Introduction
Terminology
Afferent Neuron that carries information to the central nervous system.
Allodynia Pain with a nonnoxious stimulus (e.g., touch); a common characteristic of some neuropathic pain.
Central disinhibition A central mechanism of neuropathic pain that occurs when control mechanisms along inhibitory (modulatory) pathways are lost or suppressed, leading to abnormal excitability of central neurons.
Central sensitization A key central mechanism of neuropathic pain; the abnormal hyperexcitability of central neurons in the spinal cord, which results from complex changes induced by the incoming afferent barrages of nociceptors.
Cross excitation See ephaptic conduction.
Cross talk See ephaptic conduction.
Efferent A neuron that carries information away from the central nervous system.
Ephaptic conduction Also called cross-excitation or cross-talk; a peripheral mechanism that may sustain neuropathic pain through the creation of chemically mediated connections between nerve fibers, causing the abnormal activation of nociceptive neurons, ultimately producing pain.
GABAergic Increased function of the gamma aminobutyric acid (GABA) inhibitory pathways; increased GABA (GABAergic) function may help to relieve neuropathic pain.
Hyperalgesia Increased sensation of pain in response to a normally painful stimulus.
Neuroplasticity The ability of the peripheral and central nervous systems to change both structure and function as a result of noxious stimuli.
Nociceptor Primary afferent neurons that exist throughout the body and have the intrinsic ability to respond selectively to specific noxious stimuli.
Peripheral sensitization A key peripheral mechanism of neuropathic pain that occurs when there are changes in the number and location of ion channels, in particular sodium channels, which abnormally accumulate in injured nociceptors, producing a lower nerve depolarization threshold, ectopic discharges, and an increase in the response to stimuli.
Sympathetically maintained pain Pain that is identified through a very positive response to sympathetic nerve blocks; likely to be part of the syndrome known as complex regional pain syndrome (CRPS); underlying mechanism is unclear but thought to be related to ephaptic conduction.
Wind-up The progressive increase in response of central neurons that may be induced by high-intensity activity in the peripheral nociceptors that synapse on these neurons.
There are many ways to classify pain, and clear distinctions are not always possible. Simple classifications invariably result in some omissions and overlap. General discussions of pain refer simply to three types: (1) acute pain (e.g., postoperative or trauma pain), (2) cancer pain, and (3) noncancer pain (e.g., osteoarthritis pain, postherpetic neuralgia, painful diabetic neuropathy). Pain can also be classified by its inferred pathophysiology: (1) nociceptive (physiologic) pain (normal neural processing of noxious stimuli), or (2) neuropathic pain (stimuli abnormally processed by the nervous system (Figure I-1). The types of neuropathic pain are often divided into categories on the basis of the mechanism thought to be primarily responsible for causing the pain (i.e., peripheral nervous system [PNS] or central nervous system [CNS] activity). Note that in some cases the original injury occurs in the peripheral nerves (e.g., amputation), but the mechanisms that underlie the pain (e.g., phantom pain) seem to be generated primarily in the CNS. Some patients have both nociceptive and neuropathic pain, for example, nociceptive pain resulting from tumor growth and metastasis and neuropathic pain resulting from tumor compression of neural structures (see Section II for more on classifications and assessment of types of pain).

Figure I-1 A method of classifying pain is by inferred pathophysiology: I, Nociceptive pain, stimuli from somatic and visceral structures; II, Neuropathic pain, stimuli abnormally processed by the nervous system. From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 2, St. Louis, Mosby. Data from Max, M. B., & Portenoy, R. K. (2003). Methodological challenges for clinical trials of cancer pain treatments. In C. R. Chapman, & K. M. Foley (Eds), Current and emerging issues in cancer pain: Research and practice, New York, Raven Press; Portenoy, R. K. (1996). Neuropathic pain. In R. K. Portenoy, & R. M. Kanner (Eds), Pain management: Theory and practice, Philadelphia, FA Davis. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Appropriate pharmacologic management of nociceptive and neuropathic pain requires an understanding of their complex underlying mechanisms. This section presents an overview of nociception, which is followed by a discussion of the pathophysiology of neuropathic pain. The action sites of various analgesics are presented throughout the section.
Nociception
Nociception refers to the normal functioning of physiologic systems that leads to the perception of noxious stimuli as being painful. In short, it means “normal” pain transmission and includes four specific processes: transduction, transmission, perception, and modulation (Figure I-2).

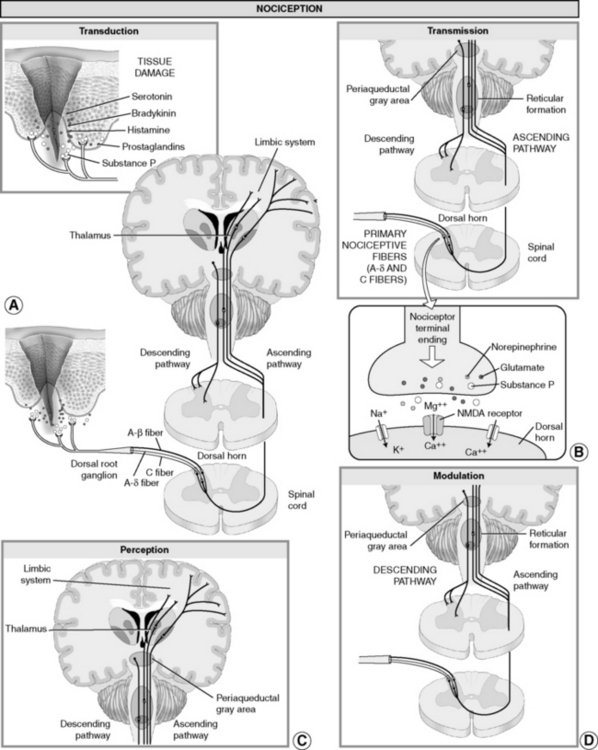
Figure I-2 Nociception: “normal” pain transmission. From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, pp. 4-5, St. Louis, Mosby. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Transduction
Transduction refers to the processes by which noxious events activate nociceptors, the primary afferent neurons that exist throughout the body (skin, subcutaneous tissue, and visceral or somatic structures) and have the intrinsic ability to respond selectively to specific noxious stimuli. Nociceptors have free nerve endings with specific channels that can respond to different kinds of stimuli. Different subtypes are activated when tissue damage or potential damage results from mechanical stimuli (e.g., incision, tumor growth); thermal stimuli (e.g., burn, frostbite); or chemical stimuli (e.g., toxins, chemotherapy) (Argoff, Albrecht, Irving, et al., 2009; Marchand, 2008). These stimuli activate nociceptors directly and also cause the release of a number of excitatory compounds (e.g., serotonin, bradykinin, histamine, substance P, and prostaglandins) that facilitate the further activation of nociceptors. These compounds, which may originate from local tissues, immune cells, or nerve endings themselves, may be collectively labeled an inflammatory soup (Marchand, 2008; see Figure I-2, A).
The prostaglandins are a particularly important group of compounds in the inflammatory soup that accompanies tissue injury. Prostaglandins are formed when the enzyme phospholipase breaks down phospholipids into arachidonic acid. The arachidonic acid, in turn, is acted upon by the enzyme cyclooxygenase (COX) to produce a set of compounds known as the prostaglandins (Figure I-3).
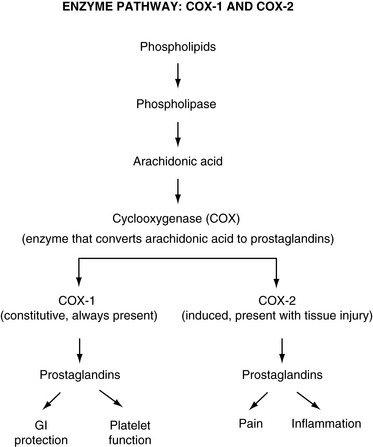
Figure I-3 Enzyme pathway: COX-1 and COX-2. From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 6, St. Louis, Mosby. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
Cyclooxygenase is actually a small family of enzymes, each one of which is known as an isoenzyme. The two best characterized COX isoenzymes are COX-1 and COX-2. These isoenzymes are widely distributed in tissues and are known to play important roles in the effects produced by the nonopioid analgesics. The existence of other COX isoenzymes is likely, including a COX-3 isoenzyme that appears to predominate in the CNS.
The nonopioid analgesics act peripherally and centrally to inhibit the COX isoenzymes; various drugs have varying selectivities (see Chapter 6 and Figure 6-1 on p. 198). Peripheral actions reduce nociception in part by diminishing the facilitory effect of inflammation-related compounds on transduction. Central actions may be involved in pain transmission and other effects such as reduction of fever.
The importance of the central effects of nonopioid analgesics is most clearly demonstrated by the efficacy of acetaminophen, which is a COX inhibitor that has minimal peripheral effect, is not antiinflammatory, and can both relieve pain and reduce fever by preventing the formation of prostaglandins in the CNS (Muth-Selbach, Tegeder, Brune, et al., 1999). The antipyretic effect of this drug and nonsteroidal antiinflammatory drugs (NSAIDs) may involve COX-3 inhibition (Botting, 2003). Research is ongoing to elucidate all of the underlying mechanisms of NSAIDs, but spinal COX-2 inhibition has been found to reduce not only prostaglandin production but also endocannabinoid breakdown (Telleria-Diaz, Schmidt, Kreusch, et al., 2010). Endocannabinoids have analgesic properties; however, they are subject to rapid breakdown, which limits their analgesic action. Recent research suggests that endocannabinoid analgesia may be enhanced by the action of COX-2 selective NSAIDs and that this may be a primary underlying mechanism of these NSAIDs (Telleria-Diaz, Schmidt, Kreusch, et al., 2010).
The ability to reduce nociception by partially blocking transduction is the mode of action of other types of analgesics as well. For example, sodium (NA+) channels are closed and inactive at rest but undergo structural changes in response to membrane depolarization. Transient channel opening leads to an influx of sodium ions and subsequent nerve conduction (Dib-Hajj, Black, Waxman, 2009). Nociceptor activation may be reduced by local anesthetics, which block sodium (Na+) channels and reduce the likelihood that the nerve will generate an action potential (see Chapter 23, Sodium Channel Blockers and Local Anesthetics). Similarly, compounds that change the flux of other ions, such as calcium (CA++) or potassium (K+), may have the potential to reduce transduction (see Chapter 23, Anticonvulsants). A variety of topical therapies, including topical NSAIDs and local anesthetics, have been developed with the goal of relieving pain through the lessening of transduction (see Chapter 7, Topical NSAIDs and Chapter 24, Topical Local Anesthetics).
Transmission
Transmission is the second process involved in nociception (see Figure I-2, B). Nociceptors are neurons that, compared with other sensory neurons, have small-diameter axons—either A-delta (δ) or C fibers (Argoff, Albrecht, Irving, et al., 2009). Effective transduction generates an action potential that is transmitted in these fibers toward the CNS. A-δ fibers are lightly myelinated, larger, and faster conducting than unmyelinated C fibers. The endings of A-δ fibers detect thermal and mechanical injury. These neurons typically transmit information that allows relatively quick localization of pain and an appropriately rapid protective response. The sensation accompanying A-δ fiber activation has been termed first pain (Dahl, Moiniche, 2004; Marchand, 2008). It is sharp and well-localized and leads to reflex withdrawal from the painful stimuli.
Unmyelinated C fibers are slow conductors and may respond to mechanical, thermal, or chemical stimuli. Activation after acute injury yields a poorly localized, typically aching or burning pain, which often is referred to as second pain (Dahl, Moiniche, 2004; Marchand, 2008). A-beta (β) fibers, the largest of the fibers, do not normally transmit pain but respond to touch, movement, and vibration.
The cell bodies of the nociceptive neurons that subserve the processes of transduction and transmission of information about noxious events are in dorsal root ganglia, which lie outside the spinal cord (see Figure I-2, A). Afferent information passes through the cell body to a central process, which synapses in the dorsal horn of the spinal cord. This synapse connects the nociceptor with the so-called second-order neuron in the dorsal horn. The second-order neuron, in turn, generates an action potential that ascends up the spinal cord and transmits information to the brain, where pain is perceived (see Figure I-2, B).
In the dorsal horn, incoming information is extensively modulated through complex neurophysiologic and neurochemical mechanisms. The primary A-δ and C afferents release a variety of transmitters, among the most important of which are excitatory amino acids, such as glutamate, neurokinins, and substance P. Glutamate binds to the N-methyl-d-aspartate (NMDA) receptor and promotes pain transmission (see inset of Figure I-2, B) (Carlton, 2009). NMDA receptors may contribute to the sensory disturbances experienced by patients with the neuropathic pain state known as complex regional pain syndrome (CRPS) (Finch, Knudsen, Drummond, 2009). Ketamine, an NMDA receptor antagonist, is an example of a drug that produces pain relief by preventing glutamate from binding to the NMDA receptor sites and thereby blocking the transmission of pain (see Chapter 23 for more on ketamine).
Dorsal horn modulation involves both local, segmental systems and descending systems. The neurochemistry is complex and not yet fully understood. Endogenous opioid compounds bind to opioid receptor sites and help to slow the transmission of pain. Opioid analgesics also bind to opioid receptor sites throughout the nervous system to produce analgesia and other effects, such as constipation, sedation, and respiratory depression (see Section IV).
Perception
The third broad process involved in nociception is termed perception. Perception, which may be viewed as the end result of the neural activity associated with transmission of information about noxious events, involves conscious awareness of pain (see Figure I-2, C). It requires the activation of higher brain structures, presumably including the cortex. It involves both awareness and related cognitions and the occurrence of emotions and drives associated with pain. These complex processes generate a network of cortical and subcortical gray matter when transmission of information about noxious events reaches the threshold for perception.
The physiology subserving the perception of pain is very poorly understood, but presumably can be targeted by therapies that activate higher cortical functions in the service of pain control or coping. Cognitive-behavioral therapy and specific approaches such as distraction and imagery have been developed based on evidence that brain processes can strongly influence pain perception.
Modulation
As noted, modulation of afferent input generated in response to noxious stimuli happens at every level, from the periphery to the cortex (see Figure I-2). Given the importance of this physiology, modulation usually is considered the last of the key processes of nociception (Marchand, 2008).
Peripheral and central systems and dozens of neurochemicals are involved in these modulatory processes. The endogenous opioids, for example, are found throughout the PNS and CNS and, like the exogenous opioids administered therapeutically, they inhibit neuronal activity through actions initiated by binding to opioid receptors. Other central inhibitory neurotransmitters important in the modulation of pain include serotonin and norepinephrine, which are released in the spinal cord and brainstem by the descending fibers of the modulatory system (see Figure I-2, D). Antidepressants relieve pain by blocking the body’s reuptake (resorption) of serotonin and norepinephrine, making them more available to fight pain (see Chapter 22 for more information about antidepressants).
Pathophysiology of Pain: Neuropathic Pain
Neuropathic pain, like nociceptive pain, is a descriptive term used to refer to pain that is believed to be sustained by a set of mechanisms that is driven by damage to, or dysfunction of, the PNS or CNS. In contrast to nociceptive pain, which reflects the clinical judgment that pain is sustained by ongoing activation of essentially normal neural systems that exist to subserve the perception of noxious events, neuropathic pain is sustained by the abnormal processing of stimuli from the PNS or CNS or both (Adler, Nico, VandeVord, et al., 2009; Argoff, Albrecht, Irving, et al., 2009; Beydoun, Backonja, 2003; Dworkin, Backonja, Rowbotham, et al., 2003). Injury to the PNS can irreversibly alter central signaling mechanisms, and disturbances of these central signaling mechanisms can alter peripheral signaling mechanisms (Argoff, Albrecht, Irving, et al., 2009). Whereas nociceptive pain involves tissue damage or inflammation (Pasero, 2004), neuropathic pain may occur in the absence of either. Acute nociceptive pain, such as that which follows trauma, serves to warn and protect individuals from further injury; neuropathic pain, even when acute, reflects a pathophysiology that is believed to serve no useful purpose (Beydoun, Backonja, 2003; Pasero, 2004). The peripheral and central mechanisms that initiate and maintain neuropathic pain are discussed subsequently and in Table I-1.
Table I-1
Mechanisms of Neuropathic Pain
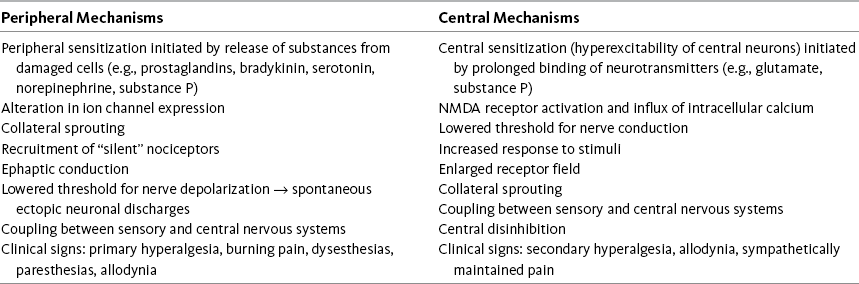
From Pasero C, McCaffery M. (2011). Pain assessment and pharmacologic management, p. 7, St. Louis, Mosby. Data from Adler, J. E., Nico, L., VandeVord, P., et al. (2009). Modulation of neuropathic pain by glial-driven factor. Pain Med, 10(7), 1229-1236; Argoff, C. E., Albrecht, P., Irving, G., et al. (2009). Multimodal analgesia for chronic pain: Rationale and future directions. Pain Med, 10(Suppl 2), S53-S66; Beydoun, A., & Backonja, M. M. (2003). Mechanistic stratification of antineuralgic agents. J Pain Symptom Manage, 25(Suppl 5), S18-S30; Bridges, D., Thompson, S. W. N., & Rice, A. S. C. (2001). Mechanisms of neuropathic pain. Br J Anaesth, 87(1), 12-26; Carlton, S. M. (2009). NMDA receptors revisted—Hope floats. Pain, 146(1-2), 1-2; Dickenson, A. H., Matthews, E. A., & Suzuki, R. (2002). Neurobiology of neuropathic pain: Mode of action of anticonvulsants. Eur J Pain, 6(Suppl A), 51-60; Mao, J., & Chen, L. L. (2000). Gabapentin in pain management. Anesth Analg, 91(3), 680-687; Pasero, C. (2004). Pathophysiology of neuropathic pain. Pain Manage Nurs, 5(4), 3-8. Pasero C. May be duplicated for use in clinical practice.
Peripheral Mechanisms
As described earlier, primary afferent neurons, called nociceptors, are distributed throughout the PNS. Nociceptors have free nerve endings that have the capacity to distinguish noxious and innocuous stimuli. They are activated when tissue damage or potential damage results from stimuli that are mechanical (e.g., surgical or nonsurgical trauma, tumor growth); thermal (e.g., burn, frostbite); or chemical (e.g., toxins, chemotherapy) (Marchand, 2008). Infection, viruses, metabolic disease, nutritional deficiencies, ischemia, and stroke are among the many other sources of tissue injury (Pasero, 2004). When noxious stimuli exceed the threshold required to activate the nociceptor (transduction), an electric signal (action potential) is set up in the nerve and is then transmitted to the CNS (see Figures I-2, A and I-2, B). In the periphery, transduction involves numerous substances (e.g., serotonin, bradykinin, histamine, substance P, prostaglandins) that may lead to activation of nociceptors. The inflammatory process that occurs with tissue damage produces these and other substances and also causes the transfer of ions (e.g., sodium, potassium, calcium) that support the creation of an action potential in the nerve.
Once an electric signal is established in the neuron, ion channels are responsible for conduction of that electric signal along the nerve axon (Beydoun, Backonja, 2003). Transmission of information about noxious events occurs along both large-diameter A-δ nerve fibers and along the slow-conducting C fibers. The largest and most rapidly conducting A-β peripheral nerve fibers do not carry information about noxious events (see Figure I-2, B).
In this normal process of transduction and transmission of information about noxious stimuli, several processes could occur that lead to the development of neuropathic pain. For example, hyperexcitable epidermal nerve endings in the periphery, which have been damaged by noxious stimuli, and nearby keratinocytes are thought to be factors in abnormal reorganization of the CNS, an underlying mechanism of some persistent neuropathic pain states (Argoff, Albrecht, Irving, et al., 2009; Fishman, 2009). Changes can occur in the number and location of ion channels. Sodium channels, in particular, abnormally accumulate in injured nociceptors (Beydoun, Backonja, 2003; Jensen, 2002). The nerve depolarization threshold is lowered, leading to ectopic discharges, and the response to stimuli is increased (Bridges, Thompson, Rice, 2001). These processes, which may lead to peripheral sensitization, may be important contributors to the maintenance of neuropathic pain (Beydoun, Backonja, 2003; Fishman, 2009). Drugs, such as anticonvulsants, antidepressants, and sodium channel blockers (including those applied locally as local anesthetics), may act in the periphery to relieve pain by modulating ion channels and suppressing ectopic discharges (see Section V).
Another peripheral mechanism that may sustain neuropathic pain is the creation of chemically mediated connections between nerve fibers. These connections, or ephapses, are the substrates of an abnormal excitatory process, which is known as ephaptic conduction (cross-excitation or cross-talk) (Bridges, Thompson, Rice, 2001). This process may cause the abnormal activation of nociceptive neurons, ultimately producing pain (Pasero, 2004). Ephaptic conduction between afferent sensory nerves that transmit information about pain and efferent sympathetic nerves that exist in the same nerve bundle has been speculated to be involved in the pathophysiology of so-called sympathetically maintained pain (Bridges, Thompson, Rice, 2001). Because sympathetically maintained pain, identified through a very positive response to sympathetic (local anesthetic) nerve blocks, is likely to be part of CRPS, the recommended early treatment of the syndrome usually includes a trial of sympathetic nerve blocks.
Hyperalgesia, which is the increased sensation of pain in response to a normally painful stimulus, is seen clinically in patients with neuropathic pain. The mechanisms underlying hyperalgesia continue to be the focus of research but are thought to be multifaceted and mediated in part by sensitization of C-fiber primary afferent neurons (Byers, Bonica, 2001).
Central Mechanisms
Central mechanisms also play a critical role in the development and maintenance of neuropathic pain. A key mechanism is called central sensitization, which is defined as abnormal hyperexcitability of central neurons, and that results from complex changes induced by the incoming afferent barrages of nociceptors (Bridges, Thompson, Rice, 2001). This can occur at any level of the CNS (Argoff, Albrecht, Irving, et al., 2009). The best-characterized mechanism linked to central sensitization involves the prolonged release and binding of excitatory neurotransmitters, such as substance P and glutamate, which activate the NMDA receptor (see inset in Figure I-2, B). Activation of this receptor causes an increase in intracellular calcium levels by moving calcium into the cell through the so-called N-type calcium channel (Jensen, 2002). The N-type calcium channel is thought to play a key role in the processing of painful stimuli; when blocked, abnormal pain sensation is inhibited (Dickenson, Matthews, Suzuki, 2002). An increase in intracellular calcium lowers the cells threshold for firing, the substrate for central sensitization.
Research also has demonstrated expression of sodium channels on dorsal horn neurons near spinal cord lesions, which were linked to central pain behaviors (Finnerup, Biering-Sorensen, Johannesen, et al., 2005; Hains, Klein, Saab, et al., 2003). An increase in the influx of sodium also could lead to a lowered threshold for activation, an increased response to stimuli, and enlargement of the receptive field served by the affected neuron (Beydoun, Backonja, 2003; see inset in Figure I-2, B). Research is ongoing, but all of these outcomes could contribute to sensitization.
Central sensitization often is considered to be closely allied, or even synonymous, with a phenomenon known as wind-up (Dickenson, Matthews, Suzuki, 2002). Wind-up refers to the progressive increase in the response of central neurons, and it may be induced by high-intensity activity in the peripheral nociceptors that synapse on these neurons (see Figure I-2) (Argoff, Albrecht, Irving, et al., 2009). The relationship between the relatively short-lived wind-up phenomenon observed in cellular modules and the persistent state of central sensitization that can occur in animals, presumably including humans, is not completely understood (Adler, Nico, VandeVord, et al., 2009; Bridges, Thompson, Rice, 2001).
Injured or intensely activated peripheral neurons may undergo synaptic reorganization or change anatomically, sprouting new processes. These processes also occur in the CNS and are thought to be sustained by an increased or inappropriate responsiveness of central neurons to relatively mild peripheral stimuli (Adler, Nico, VandeVord, et al., 2009). Loss of C-fiber afferents into the CNS, such as may follow an injury to a nerve root, leads to reorganization in the dorsal horn of the spinal cord. Large myelinated fibers sprout and invade other areas where nociceptive-specific neurons were located, and this change may correlate with the development of abnormal sensation in the area of the body served by the injured nerve (Jensen, 2002). Allodynia, or pain with a nonnoxious stimulus (e.g., touch), is one such type of abnormal sensation and is common in some patients with neuropathic pain (Pasero, 2004).
The ability of the PNS and CNS to change both structure and function as a result of noxious stimuli is called neuroplasticity (Byers, Bonica, 2001; Dickenson, Matthews, Suzuki, 2002; Jensen, 2002). Neuroplasticity is exceedingly complex but appears to be a key feature in many diseases of the nervous system, including persistent pain.
Another central mechanism, called central disinhibition, occurs when control mechanisms along inhibitory (modulatory) pathways are lost or suppressed, leading to abnormal excitability of central neurons (Pasero, 2004). Among the possible causes of disinhibition is dysfunction in gamma aminobutyric acid (GABA) pathways. GABA is the most abundant neurotransmitter in the CNS (Dickenson, Matthews, Suzuki, 2002) and composes a major inhibitory neurotransmitter system (Bridges, Thompson, Rice, 2001). When GABA receptor inhibition is suppressed, abnormal pain processing occurs (Bridges, Thompson, Rice, 2001). Conversely, increased GABA function (GABAergic) may help to relieve neuropathic pain.
Other supraspinal processes that modulate and inhibit the transmission of nociceptive impulses also may be involved in central disinhibition (Beydoun, Backonja, 2003). Abnormal functioning in the descending fibers containing the inhibitory neurotransmitters serotonin and norepinephrine may be one such process.
As mentioned, hyperalgesia, or increased pain sensation, is common in patients with neuropathic pain. Two types of hyperalgesia are described. Primary hyperalgesia is thought to be the result of peripheral changes and is seen clinically as increased pain and sensitivity at the site of the injury. Secondary hyperalgesia is the result of central neural events and is seen clinically as allodynia and increased pain and sensitivity in areas extending outside of the site of injury (Pasero, 2004).
Most of the drugs used to treat neuropathic pain produce effects through their actions on the CNS. Anticonvulsants, sodium channel blockers, and other drugs, including some antidepressants and ziconotide, produce analgesia by blocking central neuron ion channels. Local anesthetics are sodium channel blockers that can work locally when applied directly to nerves; when taken systemically, their effects appear to be related primarily to CNS actions. Antidepressants inhibit synaptic reuptake of the biogenic amines serotonin and norepinephrine, thereby increasing activity in monoaminergic pain modulating pathways that originate in the brainstem and descend to every level of the spinal cord. Ketamine produces pain relief by antagonizing activity at the NMDA receptor site. Midazolam enhances GABAergic function to produce analgesia for conditions like muscle spasm (see Section V).
Multimodal Analgesia
Multimodal therapy is a relatively new concept. It was first proposed in the early 1990s and was applied primarily to the treatment of acute pain and the prevention of persistent post-surgical pain (Gartner, Kroman, Callesen, et al., 2008; Kehlet, Jensen, Woolf, 2006; Pasero, 2003; Polomano, Rathmell, Krenzischek, et al., 2008). Multimodal pain treatment involves the use of two or more classes of analgesics to target different pain mechanisms in the PNS or CNS. It relies on the thoughtful and rational combination of analgesics to maximize pain relief and prevent analgesic gaps that may lead to worsening pain or unnecessary bouts of uncontrolled pain (Argoff, Albrecht, Irving, et al., 2009; Carr, Reines, Schaffer, et al., 2005; Tang, Evans, Chaput, et al., 2009).
A multimodal approach may allow lower doses of each of the drugs in the treatment plan, and lower doses have the potential to produce fewer adverse effects (Ashburn, Caplan, Carr, et al., 2004; Brodner, Van Aken, Hertle, et al., 2001; Tang, Evans, Chaput, et al., 2009). Further, multimodal analgesia can result in comparable or greater pain relief than can be achieved with any single analgesic (Busch, Shore, Bhandari, et al., 2006; Butterfield, Schwarz, Ries, et al., 2001; Cassinelli, Dean, Garcia, et al., 2008; Huang, Wang, Wang, et al., 2008; White, 2005). In the setting of postoperative pain, the use of combination therapy to prevent both inflammatory and neuropathic pain is likely to yield the best immediate results and also offers the promise of reducing the incidence of prolonged or persistent postsurgical pain (Kehlet, Jensen, Woolf, 2006) (see Sections III, IV, and V).
The multimodal strategy also has a role in the management of persistent pain. This is true in all of the various practice settings, including the emergency department (Baker, 2005), outpatient treatment sites (Gatchel, Okifuji, 2006), and settings providing specialist palliative care (Soares, Chan, 2007). The complex nature of the many persistent pain conditions indicates the need for appropriate combinations of analgesics to target differing underlying mechanisms (Argoff, Albrecht, Irving, et al., 2009). Broad acceptance of a role in pain management for the adjuvant analgesic drugs is premised on the growing importance of multimodal treatment for persistent pain (see Section V).
Multimodal therapy also is a useful strategy for addressing the common problem of symptom distress related to symptoms other than pain. Patients with acute or chronic pain commonly experience additional symptoms, which may influence the decision to try one analgesic rather than another. More broadly, the experience of multiple symptoms also may guide the use of multiple drugs, some of which are targeted to relieve distressing conditions such as insomnia that accompany pain and may be factors in worsening it (Gan, Meyer, Apfel, et al., 2007; Gartner, Kroman, Callesen, et al., 2008).
Multimodal Therapy versus Polypharmacy
The term polypharmacy carries a negative connotation, in contrast to multimodal therapy or combination therapy. Whereas multimodal therapy is based on rational combinations of analgesics with differing underlying mechanisms to achieve the greatest benefit in pain control, polypharmacy suggests the use of drug combinations that are irrational and less effective or less safe than would be a regimen that had fewer or different agents.
Persistent Postsurgical and Posttrauma Pain
As many as 50% of patients undergoing surgical procedures, such as inguinal hernia repair; breast, cardiac, or thoracic surgery; leg amputation; and coronary artery bypass, experience persistent pain; in 2% to 10% of these individuals, the intensity of persistent postsurgical pain is severe (Kehlet, Jensen, Woolf, 2006). A study of 90 women who underwent abdominal hysterectomy pain for noncancer conditions found that 16.7% experienced persistent postoperative pain (Brandsborg, Dueholm, Nikolajsen, et al., 2009). The incidence of persistent postmastectomy pain is reported to be as high as 65% (Smith, Bourne, Squair, et al., 1999).
Pain following traumatic injury is common as well. A multicenter study conducted in 69 hospitals in 14 states in the United States found that 62.7% of patients (N = 3047) reported injury-related pain at 12 months after a traumatic injury (Rivara, MacKenzie, Jurkovich, et al., 2008). A quarter of patients in an earlier study (N = 397) described pain that interfered with daily activity 7 years following limb-threatening lower extremity trauma; 40% reported high pain intensities (Castillo, MacKenzie, Wegener, et al., 2006).
Further research is needed, but multiple factors are thought to contribute to the likelihood of postsurgical pain, including surgical nerve injury, preexisting pain, and genetic susceptibility. For example, severe pre-amputation pain has long been associated with a higher incidence of phantom limb pain (Bach, Noreng, Tjellden, 1988; Katz, 1997; Nikolajsen, Ilkjaer, Kroner, et al., 1997). A 2-year study of 57 patients who underwent lower extremity amputation revealed that high levels of both pre-amputation pain and acute pain after amputation predicted persistent post-amputation pain (Hanley, Jensen, Smith, et al., 2006). Greater analgesic requirements during the immediate postoperative period following coronary artery bypass surgery predicted persistent pain (at multiple anatomic sites) in a study of 736 patients (Taillefer, Carrier, Belisle, et al., 2006). Older patients tend to have a lower risk of developing persistent postsurgical pain than younger patients (Poobalan, Bruce, Smith, et al., 2003; Smith, Bourne, Squair, et al., 1999). For example, one study showed that patients under the age of 40 years were at increased risk for persistent post-inguinal hernia repair pain (Poobalan, Bruce, King, et al., 2001). Another found that the prevalence of persistent chest and leg pain following cardiac surgery was 55% in patients who were less than 60 years old and 34% in those over 70 years old (Bruce, Drury, Poobalan, et al., 2003). The reader is referred to an excellent review by Perkins and Kehlet (2000) that includes predictive factors, etiology, and progression of postsurgical pain conditions.
The presence of pain at 3 months after injury was a predictive factor for both the presence and the high severity of persistent pain following major trauma (Rivara, MacKenzie, Jurkovich, et al., 2008). Although the presence of persistent pain varied with age, in this study it was more common in women and in those who had untreated depression before the traumatic injury. Another study found that multiple factors influenced the likelihood of persistent pain 7 years after major lower extremity trauma (Castillo, MacKenzie, Wegener, et al., 2006); these factors included having less than a high-school education; having less than a college education; low self-efficacy for return to usual major activities; a high level of alcohol consumption in the month prior to injury; and high pain intensity, high levels of sleep and rest dysfunction, and elevated levels of depression and anxiety at 3 months after hospital discharge. Interestingly, those who were treated with opioid analgesics during the first 3 months after discharge in this study had lower levels of persistent pain at 7 years, underscoring the importance of early initiation of aggressive pain management approaches.
The clinical presentation of persistent postsurgical or posttrauma pain is primarily a patient’s report of the features characteristic of neuropathic pain, such as continuous burning pain and pain beyond the expected time of pain resolution (see Section II for assessment of neuropathic pain). Strategies for preventing these persistent pain states are being investigated, but sustained multimodal pharmacologic approaches that target the underlying mechanisms of neuropathic pain (described earlier) are recommended (Kehlet, Jensen, Woolf, 2006). See Sections III, IV, and V for discussion of the role of the various analgesics and techniques in the prevention of persistent postsurgical and posttrauma pain. See Table I-2 for the many other harmful effects of unrelieved Pain.
Table I-2
Harmful Effects of Unrelieved Pain
| Domains Affected | Specific Responses to Pain |
| Endocrine | ↑ Adrenocorticotrophic hormone (ACTH), ↑ cortisol, ↑ antidiuretic hormone (ADH), ↑ epinephrine, ↑ norepinephrine, ↑ growth hormone (GH), ↑ catecholamines, ↑ renin, ↑ angiotensin II, ↑ aldosterone, ↑ glucagon, ↑ interleukin-1; ↓ insulin, ↓ testosterone |
| Metabolic | Gluconeogenesis, hepatic glycogenolysis, hyperglycemia, glucose intolerance, insulin resistance, muscle protein catabolism, ↑ lipolysis |
| Cardiovascular | ↑ Heart rate, ↑ cardiac workload, ↑ peripheral vascular resistance, ↑ systemic vascular resistance, hypertension, ↑ coronary vascular resistance, ↑ myocardial oxygen consumption, hypercoagulation, deep vein thrombosis |
| Respiratory | ↓ Flows and volumes, atelectasis, shunting, hypoxemia, ↓ cough, sputum retention, infection |
| Genitourinary | ↓ Urinary output, urinary retention, fluid overload, hypokalemia |
| Gastrointestinal | ↓ Gastric and bowel motility |
| Musculoskeletal | Muscle spasm, impaired muscle function, fatigue, immobility |
| Cognitive | Reduction in cognitive function, mental confusion |
| Immune | Depression of immune response |
| Developmental | ↑ Behavioral and physiologic responses to pain, altered temperaments, higher somatization, infant distress behavior; possible altered development of the pain system, ↑ vulnerability to stress disorders, addictive behavior, and anxiety states |
| Future pain | Debilitating chronic pain syndromes: postmastectomy pain, postthoracotomy pain, phantom pain, postherpetic neuralgia |
| Quality of life | Sleeplessness, anxiety, fear, hopelessness, ↑ thoughts of suicide |
From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 11, St. Louis, Mosby. Data from Cousins, M. (1994). Acute postoperative pain. In P. D. & Wall, R. Melzack (Eds.), Textbook of pain, ed 3, New York, Churchill Livingstone; Kehlet, H. (1998). Modification of responses to surgery by neural blockade. In M. J. Cousins, & P. O. Bridenbaugh (Eds.), Neural blockade, Philadelphia, Lippincott-Raven; Mcintyre, P. E., & Ready, L. B. (1996). Acute pain management: A practical guide, Philadelphia, Saunders. Pasero C, McCaffery M. May be duplicated for use in clinical practice.
References
Adler, J.E., Nico, L., VandeVord, P., et al. Modulation of neuropathic pain by a glial-driven factor. Pain Med. 2009;10(7):1229–1236.
Argoff, C.E., Albrecht, P., Irving, G., et al. Multimodal analgesia for chronic pain: Rationale and future directions. Pain Med. 2009;10(Suppl 2):S53–S66.
Ashburn, M.A., Caplan, R.A., Carr, D.B., et al. Practice guidelines for acute pain management in the perioperative setting: An updated report by the American Society of Anesthesiologists task force on acute pain management. Anesthesiology. 2004;100(6):1573–1581.
Bach, S., Noreng, M.J., Tjellden, N.U. Phantom pain in amputees during the first 12 months following limb amputation, after preoperative lumbar epidural blockade. Pain. 1988;33(3):297–301.
Baker, K. Chronic pain syndromes in the emergency department: Identifying guidelines for management. Emergency Medicine Australasia: EMA. 2005;17(1):57–64.
Beydoun, A., Backonja, M.M. Mechanistic stratification of antineuralgic agents. Journal of Pain and Symptom Management. 2003;25(Suppl 5):S18–S30.
Botting, R. COX-2 and COX-3 inhibitors. Thrombosis Research. 2003;110(5–6):269–272.
Brandsborg, B., Dueholm, M., Nikolajsen, L., et al. A prospective study of risk factors for pain persisting 4 months after hysterectomy. The Clinical Journal of Pain. 2009;25(4):263–268.
Bridges, D., Thompson, S.W.N., Rice, A.S.C. Mechanisms of neuropathic pain. British Journal of Anaesthesia. 2001;87(1):12–26.
Brodner, G., Van Aken, H., Hertle, L., et al. Multimodal perioperative management: Combining thoracic epidural analgesia, forced mobilization, and oral nutrition reduces hormonal and metabolic stress and improves convalescence after major urologic surgery. Anesthesia and Analgesia. 2001;92(6):1594–1600.
Bruce, J., Drury, N., Poobalan, A.S., et al. The prevalence of chronic chest and leg pain following cardiac surgery: A historical cohort study. Pain. 2003;104(1–2):265–273.
Busch, C.A., Shore, B.J., Bhandari, R., et al. Efficacy of periarticular multimodal drug injection in total knee arthroplasty. The Journal of Bone and Joint Surgery. 2006;88a(5):959–963.
Butterfield, N.N., Schwarz, S.K., Ries, C.R., et al. Combined pre- and post-surgical bupivacaine wound infiltrations decrease opioid requirements after knee ligament reconstruction. Canadian Journal of Anaesthesia = Journal Canadien D’anesthesie. 2001;48(3):245–250.
Byers, M.R., Bonica, J.J. Peripheral pain mechanisms and nociceptors plasticity. In: Loeser J.D., Butler S.H., Chapman R.C., Turk D.C., eds. Bonica’s management of pain. (3rd ed. Philadelphia: Lippincott, Williams & Wilkins; 2001:26–72.
Carlton, S. M. Peripheral NMDA receptors revisted—Hope floats. Pain. 2009;146(1–2):1–2.
Carr, D.B., Reines, H.D., Schaffer, J., et al. The impact of technology on the analgesic gap and quality of acute pain management. Regional Anesthesia and Pain Medicine. 2005;30(3):286–291.
Cassinelli, E.H., Dean, C.L., Garcia, R.M., et al. Ketorolac use for postoperative pain management following lumbar decompression surgery: A prospective, randomized, double-blinded, placebo-controlled trial. Spine. 2008;33(12):1313–1317.
Castillo, R.C., MacKenzie, E.J., Wegener, S.T., et al. Prevalence of chronic pain seven years following limb threatening lower extremity trauma. Pain. 2006;124(3):321–329.
Dahl, J.B., Moiniche, S. Pre-emptive analgesia. British Medical Bulletin. 2004;71(1):13–27.
Dib-Hajj, S.D., Black, J.A., Waxman, S. G. Voltage-gated sodium channels: Targets for pain. Pain Med. 2009;10(7):1260–1269.
Dickenson, A.H., Matthews, E.A., Suzuki, R. Neurobiology of neuropathic pain: Mode of action of anticonvulsants. European Journal of Pain. 2002;6(Suppl A):51–60.
Dworkin, R.H., Backonja, M., Rowbotham, M.C., et al. Advances in neuropathic pain: Diagnosis, mechanisms, and treatment recommendations. Archives of Neurology. 2003;60(11):1524–1534.
Finch, P.M., Knudsen, L., Drummond, P. D. Reduction of allodynia in patients with complex regional pain syndrome: A double-blind placebo-controlled trial of topical ketamine. Pain. 2009;146(1–2):18–24.
Finnerup, N.B., Biering-Sorensen, F., Johannesen, I.L., et al. Intravenous lidocaine relieves spinal cord injury pain. Anesthesiology. 2005;102(5):1023–1030.
Fishman, S. Opioid-based multimodal care of patients with chronic pain: Improving effectiveness and mitigating risks. Pain Med. 2009;10(Suppl 2):S49–S52.
Gan, T.J., Meyer, T.A., Apfel, C.C., et al. Society for ambulatory anesthesia guidelines for the management of postoperative nausea and vomiting. Anesthesia and Analgesia. 2007;105(6):1615–1628.
Gartner, R., Kroman, N., Callesen, T., et al. Multimodal treatment of pain and nausea in breast cancer surgery. Ugeskr Laeger. 2008;170(23):2035–2038.
Gatchel, R.J., Okifuji, A. Evidence-based scientific data documenting the treatment and cost-effectiveness of comprehensive pain programs for chronic nonmalignant pain. The Journal of Pain. 2006;7(11):779–793.
Hains, B.C., Klein, J.P., Saab, C.Y., et al. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. The Journal of Neuroscience. 2003;23(26):8881–8892.
Huang, Y.M., Wang, C.M., Wang, C.T., et al. Perioperative Celebrex administration for pain management after total knee arthroplasty: A randomized, controlled study. BMC Musculoskeletal Disorders. 2008;9:77.
Jensen, T.S. Anticonvulsants in neuropathic pain: Rationale and clinical evidence. European Journal of Pain. 2002;6(Suppl A):61–68.
Katz, J. Pain begets pain. Predictors of long-term phantom limb pain and post-thoracotomy pain. Pain Forum. 1997;6(2):140–144.
Kehlet, H., Jensen, T.S., Woolf, C.J. Persistent postsurgical pain: Risk factors and prevention. Lancet. 2006;367(9522):1618–1625.
Marchand, S. The physiology of pain mechanisms: From the periphery to the brain. Rheumatic Diseases Clinics of North America. 2008;34(2):285–309.
Muth-Selbach, U.S., Tegeder, I., Brune, K., et al. Acetaminophen inhibits spinal prostaglandin E2 release after peripheral noxious stimulation. Anesthesiology. 1999;91(1):231–239.
Nikolajsen, L., Ilkjaer, S., Kroner, K., et al. The influence of preamputation pain on postamputation stump and phantom pain. Pain. 1997;72(3):393–405.
Pasero, C. Multimodal analgesia in the PACU. Journal of Perianesthesia Nursing. 2003;18(4):265–268.
Pasero, C. Pathophysiology of neuropathic pain. Pain Management Nursing. 2004;5(4):3–8.
Perkins, F.M., Kehlet, H. Chronic pain as an outcome of surgery. Anesthesiology. 2000;93(4):1123–1133.
Polomano, R.C., Rathmell, J.P., Krenzischek, D.A., et al. Emerging trends and new approaches to acute pain management. Journal of Perianesthesia Nursing. 2008;23(Suppl 1):S43–S53.
Poobalan, A.S., Bruce, J., Smith, W.C., et al. A review of chronic pain after inguinal herniorrhaphy. The Clinical Journal of Pain. 2003;19(1):48–54.
Poobalan, A.S., Bruce, J., King, P.M., et al. Chronic pain and quality of life following open inguinal hernia repair. The British Journal of Surgery. 2001;88(8):1122–1126.
Rivara, F.P., MacKenzie, E.J., Jurkovich, G.J., et al. Prevalence of pain in patients 1 year after major trauma. Archives of Surgery. 2008;143(3):282–287.
Smith, W.C.S., Bourne, D., Squair, J., et al. A retrospective cohort study of post mastectomy pain syndrome. Pain. 1999;83(1):91–95.
Soares, L.G., Chan, V.W. The rationale for a multimodal approach in the management of breakthrough cancer pain: A review. The American Journal of Hospice & Palliative Care. 2007;24(5):430–439.
Taillefer, M.C., Carrier, M., Belisle, S., et al. Prevalence, characteristics, and predictors of chronic nonanginal postoperative pain after a cardiac operation: A cross-sectional study. The Journal of Thoracic and Cardiovascular Surgery. 2006;1131(6):1274–1280.
Tang, R., Evans, H., Chaput, A., et al. Multimodal analgesia for hip arthroplasty. The Orthopedic Clinics of North America. 2009;40(3):377–387.
Telleria-Diaz, A., Schmidt, M., Kreusch, S., et al. Spinal antinociceptive effects of cyclooxygenase inhibition during inflammation: Involvement of prostaglandins and endocannabinoids. Pain. 2010;148(1):26–35.
White, P.F. The changing role of non-opioid analgesic techniques in the management of postoperative pain. Anesthesia and Analgesia. 2005;101(Suppl 5):S5–S22.