ACTH) may be used for treating acute gout.
For many years the standard therapy for hyperuricaemia caused by urate underexcretion has been uricosuric drugs such as probenecid, which inhibit renal tubular reabsorption of urates. However, this class of drugs is ineffective when creatinine clearance is reduced, as can occur in patients over 60 years old. Aspirin inactivates the effect of uricosurics, resulting in urate retention, and should be avoided while patients are taking uricosuric drugs. Paracetamol can be used safely if analgesia is required.
Adequate urine volume with normal renal function (2–3 L/day) must be maintained to prevent the precipitation of uric acid in the renal tubules. Allopurinol, which blocks the production of uric acid, is particularly useful in patients with uric acid stones or renal impairment, in whom uricosuric drugs may be ineffective or dangerous. For patients who cannot tolerate allopurinol because of side effects, oxypurinol can be prescribed. Oxypurinol is the active metabolite of allopurinol. The angiotensin II receptor antagonist losartan may be especially useful for the treatment of elderly patients with both gout and hypertension. Losartan will promote urate diuresis and may normalise serum urate levels. Combination therapy with losartan and allopurinol may also be given. Regardless of which drugs are prescribed, serum uric acid levels must be checked regularly to monitor treatment effectiveness.
Nutritional therapy
Traditional dietary restrictions include limiting the use of alcohol and the consumption of foods high in purine (see Box 45-10). However, drugs can often control gout without necessitating these changes. Obese patients should be instructed in a carefully planned weight-reduction program.
NURSING MANAGEMENT: GOUT
Nursing interventions for the patient with acute gouty arthritis include supportive care of the inflamed joints. The nurse should avoid causing pain to an inflamed joint by careless handling. Bed rest may be appropriate with affected joints properly immobilised. Involvement of a lower extremity may require use of a cradle or footboard to protect the painful area from the weight of bedclothes. The nurse should assess the limitation of motion and degree of pain and document treatment effectiveness.
The patient and family should be helped to understand that hyperuricaemia and gouty arthritis are chronic problems that can be controlled with careful adherence to a treatment program. The nurse should offer explanations concerning the importance of drug therapy and the need for periodic determination of serum uric acid levels. The patient should be asked to identify precipitating factors that may cause an attack, including overindulgence in purine-containing foods and alcohol, starvation (fasting), drug use (e.g. niacin, aspirin, diuretics) and major medical events (e.g. surgery, myocardial infarction).
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a multisystem inflammatory autoimmune disease. It is a complex disorder of multifactorial origin resulting from interactions between genetic, hormonal, environmental and immunological factors. SLE typically affects the skin, joints and serous membranes (pleura, pericardium), along with the renal, haematological and neurological systems. The prevalence of SLE is variable internationally and is approximately 1 in 3500 in New Zealand and Australia. Most cases of SLE occur in women in their childbearing years. In New Zealand, Māori and Pacific Islanders are 3.5 times more likely to develop SLE than Europeans. A similar pattern has been noted when comparing Indigenous Australians living in northern Australia to Australians of European descent.31
SLE is characterised by variability within and among people. Its chronic unpredictable course is marked by alternating periods of exacerbations and remissions.
AETIOLOGY AND PATHOPHYSIOLOGY
The aetiology of the abnormal immune response in SLE is unknown. Based on the high prevalence of SLE among family members, a genetic influence is suspected. Multiple susceptibility genes from the HLA complex show associations with SLE, including HLA-DR3. Hormones are also known to play a role in the aetiology of SLE. Onset or exacerbation of disease symptoms sometimes occurs after the onset of menarche, with the use of oral contraceptives, and during and after pregnancy. The disease tends to worsen in the immediate postpartum period.
Environmental factors are believed to contribute to the occurrence of SLE, with sun exposure and sunburn being the most common environmental triggers. Infectious agents may serve as a stimulus for immune hyperactivity. SLE may also be precipitated or aggravated by certain drugs, such as procainamide, hydralazine and a number of antiepileptic medications.32
SLE is characterised by the production of a large variety of autoantibodies against nucleic acids (e.g. single- and double-stranded DNA), erythrocytes, coagulation proteins, lymphocytes, platelets and many other self-proteins. Autoimmune reactions characteristically are directed against constituents of the cell nucleus (antinuclear antibodies [ANAs]), particularly DNA.
Circulating immune complexes containing antibody against DNA are deposited in the basement membranes of capillaries in the kidneys, heart, skin, brain and joints. Complement is activated and inflammation occurs. The overaggressive antibody response is also related to activation of B and T cells. The specific manifestations of SLE depend on which cell types or organs are involved. (SLE is a type III hypersensitivity response [see Ch 13].)
CLINICAL MANIFESTATIONS AND COMPLICATIONS
SLE is extremely variable in its severity, ranging from a relatively mild disorder to a rapidly progressive one affecting many body systems (see Fig 64-9). No characteristic pattern occurs in the progressive involvement of SLE. Any organ can be affected by an accumulation of circulating immune complexes. The most commonly affected tissues are the skin and muscle, the lining of the lungs, the heart, nervous tissue and the kidneys. Generalised complaints such as fever, weight loss, arthralgia and excessive fatigue may precede an exacerbation of disease activity.
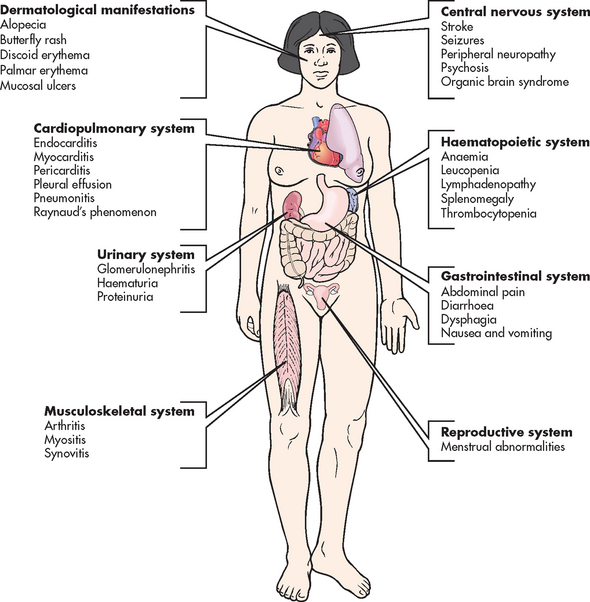
Dermatological manifestations
Cutaneous vascular lesions can appear in any location but are most likely to develop in sun-exposed areas. Severe skin reactions can occur in people who are photosensitive. The classic butterfly rash over the cheeks and bridge of the nose occurs in 50% of patients with SLE (see Fig 64-10). About 20% of patients have discoid (round, coin-shaped) lesions. A small number of patients have persistent lesions, photosensitivity and mild systemic disease in a syndrome referred to as subacute cutaneous lupus.

Ulcers of the oral or nasopharyngeal membranes occur in up to a third of patients with SLE. Transient diffuse or patchy hair loss (alopecia) is also common, with or without underlying scalp lesions. The hair may grow back during remission, but hair loss may be permanent over lesions. The scalp becomes dry, scaly and atrophied.
Musculoskeletal problems
Polyarthralgia with morning stiffness is often the patient’s first complaint and may precede the onset of multisystem disease by many years. Arthritis occurs in more than 90% of patients with SLE. Diffuse swelling is accompanied by joint and muscle pain, and some stiffness may be experienced. Lupus-related arthritis is generally non-erosive, but it may cause deformities such as swan neck deformity of the fingers (see Fig 64-4, D), ulnar deviation and subluxation with hyperlaxity of the joints. Patients with SLE have an increased risk of bone loss and fracture.
Cardiopulmonary problems
Tachypnoea and cough in patients with SLE are suggestive of restrictive lung disease. Pleurisy with or without pleural effusion is also possible. Cardiac involvement may include arrhythmias resulting from fibrosis of the sinoatrial and atrioventricular nodes. This occurrence is an ominous sign of advanced disease, contributing significantly to the morbidity and mortality seen in SLE. Pericarditis can also occur.33 Clinical factors such as hypertension and hypercholesterolaemia require aggressive therapy and careful monitoring. In addition, people with SLE are at risk for secondary antiphospholipid syndrome (APS), a disorder of coagulation that leads to clots in the arteries and veins.34
Renal problems
Lupus nephritis (LN) occurs in approximately 50% of patients with SLE.35 Manifestations of renal involvement vary from mild proteinuria to rapid, progressive glomerulonephritis.
The primary goal in treating LN is to slow the progression of nephropathy and preserve renal function by managing the underlying disease. The importance of obtaining a renal biopsy is controversial, but findings can help guide treatment, which typically includes corticosteroids, cytotoxic agents (cyclophosphamide) and immunosuppressive agents (azathioprine and cyclosporin). A newer drug, mycophenolate mofetil, may be more effective and less toxic than cyclophosphamide, which has been the standard of treatment. Oral prednisone or pulsed IV methylprednisolone may be used as an intervention for LN, especially in the initial treatment period when cytotoxic agents have not had time to take effect.
Nervous system problems
Along with renal involvement, neuropsychiatric manifestations are prevalent in SLE. Generalised or focal seizures are the most common manifestation involving the CNS and occur in as many as 15% of patients with SLE by the time of diagnosis. Seizures are generally controlled by corticosteroids or anticonvulsant drugs. Peripheral neuropathy can also occur, leading to sensory and motor deficits.
Cognitive dysfunction, recognised as a CNS manifestation of SLE, may result from the deposition of immune complexes within brain tissue. It is characterised by disordered thought processes, disorientation, memory deficits and psychiatric symptoms such as severe depression and psychosis. Various psychiatric disorders are reported in SLE, including mood disorders, anxiety and psychosis, although they may be related to the stress of having a major illness or to associated drug therapies. Occasionally a stroke or aseptic meningitis may be attributable to SLE. Headaches are also common and can become severe during a flare (exacerbation of disease).
Haematological problems
The formation of antibodies against blood cells, such as erythrocytes, leucocytes, thrombocytes and coagulation factors, is a common feature of SLE. Anaemia, mild leucopenia and thrombocytopenia are often present in SLE. Some patients develop a tendency towards coagulopathy involving either excessive bleeding or blood clot development. A manifestation of antiphospholipid antibody syndrome is a common cause of hypercoagulability in SLE patients, many of whom benefit from high-intensity treatment with warfarin.
Infection
Patients with SLE appear to have increased susceptibility to infections, possibly related to defects in the ability to phagocytise invading bacteria, deficiencies in production of antibodies, and the immunosuppressive effect of many anti-inflammatory drugs. Infection is a major cause of death, with pneumonia being the most common infection. Fever should be considered serious because it may indicate an underlying infectious process rather than lupus activity alone. Vaccinations are generally safe for patients with SLE. The exception is the need to avoid live virus vaccines for patients who are being treated with corticosteroids or cytotoxic agents.
DIAGNOSTIC STUDIES
The diagnosis of SLE is based on the presence of distinct criteria revealed through patient history, physical examination and laboratory findings (see Box 64-9). No specific test is diagnostic for SLE, but a variety of abnormalities may be present in the blood. SLE is characterised by the presence of ANA, and its identification establishes the existence of an autoimmune disease. Other antibodies include anti-DNA, anti-neuronal, anticoagulant, anti-WBC, anti-red blood cell (RBC), antiplatelet, antiphospholipid and anti-basement membrane. The tests that are most specific for SLE include the anti–double-stranded DNA and the anti-Smith (Sm). High levels of anti-DNA are rarely found in any condition other than SLE, and anti-Sm seems to be found almost exclusively in SLE. The lupus erythematosus (LE) cell prep test is a non-specific test for SLE and is positive in other rheumatic diseases. ESR and CRP levels are not diagnostic of SLE but may be used to monitor disease activity and the effectiveness of therapy.
BOX 64-9 Criteria for diagnosis of systemic lupus erythematosus*
• Arthritis: non-erosive, involvement of two or more joints characterised by tenderness, swelling and effusion, may be asymmetrical and pain may be disproportionate to swelling
• Serositis: pleuritis or pericarditis
• Renal disorder: persistent proteinuria or cellular casts in urine (3+ on dipstick)
• Neurological disorder: seizures or psychosis
• Haematological disorder: haemolytic anaemia, leucopenia, lymphopenia or thrombocytopenia
• Immunological disorder: positive lupus erythematosus preparation; anti-DNA antibody or antibody to Smith nuclear antigen; or false-positive serological tests for syphilis
Source: Tan EM, Cohen AS, Fries JF et al. The 1982 revised criteria for classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:11. Updated 2006. Available at www.rheumatology.org/publications/classification, accessed December 2010.
*A person is classified as having SLE if four or more of the criteria are present, serially or simultaneously, during any interval of observation. These criteria, developed by a subcommittee of the American College of Rheumatology, are still used internationally for the purpose of classification in population surveys, not for the diagnosis of individual patients. Each patient needs specialised medical review.
MULTIDISCIPLINARY CARE
A major challenge in the treatment of SLE is to manage the active phase of the disease while preventing complications of treatments that cause long-term tissue damage. An improving prognosis of SLE may be the result of earlier diagnosis, prompt recognition of serious organ involvement and better therapeutic regimens. Survival is influenced by several factors, including age, ethnicity, gender, socioeconomic status, coexisting conditions and severity of disease.
Drug therapy
NSAIDs continue to be an important intervention, especially for patients with mild polyarthralgias or polyarthritis. Because prolonged therapy is likely, careful patient monitoring must include the potential for GI effects from NSAID use. Antimalarial agents such as hydroxychloroquine are often used to treat fatigue and moderate skin and joint problems. A response to antimalarial therapy may not be noticed for several months.36 Exacerbation may also be prevented with these drugs. Regular funduscopic and visual field examinations must be performed by an ophthalmologist every 6–12 months when patients are on hydroxychloroquine.
Retinopathy can develop with high-dose use of these drugs, but it generally reverses when they are discontinued. If the patient cannot tolerate an antimalarial agent, an anti-leprosy drug such as dapsone may be used. Corticosteroid exposure should be limited, but tapering doses of IV methylprednisolone may be useful in controlling severe exacerbations of polyarthritis. Steroid-sparing immunosuppressants such as methotrexate can serve as an alternative treatment and are prescribed in combination with folic acid to decrease minor side effects of corticosteroids. However, high doses of corticosteroids may be especially appropriate for the patient with very severe cutaneous SLE. Immunosuppressive drugs such as azathioprine and cyclophosphamide may be prescribed to reduce the need for long-term corticosteroid therapy. Azathioprine or cyclophosphamide is also appropriate for treatment of severe organ-system disease, especially LN. Close monitoring is necessary to minimise drug toxicity and side effects. Because blood clots can be a life-threatening complication of SLE, anticoagulants such as warfarin or heparin may be prescribed.36
Clinical trials are currently investigating the effect of various medications on SLE management. These include biological/targeted therapy agents that interfere with the immune response, such as abatacept, and hormones (prasterone) to combat lupus-induced osteoporosis.
Disease management is most appropriately monitored by serial anti-DNA titres and serum complement levels (see Box 64-10). Simpler and less costly tests such as ESR or CRP may also help in monitoring treatment effectiveness. In patient teaching related to prescribed drugs, the nurse should include their indications for use, proper administration and possible side effects. The patient should also understand that abrupt cessation may precipitate exacerbation of disease activity.
BOX 64-10 Systemic lupus erythematosus
MULTIDISCIPLINARY CARE
Collaborative therapy
Steroid-sparing drugs (e.g. methotrexate)
Antimalarials (e.g. hydroxychloroquine)
Corticosteroids for exacerbations and severe disease
Immunosuppressive drugs (e.g. cyclophosphamide, mycophenolate mofetil)
ANA, antinuclear antibody; ECG, electrocardiogram; LE, lupus erythematosus; NSAIDs, non-steroidal anti-inflammatory drugs.
NURSING MANAGEMENT: SYSTEMIC LUPUS ERYTHEMATOSUS
Nursing assessment
As in the majority of rheumatic diseases, the chronic and unpredictable nature of SLE presents many challenges to the patient, their family and the multidisciplinary team. The physical, psychological and sociocultural problems associated with the long-term management of SLE require the skills and support of a multidisciplinary healthcare team.
Subjective and objective data that should be obtained from the patient with SLE are presented in Table 64-5. In particular, the extent to which pain and fatigue influence activities of daily living must be evaluated. A developmental approach focuses on age-appropriate education and counselling on issues such as personal relationships, family planning, occupational responsibilities and recreational activities.
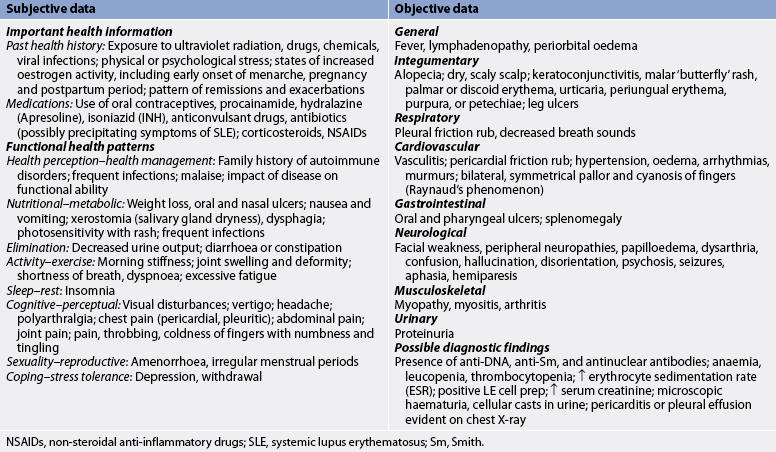
Nursing diagnoses
Nursing diagnoses for the patient with SLE may include, but are not limited to, those presented in NCP 64-2.
Planning
The overall goals are that the patient with SLE will: (1) have satisfactory pain management; (2) adhere to the therapeutic regimen to achieve maximum symptom management; (3) demonstrate awareness of and avoid activities that cause disease exacerbation; and (4) maintain optimal role function and a positive self-image.
Nursing implementation
Health promotion
Prevention of SLE is not yet possible. However, education of health professionals and the community should promote a clearer understanding of the disease and the need for early diagnosis and treatment.
Acute intervention
During an exacerbation of SLE, the patient may become abruptly and dramatically ill. Nursing interventions include accurately recording the severity of symptoms and documenting the response to therapy. The nurse should specifically assess fever pattern, joint inflammation, limitation of motion, location and degree of discomfort, and fatigue. The nurse should also monitor the patient’s weight and fluid intake and output if corticosteroids are prescribed because of the fluid-retention effect of these drugs and the possibility of renal failure. Collection of 24-hour urine samples for protein and creatinine clearance may be ordered. It is important to observe for signs of bleeding that result from drug therapy, such as pallor, skin bruising, petechiae or tarry stools.
Careful assessment of neurological status includes observing for visual disturbances, headaches, personality changes, seizures and forgetfulness. Psychosis may indicate CNS disease or may be the effect of corticosteroid therapy. Irritation of the nerves of the extremities (peripheral neuropathy) may produce numbness, tingling and weakness of the hands and feet.
The nurse should explain the nature of the disease, modes of therapy and all diagnostic procedures. Emotional support for the patient and family is also essential.
Ambulatory and home care
Nursing interventions should emphasise health teaching and the importance of patient cooperation for successful home management. The patient should understand that even strong adherence to the treatment plan is not a guarantee against exacerbation, because the course of the disease is unpredictable. However, a variety of factors may increase disease activity, such as fatigue, sun exposure, emotional stress, infection, drugs and surgery. The patient and family should be assisted to eliminate or minimise exposure to precipitating factors (see Box 64-11).
BOX 64-11 Systemic lupus erythematosus
PATIENT & FAMILY TEACHING GUIDE
Teaching related to the disease and appropriate management should include:
2. Names of drugs, actions, side effects, dosage, administration
4. Energy conservation and pacing techniques
5. Therapeutic exercise, use of heat therapy (for arthralgia)
6. Avoidance of physical and emotional stress
7. Avoidance of exposure to individuals with infection
8. Avoidance of drying soaps, powders, household chemicals
9. Use of sunscreen protection (at least SPF 15) and protective clothing, with minimal sun exposure between 11 am and 3 pm
10. Regular medical and laboratory follow-up
Lupus and pregnancy
Because SLE is most common in women of childbearing age, treatment during pregnancy must be considered. The rheumatologist and obstetrician should thoroughly discuss with the patient her desire to become pregnant. Infertility may have resulted from renal involvement and the previous use of high-dose corticosteroid and chemotherapy drugs. The patient should understand that spontaneous abortion, stillbirth and intrauterine growth retardation are common problems with pregnancy. They occur because of deposits of immune complexes in the placenta and because of inflammatory responses in the placental blood vessels. The renal, cardiovascular, pulmonary and central nervous systems (in particular) may be affected during pregnancy. Women who already demonstrate serious SLE involvement in these systems should be counselled against pregnancy. For the best outcome, pregnancy should be planned at a point when the disease activity is minimal. Exacerbation is common during the postpartum period. Therapeutic abortion offers the same risk of post-delivery exacerbation as carrying the fetus to term. Neonatal lupus erythematosus rarely occurs in infants born to women with SLE.
Psychosocial issues
The patient with SLE confronts many psychosocial issues.37 Disease onset and symptoms may be vague, and SLE may be undiagnosed for long periods. Supportive therapies may become as important as medical treatment in helping the patient to cope with the disease. The patient and family should be counselled that SLE has a good prognosis for the majority of people. Families are anxious about hereditary aspects and want to know whether their children will also have SLE. Many couples require pregnancy and sexual counselling. Individuals making decisions about marriage and careers worry about how SLE will interfere with their plans. Nurses may need to educate teachers, employers and colleagues.
The obvious physical effects of skin lesions and alopecia may cause social isolation for the patient, affecting the individual’s self-esteem and body image. Consultation with a dermatologist may be recommended for appropriate treatment and cosmetic products to conceal the rash. However, pain and fatigue are cited most frequently as interfering with quality of life. Friends and relatives are confused by the patient’s complaints of transient joint pain and overwhelming fatigue. Pacing techniques and relaxation therapy can help the patient to remain involved in day-to-day activities. The nurse should stress the importance of planning both recreational and occupational activities. Young adults find sun restrictions and physical limitations particularly difficult to follow. The patient should be assisted in developing and accomplishing reasonable goals for improving or maintaining mobility, energy levels and self-esteem.
Systemic sclerosis
Systemic sclerosis (SS), or scleroderma, is a disorder of connective tissue characterised by fibrotic, degenerative and occasionally inflammatory changes in the skin, blood vessels, synovium, skeletal muscle and internal organs. Two types of SS exist, one being the more common limited cutaneous disease (80%) and the second being diffuse cutaneous disease. Both forms are systemic, with the degree and type of organ involvement and disease progression very different.38 The disease course of SS is variable. Although symptoms may begin at any time, the usual age at onset is between 30 and 50 years. The overall incidence increases with age and 75% of sufferers are women.39
AETIOLOGY AND PATHOPHYSIOLOGY
The exact cause of SS remains unknown. Immunological dysfunction and vascular abnormalities are believed to play a role in the development of widespread systemic disease. Other risk factors associated with skin thickening include environmental or occupational exposure to coal, plastics and silica dust. In SS, collagen, the protein that gives normal skin its strength and elasticity, is overproduced (see Fig 64-11). Disruption of the cell is followed by platelet aggregation and fibrosis. Proliferation of collagen disrupts the normal functioning of internal organs, such as the lungs, kidneys, heart and GI tract.
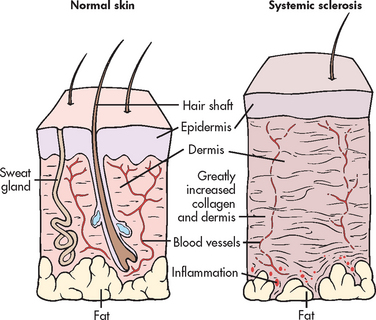
CLINICAL MANIFESTATIONS
Manifestations of SS range from a diffuse cutaneous thickening with rapidly progressive and widespread organ involvement to the more benign variant of limited cutaneous SS. The signs of limited disease appear on the face and hands, while diffuse disease initially involves the trunk and extremities. Clinical manifestations can be described by the acronym CREST:
Calcinosis—painful deposits of calcium in the skin
Raynaud’s phenomenon—abnormal blood flow in response to cold or stress (Raynaud’s phenomenon is described further in Ch 37)
(O)Esophageal dysfunction—difficulty with swallowing caused by internal scarring
Sclerodactyly—tightening of the skin on the fingers and toes
Telangiectasia—red spots on the hands, forearms, palms, face and lips.40
Raynaud’s phenomenon
Raynaud’s phenomenon (paroxysmal vasospasm of the digits) is the most common initial complaint in limited systemic sclerosis. Patients have diminished blood flow to the fingers and toes on exposure to cold (blanching or white phase), followed by cyanosis as haemoglobin releases oxygen to the tissues (blue phase) and then erythema during rewarming (red phase). The colour changes are often accompanied by numbness and tingling. Raynaud’s phenomenon may precede the onset of systemic disease by months, years or even decades.
Skin and joint changes
Symmetrical painless swelling or thickening of the skin of the fingers and hands may progress to diffuse scleroderma of the trunk. In limited disease, skin thickening generally does not extend above the elbow or above the knee, although the face can be affected in some individuals. In more diffuse disease, the skin loses elasticity and becomes taut and shiny, producing the typical expressionless facies with tightly pursed lips. Skin changes in the face may also contribute to reduced ROM in the temporomandibular joint. The hands may be affected by sclerodactyly in which the fingers are in a semi-flexed position, with tightened skin to the wrist (see Fig 64-12). Reduced peripheral joint function may occur as an early symptom of polyarthritis.
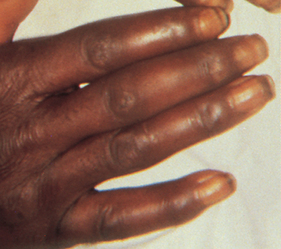
Internal organ involvement
About 20% of people with systemic sclerosis develop secondary Sjögren’s syndrome, a condition associated with dry eyes and dry mouth. Dysphagia, gum disease and dental caries can result. Frequent reflux of gastric acid also can occur as a result of oesophageal fibrosis. If swallowing becomes difficult, the patient is likely to decrease food intake and lose weight. Additional GI effects include constipation resulting from colonic hypomotility and diarrhoea caused by malabsorption from bacterial overgrowth.
Lung involvement includes pleural thickening, pulmonary fibrosis and pulmonary function abnormalities. The patient develops a cough and dyspnoea. Pulmonary artery hypertension may occur in up to 50% of patients with limited SS, and interstitial lung disease can occur additionally in another 10% of these patients. This complication has been treated successfully with medications such as extended-release nifedipine and bosentan.
Primary heart disease consists of pericarditis, pericardial effusion and cardiac arrhythmias. Myocardial fibrosis resulting in heart failure occurs most frequently in patients with diffuse SS.
Renal disease was previously a major cause of death in diffuse SS. Because malignant hypertension associated with rapidly progressive and irreversible renal insufficiency can occur, early recognition of renal involvement and initiation of therapy are critical. Recent improvements in dialysis, bilateral nephrectomy in patients with uncontrollable hypertension and kidney transplantation have offered some hope to patients with renal failure. In particular, use of angiotensin-converting enzyme (ACE) inhibitors (e.g. lisinopril) has had a marked impact on the ability to treat renal disease.
DIAGNOSTIC STUDIES
Laboratory findings are relatively normal. Blood studies may reveal a mild haemolytic anaemia as a result of RBC damage from diseased small vessels. The scleroderma antibody SCL-70 is found in about 30% of patients with diffuse disease, and serum RF is found in 30% of affected patients. An anticentromere antibody is seen in many patients with CREST. If renal involvement is present, urinalysis may show proteinuria, microscopic haematuria and casts. Serum levels of creatinine may be elevated. X-ray evidence of subcutaneous calcification, distal oesophageal hypomotility or bilateral pulmonary fibrosis is diagnostic of SS. Pulmonary function studies reveal decreased vital capacity and lung compliance.
MULTIDISCIPLINARY CARE
The multidisciplinary care of SS (see Box 64-12) offers no specific treatment with long-term effects. Supportive care is directed towards attempts to prevent or treat secondary complications of involved organs. Physiotherapy helps maintain joint mobility and preserve muscle strength. Occupational therapy assists the patient in maintaining functional abilities.
MULTIDISCIPLINARY CARE
Drug therapy
No specific drug or combination of drugs has been proven effective for the treatment of SS. Vasoactive agents are often prescribed in early disease, and calcium channel blockers (nifedipine and diltiazem) are now a common treatment choice for Raynaud’s phenomenon. Reserpine, an α-adrenergic blocking agent, increases blood flow to the fingers. Tracleer, an endothelin-receptor antagonist, and epoprostenol, a vasodilator, may assist in preventing and treating digital ulcers while improving exercise capacity and heart and lung dynamics. Losartan, an angiotensin II blocker, may be used to treat Raynaud’s phenomenon.
Corticosteroids may have little effect on SS. Topical agents may provide some relief from joint pain. Capsaicin cream may be useful, not only as a local analgesic, but also as a vasodilator. Other therapies prescribed to address specific systemic problems may include tetracycline for diarrhoea caused by bacterial overgrowth, histamine (H2)-receptor blockers (e.g. cimetidine), and proton pump inhibitors (e.g. omeprazole) for oesophageal symptoms, an antihypertensive agent (e.g. captopril, propranolol, methyldopa) for hypertension with renal involvement and immunosuppressive drugs (e.g. cyclophosphamide, mycophenolate mofetil).
NURSING MANAGEMENT: SYSTEMIC SCLEROSIS
Because prevention is not possible, nursing intervention often begins during hospitalisation for diagnostic purposes. The nurse should assist the patient to resolve feelings of helplessness by providing information about the illness and encouraging active participation in planning care. The nurse should also assess vital signs, weight, intake and output, respiratory and bowel function, and joint ROM at regular intervals as indicated by specific symptoms to plan appropriate care. Emotional stress and cold ambient temperatures may aggravate Raynaud’s phenomenon. Patients with SS should not have finger-stick blood testing done because of compromised circulation and poor healing of the fingers.
Health teaching is an important nursing intervention as the patient and family begin to live with this disease. Obvious changes in the face and hands often lead to poor self-image and the loss of mobility and function. The patient must actively complete therapeutic exercises at home to prevent skin retraction and promote vascularisation. Mouth excursion (yawning with an open mouth) is a good exercise to help with temporomandibular joint function. Isometric exercises are most appropriate if the patient has arthropathy because no joint movement occurs. The nurse should encourage the use of moist heat applications or paraffin baths to promote skin flexibility in the hands and feet, teach the patient to use assistive devices as appropriate and organise activities to preserve strength and reduce disability.
Hands and feet should be protected from cold exposure and possible burns or cuts that might heal slowly. Smoking should be avoided because of its vasoconstricting effect. Signs of infection should be reported promptly. Lotions may help alleviate skin dryness and cracking, but they must be rubbed in for an unusually long time because of the thickness of the skin.
Dysphagia may be reduced by eating small, frequent meals; chewing carefully and slowly; and drinking fluids. Heartburn may be minimised by using antacids 45–60 minutes after each meal and by sitting upright for at least 2 hours after eating. Using additional pillows or raising the head of the bed on blocks may help reduce nocturnal gastro-oesophageal reflux.
Job modifications are often necessary because stair climbing, typing, writing and cold exposure may pose particular problems. The patient may become socially withdrawn as skin tightening alters the appearance of the face and hands. Dining out may become a socially embarrassing event because the patient’s small mouth, difficulty swallowing and reflux make eating less enjoyable. Some individuals with SS wear gloves to protect fingertip ulcers and to provide extra warmth. Sensitive areas on fingertips resulting from ulcers or calcinosis may require padded utensils or special assistive devices to reduce discomfort. Daily oral hygiene must be emphasised because neglect may lead to increased tooth and gingival problems. The patient needs a dentist who is familiar with SS and can deal with a small oral aperture. Psychological support reduces stress and may positively influence peripheral motor response. Biofeedback training and relaxation techniques can reduce tension, improve sleeping habits and raise the temperature of the fingers and toes.
Sexual dysfunction resulting from body changes, pain, muscular weakness, limited mobility, decreased self-esteem, erectile dysfunction and decreased vaginal secretions may require sensitive counselling by a specialist nurse.
Polymyositis and dermatomyositis
Polymyositis (PM) and dermatomyositis (DM) are diffuse, idiopathic, inflammatory myopathies of striated muscle that produce bilateral weakness usually most severe in the proximal or limb girdle muscles. These disorders occur twice as often in women as in men but are still relatively rare.3 The diseases typically affect adults aged 45–64 years. The diseases can be similar in signs and symptoms and treatment, but they are two distinct disorders.41 Patients with PM generally have more severe disease than those with DM.
AETIOLOGY AND PATHOPHYSIOLOGY
The exact cause of PM and DM is unknown. Theories include an infectious agent, neoplasms, drugs or vaccinations and stress. Because disease severity is not well correlated with altered immune complexes, it is unclear whether the complexes occur as primary or secondary phenomena. Because T cytotoxic cells and macrophages have been found near the damaged muscle fibres of PM, this disease is believed to be caused by cell-mediated injury. In contrast, DM has been associated with B cells (humoral immunity) and destruction of the muscle microvasculature.
CLINICAL MANIFESTATIONS AND COMPLICATIONS
Muscular
Patients with DM and PM experience weight loss and increasing fatigue, with a gradually developing weakness of the muscles that leads to difficulty in performing routine activities. The most commonly affected muscles are those of the shoulders, legs, arms and pelvic girdle. The patient may have difficulty rising from a chair or bath, climbing stairs, combing their hair or reaching into a high cupboard. Neck muscles may become so weak that the patient is unable to raise their head from the pillow. Muscle discomfort or tenderness is uncommon. Muscle examination reveals an inability to move against resistance or even gravity. Weak pharyngeal muscles may produce dysphagia and dysphonia (nasal or hoarse voice).
Dermal
Skin changes of DM include a classic violet-coloured, cyanotic or erythematous symmetrical rash (heliotrope) with oedema around the eyelids. Violet-coloured or erythematous papules and small plaques can develop over the DIP or MCP areas, and at elbow or knee joints in about 70% of patients with DM (Gottron’s papules; see Fig 64-13). These early skin changes usually prompt earlier recognition of DM as compared to PM, in which a rash does not appear. Reddened, smooth or scaly patches appear with the same symmetrical distribution but sparing the interphalangeal spaces (Gottron’s sign). They can be confused with psoriasis or seborrhoeic dermatitis. An erythematous scaling rash (poikiloderma) may develop as a late finding on the back, buttocks and a V-shaped area of the anterior neck and chest. Hyperaemia and telangiectasias are often present at the nail beds. Calcium nodules (calcinosis cutis), which can develop throughout the skin, are especially common in long-standing DM.
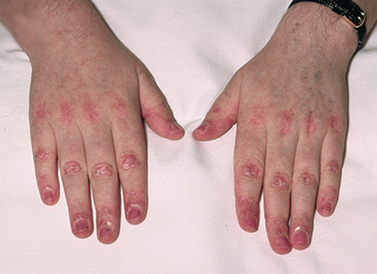
Other manifestations
Joint redness, pain and inflammation often occur and contribute to limitations in joint ROM in DM and PM. Contractures and muscle atrophy may occur with advanced disease. Weakened pharyngeal muscles can lead to a poor cough effort, difficulty swallowing and increased risk for aspiration pneumonia in both disorders. Interstitial lung disease occurs in up to 64% of all patients. People with DM also have an increased risk of an occult malignancy, which may be present at the time of diagnosis. Both diseases may be associated with other connective tissue disorders (e.g. systemic sclerosis).
DIAGNOSTIC STUDIES
A diagnosis of DM or PM is confirmed by MRI, electromyography (EMG) findings, muscle biopsy and serum muscle enzyme levels. An EMG suggestive of PM will show bizarre high-frequency discharges and spontaneous fibrillation, with positive spikes at rest. Muscle biopsy reveals necrosis, degeneration, regeneration and fibrosis with pathology findings distinct for DM or PM. Enzymes such as creatinine kinase and myoglobin are elevated. Elevation of the ESR is also expected with active disease. The typical skin rash seen with DM is not seen commonly with other disorders.
NURSING AND COLLABORATIVE MANAGEMENT: POLYMYOSITIS AND DERMATOMYOSITIS
PM and DM are treated initially with high-dose corticosteroids. Improvement generally is achieved if corticosteroid therapy is instituted promptly, and the dosage can be reduced as improvement is noted. Long-term corticosteroid therapy may be required because relapses are common when the drug is withdrawn. If corticosteroids prove ineffective and/or organ involvement is occurring, immunosuppressive drugs may also be administered (methotrexate, azathioprine, tacrolimus, cyclophosphamide) using oral or intermittent intravenous dosing. DM has been shown to improve with high doses of IV immunoglobulin. Topical corticosteroids and hydroxychloroquine may be prescribed to treat the skin rash. Ongoing clinical trials continue to explore the use of rituximab, a monoclonal antibody, in the treatment of PM and DM.
Physiotherapy can be helpful and should be tailored to the activity of the disease. Massage and passive movement are appropriate during active disease. More aggressive exercises should be reserved for periods when disease activity is minimal, as evidenced by low serum enzyme levels.
The nurse should advise the patient on the nature of the disease, the prescribed therapies, all diagnostic tests and the importance of regular medical care. It is important for the patient to understand that the benefits of therapy are often delayed. For example, weakness may increase during the first few weeks of corticosteroid therapy. Special attention is paid to patient safety and the nurse should encourage the use of assistive devices as a fall prevention strategy. To prevent aspiration, the patient should rest before meals, maintain an upright position when eating and choose a diet of easily swallowed foods.
The nurse should assist the patient to organise activities and use pacing techniques to conserve energy. Daily ROM exercises are encouraged to prevent contractures. When active inflammation is not evident, muscle-strengthening (repetitive) exercises may be started. Home care and bed rest may become necessary during the acute phase of PM because profound muscle weakness renders the patient unable to complete activities of daily living.
Mixed (overlapping) connective tissue disease
Patients having a combination of clinical features of several rheumatic diseases are described as having mixed or overlapping connective tissue disease. Although this combination was originally believed to be a distinct clinical disorder, follow-up revealed that for most patients this disorder is a stage in the progression to SLE, SS or polymyositis.34
Sjögren’s syndrome
Sjögren’s syndrome is a relatively common autoimmune disease that targets moisture-producing glands, leading to xerostomia (dry mouth) and keratoconjunctivitis sicca (dry eyes).42 The nose, throat, airways and skin can also become dry. The disease may affect other glands as well, including those in the stomach, pancreas and intestines (extraglandular involvement). The disease is usually diagnosed in people over age 40, with 90% of them being women.
In primary Sjögren’s syndrome, symptoms can be traced to problems with the lacrimal and salivary glands. The patient with primary disease is likely to have antibodies against the cytoplasmic antigens SS-A (or Ro) and SS-B (or La), as well as ANA. The patient with secondary Sjögren’s syndrome typically has had another autoimmune disease (e.g. RA, SLE) before Sjögren’s develops.
Sjögren’s syndrome appears to be caused by genetic and environmental factors. Several genes seem to be involved.42 One gene predisposes Caucasians to the disease, whereas other genes are linked to the disease in people of Japanese, Chinese and African heritage. It is estimated that 0.5% of the population in Australasia have Sjögren’s syndrome. The trigger may be a viral or bacterial infection that adversely stimulates the immune system. In Sjögren’s syndrome, lymphocytes attack and damage the lacrimal and salivary glands.
Dry eyes cause decreased tearing, which leads to a ‘gritty’ sensation in the eyes, burning, blurred vision and photosensitivity. Dry mouth produces buccal membrane fissures, altered sense of taste, dysphagia and increased frequency of mouth infections or dental caries. Dry skin and rashes, joint and muscle pain and thyroid problems may also be present. Other exocrine glands can be affected. For example, vaginal dryness may lead to dyspareunia (painful intercourse). Autoimmune thyroid disorders are common with Sjögren’s syndrome, including Graves’ disease and Hashimoto’s thyroiditis. Histological study reveals lymphocyte infiltration of salivary and lacrimal glands. The disease may become more generalised and involve the lymph nodes, bone marrow and visceral organs (pseudolymphoma). The risk of developing lymphoma is high in Sjögren’s syndrome.43
Ophthalmological examination (Schirmer’s test for tear production), measures of salivary gland function and lower lip biopsy of minor salivary glands aid in diagnosis. The treatment is symptomatic, including: (1) instillation of preservative-free artificial tears or ophthalmic anti-inflammatory drops (e.g. cyclosporin) as necessary to maintain adequate hydration and lubrication; (2) surgical punctal occlusion; and (3) increased fluids with meals. Dental hygiene is important. Pilocarpine can be used to treat symptoms of dry mouth; the use of a toothpaste designed for sensitive teeth and gums may also help. Increased humidity at home may reduce respiratory infections. Vaginal lubrication with a water-soluble product such as K-Y jelly may increase comfort during intercourse.
SOFT-TISSUE RHEUMATIC SYNDROMES
Myofascial pain syndrome, fibromyalgia syndrome and chronic fatigue syndrome are three soft-tissue disease syndromes that have many commonalities and may be related. Ongoing research continues to explore links among these three syndromes. A multidisciplinary team approach consisting of a rheumatologist, a nurse, a mental health professional and a physiotherapist may be helpful for patients with these syndromes.
Myofascial pain syndrome
Myofascial pain syndrome is a chronic form of muscle pain. It is characterised as musculoskeletal pain and tenderness, typically in the chest, neck, shoulders, hips and lower back. Referred pain from these muscle groups can also travel to the buttocks, hands and head, causing severe headaches. The pain has been shown to originate in anterior and posterior trigger points resulting from muscle trauma or chronically strained muscles (e.g. desk or computer work). Regions of pain are often within the taut bands and fascia of skeletal muscles. When activated by pressure, trigger points are thought to activate a characteristic pattern of pain that can be worse with activity or stress.44 Myofascial pain syndrome occurs more often in middle-aged adults, and in women rather than men. Patients complain of the pain as deep and aching and accompanied by a sensation of burning, stinging and stiffness.
Diagnosis of myofascial pain syndrome is done by palpation of trigger points, which reveals induration and frequently a muscle twitch in the area of a trigger point. Once a trigger point is palpated, pain will be felt locally and may also be referred to a region often at some distance away. These findings have also been noted in healthy people and in those with fibromyalgia syndrome. Similarities in myofascial pain syndrome and FMS have led to the suggestion that myofascial pain may be a form of, or evolve into, fibromyalgia syndrome. A comparison of the two syndromes is shown in Table 64-6.
TABLE 64-6 Comparison of fibromyalgia and myofascial pain
| Parameter | Fibromyalgia | Myofascial pain |
|---|---|---|
| Location | Generalised | Regional |
| Examination | Tender points | Trigger points |
| Response to local therapy | Not sustained | Curative |
| Gender | Female/male ratio: 10:1 | Equal or unknown |
Source: McCance KL, Huether se. Pathophysiology: the biologic basis for disease in adults and children. 6th edn. St Louis: Mosby; 2010.
Physiotherapy is one treatment used for myofascial pain syndrome. A typical treatment is the ‘spray and stretch’ method, in which the painful area is iced or sprayed with a coolant such as ethyl chloride and then stretched. Positive results have been seen with topical patches and injection of the trigger points with a local anaesthetic (e.g. 1% lignocaine).45 Massage, acupuncture, biofeedback and ultrasound therapy have also been shown to benefit some patients.
Patient and family teaching is an important nursing responsibility. The nurse should teach patients to prevent muscle tension in work and leisure activities, and review good posture, resting and sleeping positions. Most patients with myofascial pain syndrome are able to lead a normal and active life.
Fibromyalgia syndrome
Fibromyalgia syndrome (FMS) is a chronic disorder characterised by widespread, non-articular musculoskeletal pain and fatigue with multiple tender points. People with FMS may also experience non-restorative sleep, morning stiffness, irritable bowel syndrome and anxiety. The former name for this disorder, fibrositis, implied inflammation of the muscles and soft tissues. However, FMS is now known to be non-degenerative, non-progressive and non-inflammatory.46
Fibromyalgia is a commonly diagnosed musculoskeletal disorder and a major cause of disability. FMS affects 2–5% of the population. FMS occurs 6 times more frequently in women than in men.46 FMS and chronic fatigue syndrome share many commonalities (see Table 64-7).
TABLE 64-7 Commonalities between fibromyalgia syndrome and chronic fatigue syndrome
| Commonality | Description |
|---|---|
| Occurrence | Previously healthy, young and middle-aged women |
| Aetiology (theories) | Infectious trigger, dysfunction in HPA axis, alteration in CNS |
| Clinical manifestations | Malaise and fatigue, cognitive dysfunction, headaches, sleep disturbances, depression, anxiety, fever, generalised musculoskeletal pain |
| Course of disease | Variable intensity of symptoms, fluctuates over time |
| Diagnosis | No definitive laboratory tests or joint and muscle examinations, mainly a diagnosis of exclusion |
| Multidisciplinary care | Treatment is symptomatic and may include antidepressant drugs such as amitriptyline and fluoxetine; other measures are heat, massage, regular stretching, biofeedback, stress management and relaxation training; patient and family teaching is essential |
CNS, central nervous system; HPA, hypothalamic-pituitary-adrenal.
AETIOLOGY AND PATHOPHYSIOLOGY
Research continues to focus on identifying the underlying causes and pathophysiological mechanisms of FMS. There is general agreement that FMS is a disorder involving dysfunction in the regulation of the neuroendocrine/neurotransmitter systems.46 The pain amplification experienced by the affected patient is due to abnormal sensory processing in the CNS. Multiple physiological abnormalities include increased levels of substance P in the spinal fluid, low levels of blood flow to the thalamus, dysfunction of the hypothalamic–pituitary–adrenal (HPA) axis, low levels of serotonin and tryptophan, and abnormalities in cytokine function. Serotonin and substance P play a role in mood regulation, sleep and pain perception. Changes in the HPA axis can also negatively affect a person’s physical and mental health, leading to an increased incidence of depression and a decreased response to stress. Genetics also appear to contribute to the familial tendency to FMS.46 A recent viral illness may serve as an infectious trigger in susceptible people.
CLINICAL MANIFESTATIONS AND COMPLICATIONS
The patient complains of a widespread burning pain that worsens and improves throughout the course of a day. It is often difficult for the patient to discriminate whether pain occurs in the muscles, joints or soft tissues. Head or facial pain often results from stiff or painful neck and shoulder muscles. The pain can accompany temporomandibular joint dysfunction, which affects an estimated one-third of FMS patients. Non-restorative sleep and resulting fatigue are typical.
Physical examination characteristically reveals point tenderness at 11 or more of 18 identified sites (see Fig 64-14). Patients with FMS are sensitive to painful stimuli throughout the body and not merely at the identified tender sites. In addition, point tenderness can vary from day to day. On some occasions, the patient may respond to fewer than 11 tender points; at other times, palpation of all sites may elicit pain.
Cognitive effects range from difficulty concentrating to memory lapses and a feeling of being overwhelmed when dealing with multiple tasks. Many individuals report migraine headaches. Depression and anxiety often occur and may require drug therapy. Numbness or tingling in the hands or feet (paraesthesia) often accompanies FMS. Restless legs syndrome is also typical, with the patient describing an irresistible urge to move the legs when at rest or lying down.
Irritable bowel syndrome with manifestations of constipation and/or diarrhoea, abdominal pain and bloating is common. FMS patients may also experience difficulty swallowing, perhaps because of abnormalities in oesophageal smooth muscle function. Increased frequency of urination and urinary urgency, in the absence of a bladder infection, are typical complaints. Women with FMS may experience more difficult menstruation, with a worsening of disease symptoms during this time.
DIAGNOSTIC STUDIES
A definitive diagnosis of FMS is often difficult to establish. Lack of knowledge among health practitioners may also cause delays in diagnosis and treatment. Laboratory results in most cases serve to rule out other suspected disorders based on the patient’s history and physical examination. Occasionally a low ANA titre is seen, but it is not considered diagnostic. Muscle biopsy may reveal a non-specific moth-eaten appearance or fibre atrophy. Arthritis New Zealand uses the American College of Rheumatology classification to assist in the diagnosis of FMS. An individual is classified as having FMS if two criteria are met: (1) pain is experienced in 11 or more of the 18 tender points on palpation (see Fig 64-14); and (2) a history of widespread pain is noted for at least 3 months.47 Widespread pain is defined as occurring on both sides of the body and above and below the waist.
MULTIDISCIPLINARY CARE
The treatment of FMS is symptomatic and requires a high level of patient motivation. The nurse can play a key role in teaching the patient to be an active participant in the therapeutic regimen. Rest can help the pain, aching and tenderness. Drug treatment includes low doses of tricyclic compounds such as cyclobenzaprine and amitriptyline. Dual reuptake inhibitors (venlafaxine, milnacipran, duloxetine) and tramadol work similarly and may be effective for some patients. If amitriptyline is not well tolerated, other similar drugs can be substituted (e.g. doxepin, imipramine, trazodone). Selective serotonin reuptake inhibitor (SSRI) antidepressants (e.g. sertraline or paroxetine) tend to be reserved for patients who also have depression. SSRIs may need to be prescribed at higher doses than those used to treat depression. Both antidepressants and muscle relaxants have sedative effects that can help in improving night-time rest.
Long-acting opioids generally are not recommended unless FMS is refractory to other therapies. In some patients, pain may be managed with over-the-counter analgesics such as paracetamol, ibuprofen or naproxen. Benzodiazepines (e.g. diazepam, alprazolam, clonazepam) are often prescribed with low doses of ibuprofen to treat anxiety, as well as the muscle spasms that affect many FMS patients. The drug zolpidem is sometimes prescribed for short-term intervention in patients with severe sleep disturbances.
The antiepileptic drug gabapentin is among the newest treatments for FMS. These newer drugs may help to reduce pain and fatigue and improve sleep and daily functioning.
NURSING MANAGEMENT: FIBROMYALGIA SYNDROME
Because of the chronic nature of FMS and the necessity of maintaining an ongoing rehabilitation program, the patient with FMS needs consistent support from the whole healthcare team. Massage is often combined with ultrasound or the application of alternating heat and cold packs to soothe tense, sore muscles and increase blood circulation. Gentle stretching to relieve muscle tension and spasm can be performed by a physiotherapist or practised by the patient at home. Yoga and Tai Chi are often appropriate choices. Low-impact aerobic exercise such as walking can help prevent muscle atrophy.
Dieticians often urge FMS patients to limit their consumption of sugar, caffeine and alcohol because these substances have been shown to be muscle irritants. Vitamin and mineral supplements may be appropriate to combat stress, correct deficiencies and support the immune system. However, unproven ‘miracle diets’ or supplements should be investigated carefully by the patient and discussed with their healthcare provider before being used. The patient should understand that some foods and supplements may cause serious or even dangerous side effects when mixed with certain drugs.
Pain and the related symptoms of FMS can cause significant stress. There is some indication that patients with FMS simply do not deal with stress very effectively. Effective relaxation strategies include biofeedback, guided imagery and autogenic training. Patients need to receive initial training for these interventions, but they can then continue to practise them in their own homes. Psychological counselling (individual or group) may prove beneficial for some patients.
Chronic fatigue syndrome
Chronic fatigue syndrome (CFS), also called chronic fatigue and immune dysfunction syndrome, is a disorder characterised by debilitating fatigue and a variety of associated complaints. Immune abnormalities are also frequently present. The prevalence of CFS is difficult to establish because of the lack of validated diagnostic tests, but an estimated 0.2–0.7% of people in Australia and New Zealand have a CFS-like condition.48 Women are affected more often than men. CFS occurs in all ethnic and socioeconomic groups. It is a poorly understood condition that can have a devastating impact on the lives of patients and their families. CFS and FMS share some common features (see Table 64-7).
AETIOLOGY AND PATHOPHYSIOLOGY
Despite numerous attempts to determine the aetiology and pathology of CFS, the precise mechanisms remain unknown. However, there are many theories about the cause of CFS. Neuroendocrine abnormalities have been implicated involving a hypofunction of the HPA axis and the hypothalamic–pituitary–gonadal (HPG) axis, which together regulate the stress response and reproductive hormone levels.48 Several microorganisms have been investigated as aetiological agents, including herpes viruses (e.g. Epstein-Barr virus [EBV], cytomegalovirus [CMV]), retroviruses, enteroviruses, Candida albicans and mycoplasma. Because cognitive deficits (e.g. decreased memory attention, concentration) occur in many of these patients, it has also been proposed that CFS is due to changes in the CNS.
CLINICAL MANIFESTATIONS
It is often difficult to distinguish between CFS and FMS because many clinical features are similar (see Table 64-7). In about half of all cases, CFS develops insidiously, or the patient may have intermittent episodes that gradually become chronic. Incapacitating fatigue is the most common symptom of CFS and is usually the problem that causes the patient to seek healthcare. In other situations, CFS arises suddenly in a previously active, healthy individual. An unremarkable flu-like illness or other acute stress is often identified as a triggering event. Associated symptoms (see Box 64-13) may fluctuate in intensity over time.
BOX 64-13 Diagnostic criteria for chronic fatigue syndrome*
Source: Adapted from Centers for Disease Control and Prevention. Chronic fatigue syndrome: revised case definition. Available at www.cdc.gov/cfs/cfsdefinitionHCP.htm.
*For a diagnosis to be made, the patient must demonstrate the major criterion, plus four or more of the minor criteria for 6 months or more. These criteria were prepared by the Centers for Disease Control and Prevention, National Institutes of Health and International Chronic Fatigue Syndrome Study Group.
The patient may become angry and frustrated with the inability of healthcare providers to diagnose a problem. The disorder may have a major impact on work and family responsibilities. Some individuals may even need help with activities of daily living.
DIAGNOSTIC STUDIES
Physical examination and diagnostic studies can be used to rule out other possible causes of the patient’s symptoms. No laboratory test can diagnose CFS or measure its severity. The US Centers for Disease Control and Prevention has developed a diagnostic algorithm based on the patient’s symptoms (see Box 64-13) that can be useful as, in general, CFS remains a diagnosis of exclusion.
NURSING AND COLLABORATIVE MANAGEMENT: CHRONIC FATIGUE SYNDROME
No definitive treatment exists for CFS, so supportive management is essential. The nurse should explain to the patient what is known about the disease, and all complaints should be taken seriously. NSAIDs can be used to treat headaches, muscle and joint aches, and fever. Because many patients with CFS also have allergies and sinusitis, antihistamines and decongestants can be used to treat allergic symptoms. Tricyclic antidepressants (e.g. doxepin, amitriptyline) and SSRIs (e.g. fluoxetine, paroxetine) can improve mood and sleep problems. Clonazepam may be used to treat sleep disturbances and panic disorders. The use of low-dose hydrocortisone is being studied to decrease fatigue and disability.
Total rest is not advised because it can potentiate the self-image of being an invalid, while strenuous exertion can exacerbate the exhaustion. Therefore, it is important to plan a carefully graduated exercise program. A well-balanced diet including fibre and fresh dark-coloured fruits and vegetables for antioxidant action is essential in treatment. Behavioural therapy may be used to promote a positive outlook, as well as improve overall disability, fatigue and other symptoms.49
One of the major problems facing many patients with CFS is financial instability. When the illness strikes, they cannot work or must decrease the amount of time spent working. Obtaining disability benefits can be frustrating because of the difficulty of establishing a diagnosis of CFS.
CFS does not appear to progress. Although most patients recover or at least gradually improve over time, some do not show substantial improvement. Recovery is more common in individuals with a sudden onset of CFS. Patients with CFS may experience substantial occupational and psychosocial impairments and loss, including social pressure and isolation from being characterised as ‘lazy’ or ‘crazy’.
The patient with rheumatoid arthritis
CASE STUDY
Patient profile
Su Lee, a 36-year-old married woman, is seen at the rheumatology clinic with complaints of swelling and stiffness in the small joints of her hands.
CRITICAL THINKING QUESTIONS
1. How might you explain the pathophysiology of rheumatoid arthritis to this patient?
2. How might the recent birth of her first child have influenced the symptoms that she is currently experiencing?
3. What are some home and work modifications that you can suggest to this patient that will reduce her symptoms?
4. What suggestions can you make to this patient about coping with fatigue?
5. Based on the assessment data presented, write one or more nursing diagnoses. Are there any collaborative problems?
1. In assessing the joints of a patient with rheumatoid arthritis, the nurse understands that the joints are damaged by:
2. Assessment data noted by the nurse in the patient with osteoarthritis commonly include:
3. An important nursing intervention in caring for the patient with ankylosing spondylitis is to teach the patient:
4. When teaching the patient with gout, the nurse should instruct the patient to:
5. In teaching a patient with systemic lupus erythematosus (SLE) about the disorder, the nurse uses the knowledge that the pathophysiology of SLE includes:
6. The nurse planning teaching for the patient with rheumatoid arthritis who is prescribed multiple drug therapy includes information related to the need to:
7. In teaching a patient with chronic fatigue syndrome (CFS) about this disorder, the nurse understands that:
1 Australian Institute of Health & Welfare (AIHW). A snapshot of arthritis in Australia, 2010. Canberra: AIHW; 2010. Available at www.aihw.gov.au/publications/index.cfm/title/11154 accessed 26 December 2010.
2 New Zealand Ministry of Health (NZMOH). New Zealand health survey. Available at www.moh.govt.nz/moh.nsf/pagesmh/7601/$File/arthritis-ch3.pdf, 2006/2007. accessed 26 December 2010.
3 Roberts D. Arthritis and connective tissue disorders, National Association of Orthopaedic Nurses. NAON core curriculum for orthopaedic nursing. 6th edn. Boston: Pearson Custom Publishing; 2007.
4 Centers for Disease Control and Prevention. Arthritis basics. Available at www.cdc.gov/arthritis/arthritis/risk_factors.htm. accessed 12 December 2010.
5 Zhang Y, Jordan J. Epidemiology of arthritis. Rheum Dis Clin North Am. 2008;34:515.
6 Øistad B, Engebretsen L, Storheim K, et al. Knee osteoarthritis after anterior cruciate ligament injury: a systematic review. Am J Sports Med. 2009;37:7.
7 The Johns Hopkins white papers: arthritis. Baltimore: Johns Hopkins Medicine, 2009.
8 Mounsey A, Ewigman B. Arthroscopic surgery for knee arthritis? Just say no. J Fam Practice. 2009;58:143.
9 Guideline for the non-surgical management of hip and knee osteoarthritis. Melbourne: Royal Australian College of General Practitioners; 2009. Available at www.nhmrc.gov.au/_files_nhmrc/file/publications/synopses/cp117-hip-knee-osteoarthritis.pdf accessed 26 December 2010.
10 Zhang W, Moskowitz RW, Nuki G, et al. OARSI recommendations for the management of hip and knee osteoarthritis. Part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis and Cartilage. 2008;16:137.
11 The Australian COX-2-Specific Inhibitor (CSI) Prescribing Group. Considerations for the safe prescribing and use of COX-2-specific inhibitors, Med J Aus. 2002;176(7):328–331. Available at www.mja.com.au/public/issues/176_07_010402/bar10002_fm.html accessed 8 January 2011.
12 Nuesch E, Rutjes A, Trelle S, et al. Doxycycline for osteoarthritis of the knee or hip. Cochrane Database Syst Rev. (3):2008. CD007323.
13 Ling SM. Rehabilitation of older adult patients. Available at www.hopkins-arthritis.org/patient-corner/disease-management/rehab. accessed 22 December 2010.
14 Australian Institute of Health & Welfare (AIHW). Arthritis and musculoskeletal conditions. Canberra: AIHW; 2009. Available at www.aihw.gov.au/nhpa/arthritis/index.cfm accessed 27 December 2010.
15 Lundström E, Källberg H, Alfredson L, et al. Gene–environment interaction between the DRB1 shared epitope and smoking in the risk of anti-citrullinated protein antibody-positive rheumatoid arthritis: all alleles are important. Arthritis Rheum. 2009;60:6.
16 Donahue KE, Gartlehner G, Jonas DE, et al. Systematic review: comparative effectiveness and harms of disease-modifying medications for rheumatoid arthritis. Ann Intern Med. 2008;148:2.
17 Cleveland Clinic. New RA drugs show promise, Arthritis Advisor. 2009;8:4. Available at www.arthritis-advisor.com/pub/8_4/features/541-1.html accessed 21 June 2010.
18 Dougados M, Van Der Linden S, Juhlin R, et al. The European Spondyloarthropathy Study Group preliminary criteria for the classification of spondyloarthropathy. Arthritis Rheum. 1991;34:10. (Seminal text)
19 Kain T, Zochling J, Taylor A, et al. Evidence-based recommendations for the diagnosis of ankylosing spondylitis: results from the Australian 3E initiative in rheumatology, Med J Aus. 2008;188(4):235–237. Available at www.mja.com.au/public/issues/188_04_180208/kai10788_fm.html accessed 8 January 2011.
20 Widberg K, Karimi H, Hafström I. Self-and manual mobilization improves spine mobility in men with ankylosing spondylitis—a randomized study. Clin Rehabil. 2009;23:599.
21 National Psoriasis Foundation. The five types of psoriatic arthritis. Available at www.psoriasis.org/netcommunity/learn_psatypes. accessed 23 June 2010.
22 Carter J, Hudson A. Reactive arthritis: clinical aspects and medical management. Rheum Dis Clin North Am. 2009;35:21.
23 Mathews CJ, Coakley G. Septic arthritis: current diagnostic and therapeutic algorithm. Curr Opin Rheumatol. 2008;20:457.
24 Chen C, Wang J, Juhn R. Total hip arthroplasty for primary septic arthritis of the hip in adults. Int Orthop. 2008;32:573.
25 New South Wales Department of Health. Infectious disease factsheet: Lyme disease. Available at www.health.nsw.gov.au/factsheets/infectious/lyme_disease.html, Updated 23 July 2010. accessed 27 December 2010.
26 Centers for Disease Control and Prevention, Division of Vector-Borne Infectious Diseases. Lyme disease. Available at www.cdc.gov/ncidod/dvbid/lyme/ld_rptdLymeCasesbyState.htm. accessed 27 December 2010.
27 Lyme Info. Lyme disease vaccine. Available at www.lymeinfo.net/vaccine.html. accessed 27 December 2010.
28 Hoch BL, Klein MJ, Schiller AL. Bones and joints. In Rubin R, Strayer DS, eds.: Rubin’s pathology: clinicopathologic foundations of medicine, 5th edn., Philadelphia: Lippincott Williams & Wilkins, 2008.
29 Malik A, Schumacher H, Dinnella J, et al. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol. 2009;15:22.
30 Cada D, Levien T, Baker D. Formulary drug reviews: febuxostat. Hospital Pharmacy. 2009;44:8.
31 Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15(5):308–318.
32 Dedeoglu F. Drug-induced autoimmunity. Curr Opin Rheumatol. 2009;21:54.
33 Pullen R, Brewer S, Ballard A. Putting a face on systemic lupus erythematosus. Nursing. 2009;39:22.
34 Hellman D, Imboden J. Arthritis and musculoskeletal disorders. In McPhee S, Papadakis MA, Tierney LM, eds.: Current medical diagnosis and treatment, 47th edn., New York: McGraw-Hill, 2008.
35 Seshan S, Jennette J. Renal disease in systemic lupus erythematosus with emphasis on classification of lupus glomerulonephritis: advances and implications. Arch Pathol Lab Med. 2009;133:233.
36 Lupus Foundation of America. Medications to treat lupus symptoms. Available at www.lupus.org/webmodules/webarticlesnet/templates/new_learntreating.aspx?articleid=2246&zoneid=525. accessed 27 December 2010.
37 Danoff-Berg S, Friedberg F. Unmet needs of patients with systemic lupus erythematosus. Behav Med. 2009;35:5.
38 Vincent R. Scleroderma. Practice Nurse. 2009;38:47.
39 Khavari R, Khanna D, Clements P. Decision making in systemic sclerosis: a guide for primary care. J Musculoskeletal Med. 2009;26:247.
40 Yoon JC, Raugi GJ. CREST syndrome. eMedicine on WebMD. Available at http://emedicine.medscape.com/article/1064663-overview, Updated November 2009. accessed 8 January 2011.
41 Rellosa G, Rubin R. What’s the ‘take home’? Profound weakness in a young woman … dermatomyositis. Consultant. 2009;49:519.
42 Gálvez J, Sáiz E, López P, et al. Diagnostic evaluation and classification criteria in Sjögren’s syndrome. Joint Bone Spine. 2009;76:44.
43 Baimpa E, Dahabreh I, Voulgarelis M, et al. Hematologic manifestations and predictors of lymphoma development in primary Sjögren syndrome: clinical and pathophysiologic aspects. Medicine. 2009;88:284.
44 The Cleveland Clinic. Myofascial pain syndrome. Available at http://my.clevelandclinic.org/disorders/chronic_pain/hic_myofascial_pain_syndrome.aspx. accessed 27 December 2010.
45 Affaitati G, Fabrizio A, Savini A, et al. A randomized, controlled study comparing a lidocaine patch, a placebo patch, and anesthetic injection for treatment of trigger points in patients with myofascial pain syndrome: evaluation of pain and somatic pain thresholds. Clin Ther. 2009;31:705.
46 Russell I, Larson A. Neurophysiopathogenesis of fibromyalgia syndrome: a unified hypothesis. Rheum Dis Clin North Am. 2009;35:421.
47 Arthritis New Zealand. Fibromyalgia. Available at www.arthritis.org.nz/index.php/Fibromyalgia.html#Diagnosing_fibromyalgia. accessed January 2011.
48 Royal Australasian College of Physicians. Clinical practice guidelines—chronic fatigue syndrome, Med J Aust. 2002;176(8 suppl):S17–S55. Available at www.mja.com.au/public/guides/cfs/cfs2.html accessed 27 December 2010.
49 Santhouse AM. Review: CBT reduces fatigue in adults with chronic fatigue syndrome but effects at follow-up unclear. Evidence-Based Mental Health. 2009;12:16.
Arthritis Foundation of Australia. www.arthritisaustralia.com.au
Arthritis Foundation of Western Australia. www.arthritiswa.org.au
Arthritis New Zealand. www.arthritis.org.nz
Arthritis Queensland: self-management packages. www.arthritis.org.au/page/Our_Services/Self-Management_Courses/
Arthritis Victoria. www.arthritisvic.org.au/
Associated New Zealand Myalgic Encephalomyelitis Societies. www.anzmes.org
Australian Rheumatology Association. www.rheumatology.org.au
Garvan Institute (for research into the field). www.garvan.org.au/
Lupus Association of New South Wales. www.lupusnsw.org.au
Lupus Association of Tasmania. www.lupustas.bigpondhosting.com/home/Welcome.html
Lupus Australia, Queensland Inc. www.lupus.com.au
Lupus Trust of New Zealand. www.lupus.org.nz
Myalgic Encephalomyelitis: Chronic Fatigue Syndrome Society of New South Wales. www.me-cfs.org.au
New Zealand Rheumatology Association. www.rheumatology.org.nz
Psoriasis Association (Southland). www.psoriasis.org.nz
Scleroderma Foundation of Victoria. www.sclerodermavictoria.com.au
